

ABSTRACT
Enterococci are serious opportunistic pathogens that are resistant to many cell wall-targeting antibiotics. The CroRS two-component signaling system responds to antibiotic-mediated cell wall stress and is critical for resistance to cell wall-targeting antibiotics in Enterococcus faecalis. Here, we identify and characterize an orthologous two-component system found in Enterococcus faecium that is functionally equivalent to the CroRS system of E. faecalis. Deletion of croRS in E. faecium resulted in marked susceptibility to cell wall-targeting agents including cephalosporins and bacitracin, as well as moderate susceptibility to ampicillin and vancomycin. As in E. faecalis, exposure to bacitracin and vancomycin stimulates signaling through the CroRS system in E. faecium. Moreover, the CroRS system is critical in E. faecium for enhanced beta-lactam resistance mediated by overexpression of Pbp5. Expression of a Pbp5 variant that confers enhanced beta-lactam resistance cannot overcome the requirement for CroRS function. Thus, the CroRS system is a conserved signaling system that responds to cell wall stress to promote intrinsic resistance to important cell wall-targeting antibiotics in clinically relevant enterococci.
KEYWORDS: enterococcus, antibiotic resistance, two-component regulatory systems
INTRODUCTION
Enterococcus faecalis and Enterococcus faecium represent serious opportunistic pathogens that are responsible for many nosocomial infections. Treatment of enterococcal infections is particularly challenging due to intrinsic and acquired resistance toward many clinically relevant antibiotics, including beta-lactams, aminoglycosides, glycopeptides, and trimethoprim (1). Because all clinical isolates of E. faecalis and E. faecium are intrinsically resistant to cephalosporins (a subset of beta-lactam antibiotics), disabling cephalosporin resistance with small molecule therapeutics may be a viable strategy to overcome antibiotic-resistant enterococcal infections. Both species use transpeptidase activity of a low-affinity penicillin-binding protein (Pbp5) in cooperation with the glycosyltransferase activity of the penicillin-binding proteins (PBPs) PonA or PbpF to continue transpeptidation and transglycosylation reactions required for cell wall assembly during cephalosporin exposure (2–5). However, additional determinants contributing to cephalosporin resistance have also been explored in E. faecalis and E. faecium.
In E. faecalis, two enzymes involved in cell wall synthesis (the UDP-N-acetylglucosamine 1-carboxyvinyl transferase MurAA [6] and the alanine transferase BppA2 [7]) are known to be required for normal cephalosporin resistance. In addition, two signal transduction pathways mediate intrinsic resistance to cephalosporins and other cell wall-targeting antibiotics. One pathway includes a eukaryotic-like Ser/Thr kinase, IreK, and its cognate phosphatase, IreP, which act antagonistically to regulate a pathway leading to cephalosporin resistance (8, 9). An ortholog of IreK in E. faecium has also been implicated in cephalosporin resistance of that species (10). In E. faecalis, a substrate for phosphorylation by IreK has been described, designated IreB, which acts as a negative regulator of the pathway (11). However, the specific output of the pathway that drives cephalosporin resistance remains unknown. In addition to the IreK/IreP signaling pathway, the two-component signal transduction system (TCS) consisting of the CroS sensor kinase and its cognate response regulator CroR has a role in resistance to cell wall-targeting antibiotics. Disruption of the CroRS TCS in E. faecalis renders strains more sensitive to diverse cell wall-targeting agents such as cephalosporins, ampicillin, bacitracin, and vancomycin (12, 13). Consistent with a role for the CroRS TCS in responding to antibiotic-mediated cell wall stress, these agents can also stimulate CroR-dependent transcription (12). However, only three genes regulated by CroR have been identified (12, 14, 15), with croR itself the only of those that possesses a clear role in antimicrobial resistance. Thus, the downstream effectors in the CroR regulon that drive resistance remain to be identified.
Although E. faecium is resistant to cephalosporins, most studies have analyzed ampicillin resistance in clinical isolates. High levels of ampicillin resistance have been associated with mutations in Pbp5. However, specific variants do not always correlate with MIC values in different E. faecium lineages (16–18), implying that additional factors modulate ampicillin resistance. A genome-wide study identified several determinants required for ampicillin resistance in E. faecium, including the l,d-transpeptidase Ldtfm, the d-alanyl–d-alanine carboxypeptidase DdcP, and the glycosyltransferase Pgt (19). The Ldtfm pathway was also identified as providing high-level ampicillin resistance after successive in vitro selections for ampicillin resistance (20–23). Collectively, these studies indicate that factors involved in enterococcal cell wall remodeling, distinct from the traditional biosynthetic PBPs, modulate resistance to ampicillin in E. faecium. However, the extent to which they also influence resistance to other beta-lactams such as cephalosporins remains largely unknown. Moreover, these factors are poorly conserved in E. faecalis, which tends to be considerably less prone to development of enhanced ampicillin resistance compared to E. faecium.
Ideally, any target for new therapeutics designed to disable enterococcal resistance to cephalosporins will be conserved in both E. faecalis and E. faecium. To explore whether mechanisms mediating cephalosporin resistance in E. faecalis are conserved in E. faecium, we identified and functionally characterized a TCS encoded in the E. faecium genome that is homologous to the CroRS TCS of E. faecalis. We report that deletion of the E. faecium croRS orthologs render E. faecium more susceptible to cell wall-targeting agents, some of which were observed to stimulate CroRS signaling in a wild-type strain. Moreover, the CroRS system is critical in E. faecium for enhanced cephalosporin and ampicillin resistance mediated by overexpression of Pbp5 and by expression of a Pbp5 variant that confers enhanced beta-lactam resistance. Thus, the CroRS system represents a conserved signaling system that responds to cell wall stress to promote intrinsic resistance to important cell wall-targeting antibiotics in clinically relevant enterococci.
RESULTS
Bioinformatic identification of CroRS orthologs in E. faecium.
A BLASTn search of the E. faecium DO genome (24) with the E. faecalis croR and croS genes revealed candidates for croR and croS (HMPREF0351_12687 and HMPREF0351_12688). These genes are reciprocal best BLASTn hits with croR and croS of E. faecalis, suggesting they encode an orthologous TCS. Because functional studies (below) revealed that the E. faecium genes indeed encode an orthologous TCS, we will henceforth refer to the croRS genes and proteins using subscripts (“Efs” for E. faecalis and “Efm” for E. faecium). Figure 1A depicts the genomic architecture of the croRSEfs locus from the sequenced genome of E. faecalis OG1RF (25). The CroRS TCS is considered an “isolated” TCS, in that croREfs and croSEfs are encoded on a bicistronic message lacking the downstream gene, serS (12). The genomic architecture surrounding the croRSEfm locus is identical to that of E. faecalis. The proteins encoded by the croR and croS orthologs exhibit high similarity over their entire length: CroREfs and CroREfm are 90% identical (96% similar), and CroSEfs is 74% identical (87% similar) to CroSEfm. Alignment of the CroS protein sequences from E. faecalis and E. faecium reveal high conservation of key functional domains (e.g., the dimerization and phosphoryl acceptor domain and ATPase domain) in both (Fig. 1B). Collectively, these data led us to hypothesize that CroRSEfm is functionally orthologous to CroRSEfs.
FIG 1.

Comparison of the croRS loci in E. faecalis and E. faecium. (A) Genomic organization of the croRS locus in the sequenced genomes of E. faecalis OG1RF (top locus numbers) and E. faecium DO (bottom locus numbers). (B) CLUSTAL W alignment of CroS from E. faecalis OG1RF and E. faecium DO. Protein domains are underlined as predicted by SMART. Cylinders denote transmembrane domains, the rectangle with horizontal stripes denotes the dimerization and phosphoryl acceptor domain, and the rectangle with vertical stripes denotes the histidine kinase-like ATPase domain.
We previously identified a second TCS (CisRS), found in a subset of E. faecalis strains, that is capable of influencing the activity of CroRSEfs under certain conditions due to mutual overlap in the identity of so-called “specificity” residues of the CroS and CisS kinases that dictate response regulator specificity for TCS kinases (26–28). However, extensive studies indicated that “cross talk” between CisS and CroREfs only occurs in the absence of CroSEfs and is not physiologically relevant (26). Nevertheless, we searched for homologs of CisRS in sequenced E. faecium genomes, but none were found. To probe whether another TCS found in E. faecium had the potential to “cross talk” with CroRSEfm, we analyzed the specificity residues of all TCS histidine kinases found in the genome of E. faecium strain 1,141,733. No kinases were found that possessed substantially similar specificity residues. The closest match to the specificity residues of CroSEfm was with EFSG_00540, which shares only 4 of 9 specificity residues with CroSEfm. In comparison, CisS—the only kinase capable of “cross talk” with CroSEfs—shares 6 of 9 specificity residues with CroSEfs. Moreover, E. faecalis encodes three other TCS kinases that share 5, 3, and 3 specificity residues with CroSEfs, respectively, and yet none of those are capable of interacting with CroREfs. Given the divergence of specificity residues in TCS kinases of E. faecium 1,141,733, we conclude that E. faecium 1,141,733 does not possess another TCS kinase that is likely to interact with CroREfm. In this scenario, the phosphorylation state of CroREfm would therefore be controlled exclusively by CroSEfm.
Deletion of CroRSEfm decreases resistance to cell wall-targeting agents.
To probe the biological functions of CroRSEfm, we made an in-frame deletion of the croRSEfm locus in E. faecium 1,141,733 and performed phenotypic analyses of the resulting mutant. As with the E. faecalis ΔcroRSEfs mutant, the E. faecium ΔcroRSEfm mutant exhibited substantial loss of resistance to cell wall-targeting agents such as expanded-spectrum to “fourth-generation” cephalosporins and to bacitracin, with modest effects on resistance to ampicillin and vancomycin (Table 1). Resistance to the protein synthesis inhibitor chloramphenicol was unaltered in the absence of CroRS. To test for complementation of the ΔcroRSEfm mutation, we produced CroRSEfm from a plasmid with an inducible promoter, revealing an inducer-dependent enhancement of ceftriaxone resistance (Table 2). Although full complementation to wild-type levels of ceftriaxone resistance was not observed, we suspect this is likely due to differences in the kinetics or level of croRSEfm expression from the plasmid during the course of the MIC experiment. Consistent with this hypothesis, immunoblot analysis revealed that CroRSEfm levels from the plasmid expression platform were aberrant (elevated) compared to normal chromosomal expression in exponentially growing cells (Fig. 2). It remains unclear precisely how overexpression of CroRSEfm interferes with normal cephalosporin resistance pathway function, but improper localization of CroRSEfm in the membrane, alteration of the kinase/phosphatase balance of CroRSEfm, or adverse effects on other membrane proteins might play a role. Regardless of the mechanism, overall, these results indicate that CroRSEfm specifically influences resistance to cell wall-targeting compounds in E. faecium in a similar manner as in E. faecalis.
TABLE 1.
MICs of various antibiotics for ΔcroRS mutants
| Antibiotic | Median MIC (μg/ml)a |
|||
|---|---|---|---|---|
|
E. faecium |
E. faecalis |
|||
| Wild type | ΔcroRS mutant | Wild type | ΔcroRS mutant | |
| Cefadroxil (narrow spectrum) | 64 | 32 | 64 | 32 |
| Cefuroxime (expanded spectrum) | 512 | 64 | 32 | 4 |
| Ceftriaxone (broad spectrum) | 64 | 1 | 64 | 8 |
| Cefepime (fourth generation) | 512 | 4 | 16 | 8 |
| Ampicillin | 1 | 0.25 | 1 | 0.5 |
| Bacitracin | 64 | 8 | 64 | 8 |
| Vancomycin | 0.5 | 0.25 | 2 | 0.5 |
| Chloramphenicol | 4 | 4 | 4 | 4 |
Median MICs are reported from ≥2 biological replicates. The strains analyzed were wild-type E. faecalis OG1, E. faecalis ΔcroRS mutant SB35, wild-type E. faecium 1,141,733, and E. faecium ΔcroRS mutant JL537.
TABLE 2.
Ceftriaxone resistance of E. faecium and E. faecalis ΔcroRS mutants expressing croRSEfm
| Strain/plasmida | Median MIC (μg/ml)b |
|
|---|---|---|
| Without cCF10 | With cCF10 | |
| E. faecium | ||
| Wild type/vector | 128 | 128 |
| ΔcroRSEfm mutant/vector | 2 | 2 |
| ΔcroRSEfm mutant/croRSEfm | 2 | 16 |
| E. faecalis | ||
| Wild type/vector | 128 | 128 |
| ΔcroRSEfs mutant/vector | 8 | 8 |
| ΔcroRSEfs mutant/croRSEfm | 8 | 128 |
The strains and plasmids analyzed were as follows: wild-type E. faecium, 1,141,733; E. faecium ΔcroRS mutant, JL537, wild-type E. faecalis, OG1; E. faecalis ΔcroRS mutant, SB35; vector, pJLL105; and croRSEfm, pJLL160.
Median MICs are reported from ≥2 biological replicates.
FIG 2.
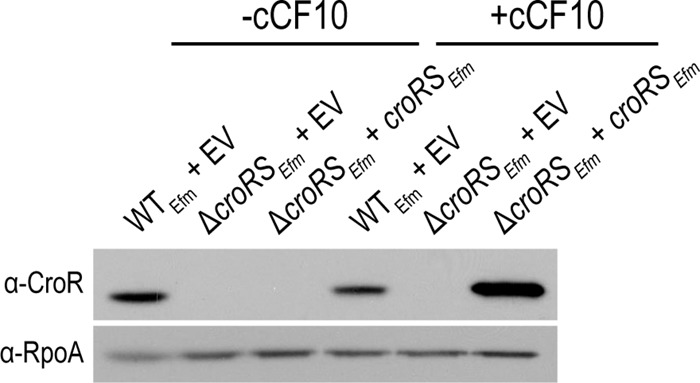
Expression of CroREfm in the E. faecium ΔcroRS mutant. Standard SDS-PAGE and immunoblot analysis of exponentially growing wild-type (WTEfm) and ΔcroRSEfm E. faecium 1,141,733 strains. Strains carry empty vector (EV) or croRS-expressing cCF10-inducible plasmids. The strains are wild-type E. faecium 1,141,733 and JL537 with pJLL105 and pJLL160.
To determine whether CroRSEfm initiates signal transduction in response to cell wall-targeting agents, we monitored the phosphorylation state of CroREfm after exposure of E. faecium cells to various antibiotics. Upon sensing their signal, histidine kinases autophosphorylate and subsequently transfer the phosphoryl group to their cognate response regulators. We monitored this process using Phos-Tag SDS-PAGE and immunoblotting for CroR (26). In E. faecalis, a slower-mobility isoform of CroREfs is observed with Phos-Tag SDS-PAGE after treatment of cells with bacitracin and vancomycin (Fig. 3A), reflecting phosphorylation of CroREfs in response to antibiotic-mediated cell wall stress. Under the conditions tested, robust phosphorylation of CroREfs is not observed after treatment with ceftriaxone or ampicillin, nor (as expected) after treatment with the ribosomal-targeting antibiotic chloramphenicol. In E. faecium, a small but detectable amount of phosphorylated CroREfm was observed even in the absence of antibiotic stress (Fig. 3B). Phosphorylation of CroREfm was markedly enhanced after treatment of cells with bacitracin and vancomycin, as observed in E. faecalis. Thus, CroRSEfm responds to antibiotic-mediated cell wall stress by enhancing phosphorylation of CroREfm, validating CroRSEfm as functional orthologs of CroRSEfs.
FIG 3.
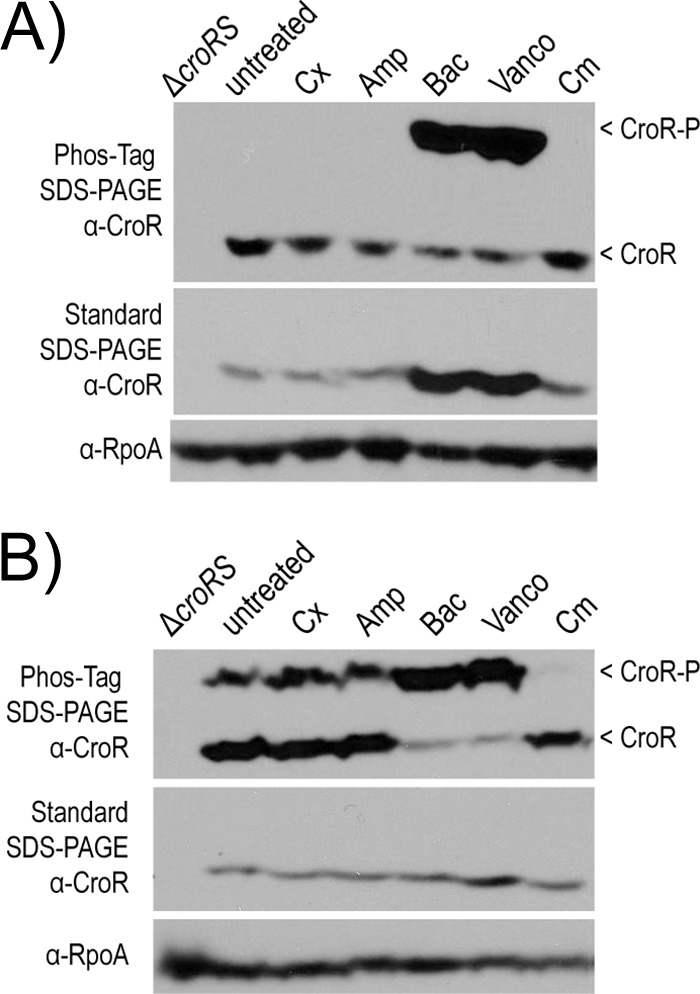
Analysis of CroR phosphorylation in whole-cell lysates. Phos-Tag SDS-PAGE and standard SDS-PAGE was followed by immunoblot analyses in response to various insults in E. faecalis OG1 (A) and E. faecium 1,141,733 (B). Exponentially growing cells were treated with ceftriaxone (Cx), ampicillin (Amp), bacitracin (Bac), vancomycin (Vanco), or chloramphenicol (Cm) for 30 min. The results are representative of ≥2 experiments. A subunit of RNA polymerase (RpoA) was used as a loading control.
The reason a fraction of CroREfm is phosphorylated in the absence of antibiotic stress is unclear. The population of CroREfm-P vanished upon treatment of cells with chloramphenicol (Fig. 3B). Similar results were obtained with the protein synthesis inhibitor fusidic acid and the DNA gyrase inhibitor norfloxacin (data not shown). Because these agents are expected to quickly arrest cellular growth by inhibiting essential cellular functions, we tested whether small amounts of CroREfm-P accumulated during cell growth itself. Exponentially growing wild-type E. faecium 1,141,733 cells were collected by centrifugation and resuspended in either growth medium (Mueller-Hinton broth [MHB], as a control for continued growth) or PBS (to halt growth). As discussed above, cells suspended in growth medium contained a small fraction of CroREfm-P. In contrast, CroREfm-P vanished in cells suspended in PBS (Fig. 4). We conclude that active growth leads to phosphorylation of a small fraction of CroREfm and that treatment of cells with chloramphenicol (or fusidic acid or norfloxacin) halts growth, leading to loss of this phosphorylated population.
FIG 4.
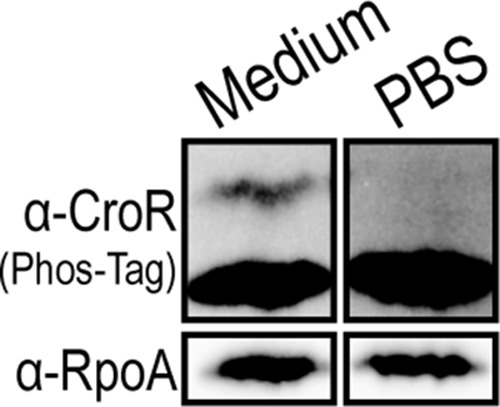
Analysis of CroR phosphorylation after growth arrest. Exponentially growing E. faecium 1,141,733 cells were collected by centrifugation and resuspended in either growth medium (for continued growth) or PBS (to halt growth). Phos-Tag SDS-PAGE and immunoblot analysis of whole-cell lysates was used to visualize CroR-P. The results are representative of ≥3 experiments. A subunit of RNA polymerase (RpoA) was used as a loading control.
E. faecium CroRS can drive resistance in both E. faecium and E. faecalis.
The molecular nature of the direct signal that triggers signaling through CroRSEfs is unknown. Similarly, the identity of the specific output of CroRSEfs signaling that mediates antibiotic resistance (i.e., the antibiotic resistance effectors in the CroREfs regulon) is unknown. To gain insight into whether CroRSEfm and CroRSEfs sense a similar direct signal and drive common molecular responses we tested for heterologous complementation. To do so, CroRSEfm was produced from an inducible promoter in an E. faecalis croRSEfs mutant. We found that CroRSEfm could enhance ceftriaxone resistance of the heterologous host in the presence of inducer (Table 2). This heterologous complementation not only confirms that CroRSEfm is a functional ortholog of CroRSEfs, but also suggests that CroRSEfm responds to the same molecular signal and drives a common molecular response in both E. faecalis and E. faecium upon activation by cell wall-targeting antibiotics.
Expression of Pbp5 is insufficient to restore beta-lactam resistance to the ΔcroRSEfm mutant.
The output of the CroRS TCS that promotes cephalosporin resistance is unknown. To test if enhanced expression of the low-affinity Pbp5 could be an output of CroRS in E. faecalis, Comenge et al. constitutively expressed Pbp5Efs in a ΔcroRSEfs mutant strain but observed no change in beta-lactam resistance (i.e., the mutant strain was still susceptible to beta-lactams) (12). Moreover, PBP labeling studies revealed no differences in the pattern or amount of PBP labeling between the wild-type and ΔcroRSEfs mutant strains (12), indicating that CroRS impacts resistance via another mechanism. To determine whether CroRSEfm influences cephalosporin resistance through Pbp5 in E. faecium, we used Bocillin-FL to label PBPs on exponential-phase wild-type and ΔcroRSEfm mutant E. faecium strains but found no differences in the pattern or amount of PBP labeling (not shown). In addition, we expressed E. faecium pbp5 in wild-type and ΔcroRSEfm mutant strains. As expected, expression of pbp5Efm derived from E. faecium 1,141,733 enhanced ceftriaxone and ampicillin resistance in wild-type E. faecium (Table 3). However, as previously observed in E. faecalis, production of Pbp5 derived from E. faecium 1,141,733 (Pbp5Efm733) was essentially unable to enhance beta-lactam resistance in the ΔcroRSEfm mutant, suggesting that a CroRSEfm-dependent function is required for Pbp5Efm to mediate resistance. To determine whether a variant form of Pbp5Efm associated with enhanced ampicillin resistance could bypass the requirement for CroRSEfm, we expressed pbp5Efm from E. faecium 1,231,408 in the ΔcroRSEfm mutant. Pbp5Efm408 carries two amino acid changes (M485A, and insertion of Ser at position 466) that have been found to result in reduced beta-lactam binding by Pbp5Efm and enhanced beta-lactam resistance (17). Despite these changes, production of Pbp5Efm408 was similarly unable to enhance beta-lactam resistance in the ΔcroRSEfm mutant (Table 3). Collectively, these data indicate that CroRSEfm is essential for Pbp5-mediated beta-lactam resistance in E. faecium via a mechanism that is independent of Pbp5Efm expression.
TABLE 3.
Resistance to ceftriaxone and ampicillin of E. faecium strains expressing pbp5 alleles from E. faecium strain 1,141,733 or strain 1,231,408
| Strain/plasmida | Median MIC (μg/ml)b |
|
|---|---|---|
| Ceftriaxone | Ampicillin | |
| Wild type/vector | 128 | 4 |
| Wild type/pbp5733 | 1,024 | 16 |
| Wild type/pbp5408 | 1,024 | 16 |
| ΔcroRSEfm mutant/vector | 1 | 1 |
| ΔcroRSEfm mutant/pbp5733 | 1 | 2 |
| ΔcroRSEfm mutant/pbp5408 | 1 | 2 |
The strains and plasmids analyzed were as follows: wild-type E. faecium, 1,141,733; E. faecium ΔcroRS mutant, JL537; vector, pJH123; pbp5733, pSLK252; and pbp5408, pSLK253.
Median MICs are reported from ≥2 biological replicates.
DISCUSSION
An understanding of antibiotic resistance mechanisms used by the opportunistic pathogens Enterococcus faecalis and Enterococcus faecium will provide an essential foundation for the development of new therapies to treat enterococcal infections or to prevent the expansion of multidrug-resistant enterococcal isolates during antibiotic therapy. With that in mind, we explored the role of the CroRSEfm TCS in intrinsic resistance to cell wall-targeting antibiotics in E. faecium. We found that the CroRSEfm signaling pathway is activated in response to cell wall-targeting antibiotics, which is consistent with a model in which CroRSEfm monitors the cell envelope for stress and initiates an adaptive biological response by enhancing the expression of (as-yet-unknown) downstream effector genes when envelope stress is detected. As a sensor kinase embedded in the cytoplasmic membrane, CroSEfm is ideally positioned to respond to insults affecting the integrity of the cell-envelope.
Our data indicate that the CroRSEfm TCS is activated during normal growth (i.e., in the absence of exogenous antimicrobials), albeit at a low level (Fig. 3 and 4). A small fraction of CroREfm exists in the phosphorylated state in growing cells, and vanishes (presumably due to phosphatase activity of CroSEfm) upon diverse treatments that halt growth (e.g., exposure to chloramphenicol, or suspension in PBS). We speculate that this reflects sensing (by CroSEfm) of low levels of cell wall stress encountered during the process of growth and/or cell division. For example, slight imbalances in peptidoglycan synthesis and degradation at sites of nascent peptidoglycan insertion could be perceived by CroSEfm as cell wall stress, leading to kinase activation and phosphorylation of CroREfm. The presence of cell wall-active antimicrobials in the environment would exacerbate the imbalance, enhancing activation of CroSEfm further and leading to robust CroREfm phosphorylation (Fig. 3).
Consistent with such a model, we found that CroRSEfm function is required for intrinsic resistance of E. faecium to a variety of cell wall-targeting antibiotics, including beta-lactams (cephalosporins and ampicillin), bacitracin, and vancomycin. These findings, in concert with our cross-species complementation study, indicate that the CroRS TCS is functionally conserved in both species of enterococci that are of the greatest clinical significance and suggest that CroRS (or its regulon members) could represent viable targets for novel adjunctive therapies to render enterococci susceptible to beta-lactam antibiotics. Moreover, because CroRSEfm is required for beta-lactam resistance even in a strain expressing a variant of Pbp5 bearing mutations that reduce beta-lactam binding affinity and enhances beta-lactam resistance (Table 3), we speculate that any such therapies will prove effective on ampicillin-resistant E. faecium clinical isolates.
Although CroRS function is required for resistance to beta-lactam antibiotics such as ceftriaxone and ampicillin in both E. faecalis and E. faecium, under our conditions ceftriaxone and ampicillin did not lead to robust activation of CroRS signaling (Fig. 3). In contrast, antibiotics that inhibit an earlier step in the peptidoglycan biosynthesis pathway (bacitracin and vancomycin) elicited robust CroRS signaling (Fig. 3). The reason for this apparent disconnect remains unknown. One possibility is that the output of CroRS signaling (i.e., CroR-dependent genes, expected to be the “effectors” of the CroRS regulon) are capable of efficiently mitigating the stress imposed by beta-lactam exposure but not that imposed by inhibitors acting earlier in the peptidoglycan pathway. In this scenario, transient CroRS activation by beta-lactams would lead to expression of effector genes that rapidly mitigate beta-lactam-imposed stress, eliminating the activation signal and leading to dampened CroRS activity. Conversely, inhibition by bacitracin or vancomycin would create a cell wall stress that cannot be efficiently mitigated by CroRS-dependent effectors, leading to sustained and robust formation of CroR-P. Future identification of the CroR regulon is necessary to explore this possibility in more detail. It is perhaps worth noting that Comenge et al. used a different experimental design, with CroREfs-dependent gene expression as a readout and observed activation upon growth in the presence of ceftriaxone, ampicillin, and other beta-lactams (12), indicating that beta-lactams can indeed function as activators of CroRS signaling. We speculate that the transcriptional reporter fusion readout of CroRS signaling is more sensitive than Phos-Tag SDS-PAGE in this case due to the ability of the β-galactosidase reporter to accumulate in cells during growth, whereas Phos-Tag provides only an “instantaneous” snapshot of CroR-P abundance.
How does CroRSEfm influence beta-lactam resistance? As noted above, previous studies indicate that CroRSEfs modulates gene expression in response to stress from cell wall-targeting antibiotics (12, 26), and the functional conservation identified here suggests this is also the case with CroRSEfm. Hence, we hypothesize that CroRSEfm enhances expression of genes that are important for resistance upon sensing cell wall stress. However, the “effector” genes under transcriptional control of CroRS that are important for resistance have not been identified in either species. Current evidence indicates that changes in the expression of the PBPs themselves (including the low-affinity Pbp5) are not responsible for enhanced resistance. For example, no changes in labeling of PBPs were observed in ΔcroRS mutants of E. faecalis (12) or E. faecium (this study). Moreover, expression of pbp5 from a constitutive promoter did not significantly enhance resistance of ΔcroRS mutants in either E. faecalis (12) or E. faecium (this study). To test the hypothesis that CroRS influences the production of substrates for Pbp5 (peptidoglycan precursors), Comenge et al. analyzed peptidoglycan precursors produced in wild-type and ΔcroRSEfs mutant strains of E. faecalis (12). However, no differences in relative abundances were detected, suggesting that the beta-lactam sensitivity of the ΔcroRSEfs mutant is not due to limitation of the substrates for Pbp5. Therefore, we hypothesize that an as-yet-unknown effector(s) within the CroR regulon influences the enzymatic activity or proper localization of Pbp5 and, in the absence of CroR-mediated expression of this effector, Pbp5 is unable to efficiently mediate peptidoglycan cross-linking. The concept that such a cofactor might be required for Pbp5 function in vivo is not without precedent: a specific modification of staphylococcal wall teichoic acid (beta-O-GlcNAcylation) is required for methicillin resistance mediated by the low-affinity Pbp2a (29), and roughly 20 distinct loci in the staphylococcal genome are required for full Pbp2a-mediated methicillin resistance (30–32). We anticipate that ongoing efforts to elucidate the composition of the CroR regulon will reveal new determinants of intrinsic antibiotic resistance that are conserved between E. faecalis and E. faecium. Analysis of the biological function of these determinants will provide new insights into the mechanisms of antibiotic resistance and cell wall homeostasis with the potential to be exploited for the development of new antimicrobials.
MATERIALS AND METHODS
Bacterial strains, growth media, and chemicals.
The bacterial strains and plasmids used in the study are listed in Table 4. E. faecalis and E. faecium strains were grown in half-strength (0.5×) brain heart infusion medium or MHB for routine maintenance. Escherichia coli strains were grown in lysogeny broth. MHB was used in experiments for data collection. Chloramphenicol was used at 10 μg/ml for plasmid selection. cCF10 was used at 0.2 ng/ml for E. faecalis and at 10 ng/ml for E. faecium. All cultures were grown aerobically with shaking.
TABLE 4.
Strains and plasmids used in this study
| Strain | Genotype or descriptiona | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| Top10 | Routine cloning host | Lab stock |
| DH5α | Routine cloning host | Lab stock |
| E. faecalis | ||
| OG1 | Wild-type laboratory strain isolated from oral sample (MLST 1) | 39 |
| SB35 | OG1 Δ(croR croS)3 | 26 |
| E. faecium | ||
| 1,141,733 | Wild-type reference strain, isolated from blood culture of hospitalized patient (MLST 327) | 40 |
| JL537 | 1,141,733 Δ(croR croS)4 | This study |
| 1,231,408 | Wild-type reference strain, isolated from blood culture of hospitalized patient (MLST 582) | 40 |
| Plasmids | ||
| pJH123 | Expression vector, constitutive P23S promoter (Cmr) | This study |
| pSLK252 | pbp5 from E. faecium 1,141,733 in pJH123 | This study |
| pSLK253 | pbp5 from E. faecium 1,231,408 in pJH123; carries M485A and Ser466 insertion | This study |
| pBK2 | cCF10-inducible expression vector with lacZ in MCS (Cmr) | 34 |
| pJLL105 | pBK2 with lacZ removed (Cmr) | This study |
| pJLL160 | croRS from E. faecium 1,141,733 in pBK2 (replaces lacZ) | This study |
| pJH086 | E. faecalis allelic-exchange vector (Cmr, repA V71G, lacZ pheS*) | This study |
| pJLL150 | Δ(croREfm croSEfm)4 deletion allele in pJH086 | This study |
Cmr, chloramphenicol resistance.
Plasmid construction.
All plasmids were constructed using Gibson assembly (33) to express native croRSEfm or pbp5Efm alleles. For all recombinant plasmids, sequencing of the full insert was performed to verify the absence of errors. Ectopic expression of croRSEfm was accomplished using the cCF10-inducible promoter in the enterococcal expression vector pBK2 (34). E. faecium pbp5 alleles were expressed from pJH123, a modified version of the enterococcal expression vector pJRG9 containing the constitutive P23s promoter (35). pJH123 was generated to have a versatile multiple cloning site for which N-terminal or C-terminal hemagglutinin (HA) epitope fusions could be generated if desired. A ribosome-binding site was also included (AGGAGG) for consistency and convenience. The multiple cloning site (MCS) of pJRG9 was removed by restriction digest and the newly constructed MCS was inserted. The resulting MCS contains features in the following order downstream of the P23s promoter: EcoRI site, ribosome binding site (RBS), PstI site, HA epitope, AgeI site, and XhoI site. The E. faecium pbp5 alleles were cloned into pJH123 at the PstI and XhoI restriction sites, resulting in no HA fusion.
To improve the temperature-sensitive allelic-exchange vector for E. faecalis, pCJK218 (36), RepA was analyzed for hydrophobic residues that may be disrupted to generate a more temperature-sensitive phenotype. The method described by Varadarajan et al. (37, 38) was used to identify V71G as a candidate substitution to provide temperature sensitivity in RepA. Inverse PCR on pCJK218 was used to introduce the V71G substitution into repA. During sequence confirmation of the V71G substitution, we discovered that the clone of pCJK218 used did not carry the original temperature-sensitive repA substitutions. Therefore, the resulting plasmid, pJH086, carries a repA allele with only the V71G substitution but otherwise retains the features of pCJK218 (chloramphenicol resistance [Cmr], lacZ, pheS*). E. coli, E. faecalis, and E. faecium carrying pJH086 grow at 30°C but are significantly impaired in growth at 42°C, confirming that the V71G substitution provides temperature sensitivity.
Construction of E. faecium ΔcroRS mutant.
An in-frame deletion of croRS in E. faecium was constructed using markerless allelic exchange as previously described for E. faecalis (36) with a derivative of pJH086. Although no other genes are expected to be cotranscribed with croRSEfm, the deletion allele retains 126 codons at the 5′ end of croREfm (the entire DNA binding domain of CroREfm is deleted) and the final 9 codons at the 3′ end of croSEfm in an attempt to avoid perturbing the expression of adjacent genes.
Antibiotic susceptibility determinations.
The MICs of antibiotics were determined as described previously (26). Briefly, bacteria from stationary-phase cultures in MHB (plus 10 μg/ml chloramphenicol for plasmid carrying strains) were inoculated at a cell density of ∼105 CFU/ml into microplate wells containing 2-fold serial dilutions of antibiotic. Plates were incubated in a Bioscreen C plate reader at 37°C for 24 h with brief shaking before each measurement. The optical density at 600 nm (OD600) was read every 15 min, and the lowest concentration of antibiotic that prevented growth was recorded as the MIC.
Phos-Tag SDS-PAGE and immunoblot analysis of CroR.
Acrylamide-pendant Phos-Tag is a dinuclear metal complex that is polymerized directly into polyacrylamide gels and acts as a selective phosphate-binding tag to retard the migration of phosphorylated protein isoforms. Analysis of CroR phosphorylation by Phos-Tag-Mn2+ SDS-PAGE was performed as described previously (26). Wild-type E. faecalis (OG1) and E. faecium (strain 1,141,733) were grown to exponential phase (OD600 ∼ 0.2 in MHB) and treated with 2× the MIC of ampicillin, bacitracin, vancomycin, or chloramphenicol for 30 min. Due to the increase in cephalosporin MIC with high cell density (inoculum effect), 1.5 mg/ml ceftriaxone was used for E. faecalis and E. faecium. Samples were collected by mixing with an equal volume of cold ethanol-acetone (1:1) mixture to rapidly kill the bacteria and prevent any further signaling events. CroR from E. faecalis and E. faecium was detected using custom rabbit polyclonal antiserum raised against E. faecalis CroR protein.
ACKNOWLEDGMENTS
This study was supported by grants R01 AI081692 and OD006447 from the National Institutes of Health (NIH). The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Hollenbeck BL, Rice LB. 2012. Intrinsic and acquired resistance mechanisms in enterococcus. Virulence 3:421–433. doi: 10.4161/viru.21282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Canepari P, Lleò MM, Cornaglia G, Fontana R, Satta G. 1986. In Streptococcus faecium penicillin-binding protein 5 alone is sufficient for growth at submaximal but not at maximal rate. J Gen Microbiol 132:625–631. [DOI] [PubMed] [Google Scholar]
- 3.Signoretto C, Boaretti M, Canepari P. 1994. Cloning, sequencing and expression in Escherichia coli of the low-affinity penicillin binding protein of Enterococcus faecalis. FEMS Microbiol Lett 123:99–106. doi: 10.1111/j.1574-6968.1994.tb07207.x. [DOI] [PubMed] [Google Scholar]
- 4.Arbeloa A, Segal H, Hugonnet JE, Josseaume N, Dubost L, Brouard JP, Gutmann L, Mengin-Lecreulx D, Arthur M. 2004. Role of class A penicillin-binding proteins in PBP5-mediated beta-lactam resistance in Enterococcus faecalis. J Bacteriol 186:1221–1228. doi: 10.1128/JB.186.5.1221-1228.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rice LB, Carias LL, Rudin S, Hutton R, Marshall S, Hassan M, Josseaume N, Dubost L, Marie A, Arthur M. 2009. Role of class A penicillin-binding proteins in the expression of beta-lactam resistance in Enterococcus faecium. J Bacteriol 191:3649–3656. doi: 10.1128/JB.01834-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vesić D, Kristich CJ. 2012. MurAA is required for intrinsic cephalosporin resistance of Enterococcus faecalis. Antimicrob Agents Chemother 56:2443–2451. doi: 10.1128/AAC.05984-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouhss A, Josseaume N, Severin A, Tabei K, Hugonnet JE, Shlaes D, Mengin-Lecreulx D, Van Heijenoort J, Arthur M. 2002. Synthesis of the l-alanyl-l-alanine cross-bridge of Enterococcus faecalis peptidoglycan. J Biol Chem 277:45935–45941. doi: 10.1074/jbc.M207449200. [DOI] [PubMed] [Google Scholar]
- 8.Kristich CJ, Wells CL, Dunny GM. 2007. A eukaryotic-type Ser/Thr kinase in Enterococcus faecalis mediates antimicrobial resistance and intestinal persistence. Proc Natl Acad Sci U S A 104:3508–3513. doi: 10.1073/pnas.0608742104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kristich CJ, Little JL, Hall CL, Hoff JS. 2011. Reciprocal regulation of cephalosporin resistance in Enterococcus faecalis. mBio 2:e00199–. doi: 10.1128/mBio.00199-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Desbonnet C, Tait-Kamradt A, Garcia-Solache M, Dunman P, Coleman J, Arthur M, Rice LB. 2016. Involvement of the eukaryote-like kinase-phosphatase system and a protein that interacts with penicillin-binding protein 5 in emergence of cephalosporin resistance in cephalosporin-sensitive class A penicillin-binding protein mutants in Enterococcus faecium. mBio 7:e02188–. doi: 10.1128/mBio.02188-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hall CL, Tschannen M, Worthey EA, Kristich CJ. 2013. IreB, a Ser/Thr kinase substrate, influences antimicrobial resistance in Enterococcus faecalis. Antimicrob Agents Chemother 57:6179–6186. doi: 10.1128/AAC.01472-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Comenge Y, Quintiliani R, Li L, Dubost L, Brouard JP, Hugonnet JE, Arthur M. 2003. The CroRS two-component regulatory system is required for intrinsic beta-lactam resistance in Enterococcus faecalis. J Bacteriol 185:7184–7192. doi: 10.1128/JB.185.24.7184-7192.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hancock LE, Perego M. 2004. Systematic inactivation and phenotypic characterization of two-component signal transduction systems of Enterococcus faecalis V583. J Bacteriol 186:7951–7958. doi: 10.1128/JB.186.23.7951-7958.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muller C, Le Breton Y, Morin T, Benachour A, Auffray Y, Rincé A. 2006. The response regulator CroR modulates expression of the secreted stress-induced SalB protein in Enterococcus faecalis. J Bacteriol 188:2636–2645. doi: 10.1128/JB.188.7.2636-2645.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le Breton Y, Muller C, Auffray Y, Rincé A. 2007. New insights into the Enterococcus faecalis CroRS two-component system obtained using a differential-display random arbitrarily primed PCR approach. Appl Environ Microbiol 73:3738–3741. doi: 10.1128/AEM.00390-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sifaoui F, Arthur M, Rice L, Gutmann L. 2001. Role of penicillin-binding protein 5 in expression of ampicillin resistance and peptidoglycan structure in Enterococcus faecium. Antimicrob Agents Chemother 45:2594–2597. doi: 10.1128/AAC.45.9.2594-2597.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rice LB, Bellais S, Carias LL, Hutton-Thomas R, Bonomo RA, Caspers P, Page MG, Gutmann L. 2004. Impact of specific pbp5 mutations on expression of beta-lactam resistance in Enterococcus faecium. Antimicrob Agents Chemother 48:3028–3032. doi: 10.1128/AAC.48.8.3028-3032.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galloway-Peña JR, Rice LB, Murray BE. 2011. Analysis of PBP5 of early U.S. isolates of Enterococcus faecium: sequence variation alone does not explain increasing ampicillin resistance over time. Antimicrob Agents Chemother 55:3272–3277. doi: 10.1128/AAC.00099-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X, Paganelli FL, Bierschenk D, Kuipers A, Bonten MJ, Willems RJ, van Schaik W. 2012. Genome-wide identification of ampicillin resistance determinants in Enterococcus faecium. PLoS Genet 8:e1002804. doi: 10.1371/journal.pgen.1002804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mainardi JL, Legrand R, Arthur M, Schoot B, van Heijenoort J, Gutmann L. 2000. Novel mechanism of beta-lactam resistance due to bypass of d,d-transpeptidation in Enterococcus faecium. J Biol Chem 275:16490–16496. doi: 10.1074/jbc.M909877199. [DOI] [PubMed] [Google Scholar]
- 21.Mainardi JL, Fourgeaud M, Hugonnet JE, Dubost L, Brouard JP, Ouazzani J, Rice LB, Gutmann L, Arthur M. 2005. A novel peptidoglycan cross-linking enzyme for a beta-lactam-resistant transpeptidation pathway. J Biol Chem 280:38146–38152. doi: 10.1074/jbc.M507384200. [DOI] [PubMed] [Google Scholar]
- 22.Sacco E, Hugonnet JE, Josseaume N, Cremniter J, Dubost L, Marie A, Patin D, Blanot D, Rice LB, Mainardi JL, Arthur M. 2010. Activation of the l,d-transpeptidation peptidoglycan cross-linking pathway by a metallo-d,d-carboxypeptidase in Enterococcus faecium. Mol Microbiol 75:874–885. doi: 10.1111/j.1365-2958.2009.07014.x. [DOI] [PubMed] [Google Scholar]
- 23.Sacco E, Cortes M, Josseaume N, Rice LB, Mainardi JL, Arthur M. 2014. Serine/threonine protein phosphatase-mediated control of the peptidoglycan cross-linking l,d-transpeptidase pathway in Enterococcus faecium. mBio 5:e01446–. doi: 10.1128/mBio.01446-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qin X, Galloway-Peña JR, Sillanpaa J, Roh JH, Nallapareddy SR, Chowdhury S, Bourgogne A, Choudhury T, Muzny DM, Buhay CJ, Ding Y, Dugan-Rocha S, Liu W, Kovar C, Sodergren E, Highlander S, Petrosino JF, Worley KC, Gibbs RA, Weinstock GM, Murray BE. 2012. Complete genome sequence of Enterococcus faecium strain TX16 and comparative genomic analysis of Enterococcus faecium genomes. BMC Microbiol 12:135. doi: 10.1186/1471-2180-12-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bourgogne A, Garsin DA, Qin X, Singh KV, Sillanpaa J, Yerrapragada S, Ding Y, Dugan-Rocha S, Buhay C, Shen H, Chen G, Williams G, Muzny D, Maadani A, Fox KA, Gioia J, Chen L, Shang Y, Arias CA, Nallapareddy SR, Zhao M, Prakash VP, Chowdhury S, Jiang H, Gibbs RA, Murray BE, Highlander SK, Weinstock GM. 2008. Large-scale variation in Enterococcus faecalis illustrated by the genome analysis of strain OG1RF. Genome Biol 9:R110. doi: 10.1186/gb-2008-9-7-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kellogg SL, Kristich CJ. 2016. Functional dissection of the CroRS two-component system required for resistance to cell wall stressors in Enterococcus faecalis. J Bacteriol 198:1326–1336. doi: 10.1128/JB.00995-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Skerker JM, Perchuk BS, Siryaporn A, Lubin EA, Ashenberg O, Goulian M, Laub MT. 2008. Rewiring the specificity of two-component signal transduction systems. Cell 133:1043–1054. doi: 10.1016/j.cell.2008.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Capra EJ, Perchuk BS, Lubin EA, Ashenberg O, Skerker JM, Laub MT. 2010. Systematic dissection and trajectory-scanning mutagenesis of the molecular interface that ensures specificity of two-component signaling pathways. PLoS Genet 6:e1001220. doi: 10.1371/journal.pgen.1001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown S, Xia G, Luhachack LG, Campbell J, Meredith TC, Chen C, Winstel V, Gekeler C, Irazoqui JE, Peschel A. 2012. Methicillin resistance in Staphylococcus aureus requires glycosylated wall teichoic acids. Proc Natl Acad Sci U S A 109:18909–18914. doi: 10.1073/pnas.1209126109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berger-Bächi B, Strässle A, Gustafson JE, Kayser FH. 1992. Mapping and characterization of multiple chromosomal factors involved in methicillin resistance in Staphylococcus aureus. Antimicrob Agents Chemother 36:1367–1373. doi: 10.1128/AAC.36.7.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Lencastre H, Wu SW, Pinho MG, Ludovice AM, Filipe S, Gardete S, Sobral R, Gill S, Chung M, Tomasz A. 1999. Antibiotic resistance as a stress response: complete sequencing of a large number of chromosomal loci in Staphylococcus aureus strain COL that impact on the expression of resistance to methicillin. Microb Drug Resist 5:163–175. doi: 10.1089/mdr.1999.5.163. [DOI] [PubMed] [Google Scholar]
- 32.de Lencastre H dJB, Matthews PR, Tomasz A. 1994. Molecular aspects of methicillin resistance in Staphylococcus aureus. J Antimicrob Chemother 33:18. [DOI] [PubMed] [Google Scholar]
- 33.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 34.Shokeen S, Johnson CM, Greenfield TJ, Manias DA, Dunny GM, Weaver KE. 2010. Structural analysis of the anti-Q-Qs interaction: RNA-mediated regulation of Enterococcus faecalis plasmid pCF10 conjugation. Plasmid 64:26–35. doi: 10.1016/j.plasmid.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Snyder H, Kellogg SL, Skarda LM, Little JL, Kristich CJ. 2014. Nutritional control of antibiotic resistance via an interface between the phosphotransferase system and a two-component signaling system. Antimicrob Agents Chemother 58:957–965. doi: 10.1128/AAC.01919-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vesić D, Kristich CJ. 2013. A Rex family transcriptional repressor influences H2O2 accumulation by Enterococcus faecalis. J Bacteriol 195:1815–1824. doi: 10.1128/JB.02135-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Varadarajan R, Nagarajaram HA, Ramakrishnan C. 1996. A procedure for the prediction of temperature-sensitive mutants of a globular protein based solely on the amino acid sequence. Proc Natl Acad Sci U S A 93:13908–13913. doi: 10.1073/pnas.93.24.13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chakshusmathi G, Mondal K, Lakshmi GS, Singh G, Roy A, Ch RB, Madhusudhanan S, Varadarajan R. 2004. Design of temperature-sensitive mutants solely from amino acid sequence. Proc Natl Acad Sci U S A 101:7925–7930. doi: 10.1073/pnas.0402222101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gold OG, Jordan HV, van Houte J. 1975. The prevalence of enterococci in the human mouth and their pathogenicity in animal models. Arch Oral Biol 20:473–477. doi: 10.1016/0003-9969(75)90236-8. [DOI] [PubMed] [Google Scholar]
- 40.Palmer KL, Carniol K, Manson JM, Heiman D, Shea T, Young S, Zeng Q, Gevers D, Feldgarden M, Birren B, Gilmore MS. 2010. High-quality draft genome sequences of 28 Enterococcus sp. isolates. J Bacteriol 192:2469–2470. doi: 10.1128/JB.00153-10. [DOI] [PMC free article] [PubMed] [Google Scholar]