

 This work is licensed under a
This work is licensed under a Abstract
Summary
Diabetic ketoacidosis (DKA) is commonly encountered in clinical practice. The current case is a unique and rare presentation of DKA as the initial manifestation of Cushing’s disease secondary to ACTH-secreting pituitary adenoma. Appropriate management as elaborated in the article led to total remission of diabetes as well as the Cushing’s disease.
Learning points:
DKA is a serious and potentially life-threatening metabolic complication of diabetes mellitus.
Some well-known precipitants of DKA include new-onset T1DM, insulin withdrawal and acute illness.
In a patient presenting with DKA, the presence of a mixed acid–base disorder warrants further evaluation for precipitants of DKA.
We present a rare case of DKA as an initial manifestation of Cushing’s disease secondary to ACTH-producing pituitary adenoma.
Background
Diabetic ketoacidosis (DKA) is an acute metabolic disorder characterized by markedly increased circulating ketone bodies e.g. beta-hydroxybutyrate, aceto-acetate and acetone in the presence of hyperglycemia. DKA is a serious and potentially life-threatening metabolic complication of diabetes mellitus. Some well-known precipitants of DKA include new-onset T1DM, insulin withdrawal and acute illness. In this report, we present a subject who presented with DKA as an initial manifestation of Cushing’s disease secondary to ACTH-producing pituitary adenoma.
Case presentation
A 48-year-old Caucasian woman was admitted with fever, cough, left-sided chest pain, shortness of breath and hemoptysis. She reported amenorrhea of one-year duration. She was a chronic smoker for over 20 years. Patient was unable to provide a detailed history at the time of admission due to acuteness of her illness and respiratory distress. On physical examination, patient was alert, oriented and in moderate respiratory distress with Kussmaul breathing, temperature: 102 F; respiratory rate: 20/min; pulse: 110/min and blood pressure: 148/98 mmHg; body weight: 158 lbs as well as round flushed face with acne, hirsutism and dark purple striae of the abdominal wall (Fig. 1). Lung examination revealed bronchial breath sounds with crackles in the lower left field. Heart evaluation showed normal heart sounds with tachycardia without a murmur, and neurological assessment was unremarkable.
Figure 1.
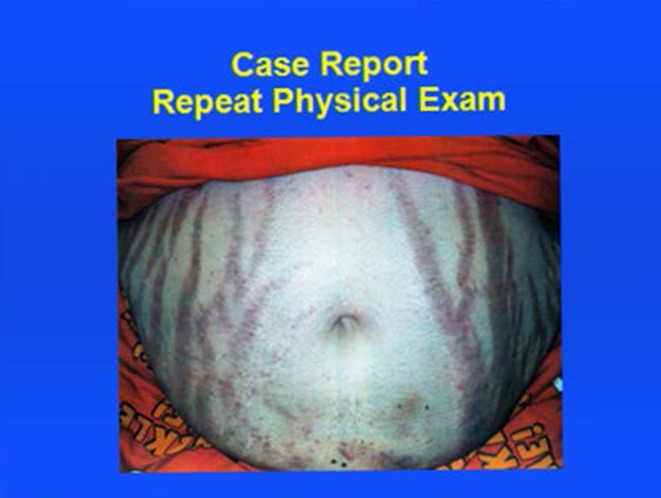
Purple striae.
Investigation
Complete blood count was significant for WBC 21.600/mL with segmented neutrophils 69%. Serum chemistries showed sodium: 134 mM/L (normal: 135–146); potassium: 3.5 mM/L (normal: 3.5–5.3); chloride: 78 mM/L (normal: 98–110); HCO3−: <10 mM/L (normal: 20–31); anion gap: 50 mM/L (normal: 8–16); serum urea nitrogen: 36 mg/dL (normal: 7–20); serum creatinine: 2.7 mg/dL (normal: 0.6–1.2); random serum glucose: 255 mg/dL (normal: 79–139, diabetes: >200); serum osmolarity: 308 mM/kg (normal: 275–295); serum beta-hydroxybutyrate: 27 mg/dL (normal: <5); lactic acid: 1.2 mM/L (normal: <2.5) and HbA1c: 8% (normal: 4–5.6%). Arterial blood gases obtained at admission simultaneously with aforementioned laboratory tests showed pH: 7.12; PO2, 80 mmHg and PCO2: 18 mmHg. Electrocardiogram showed sinus tachycardia with no other abnormality. Chest X-ray revealed left lower lobe pneumonia. The presence of hyperglycemia, elevated serum beta-hydroxybutyrate and anion gap metabolic acidosis established the diagnosis of DKA.
Treatment
The subject was promptly administered intravenous fluids and insulin infusion as per the treatment protocol for DKA previously established by the institution. Intravenous antibiotic administration was also simultaneously initiated along with nasal oxygen. Over the next 48 h, symptomatology improved after resolution of ketoacidosis as well as hypoxemia. At this time, oral feeding was initiated with subcutaneous insulin administration in basal bolus pattern. Moreover, further evaluation of laboratory data specifically arterial blood gases and serum electrolytes at admission indicated the presence of both metabolic alkalosis and respiratory alkalosis concurrently with DKA as indicated by ∆HCO3− (normal serum HCO3− 25 mM/L minus patient’s serum HCO3−) and ∆anion gap (patient’s anion gap minus normal anion gap, 12 mM/L). Respiratory alkalosis was attributed to hypoxemia secondary to pneumonia. However, the causative disorder for the presence of metabolic alkalosis was not promptly determined, and therefore, was deemed to require additional evaluation.
On further inquiry after improvement in symptoms at 48 h, the patient reported polyuria, polydipsia, nocturia, fatigue, muscle weakness, easy bruising, back ache and epigastric discomfort including burning for approximately 6–8 months prior to admission. She denied family history of diabetes or use of any drugs or supplements. Detailed physical examination at this time revealed central obesity, dorsocervical fat pad, proximal myopathy and persistent hypertension indicating the presence of Cushing’s syndrome. Arterial blood gases and serum electrolytes at this time confirmed the presence of metabolic alkalosis and enhanced the necessity of laboratory testing for the assessment for diagnosis of Cushing’s syndrome. Therefore, serum cortisol concentration was determined using the blood sample withdrawn at the time of admission. Serum creatinine kinase (CK) was also determined for the assessment of myopathy. Serum cortisol was markedly elevated (65 μg/dL) and was considered greater than a usual concentration noted during most stressful states. CK was elevated at 256 units with MM fraction, 95% (normal: 45–100) and thus was consistent with the presence of myopathy. Patient was discharged on the fifth day with instructions to continue oral antibiotic therapy for another 2 weeks and subcutaneous basal/bolus insulin until the scheduled appointment at 3 weeks in endocrinology clinic.
Additional laboratory testing conducted a week prior to the appointment after an overnight fast showed hypokalemic hypochloremic metabolic alkalosis, ACTH: 75 pg/mL (normal: 15–55); 1130 pm salivary cortisol X 2, 0.19 and 0.2 ng/dL (normal: 0.0); and 24-h urine free cortisol: 130 μg (normal: 15–90 μg) confirmed the presence of ACTH-dependent Cushing’s syndrome. At this time, laboratory tests to assess the function of the rest of the anterior pituitary gland were normal as expressed by normal serum levels of prolactin, IGF-1, estradiol, FSH, LH and free T4, the exception being low serum TSH level. Diabetes insipidus was excluded by urine specific gravity of 1.020. The diagnosis of non-insulin-dependent diabetes was also confirmed by normal serum c-peptide concentration and the absence of GAD antibody. Therefore, subcutaneous insulin therapy was discontinued and oral metformin, 500 mg daily after dinner was initiated with a request to increase the daily dose to 1000 mg after a week if none of the gastrointestinal symptoms, e.g. abdominal pain, nausea, vomiting or diarrhea occurred. Patient was also advised to perform premeal self-blood glucose monitoring and inject subcutaneous insulin lispro in a correction dose of 5 units for each 50 mg/dL over 200 mg/dL premeal blood glucose level. MRI of pituitary gland was ordered, and patient was scheduled for follow-up in 2 weeks.
At this visit, the diagnosis of Cushing’s disease was suspected due to the presence of a 6 mm pituitary microadenoma. Patient was referred for an inferior petrosal sinus sampling at a tertiary care medical center, which showed a markedly elevated serum ACTH concentration (224 pg/mL) on the left side in comparison to right side (15 pg/mL) and the peripheral arm vein (146 pg/mL) indicating the localization of microadenoma to the left half of the pituitary gland confirming the diagnosis of Cushing’s disease. Patient tolerated metformin well without side effects and denied requiring any correction dose insulin prior to follow-up. Therefore, subcutaneous insulin therapy was discontinued.
Patient was hospitalized and underwent pituitary microadenectomy. During hospitalization, dexamethasone 1.0 mg was administered intravenously at an interval of 6 h for the first 48 h and then changed to oral administration 1 mg twice daily. Simultaneously, metformin was discontinued, and insulin infusion was administered until patient resumed oral feeding within 48 h. Metformin was then restarted in the same prehospitalization daily dose, 1000 mg daily with premeal lispro insulin administration in the correction dose as described previously. The patient was discharged on day 7 with instructions to continue oral dexamethasone 0.5 mg twice daily as well as metformin and insulin lispro as recommended during hospitalization with a follow-up appointment in 2 weeks.
Outcome and follow-up
At this visit, further laboratory data were obtained. Serum electrolytes were normal. ACTH stimulation test showed baseline cortisol of 2 μg/dL and 21 μg/dL at 30 min. Serum prolactin, free T4, TSH, FSH, LH and IGF-1 concentrations were normal along with urine-specific gravity of 1.024 indicating normal pituitary function. Therefore, dexamethasone and metformin were discontinued. Histopathology of the resected pituitary adenoma revealed hypertrophic basophilic cells with positive immunostaining for ACTH and thus confirmed the diagnosis of Cushing’s disease.
At 12-week follow-up appointment, serum concentrations of glucose, electrolytes, ACTH and cortisol were normal and HbA1c improved to 5.2% indicating normalization of glycemia and hypothalamic–pituitary adrenal axis. During the follow-up period of 30 months, clinical manifestations of Cushing’s disease resolved, body weight declined to 127 lbs and the laboratory tests including fasting plasma glucose, HbA1c, serum electrolytes, AM serum cortisol and ACTH remained normalized. Thus, the subject remained totally free from diabetes mellitus without any therapeutic lifestyle or drug intervention during the follow-up period of 30 months.
Discussion
DKA is often a presenting manifestation of type 1 diabetes mellitus (T1DM) at the time of diagnosis (1). Among patients with prior presence of diabetes, withdrawal or omission of insulin due to psychiatric, social or economic reasons is a common trigger (1, 2). DKA also occurs in subjects with both type 1 and 2 diabetes due to relative insulin deficiency at the onset of infections (3), myocardial infarction (1, 2), congestive heart failure (1, 2), lack of appropriate adjustment of insulin regimen in pregnancy (4), as well as after steroid therapy (5).
The presentation of a mixed acid–base disorder in our subject is consistent with several patients manifesting DKA concurrently with other acid–base disorders established by examination of ∆AG and ∆HCO3− (Table 1) (1, 2, 3, 4, 5). In the presence of a pure or lone DKA, ∆AG is equal to ∆HCO3−. ∆AG < ∆HCO3− indicates a simultaneous occurrence of hyperchloremic acidosis with anion gap metabolic acidosis due to a greater fall in serum HCO3− in relation to the increase in the anion gap. This mixed acidosis is often present with DKA and may be attributed to the prior presence of diabetic nephropathy and/or acute renal injury due to decreased renal perfusion resulting from prolonged period of dehydration. The additional fall in HCO3− is due to further buffering of an acid that does not contribute to the anion gap. In a few instances, multiple types of anion gap acidosis are present, e.g. lactic acidosis concurrent with DKA due to inadequate tissue perfusion secondary to severe dehydration as well as a concurrent acute disorder such as septic shock or acute myocardial infarction. On the other hand, the presence of metabolic alkalosis is established by ∆AG > ∆HCO3−, indicating a lesser fall in HCO3− corresponding to the rise in anion gap. Metabolic alkalosis is induced by dehydration often present in DKA secondary to polyuria, nausea and vomiting. Alternatively, other disorders may contribute to metabolic alkalosis, i.e., a compensation for acute or chronic respiratory acidosis induced by a primary lung disorder or rarely secondary to primary hyperaldosteronism as well as Cushing’s disease or syndrome.
Table 1.
High anion gap metabolic acidosis and mixed acid–base disorders.
| ΔAG = ΔHCO3− | Pure anion gap acidosis |
| ΔAG < ΔHCO3− | Anion gap acidosis + normal gap metabolic acidosis (renal tubular acidosis) |
| ΔAG > ΔHCO3− | Anion gap acidosis + metabolic alkalosis (primary aldosteronism, hypercortisolism, contraction alkalosis and diuretic use) |
| ΔAG >> ΔHCO3− | Anion gap acidosis + metabolic alkalosis + primary respiratory alkalosis (hypoxia) |
Δ, change; AG, anion gap; HCO3−, bicarbonate.
The diagnosis of ACTH-dependent Cushing’s syndrome was entertained in our patient because of the presence of typical clinical manifestations as well as hypertension despite hypovolemia, concurrent metabolic alkalosis with DKA as expressed by a markedly higher ∆AG (38 meq/L) as compared to ∆HCO3− (25 meq/dL), relatively low potassium, eosinopenia and markedly elevated random serum cortisol. The diagnosis of Cushing’s disease was later confirmed by elevated levels of AM serum cortisol and ACTH, 24-h urine free cortisol and midnight salivary cortisol as well as documentation of pituitary microadenoma on MRI as well as its lateralization to the left half of the pituitary by inferior petrosal sinus sampling.
Diabetic ketoacidosis as an initial presentation of Cushing’s syndrome or disease documented in our subject is rarely reported in the literature (6, 7, 8, 9). In all these reports, the presence of Cushing’s disease or syndrome was suspected because of the presentation of concurrent metabolic alkalosis based on electrolyte abnormalities similar to that noted in our subject (6, 7, 8, 9). In three of these subjects, Cushing’s disease secondary to pituitary disorder was established (6, 7, 9), whereas the remaining report described a unique subject with Cushing’s syndrome secondary to ectopic ACTH-secreting carcinoid tumor of the duodenum (8). In another recent case report, DKA as an initial manifestation of Cushing’s disease in a child is reported (9). However, in none of these reports, did authors describe a remission of diabetes after resolution of hypercortisolemia. In contrast, diabetes remitted in our patient soon after the removal of pituitary adenoma and the remission is sustained for 30 months so far. Thus, the presentation in our patient is distinctly more unique in comparison to other reports in the literature.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Patient consent
Written consent has been obtained for the patient for publication of the submitted article and accompanying image.
Author contribution statement
Dr Runa Acharya: First author involved in writing the case report. Dr Udaya Kabadi: Corresponding author and mentor to Dr Runa Acharya. Dr Kabadi: Obtained consent for the case report.
References
- 1.Yong KW, Moore PM, Lunt H. 2014. Triggers for the development of diabetic ketoacidosis. New Zealand Medical Journal 127 86–94. [PubMed] [Google Scholar]
- 2.Randall L, Begovic J, Hudson M, Smiley D, Peng L, Pitre N, Umpierrez D, Umpierrez G. 2011. Recurrent diabetes ketoacidosis in inner-city minority patients behavioral, social and psychological factors. Diabetes Care 34 1891–1896. ( 10.2337/dc11-0701) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azoulay E, Chevret S, Didier J, Neuville S, Barboteu M, Bornstain C, Darmon M, Le Gall J-R, Vexiau P, Schlemmer B. 2001. Infection as a trigger of diabetes ketoacidosis in intensive care unit patients. Clinical Infectious Disease 32 30–35. ( 10.1086/317554) [DOI] [PubMed] [Google Scholar]
- 4.Inagaki T, Nishii Y, Suzuki N, Suzuki S, Koizumi Y, Aizawa T, Hashizume K. 2002. Fulminant diabetes mellitus associated with pregnancy: case reports and literature review. Endocrine Journal 49 319–322. ( 10.1507/endocrj.49.319) [DOI] [PubMed] [Google Scholar]
- 5.Alavi IA, Sharma BK, Pillay VK. 1971. Steroid-induced diabetic ketoacidosis. American Journal of the Medical Sciences 262 15–23. ( 10.1097/00000441-197107000-00003) [DOI] [PubMed] [Google Scholar]
- 6.Sugino I, Hiroi N, Yoshihara A. 2011. Diabetic ketoacidosis associated with adrenocorticotropic hormone-producing pituitary adenoma. American Journal of Case Reports 12 31–34. ( 10.12659/AJCR.881696) [DOI] [Google Scholar]
- 7.Kahara T, Chikashi S, Uchiyama A, Usuda D, Akahori H, Tajika E, Miwa A, Usuda R, Suzuki T, Sasano H. 2007. Preclinical Cushing’s syndrome resulting from adrenal black adenoma diagnosed with diabetic ketoacidosis. Endocrine Journal 54 543–551. ( 10.1507/endocrj.K06-071) [DOI] [PubMed] [Google Scholar]
- 8.Uecker JM, Janzow MT. 2005. A case of Cushing syndrome secondary to ectopic adrenocorticotropic hormone producing carcinoid of the duodenum. American Surgeon 71 445–446. [PubMed] [Google Scholar]
- 9.Catli G, Abaci A, Tanrisever O, Kocyigit C, Sule Can P, Dundar B N. 2015. An unusual presentation of pediatric Cushing disease: diabetic ketoacidosis. AACE Clinical Case Reports: Winter 1 e53–e58. ( 10.4158/EP14284.CR) [DOI] [Google Scholar]