

ABSTRACT
Expanding echinocandin use to prevent or treat invasive fungal infections has led to an increase in the number of breakthrough infections due to resistant Candida species. Although it is uncommon, echinocandin resistance is well documented for Candida albicans, which is among the most prevalent bloodstream organisms. A better understanding is needed to assess the cellular factors that promote tolerance and predispose infecting cells to clinical breakthrough. We previously showed that some mutants that were adapted to growth in the presence of toxic sorbose due to loss of one chromosome 5 (Ch5) also became more tolerant to caspofungin. We found here, following direct selection of mutants on caspofungin, that tolerance can be conferred by at least three mechanisms: (i) monosomy of Ch5, (ii) combined monosomy of the left arm and trisomy of the right arm of Ch5, and (iii) an aneuploidy-independent mechanism. Tolerant mutants possessed cell walls with elevated chitin and showed downregulation of genes involved in cell wall biosynthesis, namely, FKS, located outside Ch5, and CHT2, located on Ch5, irrespective of Ch5 ploidy. Also irrespective of Ch5 ploidy, the CNB1 and MID1 genes on Ch5, which are involved in the calcineurin signaling pathway, were expressed at the diploid level. Thus, multiple mechanisms can affect the relative expression of the aforementioned genes, controlling them in similar ways. Although breakthrough mutations in two specific regions of FKS1 have previously been associated with caspofungin resistance, we found mechanisms of caspofungin tolerance that are independent of FKS1 and thus represent an earlier event in resistance development.
KEYWORDS: Candida albicans, chromosome 5, molecular mechanisms, caspofungin tolerance
INTRODUCTION
The fungus Candida albicans is part of the normal mycobiota of mucosal surfaces in the human gut and genitals. However, in severely immunocompromised individuals, C. albicans can become an invading pathogen that can cause various degrees of infection, including systemic candidiasis leading to mortality. The echinocandin class drugs are now recommended as first-line therapy for candidiasis because of their low toxicity and high efficacy, especially against Candida isolates resistant to azole drugs (1). They act by interfering with the biosynthesis of the cell wall by inhibiting 1,3-β-d-glucan synthase.
Caspofungin was introduced in 2001 as the first member of the echinocandin drugs. Since that time, clinical resistance of C. albicans to caspofungin has remained low despite increasing reports of cases of resistance. For example, a recent surveillance study revealed an increase in breakthrough infections by resistant strains, from 0.5% in 2001 to 3.1% (6-fold) in 2009 (2), and additional studies also reported low-level resistance among C. albicans strains (3–6). However, C. glabrata showed higher levels of breakthrough infections in some clinical settings (6–9). As echinocandin usage continues to broaden for therapy and prophylaxis, there is a need to better understand the cellular processes that promote resistance.
The only known mechanism of C. albicans clinical resistance to caspofungin and other echinocandins is the occurrence of point mutations in the FKS1 (GSC1; orf19.2929) gene on chromosome 1 (Ch1), which encodes a catalytic subunit of the drug target 1,3-β-d-glucan synthase (10). The FKS1 mutations, clustered in two hot spot regions, HS1 and HS2, reduce glucan synthase sensitivity to echinocandins, elevate MICs, and confer reduced pharmacodynamic responses (reviewed in references 3, 11, and 12). Mutations in FKS1 are also known to cause expression changes of FKS1 relative to that of FKS2 (GSL2; orf19.3269) and FKS3 (GSL1; orf19.2495) (13), as well as enhanced expression of chitin genes, leading to elevated amounts of chitin in the cell wall in some resistant clinical isolates (5, 14).
Clinical isolates may show a range of MIC values that are below the clinical breakpoint without the presence of an FKS mutation (15–17). Many of these strains are conditionally drug adapted or tolerant of drug. However, the molecular bases of the tolerance mechanism(s) are not fully resolved.
There is a large body of evidence relating caspofungin tolerance in laboratory strains to the cell wall salvage mechanism. This is a reversible mechanism that involves an increase in chitin synthesis that compensates for a diminished amount of cell wall 1,3-β-d-glucan when glucan synthase is inhibited by caspofungin. The increased chitin level serves to fortify the cell wall, which would otherwise be weakened by the reduced glucan content (14). For example, when C. albicans cells were treated with calcium and calcofluor white to induce elevated cell wall chitin levels and then injected into mice, these cells exhibited reduced susceptibility to echinocandins compared to that of cells with normal levels of chitin (18). In another study involving the paradoxical effect, one C. albicans isolate increased its chitin content ∼900% and decreased its 1,3-β-d-glucan and 1,6-β-d-glucan contents 81% and 73%, respectively (19). In other studies, laboratory mutants were used to demonstrate that susceptibility to caspofungin decreases due to increases in cell wall chitin (20, 21). The cell wall salvage mechanism is activated in clinical isolates with increased caspofungin MICs as determined with a broth microdilution method (10, 14). Despite the fact that overexpression of chitin has not been observed in resistant clinical strains (14), it seems to promote tolerance to drug that can ultimately lead to clinical resistance in the form of a stable FKS1 mutant.
Using our C. albicans model system, we recently discovered that the Ch5 copy number can control cell wall remodeling and susceptibility to caspofungin (22). We analyzed a large number of mutants that arose independently due to the loss of one Ch5 on medium containing the toxic sugar l-sorbose, which kills C. albicans in a manner similar to that for echinocandins (reviewed in reference 22). All Ch5 monosomic mutants exhibited significant increases in cell wall chitin, and all analyzed mutants exhibited significant decreases in 1,3-β-d-glucan. In contrast, strains with a duplicated Ch5 and control strains had levels of chitin or glucan similar to the levels in the parental strains (22). Many of these mutants also acquired tolerance to caspofungin, which we now explain as being due to a diminished gene dose of at least three Ch5-carried genes, PGA4, CHT2, and CSU51, which encode negative regulators of caspofungin susceptibility (23).
In the present study, we generated caspofungin-tolerant mutants by direct selection on medium supplemented with caspofungin and found several distinctive mechanisms of tolerance, including (i) Ch5 monosomy similar to that of the sorbose-generated mutants (22), (ii) a combination of one normal Ch5 and one iso-Ch5 with two right arms (iso-Ch5R), and (iii) an aneuploidy-independent mechanism.
We used representative mutants of different kinds to further demonstrate, in accordance with our earlier data, that mechanisms of caspofungin tolerance are coupled with cell wall remodeling. In these mutants, we also showed changes of expression of all three FKS genes that are found in the C. albicans genome. We also showed changes of expression of the following three genes residing on the right arm of Ch5: CHT2 (orf19.3895), which is implicated in cell remodeling, and CNB1 (orf19.4009) and MID1 (orf19.3212), which belong to the calcineurin stress response signaling pathway.
RESULTS
Generation of caspofungin-adapted mutants by direct selection on caspofungin.
Exposure in vitro to lethal doses of caspofungin causes the vast majority of C. albicans cells to die. However, some cells survive and form colonies on plates (Fig. 1). We used two parental strains, JRCT1 and SC5314, to generate caspofungin-adapted mutants that arose with frequencies of 0.01% and 0.04%, respectively, following direct selection on medium supplemented with caspofungin, as exemplified in Fig. 1 and described in Materials and Methods.
FIG 1.

Generation of caspofungin-tolerant mutants. (Left) Confluent growth was observed after plating cells of the parental strain on control SD medium with no caspofungin. (Right) Caspofungin-tolerant colonies arose after plating of the parental cells on SD medium supplemented with caspofungin.
Briefly, cells of strain JRCT1 were plated on synthetic dextrose (SD) plates supplemented with three different concentrations of caspofungin (120 ng/ml, 160 ng/ml, and 200 ng/ml) and then incubated at 37°C. When colonies appeared, a total of 54 colonies were randomly picked up. When those mutants were streaked on yeast extract-peptone-dextrose (YPD) plates (i.e., in the absence of selection), 42 of the mutants grew as uniform, relatively small colonies, but 12 of the mutants gave rise to a small percentage of large colonies versus the majority of smaller colonies. This was reminiscent of sorbose-generated Ch5 monosomic mutants that acquired a spontaneously duplicated Ch5 in the absence of selection and subsequently grew on plates as large colonies (22, 24). All 12 mutants producing large colonies, as well as 6 representative mutants not producing large colonies, were further characterized (see below).
Another collection of 15 mutants that arose from SC5314 were obtained on YPD medium supplemented with 60 ng/ml of caspofungin, and 3 of the 15 mutants produced large colonies versus the majority of small colonies when streaked on YPD medium. All 15 mutants were further characterized (see below).
Chromosomal condition of caspofungin-adapted mutants.
We set about analyzing the copy number of Ch5 in our mutants because in our previous study many sorbose-generated mutants that became resistant to toxic sorbose due to loss of one Ch5 also showed decreased susceptibility to caspofungin (22). Also, the appearance of large colonies (see above) was reminiscent of reduplication of the monosomic Ch5. We started characterization of the mutants by screening with a PCR method using primers for the MTL locus residing on Ch5, as in previous work by our group (22). We tested 18 mutants derived from JRCT1 and 15 mutants derived from SC5314 (see above for more details). We found that the majority of mutants producing large colonies were hemizygous for the MTL locus, carrying either MTLa or MTLα (10 of 12 JRCT1 mutants and 2 of 3 SC5314 mutants), thus implying the loss of one Ch5. The remaining 20 mutants from both genetic backgrounds showed normal heterozygosity (MTLa/MTLα). (See Fig. 2 for PCR results for all 33 mutants.)
FIG 2.

Products of PCR amplification obtained with total DNA of each strain and primers specific for the MTLa or MTLα locus. Data are presented for the parental strains JRCT1 and SC5314 and their mutants, as indicated. The approximately 342-bp band corresponds to MTLa, and the approximately 409-bp band corresponds to MTLα.
We next used a pulsed-field gel electrophoresis (PFGE) method to characterize the chromosomes of 13 JRCT1 mutants (10 MTL locus hemizygous and 3 MTL locus heterozygous) as well as 3 SC5314 mutants (2 MTL hemizygous and 1 MTL heterozygous). We found no ploidy change or gross chromosome rearrangements in the mutants with normal heterozygous MTL loci (see Fig. S2E in the supplemental material), but we found two classes of chromosome changes in the mutants with a hemizygous MTL locus. Approximately half of the latter mutants lost one Ch5 but kept the other chromosomes intact (see representative mutant SMC60-2-5 in Fig. 3A, left panel). Unexpectedly, the other half carried one normal Ch5 and also showed an unknown band with a size intermediate between those of Ch5 and Ch4 instead of the band for another normal Ch5 (see representative mutant JMC120-2-5 in Fig. 3A, right panel). (For data on more mutants with a hemizygous MTL locus that were analyzed by PFGE, see Fig. S2A to D.)
FIG 3.
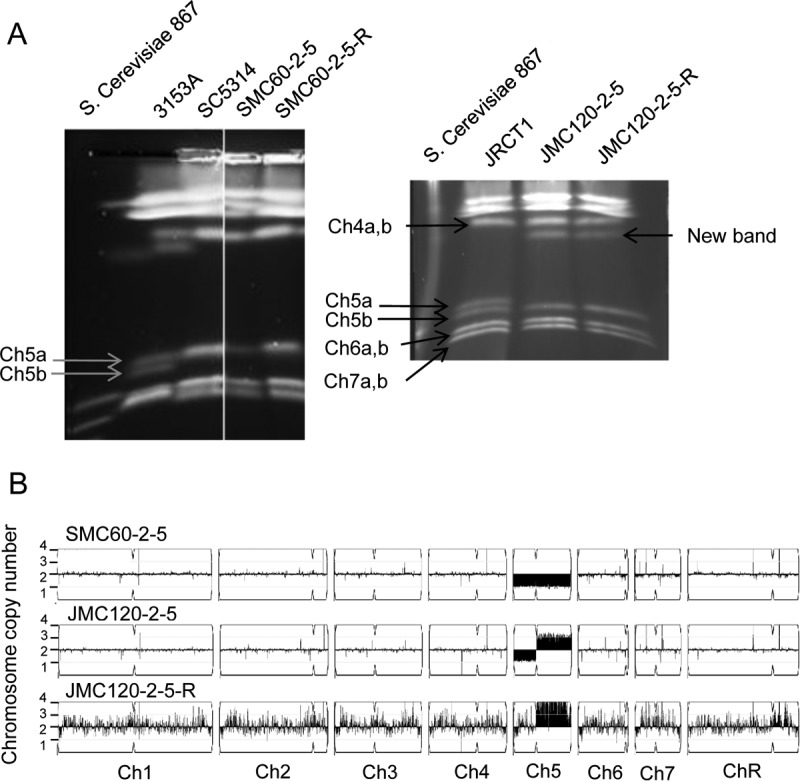
Chromosome patterns of two representative mutants, SMC60-2-5 (with Ch5 monosomy) and JMC120-2-5 (with one Ch5 converted into iso-Ch5R), as well as the derivative strain JMC120-2-5-R (with a duplicated Ch5). (A) Electrophoretic karyotyping of strains by the PFGE variation orthogonal field alternating gel electrophoresis (OFAGE). Strains are indicated, and Ch5 is indicated with arrows. Note that Ch5 is represented by a single band for SC5314 and its derivatives and by two bands for JRCT1 and its derivatives. Also shown are data for the C. albicans reference strain 3153A, in which Ch5 is represented by two bands, and for Saccharomyces cerevisiae reference strain 867, for the markers of Candida chromosomes. Note that Ch5 exhibited weaker intensity in the mutant SMC60-2-5, as expected for a single chromosome. Also note that OFAGE separations present precisely separated Ch7, Ch6, Ch5, and Ch4, whereas the remaining, longer chromosomes are compressed at the top of the gel. (B) Graphic presentation by the YMAP pipeline (41) of the whole-genome sequencing data for the above strains (see Materials and Methods for more explanations). The names of the strains are indicated above the panels, and the chromosome copy number is indicated on the y axis. The x axis indicates the positions of the reads on each chromosome for the reference strain SC5314, as annotated in genomic assembly 22 in CGD. Chromosomes are designated Ch1 to Ch7 and ChR. ChR refers to the chromosome containing a cluster of tandemly repeated ribosomal DNA (rDNA) units (42).
To better characterize the DNA changes in the mutants, we next used DNA sequencing (DNA-seq) to analyze 3 Ch5 monosomic mutants, 3 mutants showing an unknown band in addition to the normal Ch5 band, and 2 mutants with no ploidy change relative to the reference parental strain JRCT1 or SC5314. We confirmed, as expected from the PFGE results, the Ch5 monosomy change in the mutants SMC60-2-5, JMC200-3-3, and JMC200-3-4 (see Fig. 3B for the representative mutant SMC60-2-5 and Fig. S3 for more mutants). All three mutants showing the unknown band, i.e., JMC160-3-5, JMC120-2-5, and JMC200-3-3, contained one Ch5 left arm and three Ch5 right arms (see Fig. 3B for the representative mutant JMC120-2-5 and Fig. S3 for other mutants). By combining analyses of MTL locus hemizygosity, which implied the loss of one Ch5, PFGE analyses of the chromosome banding patterns, which showed an unknown band migrating between Ch5 (∼1,295 kbp) and Ch4 (∼1,883 kbp) (25), and DNA-seq analyses, we concluded that the unknown band was a chromosome carrying two right arms of Ch5 (each of ∼825.6 kbp), thus resulting in an iso-Ch5R with a total length of ∼1,651 kbp. Because the MTL locus resides on the left arm of Ch5, iso-Ch5R has no MTL locus, and the corresponding mutants are hemizygous for the MTL locus. Finally, no ploidy change or chromosome rearrangement(s) was found in mutants JMC160-2-5 and JMC200-2-5, which remained normal diploids heterozygous for the MTL locus according to PCR or PFGE analyses (data not shown).
Generation of sequential derivatives with alternating Ch5 ploidy.
To study the effects of the copy number of Ch5 on caspofungin susceptibility and other phenotypes, we created 9 series of strains, with each series containing 3 sequential strains in which Ch5 alternated between disomy and aneuploidy: parental Ch5 disomy → Ch5 aneuploidy → Ch5 duplication. We used 7 JRCT1 mutants and 2 SC5314 mutants that had become hemizygous for the MTL locus either due to Ch5 monosomy or due to the combination of Ch5 monosomy and iso-Ch5R and that also produced large colonies in the absence of selection (see “Chromosomal condition of caspofungin-adapted mutants”). PFGE demonstrated that the large colonies were derivatives with a spontaneously duplicated normal Ch5 in cells with iso-Ch5R. In derivatives with duplicated Ch5, other chromosomes, including iso-Ch5R, remained unchanged (Fig. 3; Fig. S2A and D). We previously used the same approach to generate derivatives with spontaneously duplicated Ch5 in sorbose-generated mutants carrying monosomic Ch5 (22, 24).
We also used DNA-seq to confirm the duplication of Ch5 and no other change in representative derivatives with iso-Ch5R, i.e., JMC200-3-3-R, JMC160-3-5-R, and JMC120-2-5-R (see Fig. 3B for the representative mutant JMC120-2-5-R and Fig. S3 for the other strains).
Caspofungin tolerance in caspofungin-adapted mutants.
To characterize caspofungin susceptibility in our mutants, we first used an agar-based spot assay method (see Materials and Methods) to test for the comparative growth of various mutants versus parents on medium supplemented with caspofungin. We conducted pilot experiments in order to find optimal caspofungin concentrations that revealed growth differences based on the differences in numbers of growing spots between the parental strain and mutants. For example, no growth of parental JRCT1 versus its caspofungin-tolerant mutants that remained normal diploids or contained iso-Ch5R in any of the four spots at a caspofungin concentration of 70 ng/ml or no growth of parental SC5314 versus its Ch5 monosomic mutant at a caspofungin concentration of 54 ng/ml (Fig. 4) was considered suppressed growth of the parental strain compared to that of the mutants.
FIG 4.
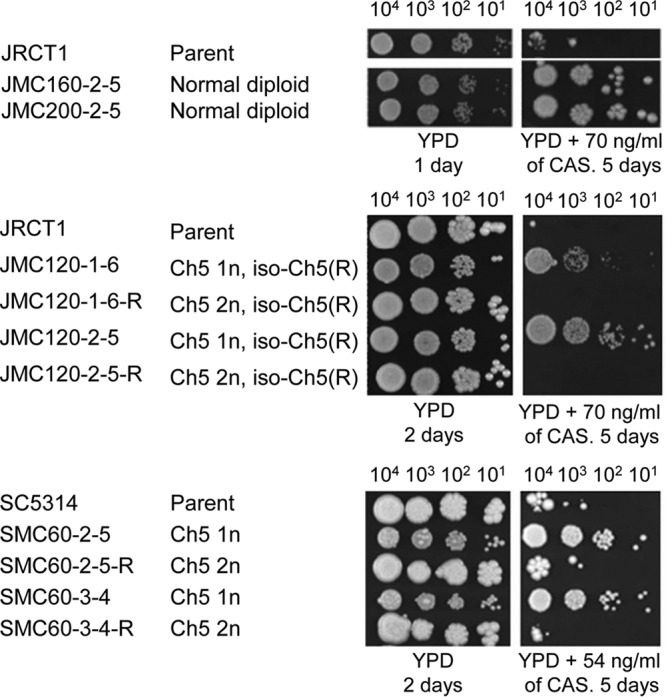
Susceptibility of mutants to caspofungin (CAS) compared to that of parental strains and strains with duplicated Ch5. The results shown are for spot assays of growth on control YPD medium as well as YPD medium supplemented with caspofungin, as indicated. The time of incubation at 37°C is indicated. Strains are indicated on the left. Names of the derivatives are shown under the names of the corresponding parental strain (JRCT1 or SC5314). The monosomic (1n), duplicated (2n), or iso-Ch5R condition of Ch5 is indicated. Note that Ch5 monosomic mutants demonstrated less growth than related disomic strains on YPD control medium. However, this pattern was reversed in the presence of caspofungin.
We found that every mutant with either a single Ch5 or one Ch5 and one iso-Ch5R showed better growth than that of its parental strain or its derivative with duplicated Ch5; the growth of the latter usually matched the growth of the parental strain. Similarly, all tolerant mutants that remained normal diploids grew better than the parental strains (see data for representative mutants in Fig. 4 and for more mutants of all kinds in Fig. S4).
Strains from 3 representative series as well as 3 mutants that remained normal diploids were further validated with a standard broth microdilution method (see Materials and Methods). We found a good correspondence between the two methods, as expected (22, 23, 26), as all caspofungin-tolerant mutants exhibited better growth than that of the strains with parental disomic or duplicated Ch5 (see data for one representative series in Fig. 5 and for all 3 series in Fig. S5). These experiments established a close correlation between the reversible caspofungin tolerance phenotype and reversible rearrangements of Ch5 monosomy, as well as reduced caspofungin susceptibility in the tolerant mutants with no aneuploidy.
FIG 5.

Growth in the presence of the indicated concentrations of caspofungin of the representative caspofungin-tolerant mutant JMC200-2-5, which is a normal diploid (○), or the caspofungin-tolerant mutant JMC200-3-4, which is monosomic for Ch5 (×), versus that of the parental strain JRCT1 (■) or the derivative JMC200-3-4-R, with a duplicated Ch5 (▲). Note that the amount of drug that completely inhibited growth of the tolerant mutants, either 2,000 ng/ml or 8,000 ng/ml, was higher than the amount of drug, 1,000 ng/ml, that completely inhibited the growth of the parental strain or the strain with duplicated Ch5. This is consistent with the better growth of the tolerant mutants on agar-based plates shown in Fig. 4.
Chitin and 1,3-β-glucan levels in the cell walls of caspofungin-tolerant mutants.
Guided by our previous data obtained with the sorbose resistance model, we investigated whether caspofungin-generated mutants possess remodeled cell walls similar to those of sorbose-generated mutants monosomic for Ch5 (22). For this purpose, we determined the chitin levels in strains from 9 representative sequential series (parental Ch5 disomy → Ch5 aneuploidy → Ch5 duplication) as well as in 3 caspofungin-tolerant mutants that remained normal diploids. We also determined the 1,3-β-d-glucan levels in strains from 1 representative series as well as in a representative mutant that remained diploid. We found that both types of caspofungin-tolerant mutants, those with aneuploid Ch5 and those with no aneuploidy, but not the revertant derivatives with reduplicated Ch5, contained increased levels of chitin (Fig. 6) and slight but statistically significant decreases in 1,3-β-d-glucan content (Fig. 7). The differences in chitin content between parental JRCT1 or SC5314 and its derivatives and in 1,3-β-d-glucan content between parental JRCT1 and its derivatives were evaluated by use of Student's t test, and P values of the tolerant mutants were <0.05. These results established that cell wall remodeling accompanies the increase of tolerance either due to ploidy changes of Ch5 or due to other, still unknown mechanisms.
FIG 6.
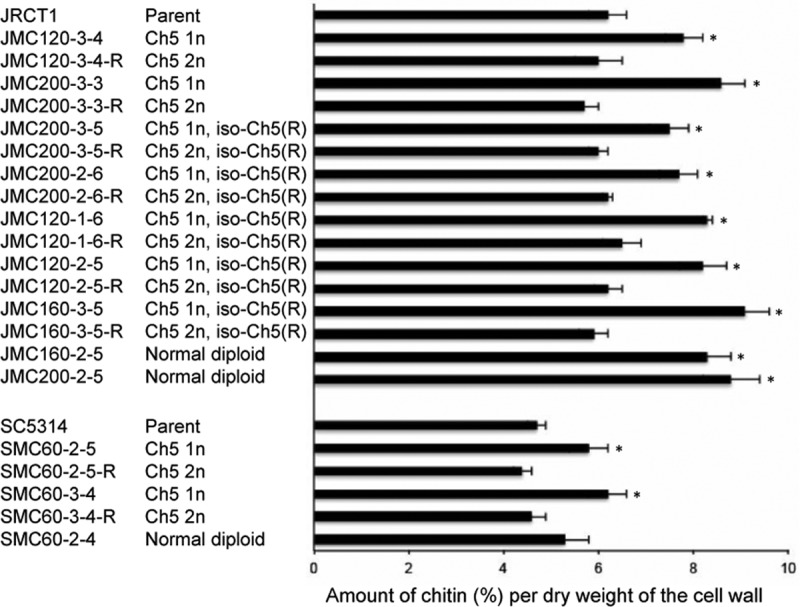
Comparative levels of cell wall chitin in various strains. The data shown are for 9 representative sets of sequential derivatives, i.e., parental disomic Ch5 → aneuploid Ch5 → duplicated Ch5 (marked with “R”), as well as 3 representative mutants that kept a normal diploid genome. In some series, one Ch5 was combined with one iso-Ch5R, as indicated on the left. The Ch5 condition is also indicated on the left (2n or 1n for two or one Ch5 copy, respectively). Strains are indicated on the left. The test was conducted in triplicate. Error bars are shown. The differences between parental strains and their derivatives were evaluated by use of Student's t test. Derivatives showing statistically significant differences, with P values of <0.05, are marked with asterisks.
FIG 7.
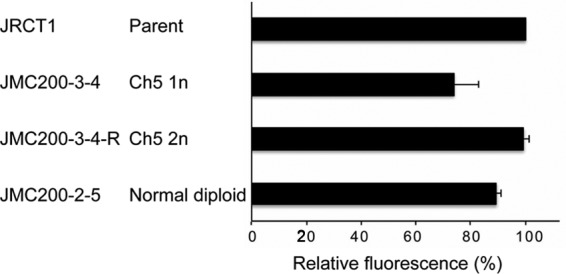
Comparative levels of cell wall 1,3-β-glucan in representative caspofungin-tolerant mutants versus the parental strain or the derivative with duplicated Ch5. The differences between parental JRCT1 and its derivatives were evaluated by use of Student's t test, and P values for JMC200-3-4 and JMC200-2-5 were <0.05. For more explanations, see the legend to Fig. 6.
Expression changes in FKS genes, residing outside Ch5, as well as in the CHT2, CNB1, and MID1 genes, residing on Ch5.
We addressed the question of whether expression of the clinically important FKS1 gene and its two paralogous genes, FKS2 and FKS3, changed in our mutants. Using a semiquantitative reverse transcription-PCR (RT-PCR) method, we found that FKS genes were downregulated over the range of 0.1- to 0.7-fold (Table 1). FKS3 was downregulated in all three kinds of mutants, while FKS1 was downregulated in mutants with iso-Ch5R or in mutants with no ploidy change. FKS2 was downregulated in only two instances. Downregulation of FKS genes is consistent with the decreased amount of 1,3-β-d-glucan content in the cell wall.
TABLE 1.
Expression changes of FKS1, FKS2, FKS3, CHT2, CNB1, and MID1 in mutants carrying monosomic Ch5, duplicated Ch5, iso-Ch5R, or normal disomic Ch5a
| Gene | Chromosome | Expression change |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Monosomic Ch5 |
Duplicated Ch5 |
Iso-Ch5R |
No ploidy change |
||||||
| JMC200-3-3/JRCT1 | SMC60-2-5/SC5314 | JMC200-3-3R/JRCT1 | SMC60-2-5R/SC5314 | JMC120-2-5/JRCT1 | JMC160-3-5/JRCT1 | JMC160-2-5/JRCT1 | JMC200-2-5/JRCT1 | ||
| FKS1 | 1 | 0.8 ± 0.07 | 0.9 ± 0.12 | 0.7 ± 0.06 | 0.9 ± 0.09 | 0.4 ± 0.12 | 0.3 ± 0.11 | 0.6 ± 0.09 | 0.3 ± 0.10 |
| FKS2 | R | 0.7 ± 0.04 | 1.0 ± 0.02 | 1.1 ± 0.05 | 0.8 ± 0.35 | 0.9 ± 0.01 | 0.6 ± 0.01 | 1.2 ± 0.01 | 0.9 ± 0.07 |
| FKS3 | 1 | 0.5 ± 0.07 | 0.7 ± 0.07 | 0.7 ± 0.16 | 0.8 ± 0.08 | 0.4 ± 0.03 | 0.1 ± 0.01 | 0.2 ± 0.08 | 0.7 ± 0.06 |
| CHT2 | 5 | 0.6 ± 0.08 | 0.1 ± 0.03 | 1.45 ± 0.15 | 1.0 ± 0.15 | 0.4 ± 0.27 | 0.1 ± 0.01 | 0.5 ± 0.06 | 0.6 ± 0.19 |
| CNB1 | 5 | 1.0 ± 0.05 | 1.0 ± 0.12 | 1.1 ± 0.05 | 1.4 ± 0.12 | 1.1 ± 0.06 | 1.1 ± 0.09 | 1.1 ± 0.08 | 1.2 ± 0.09 |
| MID1 | 5 | 1.2 ± 0.09 | 1.0 ± 0.08 | 0.8 ± 0.06 | 1.1 ± 0.08 | 1.1 ± 0.09 | 1.2 ± 0.04 | 1.1 ± 0.06 | 1.0 ± 0.04 |
As determined by semi-quantitative RT-PCR analysis of three independent RNA preparations. The expression change for each gene was determined as the mutant/parent expression ratio, using two series of sequential derivatives: (i) JRCT1 parent strain → JMC200-3-3 (monosomic Ch5) → JMC200-3-3R (duplicated Ch5) and (ii) SC5314 parent strain → SMC60-2-5 (monosomic Ch5) → SMC60-2-5R (duplicated Ch5). Two representative mutants with iso-Ch5R and two representative mutants with no ploidy change were also used, as indicated. The FKS and CHT2 genes were measured using REX2 as a control. CNB1 and MID1 were measured using VPH1 as a control. Significant expression changes are shown in bold. Note that for the single or three copies of a gene on monosomic Ch5 or on Ch5 in cells with iso-Ch5R, respectively, an expression ratio of around 1.0 indicates normalization to the disomic level. Note that the data for CHT2 expression in mutants JMC200-3-3 and SMC60-2-5, with monosomic Ch5, and in derivatives JMC200-3-3R and SMC60-2-5R, with duplicated Ch5, were adopted from reference 23.
Among the genes residing on Ch5 (Fig. 8), we determined the expression of the representative gene CHT2, encoding a factor for chitin hydrolysis. We previously demonstrated downregulation of CHT2 on the monosomic Ch5 generated by exposure to sorbose (22) or to caspofungin (23). Apparently, CHT2 is downregulated in all kinds of mutants (Table 1).
FIG 8.

Map of Ch5 showing genes residing on the Ch5 right arm that encode negative (blue) or positive (black) regulators of caspofungin susceptibility, as well as two genes on the left arm that were previously shown to be involved in fluconazole resistance.
We also determined the expression of two other Ch5 residents that are part of the calcineurin stress signaling pathway: CNB1, encoding a regulatory subunit of calcineurin B, and MID1, encoding a putative stretch-activated Ca2+ channel of the high-affinity calcium uptake system (27). The measurements were done with two different genes for internal control (REX2 and VPH1). We found that expression of these genes was close to the diploid level in cells carrying either monosomic Ch5, with one copy of each gene, or iso-Ch5R, with three copies of each gene (Fig. 8), independent of the control gene used. See Table 1 for the data generated using VPH1 and Table S2 for the data generated using REX2.
Analysis of HS regions of FKS genes.
We next addressed the question of whether clinically relevant mutations in the HS1 and HS2 regions of FKS1 are implicated in the in vitro resistance. We analyzed HS regions in all three biological repeats of DNA-seq data for all mutants, either carrying the rearranged Ch5 or remaining normal diploids. The sequence of the HS1 or HS2 region in the mutants was compared to the corresponding sequence in the parental strain, JRCT1 or SC5314. We also analyzed HS regions of FKS2 and FKS3. We found no mutations in HS regions of FKS genes, similar to the results for previously reported mutants that became resistant to sorbose by losing one Ch5 (22).
DISCUSSION
In this work, we demonstrate that caspofungin tolerance in C. albicans mutants that arose independently by adaptation to caspofungin is controlled by at least three different mechanisms that are independent of FKS1 mutations. A clear distinction between these mechanisms appears to be whether there is or is not a rearrangement of Ch5. The former case includes either a loss of one Ch5, resulting in Ch5 monosomy, or a combination of one normal Ch5 and one iso-Ch5R. The third class of mutants includes mutants that remain normal diploids. These mechanisms are still being elucidated, their final number is still being established, and a full understanding of the genes involved in the already elucidated mechanisms is in its early stages.
Decreased caspofungin susceptibility by the loss of one copy of Ch5 can be explained by a diminished gene dose of at least three Ch5 genes encoding negative regulators of caspofungin susceptibility that we recently found on the right arm of Ch5 (Fig. 8) (23): CHT2, encoding a glycosylphosphatidylinositol (GPI)-dependent chitinase which is a covalently bound cell wall protein; PGA4 (orf19.4035), encoding a GPI-anchored cell surface 1,3-β-d-glucanosyltransferase; and CSU51 (orf19.1105.2), encoding another putative GPI-anchored protein. The final number of Ch5 genes for negative regulation of caspofungin susceptibility has yet to be determined (23).
We previously showed that CHT2, PGA4, and CSU51 are downregulated on the monosomic Ch5 but not on the spontaneously reduplicated Ch5 (23) (see Table 1 for CHT2 data). Here we show that a similar significant downregulation of the representative CHT2 gene involved in cell wall biosynthesis occurs on three right arms in mutants with iso-Ch5R or on two right arms in normal diploid mutants. Hence, CHT2 downregulation in these mutants cannot be explained by a decreased gene dose as for monosomic Ch5. Other examples of gene regulation due to different mechanisms are expression of MID1 and CNB1 at approximately the disomic level on either monosomic or trisomic right arms of Ch5. Presumably, in mutants with the monosomic Ch5, a single copy of either MID1 or CNB1 is upregulated to the disomic level, whereas in mutants carrying iso-Ch5R, three copies of each of these genes are downregulated to the disomic level. We previously reported transcriptional dosage compensation on the monosomic Ch5 (28). Transcriptional dosage compensation on a trisomic chromosome will be reported elsewhere (E. Rustchenko, unpublished data). It is important that MID1 and CNB1, which are involved in calcineurin signaling, encode positive regulators of caspofungin susceptibility (Candida Genome Database [http://www.candidagenome.org/]).
Thus, the regulation of genes relevant to caspofungin susceptibility may or may not be based on gene copy number. We previously reported regulation of the same phenotype in C. albicans by two different mechanisms, one of which is associated with Ch5 rearrangement and the other with gene dose. The SOU1 gene, carried on the trisomic Ch4/7b in the mutant Sor125(55), which also possesses the monosomic Ch5, can be upregulated either due to an additional copy or due to the monosomic condition of Ch5 (25).
It appears that the tolerance phenotype can depend on the ratio between Ch5 genes for negative and positive regulation. We suggest that the loss of one Ch5 shifts the ratio such that a diminished dose of genes for negative regulation determines tolerance to caspofungin. On the other hand, the formation of iso-Ch5R may involve negative (monosomy of the left arm) and positive (trisomy of the right arm) regulation. Importantly for this model, duplication of a normal Ch5 in mutants with iso-Ch5R changes the total cellular complement from a ratio of 3 right arms to 1 left arm to a ratio of 4 right arms to 2 left arms, which abrogates tolerance, indicating the importance of gene ratios. It is known that duplication of the left arm of Ch5, resulting in formation of iso-Ch5L, leads to the acquisition of fluconazole resistance in clinical and laboratory strains. This is because of the increasing copy number of the two genes implicated in fluconazole resistance, namely, the fluconazole target gene ERG11, whose increased expression confers increased resistance to fluconazole, and the gene TAC1, encoding a transcription factor which is an activator of the CDR1 and CDR2 genes residing on Ch3, which encode multidrug transporters of the ABC superfamily for the drug efflux arm (Fig. 8) (29). Importantly, numerous rearrangements of Ch5, including trisomy of the entire Ch5 or large portions of Ch5 as well as duplication of the left arm, were recently found in clinical isolates (30–32).
The commonalities between all three classes of mutants include downregulation of FKS genes that reside outside Ch5 and control the synthesis of cell wall 1,3-β-d-glucan. Specifically, FKS3 is downregulated in all kinds of mutants, FKS1 is downregulated in mutants carrying iso-Ch5R and in normal diploids, and FKS2 is downregulated in two instances. The roles of FKS2 and FKS3 in cell wall stress require further investigation. Consistent with downregulation of FKS genes, we detected a decrease of 1,3-β-d-glucan in the cell walls of our representative mutants.
Another common feature of our caspofungin-tolerant mutants is an increase of cell wall chitin in accordance with the cell wall salvage concept (14). Overall, this result is consistent with earlier reports that treatment with caspofungin stimulates expression of chitin synthases or that inducing larger amounts of cell wall chitin confers caspofungin resistance in animal models (18, 20, 33). We believe that multiple genes are regulated in concert in order to increase the amount of chitin. This regulation may include, for example, the downregulation of CHT2, PGA4, and CSU51 on the monosomic Ch5 that we recently found in caspofungin-generated mutants (23) (Table 1), as well as upregulation of CHS2 and CHS3 for the chitin synthases encoded outside Ch5 (34).
In summary, Ch5 carries genes that encode negative and positive regulators of caspofungin susceptibility, whose final number still needs to be determined. This work strengthens our earlier observation that reversible monosomy of Ch5 confers reversible caspofungin tolerance which is independent of point mutations in HS regions of the FKS1 gene and is associated with cell wall remodeling. We further identified two novel mechanisms of tolerance to caspofungin that are also independent of point mutations in HS regions of FKS1 and are associated with cell wall remodeling. One of these mechanisms involves formation of iso-Ch5R, resulting in monosomy of the left arm and trisomy of the right arm of Ch5, while another mechanism is independent of chromosome aneuploidy. For the three kinds of mutants, we demonstrated overall consistent expression changes in genes residing on or outside Ch5 that are responsible for cell wall remodeling as well as for signaling. Apparently, these genes can be controlled in similar ways by different mechanisms, one of which is gene dose, while other mechanisms still remain to be determined.
We also obtained evidence relating the caspofungin tolerance of our mutants to the calcineurin pathway; calcineurin is a known multifunctional regulator in fungi that influences, for example, FKS2, chitin synthases, and caspofungin resistance (35).
As shown with the clinical isolates, evolution of clinical resistance to caspofungin can involve mutations in genes that are different from FKS genes and that confer a small increase in resistance or promote increased fitness (36). We believe that the mechanisms of caspofungin tolerance that are independent of stable resistance mutations in the HS regions of FKS1 can contribute to evolution of clinical drug resistance, which is based on formation of stable FKS1 mutations. The increased chitin in our mutants may be relevant to the increased chitin observed in some clinical isolates that show increased MICs but no FKS1 mutations.
MATERIALS AND METHODS
Strains, media, growth conditions, and primers.
The C. albicans clinical isolate JRCT1 (22) and strain SC5314, originally a clinical isolate but currently serving as a reference sequencing strain (Candida Genome Database [http://www.candidagenome.org/]), were used in this work to generate caspofungin-tolerant mutants. All strains were stored in frozen vials at −70°C, which interrupts cellular metabolism, thus preventing induction and propagation of genetic instability (37).
The preparation of yeast extract-peptone-dextrose (YPD), synthetic dextrose (SD), and sorbitol medium in which glucose was substituted for sorbitol was described previously (38, 39). YPD and SD were supplemented with caspofungin as previously described (22). In order to prepare solid medium, 2% (wt/vol) agar was added. Cells were routinely incubated at 37°C.
Primers used in this study are presented in Table S1 in the supplemental material.
Semiquantitative RT-PCR.
For the determination of expression levels, we standardized the growth of C. albicans cells. Briefly, petri dishes with synthetic medium in which glucose was substituted for sorbitol were seeded with ∼3,000 CFU per plate and incubated at 37°C for 20 to 40 h until colonies contained ∼105 cells/colony. RNA extraction and reverse transcription were conducted at the University of Rochester Genomics Research Center. PCRs were performed using Dream Taq DNA polymerase (Thermo Scientific, Rockford, IL) according to the manufacturer's recommendations. The REX2 (orf19.1466) and VPH1 (orf19.6863) genes were used as internal controls. Pilot PCRs were conducted and showed that 26 to 29 amplification cycles produced amplicons with a linear increase of DNA. For the final PCRs, approximately 20- to 80-ng (each) aliquots of cDNAs of a gene of interest and a control gene were used as templates in order to amplify the two genes in the same tube for the above-mentioned numbers of cycles (Fig. S1). Each transcript was examined in several independent RNA batches. PCR products from several consecutive cycles in exponential phase were electrophoresed in 1% agarose at 95 V for 45 min and stained with 1 μg/ml ethidium bromide for 10 min. Intensities of stained DNA bands were detected using a Molecular Imager Gel Doc XR+ system (Bio-Rad, Hercules, CA). The gene of interest was normalized against the control gene by calculating the mean ratio of densitometry values.
Generation of caspofungin-tolerant mutants.
C. albicans cells were removed from a −70°C stock and streaked for independent colonies on a YPD or SD master plate. Colonies were collected, cells were counted with the aid of a hemocytometer, and approximately 1 × 106 cells per plate were plated on solid medium containing a lethal concentration of caspofungin. The smallest lethal concentration was determined for each of two parental strains in pilot experiments as previously described (22). Colonies growing in the presence of the lethal dose of caspofungin were randomly chosen and purified by streaking for independent colonies on caspofungin-supplemented medium, plates were incubated, and cell mass was deposited at −70°C.
Spot assay.
Cells from −70°C freezer stocks were streaked on YPD plates and incubated at 37°C until young colonies (approximately 2 × 105 cells per colony) appeared. Colonies then were collected and counted with a hemocytometer, and serial 10-fold dilutions of cells were prepared in sterile distilled water. The corresponding suspensions were plated at 104, 103, 102, and 101 CFU per spot on plates that were supplemented with caspofungin in amounts that facilitated discrimination of growth rates between the parental strain and its caspofungin-tolerant derivatives.
Broth microdilution assay.
We performed a broth microdilution assay in accordance with the CLSI reference M27-A3 broth microdilution method for yeasts (40; see reference 23 for a detailed description).
DNA profiling by DNA-seq and analysis.
Chromosome ploidy was determined by DNA-seq. Genomic DNA was prepared by glass bead beating followed by phenol-chloroform extraction (22). The DNA concentration was determined by use of Qubit HS DNA reagent (Invitrogen). The DNA quality was determined by gel imaging and absorbance ratios (260/280 and 260/230 ratios of 1.8 to 2.2). The genomic DNA was fragmented by sonication. A VAHTS Nano DNA library prep kit for Illumina (Vazyme) was used for library preparation, including blunt ending, dA tail addition, ligation, and library amplification. The genomes were sequenced by the Yun Ang Biotech Company (Shanghai, China) on an Illumina HiSeq 4000 instrument using paired-end sequencing libraries with 350-bp inserts.
DNA-seq data were analyzed with the YMAP pipeline (41). Copy number analysis was based upon read depth across the genome. The sequencing data for SMC60-2-5 were aligned to the sequence of the parental strain SC5314. For JRCT1-derived mutants, the sequencing data for JRCT1 were first aligned to the reference sequence of SC5314 in order to confirm the diploid condition of JRCT1 chromosomes. The sequencing data for JMC120-2-5 and JMC120-2-5-R, JMC160-3-5 and JMC160-3-5-R, or JMC200-3-3 and JMC200-3-3-R were then aligned to the genome of the parent strain JRCT1.
Miscellaneous methods.
Spot assay, colony PCR, PFGE, determinations of 1,3-β-glucan and chitin levels in the cell wall, and CLSI broth microdilution were performed as previously described (22). Plates were photographed with a Molecular Imager Gel Doc XR+ system (Bio-Rad, Hercules, CA).
Supplementary Material
ACKNOWLEDGMENTS
We thank Robert Bets for important discussions and Mark Dumont and Jeffrey Hayes for reading the manuscript and for comments. We thank our reviewers for important suggestions that improved our manuscript. We thank Merck and Co., Inc., for donating caspofungin.
This work was supported in part by National Institutes of Health grant AI110764 to E.R. and grant AI109025 to D.S.P., Merck and Co., Inc. MISP award 51184, The University of Rochester funds to E.R., and National Natural Science Foundation of China grants (grants 81173100 and 81273556) to Y.C.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00071-17.
REFERENCES
- 1.Pappas PG, Kauffman CA, Andes DR, Clancy CJ, Marr KA, Ostrosky-Zeichner L, Reboli AC, Schuster MG, Vazquez JA, Walsh TJ, Zaoutis TE, Sobel JD. 2016. Clinical practice guideline for the management of candidiasis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis 62:e1–. doi: 10.1093/cid/civ933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pfaller M, Boyken L, Hollis R, Kroeger J, Messer S, Tendolkar S, Diekema D. 2011. Use of epidemiological cutoff values to examine 9-year trends in susceptibility of Candida species to anidulafungin, caspofungin, and micafungin. J Clin Microbiol 49:624–629. doi: 10.1128/JCM.02120-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beyda ND, Lewis RE, Garey KW. 2012. Echinocandin resistance in Candida species: mechanisms of reduced susceptibility and therapeutic approaches. Ann Pharmacother 46:1086–1096. doi: 10.1345/aph.1R020. [DOI] [PubMed] [Google Scholar]
- 4.Dannaoui E, Desnos-Ollivier M, Garcia-Hermoso D, Grenouillet F, Cassaing S, Baixench MT, Bretagne S, Dromer F, Lortholary O. 2012. Candida spp. with acquired echinocandin resistance, France, 2004–2010. Emerg Infect Dis 18:86–90. doi: 10.3201/eid1801.110556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Imtiaz T, Lee KK, Munro CA, Maccallum DM, Shankland GS, Johnson EM, Macgregor MS, Bal AM. 2012. Echinocandin resistance due to simultaneous FKS mutation and increased cell wall chitin in a Candida albicans bloodstream isolate following brief exposure to caspofungin. J Med Microbiol 61:1330–1334. doi: 10.1099/jmm.0.045047-0. [DOI] [PubMed] [Google Scholar]
- 6.Maubon D, Garnaud C, Calandra T, Sanglar D, Cornet M. 2014. Resistance of Candida spp. to antifungal drugs in the ICU: where are we now? Intensive Care Med 40:1241–1255. doi: 10.1007/s00134-014-3404-7. [DOI] [PubMed] [Google Scholar]
- 7.Alexander BD, Johnson MD, Pfeifer CD, Jimenez-Ortigosa C, Castania J, Booker R, Castanheira M, Messer SA, Perlin DS, Pfaller M. 2013. Increasing echinocandin resistance in Candida glabrata: clinical failure correlates with presence of FKS mutations and elevated minimum inhibitory concentrations. Clin Infect Dis 56:1724–1732. doi: 10.1093/cid/cit136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arendrup MC, Perlin DS. 2014. Echinocandin resistance: an emerging clinical problem? Curr Opin Infect Dis 27:484–492. doi: 10.1097/QCO.0000000000000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shields RK, Nguyen MH, Press EG, Cumbie R, Driscoll E, Pasculle AW, Clancy CJ. 2015. Rate of FKS mutations among consecutive Candida isolates casing bloodstream infection. Antimicrob Agents Chemother 59:7465–7470. doi: 10.1128/AAC.01973-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perlin DS. 2011. Current perspectives on echinocandin class drugs. Future Microbiol 4:441–457. doi: 10.2217/fmb.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shapiro RS, Robbins N, Cowen LE. 2011. Regulatory circuitry governing fungal development, drug resistance, and disease. Microbiol Mol Biol Rev 75:213–267. doi: 10.1128/MMBR.00045-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perlin DS. 2015. Mechanisms of echinocandin antifungal drug resistance. Ann N Y Acad Sci 1354:1–11. doi: 10.1111/nyas.12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garcia-Effron G, Park S, Perlin DS. 2009. Correlating echinocandin MIC and kinetic inhibition of fks1 mutant glucan synthases for Candida albicans: implications for interpretive breakpoints. Antimicrob Agents Chemother 53:112–122. doi: 10.1128/AAC.01162-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walker LA, Gow NA, Munro CA. 2010. Fungal echinocandin resistance. Fungal Genet Biol 47:117–126. doi: 10.1016/j.fgb.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castanheira M, Woosley LN, Diekema DJ, Messer SA, Jones RN, Pfaller MA. 2010. Low prevalence of fks1 hot spot 1 mutations in a worldwide collection of Candida strains. Antimicrob Agents Chemother 54:2655–2659. doi: 10.1128/AAC.01711-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desnos-Ollivier M, Bretagne S, Raoux D, Hoinard D, Dromer F, Dannaoui E. 2008. Mutations in the fks1 gene in Candida albicans, C. tropicalis, and C. krusei correlate with elevated caspofungin MICs uncovered in AM3 medium using the method of the European Committee on Antibiotic Susceptibility Testing. Antimicrob Agents Chemother 52:3092–3098. doi: 10.1128/AAC.00088-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pham CD, Iqbal N, Bolden CB, Kuykendall RJ, Harrison LH, Farley MM, Schaffner W, Beldavs ZG, Chiller TM, Park BJ, Cleveland AA, Lockhart SR. 2014. Role of FKS mutations in Candida glabrata: MIC values, echinocandin resistance, and multidrug resistance. Antimicrob Agents Chemother 58:4690–4696. doi: 10.1128/AAC.03255-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee KK, Maccallum DM, Jacobsen MD, Walker LA, Odds FC, Gow NA, Munro CA. 2012. Elevated cell wall chitin in Candida albicans confers echinocandin resistance in vivo. Antimicrob Agents Chemother 56:208–217. doi: 10.1128/AAC.00683-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stevens DA, Ichinomiya M, Koshi Y, Horiuchi H. 2006. Escape of Candida from caspofungin inhibition at concentrations above the MIC (paradoxical effect) accomplished by increased cell wall chitin; evidence for beta-1,6-glucan synthesis inhibition by caspofungin. Antimicrob Agents Chemother 9:3160–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walker LA, Munro CA, de Bruijn I, Lenardon MD, McKinnon A, Gow NA. 2008. Stimulation of chitin synthesis rescues Candida albicans from echinocandins. PLoS Pathog 4:e1000040. doi: 10.1371/journal.ppat.1000040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Plaine A, Walker L, Da Costa G, Mora-Montes HM, McKinnon A, Gow NA, Gaillardin C, Munro CA, Richard ML. 2008. Functional analysis of Candida albicans GPI-anchored proteins: roles in cell wall integrity and caspofungin sensitivity. Fungal Genet Biol 45:1404–1414. doi: 10.1016/j.fgb.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang F, Kravets A, Bethlendy G, Welle S, Rustchenko E. 2013. Chromosome 5 monosomy of Candida albicans controls susceptibility to various toxic agents, including major antifungals. Antimicrob Agents Chemother 57:5026–5036. doi: 10.1128/AAC.00516-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suwunnakorn S, Wakabayashi H, Rustchenko E. 2016. Chromosome 5 of human pathogen Candida albicans carries multiple genes for negative control of caspofungin and anidulafungin susceptibility. Antimicrob Agents Chemother 60:7457–7467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janbon G, Sherman F, Rustchenko E. 1998. Monosomy of a specific chromosome determines l-sorbose utilization: a novel regulatory mechanism in Candida albicans. Proc Natl Acad Sci U S A 9:5150–5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kravets A, Yang F, Bethlendy G, Sherman F, Rustchenko E. 2014. Adaptation of Candida albicans to growth on sorbose via monosomy of chromosome 5 accompanied by duplication of another chromosome carrying a gene responsible for sorbose utilization. FEMS Yeast Res 14:708–713. doi: 10.1111/1567-1364.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cornet M, Gaillardin C, Richard ML. 2006. Deletions of the endocytic components VPS28 and VPS32 in Candida albicans lead to echinocandin and azole hypersensitivity. Antimicrob Agents Chemother 10:3492–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reedy JL, Filler SG, Heitman J. 2010. Elucidating the Candida albicans calcineurin signaling cascade controlling stress response and virulence. Fungal Genet Biol 47:107–116. doi: 10.1016/j.fgb.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kravets A, Qin H, Ahmad A, Bethlendy G, Gao Q, Rustchenko E. 2010. Widespread occurrence of dosage compensation in Candida albicans. PLoS One 5:e10856. doi: 10.1371/journal.pone.0010856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Selmecki A, Gerami-Nejad M, Paulson C, Forche A, Berman J. 2008. An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Mol Microbiol 68:624–641. doi: 10.1111/j.1365-2958.2008.06176.x. [DOI] [PubMed] [Google Scholar]
- 30.Hirakawa MP, Martinez DA, Sakthikumar S, Anderson MZ, Berlin A, Gujja S, Zeng Q, Zisson E, Wang JM, Greenberg JM, Berman J, Bennett RJ, Cuomo CA. 2015. Genetic and phenotypic intra-species variation in Candida albicans. Genome Res 25:413–425. doi: 10.1101/gr.174623.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ford CB, Funt JM, Abbey D, Issi L, Guiducci C, Martinez DA, Delorey T, Li BY, White TC, Cuomo C, Rao RP, Berman J, Thompson DA, Regev A. 2014. The evolution of drug resistance in clinical isolates of Candida albicans. eLife 4:e00662. doi: 10.7554/eLife.00662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wertheimer NB, Stone N, Berman J. 2016. Ploidy dynamics and evolvability in fungi. Philos Trans R Soc Lond B Biol Sci 371:20150461. doi: 10.1098/rstb.2015.0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walker LA, Gow NAR, Munro C. 2013. Elevated chitin content reduces the susceptibility of Candida species to caspofungin. Antimicrob Agents Chemother 57:146–154. doi: 10.1128/AAC.01486-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Munro CA, Selvaggini S, de Bruijn I, Walker L, Lenardon MD, Gerssen B, Milne S, Brown AJ, Gow NA. 2007. The PKC, HOG and Ca2+ signalling pathways co-ordinately regulate chitin synthesis in Candida albicans. Mol Microbiol 63:1399–1413. doi: 10.1111/j.1365-2958.2007.05588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Juvvadi PR, Lamoth F, Steinbach WJ. 2014. Calcineurin as a multifunctional regulator: unraveling novel functions in fungal stress responses, hyphal growth, drug resistance, and pathogenesis. Fungal Biol Rev 28:56–69. doi: 10.1016/j.fbr.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh-Babak SD, Babak T, Diezmann S, Hill JA, Xie JL, Chen YL, Poutanen SM, Rennie RP, Heitman J, Cowen LE. 2012. Global analysis of the evolution and mechanism of echinocandin resistance in Candida glabrata. PLoS Pathog 8:e1002718. doi: 10.1371/journal.ppat.1002718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahmad A, Kabir MA, Kravets A, Andaluz E, Larriba G, Rustchenko E. 2008. Chromosome instability and unusual features of some widely used strains of Candida albicans. Yeast 25:433–448. doi: 10.1002/yea.1597. [DOI] [PubMed] [Google Scholar]
- 38.Sherman F. 2002. Getting started with yeast. Methods Enzymol 350:3–41. doi: 10.1016/S0076-6879(02)50954-X. [DOI] [PubMed] [Google Scholar]
- 39.Ahmad A, Kravets A, Rustchenko E. 2012. Transcriptional regulatory circuitries in the human pathogen Candida albicans involving sense-antisense interactions. Genetics 190:537–547. doi: 10.1534/genetics.111.136267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clinical and Laboratory Standards Institute. 2008. M27-A3 reference method for broth dilution antifungal susceptibility testing of yeasts: approved standard, 3rd ed Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 41.Abbey DA, Funt J, Lurie-Weinberg MN, Thompson DA, Regev A, Myers CL, Berman J. 2014. YMAP: a pipeline for visualization of copy number variation and loss of heterozygosity in eukaryotic pathogens. Genome Med 6:100. doi: 10.1186/s13073-014-0100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rustchenko EP, Curran TM, Sherman F. 1993. Variations in the number of ribosomal DNA units in morphological mutants and normal strains of Candida albicans and in normal strains of Saccharomyces cerevisiae. J Bacteriol 175:7189–7199. doi: 10.1128/jb.175.22.7189-7199.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.