

ABSTRACT
The evolutionary selection of malaria parasites within an individual host plays a critical role in the emergence of drug resistance. We have compared the selection of atovaquone resistance mutants in mouse models reflecting two different causes of failure of malaria treatment, an inadequate subtherapeutic dose and an incomplete therapeutic dose. The two models are based on cycles of insufficient treatment of Plasmodium berghei-infected mice: repeated inadequate treatment associated with a subtherapeutic dose (RIaT) (0.1 mg kg−1 of body weight) and repeated incomplete treatment with a therapeutic dose (RIcT) (14.4 mg kg−1 of body weight). The number of treatment cycles for the development of a stable resistance phenotype during RIaT was 2.00 ± 0.00 cycles (n = 9), which is not statistically different from that during RIcT (2.57 ± 0.85 cycles; combined n = 14; P = 0.0591). All mutations underlying atovaquone resistance selected by RIaT (M133I, T142N, and L144S) were found to be in the Qo1 (quinone binding 1) domain of the mitochondrial cytochrome b gene, in contrast to those selected by RIcT (Y268N/C, L271V, K272R, and V284F) in the Qo2 domain or its neighboring sixth transmembrane region. Exposure of mixed populations of resistant parasites from RIaT to the higher therapeutic dose of RIcT revealed further insights into the dynamics of within-host selection of resistance to antimalarial drugs. These results suggest that both inadequate subtherapeutic doses and incomplete therapeutic doses in malaria treatment pose similar threats to the emergence of drug resistance. RIcT and RIaT could be developed as useful tools to predict the potential emergence of resistance to newly introduced and less-understood antimalarials.
KEYWORDS: dose-dependent selection, mouse malaria model, repeated inadequate treatment, repeated incomplete treatment, within-host selection of atovaquone resistance
INTRODUCTION
The evolutionary selection of malaria parasites within an individual host plays a critical role in the emergence of antimalarial drug resistance, a major problem in malaria control. The study of within-host selection of drug resistance benefits from animal models of malaria infection, as it allows pharmacological manipulations in vivo. We recently reported such a model, based on cycles of repeated incomplete treatment (RIcT) of Plasmodium berghei-infected mice with a constant therapeutic dose of an antimalarial drug, which mimics treatment failure in the human field situation (1). We showed that under these conditions, stable resistance of P. berghei to the antimalarial atovaquone developed rapidly, after only 2.5 cycles of treatment.
Atovaquone, a hydroxy-1,4,-naphtho-quinone, is an antimalarial for which the biochemical mechanism of action is well established (2, 3). It is an analog of coenzyme Q (CoQ) (ubiquinone), a coenzyme involved in the mitochondrial respiratory chain, which inhibits mitochondrial respiratory complex III (bc1 complex; quinol-cytochrome c reductase) activity in Plasmodium spp. by competitive binding with the quinone binding domain of the enzyme complex (4). Resistance to atovaquone is associated with genetic lesions in the mitochondrial cytochrome b gene (3–5). The rapid development of resistance to atovaquone is consistent with the general observation that the rate of mutation in mitochondrial DNA (mtDNA) is significantly higher than that in nuclear DNA; the mutation rate of animal mtDNA is 5 to 10 times higher than that of single-copy nuclear DNA (6), including the mitochondrion-encoded cytochrome b gene (7).
Almost all mutations underlying atovaquone resistance of P. berghei resulting from RIcT selection were found to be in the Qo2 (quinone binding 2) domain of the cytochrome b gene (Y268C/N/S, L271V, and K272R), associated with relatively high degrees of resistance in vivo as well as in vitro (7, 8). Previous attempts to isolate atovaquone-resistant mutants of P. berghei by the serial technique (ST) method, which involves serial passages of parasites in mice treated with gradually increasing drug doses (5), on the other hand, resulted in atovaquone resistance associated with mutations that are predominantly in the transmembrane (V284F) or in the Qo1 (M133I and L144S) domain of the cytochrome b gene.
To examine the dynamics of within-host selection of antimalarial drug resistance that might be responsible for the above-described observations, we compared the selection of atovaquone resistance mutants in two different animal models: (i) previously reported RIcT (1) and (ii) a new repeated inadequate treatment associated with a subtherapeutic dose of antimalarial drugs (RIaT). These animal models mimic two different causes of treatment failure in cases of human malaria.
RESULTS
Rapid development of resistance during RIcT and RIaT.
Typical experiments, as shown in Fig. 1, suggest that resistance to atovaquone developed rapidly during both RIcT (therapeutic dose of 14.4 mg kg−1 of body weight) (Fig. 1b) and RIaT (subtherapeutic dose of 0.1 mg kg−1 of body weight) (Fig. 1a), during which the stable resistance phenotype was already established in the second treatment cycle. The treatment time required to decrease the parasitemia level to <10% of that at the start of treatment, however, is much longer for RIaT (5 days) than for RIcT (2 days). In addition, RIaT showed a significantly shorter recovery time following the first treatment cycle (4 days) than did RIcT (12 days).
FIG 1.
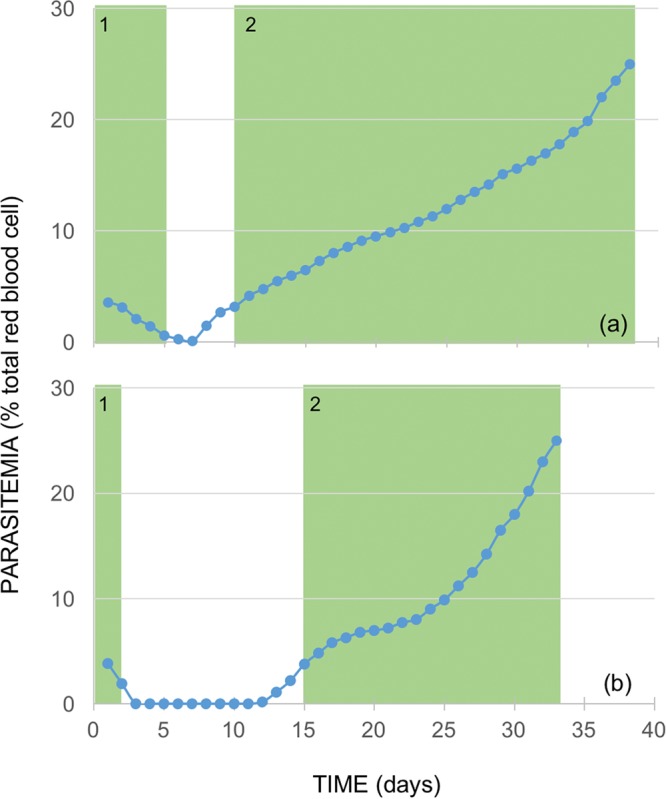
The two experimental systems employed in this study, based on cycles of incomplete treatment of P. berghei-infected mice. The new RIaT model is basically the same as the previously described RIcT model (1), except that the dose of atovaquone used for treatment was a subtherapeutic dose instead of a therapeutic dose. The number of treatment cycles for the development of a stable resistance phenotype during RIaT (a) is not statistically different from that for RIcT (b).
To establish the generality and reproducibility of the above-described observations, we repeated the experiments with larger numbers of mice (Table 1). The number of treatment cycles that led to parasite resistance to atovaquone in RIcT was found to be 2.40 ± 0.89 cycles (mean ± standard deviation [SD]), similar to the values in our previous report (2.67 ± 0.87 cycles; P = 0.5951) (1). The number of treatment cycles for the development of a stable resistance phenotype during RIaT was 2.00 ± 0.00 cycles (n = 9), which is not statistically different from that of RIcT (2.57 ± 0.85 cycles; combined n = 14; P = 0.0591).
TABLE 1.
Rapid development of resistance of P. berghei to atovaquone with RIcT and RIaT
| Treatment | Isolate | No. of days of treatment to bring parasitemia level down to <0.4%-no. of days of recovery for parasitemia level to return to 3–5% during cyclea: |
No. of cycles to resistance | Mutation(s) |
||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | Qo1 | Qo2 | |||
| RIcT | PbLASN8b | 1-13 | 8-● | 2 | Y268C | |||
| PbLASN9b | 2-15 | 6-● | 2 | L271V + K272R | ||||
| PbLAJR1b | 1-13 | 8-● | 2 | L271V + V284F | ||||
| PbLAJR2b | 1-12 | 4-20 | 3-3 | 2-● | 4 | L271V + K272R | ||
| PbLAJR3b | 2-15 | 6-● | 2 | Y268N | ||||
| PbLJS1c | 1-7 | 3-13 | 2-18 | 3-● | 4 | Y268N | ||
| PbLJS2c | 3-18 | 1-32 | 5-● | 3 | L271V + K272R | |||
| PbLASN1c | 2-13 | 3-4 | 18-● | 3 | Y268C | |||
| PbLASN2c | 2-12 | 18-● | 2 | Y268C | ||||
| PbLASN3c | 2-6 | 10-● | 2 | Y268C | ||||
| PbLASN4c | 3-8 | 8-● | 2 | Y268N | ||||
| PbLASN5c | 3-9 | 6-● | 2 | L271V + K272R | ||||
| PbLASN6c | 2-10 | 2-● | 2 | L271V + K272R | ||||
| PbLASN7c | 1-12 | 4-20 | 3-3 | 2-● | 4 | Y268C | ||
| RIaT | PbLASNL1 | 6-2 | 23-● | 2 | M133Id | |||
| PbLASNL2 | 4-7 | 27-● | 2 | M133Id | ||||
| PbLASNL3 | 4-6 | 28-● | 2 | M133Id | ||||
| PbLASNL4 | 5-3 | 31-● | 2 | M133Ie | ||||
| PbLASNL5 | 5-4 | 28-● | 2 | M133Ie | ||||
| PbLASNL6 | 4-4 | 29-● | 2 | M133Ie | ||||
| PbLASNL7 | 5-5 | 23-● | 2 | M133Ie | ||||
| PbLASNL8 | 4-2 | 25-● | 2 | T142Ne | ||||
| PbLASNL9 | 4-3 | 22-● | 2 | M133I, L144Se | ||||
-●, termination of treatment cycles due to the development of stable resistance (parasitemia level of ±25%).
This study (mean number of cycles ± SD of 2.40 ± 0.89).
See reference 1.
Homogenous populations of M133I-carrying mutants.
Mixed populations of M133I-, T142N-, and L144S-carrying mutants and the wild type. The most common mutation leading to M133I was nt399G→T (87.5%), but 399G→A and 399G→C mutations were also observed.
The dynamics of within-host selection of resistance to atovaquone under RIcT and RIaT deduced from the data described above are summarized in Table 2. The number of days needed to develop a stable resistance phenotype for RIaT (34.78 ± 3.77 days) was similar to that for RIcT (30.50 ± 14.06 days; P = 0.3859). However, a significant difference was observed in the time required to decrease the parasitemia level in the first cycle of treatment between RIcT (1.86 ± 0.77 days) and RIaT (4.56 ± 0.73 days; P = 0.0001). The recovery time needed following the first cycle of RIcT (11.64 ± 3.34 days), on the other hand, was significantly longer than that for RIaT (4.00 ± 1.73 days; P = 0.0001).
TABLE 2.
Dynamics of atovaquone resistance selection under RIcT and RIaT
| Parameter | Mean value for treatment ± SD |
P valuea | |
|---|---|---|---|
| RIcT | RIaT | ||
| Total no. of cycles to resistance | 2.57 ± 0.85 | 2.00 ± 00.00 | 0.0591 |
| Total no. of days to resistance | 30.50 ± 14.06 | 34.78 ± 3.77 | 0.3859 |
| Treatment time (days) | 1.86 ± 0.77 | 4.56 ± 0.73 | 0.0001 |
| Recovery time (days) | 11.64 ± 3.34 | 4.00 ± 1.73 | 0.0001 |
Determined by a Student t test.
Mutations underlying resistance to atovaquone resulting from RIcT and RIaT indicate a dose-dependent effect.
The four mutations in the Qo2 domain of the cytochrome b gene (Y268N/C, L271V, and K272R) that we recently reported were associated with RIcT (1, 4) were also observed in the present study (Table 1). In addition, a mutation, V284F, in the neighboring sixth transmembrane region was found. In contrast, all mutations selected by RIaT (M133I, T142N, and L144S) were located in the Qo1 domain, indicating a dose-dependent selection process in the development of resistance to atovaquone.
Furthermore, while the resistant parasites resulting from RIcT were in all cases found to be associated with homogenous populations of the resistant mutants, in most cases of RIaT, mixed populations of parasites were observed when the last treatment cycles were terminated at a parasitemia level of 25%. Thus, only three of the nine RIaT experiments resulted in homogenous populations of M133I-carrying mutants. In two of these populations, the M133I amino acid change resulted from a G-to-T mutation at nucleotide 399 (nt399G→T), but in one population, two resistant mutations (nt399A and -C) coexisted. In four cases, mixed populations of M133I-carrying mutants and the wild-type parental strain were observed, while in one case, two resistant M133I strains with nt399T and -C mutations coexisted with the parental strain. One of the nine RIaT experiments resulted in a new Qo1 mutation, T142N, which coexisted with the parental strain.
P. berghei carrying an atovaquone resistance Qo1 mutation after RIaT develops additional mutations following further exposure to RIcT.
To examine the development of resistance to a high dose of atovaquone in a strain already carrying a mutation in the Qo1 domain of the cytochrome b gene, associated with low-level resistance to the antimalarial, we subjected strain PbLASNL1 (homogenous population of the M133I-carrying resistance mutant selected by RIaT) to RIcT. In all seven separate experiments, the M133I mutation was still found at the end of RIcT selection (Table 3). Two isolates were found to carry additional mutations in the Qo2 domain, T273P and K272R. The other five isolates carried a G280D mutation in the sixth transmembrane region close to the Qo2 domain. The M133I-carrying strain needed 2.14 ± 3.08 cycles of RIcT to develop high-level resistance to atovaquone, similar to the values for the wild-type parents (2.57 ± 0.85 cycles; n = 14; P = 0.2235) (Table 2).
TABLE 3.
Exposure to RIcT of atovaquone-resistant P. berghei isolates carrying the M133I Qo1 mutation selected by RIaT
| M133I-carrying mutant population | Isolate | No. of cycles to resistance | Mutation(s) detected after RIcTa |
||
|---|---|---|---|---|---|
| Qo1 | Qo2 | Tm VI | |||
| Homogenous mutant population | PbLASNL1.1 | 2 | M133I | T273P | |
| PbLASNL1.2 | 2 | M133I | K272R | ||
| PbLASNL1.3 | 3 | M133I | G280D | ||
| PbLASNL1.4 | 2 | M133I | G280D | ||
| PbLASNL1.5 | 2 | M133I | G280D | ||
| PbLASNL1.6 | 2 | M133I | G280D | ||
| PbLASNL1.7 | 2 | M133I | G280D | ||
| Mixed mutant–wild-type population | PbLASNL4.1 | 3 | M133I + T139N | ||
| PbLASNL4.2 | 4 | M133I + V140A | |||
| PbLASNL4.3 | 3 | M133I + V140A | |||
| PbLASNL4.4 | 5 | M133I + L144Y | |||
| PbLASNL4.5 | 5 | M133I | T273P | ||
| PbLASNL4.6 | 3 | M133I | L271V + K272V | ||
| PbLASNL4.7 | 4 | T139N | V284F | ||
| PbLASNL4.8 | 5 | L271F | V284F | ||
| PbLASNL4.9 | 2 | L271V + K272R | |||
| PbLASNL4.10 | 3 | L271V + K272R | |||
Tm, transmembrane.
To examine the development of high-level resistance to atovaquone in mixed M133I-carrying and wild-type populations, we similarly subjected PbLASNL4 to RIcT. In 6 of 10 separate experiments, the original M133I Qo1 mutation was still present at the conclusion of RIcT. In two of these experiments, additional mutations in the Qo2 domain were again detected albeit in different sites (L271V plus K272V and T273P). The secondary mutations in the other four experiments, however, were found to be in the Qo1 domain (T139N, V140A, and L144Y).
In the other experiments, the high-resistance isolates did not carry the original M133I mutation and thus seem to have developed from the wild-type parents. Three of these resistant isolates carried mutations in the Qo2 domain, as expected (two L271V-plus-K272R mutations and one L271F-plus-V284F mutation), but one had a mutation in the Qo1 domain (T139N) in addition to mutation V284F in transmembrane VI near the Qo2 domain. The mixed M133I-carrying–wild-type populations needed 3.70 ± 1.06 cycles of RIcT to develop high-level resistance to atovaquone (Table 3), which was significantly longer than that for the wild-type parents (2.57 ± 0.85 cycles; n = 14; P = 0.0084) (Table 2).
Atovaquone resistance mutations affect growth fitness of the mutant strains.
To better understand the dynamics of antimalarial resistance selection in the two models, we examined the fitness of P. berghei isolates carrying atovaquone resistance mutations by determining their growth rate in mice, as illustrated in Fig. 2. The three mutations at codon 268 (Y268C/N/S) in the Qo2 domain were found to have affected growth fitness differently (Table 4). Mutant isolate PbLACSN4, carrying the Y268S mutation, had the best growth fitness (growth rate of 5.26 ± 0.52 μg mtDNA/ml blood/24 h) compared to the parental strain (9.46 ± 1.10 μg mtDNA/ml blood/24 h) and, thus, the lowest fitness cost of resistance (44.4%). This result suggests that the functional defect in respiratory complex III associated with an amino acid change from tyrosine to serine is less severe than changes to cysteine (isolate PbLSJ2) or asparagine (isolate PbLSJ6), with fitness costs of resistance of 69.1% and 78.3%, respectively. Interestingly, the fitness cost of isolate PbLALSN1 (3.21 ± 0.30 µg mtDNA/ml/blood/24 h), associated with mutation M133I in the Qo1 domain (isolate PbLASNL1, with a fitness cost of 66.1%) is similar to those of the Y268N and Y268C mutants. The severe functional defect associated with the M133I mutation seems to be partially compensated for by the L271V mutation in isolate PbSK2A1Tb (cost of fitness of 39.0%).
FIG 2.
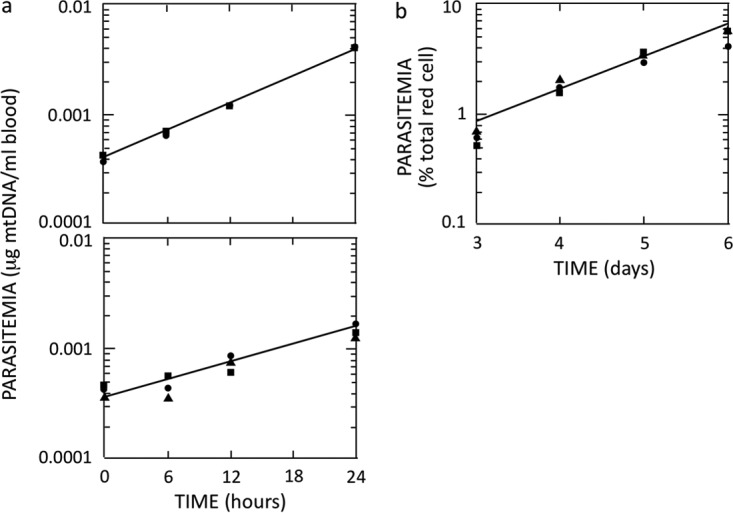
Atovaquone resistance affects the fitness of growth of mutant strain Y268C. Parasite growth rates were determined by qRT-PCR as described in Materials and Methods. (a) Growth measurements for the wild-type strain (9.46 ± 1.10 per 24 h) (top) and an atovaquone mutant strain with a Y268C amino acid substitution (2.92 ± 0.61 per 24 h) (bottom). (b) Data from qRT-PCR were validated by microscopic measurement of parasitemia levels for the wild-type strain over a period of 6 days.
TABLE 4.
Correlation between site of mutation, degree of resistance, and fitness cost of various atovaquone resistance mutations
| Isolate | Mutation | Mean IC50 (nM) ± SDa | Mean growth rate (μg mtDNA/ml blood/24 h) ± SD | Fitness cost of resistance (%)b |
|---|---|---|---|---|
| PbL | Wild type | 0.327 ± 0.141 | 9.46 ± 1.10 | Control |
| PbLSJ2.1 | Y268C | 42.5 ± 5.9 | 2.92 ± 0.61 | 69.13 |
| PbLSJ6 | Y268N | 43.5 ± 3.9 | 2.05 ± 0.54 | 78.33 |
| PbLACSN4 | Y268S | ND | 5.26 ± 0.42 | 44.40 |
| PbLASNL1 | M133I | ND | 3.21 ± 0.30 | 66.07 |
| PbSK2A1Tb | M133I/L271V | 20.0 | 5.77 ± 1.45 | 39.01 |
IC50s (50% inhibitory concentrations) reported previously (8). ND, not determined.
Percent loss of fitness of the mutant strains relative to the wild type calculated from the growth rates; in vivo growth rates were determined by measuring the increase in the level of parasite mtDNA per milliliter of blood over a period of 24 h, as described in Materials and Methods.
DISCUSSION
The present study reveals two interesting observations that inform the dynamics of within-host mutation selection. First, our results clearly demonstrate a dose-dependent phenomenon in the selection of mutations that confer resistance to atovaquone. All resistant mutants selected as a result of repeated inadequate (subtherapeutic dose) treatment (RIaT) with atovaquone were found to be associated with mutations in the Qo1 domain of the mitochondrial cytochrome b gene, coding for apocytochrome b of respiratory complex III, as depicted in Fig. 3, showing the transmembrane arrangement of the apoprotein, while a similar repeated incomplete treatment (RIcT) but with a higher, therapeutic dose of the drug led to resistance mutations mostly in the Qo2 domain. The sites correspond to the degree of resistance conferred by the mutations; the level of atovaquone resistance associated with mutations in the Qo1 domain has been shown to be at least 20 times lower than that associated with mutations in the Qo2 domain in P. berghei (8). Atovaquone resistance mutations in both Qo1 (M133I) and Qo2 (Y268S) have been reported for the human malaria parasite Plasmodium falciparum. In this case, parasites carrying the M133I mutation were found to be around 375 times less resistant than those with Y268S mutation. The degree of resistance associated with the M133I mutation is 8.8 to 36 times higher when combined with Qo2 mutations (9).
FIG 3.

Sites of atovaquone resistance mutations in the Qo1 and Qo2 domains of apocytochrome b of P. berghei mitochondria. The secondary structure of the P. berghei cytochrome b protein (367 amino acid residues) was derived on the basis of the eight-transmembrane-helix model for yeast (16, 17) (burgundy boxes) and three extramembranous amphipathic helices (18) (boxes with a yellow outline). Amino acid residues suggested to be critical for the function of center o (Qo) and center i (Qi) in the cytoplasmic and matrix sides of the inner mitochondrial membrane, respectively, were deduced from mutations in equivalent sites in yeast that confer resistance to Qo inhibitors (G131, N245, and F264 for mucidin; I141 and T142 for stigmatellin; and F123, G131, and G137 for myxothiazol) or Qi inhibitors (N27, G33, and L217 for antimycin A and I14 and L215 for diuron) (17, 19–21). Atovaquone is a competitive Qo site-specific inhibitor (2, 22). Atovaquone resistance mutations found in P. berghei (1; this study) are indicated and shaded in green in the Qo1 domain (M133I, T139N, V140A, T142N, and L144S), the Qo2 domain (Y268C/N/S, K271F/V, L272R, and T273P), and the sixth transmembrane domain (G280D and V284F). Atovaquone resistance mutations reported for P. falciparum (9) are highlighted by a red outline.
Second, in contrast to RIcT, RIaT resulted in mixtures of various mutant and wild-type parasites, presumably reflecting the interplay between various factors contributing to the dynamics of resistance selection. Only three of the nine RIaT experiments resulted in homogenous populations of M133I-carrying mutants. In the other six cases, mixed populations of either M133I-carrying mutants (four cases), an M133I- and L144S-carrying mutant (one case), or a novel T142N-carrying mutant (one case) and the wild-type parental strain were observed.
The various outcomes of RIcT and RIaT selections of atovaquone resistance mutations are illustrated in Fig. 4. In the presence of a therapeutic (high) dose of atovaquone, only mutants with lesions in the Qo2 domain, associated with a higher degree of resistance, survived and were selected out (Fig. 4, top). At a subtherapeutic (low) dose, however, only mutants carrying mutations in the Qo1 domain were selected, detected as homogenous populations of the mutants or still in a mixture with the parental wild-type strain (Fig. 4, bottom); no mutation associated with a high degree of resistance in the Qo2 domain was detected.
FIG 4.
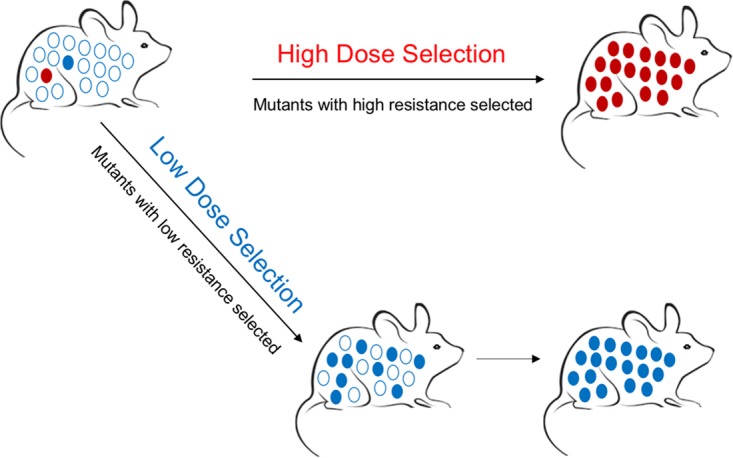
Within-host selection of atovaquone-resistant mutations with a therapeutic high dose (top) or a subtherapeutic low dose (bottom) of atovaquone. Mutants with lesions in the Qo2 domain, associated with a higher degree of resistance (red circles), or in the Qo1 domain, associated with lower degree of resistance (blue circles), were selected independently.
The probability of fixation of a resistant mutant is determined by the interplay among three variables: the rate of formation of mutants, the level of drug selection, and the relative fitness of the mutant strain (10, 11). Interestingly, in the case of atovaquone resistance, we found that there is no direct correlation between the fitness cost and the drug selection dose as well as the degree of resistance. Mutations in the Qo1 and Qo2 domains could affect fitness to the same degree depending on the nature of the amino acid substitutions. Thus, mutation M133I in the Qo1 domain (mutant isolate PbLASNL1) affected fitness as severely as did mutation Y268N in the Qo2 domain (mutant isolate PbLSJ2.1) (Table 4).
Is the growth of Y268N/C/S mutants inhibited in the presence of a low dose of atovaquone? To test whether this is the case and to gain further insight into the dynamics of mutant selection in P. berghei, we examined the development of resistance to a high dose of atovaquone in a strain already carrying a mutation in the Qo1 domain. RIcT of a homogenous population of M133I-carrying parasites resulted in double mutants, with additional mutations in the Qo2 region (K272R and T273P) or the neighboring transmembrane VI domain (G280D).
The same treatment of mixed populations of M133I-carrying parasites and the parental wild type resulted in outcomes that were more complex. In 6 of 10 independent experiments, mutations in addition to M133I, in either the Qo2 region (L271V plus K272V and T273P), as described above, or the Qo1 region (T139N, V140A, and L144Y) were observed; in all cases, the wild type disappeared from the populations. In the other four experiments, however, parasites carrying the M133I mutation disappeared and were replaced by homogenous populations of resistance strains with a range of double mutants: T139N/V284F (Qo1 and transmembrane VI), L271F/V284F (Qo2 and transmembrane VI), and L271V/K272R (Qo2). In these cases, the M133I-carrying parasites were probably selected out under RIcT, and new double mutants arose from the wild type in the mixed populations.
In none of the above-described cases, however, were resistant mutants associated with the Y268N/C/S mutations observed, indicating that they were inhibited and disappeared during earlier RIaT with the subtherapeutic (low) dose of atovaquone. Resistances to a therapeutic or subtherapeutic dose of atovaquone appear to be independent events.
The mechanism by which mutations in the cytochrome b gene contribute to atovaquone resistance has been quite extensively investigated. The relationship between mutations in the Qo1 and Qo2 domains of the cytochrome b protein and the degree of resistance to atovaquone has been reported (4, 8). A structural interaction between atovaquone and the cytochrome b protein has also been proposed (9). The atovaquone resistance mutations observed following RIcT and RIaT of P. berghei confirmed the putative drug binding sites suggested for P. falciparum. Notably, the M133I, V140A, L144Y, Y268C/N, L271F/V, and V284F mutations observed in this murine malaria parasite occurred at the putative contact residues of the atovaquone binding sites (I119, F123, Y126, M133, V140, I141, L144, I258, P260, F264, F267, Y268, L271, V284, L285, and L288) (9).
The rapid within-host selection of atovaquone resistance observed in the present study suggests that the two different causes of failure in malaria treatment, an inadequate subtherapeutic dose and an incomplete therapeutic dose, present similar threats to the emergence of drug resistance. RIcT and RIaT therefore could be developed as useful tools to predict the potential emergence of resistance to antimalarials. In the case of atovaquone, the rapid within-host selection of resistance had not been followed by its spread in the population, presumably because parasites that are resistant to atovaquone are not transmissible by mosquitoes due to the resulting defective sexual cycle (12). However, information regarding the potential emergence of resistance to other antimalarials currently used, as well as newly introduced compounds, could provide knowledge essential for planning malaria control and devising strategies to delay the emergence of resistance. It is thus important to establish the generality of the phenomenon observed in present study by examining within-host selection of resistance to the antimalarial drugs in other species of malaria parasites, notably Plasmodium yoelii, which, together with P. berghei, was the first murine model of malaria developed to test drug efficacy (13, 14). RIcT and RIaT also turn out to be the most rapid, efficient, and reproducible ways to raise antimalaria resistance mutants and thus could be critical in raising mutants against less-understood antimalarials such as doxycycline and artemisinin.
MATERIALS AND METHODS
Malarial parasites and mice.
Plasmodium berghei ANKA strain Leiden was obtained from Andy Waters, Leiden University Medical Center, Netherlands, and maintained by intraperitoneal passages in pathogen-free 10- to 12-week-old BALB/c mice (originally obtained from the Animal Resources Centre, Murdoch, Western Australia). The P. berghei stocks were screened for epizootic diarrhea of infant mice (EDIM), Mycoplasma pulmonis, mouse hepatitis virus (MHV), murine norovirus (MNV), mouse parvovirus (MPV), minute virus of mice (MVM), Theiler's encephalomyelitis virus (TMEV), and Sendai virus, as previously described (1). Approximately 106 parasitized red blood cells were inoculated per mouse at every passage. During the study, mice were maintained in the pathogen-free animal house facility of the Eijkman Institute. This study was approved by the institutional review board of the Eijkman Institute (EIREC no. 41).
Drug and doses.
Atovaquone was kindly provided by Mary Pudney of the Wellcome Research Laboratories, UK. Atovaquone was dissolved in dimethyl sulfoxide (DMSO) as 28.8-mg/ml and 0.2-mg/ml stock solutions, stored at −20°C, and diluted to the required concentration with sterilized double-distilled water freshly before use.
Monitoring of infections.
Peripheral blood smears were prepared daily from tail vein bleeds. Thin films were fixed in methanol and stained with 10% Giemsa stain. The parasitemia level was determined microscopically with at least 5,000 erythrocytes.
In vivo selection of resistant P. berghei clones under repeated incomplete treatment and inadequate treatment.
The development of the RIcT model was reported in detail previously (1). In this model, P. berghei-infected mice are intraperitoneally treated with a therapeutic dose of antimalarial drugs, which is interrupted every time the parasitemia level is reduced from 3 to 5% at the beginning of treatment to <0.4%, allowing the recovery of the parasitemia level before another treatment is administered. This incomplete regimen is repeated for several cycles until resistance is observed, indicated by a continuing increase of parasitemia during treatment. The RIaT model has been developed in this study basically according to the RIcT model, except that the dose of atovaquone used for treatment was only 0.01 mg kg−1 of body weight (subtherapeutic) instead of 14.4 mg kg−1 of body weight (therapeutic).
Determination of mutations in the cytochrome b gene.
Parasite DNA was isolated from 50 μl of saponin-lysed infected blood by treatment with Chelex-100 (Sigma-Aldrich, St. Louis, MO, USA) as described previously by Wooden et al. (15). DNA was used for PCR amplification immediately or stored at −20°C before use.
Fragments of the mitochondrial cytochrome b gene were amplified by PCR as described previously (4), employing forward primer PbF-3368 5′-CCTTTAGGGTATGATACAGC and reverse primer PbR-4103 5′-GTTTGCTTGGGAGCTGTAATC for the Qo2 domain or forward primer PbF-4086 5′-TGCCTAGACGTATTCCTGAT and reverse primer PbR-4615 5′-TGATGTATCATACCCTAAAG for the Qo1 domain. PCR was carried out for 34 cycles of 15 s of denaturation at 94°C (first cycle, 5 min), 15 s of annealing at 55°C (first primer pair) or 52°C (second primer pair), and a 2-min extension step at 72°C (final extension, 5 min). The PCR products obtained were directly sequenced by using the forward primers to generate sequences in an ABI 377 automatic sequencer. DNA sequences were aligned by using the BioEdit program (Ibis Biosciences, Carlsbad, CA).
Quantitative real-time PCR using SYBR green I.
To examine the growth fitness of the atovaquone-resistant mutants carrying amino acid changes Y268C/N/S, M133I, and L271V/K272R, parasites were inoculated intraperitoneally into BALB/c mice. Tail blood samples (50 μl) were collected every 6 h for 24 h on day 2 after inoculation for DNA extraction. The growth rate of the various mutants was determined from the increase in the level of parasite mtDNA over the 24-h period, as measured by quantitative real-time PCR (qRT-PCR) using the Light Cycler 2.0 system and a LightCycler FastStart DNA MasterPLUS SYBR green I kit (Roche Diagnostic GmbH, Mannheim, Germany). The primer pairs employed that were designed to amplify a fragment of the P. berghei cytochrome b gene were PbF-765 (5′-ATTACAAATAGTTCCAGAAT) and PbR-864 (5′-TAAAGATGCTATAACAATGA). PCR was carried out for 35 cycles of 10 s of denaturation at 95°C (first cycle, 10 min); 10 s of annealing at 46°C and extension at 72°C, respectively; 1 cycle of melting at 65°C for 1 min; and 1 cycle of cooling at 40°C for 30 s.
ACKNOWLEDGMENTS
This work was supported by an annual institutional grant to the Eijkman Institute from the Ministry of Research, Technology, and Higher Education of the Republic of Indonesia. We gratefully acknowledge the support of various research grants to D.S. at the Eijkman Institute Malaria Laboratory.
REFERENCES
- 1.Nuralitha S, Siregar JE, Syafruddin D, Roelands J, Verhoef J, Hoepelman IM, Marzuki S. 2016. Within-host selection of drug resistance in a mouse model of repeated incomplete malaria treatment: comparison between atovaquone and pyrimethamine. Antimicrob Agents Chemother 60:258–263. doi: 10.1128/AAC.00538-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fry M, Pudney M. 1992. Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4′-clhorophenyl)cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566C80). Biochem Pharmacol 43:1545–1553. doi: 10.1016/0006-2952(92)90213-3. [DOI] [PubMed] [Google Scholar]
- 3.Srivastava IK, Rottenberg H, Vaidya AB. 1997. Atovaquone, a broad spectrum antiparasitic drug, collapses mitochondrial membrane potential in a malarial parasite. J Biol Chem 272:3961–3966. doi: 10.1074/jbc.272.7.3961. [DOI] [PubMed] [Google Scholar]
- 4.Siregar JE, Syaffruddin D, Matsuoka H, Kita K, Marzuki S. 2008. Mutation underlying resistance of Plasmodium berghei to atovaquone in the quinone binding domain 2 (Qo2) of the cytochrome b gene. Parasitol Int 57:229–232. doi: 10.1016/j.parint.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Syafruddin D, Siregar JE, Marzuki S. 1999. Mutations in the cytochrome b gene of Plasmodium berghei conferring resistance to atovaquone. Mol Biochem Parasitol 104:185–194. doi: 10.1016/S0166-6851(99)00148-6. [DOI] [PubMed] [Google Scholar]
- 6.Brown WM, George M Jr, Wilson AC. 1979. Rapid evolution of animal mitochondrial DNA. Proc Natl Acad Sci U S A 76:1967–1971. doi: 10.1073/pnas.76.4.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Comtamine V, Picard M. 2000. Maintenance and integrity of the mitochondrial genome: a plethora of nuclear genes in the budding yeast. Microbiol Mol Biol. Rev 64:281–315. doi: 10.1128/MMBR.64.2.281-315.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Siregar JE, Kurisu G, Kobayashi T, Matsuzaki M, Sakamoto K, Mi-ici F, Watanabe Y, Hirai M, Matsuoka H, Syafruddin D, Marzuki S, Kita K. 2015. Direct evidence for the atovaquone action on the Plasmodium cytochrome bc1 complex. Parasitol Int 64:295–300. doi: 10.1016/j.parint.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 9.Korsinczky M, Chen N, Kotecka B, Saul A, Rieckmann K, Cheng Q. 2000. Mutations in Plasmodium falciparum cytochrome b that are associated with atovaquone resistance are located at a putative drug-binding site. Antimicrob Agents Chemother 44:2100–2108. doi: 10.1128/AAC.44.8.2100-2108.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andersson DI, Hughes D, Roth JR. 17 March 2011. The origin of mutants under selection: interactions of mutation, growth, and selection. EcoSal Plus 2011 doi: 10.1128/ecosalplus.5.6.6. [DOI] [PubMed] [Google Scholar]
- 11.Andersson DI, Hughes D. 2015. Evolutionary consequences of drug resistance: shared principles across diverse targets and organisms. Nat Rev Genet 16:459–471. doi: 10.1038/nrg3922. [DOI] [PubMed] [Google Scholar]
- 12.Goodman CD, Siregar JS, Mollard V, Vega-Rodríguez J, Syaffruddin D, Matsuoka H, Matsuzaki M, Toyama T, Sturm A, Cozijnsen A, Jacobs-Loreno M, Kita K, Marzuki S, McFadden GI. 2016. Parasite resistance to the antimalarial atovaquone is not transmissible by mosquitoes. Science 352:349–353. doi: 10.1126/science.aad9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peters W. 1999. The chemotherapy of rodent malaria. LVII. Drug combinations to impede the selection of drug resistance, part 1: which model is appropriate? Ann Trop Med Parasitol 93:569–587. [DOI] [PubMed] [Google Scholar]
- 14.Jambou R, El-Assaad F, Combes V, Grau GE. 2011. In vitro culture of Plasmodium berghei-ANKA maintains infectivity of mouse erythrocytes inducing cerebral malaria. Malar J 10:346. doi: 10.1186/1475-2875-10-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wooden J, Kyes S, Sibley CH. 1993. PCR and strain identification in Plasmodium falciparum. Parasitol Today 9:303–305. doi: 10.1016/0169-4758(93)90131-X. [DOI] [PubMed] [Google Scholar]
- 16.Brasseur R. 1988. Calculation of the three-dimensional structure of Saccharomyces cerevisiae cytochrome b inserted in a lipid matrix. J Biol Chem 263:12571–12575. [PubMed] [Google Scholar]
- 17.di Rago JP, Colson AM. 1988. Molecular basis for resistance to antimycin and diuron, Q cycle inhibitors acting at the Qi site in the mitochondrial ubiquinol-cytochrome c reductase in Saccharomyces cerevisiae. J Biol Chem 263:12564–12570. [PubMed] [Google Scholar]
- 18.Birth D, Kao WC, Hunte C. 2014. Structural analysis of atovaquone-inhibited cytochrome bc1 complex reveals the molecular basis of antimalarial drug action. Nat Commun 5:4029. doi: 10.1038/ncomms5029. [DOI] [PubMed] [Google Scholar]
- 19.Vaidya AB, Lashgari MS, Pologe LG, Morrisey J. 1993. Structural features of Plasmodium cytochrome b that may underlie susceptibility to 8-aminoquinolines and hydroxynaphthoquinones. Mol Biochem Parasitol 58:33–42. doi: 10.1016/0166-6851(93)90088-F. [DOI] [PubMed] [Google Scholar]
- 20.di Rago JP, Coppee JY, Colson AM. 1989. Molecular basis for resistance to myxothiazol, mucidin (strobilurin A), and stigmatellin. Cytochrome b inhibitors acting at the center of the mitochondrial ubiquinol-cytochrome c-reductase in Saccharomyces cerevisiae. J Biol Chem 264:14543–14548. [PubMed] [Google Scholar]
- 21.Howell N, Gilbert K. 1988. Mutational analysis of the mouse mitochondrial cytochrome b gene. J Mol Biol 203:607–618. doi: 10.1016/0022-2836(88)90195-7. [DOI] [PubMed] [Google Scholar]
- 22.Kessl JJ, Lange BB, Merbitz-Zaharadnik T, Zwicker K, Hill P, Meunier B, Palsdottir H, Hunte C, Meshnick S, Trumpower BL. 2003. Molecular basis for atovaquone binding to the cytochrome bc1 complex. J Biol Chem 278:31312–31318. doi: 10.1074/jbc.M304042200. [DOI] [PubMed] [Google Scholar]