

 This work is licensed under a
This work is licensed under a Abstract
Summary
Poland syndrome (PS) is a rare congenital condition, affecting 1 in 30 000 live births worldwide, characterised by a unilateral absence of the sternal head of the pectoralis major and ipsilateral symbrachydactyly occasionally associated with abnormalities of musculoskeletal structures. A baby girl, born at 40 weeks’ gestation with birth weight of 3.33 kg (−0.55 SDS) had typical phenotypical features of PS. She had recurrent hypoglycaemic episodes early in life requiring high concentration of glucose and glucagon infusion. The diagnosis of congenital hyperinsulinism (CHI) was biochemically confirmed by inappropriately high plasma concentrations of insulin and C-peptide and low plasma free fatty acids and β-hydroxyl butyrate concentrations during hypoglycaemia. Sequencing of ABCC8, KCNJ11 and HNF4A did not show any pathogenic mutation. Microarray analysis revealed a novel duplication in the short arm of chromosome 10 at 10p13–14 region. This is the first reported case of CHI in association with PS and 10p duplication. We hypothesise that the HK1 located on the chromosome 10 encoding hexokinase-1 is possibly linked to the pathophysiology of CHI.
Learning points:
Congenital hyperinsulinism (CHI) is known to be associated with various syndromes.
This is the first reported association of CHI and Poland syndrome (PS) with duplication in 10p13–14.
A potential underlying genetic link between 10p13–14 duplication, PS and CHI is a possibility.
Background
Congenital hyperinsulinism (CHI) is a rare disorder of dysregulated insulin secretion from pancreatic β-cells with a reported incidence of 1 in 50 000 live births. CHI is the commonest cause of recurrent and persistent hypoglycaemia during infancy. Mutations in 11 different genes (ABCC8, KCNJ11, GLUD1, GCK, HADH1, UCP2, MCT1, HNF4A, HNF1A, HK1, PGM1) have been implicated in monogenic forms of CHI (1). Hyperinsulinism can also be associated with many overgrowth syndromes and secondary to conditions like birth asphyxia, intra-uterine growth retardation, Rh isoimmunisation and maternal diabetes mellitus (2).
Poland syndrome (PS) was originally described in 1841 by Sir Alfred Poland (1822–1872), an anatomist at Guy’s Hospital in London on a 27-year-old patient who had complete unilateral absence of the sternal head of pectoralis major muscle and ipsilateral symbrachydactyly (3). PS is three times more common in boys than girls, and affects the right side of the body twice as often as the left. The spectrum of thoracic defects varies from subtle hypoplasia to aplasia of the breasts and ribs, chest wall depression, subcutaneous fat, partial agenesis of the ribs (usually II–IV or II–V) (4).
We report a rare association of CHI in PS with a novel 10p13–14 duplication that has not been previously reported in the literature.
Case presentation
A baby girl first by birth order was born to non-consanguineous parents of mixed ethnic origin (British mother and south Asian father). The antenatal period was uneventful. She was delivered at 40 weeks’ gestation by normal vaginal delivery with a birth weight of 3.33 kg (−0.55 SDS). Baby was noted to have left-sided chest wall hypoplasia along with sprengel anomaly of left scapula with restriction of movements on the left arm (Fig. 1). She was noted to have recurrent and persistent hypoglycaemia in the immediate newborn period.
Figure 1.
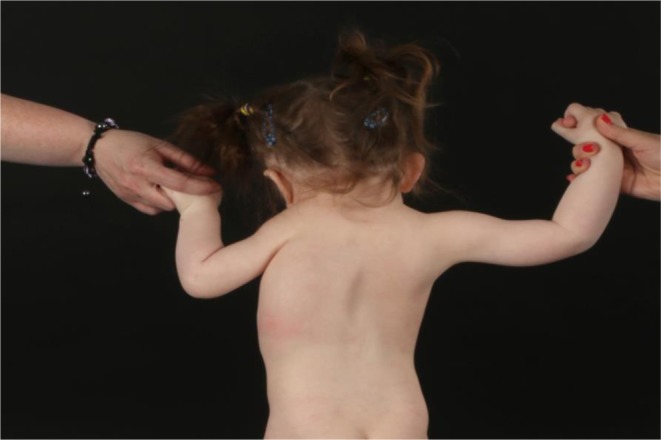
Sprengel’s anomaly.
Investigations
The plasma insulin concentration was inappropriately elevated at 12.8 µIU/L and C-peptide of 220 ng/mL during hypoglycaemia (blood glucose 1.0 mmol/L). The plasma free fatty acids (447 μmol/L) and 3-hydroxybutyrate (29 μmol/L) were inappropriately suppressed confirming a diagnosis of CHI. Chest X-ray showed the absence of upper four thoracic ribs (Fig. 2). MRI of spine was suggestive of complex multiple fusion abnormalities of spine and spina bifida occulta in upper thoracic region with cervical meningocele.
Figure 2.

Chest X-ray showing the absence of upper left ribs.
Microarray analysis of the genomic DNA using Bluegnome ISCA 60K Oligo Cytochip revealed 1.29–1.41 Mb duplication of chromosome 10 between 10p13 and 10p14. Genetic testing for CHI did not identify mutation in ABCC8, KCNJ11 and HNF4A.
Treatment
Normoglycaemia was achieved with total glucose requirement of 18 mg/kg/min and glucagon infusion at a dose of 10 µg/kg/day. Diazoxide was commenced at a dose of 5 mg/kg/day along with chlorothiazide of 7 mg/kg/day. The intravenous fluids and glucagon were gradually weaned and stopped as the baby responded well to diazoxide therapy. The baby’s glucose requirement gradually reduced and normoglycaemia was maintained on enteral feeds and medications (diazoxide and chlorothiazide). Diazoxide was tolerated without any side effect. Based on the phenotypical features, a diagnosis of PS was considered. The fundi examination was normal, making Moebius syndrome, a common association with PS unlikely. A small atrial and ventricular septal defect with normal ventricular function was noted on cardiac echocardiography. There were no phenotypic features suggestive of Poland syndrome in either of the parents, however there was a strong family history of similar structural abnormalities in the mother’s siblings who were estranged.
Outcome and follow-up
Currently, the patient is 18 months old with normal blood glucose concentrations and continues to require diazoxide to maintain normoglycaemia. She is in spinal braces and her left-sided chest movements are improving with physiotherapy. She is being closely monitored by the neurosurgical team.
Discussion
PS is a rare developmental anomaly characterised by the absence of pectoral major muscle with chest wall hypoplasia and brachysyndactyly (short and/or fused). The spectrum of clinical features of PS can be extremely variable and can involve adjacent muscles like latissimus dorsi, serratus anterior, hypoplastic/absent nipples, rhizomelic micromelia and scapular vertebral segmentation anomalies.
CHI has been described in association with various syndromes like Beckwith–Wiedemann syndrome, Usher syndrome, Sotos syndrome and Costello syndrome (Table 1). However the association of CHI with PS has not been previously reported in the literature.
Table 1.
Syndromes associated with CHI.
| Beckwith–Wiedemann |
| Kabuki |
| Simpson–Golabi syndrome |
| Trisomy 13 |
| Mosaic Turner |
| Perlman syndrome |
| Soto |
| Usher |
| Timothy |
| Costello |
| Central hypoventilation syndrome |
The exact pathogenesis of PS is unknown but many theories have been postulated. The most common mechanism cited is ‘Vascular Theory’ which proposes that a critical vascular event, which occurs during 6th week of intra-uterine life, also known as subclavian artery supply disruption sequence (SASDS), causes hypoplasia of the subclavian artery leading to musculoskeletal malformations. Development of subclavian vessels is interrupted due to the medial and forward growth of the ribs (5). Most cases of Poland syndrome are sporadic, and the underlying genetic aetiology of PS is not known. PS has been reported in association with de novo deletion of 11q12.3 in monozygotic twins (6).
Our index case had duplication in the region between 10p13 and 10p14 spanning 1.29–1.41 Mb involving 16 genes. The duplicated region involves UCMA (upper zone of growth plate and cartilage matrix associated). UCMA (HGNC:25205) encodes a chondrocyte-specific, highly charged protein that is abundantly expressed in the upper immature zone of foetal and juvenile epiphyseal cartilage (7). UCMA has also been thought to play an important role in control of osteogenic differentiation of osteochondrogenic precursor cells in peripheral zones of foetal cartilage and at the cartilage–bone interface (8). We hypothesise a potential involvement of this gene contributing to PS phenotype in our patient. It is possible that UCMA influences the musculoskeletal development during the critical stage of foetal development and duplication in the region interrupts its function.
Our index case also has persistent CHI. The patient did not have any other risk factors for neonatal hyperinsulinism like prematurity, intra-uterine growth retardation or maternal diabetes mellitus. This increases the possibility of a genetic link between the detected duplication and CHI. Dallapiccola et al. determined hexokinase activity in the red blood cells of 5 patients with various partial duplications of 10p and also demonstrated an increase in HK1 activity in patients with 10p duplications (9, 10). Although Daniele et al. mapped HK1 to 10q, the possibility of 2 or more HK loci on chromosome 10 was acknowledged (11). The presence of 10p13–14 duplication in our patient suggests the possibility of HK1 loci in the duplicated region that contributes to the pathogenesis of CHI in our patient. HK1 encodes hexokinase-1, which catalyses the first step in glucose metabolism using ATP for the phosphorylation of glucose to glucose-6-phosphate. HK1 has been recently included as one of the causative genes of CHI (12, 13). HK1 has high affinity for glucose and is normally not expressed in beta-cells, as the expression would allow the insulin release at very low levels of glucose resulting in hypoglycaemia (14). HK1 mutations have been described to be diazoxide sensitive. Thus the CHI in our patient could be due to the increased hexokinase activity, potentially due to the duplication resulting in the increased activity of this enzyme.
Conclusion
PS is a rare multisystem disorder reported in the literature. This is the first reported case of PS in association with CHI and a novel 10p duplication. We hypothesise the possible role of UCMA contributing to the phenotype of PS and a potential role of HK1 in the pathogenesis of CHI. Further genetic studies are needed to establish the link between 10p13 and 10p14 duplication, PS and CHI.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Patient consent
A written informed consent has been obtained from the patient’s parent for publication of the submitted article and the accompanying images.
Author contribution statement
D Giri and P Patil wrote the manuscript draft. R Hart helped with the genetic analysis. M Didi helped with the revision of the manuscript. S Senniappan is the lead clinician for the patient and supervised the manuscript writing and helped with further revision. All authors read and approved the final draft of the manuscript.
References
- 1.Stanley CA. 2016. Perspective on the genetics and diagnosis of congenital hyperinsulinism disorders. Journal of Clinical Endocrinology and Metabolism 101 815–826. ( 10.1210/jc.2015-3651) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Senniappan S, Arya VB, Hussain K. 2013. The molecular mechanisms, diagnosis and management of congenital hyperinsulinism. Indian Journal of Endocrinology and Metabolism 17 19–30. ( 10.4103/2230-8210.107822) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lord MJ, Laurenzano KR, Hartmann RW., Jr 1990. Poland’s syndrome. Clinical Pediatrics 29 606–609. ( 10.1177/000992289002901013) [DOI] [PubMed] [Google Scholar]
- 4.Stylianos K, Constantinos P, Alexandros T, Aliki F, Nikolaos A, Demetriou M, Petros P. 2010. Muscle abnormalities of the chest in Poland’s syndrome: variations and proposal for a classification. Surgical and Radiologic Anatomy 34 5763 ( 10.1007/s00276-011-0851-5) [DOI] [PubMed] [Google Scholar]
- 5.Bavinck JNB, Weaver DD. 1986. Subclavian artery supply disruption sequence hypothesis of a vascular etiology for Poland, Klippel- Feil, and Mobius anomalies. American Journal of Medical Genetics 23 903–918. ( 10.1002/ajmg.1320230405) [DOI] [PubMed] [Google Scholar]
- 6.Vaccari CM, Romanini MV, Musante I, Tassano E, Gimelli S, Divizia MT, Torre M, Morovic CG, Lerone M, Ravazzolo R, et al. 2014. De novo deletion of chromosome 11q12.3 in monozygotic twins affected by Poland syndrome. BMC Medical Genetics 15 63 ( 10.1186/1471-2350-15-63) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tagariello A, Luther J, Streiter M, Didt-Koziel L, Wuelling M, Surmann-Schmitt C, Stock M, Adam N, Vortkamp A, Winterpacht A. 2008. UCMA – a novel secreted factor represents a highly specific marker for distal chondrocytes. Matrix Biology 27 3–11. ( 10.1016/j.matbio.2007.07.004) [DOI] [PubMed] [Google Scholar]
- 8.Surmann-Schmitt C, Dietz U, Kireva T, Adam N, Park J, Tagariello A, Onnerfjord P, Heinegård D, Schlötzer-Schrehardt U, Deutzmann R, et al. 2008. UCMA, a novel secreted cartilage-specific protein with implications in osteogenesis. Journal of Biological Chemistry 283 7082–7093. ( 10.1074/jbc.M702792200) [DOI] [PubMed] [Google Scholar]
- 9.Dallapiccola B, Novelli G, Micara G, Delaroche I, Moric-Petrovic S, Magnani M. 1984. Regional mapping of hexokinase-1 within the short arm of chromosome 10. Human Heredity 34 156–160. ( 10.1159/000153453) [DOI] [PubMed] [Google Scholar]
- 10.Dallapiccola B, Chessa L, Vignetti P, Ferrante E, Gandini E. 1979. Increased HK1 activity levels in the red cells of a patient with a de novo trisomy 10p: t(Y;10)(p11;p12). Human Genetics 50 45–49. ( 10.1007/BF00295588) [DOI] [PubMed] [Google Scholar]
- 11.Daniele A, Altruda F, Ferrone M, Silengo L, Romeo G, Archidiacono N, Rocchi M. 1992. Mapping of human hexokinase 1 gene to 10q11-qter. Human Heredity 42 107–110. ( 10.1159/000154049) [DOI] [PubMed] [Google Scholar]
- 12.Pinney SE, Ganapathy K, Bradfield J, Stokes D, Sasson A, Mackiewicz K, Boodhansingh K, Hughes N, Becker S, Givler S, et al. 2013. Dominant form of congenital hyperinsulinism maps to HK1 region on 10q. Hormone Research in Paediatrics 80 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quintens R, Hendrickx N, Lemaire K, Schuit F. 2008. Why expression of some genes is disallowed in beta-cells. Biochemical Society Transactions 36 300–305. ( 10.1042/BSbib360300) [DOI] [PubMed] [Google Scholar]
- 14.Henquin J-C, Sempoux C, Marchandise J, Godecharles S, Guiot Y, Nenquin M, Rahier J. 2013. Congenital hyperinsulinism caused by hexokinase I expression or glucokinase-activating mutation in a subset of β-cells. Diabetes 62 1689–1696. ( 10.2337/db12-1414) [DOI] [PMC free article] [PubMed] [Google Scholar]