

ABSTRACT
Clarithromycin (CLA) is a commonly recommended drug for Helicobacter pylori eradication. However, the prevalence of CLA-resistant H. pylori is increasing. Although point mutations in the 23S rRNA are key factors for CLA resistance, other factors, including efflux pumps and regulation genes, are also involved in the resistance of H. pylori to CLA. Guanosine 3′-diphosphate 5′-triphosphate and guanosine 3′,5′-bispyrophosphate [(p)ppGpp)], which are synthesized by the bifunctional enzyme SpoT in H. pylori, play an important role for some bacteria to adapt to antibiotic pressure. Nevertheless, no related research involving H. pylori has been reported. In addition, transporters have been found to be related to bacterial drug resistance. Therefore, this study investigated the function of SpoT in H. pylori resistance to CLA by examining the shifts in the expression of transporters and explored the role of transporters in the CLA resistance of H. pylori. A ΔspoT strain was constructed in this study, and it was shown that SpoT is involved in H. pylori tolerance of CLA by upregulating the transporters HP0939, HP1017, HP0497, and HP0471. This was assessed using a series of molecular and biochemical experiments and a cDNA microarray. Additionally, the knockout of genes hp0939, hp0471, and hp0497 in the resistant strains caused a reduction or loss (the latter in the Δhp0497 strain) of resistance to CLA. Furthermore, the average expression levels of these four transporters in clinical CLA-resistant strains were considerably higher than those in clinical CLA-sensitive strains. Taken together, our results revealed a novel molecular mechanism of H. pylori adaption to CLA stress.
KEYWORDS: Helicobacter pylori, SpoT, transporters, clarithromycin, antibiotic resistance
INTRODUCTION
Helicobacter pylori is a helical or spiral-shaped Gram-negative bacterium that selectively colonizes the gastric mucosa. Most people worldwide are infected in early childhood and carry the organism throughout their lives (1). To date, H. pylori infection has been linked to human gastritis and gastric ulcers; moreover, long-term chronic infection by this pathogen increases the risk of gastric adenocarcinoma and mucosa-associated lymphoid tissue lymphoma (1, 2). Because H. pylori infection severely endangers human health, the treatment and eradication of H. pylori infection should be urgently addressed. Currently, the first-line treatment for H. pylori infection is a triple therapy that includes colloidal bismuth, proton pump inhibitors, and two antibiotics that are macrolides, nitroimidazoles, or β-lactam (3, 4). However, this traditional therapy does not work as effectively as before because of complex factors, such as the bacterium itself, host, treatment environment, and treatment methods (5). Currently, the antibiotic resistance of H. pylori is the principal reason for eradication failure (6). Nevertheless, the mechanism of resistance acquirement is not fully understood.
Clarithromycin (CLA) is the new generation of macrolide antibiotics. This drug is stable in acidic environments and well absorbed in the stomach (7). Consequently, CLA-based triple therapy has been recommended in children and adults by the latest Maastricht Consensus (8). One mechanism of H. pylori eradication is through CLA interference with protein elongation by binding reversibly to the peptidyl transferase loop of domain V in the 23S rRNA molecule, which could be induced to block bacterial protein synthesis (9, 10).
Recently, the drug resistance rate of H. pylori against CLA has been increasing worldwide because of the excessive and repeated use of CLA (4). The current research for CLA tolerance focuses on point mutations in the peptidyl transferase domain of the 23S rRNA ribosomal subunit (10). Recently, Hirata et al. reported that efflux pumps play a vital role in CLA resistance (11), and Smiley et al. completed a proteomic analysis and found that alterations in the outer membrane protein profile might be a novel mechanism conferring CLA resistance in H. pylori (12). Therefore, some other factors must be involved in CLA resistance in H. pylori.
SpoT regulates the stringent response in Escherichia coli (13). The stringent response is a bacterial adaptation that affects global gene expression during nutrient limitation and under other stress conditions (14). In E. coli, the stringent response is regulated by the small molecules guanosine 3′-diphosphate 5′-triphosphate and guanosine 3′,5′-bispyrophosphate [(p)ppGpp], which are synthesized by the enzymes RelA and SpoT. RelA and SpoT synthesize a considerable amount of (p)ppGpp using the substrates GDP and GTP and use ATP as a phosphorus source under nutrient limitation and other stressful conditions (13). These small-molecular effectors can combine with the RNA polymerase promoter (13–15) and subsequently change the promoter specificity and the efficiency of transcription (16).
Different from E. coli, the H. pylori genome contains only SpoT and lacks RelA (17, 18). SpoT is a bifunctional enzyme that has both synthetase and hydrolase activities, which are principal regulators for various stress responses in this bacterium (19–21). Recent studies have shown that (p)ppGpp mediates antibiotic tolerance in bacteria (22–24), which implies that antibiotic stress also activates the stringent response. For many gammaproteobacteria, microarray analyses of strains lacking ppGpp-synthesizing enzymes have revealed the deregulation of several hundred genes (25, 26). Thus, (p)ppGpp may be the universal signaling molecule regulator causing antibiotic resistance in H. pylori.
Multidrug efflux transporters are formed from the combination of a series of proteins, which can pump noxious compounds out of cells and protect cells from environmental stresses (27, 28). On the basis of sequence similarity, multidrug efflux transporters are classified into five families: the resistance-nodulation-cell division (RND) family, the major facilitator superfamily, the small multidrug resistance family, the multidrug toxic compound extrusion family, and the ATP-binding cassette (ABC) family (27, 28). Some identified multidrug efflux transporters are the AcrAB-TolC system in E. coli (29) and the MexAB-OprM system in Pseudomonas aeruginosa (30). H. pylori has four RND efflux transporters, and their participation in multidrug resistance has been confirmed (31–33). Nonetheless, many transporters are present in the H. pylori genome, and whether they participate in antibiotic tolerance should be determined.
Considering the important function of (p)ppGpp in the regulation of bacterial antibiotic resistance, the gradually increasing resistance to CLA in H. pylori, and the importance of transporters as factors in bacterial drug resistance, this study aimed to investigate whether (p)ppGpp is involved in the regulation of CLA resistance in H. pylori and to identify new transporters regulated by (p)ppGpp that participate in CLA resistance in H. pylori.
RESULTS
SpoT is involved in H. pylori resistance to CLA.
The bifunctional enzyme SpoT and (p)ppGpp are the key regulators in the stringent response of bacteria (16, 25). Moreover, the presence of CLA is unfavorable for H. pylori survival and growth. SpoT is involved in some bacterial antibiotic resistance. To determine whether SpoT participates in the regulation of the adaptation of H. pylori to CLA stress, the mRNA expression level of spoT was detected by quantitative real-time PCR (qRT-PCR). The expression of spoT in H. pylori strain 26695 (wild type [WT]) treated with CLA (1× MIC) was considerably higher (P < 0.01) than that in the control strain (Fig. 1A). SpoT is required for the production of (p)ppGpp. We subjected the H. pylori 26695 (WT), ΔspoT mutant, and spoT-complemented (spoT*; see Materials and Methods) strains to CLA stress, and they were incubated in minimal medium with 32P for 1 h. As expected, the WT and spoT* strains accumulated significant amounts of (p)ppGpp upon exposure to this stress. In contrast, (p)ppGpp was absent in the ΔspoT strain (Fig. 1B). We further examined the MIC of the WT, ΔspoT, and spoT* strains in response to CLA. The results showed that the MIC of the ΔspoT strain was only one-half of that of the WT strain (Fig. 1C). The viability of the ΔspoT strain was weaker than that of the WT strain after the stationary phase. In contrast, there was no significant difference in the exponential phase between these two groups (Fig. 1D). In time-kill experiments, we used cells in the exponential phase, and the results showed that the number of living cells of the ΔspoT strain decreased faster than the number of living cells of H. pylori 26695 and spoT* strains after treatment with 5× MIC of CLA for 2 h. After treatment for 8 h, the living cells of the ΔspoT strain decreased to 1% or less, but those of H. pylori 26695 and spoT* strains still made up more than 10%, which indicated that the ΔspoT strain is susceptible to CLA (Fig. 1E). Furthermore, we examined the cell membrane integrity of H. pylori cells and the morphological features of the WT and ΔspoT strains under CLA stress condition by applying membrane-permeable (Syto 9) and membrane-impermeable (propidium iodide [PI]) fluorescent dyes to stain living and dead cells, respectively. A larger number of cells of the ΔspoT strain than of wild-type cells died and lost cytoplasmic membrane integrity. Additionally, the ΔspoT strain transformed from its normal helical bacillary morphological features to coccoid features (Fig. 1F).
FIG 1.

SpoT is involved in the resistance of H. pylori to clarithromycin (CLA). (A) CLA induces the high expression of the mRNA for SpoT in wild-type (WT) and SpoT-complemented (spoT*) strains. (B) H. pylori SpoT mutant strain (ΔspoT) is deficient in guanosine 3′-diphosphate 5′-triphosphate and guanosine 3′,5′-bispyrophosphate [(p)ppGpp] production when exposed to CLA (2× MIC) for 20 min. (C) MICs of WT, ΔspoT, and spoT* strains. The H. pylori ΔspoT mutant strain is sensitive to CLA. Complementation restores the MIC values. (D) Growth curves of the WT and ΔspoT strains. (E) Time-kill curves of WT, ΔspoT, and spoT* strains in the presence of 2× MIC of CLA. (F) Exposure to CLA (2× MIC, 2 h) induces coccoid transformation and the death of H. pylori cells. Cells stained with membrane-permeable Syto 9 (green) and membrane-impermeable PI (red) were visualized by confocal microscopy. The data are representative of three independent experiments and are shown as the means ± SEM from three independent experiments. Significance by t test: **, P < 0.01; ***, P < 0.001.
The inactivation of SpoT decreases efflux activity.
The results reported above indicated that SpoT contributes to the CLA stress response in H. pylori. Thus, we investigated its regulatory mechanism. Because previous studies have shown that SpoT regulates bacterial drug resistance via many aspects and efflux pumps are an important factor in multidrug-resistant bacteria, we assessed whether SpoT was involved in bacterial drug resistance by regulating the expression of efflux pumps in this study. The efflux activities of the ΔspoT and WT strains were assessed by determining the accumulation of the fluorescent dye Hoechst 33342 (H33342), and the results showed that SpoT inactivation caused an obvious increase in the accumulation of Hoechst 33342 (Fig. 2). These results indicated that some efflux pumps can be regulated by SpoT in H. pylori.
FIG 2.
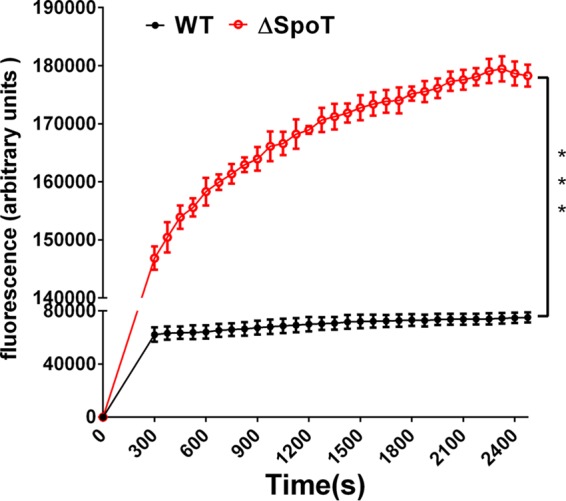
The accumulation of H33342 (2.5 μM) in H. pylori WT and ΔspoT strains. The fluorescence intensity was recorded at the excitation and emission wavelengths of 350 and 460 nm, respectively, over a 30-min incubation period. The data presented are the means ± SEM from three separate experiments. Student's t test was performed to compare the accumulation of Hoechst 33342 between the WT and ΔspoT mutant strains. ***, P < 0.001.
cDNA microarray analysis was performed to detect the efflux pumps involved in H. pylori CLA resistance.
In general, because antibiotics are immediately drained, the genes encoding efflux pumps (proteinaceous transporters localized in the cytoplasmic membrane of all cells) in bacteria will be highly expressed under antibiotic pressure (34). To determine which efflux pump genes are involved in the resistance of H. pylori to CLA, we analyzed the differential expression of transporters between H. pylori strain 26695 cells treated with 2× MIC of CLA for 20 min and untreated cells (control) (Fig. 3A). The transporter genes that showed >3-fold-higher expression than the control were selected as potential target genes (Fig. 3B).
FIG 3.

Microarray assay of the gene expression in H. pylori treated with CLA (0.125 μg/ml) for 2 h. (A) Scatter plot of the differentially expressed genes. The values on the x and y axes are the normalized signal values of the samples (log2 scaled). The green lines are the fold change lines. The default fold change value given is 1.5. The genes above the top green line and below the bottom green line indicated >1.5-fold change between the two compared samples. (B) Genes with >3-fold changes in H. pylori with CLA treatment in the microarray analysis. The microarray results represent a single experiment.
qRT-PCR analysis was performed to identify the transporters that participate in H. pylori CLA tolerance.
On the basis of microarray analysis, the selected transporters were further confirmed by qRT-PCR in the H. pylori 26695 WT strain exposed to 2× MIC of CLA for 2 h. We selected the transporters that were expressed >10-fold relative to those of the control (Fig. 4A). They were identified in a CLA-resistant strain (ST, selected artificially; MIC = 8 μg/ml). Sequence analysis of the 23S rRNA gene of the ST revealed mutations (data not shown), as shown by qRT-PCR. Only four genes (hp0939, hp1017, hp0471, and hp0497) were expressed significantly higher than in the control group (Fig. 4B). According to the analysis above, these four transporters are important for H. pylori adaptation to the CLA stress.
FIG 4.
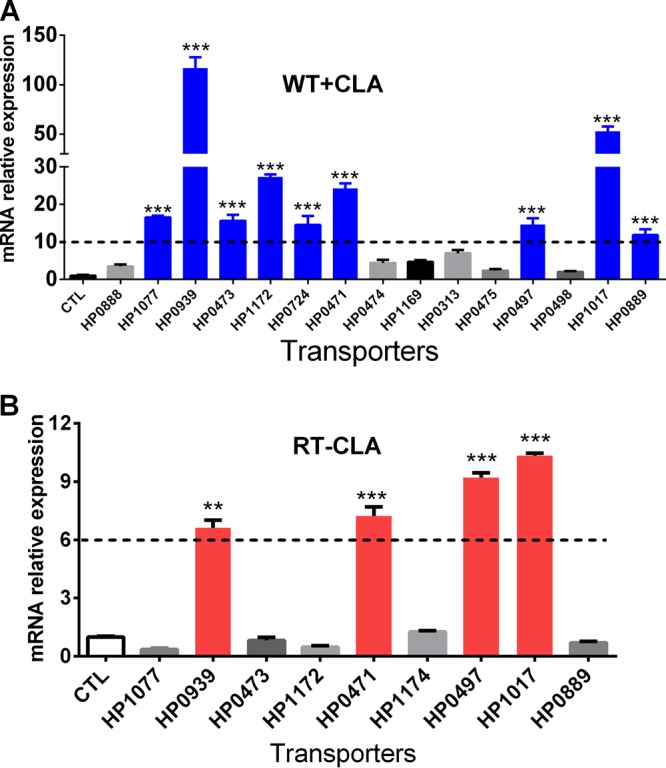
qRT-PCR analysis of the mRNA levels of transporters in H. pylori CLA-sensitive and -resistant strains. (A) qRT-PCR was used to confirm the expression of the transporters using cDNA microarray analysis (Fig. 3B) in a CLA-sensitive strain exposed to CLA for 2 h. The results were compared to those obtained in the absence of CLA treatment (CTL). Transporter expressions that increased >10-fold are in blue. (B) The transporters marked in blue in panel A were further analyzed by qRT-PCR in a CLA-resistant strain (ST, selected artificially) and compared to those of the CLA-sensitive strain without CLA treatment. Transporter expressions that increased more than 6-fold are in red (encoded by hp1017, hp0939, hp0497, and hp0471). The signals were normalized to the 16S rRNA levels. Data are means ± SEM from three independent experiments. Significance by t test: **, P < 0.01; ***, P < 0.001.
The transporters HP0939, HP1017, HP0497, and HP0471 are involved in H. pylori resistance to CLA.
The qRT-PCR data suggested that the transporters HP0939, HP1017, HP0497, and HP0471 may be involved in the resistance of H. pylori to CLA. Therefore, we constructed mutant strains for further study and examined the functions of these four transporters in drug resistance. However, we could not acquire a mutant strain for HP1017, which indicates that this transporter is essential for H. pylori survival. We successfully constructed hp0939, hp0497, and hp0471 mutant strains in the WT and CLA-resistant strains (ST, artificially selected.). To test whether these transporters were required for H. pylori growth, we assayed the viability of the WT and mutant strains of these three genes (Δhp0939, Δhp0497, and Δhp0471 strains) in liquid cultures over 144 h under standard laboratory growth conditions. All strains exhibited similar population dynamics (Fig. 5A). In addition, the time-kill assays showed that the living cells of these three mutant strains exhibited a considerably sharper decrease than H. pylori (WT) after treatment with 5× MIC of CLA for 2 h. After 8 h of treatment, the living cells of these three mutant strains dropped to 0.1% or less, but those of the WT strain were still more than 10%. This observation indicated that these three mutant strains, and specifically Δhp0939 and Δhp0497 strains, were more susceptible to CLA than the WT strain (Fig. 5B). We further examined the MICs of these three mutant strains; the results showed that the MIC of the Δhp0471 mutant strain was only one-half that of the WT strain and the MICs of the Δhp0939 and Δhp0497 mutant strains were hardly one-quarter of that of the WT strain (Fig. 5C). Additionally, the MICs of ST and its Δhp0939, Δhp0497, and Δhp0471 mutant strains were also significantly decreased compared with those of the ST group; particularly, the MIC of the Δhp0497 strain was close to that of the WT strain (Fig. 5C). Moreover, we also examined the cell membrane integrity of H. pylori and the morphological features of the WT and these three mutant strains under CLA stress by applying the membrane-permeable (Syto 9) and membrane-impermeable (PI) fluorescent dyes to stain living and dead cells, respectively; a larger number of mutant cells than WT cells died and lost their cytoplasmic membrane integrity (Fig. 5D). As transporters, hp0939, hp0497, and hp0471 were highly expressed after treatment with CLA, so we hypothesized that the transporters may discharge antibiotics. Subsequently, we assessed the efflux activities of these three mutant strains, the ST strain, and the WT strain by detecting the accumulation of the fluorescent dye Hoechst 33342. The result displayed that the inactivation of these three genes caused a distinct increase in the accumulation of Hoechst 33342. Notably, the fluorescence values of the Δhp0939 and Δhp0497 mutant strains were >4-fold greater than that of the WT (Fig. 5E). The relative mRNA expression levels of hp0939, hp1017, hp0497, and hp0471 were assessed by qRT-PCR in the clinical CLA-resistant strains and the clinical CLA-sensitive strains. The average expression levels of these four genes in the clinical CLA-resistant strains were significantly higher than those in the clinical CLA-sensitive ones (Fig. 5F).
FIG 5.

HP0939, HP0497, and HP0471 are involved in the resistance of H. pylori to CLA. (A) Growth curves of the WT strain and the Δhp0939, Δhp0497, and Δhp0471 mutants. (B) Time-kill assays in WT, Δhp0939, Δhp0497, and Δhp0471 strains with 2× MIC of CLA. (C) MICs of WT, ST, and their Δhp0939, Δhp0497, and Δhp0471 mutant strains. Compared with nonmutant strains, H. pylori transporter mutant strains are more sensitive to CLA. In ST mutant strains, the MICs of Δhp0939, Δhp0497, and Δhp0471 strains decreased considerably compared with those of the ST group. (D) Exposure to CLA (2× MIC, 2 h) induces coccoid transformation and the death of H. pylori cells. Cells stained with membrane-permeable Syto 9 (green) and membrane-impermeable PI (red) were visualized by confocal microscopy. (E) Accumulation of H33342 (2.5 mM) in the WT, ST, Δhp0939, Δhp0497, and Δhp0471 strains. The fluorescence intensity was recorded at the excitation and emission wavelengths of 350 and 460 nm, respectively, over a 30-min incubation period. The data presented are the means ± SEM from three separate experiments. Student's t test was performed to compare the accumulation of Hoechst 33342 in each strain with that in the WT. (F) qRT-PCR analysis of the mRNA levels of the H. pylori transporters (hp1017, hp0939, hp0497, and hp0471) between clinical CLA-resistant and -sensitive strains. Data are the means ± SEM from three independent experiments. Significance by t test: **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
The bifunctional SpoT significantly mediates the transcriptional levels of transporters HP0939, HP1017, HP0497, and HP0471.
The data reported above indicated that the transporters HP0939, HP1017, HP0497, and HP0471 were important for the adaption of H. pylori to CLA stress. Whether these transporters are regulated by the bifunctional SpoT was subsequently determined. First, we stimulated the WT and ΔspoT strains with 1.5× MIC of CLA along a time gradient (Fig. 6A). Second, we stimulated the WT and ΔspoT strains with different CLA concentrations (1.5× and 2× MIC) for 30 min (Fig. 6B). Compared with the control, the expression of all four transporters was highly induced by CLA in the WT strain in a concentration- and time-dependent manner. However, CLA could hardly induce their expression in the ΔspoT strain (Fig. 6A and B). These results suggested that these four transporters might be upregulated by SpoT in response to CLA stress.
FIG 6.

qRT-PCR analysis of the mRNA levels of transporters (hp1017, hp0939, hp0497, and hp0471) in WT and ΔspoT mutant strains exposed to different CLA concentrations for 30 min (A) and to 2× MIC CLA for different time periods (B). The results were compared to WT without CLA treatment (CTL). The signal was normalized to the 16S rRNA levels. *, P < 0.05 by t test.
qRT-PCR was performed to analyze the expression of the efflux pumps (HefF and HefC) in the WT, Δhp0939, Δhp0497, Δhp0471, and ΔspoT strains left untreated or treated with CLA.
The accumulation of H33342 indicated that both SpoT (Fig. 2) and the transporter proteins (Fig. 5E) could affect the activity of the efflux pumps. However, we did not screen out efflux pumps by cDNA microarray analysis. Previous studies have shown that the efflux pumps of the RND family (HefF and HefC) are involved in H. pylori resistance to CLA (35). Therefore, we examined the expression of these two efflux pumps in the WT, Δhp0939, Δhp0497, Δhp0471, and ΔspoT strains treated with 2× MIC of CLA or without CLA. The results showed that their expression levels in the mutant strains (Δhp0939, Δhp0471, and Δhp0497 mutants) were significantly lower than in the WT strain in both groups (Fig. 7). In the SpoT strain, the expression levels of these two efflux pumps were reduced in the absence of the antibiotic, but they were still induced by the addition of antibiotics (Fig. 7).
FIG 7.
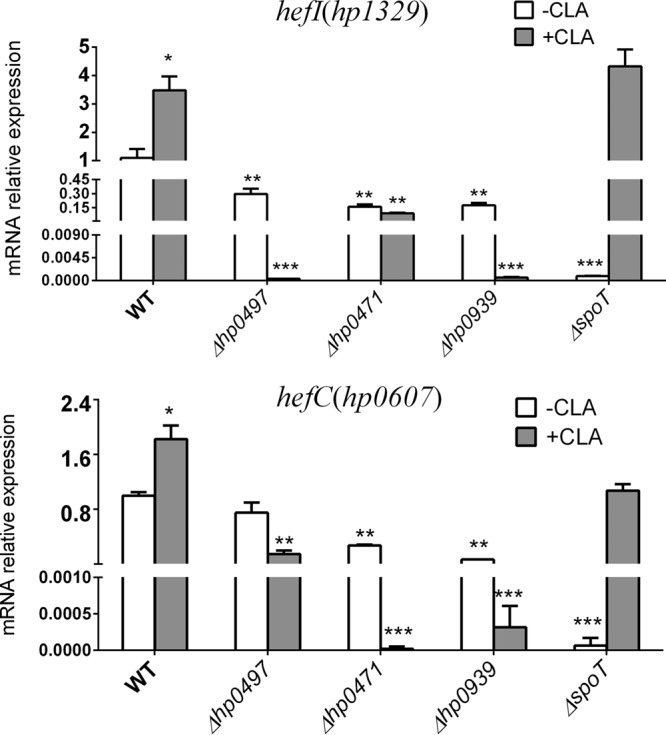
qRT-PCR analysis of the mRNA levels of efflux pumps (HefC and HefF) in WT, Δhp0939, Δhp0497, Δhp0471, and ΔspoT mutant strains treated with or without CLA. The signal was normalized to the 16S rRNA levels. Significance by t test: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
DISCUSSION
In this study, SpoT, which participates in CLA resistance of H. pylori, was shown to be a global regulatory factor that might be involved in H. pylori antibiotic resistance in many ways. Our present study focused on its regulatory action on transporter proteins, and the results demonstrated that CLA stress could induce H. pylori to produce a large amount of (p)ppGpp, which contributes to the high expression of transporter protein genes. The regulation of transporter protein genes by SpoT might be one of the CLA-resistant mechanisms of H. pylori.
CLA is a common drug used in the first-line eradication therapy for H. pylori infection. Epidemiological studies have shown that the prevalence of CLA-resistant H. pylori has been increasing annually (9). Tolerance to CLA in H. pylori strains is often related to point mutations in two adjacent 23S rRNA nucleotides (A2143G or A2144G) that change the target region of CLA and decrease the affinity for 23S rRNA (10). The existing clinical data indicate that almost all CLA-resistant strains had 23S rRNA mutations (4, 11), including the strains used in the present study. However, our data demonstrate that transporter proteins (encoded by hp0939, hp0471, hp0497, and hp1017) are also important in H. pylori resistance to CLA, even in the resistant strains with 23S rRNA mutations. After knocking out the transporter genes (hp0939, hp0471, and hp0497), all strains exhibited a growth curve similar to that of the WT (Fig. 5A), but their sensitivity to CLA obviously increased. In our MIC determination study for the ST and its Δhp0497 mutant strain, the MIC of the Δhp0497 mutant was similar to that of the WT strain. This manifestation indicated that transporter proteins perform a crucial role in this drug resistance process in addition to point mutations.
Several efflux pump genes have been confirmed to participate in CLA tolerance (11, 35, 36). To avoid being killed by antibiotics, the bacteria achieve a lower intracellular drug concentration by lowering the rate of drug uptake or increasing the rate of drug export or both (34). In these experiments, the inactivation of these three genes (hp0939, hp0471, and hp0497) caused a distinctly increased accumulation of the dye Hoechst 33342. However, these four transporter proteins (HP0939, HP0471, HP0497, and HP1017) are not efflux pumps, and their predicted functions are the uptake of nutrients and the transport of ions. HP0939-0940 belongs to the YckJ amino acid ABC transporter family and may be related to amino acid uptake. The gene encoding HP1017 is similar to the rocE gene from Bacillus subtilis and encodes an arginase transporter (17). HP0497 is a sodium- and chloride-dependent transporter, and HP0471 (KefB) is a glutathione-regulated potassium efflux system protein (18). In short, the above-mentioned genes are involved mainly in the metabolism of H. pylori, and the physiological and metabolic states of bacteria can affect the efficacy of antibiotics (37, 38). Taking into account that these three transporters are not efflux pumps, the increased accumulation of the dye Hoechst 33342 in mutant strains may be attributed to either efflux or the uptake of the outer membrane affected by these mutants. A previous study reported that H. pylori had a less efficient self-promoted uptake pathway (31). In order to verify this, we examined the expression of the efflux pumps of hefC and hefI (RND family) with and without CLA treatment. The results showed that their expressions in the mutant strains (Δhp0939, Δhp0471, and Δhp0497 strains) were significantly lower than those in the WT strain (Fig. 7). It has been shown that these two efflux pumps are involved in H. pylori resistance to CLA (11, 35), which might be one of the most important reasons why these three mutants (hp0939, hp0471, and hp0497 mutants) are more sensitive to CLA.
It can be seen in Fig. 5 that the mutation of any of the three transporters causes H. pylori to be sensitive to CLA. This finding suggested that the three gene products interacted in some way. To test this hypothesis, we analyzed the interactions among these transporters by qRT-PCR and found that after mutating one of these transporters, the expressions of the other transporters were suppressed compared with those in the WT strain (data not shown). This suggested that these four transporters might be regulated by similar mechanisms. In this study, we found that they were both regulated by SpoT (Fig. 6). Because SpoT is primarily involved in the stringent response of bacteria (23, 26), these four transporters might be important for the bacterial adaptation to environment stresses as well. Studies have shown that HP0497 is involved in the adaption of H. pylori to salt (39) and acid (40) stresses. According to the published H. pylori genome sequence of strain 26695, the HP1017 gene is an important part of the htrA locus (18). HtrA proteases and chaperones exhibit important roles in the stress responses (41). Moreover, there is evidence that HP0471 is involved in the H. pylori tolerance to CLA (42). Considering that the deletion of spoT did not increase the susceptibility of the resistant ST strain to CLA (data not shown) and these four transporters were highly expressed in CLA-resistant clinical isolates, genes encoding these four transporters might be potential candidate genes for H. pylori resistance to CLA.
In a comparison of the growth curves of the WT strain and SpoT resistant strain, the ΔspoT strain showed growth defects and was more likely to enter the decline phase (Fig. 1D), while no significant differences were found between the growth curves of the transporter mutant strains (Δhp0939, Δhp0471, and Δhp0497 strains) and the WT strain (Fig. 5A). This suggested that the ΔspoT strain was more sensitive to CLA than were the transporter mutants. However, the transporter mutant strains were more sensitive to CLA. In the transporter mutants, whenever they were exposed to CLA, the expressions of both of the efflux pumps (HefC and HefI) were inhibited (Fig. 7). However, in the ΔspoT strain, although the expression levels of these two efflux pumps were reduced in the absence of the antibiotic, they were still induced by the addition of antibiotics, which suggested that these two efflux pumps not only were regulated by SpoT under antibiotic stress but might also be controlled by other genes. This might be the reason why the transporter mutants were more sensitive to CLA than the ΔspoT strain. Alternatively, SpoT may affect the productions of HefC and HefI by regulating the expressions of these transporter genes (hp0939, hp0471, and hp0497), but this requires further study.
Acting as a global transcriptional regulator, (p)ppGpp enhances the bacterial ability to adapt to environmental pressures by controlling replication, transcription, translation, and metabolism (43, 44). Antibiotics are the most powerful weapons in the fight against bacteria, but they induce the stringent response and then result in (p)ppGpp accumulation. (p)ppGpp influences the transcription of genes associated with important metabolic pathways, which consequently affects various bacterial physiological processes, including resistance to antibiotics. (p)ppGpp is implicated in the resistance of bacteria to vancomycin (22), penicillin (45), and amdinocillin (46). A series of experiments demonstrated that (p)ppGpp affected bacterial resistance to drugs through diverse mechanisms (47). During the stringent response, (p)ppGpp inhibits peptidoglycan metabolism and induces bacteria resistant to penicillin (48, 49). (p)ppGpp can also mediate the upregulation of the YojI efflux pump and then enhance microcin resistance in E. coli (50). (p)ppGpp was synthesized by SpoT in H. pylori (17, 18), and SpoT is involved in H. pylori adaptation to nutritional deficiencies, oxygen, and acid stress (19–21). Our work extended those studies to CLA, a completely different kind of stress environment, and showed that the role of ppGpp in CLA resistance included the stimulation of the high expression of hp0939, hp0471, hp0497, and hp1017 transporters, which would reduce the intracellular levels of the antibiotic by exporting CLA. However, the alarmone effect of ppGpp dependency in vivo is still unknown.
Although ppGpp activates the expression of transporters, such stimulation is only partial and perhaps insufficient to explain the function of ppGpp in the resistance of H. pylori to CLA. Therefore, more mechanisms need to be found to interpret all of ppGpp's effects. Thus, it can be suggested that mechanisms unrelated to efflux could potentially contribute to CLA resistance. CLA belongs to a group of macrolides that bind to the peptidyl transferase loop of domain V of the 23S rRNA molecule. This binding interferes with protein elongation and effectively blocks bacterial protein synthesis (9, 10). (p)ppGpp indirectly regulates translation by reducing the transcription of rRNA and protein genes, subsequently restraining the production of the building blocks for ribosome assembly (46). Additionally, rRNA is the target of CLA; thus, the inhibition of rRNA synthesis by (p)ppGpp may reduce the target of CLA, which is probably the reasons why (p)ppGpp is involved in the resistance of H. pylori to CLA.
In conclusion, this study discovered a new mechanism of H. pylori resistance to CLA. Admittedly, the specific regulatory mechanisms of (p)ppGpp for transporters still need further studies. With the present evidence, our study provides support for the clinical treatment and epidemiological investigation of drug resistance in H. pylori.
MATERIALS AND METHODS
Bacterial strains, media, growth conditions, clinical isolation of H. pylori, determination of the MIC, and detection of mutations.
The H. pylori strains used in this study were reference strain 26695, which was kindly provided by Zhang Jianzhong (Chinese Disease Control and Prevention Center), and its isogenic spoT mutant, which was used in our previous report and comes from the collection of our laboratory (52). All E. coli strains used were DH5α and TOP10 (TransGen Biotech, Beijing, China). H. pylori strains were cultivated on Skirrow agar plates with 5% (vol/vol) sheep blood at 37°C and in a microaerobic environment (5% O2, 10% CO2, and 85% N2). The liquid culture medium for H. pylori consisted of brucella broth (BB) containing 10% fetal bovine serum (FBS), and the cells were incubated at 37°C in a shaker set at 120 rpm. The mutant strains were supplemented with kanamycin (Sigma-Aldrich, St. Louis, MO) to 25 μg/ml. E. coli strains were grown in Luria-Bertani medium at 37°C. Ampicillin and CLA were obtained from Sigma-Aldrich (St. Louis, MO).
Twenty-two CLA-resistant and 22 CLA-sensitive clinical isolates were obtained from patients, including those with gastritis, gastric ulcers, and duodenal ulcers and gastric cancer patients, at Qiannan People's Hospital (Guizhou Province). All patients provided informed consent before examination. The MICs of all the clinical and standard strains for CLA were determined by Etest as well as by the agar dilution method reported by Osato et al. (51). The bacteria (optical density at 600 nm [OD600], 0.8) were inoculated on an agar plate containing 2-fold dilutions of CLA (0.0156 to 1 μg/ml). All the plates were incubated at 37°C under microaerobic conditions, and the MIC values were determined. For resistant strains, PCR amplification of the 23S rRNA and sequence analysis were performed to examine the genetic basis of these resistant phenotypes. The above-described experiment details have been previously reported in a published article (53).
Construction of hp0939, hp1017, hp0497, and hp0471 mutant and spoT-complemented strains.
The plasmids pILL570 and pUC18K2 were kindly provided by Agnès Labigne (Unité de Pathogénie Bactérienne des Muqueuses, Institut Pasteur). The construction of hp0939, hp1017, hp0497, and hp0471 mutant strains was identical to the construction of the ΔspoT strain, as described in the literature (52). Briefly, these four genes from the genome of H. pylori 26695 (wild-type and artificially selected resistance strains) were destroyed with the insertion of the nonpolar aphA-3 gene encoding a kanamycin resistance cassette (54). All primers used in these studies are listed in Table 1.
TABLE 1.
Primers used in this studya
| Forward primers |
Reverse primers |
||
|---|---|---|---|
| Name | Sequence (5′–3′) | Name | Sequence (5′–3′) |
| hp1017XF | AACTGCAGAACCGTTTCTCAAGTCGTGG | hp1017XR | CGGAATTCGCGATGTATTCTAACGCCACC |
| hp1017SF | CGGGATCCAGCCCTTTTGTGAGCGTTTT | hp1017SR | CCATCGATTTGGGCATGCTCTGGATTTG |
| hp0939SF | CCATCGATGCCCCATGTTTGAAAAGCCT | hp0939SR | CGGGATCCACGCCAAGCTTGAGTAACAC |
| hp0939XF | CGGAATTCGGCGCGATTTTAATGAGAGCT | hp0939XR | AACTGCAGTGATGTGGAAGTGGCTAGGG |
| hp0471SF | CCATCGATATTGAGCGTAAGCCATCACC | hp0471SR | CGGGATCCACCTCTTCTTGGCCCTTTTT |
| hp0471XF | CGGAATTCTGGGAATGGCTGCAATATCT | hp0471XR | AACTGCAGCCATTCGCTCTTCTTCATCC |
| hp0497XF | AACTGCAGCGCCTTCAGAGTCGGTATCT | hp0497XR | CGGAATTCGCCCTATGGATTGTAGCCCT |
| hp0497SF | CGGGATCCTATGGGGCGAATGTCTCACA | hp0497SR | CCATCGATATGAGAGGGCCGCCTAAAAT |
| hp1172F | AGTCGCTTTAGGGGTGGTTT | hp1172R | AGGGTGTTGTTTCGTCCAAG |
| hp1169F | ATCGGTTTGAGCGCTTTAGA | hp1169R | AAATCACATTCGCCCCTATG |
| hp0473F | GAAAAGCCAGCATGGAAGTC | hp0473R | GGGTTGTTAGCCCCATTTTT |
| hp0888F | CGTTCAATTTCAGCGTGCTA | hp0888R | AAGCACCATTTGCCTTTGAC |
| hp0724F | CGCTCATTCAAGCGTGTTTA | hp0724R | GGATGAGCGCTTTAGAGGTG |
| hp1077F | TTTCTATGGGGCATTCAAGC | hp1077R | TTGTTGCTGGCTTAGGCTTT |
| hp0939F | CACGCCAAGCTTGAGTAACA | hp0939R | ATCAAAGCGGCTTCCAAATA |
| hp0475F | CGAAAAATCGGCTTTGTGTT | hp0475R | GGCTGCGATTAAAGCTCTTG |
| hp0498F | GCCGACAATTTGGTTTGTTT | hp0498R | TGCCCTAAAGCGTCCATAAC |
| hp0313F | CGCATGCTTTTTACCCATTT | hp0313R | AGAAAACACCACCCACGAAG |
| hp0724F | CACCTCTAAAGCGCTCATCC | hp0724R | AAACCCCAGGGATCAAAAAC |
| hp0471F | TGGGGATTTTGATTTTCCAA | hp0471R | GCGCTGCAAACAATCACTAA |
| hp0497F | TTGCTGGCATCACTTCTACG | hp0497R | TGCAAAATCCAACCAATCAA |
| hp1017F | ATGGGCTGTCCAAACAAAAG | hp1017R | TGCGAAACAGACACGCTAAC |
| hp0889F | TAATTCCGTCCTTTCGTTGG | hp0889R | AATTGATGCGCCACCTTAAC |
| hp0607F | GTTCGCCCTTCCAAACCTTT | hp0607R | CGCTCACGCCGTATTTGTTA |
| hp1329F | TGCCCCTTTAGCTTACACCA | hp1329R | TAAACCCAGGACGCTTGCTA |
Underlining indicates nucleotides that were added at the 5′ end to create a restriction site.
The spoT complement was constructed using the chloramphenicol resistance cassette from pMcagA, kindly provided by Wei Hong (Department of Microbiology, Guizhou Medical University, China). Full-length spoT was cloned into pMcagA, and the resulting plasmid was inserted into the middle of the hp0547 (cagA) gene, which provided homologous recombination sites in H. pylori. The vector-transformed spoT mutant strain was constructed by electroporation to obtain the spoT-complemented strain (spoT*). The genotype of the complemented spoT* transformant was verified by PCR and the sequencing of the genomic loci.
Detection of (p)ppGpp accumulation patterns.
The (p)ppGpp production was assayed as previously described (20, 22). Briefly, H. pylori strains were grown overnight in brucella broth containing 10% FBS to the early exponential phase (OD600, approximately 0.4), diluted back to an OD600 of 0.2, and incubated for an additional 2 h. When all strains reached an OD600 of approximately 0.3, samples (OD600, approximately 0.3) of each culture were removed and pelleted via centrifugation at 10,000 rpm for 5 min, after which samples were resuspended in 250 μl of liquid culture medium. 32P (Amersham) was added to 100 μCi ml−1, and the cultures were labeled for 2 h at 37°C. Subsequently, the experimental cells were treated with CLA (0.12 μg ml−1) for 1 h. Aliquots that were labeled for the duration of the experimental cultures were used as controls. After treatment, 50 μl of samples was added to an equal volume of 2 M formic acid. Afterward, at least four freeze-thaw cycles were conducted. The acid extracts were centrifuged briefly, and the supernatant fluids were spotted onto polyethyleneimine-cellulose plates (Sigma-Aldrich), dried, and developed in 1.5 M KH2PO4 (pH 3.4) for approximately 2.5 h. The results were obtained using phosphor screen scanning (Bio-Rad).
Time-kill assay and growth curves.
Time-kill curve analyses were performed by culturing H. pylori in brucella broth medium. Plate-grown bacteria were cultured for 48 h under microaerobic conditions, inoculated into brucella broth with a preliminary OD600 of 0.08, and cultured for another 36 h with shaking at 120 rpm. CLA was added to the broth to a final concentration of 0.6 μg/ml. The CFU were counted at different time intervals (0, 2, 4, 6, and 8 h). Each experiment was repeated at least three times, and the raw data were analyzed using Excel.
To determine the involvement of knockout genes (hp0939, hp0471, hp0497, and spoT) in growth when strains were subjected to the same conditions, the growth kinetics of mutant strains were compared with those of the WT. The growth profiles were monitored in brucella broth with a preliminary OD600 of 0.08 and then cultured for another 144 h at 37°C with shaking. The records were taken every 12 h by determining the OD600 of the test strains. The values stated are the mode values from at least three biological replicates performed in at least three independent occasions.
H33342 accumulation assay.
Accumulation assays were performed as described previously with slight modifications (55). Strains were inoculated in fresh medium and cultured under microaerobic conditions for 48 h at 37°C. The cells were harvested by centrifugation at 6,000 × g for 5 min at room temperature and subsequently suspended in phosphate-buffered saline (PBS). The final OD600 of the suspensions was adjusted to 0.1, and aliquots (180 μl) were transferred into a 96-well plate. The excitation and emission were measured at 355 and 460 nm, respectively, using FLUOstar Optima (Aylesbury, UK). Recordings were started 5 min after the addition of H33342 (25 μM, 20 μl). Readings were taken every 75 s for 30 cycles, and the raw data were analyzed using Excel. Each experiment was repeated at least three times.
Microarray analysis.
The total RNA was extracted from H. pylori left untreated or treated with CLA (0.25 μg/ml) for 20 min using TRIzol reagent (Invitrogen, Carlsbad, CA). For microarray analysis, an Agilent array platform was employed by Shanghai Kangcheng Sheng Biological Engineering Co., Ltd. (China). The sample preparation and microarray hybridization were performed based on the manufacturer's standard protocols. Briefly, 1 μg of total RNA from each sample was amplified and transcribed into fluorescent cRNA using the manufacturer's Agilent's Quick Amp Labeling protocol (version 5.7; Agilent Technologies). The labeled cRNAs (Cy3) were hybridized onto the whole H. pylori 26695 Genome Oligo Array (8×15K; Agilent Technologies). After the slides were washed, the arrays were scanned by the Agilent Scanner G2505B. The Agilent Feature Extraction software (version 10.7.3.1) was used to analyze acquired array images. Quantile normalization and subsequent data processing were performed using the GeneSpring GX v11.5.1 software package (Agilent Technologies). After quantile normalization of the raw data, the genes with at least 2 of 2 samples having flags in “Present or marginal” (“All Targets Value”) were used for further data analysis. Differentially expressed genes between the two samples were identified through fold change filtering (fold change ≥ 1.5). The microarray results represent an independent single experiment.
qRT-PCR.
The total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA). The total RNA (1 μg) was reverse transcribed using the PrimeScript RT Reagent kit with gDNA Eraser (TaKaRa). The primers are shown in Table 1. The resulting cDNA was diluted, and 5 μl of this dilution was used in 20-μl qPCR mixtures containing 0.8 μl of the primer mixtures, 10 μl of SYBR Premix Ex TaqTM (TaKaRa, Otsu, Shiga, Japan), and 4.2 μl of double-distilled water. RT-PCR was done using the ABI Prism 7000 Sequence detection system (Applied Biosystems, Carlsbad, CA) for 1 cycle at 95°C for 30 s and 40 cycles at 95°C for 5 s and 60°C for 31 s. Dissociation curve analysis was performed to verify product homogeneity. The 16S rRNA amplicon was used as an internal control for data normalization. The changes in transcript level were determined by applying the relative quantitative method (ΔΔCT). The threshold cycle (CT) values from all three biological replicates for each strain were compiled. The primers used are listed in Table 1.
Confocal microscopy.
For confocal microscopy, we used the same method as the one described in our previous work (56). To determine bacterial shape and viability, bacterial cells were stained with membrane-permeable and membrane-impermeable fluorescent dyes from the Live/Dead BacLight Bacterial Viability kits (Molecular Probes, Invitrogen, USA) and were observed by confocal microscopy. The H. pylori cells were collected, washed, resuspended in BB liquid medium, and inoculated to the desired optical density at OD600 into BB liquid medium buffered with 10 mM sodium phosphate (pH 6.3). In addition, the cells were supplemented with 10% newborn calf serum or pure water (preliminary OD600, 0.05) and grown under microaerobic conditions. Aliquots were taken at different time points. They were stained with Syto 9 and propidium iodide (PI) for 15 min and washed twice with PBS. The cells were spread on glass slides, covered with mounting medium and coverslips, and visualized by confocal microscopy (Leica TCS SP5; Leica Microsystems GmbH, Wetzlar, Germany). Syto 9 is a green fluorescent membrane-permeable dye that labels all bacteria by staining nucleic acid, whereas PI is a red fluorescent membrane-impermeable dye that labels only bacteria with damaged membranes.
Statistical analysis.
The data are presented as the means ± standard errors of the means (SEM). Statistical significance was determined using an unpaired Student's t test, and the P values were corrected by the Sidak-Bonferroni method for multiple comparisons. P values of <0.05 were considered statistically significant. The results were analyzed using the GraphPad Prism software (GraphPad Software Inc., La Jolla, CA, USA).
Sequence accession number(s).
The microarray data obtained in this study were deposited in the Gene Expression Omnibus database (accession no. GSE84501).
ACKNOWLEDGMENTS
The present research was supported by the National Natural Science Foundation of China (No. 81471991, 81671978, 81401696, 81372680, and 81571960) and the National Basic Research Program of China (973 Program 2012CB911202).
We declare that we have no conflicts of interest.
REFERENCES
- 1.Blaser MJ, Atherton JC. 2004. Helicobacter pylori persistence: biology and disease. J Clin Invest 113:321–333. doi: 10.1172/JCI20925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suzuki H, Hibi T, Marshall BJ. 2007. Helicobacter pylori: present status and future prospects in Japan. J Gastroenterol 42:1–15. doi: 10.1007/s00535-006-1990-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chey WD, Wong BC. 2007. American College of Gastroenterology guideline on the management of Helicobacter pylori infection. Am J Gastroenterol 102:1808–1825. doi: 10.1111/j.1572-0241.2007.01393.x. [DOI] [PubMed] [Google Scholar]
- 4.Malfertheiner P, Megraud F, O'Morain C, Bazzoli F, El-Omar E, Graham D, Hunt R, Rokkas T, Vakil N, Kuipers EJ. 2007. Current concepts in the management of Helicobacter pylori infection: the Maastricht III Consensus Report. Gut 56:772–781. doi: 10.1136/gut.2006.101634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uotani T, Miftahussurur M, Yamaoka Y. 2015. Effect of bacterial and host factors on Helicobacter pylori eradication therapy. Expert Opin Ther Targets 19:1637–1650. doi: 10.1517/14728222.2015.1073261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang M. 2015. High antibiotic resistance rate: a difficult issue for Helicobacter pylori eradication treatment. World J Gastroenterol 21:13432–13437. doi: 10.3748/wjg.v21.i48.13432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pereira JM, Mejia-Ariza R, Ilevbare GA, McGettigan HE, Sriranganathan N, Taylor LS, Davis RM, Edgar KJ. 2013. Interplay of degradation, dissolution and stabilization of clarithromycin and its amorphous solid dispersions. Mol Pharm 10:4640–4653. doi: 10.1021/mp400441d. [DOI] [PubMed] [Google Scholar]
- 8.Xiong LJ, Tong Y, Wang Z, Mao M. 2013. Detection of clarithromycin-resistant Helicobacter pylori by stool PCR in children: a comprehensive review of literature. Helicobacter 18:89–101. doi: 10.1111/hel.12016. [DOI] [PubMed] [Google Scholar]
- 9.Gerrits MM, van Vliet AH, Kuipers EJ, Kusters JG. 2006. Helicobacter pylori and antimicrobial resistance: molecular mechanisms and clinical implications. Lancet Infect Dis 6:699–709. doi: 10.1016/S1473-3099(06)70627-2. [DOI] [PubMed] [Google Scholar]
- 10.Versalovic J, Shortridge D, Kibler K, Griffy MV, Beyer J, Flamm RK, Tanaka SK, Graham DY, Go MF. 1996. Mutations in 23S rRNA are associated with clarithromycin resistance in Helicobacter pylori. Antimicrob Agents Chemother 40:477–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirata K, Suzuki H, Nishizawa T, Tsugawa H, Muraoka H, Saito Y, Matsuzaki J, Hibi T. 2010. Contribution of efflux pumps to clarithromycin resistance in Helicobacter pylori. J Gastroenterol Hepatol 25(Suppl 1):S75–S79. doi: 10.1111/j.1440-1746.2009.06220.x. [DOI] [PubMed] [Google Scholar]
- 12.Smiley R, Bailey J, Sethuraman M, Posecion N, Showkat AM. 2013. Comparative proteomics analysis of sarcosine insoluble outer membrane proteins from clarithromycin resistant and sensitive strains of Helicobacter pylori. J Microbiol 51:612–618. doi: 10.1007/s12275-013-3029-5. [DOI] [PubMed] [Google Scholar]
- 13.O'Farrell PH. 1978. The suppression of defective translation by ppGpp and its role in the stringent response. Cell 14:545–557. doi: 10.1016/0092-8674(78)90241-6. [DOI] [PubMed] [Google Scholar]
- 14.Chatterji D, Fujita N, Ishihama A. 1998. The mediator for stringent control, ppGpp, binds to the beta-subunit of Escherichia coli RNA polymerase. Genes Cells 3:279–287. doi: 10.1046/j.1365-2443.1998.00190.x. [DOI] [PubMed] [Google Scholar]
- 15.Toulokhonov II, Shulgina I, Hernandez VJ. 2001. Binding of the transcription effector ppGpp to Escherichia coli RNA polymerase is allosteric, modular, and occurs near the N terminus of the beta'-subunit. J Biol Chem 276:1220–1225. doi: 10.1074/jbc.M007184200. [DOI] [PubMed] [Google Scholar]
- 16.Magnusson LU, Farewell A, Nystrom T. 2005. ppGpp: a global regulator in Escherichia coli. Trends Microbiol 13:236–242. doi: 10.1016/j.tim.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 17.Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, Smith DR, Noonan B, Guild BC, DeJonge BL, Carmel G, Tummino PJ, Caruso A, Uria-Nickelsen M, Mills DM, Ives C, Gibson R, Merberg D, Mills SD, Jiang Q, Taylor DE, Vovis GF, Trust TJ. 1999. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397:176–180. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- 18.Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 19.Mouery K, Rader BA, Gaynor EC, Guillemin K. 2006. The stringent response is required for Helicobacter pylori survival of stationary phase, exposure to acid, and aerobic shock. J Bacteriol 188:5494–5500. doi: 10.1128/JB.00366-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wells DH, Gaynor EC. 2006. Helicobacter pylori initiates the stringent response upon nutrient and pH downshift. J Bacteriol 188:3726–3729. doi: 10.1128/JB.188.10.3726-3729.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou YN, Coleman WJ, Yang Z, Yang Y, Hodgson N, Chen F, Jin DJ. 2008. Regulation of cell growth during serum starvation and bacterial survival in macrophages by the bifunctional enzyme SpoT in Helicobacter pylori. J Bacteriol 190:8025–8032. doi: 10.1128/JB.01134-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abranches J, Martinez AR, Kajfasz JK, Chavez V, Garsin DA, Lemos JA. 2009. The molecular alarmone (p)ppGpp mediates stress responses, vancomycin tolerance, and virulence in Enterococcus faecalis. J Bacteriol 191:2248–2256. doi: 10.1128/JB.01726-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corrigan RM, Bellows LE, Wood A, Grundling A. 2016. ppGpp negatively impacts ribosome assembly affecting growth and antimicrobial tolerance in Gram-positive bacteria. Proc Natl Acad Sci U S A 113:E1710–E1719. doi: 10.1073/pnas.1522179113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu J, Tozawa Y, Lai C, Hayashi H, Ochi K. 2002. A rifampicin resistance mutation in the rpoB gene confers ppGpp-independent antibiotic production in Streptomyces coelicolor A3(2). Mol Genet Genomics 268:179–189. doi: 10.1007/s00438-002-0730-1. [DOI] [PubMed] [Google Scholar]
- 25.Durfee T, Hansen AM, Zhi H, Blattner FR, Jin DJ. 2008. Transcription profiling of the stringent response in Escherichia coli. J Bacteriol 190:1084–1096. doi: 10.1128/JB.01092-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nascimento MM, Lemos JA, Abranches J, Lin VK, Burne RA. 2008. Role of RelA of Streptococcus mutans in global control of gene expression. J Bacteriol 190:28–36. doi: 10.1128/JB.01395-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Higgins CF. 2007. Multiple molecular mechanisms for multidrug resistance transporters. Nature 446:749–757. doi: 10.1038/nature05630. [DOI] [PubMed] [Google Scholar]
- 28.Li XZ, Nikaido H. 2009. Efflux-mediated drug resistance in bacteria: an update. Drugs 69:1555–1623. doi: 10.2165/11317030-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. 1995. Genes acrA and acrB encode a stress-induced efflux system of Escherichia coli. Mol Microbiol 16:45–55. doi: 10.1111/j.1365-2958.1995.tb02390.x. [DOI] [PubMed] [Google Scholar]
- 30.Li XZ, Nikaido H, Poole K. 1995. Role of mexA-mexB-oprM in antibiotic efflux in Pseudomonas aeruginosa. Antimicrob Agents Chemother 39:1948–1953. doi: 10.1128/AAC.39.9.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bina JE, Alm RA, Uria-Nickelsen M, Thomas SR, Trust TJ, Hancock RE. 2000. Helicobacter pylori uptake and efflux: basis for intrinsic susceptibility to antibiotics in vitro. Antimicrob Agents Chemother 44:248–254. doi: 10.1128/AAC.44.2.248-254.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu ZQ, Zheng PY, Yang PC. 2008. Efflux pump gene hefA of Helicobacter pylori plays an important role in multidrug resistance. World J Gastroenterol 14:5217–5222. doi: 10.3748/wjg.14.5217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Amsterdam K, Bart A, van der Ende A. 2005. A Helicobacter pylori TolC efflux pump confers resistance to metronidazole. Antimicrob Agents Chemother 49:1477–1482. doi: 10.1128/AAC.49.4.1477-1482.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alekshun MN, Levy SB. 2007. Molecular mechanisms of antibacterial multidrug resistance. Cell 128:1037–1050. doi: 10.1016/j.cell.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 35.Kutschke A, de Jonge BL. 2005. Compound efflux in Helicobacter pylori. Antimicrob Agents Chemother 49:3009–3010. doi: 10.1128/AAC.49.7.3009-3010.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iwamoto A, Tanahashi T, Okada R, Yoshida Y, Kikuchi K, Keida Y, Murakami Y, Yang L, Yamamoto K, Nishiumi S, Yoshida M, Azuma T. 2014. Whole-genome sequencing of clarithromycin resistant Helicobacter pylori characterizes unidentified variants of multidrug resistant efflux pump genes. Gut Pathog 6:27. doi: 10.1186/1757-4749-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martinez JL, Fajardo A, Garmendia L, Hernandez A, Linares JF, Martinez-Solano L, Sanchez MB. 2009. A global view of antibiotic resistance. FEMS Microbiol Rev 33:44–65. doi: 10.1111/j.1574-6976.2008.00142.x. [DOI] [PubMed] [Google Scholar]
- 38.Martinez JL, Rojo F. 2011. Metabolic regulation of antibiotic resistance. FEMS Microbiol Rev 35:768–789. doi: 10.1111/j.1574-6976.2011.00282.x. [DOI] [PubMed] [Google Scholar]
- 39.Voss BJ, Loh JT, Hill S, Rose KL, McDonald WH, Cover TL. 2015. Alteration of the Helicobacter pylori membrane proteome in response to changes in environmental salt concentration. Proteomics Clin Appl 9:1021–1034. doi: 10.1002/prca.201400176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wen Y, Marcus EA, Matrubutham U, Gleeson MA, Scott DR, Sachs G. 2003. Acid-adaptive genes of Helicobacter pylori. Infect Immun 71:5921–5939. doi: 10.1128/IAI.71.10.5921-5939.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tegtmeyer N, Moodley Y, Yamaoka Y, Pernitzsch SR, Schmidt V, Traverso FR, Schmidt TP, Rad R, Yeoh KG, Bow H, Torres J, Gerhard M, Schneider G, Wessler S, Backert S. 2016. Characterisation of worldwide Helicobacter pylori strains reveals genetic conservation and essentiality of serine protease HtrA. Mol Microbiol 99:925–944. doi: 10.1111/mmi.13276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Binh TT, Shiota S, Suzuki R, Matsuda M, Trang TT, Kwon DH, Iwatani S, Yamaoka Y. 2014. Discovery of novel mutations for clarithromycin resistance in Helicobacter pylori by using next-generation sequencing. J Antimicrob Chemother 69:1796–1803. doi: 10.1093/jac/dku050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Potrykus K, Cashel M. 2008. (p) ppGpp: still magical? Annu Rev Microbiol 62:35–51. doi: 10.1146/annurev.micro.62.081307.162903. [DOI] [PubMed] [Google Scholar]
- 44.Srivatsan A, Wang JD. 2008. Control of bacterial transcription, translation and replication by (p)ppGpp. Curr Opin Microbiol 11:100–105. doi: 10.1016/j.mib.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 45.Vinella D, D'Ari R, Jaffe A, Bouloc P. 1992. Penicillin binding protein 2 is dispensable in Escherichia coli when ppGpp synthesis is induced. EMBO J 11:1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bouloc P, Vinella D, D'Ari R. 1992. Leucine and serine induce mecillinam resistance in Escherichia coli. Mol Gen Genet 235:242–246. doi: 10.1007/BF00279366. [DOI] [PubMed] [Google Scholar]
- 47.Wu J, Long Q, Xie J. 2010. (p)ppGpp and drug resistance. J Cell Physiol 224:300–304. doi: 10.1002/jcp.22158. [DOI] [PubMed] [Google Scholar]
- 48.Eymann C, Homuth G, Scharf C, Hecker M. 2002. Bacillus subtilis functional genomics: global characterization of the stringent response by proteome and transcriptome analysis. J Bacteriol 184:2500–2520. doi: 10.1128/JB.184.9.2500-2520.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hesketh A, Chen WJ, Ryding J, Chang S, Bibb M. 2007. The global role of ppGpp synthesis in morphological differentiation and antibiotic production in Streptomyces coelicolor A3(2). Genome Biol 8:R161. doi: 10.1186/gb-2007-8-8-r161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pomares MF, Vincent PA, Farias RN, Salomon RA. 2008. Protective action of ppGpp in microcin J25-sensitive strains. J Bacteriol 190:4328–4334. doi: 10.1128/JB.00183-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Osato MS, Reddy R, Reddy SG, Penland RL, Graham DY. 2001. Comparison of the Etest and the NCCLS-approved agar dilution method to detect metronidazole and clarithromycin resistant Helicobacter pylori. Int J Antimicrob Agents 17:39–44. doi: 10.1016/S0924-8579(00)00320-4. [DOI] [PubMed] [Google Scholar]
- 52.Sun Y, Li X, Li W, Zhao M, Wang L, Liu S, Zeng J, Liu Z, Jia J. 2012. Proteomic analysis of the function of spot in Helicobacter pylori anti-oxidative stress in vitro and colonization in vivo. J Cell Biochem 113:3393–3402. doi: 10.1002/jcb.24215. [DOI] [PubMed] [Google Scholar]
- 53.Qi T, Pan K, Wang F, Wei G, Jiang Y, Zhang R, Zheng J, Zhang D, Chen Z. 2015. Clarithromycin resistance and 23S rRNA gene mutation characteristics of Helicobacter pylori isolates from patients in Qiannan Autonomous Prefecture. World Chinese J Digestol 2015(18):2901–2906. doi: 10.11569/wcjd.v23.i18.2901. [DOI] [Google Scholar]
- 54.Contreras M, Thiberge JM, Mandrand-Berthelot MA, Labigne A. 2003. Characterization of the roles of NikR, a nickel-responsive pleiotropic autoregulator of Helicobacter pylori. Mol Microbiol 49:947–963. doi: 10.1046/j.1365-2958.2003.03621.x. [DOI] [PubMed] [Google Scholar]
- 55.Coldham NG, Webber M, Woodward MJ, Piddock LJ. 2010. A 96-well plate fluorescence assay for assessment of cellular permeability and active efflux in Salmonella enterica serovar Typhimurium and Escherichia coli. J Antimicrob Chemother 65:1655–1663. doi: 10.1093/jac/dkq169. [DOI] [PubMed] [Google Scholar]
- 56.Shan Y, Lu X, Han Y, Li X, Wang X, Shao C, Wang L, Liu Z, Tang W, Sun Y, Jia J. 2015. Helicobacter pylori outer membrane protein 18 (Hp1125) is involved in persistent colonization by evading interferon-gamma signaling. Biomed Res Int 2015:571280. doi: 10.1155/2015/571280. [DOI] [PMC free article] [PubMed] [Google Scholar]