

ABSTRACT
While azole drugs targeting the biosynthesis of ergosterol are effective antifungal agents, their extensive use has led to the development of resistant organisms. Infections involving azole-resistant forms of the filamentous fungus Aspergillus fumigatus are often associated with genetic changes in the cyp51A gene encoding the lanosterol α14 demethylase target enzyme. Both a sequence duplication in the cyp51A promoter (TR34) and a substitution mutation in the coding sequence (L98H) are required for the full expression of azole resistance. A mechanism commonly observed in pathogenic yeast such as Candida albicans involves gain-of-function mutations in transcriptional regulatory proteins that induce expression of genes encoding ATP-binding cassette (ABC) transporters. We and others have found that an ABC transporter protein called Cdr1B (here referred to as AbcG1) is required for wild-type azole resistance in A. fumigatus. Here, we test the genetic relationship between the TR34 L98H allele of cyp51A and an abcG1 null mutation. Loss of AbcG1 from a TR34 L98H cyp51A-containing strain caused a large decrease in the azole resistance of the resulting double-mutant strain. We also generated antibodies that enabled the detection of both the wild-type and L98H forms of the Cyp51A protein. The introduction of the L98H lesion into the cyp51A gene led to a decreased production of immunoreactive enzyme, suggesting that this mutant protein is unstable. Our data confirm the importance of AbcG1 function during azole resistance even in a strongly drug-resistant background.
KEYWORDS: ATP-binding cassette transporter, Aspergillus fumigatus, genetic analysis, Western blotting, azole resistance, cyp51A
INTRODUCTION
Azole drugs are the most common clinical treatment for infections caused by the major human filamentous fungal pathogen Aspergillus fumigatus (see reference 1 for a recent review). While azole drugs still retain significant efficacy in limiting the growth of A. fumigatus, the continued reliance on this drug has led to the emergence of resistant isolates. In fact, an infection with azole-resistant A. fumigatus leads to a dramatic increase in mortality, and resistance is a major problem in the treatment of this fungal disease (2–4).
The increasing incidence of azole-resistant isolates causing A. fumigatus infections has led to intensive analyses of the molecular basis of azole resistance. Important early work indicated that, at least in Western Europe, the primary genetic alteration responsible for azole resistance lay within the cyp51A locus (5). The target enzyme of azole drugs, lanosterol α14 demethylase, is encoded in A. fumigatus by the cyp51A and cyp51B genes (6). Surprisingly, two linked mutations were found that mapped exclusively to cyp51A, namely, a change in the promoter sequence for this gene and a single-amino-acid substitution mutation. The promoter mutation consists of a duplication of a 34-bp region and was designated TR34, while the coding sequence mutation corresponds to the replacement of a leucine residue at position 98 with a histidine (L98H) (7, 8). This compound mutant (TR34 L98H cyp51A) allele was detected in the vast majority of resistant isolates and has spread from Europe to Asia (9). Aspergillosis associated with azole resistance has a mortality rate that can approach 90%, making the treatment of infections with fungi containing this drug-resistant allele a high priority (10).
The preponderance of TR34 L98H cyp51A alleles was unusual and complicated by the fact that many resistant isolates had no discernible change in the DNA sequence of this gene (recently reviewed in reference 1). The major human fungal pathogen, Candida albicans, also develops azole resistance. However, these isolates do not routinely have alterations in ERG11 (the sole gene encoding lanosterol α14 demethylase in C. albicans) but rather overexpress ATP-binding cassette (ABC) transporter-encoding genes (11). The major ABC transporter that is produced at levels far higher than those in the wild-type yeast is called Cdr1 (12). The overexpression of the CDR1 transcript is most often caused by single-amino-acid substitution mutations in a Zn2Cys6 zinc cluster-containing transcription factor called Tac1 (13). The link between the overexpression of ABC transporter proteins and azole resistance is also observed in another Candida species, Candida glabrata, as well as in the typically nonpathogenic yeast Saccharomyces cerevisiae (14–16).
Given that at least two pathogenic yeast acquire azole tolerance by increasing the expression ABC transporters, we wondered if A. fumigatus might acquire resistance by a similar mechanism. Evidence has accumulated that the organisms in patients exposed chronically to azole drug therapy can develop resistance to this important antifungal drug yet still maintain a wild-type version of cyp51A (2, 17, 18), and evidence exists for the involvement of ABC transporters in azole resistance (19). The analyses of representative patient isolates that exhibited an unaltered copy of cyp51A yet were highly itraconazole resistant led to the discovery that these strains overexpressed an ABC transporter-encoding gene designated cdr1B (20). This locus has been assigned multiple names and, in this article, we will refer to the gene as abcG1 using a systematic nomenclature suggested by others (21). The expression of abcG1 was found to be required for wild-type voriconazole resistance in several laboratory strains and localized to the plasma membrane of A. fumigatus (22). Together, these data suggested that many of the properties of AbcG1 resemble those of C. albicans Cdr1. We wanted to determine if a role for abcG1 in azole resistance could be assessed in A. fumigatus in the presence of the TR34 L98H cyp51A allele. Our data indicate that abcG1 expression is required for the elevated azole tolerance typically seen in the presence of the compound resistance allele of cyp51A. We also prepared antipeptide antibodies directed against Cyp51A-specific epitopes and used these to demonstrate that the L98H form of Cyp51A is produced at a lower level than the wild-type protein, although this mutant enzyme supports more azole resistance than the wild type. These data provide new insight into the molecular basis of azole resistance in A. fumigatus.
RESULTS
Epistasis analysis of abcG1 and cyp51A.
Previous work on azole resistance in A. fumigatus had focused on the analysis of either cyp51A or abcG1, but the relationship between these two resistance determinants had not been investigated. To explore the possible epistasis between mutant alleles of these two genes, we obtained a set of mutant cyp51A-containing plasmids to construct an isogenic series of cyp51A mutant strains containing or lacking the abcG1 locus. These cyp51A clones corresponded to single mutants containing either the TR34 or L98H lesion individually or both lesions present together in the same cyp51A clone. These plasmids were integrated using a previously described strategy (23) at the wild-type cyp51A locus in isogenic strains containing wild-type abcG1 or a disruption mutant form of this gene (22). Two isolates of each mutant strain were tested to ensure consistent behaviors. A representative experiment is shown in Fig. 1.
FIG 1.
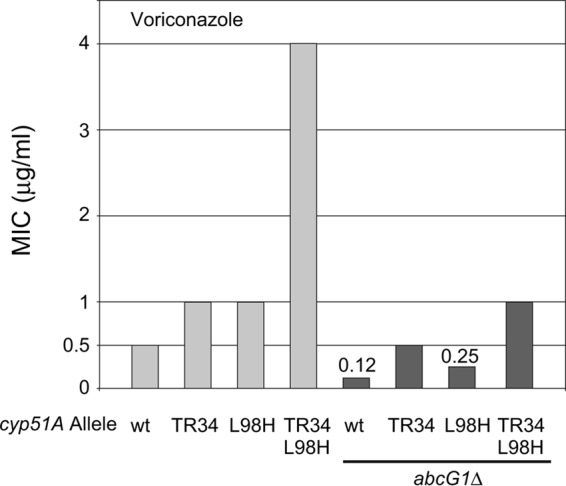
AbcG1 is required for wild-type azole resistance. Cells containing or lacking (abcG1Δ) the abcG1 ABC transporter-encoding genes and the indicated cyp51A alleles were tested for their MIC for voriconazole as previously described (33). Strains with MIC values different than those on the ordinate are indicated on the plot above the bars of interest.
Strikingly, the loss of abcG1 in each of the strains led to significant reductions in the MICs to voriconazole. The presence of either the L98H or TR34 form of cyp51A elevated the voriconazole MIC, even in the absence of abcG1. However, the degree of elevation was 25% (L98H) or 50% (TR34) of that seen in the presence of the ABC transporter. Combining these two mutant alleles of cyp51A increased the voriconazole MIC to 400% of that seen in either of the single mutant forms in the presence of the wild-type abcG1 gene. The synergistic increase in azole MIC upon the combination of the two single TR34 and L98H alleles was previously described (7). However, there was a 4-fold decrease in the MIC value when the two single mutant cyp51A alleles were combined in the absence of abcG1. These data clearly indicate a necessity for the ABC transporter AbcG1 for acquiring the elevated MIC normally seen in the presence of the TR34 L98H cyp51A allele.
Steady-state level of Cyp51A is reduced by replacing leucine 98 with histidine.
Although the importance of Cyp51A in azole resistance has been clear for some time, very limited information is available concerning the expression of the Cyp51A protein. Previous experiments have focused on transcriptional measurements of cyp51A mRNA. To directly examine the expression of the cyp51A-encoded enzyme, we prepared two different antipeptide antibodies that recognize either a region of Cyp51A from residues 95 to 108 (referred to as peptide 3) or residues 478 to 491 (peptide 2). Note that the antipeptide 3 antibody contains the L98 position that is altered in the L98H mutant form of Cyp51A. This makes data obtained using the antipeptide 3 antibody suspect when used with this mutant form of Cyp51A, although this antibody works well with wild-type Cyp51A.
We validated both antibody preparations by comparing the production of cyp51A-dependent proteins in the wild-type and cyp51AΔ strains. These strains were left untreated or were challenged with azole drug (Fig. 2). For each antibody preparation, only one cyp51A-specific polypeptide was detected and only in the presence of azole induction. Both of these antibodies detect the wild-type form of Cyp51A.
FIG 2.
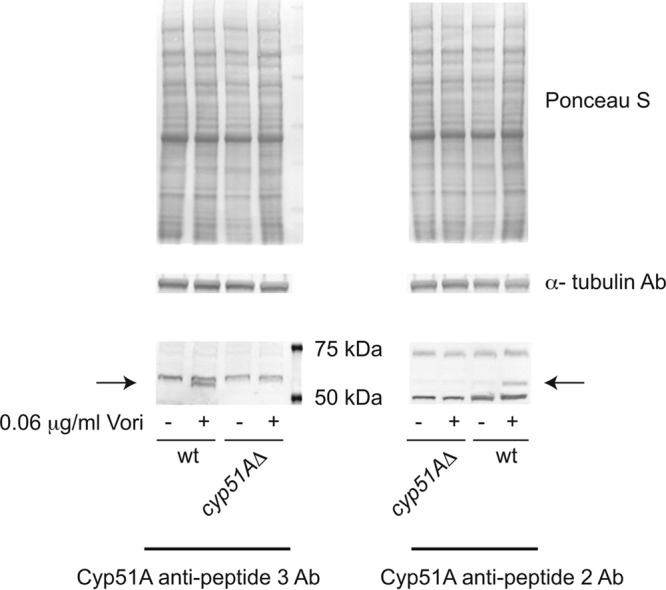
Validation of anti-Cyp51A antibodies. Whole-cell protein extracts were prepared from untreated (−) or voriconazole-challenged (+) cells. The two strains used were wild-type AfS35 cells or an isogenic derivative lacking the cyp51A gene. The molecular weight standards are indicated on the right-hand side of the antipeptide 3 Cyp51A Western blot. The arrows denote the location of the specific Cyp51A polypeptide with both antibodies. The top panels represent loading controls in which the blot was stained for total protein (Ponceau S) or subjected to Western blotting using an anti-α tubulin antibody.
We then used these antibody preparations to carry out Western blot detection of the various forms of Cyp51A produced in transformants containing different mutant forms of the cyp51A gene. Cells were grown either without or with voriconazole and were then processed for protein extraction. Equal amounts of protein were resolved on SDS-PAGE and were probed with different antibodies, including the two different anti-Cyp51A peptide antibodies, and anti-tubulin as the loading control (Fig. 3).
FIG 3.

Western blot analysis of Cyp51A protein levels. (A) Whole-cell protein extracts were prepared by grinding hyphae in liquid nitrogen. Equal amounts of protein extracts were resolved by SDS-PAGE and were transferred to nitrocellulose membranes. Cultures were grown in the absence (−) or presence (+) of 0.06 μg/ml voriconazole in minimal medium. Equal loading and transfer were ensured via staining the membranes with Ponceau S dye. Membranes were blocked and then probed with rabbit polyclonal antipeptide 3 antibodies prepared against a peptide of Cyp51A corresponding to residues 95 to 108 of the enzyme. Extracts were prepared from strains corresponding to an isogenic wild-type (wt, Afs35) or the same strain containing a TR34 promoter mutation, an L98H substitution form of cyp51A, or the corresponding double mutant strain (TR34 L98H) (left). Extracts were prepared from wild-type cells or an isogenic strain carrying multiple copies of the TR34 version of cyp51A (mcTR34) (right). Estimates by qPCR of the copy number of the cyp51A gene in this multiple copy strain suggest that at least 12 copies of the gene were introduced. These strains were grown as above, either in the absence or presence of voriconazole. (B) The same strains as in panel A were grown in the absence or presence of azole drug and analyzed by Western blotting but with two different antibodies. The top shows blotting with anti-tubulin antibody (α-tubulin Ab) as a loading control. The bottom denotes utilization of the rabbit polyclonal antipeptide 2 antibodies prepared against a peptide of Cyp51A corresponding to residues 478 to 491 of the enzyme. Use of this antibody avoids reactivity issues that might be caused by the L98H form of Cyp51A that is contained within the antigenic region of the antipeptide 3 antibody (see the text for details).
Western analysis showed that the anti-Cyp51A peptide 3 antibody (Fig. 3A) detected a polypeptide consistent with the 58-kDa predicted molecular mass of native Cyp51A in extracts from wild-type cells. The expression of Cyp51A was strongly induced after exposure to voriconazole (>10-fold). The steady-state level of Cyp51A produced in the absence of an azole challenge was quite low but the strong induction after drug treatment produced an easily detectable signal. As expected from previous analyses of cyp51A transcription (7, 24), the presence of the TR34 promoter mutation led to elevated expression of Cyp51A protein.
These Western blot patterns were affected by the introduction of the L98H allele of cyp51A, especially in the presence of the wild-type promoter. In the absence of voriconazole induction, the L98H Cyp51A protein was difficult to detect. After a drug challenge, the mutant protein could easily be seen, albeit at lower levels than the wild-type enzyme. The TR34-driven L98H Cyp51A was found at levels very similar to those seen in the presence of the TR34 promoter lesion alone, in both the drug-treated and untreated samples. The levels of wild-type Cyp51A were highest when produced from a multicopy integrant of the TR34 cyp51A gene, consistent with the presence of roughly 10 copies of the gene.
As mentioned above, the interpretation of the Western blot results from the L98H form of Cyp51A using the antipeptide 3 antibody was complicated by the overlapping nature of the epitope and the L98 position. To address this concern, we used the antipeptide 2 antibody that recognizes a C-terminal epitope located 389 residues away from the peptide 3 antigen, and we performed Western blotting as per the method described above.
Use of the antipeptide 2 antibody produced results very similar to those seen with the previous antipeptide 3 Western blotting (Fig. 3B). Using densitometry, we quantitated the levels of Cyp51A seen in all of these samples. The steady-state levels of the wild-type and L98H forms of Cyp51A were very similar in the absence of drug, but the wild-type protein was induced 3-fold more than L98H Cyp51A upon voriconazole treatment. Interestingly, we did not detect a difference in the steady-state levels of wild-type and L98H Cyp51A proteins when expressed from the hyperactive TR34 promoter. A comparison of the results obtained using the two different antibodies suggests that the L98H alteration does not dramatically influence Cyp51A recognition by the antipeptide 3 antibody. Tubulin levels were consistent across all samples, confirming that equivalent amounts of protein were loaded in each lane.
Transcription level of the cyp51A gene.
To correlate the extent of expression of the Cyp51A protein with its mRNA level, we analyzed cyp51A transcription using reverse transcription coupled with quantitative PCR (RT-qPCR) as described previously (24). We prepared total RNA from a wild-type strain carrying an unaltered cyp51A gene or a mutant locus encoding the L98H form of Cyp51A, along with isogenic derivatives containing the TR34 mutant form of the cyp51A promoter driving either the wild-type or the L98H form of the enzyme. We also prepared RNA from a strain containing the multicopy version of TR34 cyp51A (mcTR34). Levels of mRNA were measured by RT-qPCR and then normalized to the expression of the wild-type cyp51A mRNA (Fig. 4A).
FIG 4.
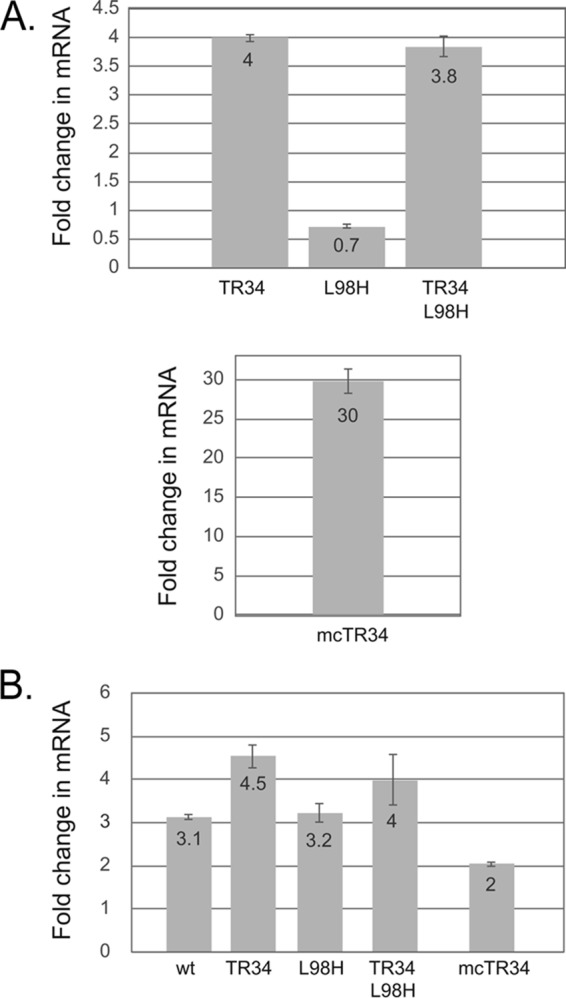
Levels of cyp51A mRNA. Steady-state levels of cyp51A mRNA were analyzed using reverse transcription-qPCR from total RNA recovered from strains containing the indicated forms of the cyp51A gene. (A) Strains were grown to mid-log phase and analyzed for cyp51A mRNA levels. Data are presented after normalization to cyp51A expression in wild-type cells. The bottom shows cyp51A mRNA produced in the strain containing multiple copies of cyp51A containing the TR34 promoter mutation (mcTR34). (B) Voriconazole induction of cyp51A. The strains used above were grown in parallel cultures with voriconazole added to one flask for the final 8 h of growth. Total RNA was prepared from cultures grown in the absence and presence of voriconazole. Data are presented as the ratio of mRNA expression in the presence of voriconazole normalized to expression in the absence of drug for each cyp51A gene.
As reported (7), we found a roughly 4-fold elevation in the amount of mRNA encoding either the wild-type or the L98H form of Cyp51A expressed from the promoter containing the TR34 element in comparison to the expression of the wild-type cyp51A gene. The L98H cyp51A allele was expressed at 70% of the level of the wild-type gene. Strikingly, mRNA levels for the multicopy TR34 allele were the highest observed, fully 30-fold greater than those of the wild type.
The mRNA levels produced after a voriconazole challenge were also determined to evaluate the effect of azole drug treatment on the expression of these different cyp51A alleles. Strains were cultured with or without 0.06 μg/ml voriconazole for 8 h, and then total RNA was isolated and analyzed by RT-qPCR. In this experiment, expression levels were determined as the ratios of the amounts of mRNA produced in the presence of azole drug to the amounts of transcription in the absence of drug (Fig. 4B).
The induction of cyp51A transcription ranged from 3-fold when the wild-type promoter was present to 4- to 4.5-fold in the presence of the TR34 promoter. The multicopy TR34 cyp51A locus exhibited the lowest induction, only 2-fold, likely due to the high levels of the wild-type enzyme preventing voriconazole at this dose from causing an upregulation of cyp51A. Note that the level of Cyp51A protein expression seen with the multicopy TR34 cyp51A locus in the absence of a drug challenge exceeded that of the wild-type cyp51A even after voriconazole treatment.
DISCUSSION
This work has revealed several new features of azole drug resistance in A. fumigatus. Our primary goal was to determine if the major drug resistance allele found in clinical isolates (TR34 L98H cyp51A) was sufficient for high-level azole resistance. Clearly, even in the presence of this strong resistance allele, AbcG1 still has a significant impact on the resistance of the mutant strain. The elimination of AbcG1 caused large reductions in azole MIC values in every genetic background examined. These data support the view that interventions that lower the activity of AbcG1 might also reduce drug resistance even in the presence of the TR34 L98H cyp51A mutation.
Our development of two different antibodies that recognize the Cyp51A enzyme has provided the first information about the levels of this important protein in A. fumigatus. The only previous examination of Cyp51A protein levels we are aware of involved the heterologous expression of the A. fumigatus proteins in Saccharomyces cerevisiae, and no data were shown (25). Here, we establish that Cyp51A protein levels were as expected on the basis of previous measurements of mRNA with azole induction and with the presence of the TR34 promoter mutation. However, these antisera have led to the unexpected determination that the level of the L98H form of Cyp51A is reduced compared with that of the wild-type enzyme. The direct detection of the protein is the only manner in which data such as these could be obtained. Surprisingly, the combination of the TR34 promoter mutation with the L98H form of Cyp51A appeared to return protein levels to those of the wild-type enzyme driven by this promoter mutation.
These data suggest an underlying complexity to both the azole resistance and to Cyp51A function. Even though the level of L98H Cyp51A was lower than the wild-type enzyme, this protein still supported a higher level of azole resistance. Molecular modeling studies have indicated that the L98H mutation in Cyp51A may change the structure of the enzyme, possibly altering the access of the drug to the active site (23). A more recent analysis using the crystal structure of S. cerevisiae Erg11 as a scaffold on which to model the A. fumigatus Cyp51A structure suggested that the L98H mutation may also destabilize the protein (26). Our antibody data directly confirm this prediction, as levels of the L98H Cyp51A protein were found to be reduced compared with those of the wild type.
As mentioned above, the equivalency of the expressions of the wild-type and L98H Cyp51A proteins when driven by the TR34 promoter is remarkable. We speculate this may be due to interactions of Cyp51A with other enzymatic components. Data from S. cerevisiae, as well as from plants, indicate that there are extensive protein-protein interactions involved in the sterol biosynthetic pathways for forming enzymatic complexes (27, 28). It is possible that the instability of the L98H form of the enzyme is suppressed by associations with other proteins involved in ergosterol biosynthesis. This suppression may only happen when the L98H form is produced to a sufficiently high level, such as that provided by the TR34 promoter. Further experiments are required to test this hypothesis.
We hypothesize that wild-type azole resistance requires the expression of both cyp51A and abcG1. The recent discovery of a transcriptional regulatory protein called AtrR serves to directly link these two genes at the level of gene transcription. AtrR was originally detected as a positive regulator of azole resistance and was then demonstrated to regulate cyp51A gene transcription in addition to abcG1 (29). This link suggests the intriguing possibility that the finding that ABC transporters such as AbcG1 and ergosterol biosynthetic enzymes both affect azole drug resistance is not serendipitous but rather illustrates a physiological connection between these two different groups of proteins. Understanding the biology behind the linkage between ABC transporters and ergosterol biosynthesis in A. fumigatus may enable a better and more sustainable utilization of important antifungal drugs, such as the azole compounds.
MATERIALS AND METHODS
A. fumigatus strains and media.
All of the strains used in this study were derived from the AfS35 (FGSC A1159) strain and are listed in Table 1. A. fumigatus strains were typically grown at 37°C in rich medium (Sabouraud dextrose containing 0.5% tryptone, 0.5% peptone, 2% dextrose, pH 5.6 ± 0.2). Transformants were selected using minimal medium (1% glucose, pH 6.5, with nitrate salts, trace elements, and vitamins as described in the appendix of reference 30), supplemented with 1% sorbitol and 200 mg/liter hygromycin gold (Invivogen, San Diego, CA). For solid medium, 1.5% agar was added.
TABLE 1.
A. fumigatus strains used in this study
| Strain | Parent | Genotype | Source or reference |
|---|---|---|---|
| AfS35 | D141 | akuAΔ::loxP | Fungal Genetic Stock Center |
| SPF92 | AfS35 | wt cyp51A hph | This study |
| SPF94 | AfS35 | TR34 cyp51A hph | This study |
| SPF95 | AfS35 | TR34 cyp51A hph, multicopy | This study |
| SPF96 | AfS35 | L98H cyp51A hph | This study |
| SPF98 | AfS35 | TR34 L98H cyp51A hph | This study |
| SPF84 | SPF84 | abcG1Δ::loxP | 22 |
| SPF100 | SPF84 | wt cyp51A hph | This study |
| SPF102 | SPF84 | TR34 cyp51A hph | This study |
| SPF102A | SPF84 | TR34 cyp51A hph, multicopy | This study |
| SPF106 | SPF84 | L98H cyp51A hph | This study |
| SPF106 | SPF84 | TR34 L98H cyp51A hph | This study |
| SPF129 | AfS35 | cyp51AΔ::ptrA | This study |
Transformation and generation of A. fumigatus cyp51A mutants.
Plasmids for generating A. fumigatus cyp51A mutants were provided by Eveline Snelders and colleagues. Plasmids containing wild-type, TR34, L98H, or TR34 L98H versions of the cyp51A gene (23) were digested with MluI and SpeI for transformation into the A. fumigatus AfS35 strain or into an isogenic derivative lacking abcG1 (SPF84; AfS35 abcG1Δ::loxP). Transformation was carried out by generating protoplasts as described elsewhere (31). Hygromycin-resistant transformants were regrown on Sabouraud dextrose medium supplemented with 200 mg/liter hygromycin. DNA was isolated and used as the template to detect the novel junctions formed upon correctly targeted integration of the cyp51A alleles. Primers Hph-DF and cyp51A-DR were used to detect the downstream novel junction. Once transformants with targeted integration were identified by PCR, the cyp51A promoter and gene were PCR amplified using the oligonucleotides cyp51A-Seq-F1 and cyp51A-Seq-R1. The PCR products were then used as the templates to sequence the cyp51A promoter and gene to confirm the presence of the TR34 element and/or the L98H mutation using the following primer sets: cyp51A-Seq-F to confirm the TR34 lesion, cyp51A-Seq-R2 to confirm the L98H mutation, and the primers cyp51A-Seq-F2 and cyp51A-Seq-R to sequence the rest of the cyp51A gene downstream of the L98H mutation. Two independent cyp51A mutants each carrying the wild-type (having a silent mutation for Val immediately downstream of translation initiation codon AUG), TR34, L98H, or TR34 L98H alleles were used for further assays.
Determination of the copy number of cyp51A alleles was accomplished using genomic DNA of targeted integrants as the templates. Quantitative real-time PCR was performed using iTaq universal SYBR green mix (Bio-Rad) along with the cyp51A-specific primers cyp51A-RT-F and cyp51A-RT-R or reference gene (act1)-specific primers act1-RT-F and act1-RT-R. The CT values were calculated for cyp51A versus act1 to determine single or multicopy status of the cyp51A gene. Amplification was carried out in the iCycler apparatus (Bio-Rad, Hercules, CA) in a two-step process as follows: a denaturation step of 30 s at 95°C and 40 cycles of 95°C for 10 s each, with annealing and extension at 60°C for 30 s each. The reporter signals were analyzed using the iCycler iQ software (Bio-Rad). The relative copy numbers of the cyp51A gene with respect to act1 were determined according to the 2−ΔΔCT method (32), where ΔΔCT = ΔCT of cyp51A − ΔCT of act1.
A cyp51A deletion strain was generated using ptrA split marker cassettes. The split marker cassettes were constructed by fusion PCR as described below. The upstream split marker had a 1.2-kb region corresponding to the DNA segment immediately upstream of the cyp51A gene and was PCR amplified using cyp51Ako-US-F1 and cyp51Ako-US-R1 primers. The partial ptrA gene in the upstream split marker was amplified using ptrA-US-F1 and ptrA-US-R1 primers. The fusion PCR was performed using nested primers cyp51Ako-US-F1a and ptrA-US-R1a. Similarly, the downstream split marker had a 1.3-kb region corresponding to a region immediately downstream of the cyp51A gene and was PCR amplified using cyp51Ako-DS-F1 and cyp51Ako-DS-R1 primers. The partial ptrA gene in the downstream split marker was amplified using ptrA-DS-F2 and ptrA-DS-R2 primers. The fusion PCR was performed using nested primers ptrA-US-F2a and cyp51Ako-DS-R2. The upstream and downstream ptrA split markers were cotransformed into the AfS35 strain, and pyrithiamine-resistant transformants were verified for targeted integration by detecting the presence of novel junctions formed upstream and downstream of the cyp51A locus by PCR as well as by the sensitivity to azole drugs. The representative cyp51AΔ strain used in the study was designated SPF129.
MIC assay.
MICs of the azoles itraconazole and voriconazole against Aspergillus were determined using a broth microdilution (BMD) method. Itraconazole (Janssen), posaconazole, and voriconazole (Pfizer) were obtained as reagent-grade powders from their respective manufacturers. The BMD method was performed according to the CLSI M38-A2 standard (33). Trays containing a 0.1-ml aliquot of the appropriate drug solution (2× the final drug concentration) in each well of a 96-well plate were sealed and stored at −70°C until they were used in the study. A stock conidial suspension (106 spores/ml) was diluted to a final inoculum concentration of 0.4 × 104 to 5 × 104 CFU/ml and was dispensed into the microdilution wells. The final concentrations of drugs in the wells ranged from 0.007 to 8 g/ml. The inoculated microdilution trays were incubated at 35°C and read at 24 and 48 h. The MIC endpoint was defined as the lowest concentration that produced complete inhibition of growth. Quality control was ensured by testing the following strains recommended in CLSI standard M38-A2: Candida parapsilosis ATCC 22019, Candida krusei ATCC 6258, and Aspergillus flavus ATCC 204304.
Measurement of mRNA level.
Reverse transcription quantitative PCR (RT-qPCR) was performed as described in reference 24, with the following modifications. Approximately 106 spores were inoculated in a petri dish containing 20 ml of Sabouraud dextrose broth overnight at 37°C and either were left untreated or were treated with 0.0625 mg/liter voriconazole for 8 h. Mycelium that formed as a biofilm on the top was collected (∼100 mg) and was ground into a fine powder in liquid nitrogen using a mortar and pestle. Total RNA was then isolated from the ground mycelium using the Trizol reagent (Ambion) according to the manufacturer's instructions. Total RNA was isolated and purified using an RNeasy minikit (Qiagen). Traces of contaminating genomic DNA were eliminated by treating the RNA with turbo DNase followed by a DNase inactivation reagent (both reagents from Ambion). Synthesis of cDNA was performed using an iScript cDNA synthesis kit (Bio-Rad) with 500 μg of RNA as the template. For the quantitative PCR, primers were designed using the Primer3 software (http://bioinfo.ut.ee/primer3/). Primer concentrations were optimized for each gene, and annealing profiles were analyzed to evaluate nonspecific amplification by primer dimers. Control reaction mixtures including RNA instead of cDNA were made for each gene. The threshold cycle (CT) values were determined in the logarithmic phase of amplification for all of the genes, and the average CT value for each sample was calculated from three replicates. The CT value of the gene coding for actin (act1) was used for normalization of variable cDNA levels, and the fold difference in transcript levels was determined with respect to cyp51A or abcG1. Amplification was carried out in the iCycler apparatus from Bio-Rad in a two-step process as follows: a denaturation step of 30 s at 95°C and 40 cycles of 95°C for 10 s each, with annealing and extension at 60°C for 30 s each. The reporter signals were analyzed using the iCycler iQ software (Bio-Rad).
Generation of Cyp51 peptide antibodies.
Immunoreactive peptides against Cyp51A were shortlisted using the OptimumAntigen design tool algorithm (GenScript). Three of the predicted most-immunoreactive peptides were synthesized and conjugated to a carrier protein, and each peptide was used to immunize 2 rabbits. The antiserum obtained was then affinity purified. All of the above steps were carried out at GenScript, Inc. The two antipeptide antibodies that showed Cyp51A-specific immunoreactivity in A. fumigatus cell lysates were raised against FNVDGKKGVPETDY (peptide 2) and NGKLKDVNAEEVYS (peptide 3), while a third peptide failed to show any specific immunoreactivity.
Western blot analysis.
Approximately 106 spores were inoculated in a petri dish containing 20 ml of Sabouraud dextrose broth at 37°C for 6 h, and either were left untreated or were treated with 0.0625 mg/liter voriconazole for 16 h. Mycelium that formed as a biofilm on the top was collected (∼100 mg) and was ground into fine powder in liquid nitrogen using a mortar and pestle. The ground mycelium was resuspended in 0.5 ml of 10% trichloroacetic acid (TCA), thoroughly vortexed, incubated for 5 min at room temperature, and then washed 3 times in 90% acetone with 20 mM HCl and air dried. The TCA precipitate was then extracted with 8 M urea sample buffer (8 M urea, 5% SDS, 1% β-ME, 40 mM Tris-HCl [pH 8], with traces of bromophenol blue). Aliquots (5 μl) of this suspension were electrophoresed by 10% SDS-PAGE and then transferred to a nitrocellulose membrane. The membrane was blocked with 5% nonfat dry milk in tris-buffered saline with 0.1% Tween 20 (TBST), washed three times with TBST, and then probed with the appropriate polyclonal antibody raised in rabbit. Cyp51A peptide 2 antibody was used at a 1/75 dilution, while the Cyp51A peptide 3 antibody was used at a 1/750 dilution. Membranes were incubated for 40 min in IRDye 680RD goat anti-rabbit (LI-COR) secondary antibody at a 1:15,000 dilution. After 4 washes with TBST, the infrared signal was detected and analyzed using the Odyssey infrared imager 9120 (LI-COR). The bands were quantitated using the Image Studio Lite 4.0 software (LI-COR).
ACKNOWLEDGMENTS
We thank Eveline Snelders, Jan Zoll, and Willem Melchers for providing the cloned alleles of cyp51A and Linda Boyken for performing the MIC determinations. We thank Fabio Gsaller, Mike Bromley, and Paul Bowyer for important discussions.
REFERENCES
- 1.Hagiwara D, Watanabe A, Kamei K, Goldman GH. 2016. Epidemiological and genomic landscape of azole resistance mechanisms in Aspergillus fungi. Front Microbiol 7:1382. doi: 10.3389/fmicb.2016.01382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Howard SJ, Cerar D, Anderson MJ, Albarrag A, Fisher MC, Pasqualotto AC, Laverdiere M, Arendrup MC, Perlin DS, Denning DW. 2009. Frequency and evolution of azole resistance in Aspergillus fumigatus associated with treatment failure. Emerg Infect Dis 15:1068–1076. doi: 10.3201/eid1507.090043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Linden JW, Snelders E, Kampinga GA, Rijnders BJ, Mattsson E, Debets-Ossenkopp YJ, Kuijper EJ, Van Tiel FH, Melchers WJ, Verweij PE. 2011. Clinical implications of azole resistance in Aspergillus fumigatus, The Netherlands, 2007–2009. Emerg Infect Dis 17:1846–1854. doi: 10.3201/eid1710.110226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van der Linden JW, Camps SM, Kampinga GA, Arends JP, Debets-Ossenkopp YJ, Haas PJ, Rijnders BJ, Kuijper EJ, van Tiel FH, Varga J, Karawajczyk A, Zoll J, Melchers WJ, Verweij PE. 2013. Aspergillosis due to voriconazole highly resistant Aspergillus fumigatus and recovery of genetically related resistant isolates from domiciles. Clin Infect Dis 57:513–520. doi: 10.1093/cid/cit320. [DOI] [PubMed] [Google Scholar]
- 5.Snelders E, van der Lee HA, Kuijpers J, Rijs AJ, Varga J, Samson RA, Mellado E, Donders AR, Melchers WJ, Verweij PE. 2008. Emergence of azole resistance in Aspergillus fumigatus and spread of a single resistance mechanism. PLoS Med 5:e219. doi: 10.1371/journal.pmed.0050219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mellado E, Diaz-Guerra TM, Cuenca-Estrella M, Rodriguez-Tudela JL. 2001. Identification of two different 14-alpha sterol demethylase-related genes (cyp51A and cyp51B) in Aspergillus fumigatus and other Aspergillus species. J Clin Microbiol 39:2431–2438. doi: 10.1128/JCM.39.7.2431-2438.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mellado E, Garcia-Effron G, Alcazar-Fuoli L, Melchers WJ, Verweij PE, Cuenca-Estrella M, Rodriguez-Tudela JL. 2007. A new Aspergillus fumigatus resistance mechanism conferring in vitro cross-resistance to azole antifungals involves a combination of cyp51A alterations. Antimicrob Agents Chemother 51:1897–1904. doi: 10.1128/AAC.01092-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verweij PE, Snelders E, Kema GH, Mellado E, Melchers WJ. 2009. Azole resistance in Aspergillus fumigatus: a side-effect of environmental fungicide use? Lancet Infect Dis 9:789–795. doi: 10.1016/S1473-3099(09)70265-8. [DOI] [PubMed] [Google Scholar]
- 9.Chowdhary A, Kathuria S, Xu J, Meis JF. 2013. Emergence of azole-resistant aspergillus fumigatus strains due to agricultural azole use creates an increasing threat to human health. PLoS Pathog 9:e1003633. doi: 10.1371/journal.ppat.1003633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verweij PE, Chowdhary A, Melchers WJ, Meis JF. 2016. Azole resistance in Aspergillus fumigatus: can we retain the clinical use of mold-active antifungal azoles? Clin Infect Dis 62:362–368. doi: 10.1093/cid/civ885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanglard D, Kuchler K, Ischer F, Pagani JL, Monod M, Bille J. 1995. Mechanisms of resistance to azole antifungal agents in Candida albicans isolates from AIDS patients involve specific multidrug transporters. Antimicrob Agents Chemother 39:2378–2386. doi: 10.1128/AAC.39.11.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prasad R, Dewergifosse P, Goffeau A, Balzi E. 1995. Molecular cloning and characterization of a novel gene of Candida albicans, CDR1, conferring multiple resistance to drugs and antifungals. Curr Genet 27:320–329. doi: 10.1007/BF00352101. [DOI] [PubMed] [Google Scholar]
- 13.Coste AT, Karababa M, Ischer F, Bille J, Sanglard D. 2004. TAC1, transcriptional activator of CDR genes, is a new transcription factor involved in the regulation of Candida albicans ABC transporters CDR1 and CDR2. Eukaryot Cell 3:1639–1652. doi: 10.1128/EC.3.6.1639-1652.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vermitsky JP, Edlind TD. 2004. Azole resistance in Candida glabrata: coordinate upregulation of multidrug transporters and evidence for a Pdr1-like transcription factor. Antimicrob Agents Chemother 48:3773–3781. doi: 10.1128/AAC.48.10.3773-3781.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsai HF, Krol AA, Sarti KE, Bennett JE. 2006. Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrob Agents Chemother 50:1384–1392. doi: 10.1128/AAC.50.4.1384-1392.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balzi E, Wang M, Leterme S, Van Dyck L, Goffeau A. 1994. PDR5: a novel yeast multidrug resistance transporter controlled by the transcription regulator PDR1. J Biol Chem 269:2206–2214. [PubMed] [Google Scholar]
- 17.Camps SM, Dutilh BE, Arendrup MC, Rijs AJ, Snelders E, Huynen MA, Verweij PE, Melchers WJ. 2012. Discovery of a HapE mutation that causes azole resistance in Aspergillus fumigatus through whole genome sequencing and sexual crossing. PLoS One 7:e50034. doi: 10.1371/journal.pone.0050034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei X, Chen P, Gao R, Li Y, Zhang A, Liu F, Lu L. 2016. Screening and characterization of a non-cyp51A mutation in an Aspergillus fumigatus cox10 strain conferring azole resistance. Antimicrob Agents Chemother 61:e02101-16. doi: 10.1128/AAC.02101-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nascimento AM, Goldman GH, Park S, Marras SA, Delmas G, Oza U, Lolans K, Dudley MN, Mann PA, Perlin DS. 2003. Multiple resistance mechanisms among Aspergillus fumigatus mutants with high-level resistance to itraconazole. Antimicrob Agents Chemother 47:1719–1726. doi: 10.1128/AAC.47.5.1719-1726.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fraczek MG, Bromley M, Buied A, Moore CB, Rajendran R, Rautemaa R, Ramage G, Denning DW, Bowyer P. 2013. The cdr1B efflux transporter is associated with non-cyp51a-mediated itraconazole resistance in Aspergillus fumigatus. J Antimicrob Chemother 68:1486–1496. doi: 10.1093/jac/dkt075. [DOI] [PubMed] [Google Scholar]
- 21.Kovalchuk A, Driessen AJ. 2010. Phylogenetic analysis of fungal ABC transporters. BMC Genomics 11:177. doi: 10.1186/1471-2164-11-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paul S, Diekema D, Moye-Rowley WS. 2013. Contributions of Aspergillus fumigatus ATP-binding cassette transporter proteins to drug resistance and virulence. Eukaryot Cell 12:1619–1628. doi: 10.1128/EC.00171-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Snelders E, Karawajczyk A, Verhoeven RJ, Venselaar H, Schaftenaar G, Verweij PE, Melchers WJ. 2011. The structure-function relationship of the Aspergillus fumigatuscyp51A L98H conversion by site-directed mutagenesis: the mechanism of L98H azole resistance. Fungal Genet Biol 48:1062–1070. doi: 10.1016/j.fgb.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 24.Paul S, Klutts JS, Moye-Rowley WS. 2012. Analysis of promoter function in Aspergillus fumigatus. Eukaryot Cell 11:1167–1177. doi: 10.1128/EC.00174-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Albarrag AM, Anderson MJ, Howard SJ, Robson GD, Warn PA, Sanglard D, Denning DW. 2011. Interrogation of related clinical pan-azole-resistant Aspergillus fumigatus strains: G138C, Y431C, and G434C single nucleotide polymorphisms in cyp51A, upregulation of cyp51A, and integration and activation of transposon Atf1 in the cyp51A promoter. Antimicrob Agents Chemother 55:5113–5121. doi: 10.1128/AAC.00517-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu M, Zheng N, Li D, Zheng H, Zhang L, Ge H, Liu W. 2016. cyp51A-based mechanism of azole resistance in Aspergillus fumigatus: illustration by a new 3D structural model of Aspergillus fumigatus CYP51A protein. Med Mycol 54:400–408. doi: 10.1093/mmy/myv102. [DOI] [PubMed] [Google Scholar]
- 27.Mo C, Bard M. 2005. A systematic study of yeast sterol biosynthetic protein-protein interactions using the split-ubiquitin system. Biochim Biophys Acta 1737:152–160. doi: 10.1016/j.bbalip.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 28.Mialoundama AS, Jadid N, Brunel J, Di Pascoli T, Heintz D, Erhardt M, Mutterer J, Bergdoll M, Ayoub D, Van Dorsselaer A, Rahier A, Nkeng P, Geoffroy P, Miesch M, Camara B, Bouvier F. 2013. Arabidopsis ERG28 tethers the sterol C4-demethylation complex to prevent accumulation of a biosynthetic intermediate that interferes with polar auxin transport. Plant Cell 25:4879–4893. doi: 10.1105/tpc.113.115576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hagiwara D, Miura D, Shimizu K, Paul S, Ohba A, Gonoi T, Watanabe A, Kamei K, Shintani T, Moye-Rowley WS, Kawamoto S, Gomi K. 2017. A novel Zn2-Cys6 transcription factor AtrR plays a key role in an azole resistance mechanism of Aspergillus fumigatus by coregulating cyp51A and cdr1B expressions. PLoS Pathog 13:e1006096. doi: 10.1371/journal.ppat.1006096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kafer E. 1977. Meiotic and mitotic recombination in Aspergillus and its chromosomal aberrations. Adv Genet 19:33–131. [DOI] [PubMed] [Google Scholar]
- 31.Osmani SA, May GS, Morris NR. 1987. Regulation of the mRNA levels of nimA, a gene required for the G2-M transition in Aspergillus nidulans. J Cell Biol 104:1495–1504. doi: 10.1083/jcb.104.6.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 33.CLSI. 2008. Reference method for broth dilution antifungal susceptibility testing of filamentous fungi; approved standard, 2nd ed, vol M38-A2 CLSI, Wayne, PA. [Google Scholar]