

ABSTRACT
Synthesis and integrity of the cytoplasmic membrane are fundamental to cellular life. Experimental evolution studies have hinted at unique physiology in the Gram-positive bacteria Streptococcus mitis and S. oralis. These organisms commonly cause bacteremia and infectious endocarditis (IE) but are rarely investigated in mechanistic studies of physiology and evolution. Unlike in other Gram-positive pathogens, high-level (MIC ≥ 256 μg/ml) daptomycin resistance rapidly emerges in S. mitis and S. oralis after a single drug exposure. In this study, we found that inactivating mutations in cdsA are associated with high-level daptomycin resistance in S. mitis and S. oralis IE isolates. This is surprising given that cdsA is an essential gene for life in commonly studied model organisms. CdsA is the enzyme responsible for the synthesis of CDP-diacylglycerol, a key intermediate for the biosynthesis of all major phospholipids in prokaryotes and most anionic phospholipids in eukaryotes. Lipidomic analysis by liquid chromatography-mass spectrometry (LC-MS) showed that daptomycin-resistant strains have an accumulation of phosphatidic acid and completely lack phosphatidylglycerol and cardiolipin, two major anionic phospholipids in wild-type strains, confirming the loss of function of CdsA in the daptomycin-resistant strains. To our knowledge, these daptomycin-resistant streptococci represent the first model organisms whose viability is CdsA independent. The distinct membrane compositions resulting from the inactivation of cdsA not only provide novel insights into the mechanisms of daptomycin resistance but also offer unique opportunities to study the physiological functions of major anionic phospholipids in bacteria.
KEYWORDS: daptomycin, Streptococcus, lipidomics
INTRODUCTION
Streptococcus mitis and S. oralis are human oral colonizers, opportunistic pathogens, and species of the viridans group streptococci (VGS). VGS are associated with ∼23% of Gram-positive bacteremia in immunocompromised patients (1, 2) and ∼17% of infective endocarditis (IE) cases (3). The mitis group VGS are difficult to accurately identify to the species level in clinical settings (4, 5). However, retrospective molecular studies have determined that S. mitis and S. oralis are major causative agents of VGS bacteremia and IE (6–9). Although these organisms are unquestionably significant for human health, very little is known about the physiology and virulence of S. mitis and S. oralis. This is in part due to their close phylogenetic relationship with S. pneumoniae (10–15), to which S. mitis and S. oralis are often compared; they are rarely considered in their own right.
Recent research hints at unique physiology in S. mitis and S. oralis (16–18). Daptomycin (DAP) is a cyclic lipopeptide antibiotic with potent activity against Gram-positive bacterial pathogens, including vancomycin-resistant enterococci (VRE) and methicillin-resistant Staphylococcus aureus (MRSA) (19, 20). Akins et al. reported that high-level DAP resistance (defined as a MIC of ≥256 μg/ml) emerged in S. mitis and S. oralis IE isolates after a single DAP exposure in a simulated endocardial vegetation model (17). High-level DAP resistance after a single drug exposure was also reported by García-de-la-Mària et al. (16) for a collection of mitis group VGS. These results are surprising because DAP resistance in VRE, MRSA, and other model Gram-positive organisms typically emerges in a stepwise fashion by mutation accretion over days or weeks of DAP exposure (21, 22). We infer that DAP resistance in S. mitis and S. oralis proceeds through a novel genetic mechanism compared to those in other Gram-positive bacteria.
In this investigation, we use experimental evolution studies and lipidomics to show that one-step high-level DAP resistance in S. mitis and S. oralis is associated with loss-of-function mutations in cdsA, a gene for phospholipid biosynthesis that is essential for Escherichia coli (23), Bacillus subtilis (24), Staphylococcus aureus (25), better-studied streptococci (including S. pneumoniae [26–29]), and an organism with an engineered minimal genome (30). We conclude that S. mitis and S. oralis possess unique physiology, allowing DAP resistance to emerge by a novel mechanism.
RESULTS
Emergence of high-level DAP resistance in S. mitis and S. oralis.
The IE clinical isolates S. mitis strain 1643 and S. oralis strains 1647 and 1648 achieved high-level DAP resistance after a single DAP exposure in both simulated endocardial vegetation and bacteremia models (17). We performed serial passage experiments to determine whether DAP resistance would similarly emerge under standard laboratory culture conditions. Parental strains were cultured with DAP (2, 4, 8, and 16 μg/ml). After 24 h of incubation, growth was observed in each inoculated well, regardless of the DAP concentration. Resistant strains obtained from these cultures are referred to as S. mitis 1643-HA04 (derived from parental strain 1643), S. oralis 1647-HA06 (derived from 1647), and S. oralis 1648-HA08 (derived from 1648) (Table 1). We conclude that high-level DAP resistance emerges in S. mitis and S. oralis IE isolates after a single DAP exposure, regardless of the incubation or environmental conditions.
TABLE 1.
DAP MICs and CdsA details for S. mitis and S. oralis strains used in this study
| Strain | Strain descriptiona | DAP MIC (μg/ml)b | CdsA changec |
|---|---|---|---|
| S. mitis 1643 | Wild-type IE isolate | 0.75 | (Q31) |
| S. mitis 1643-HA04 | DAP resistant | >256 | *31 |
| S. mitis 1643-HA14 | DAP revertant | 0.75 | W31 |
| S. oralis 1647 | Wild-type IE isolate | 1 | (G246) |
| S. oralis 1647-HA06 | DAP resistant | >256 | C246 |
| S. oralis 1647-HA16 | DAP revertant | 1.5 | S246 |
| S. oralis 1648 | Wild-type IE isolate | <0.5 | (D249) |
| S. oralis 1648-HA08 | DAP resistant | >256 | N249 |
| S. oralis 1648-HA18 | DAP revertant | <0.5 | D249 |
DAP-resistant strains were obtained after wild-type strains were exposed to DAP. DAP revertants were obtained by passage of DAP-resistant strains in drug-free medium.
MICs were determined by Etest on MHA plates.
Wild-type amino acids are shown in parentheses. * indicates a stop codon.
DAP resistance confers a modest growth defect on S. mitis and S. oralis. In standard laboratory medium, the resistant strains grew with generation times ≥20% longer than those of parental strains (see Fig. S1 in the supplemental material).
Mutations in cdsA occur in DAP-resistant strains.
We used genome sequencing to identify mutations in the DAP-resistant strains (Table 2). Nonsynonymous mutations in cdsA occurred in all three strain pairs. CdsA catalyzes the conversion of phosphatidic acid (PA) into CDP-diacylglycerol (CDP-DAG), which is the precursor for synthesis of the major membrane phospholipids, including phosphatidylglycerol (PG), cardiolipin (CL), phosphatidylethanolamine (PE), and phosphatidylserine (PS) (Fig. 1).
TABLE 2.
Mutations occurring in DAP-resistant strains
| Strain | Description of gene | Nucleotide variation in genea | Amino acid change | Locusb |
|---|---|---|---|---|
| S. mitis 1643-HA04 | Hypothetical | G531T (98) | S177R | SK608_0120 (100, 100) |
| Serine/threonine kinase | A526C (90) | K176Q | smi_1622 (45, 29) | |
| Ribosomal S5p alanine acyltransferase | G502T (98) | H168N | TZ92_01497 (100, 99) | |
| Phosphatidate cytidylyltransferase (CdsA)c | G91A (99) | Q31Stop | smi_1854 (100, 81) | |
| S. oralis 1647-HA06 | Intergenic region 13 | C to T (99) | N/A | 9 bp between SOR_1153 and SOR_1154 |
| Intergenic region 17 | A to C (99) | N/A | 47 bp upstream of SOR_0277 | |
| Phosphatidate cytidylyltransferase (CdsA) | C736A (97) | G246C | SOR_1730 (100, 96) | |
| S. oralis 1648-HA08 | ppGpp synthetase | A2075C (96) | V692G | SOR_1513 (100, 99) |
| Helicase PriA | T77G (93) | E26A | SOR_1544 (100, 96) | |
| Ribosomal SSU methyltransferase | C454A (96) | V152F | SOR_1542 (100, 95) | |
| Phosphatidate cytidylyltransferase (CdsA) | C745T (97) | D249N | SOR_1730 (100, 96) |
The frequency of the mutation in the read assembly is shown in parentheses.
Loci were identified using protein BLAST. Query coverage and percent amino acid identity are shown in parentheses. References used were S. oralis Uo5 (SOR_) (53), S. mitis B6 (smi_) (13), S. mitis SK608 (SK608_) (54), and S. oralis SK141 (TZ92_) (55).
Bold type indicates that the mutation reverted after serial passage without DAP.
FIG 1.

Biosynthetic pathway of the major phospholipids in bacteria. Abbreviations: PA, phosphatidic acid; CDP-DAG, CDP-diacylglycerol; PS, phosphatidylserine; PE, phosphatidylethanolamine; G-3-P, glycerol-3-phosphate; PG-P, phosphatidylglycerol-3-P; PG, phosphatidylglycerol; CL, cardiolipin.
We analyzed the cdsA mutations to predict their impacts on CdsA function. S. mitis 1643-HA04 possesses a G91A transition that generates a premature stop codon after the 31st amino acid in the polypeptide chain. We predict that this mutation results in a complete loss of CdsA function in S. mitis 1643-HA04. S. oralis 1648-HA08 possesses a C745T transition that alters the 249th residue from aspartic acid to asparagine. A recently determined crystal structure of CdsA from Thermotoga maritima (TmCdsA) identified residues necessary for TmCdsA function (31). D249 of the streptococcal CdsA aligns to an active-site residue (D246) in TmCdsA which was shown to be required for CdsA activity (31). Finally, S. oralis 1647-HA06 possesses a C736A transversion that converts the 246th residue from a glycine to a cysteine. Analysis of putative secondary structures for the peptide chain using the garnier tool in EMBOSS (32) revealed that G246 in 1647-HA06 occurs in a bend in the peptide (data not shown). Altering the flexible glycine residue to a rigid cysteine residue may prevent the polypeptide chain from folding properly. In summary, cdsA mutations in the DAP-resistant strains likely result in CdsA proteins that have loss of function by altering an active site (S. oralis 1648-HA08), preventing proper protein folding (S. oralis 1647-HA06), or by preventing synthesis of the full polypeptide (S. mitis 1643-HA04).
Lipidomic analysis confirms CdsA loss of function in DAP-resistant S. mitis and S. oralis.
To assess the functional consequences of cdsA mutations on the synthesis of phospholipids in S. mitis and S. oralis, we carried out lipidomic analysis. The total lipids from DAP-susceptible and DAP-resistant strains of S. mitis and S. oralis were subjected to normal-phase liquid chromatography-electrospray ionization mass spectrometry (LC-ESI/MS) using a silica column for lipid separation. As shown by the total negative-ion chromatograms (Fig. 2 and 3; see also Fig. S2), PG and CL are the two major anionic phospholipids detected in all three DAP-susceptible strains. They appear at the retention time windows of ∼12.5 to 13.5 min and 13.5 to 14.5 min, respectively. In contrast, PG and CL are completely absent in three DAP-resistant strains. The results confirm cdsA inactivation in the DAP-resistant strains as well as the essential role of cdsA for PG and CL synthesis in S. mitis and S. oralis under the conditions tested.
FIG 2.

Normal-phase LC-ESI/MS analysis of the total lipid extracts of DAP-susceptible and DAP-resistant strains of S. mitis. The y-axis numbers represent ion intensities of arbitrary units. PG and CL are major lipids found in S. mitis 1643. PG and CL are absent in the DAP-resistant derivative 1643-HA04. After passage without selection, DAP susceptibility was restored in 1643-HA14, as were PG and CL levels.
FIG 3.

Normal-phase LC-ESI/MS analysis of the total lipid extracts of DAP-susceptible and DAP-resistant strains of S. oralis. The y-axis numbers represent ion intensities of arbitrary units. PG and CL are major lipids found in DAP-susceptible S. oralis 1647 and 1648. PG and CL are missing from DAP-resistant derivatives 1647-HA06 and 1648-HA08. Upon restoration of DAP susceptibility in 1647-HA16 and 1648-HA18, PG levels return to normal. CL levels are restored in 1648-HA18 but not 1647-HA16.
Other species identified in both DAP-susceptible and DAP-resistant strains are diacylglycerol (DAG), monohexosyldiacylglycerol (MHDAG), fatty acid (FA), dihexosyldiacylglycerol (DHDAG), undecaprenyl phosphate (C55-P), and PA. The levels of PA are consistently increased in all DAP-resistant strains (Fig. 2 and 3). PA is a substrate of CdsA for the synthesis of CDP-DAG (Fig. 1); thus, its accumulation is an expected consequence of the inactivation of cdsA. The acyl compositions of PA in the DAP-sensitive strains are slightly different from those in the DAP-resistant strains (Fig. S3). For reference, the total lipid content of S. mitis 1643 and its derivatives is displayed in Table S3 in the supplemental material.
An unknown species, appearing at 14.8 to 15.3 min, is significantly elevated in the DAP-resistant S. mitis 1643-HA04. Exact mass measurement and collision-induced dissociation (CID) tandem MS identified this unknown lipid as phosphatidyl-N-acetylhexosamine (PAHN), a PA-derived glycolipid (Fig. S4). A small amount of PAHN was detected in the DAP-sensitive strain S. mitis 1643-RA03 but was not detected in any of the S. oralis strains.
Phosphatidylcholines (PC) were detected by positive ion ESI/MS in all strains used in this study. Shown in Fig. S3 are representative mass spectra of PCs in S. mitis 1643 and 1643-HA04. The level of PC is lower (3- to 10-fold) in the DAP-resistant strains than in the DAP-sensitive strains (Fig. S5).
Mutations in cdsA revert concurrently with phenotypic reversion to DAP susceptibility.
Resistant strains were passaged in CA-SMHB until DAP susceptibility was restored. S. mitis 1643-HA04 began showing a zone of inhibition, indicating reversion, after the 4th passage, while S. oralis 1647-HA06 and S. oralis 1648-HA08 reverted after the 12th passage. Revertant populations recovered from the 5th overnight for S. mitis 1643-HA04 and the 13th overnight for S. oralis 1647-HA06 and S. oralis 1648-HA08 are referred to as S. mitis 1643-HA14, S. oralis 1647-HA16, and S. oralis 1648-HA18, respectively. These populations have MICs identical or comparable to those of the original susceptible parental strains (Table 1) and growth rates similar to those of the parental strains (Fig. S1).
Mutations listed in Table 2 were queried in the revertant populations using PCR and Sanger sequencing. All mutations present in the DAP-resistant strains were also present in the revertant populations, except for the cdsA mutations. Analysis of cdsA showed that the premature stop codon in S. mitis 1643-HA04 was replaced with a tryptophan codon in S. mitis 1643-HA14, allowing for proper read-through of the coding region; the C246 of S. oralis 1647-HA06 was replaced with a serine in S. oralis 1647-HA16, restoring flexibility to the region, and the active site of S. oralis 1648-HA08 was restored in S. oralis 1648-HA18, as the mutation reverted to wild type. Table 1 displays the DAP MICs for the susceptible, resistant, and revertant populations of each lineage, as well as the corresponding amino acid changes in CdsA. Our data indicate that restoration of CdsA function results in DAP susceptibility.
To confirm CdsA functionality in our revertants, we subjected them to lipidomic analyses as described above. All three revertant strains synthesize PG, indicating the synthesis of CDP-DAG and therefore confirming the restored function of CdsA (Fig. 2 and 3). Interestingly, 1647-HA16 does not possess any CL and continues to have a small accumulation of PA (Fig. 3). It is possible that 1647-HA16 acquired a loss-of-function mutation in a cardiolipin synthase gene over the course of the reversion passage, thereby preventing formation of CL. Alternatively, and not mutually exclusively, 1647-HA16 might possess only a partial restoration of CdsA function, resulting in an accumulation of PA. That 1647-HA16 is DAP sensitive indicates that PG, and not CL, is required for DAP susceptibility in S. oralis.
Spontaneous DAP resistance is always associated with cdsA mutation.
To assess the frequency and diversity of cdsA mutations, we plated S. mitis and S. oralis parental strains on DAP-containing agar and screened colonies for cdsA mutation. In addition, we compared the frequency of spontaneous DAP resistance to that of spontaneous resistance to rifampin, a commonly used antibiotic for mutation frequency studies. S. oralis 1648 was not analyzed due to its preexisting rifampin resistance (17). Spontaneous DAP resistance arose at an average frequency of 1 × 10−6 for both strains on 10 μg/ml of DAP and at an average frequency of 7 × 10−7 for both strains on 128 μg/ml of DAP. Rifampin resistance arose at frequencies of 1 × 10−7 for S. mitis 1643 and 8 × 10−9 for S. oralis 1647. Of colonies arising on the 10-μg/ml and 128-μg/ml DAP plates that were screened (n = 15 and 14, respectively), all possessed nonsynonymous mutations in cdsA. In two colonies, two separate polymorphisms were found. Polymorphisms were observed that resulted in amino acid substitutions (n = 20; 15 unique), premature stop codons (n = 6; 4 unique), large-scale (>200-bp) deletions (n = 2), or frameshifts due to small insertions or deletions (n = 3). The two large deletions (268 bp and 252 bp) encompassed approximately the same region, but only one of the deletions resulted in a frameshift. Figure 4 and Table S4 in the supplemental material catalog all mutations detected in cdsA in this study. We conclude that diverse cdsA mutants rapidly emerge in S. mitis and S. oralis populations under DAP selection.
FIG 4.
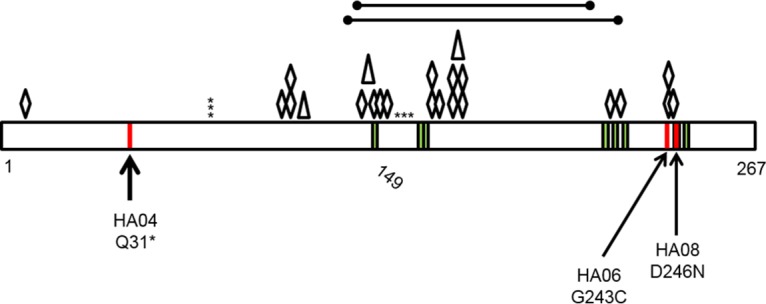
Summary of CdsA alterations identified in S. mitis and S. oralis after DAP selection. CdsA is depicted as a black rectangle, with amino acid positions indicated at the bottom. Green stripes represent active sites identified previously in Thermotoga maritima (31). Red stripes represent the original mutations identified in the broth-derived DAP-resistant strains. Asterisks depict premature stop codons, diamonds depict amino acid substitutions, triangles depict a nucleotide coding region deletion event of 1 to 2 bp, and the large lines at the top depict large areas of deletion identified in spontaneous-resistance studies on DAP agar. HA04, S. mitis 1643-HA04; HA06, S. oralis 1647-HA06; HA08, S. oralis 1648-HA08.
DISCUSSION
CdsA catalyzes the synthesis of CDP-DAG, a key intermediate in phospholipid biosynthesis in all cells. CDP-DAG is the source of the phosphatidyl group for all major phospholipids in prokaryotes and for most anionic phospholipids in eukaryotes. Historical efforts to elucidate the biochemical pathways for membrane phospholipid biosynthesis used chemical mutagenesis to isolate E. coli mutants with reduced CDP-DAG synthase activity (33, 34). Those mutants were observed to accumulate PA but were not complete-loss-of-function mutants due to the continued presence of PG, CL, and PE in the E. coli membrane (33, 34). More recently, E. coli partial-loss-of-function cdsA mutants were found to accumulate PA and have decreased vancomycin susceptibility, presumably by altering outer membrane structure and access of the drug to the periplasm (35). Efforts to define the essential gene cohorts of E. coli, B. subtilis, S. pneumoniae, and other bacteria have identified cdsA as being essential (23–29). Crucially, efforts to define the minimum cohort of genes required to sustain cellular life also identified cdsA as being essential (30). In contrast to these observations, S. mitis 1643-HA04, S. oralis 1647-HA06, and S. oralis 1648-HA08 lack CdsA function, as confirmed by lipidomics analysis. Furthermore, lipidomics results demonstrate that a CdsA-independent pathway for anionic phospholipid biosynthesis does not exist or is not active in these bacteria under the conditions tested in this study.
Why is cdsA essential in other bacteria but not in S. mitis and S. oralis? Presumably, cdsA is essential in other bacteria because the lipids synthesized from the CdsA product, CDP-DAG, are essential for cytoplasmic membrane integrity, fluidity, and/or the localization of key proteins for cellular maintenance and replication. Alternatively, the substrate of CdsA, phosphatidic acid, could be toxic at high levels. Potential physiological mechanisms for growth of S. mitis and S. oralis cdsA mutants include altered cell wall structure and/or altered distribution or perhaps a lack of necessity for key membrane proteins involved in cell division. These mechanisms would be expected to be absent from model bacteria for which cdsA is essential. Unfortunately, little is known about the physiology and genetics of S. mitis and S. oralis. Characterization of peptidoglycan structure, membrane proteomics, and the transcriptomes of the wild type and cdsA mutants will be informative to understand the mechanism of survival of S. mitis and S. oralis cdsA mutants. In addition, studies which assess the impact of altered membrane structure and altered growth rates on the virulence and in vivo physiology of DAP-resistant S. mitis and S. oralis are warranted.
PC is a relatively rare membrane phospholipid in the bacterial domain, with only ∼15% of bacteria encoding the necessary biosynthetic pathway (36). Eukaryotes synthesize PC using the Kennedy pathway (37). A homologous pathway has been discovered in the bacterium Treponema denticola (38, 39). The first two genes in the pathway are licA and licC. BioCyc is a pathway and genome database which predicts metabolic networks for various organisms (40). S. mitis and S. oralis are predicted to possess homologs of the first two genes in the Kennedy pathway; however, the third gene has yet to be elucidated. S. pneumoniae uses licA and licC to incorporate choline into the cell wall to aid in adhesion to human epithelial cells (41). It is possible that the cdsA mutants repurpose the lic pathway for PC synthesis under certain conditions. Other Gram-positive model organisms, such as Enterococcus faecalis, S. aureus, and B. subtilis, do not carry licA and licC. This is further evidence of S. mitis and S. oralis possessing unique physiological characteristics that allow them to tolerate CdsA loss of function.
Studies with other Gram-positive bacteria have associated a wide range of mutations with reduced DAP susceptibility. A common feature identified by some of these studies is a bacterial cell membrane with reduced PG content. Mutations in mprF were shown to increase lysyl-PG content in DAP-nonsusceptible MRSA, concomitantly reducing the PG content of the membrane (42). In B. subtilis, an evolved DAP-nonsusceptible strain possessed reduced function mutations in pgsA, which is responsible for the synthesis of PG (43). The evolved B. subtilis strain possessed PG levels >5-fold lower than its susceptible parental strain and a DAP MIC nearly 30-fold higher. In this study, we have identified a novel mechanism by which S. mitis and S. oralis purge their membranes of PG and, as previously shown, DAP MICs increase up to 512-fold higher than DAP-sensitive parental strains (17, 18).
Several models for DAP's mechanism of action against Gram-positive bacteria have been proposed (21, 22). These mechanisms generally implicate membrane pore formation by DAP or recruitment of DAP to sites of membrane curvature. More recently, a revised mechanism of DAP action was proposed for the model Gram-positive bacterium B. subtilis (44). In this model, DAP's lipid tail interacts with domains of increased membrane fluidity, allowing DAP to oligomerize at those sites. This causes rapid delocalization of membrane proteins essential for peptidoglycan biosynthesis (MurG) and lipid biosynthesis (PlsX), which are also associated with the membrane fluid domains. The displacement of MurG interrupts peptidoglycan biosynthesis, ultimately leading to cell lysis. DAP-resistant S. mitis and S. oralis may synthesize a membrane for which DAP has low affinity, and loss of CdsA activity is required for synthesis of this membrane. Our data indicate that PG is a critical phospholipid associated with DAP susceptibility. Alternatively, S. mitis and S. oralis may not organize essential membrane proteins in membrane fluid domains. The biophysical mechanism for DAP resistance in these strains remains to be determined. It will be interesting to determine whether S. mitis and S. oralis cdsA mutants emerge in humans treated with DAP and whether these cdsA mutations are as permissive for S. mitis and S. oralis outgrowth under human in vivo conditions as they are under in vitro conditions.
MATERIALS AND METHODS
Bacterial strains, media, and susceptibility testing.
Bacterial strains used in this study are shown in Table 1. All testing was conducted in Mueller-Hinton broth or agar supplemented with 50 μg/ml of calcium and 12.5 μg/ml of magnesium (CA-SMHB and CA-SMHA, respectively) as required for DAP (45, 46), unless otherwise stated. Cultures were incubated at 37°C in a GasPak EZ Campy container system (BD) unless otherwise stated. Etest (bioMérieux, Inc.) susceptibilities were determined on tryptic soy agar (TSA) supplemented with 5% defibrinated horse blood (Remel) or on MHA.
Serial passage experiments with DAP.
Parental strains (S. mitis 1643, S. oralis 1647, and S. oralis 1648) were cultured in 2 ml of CA-SMHB with DAP (2, 4, 8, and 16 μg/ml) overnight at 37°C. After 24 h of incubation, growth was observed in each inoculated well, regardless of the DAP concentration. Strains obtained from these cultures (referred to as HA0X [Table 1]) were confirmed to be DAP resistant and used for genome sequencing.
Genome sequencing and analysis.
Genomic DNA from parental strains was isolated using Roche MagNA Pure per the manufacturer's instructions and sequenced with Illumina technology at GENEWIZ, Inc. Single end reads of 50 bp were obtained. Genomic DNA from resistant strains was isolated using a modified Qiagen blood and tissue DNeasy kit protocol as previously described (47) and sequenced with Illumina technology at Molecular Research LP. Paired end, 2 × 300-bp reads were obtained. DAP resistance of the cultures was confirmed via Etest prior to genomic DNA extraction.
De novo draft genomes were assembled using CLC Genomics Workbench with default parameters (Table S1) and annotated using Rapid Annotation using Subsystem Technology (48). The taxonomic identification for the parental strains were confirmed using GyrB typing (49). Reads from the resistant strains were mapped to the draft parent genomes, and polymorphisms were detected using default parameters. Polymorphisms were manually curated. Polymorphisms occurring on contigs of <500 bp, within 300 bp from a contig end, in rRNA or tRNA regions, or in polymorphic regions with sequence variation in both the parental de novo assembly and the resistant strain read mapping were removed from further analysis. This screening process generated a list of candidate mutations that were further curated by independent confirmation with Sanger sequencing. Primers were designed to amplify approximately 500 bp surrounding putative mutations (Table S2). PCR with Taq polymerase (New England BioLabs [NEB]) was used to amplify these regions from both parental and resistant strains. Products were sequenced at the Massachusetts General Hospital DNA Sequencing Core. Nucleotide sequences for wild-type and mutant loci of interest are in Text S1 in the supplemental material.
Bacterial growth curves.
To quantify growth, brain heart infusion broth (BHI) was inoculated to an optical density at 600 nm (OD600) of 0.05 from overnight cultures. The OD600 was monitored for parental and resistant strains every 60 to 75 min until cultures reached stationary phase. The experiments were performed independently three times. Revertant strains were monitored as described above at 60-min intervals for two independent trials.
Reversion passage experiments.
Resistant strains (S. mitis 1643-HA04, S. oralis 1647-HA06, and S. oralis 1648-HA08) were inoculated into 1 ml of CA-SMHB in 1.5-ml Eppendorf tubes and cultured overnight. After each passage, 100 μl of culture was used as the inoculum for the next passage, 500 μl was used to generate a frozen stock, and ∼400 μl was spread on MHA for DAP Etest. Passaging was terminated when a zone of inhibition was observed on DAP Etest.
Spontaneous resistance incidence.
Overnight cultures were used to inoculate BHI to an OD600 of 0.05. Cultures were incubated until mid-exponential phase and serially diluted, and 100 μl of culture or dilutions were plated on BHI agar, BHI agar with 50 μg/ml of rifampin (n = 2 independent trials), CA-SMHA, and CA-SMHA with either 10 μg/ml of DAP (n = 5 independent trials) or 128 μg/ml of DAP (n = 3 independent trials). Plates were incubated for 20 to 24 h at 37°C in a chamber with a CO2 GasPak prior to colony counting. For DAP tests, individual colonies were picked, and PCR and Sanger sequencing (Table S2) were used to query the entire cdsA coding region as well as its putative promoter for mutations.
Lipidomic analysis.
A single colony was picked into 50 ml of Todd-Hewitt broth (THB) and incubated at 37°C with 5% CO2 overnight. The 50-ml cultures were added to 250 ml of prewarmed THB and incubated until an OD600 of ∼0.6 was obtained. A total of 500 μl was removed for DAP Etest on 5% horse blood TSA plates. The remaining culture was pelleted at 10,000 rpm and 4°C. Cell pellets were stored at −80°C prior to lipid extraction by the Bligh and Dyer method (50). Normal-phase LC was performed on an Agilent 1200 quaternary LC system equipped with an Ascentis silica high-performance liquid chromatography (HPLC) column (5 μm; 25 cm by 2.1 mm; Sigma-Aldrich) as published previously (51, 52). Briefly, mobile phase A consisted of chloroform-methanol-aqueous ammonium hydroxide (800:195:5, vol/vol), mobile phase B consisted of chloroform-methanol-water-aqueous ammonium hydroxide (600:340:50:5, vol/vol), and mobile phase C consisted of chloroform-methanol-water-aqueous ammonium hydroxide (450:450:95:5, vol/vol/vol/vol). The elution program consisted of the following: 100% mobile phase A was held isocratically for 2 min, then linearly increased to 100% mobile phase B over 14 min, and held at 100% mobile phase B for 11 min. The LC gradient was then changed to 100% mobile phase C over 3 min, held at 100% mobile phase C for 3 min, and, finally, returned to 100% mobile phase A over 0.5 min and held at 100% mobile phase A for 5 min. The LC eluent (with a total flow rate of 300 ml/min) was introduced into the ESI source of a high-resolution TripleTOF5600 mass spectrometer (Sciex, Framingham, MA). Instrumental settings for negative-ion ESI and MS/MS analysis of lipid species were as follows: IS = −4,500 V, CUR = 20 lb/in2, GSI = 20 lb/in2, DP = −55 V, and FP = −150 V. The MS/MS analysis used nitrogen as the collision gas. Data analysis was performed using Analyst TF1.5 software (Sciex).
Accession number(s).
Illumina sequence reads generated in this study have been deposited in the Sequence Read Archive under the accession number PRJNA354070.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by GM069338 and EY023666 to Z.G. and University of Texas at Dallas start-up funds to K.L.P. R.L.A. has received research funding from Merck & Co. and Theravance Biopharma. K.L.P. has received research funding from Merck & Co. and Synereca Pharmaceuticals.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02552-16.
REFERENCES
- 1.Gudiol C, Bodro M, Simonetti A, Tubau F, Gonzalez-Barca E, Cisnal M, Domingo-Domenech E, Jimenez L, Carratala J. 2013. Changing aetiology, clinical features, antimicrobial resistance, and outcomes of bloodstream infection in neutropenic cancer patients. Clin Microbiol Infect 19:474–479. doi: 10.1111/j.1469-0691.2012.03879.x. [DOI] [PubMed] [Google Scholar]
- 2.Marin M, Gudiol C, Garcia-Vidal C, Ardanuy C, Carratala J. 2014. Bloodstream infections in patients with solid tumors: epidemiology, antibiotic therapy, and outcomes in 528 episodes in a single cancer center. Medicine (Baltimore) 93:143–149. doi: 10.1097/MD.0000000000000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murdoch DR, Corey GR, Hoen B, Miro JM, Fowler VG Jr, Bayer AS, Karchmer AW, Olaison L, Pappas PA, Moreillon P, Chambers ST, Chu VH, Falco V, Holland DJ, Jones P, Klein JL, Raymond NJ, Read KM, Tripodi MF, Utili R, Wang A, Woods CW, Cabell CH, International Collaboration on Endocarditis-Prospective Cohort Study. 2009. Clinical presentation, etiology, and outcome of infective endocarditis in the 21st century: the International Collaboration on Endocarditis-Prospective Cohort Study Arch Intern Med 169:463–473. doi: 10.1001/archinternmed.2008.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Teles C, Smith A, Ramage G, Lang S. 2011. Identification of clinically relevant viridans group streptococci by phenotypic and genotypic analysis. Eur J Clin Microbiol Infect Dis 30:243–250. doi: 10.1007/s10096-010-1076-y. [DOI] [PubMed] [Google Scholar]
- 5.Hoshino T, Fujiwara T, Kilian M. 2005. Use of phylogenetic and phenotypic analyses to identify nonhemolytic streptococci isolated from bacteremic patients. J Clin Microbiol 43:6073–6085. doi: 10.1128/JCM.43.12.6073-6085.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sahasrabhojane P, Galloway-Pena J, Velazquez L, Saldana M, Horstmann N, Tarrand J, Shelburne SA. 2014. Species-level assessment of the molecular basis of fluoroquinolone resistance among viridans group streptococci causing bacteraemia in cancer patients. Int J Antimicrob Agents 43:558–562. doi: 10.1016/j.ijantimicag.2014.01.031. [DOI] [PubMed] [Google Scholar]
- 7.Shelburne SA, Sahasrabhojane P, Saldana M, Yao H, Su X, Horstmann N, Thompson E, Flores AR. 2014. Streptococcus mitis strains causing severe clinical disease in cancer patients. Emerg Infect Dis 20:762–771. doi: 10.3201/eid2005.130953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitten T, Munro CL, Zollar NQ, Lee SP, Patel RD. 2012. Oral streptococcal bacteremia in hospitalized patients: taxonomic identification and clinical characterization. J Clin Microbiol 50:1039–1042. doi: 10.1128/JCM.06438-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chun S, Huh HJ, Lee NY. 2015. Species-specific difference in antimicrobial susceptibility among viridans group streptococci. Ann Lab Med 35:205–211. doi: 10.3343/alm.2015.35.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donati C, Hiller NL, Tettelin H, Muzzi A, Croucher NJ, Angiuoli SV, Oggioni M, Dunning Hotopp JC, Hu FZ, Riley DR, Covacci A, Mitchell TJ, Bentley SD, Kilian M, Ehrlich GD, Rappuoli R, Moxon ER, Masignani V. 2010. Structure and dynamics of the pan-genome of Streptococcus pneumoniae and closely related species. Genome Biol 11:R107. doi: 10.1186/gb-2010-11-10-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnston C, Hinds J, Smith A, van der Linden M, Van Eldere J, Mitchell TJ. 2010. Detection of large numbers of pneumococcal virulence genes in streptococci of the mitis group. J Clin Microbiol 48:2762–2769. doi: 10.1128/JCM.01746-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richards VP, Palmer SR, Pavinski Bitar PD, Qin X, Weinstock GM, Highlander SK, Town CD, Burne RA, Stanhope MJ. 2014. Phylogenomics and the dynamic genome evolution of the genus Streptococcus. Genome Biol Evol 6:741–753. doi: 10.1093/gbe/evu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Denapaite D, Bruckner R, Nuhn M, Reichmann P, Henrich B, Maurer P, Schahle Y, Selbmann P, Zimmermann W, Wambutt R, Hakenbeck R. 2010. The genome of Streptococcus mitis B6—what is a commensal? PLoS One 5:e9426. doi: 10.1371/journal.pone.0009426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kilian M, Riley DR, Jensen A, Bruggemann H, Tettelin H. 2014. Parallel evolution of Streptococcus pneumoniae and Streptococcus mitis to pathogenic and mutualistic lifestyles. mBio 5:e01490–. doi: 10.1128/mBio.01490-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Denapaite D, Rieger M, Kondgen S, Bruckner R, Ochigava I, Kappeler P, Matz-Rensing K, Leendertz F, Hakenbeck R. 2016. Highly variable Streptococcus oralis strains are common among viridans streptococci isolated from primates. mSphere 1:e00041–. doi: 10.1128/mSphere.00041-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.García-de-la-Mària C, Pericas JM, Del Rio A, Castaneda X, Vila-Farres X, Armero Y, Espinal PA, Cervera C, Soy D, Falces C, Ninot S, Almela M, Mestres CA, Gatell JM, Vila J, Moreno A, Marco F, Miro JM, Hospital Clinic Experimental Endocarditis Study Group. 2013. Early in vitro and in vivo development of high-level daptomycin resistance is common in mitis group streptococci after exposure to daptomycin. Antimicrob Agents Chemother 57:2319–2325. doi: 10.1128/AAC.01921-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akins RL, Katz BD, Monahan C, Alexander D. 2015. Characterization of high-level daptomycin resistance in viridans group streptococci developed upon in vitro exposure to daptomycin. Antimicrob Agents Chemother 59:2102–2112. doi: 10.1128/AAC.04219-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akins RL, Barber KE, Palmer KL. 2016. Pronounced heterogeneity observed in high-level daptomycin-resistant viridans group streptococci. J Glob Antimicrob Resist 7:159–166. doi: 10.1016/j.jgar.2016.09.005. [DOI] [PubMed] [Google Scholar]
- 19.Akins RL, Rybak MJ. 2001. Bactericidal activities of two daptomycin regimens against clinical strains of glycopeptide intermediate-resistant Staphylococcus aureus, vancomycin-resistant Enterococcus faecium, and methicillin-resistant Staphylococcus aureus isolates in an in vitro pharmacodynamic model with simulated endocardial vegetations. Antimicrob Agents Chemother 45:454–459. doi: 10.1128/AAC.45.2.454-459.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sader HS, Jones RN. 2009. Antimicrobial susceptibility of Gram-positive bacteria isolated from US medical centers: results of the Daptomycin Surveillance Program (2007-2008). Diagn Microbiol Infect Dis 65:158–162. doi: 10.1016/j.diagmicrobio.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 21.Humphries RM, Pollett S, Sakoulas G. 2013. A current perspective on daptomycin for the clinical microbiologist. Clin Microbiol Rev 26:759–780. doi: 10.1128/CMR.00030-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tran TT, Munita JM, Arias CA. 2015. Mechanisms of drug resistance: daptomycin resistance. Ann N Y Acad Sci 1354:32–53. doi: 10.1111/nyas.12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kobayashi K, Ehrlich SD, Albertini A, Amati G, Andersen KK, Arnaud M, Asai K, Ashikaga S, Aymerich S, Bessieres P, Boland F, Brignell SC, Bron S, Bunai K, Chapuis J, Christiansen LC, Danchin A, Debarbouille M, Dervyn E, Deuerling E, Devine K, Devine SK, Dreesen O, Errington J, Fillinger S, Foster SJ, Fujita Y, Galizzi A, Gardan R, Eschevins C, Fukushima T, Haga K, Harwood CR, Hecker M, Hosoya D, Hullo MF, Kakeshita H, Karamata D, Kasahara Y, Kawamura F, Koga K, Koski P, Kuwana R, Imamura D, Ishimaru M, Ishikawa S, Ishio I, Le Coq D, Masson A, Mauël C, Meima R, Mellado RP, Moir A, Moriya S, Nagakawa E, Nanamiya H, Nakai S, Nygaard P, Ogura M, Ohanan T, O'Reilly M, O'Rourke M, Pragai Z, Pooley HM, Rapoport G, Rawlins JP, Rivas LA, Rivolta C, Sadaie A, Sadaie Y, Sarvas M, Sato T, Saxild HH, Scanlan E, Schumann W, Seegers JF, Sekiguchi J, Sekowska A, Séror SJ, Simon M, Stragier P, Studer R, Takamatsu H, Tanaka T, Takeuchi M, Thomaides HB, Vagner V, van Dijl JM, Watabe K, Wipat A, Yamamoto H, Yamamoto M, Yamamoto Y, Yamane K, Yata K, Yoshida K, Yoshikawa H, Zuber U, Ogasawara N. 2003. Essential Bacillus subtilis genes. Proc Natl Acad Sci U S A 100:4678–4683. doi: 10.1073/pnas.0730515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forsyth RA, Haselbeck RJ, Ohlsen KL, Yamamoto RT, Xu H, Trawick JD, Wall D, Wang L, Brown-Driver V, Froelich JM, Kedar GC, King P, McCarthy M, Malone C, Misiner B, Robbins D, Tan Z, Zhu ZY, Carr G, Mosca DA, Zamudio C, Foulkes JG, Zyskind JW. 2002. A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol Microbiol 43:1387–1400. doi: 10.1046/j.1365-2958.2002.02832.x. [DOI] [PubMed] [Google Scholar]
- 26.van Opijnen T, Bodi KL, Camilli A. 2009. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods 6:767–772. doi: 10.1038/nmeth.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Le Breton Y, Belew AT, Valdes KM, Islam E, Curry P, Tettelin H, Shirtliff ME, El-Sayed NM, McIver KS. 2015. Essential genes in the core genome of the human pathogen Streptococcus pyogenes. Sci Rep 5:9838. doi: 10.1038/srep09838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu P, Ge X, Chen L, Wang X, Dou Y, Xu JZ, Patel JR, Stone V, Trinh M, Evans K, Kitten T, Bonchev D, Buck GA. 2011. Genome-wide essential gene identification in Streptococcus sanguinis. Sci Rep 1:125. doi: 10.1038/srep00125,10.1038/srep00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hooven TA, Catomeris AJ, Akabas LH, Randis TM, Maskell DJ, Peters SE, Ott S, Santana-Cruz I, Tallon LJ, Tettelin H, Ratner AJ. 2016. The essential genome of Streptococcus agalactiae. BMC Genomics 17:406. doi: 10.1186/s12864-016-2741-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hutchison CA III, Chuang RY, Noskov VN, Assad-Garcia N, Deerinck TJ, Ellisman MH, Gill J, Kannan K, Karas BJ, Ma L, Pelletier JF, Qi ZQ, Richter RA, Strychalski EA, Sun L, Suzuki Y, Tsvetanova B, Wise KS, Smith HO, Glass JI, Merryman C, Gibson DG, Venter JC. 2016. Design and synthesis of a minimal bacterial genome. Science 351:aad6253. doi: 10.1126/science.aad6253. [DOI] [PubMed] [Google Scholar]
- 31.Liu X, Yin Y, Wu J, Liu Z. 2014. Structure and mechanism of an intramembrane liponucleotide synthetase central for phospholipid biosynthesis. Nat Commun 5:4244. doi: 10.1038/ncomms5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 33.Ganong BR, Leonard JM, Raetz CR. 1980. Phosphatidic acid accumulation in the membranes of Escherichia coli mutants defective in CDP-diglyceride synthetase. J Biol Chem 255:1623–1629. [PubMed] [Google Scholar]
- 34.Ganong BR, Raetz CR. 1982. Massive accumulation of phosphatidic acid in conditionally lethal CDP-diglyceride synthetase mutants and cytidine auxotrophs of Escherichia coli. J Biol Chem 257:389–394. [PubMed] [Google Scholar]
- 35.Sutterlin HA, Zhang S, Silhavy TJ. 2014. Accumulation of phosphatidic acid increases vancomycin resistance in Escherichia coli. J Bacteriol 196:3214–3220. doi: 10.1128/JB.01876-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geiger O, Lopez-Lara IM, Sohlenkamp C. 2013. Phosphatidylcholine biosynthesis and function in bacteria. Biochim Biophys Acta 1831:503–513. doi: 10.1016/j.bbalip.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 37.Kennedy EP, Weiss SB. 1956. The function of cytidine coenzymes in the biosynthesis of phospholipides. J Biol Chem 222:193–214. [PubMed] [Google Scholar]
- 38.Kent C, Gee P, Lee SY, Bian X, Fenno JC. 2004. A CDP-choline pathway for phosphatidylcholine biosynthesis in Treponema denticola. Mol Microbiol 51:471–481. doi: 10.1046/j.1365-2958.2003.03839.x. [DOI] [PubMed] [Google Scholar]
- 39.Vences-Guzman MA, Goetting-Minesky MP, Guan Z, Castillo-Ramirez S, Cordoba-Castro LA, Lopez-Lara IM, Geiger O, Sohlenkamp C, Fenno JC. 2016. 1,2-Diacylglycerol choline phosphotransferase catalyzes the final step in the unique Treponema denticola phosphatidylcholine biosynthesis pathway. Mol Microbiol doi: 10.1111/mmi.13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caspi R, Altman T, Billington R, Dreher K, Foerster H, Fulcher CA, Holland TA, Keseler IM, Kothari A, Kubo A, Krummenacker M, Latendresse M, Mueller LA, Ong Q, Paley S, Subhraveti P, Weaver DS, Weerasinghe D, Zhang P, Karp PD. 2014. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res 42:D459–D471. doi: 10.1093/nar/gkt1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cundell DR, Gerard NP, Gerard C, Idanpaan-Heikkila I, Tuomanen EI. 1995. Streptococcus pneumoniae anchor to activated human cells by the receptor for platelet-activating factor. Nature 377:435–438. doi: 10.1038/377435a0. [DOI] [PubMed] [Google Scholar]
- 42.Mishra NN, Bayer AS. 2013. Correlation of cell membrane lipid profiles with daptomycin resistance in methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 57:1082–1085. doi: 10.1128/AAC.02182-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hachmann AB, Sevim E, Gaballa A, Popham DL, Antelmann H, Helmann JD. 2011. Reduction in membrane phosphatidylglycerol content leads to daptomycin resistance in Bacillus subtilis. Antimicrob Agents Chemother 55:4326–4337. doi: 10.1128/AAC.01819-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muller A, Wenzel M, Strahl H, Grein F, Saaki TN, Kohl B, Siersma T, Bandow JE, Sahl HG, Schneider T, Hamoen LW. 2016. Daptomycin inhibits cell envelope synthesis by interfering with fluid membrane microdomains. Proc Natl Acad Sci U S A 113:E7077–E7086. doi: 10.1073/pnas.1611173113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clinical and Laboratory Standards Institute. 2015. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically: approved standard, 10th ed CLSI document M07-A10. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 46.Clinical and Laboratory Standards Institute. 2016. Performance standards for antimicrobial susceptibility testing, 26th ed CLSI supplement M100S. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 47.Adams HM, Li X, Mascio C, Chesnel L, Palmer KL. 2015. Mutations associated with reduced surotomycin susceptibility in Clostridium difficile and Enterococcus species. Antimicrob Agents Chemother 59:4139–4147. doi: 10.1128/AAC.00526-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, Vonstein V, Wattam AR, Xia F, Stevens R. 2014. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res 42:D206–D214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Galloway-Peña J, Sahasrabhojane P, Tarrand J, Han XY, Shelburne SA. 2014. GyrB polymorphisms accurately assign invasive viridans group streptococcal species. J Clin Microbiol 52:2905–2912. doi: 10.1128/JCM.01068-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 51.Tan BK, Bogdanov M, Zhao J, Dowhan W, Raetz CR, Guan Z. 2012. Discovery of a cardiolipin synthase utilizing phosphatidylethanolamine and phosphatidylglycerol as substrates. Proc Natl Acad Sci U S A 109:16504–16509. doi: 10.1073/pnas.1212797109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li C, Tan BK, Zhao J, Guan Z. 2016. In vivo and in vitro synthesis of phosphatidylglycerol by an Escherichia coli cardiolipin synthase. J Biol Chem 291:25144–25153. doi: 10.1074/jbc.M116.762070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reichmann P, Nuhn M, Denapaite D, Bruckner R, Henrich B, Maurer P, Rieger M, Klages S, Reinhard R, Hakenbeck R. 2011. Genome of Streptococcus oralis strain Uo5. J Bacteriol 193:2888–2889. doi: 10.1128/JB.00321-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kilian M, Poulsen K, Blomqvist T, Havarstein LS, Bek-Thomsen M, Tettelin H, Sorensen UB. 2008. Evolution of Streptococcus pneumoniae and its close commensal relatives. PLoS One 3:e2683. doi: 10.1371/journal.pone.0002683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sabharwal A, Liao YC, Lin HH, Haase EM, Scannapieco FA. 2015. Draft genome sequences of 18 oral Streptococcus strains that encode amylase-binding proteins. Genome Announc 3:e00510–. doi: 10.1128/genomeA.00510-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.