

ABSTRACT
para-Aminosalicylic acid (PAS) is a second-line antituberculosis drug that has been used to treat multidrug-resistant and extensively drug-resistant tuberculosis for more than 60 years. Renal secretion and glomerular filtration are the major pathways for the elimination of PAS. We comprehensively studied PAS transport by using cell lines that overexpressed various transporters and found that PAS acts as a novel substrate of an organic anionic polypeptide (OATP1B1), organic cationic transporters (OCT1 and OCT2), and organic anion transporters (OAT1 and OAT3) but is not a substrate of any ATP-binding cassette (ABC) transporters. Net PAS uptake was measured, and the transport affinities (Km values) for OATP1B1, OCT1, OCT2, OAT1, and OAT3 were found to be 50.0, 20.3, 28.7, 78.1, and 100.1 μM, respectively. The net uptake rates suggested that renal OAT1 and OAT3 play relatively major roles in PAS elimination. The representative inhibitors rifampin for OATP1B1, probenecid for OAT1 and OAT3, and verapamil for OCT1 and OCT2 greatly inhibited PAS uptake, suggesting that PAS is dependent on multiple transporters for uptake. We also evaluated nonsteroidal anti-inflammatory drugs (NSAIDs), proton pump inhibitors (PPIs), and metformin for the inhibition of PAS uptake via these transporters. Half-maximal (50%) inhibitory concentrations (IC50s) were kinetically determined and used to predict the drug-drug interactions (DDIs) affecting these transporters' activity toward PAS. We found that rifampin, probenecid, ibuprofen, naproxen, cimetidine, and quinidine each exhibited a significant potential for in vivo DDIs with PAS. In this study, PAS was found to be a novel substrate of several transporters, and drugs that inhibit these transporters can reduce PAS elimination.
KEYWORDS: antituberculosis drug, membrane transporter, drug-drug interactions
INTRODUCTION
The second-line antituberculosis drug para-aminosalicylic acid (PAS) has been used to treat multidrug-resistant and extensively drug-resistant tuberculosis for more than 60 years (1). PAS was widely used in combination chemotherapy against Mycobacterium tuberculosis. As better-tolerated antibiotics became available, PAS usage diminished considerably. Later, the widespread appearance of multidrug-resistant (MDR) tuberculosis (MDR-TB) necessitated the addition of PAS to the current list of SLDs. Gastrointestinal toxicity, which leads to poor patient compliance, is a significant drawback to PAS usage. Despite this drawback, PAS has become one of the principal SLDs for the treatment of MDR-TB (2). An improved (granule) formulation of PAS has been adopted to treat patients with MDR-TB and patients who are intolerant to first-line anti-TB medications.
Despite the long history of PAS usage, it has received little attention, and little is known about its disposition and other factors that may influence its pharmacokinetics (PK) and pharmacodynamics (PD). Pharmacokinetic data are key to achieving a PAS dosage in children that avoids high serum concentrations (which could be toxic) or concentrations too low to complete the eradication of M. tuberculosis. PAS is an amphoteric compound which may act as either a base or an acid, depending on its environment, and it has been reported that food increases PAS absorption, although orange juice and antacids showed a minor effect (3). The half-life of PAS is about 1 h, and plasma concentrations after 4 to 5 h are minimal. Its short half-life justifies the use of relatively high doses of 10 to 12 g in order to maintain bacteriostatic activity. More than 80% of the drug (parent and metabolite) is excreted by the kidney through glomerular filtration and tubular secretion (4). PAS is metabolized in the intestines and the liver via acetylation. The two main products of PAS metabolism are N-acetyl-p-aminosalicylate (APAS), formed during PAS acetylation by N-acetyltransferase 1 (NAT1), and p-aminosalicyluric acid (PAA), formed by conjugation with glycine. These major metabolites account for approximately 70% of the absorbed dose for PAS and 56.4% for the PAS Na formulation, 57.7% for the PAS Ca formulation, and 54.5% for the PAS K formulation (5, 6). A considerable proportion of drug metabolism occurs in the gut and liver before the drug reaches its target. This first-pass effect can considerably affect achievable blood concentrations. Understanding the molecular mechanism(s) of action of current drugs (and the mechanisms of drug resistance) will provide new perspectives and approaches to drug discovery. PAS is a prodrug that targets dihydrofolate reductase (DHFR) through an unusual and novel mechanism of action. Recently reported evidence shows that PAS is incorporated into the folate biosynthesis pathway by dihydropteroate synthase (DHPS) and dihydrofolate synthase (DHFS) to generate a hydroxyl dihydrofolate antimetabolite, which in turn inhibits DHFR enzymatic activity. Interestingly, PAS is recognized by DHPS as efficiently as its natural substrate, para-aminobenzoic acid is. Chemical inhibition of DHPS or a mutation in DHFS prevents the formation of the antimetabolite, thereby conferring resistance to PAS (7).
PAS causes several adverse effects, including gastrointestinal symptoms (anorexia, diarrhea, nausea, and vomiting) and hypothyroidism, with the latter occurring more frequently when PAS is administered concomitantly with ethionamide, although thyroid function returns to normal when the drug is discontinued. PAS can cause hypothyroidism by inhibiting iodine organification, a necessary step in thyroid hormone synthesis (8). PAS therapy was initially discontinued after the introduction of rifampin and pyrazinamide. PAS was reintroduced in the United States in 1992, following several outbreaks caused by MDR isolates (9). The use of controlled and targeted release may be the best choice to minimize side effects and increase the intrinsic half-life of the drug (10, 11).
Since polypharmacy (the practice of the simultaneous prescription of more than one drug to treat one or more conditions in a single patient) has become more common, drug interactions have been cited as a major reason for hospitalization and death (12). Multiple drugs may be coadministered to a patient to effectively treat a single disease (e.g., TB, HIV infection) or to treat multiple diseases or disease symptoms. It is known that drug-drug interactions (DDIs) may have serious and sometimes fatal consequences (13). Strains causing MDR-TB are resistant to at least two first-line drugs, isoniazid and rifampin, while strains causing extensively drug-resistant tuberculosis (XDR-TB) are additionally resistant to fluoroquinolones and at least one of three second-line injectable drugs. XDR-TB is associated with high rates of early mortality among HIV-infected individuals, and the scarcity of effective treatment options against MDR-TB/XDR-TB prompted the reintroduction of second-line TB drugs, including PAS (14).
Transporter activity is significantly associated with the clinical phenotype and toxic events (15). The uptake and efflux of drugs and endogenous substrates are facilitated by the solute carrier (SLC) and ATP-binding cassette (ABC) family transporters. To date, approximately 400 human transporter genes have been classified into two major superfamilies (16, 17). Among the members of the SLC superfamily, the organic anionic transporter polypeptides (OATPs), organic cation transporters (OCTs), and organic anion transporters (OATs) play major roles in the disposition of drugs (18). Among the ABC transporters, permeability glycoprotein (P-gp), breast cancer resistance protein (BCRP), and multidrug resistance proteins (MRPs) also play major roles in drug disposition (17). Among TB patients, the coadministration of probenecid increased the PAS concentration in plasma approximately 2- to 3-fold by decreasing renal clearance compared with that in patients who did not take probenecid. The coadministration of diclofenac also increased the level of PAS exposure and the numbers of adverse events (19–21). The mechanism of these DDIs has yet to be elucidated. It has been hypothesized that hepatic and renal transporters may play a role in PAS disposition, but to date, no data on the involvement of membrane transporters in PAS transport have been reported. Therefore, we aimed to characterize the involvement of transporters in the ABC and SLC families in the disposition of PAS and mechanistically study the possibility of DDIs with concomitantly administered modulators of transporter activity in vitro in cell lines.
RESULTS
Uptake of [3H]ES, [3H]E2G, [3H]MPP+, and [3H]ES by HEK cells overexpressing various transporters.
The uptake of the prototype substrates [3H]estrone-3-sulfate ([3H]ES) for OATP1B1, OATP2B1, and OAT3, [3H]estradiol 17β-d-glucuronide ([3H]E2G) for OATP2B1, [3H]para-aminohippurate ([3H]PAH) for OAT1, and [3H]N-methyl-4-phenylpyridinium acetate ([3H]MPP+) for OCT1 and OCT2 was determined. The levels of uptake of the prototype substrates by human embryonic kidney 293 (HEK) cells overexpressing the transporters OATP1B1, OATP2B1, OATP1B3, OAT1, OAT3, OCT1, and OCT2 (HEK-OATP1B1, HEK-OATP2B1, HEK-OATP1B3, HEK-OAT1, HEK-OAT3, HEK-OCT1, and HEK-OCT2 cells, respectively) were 48-, 41-, 44-, 10-, 13-, 17.7-, and 16-fold higher, respectively, than those by mock-transfected cells (see Fig. S1 in the supplemental material). Furthermore, the uptake kinetics of each prototype substrate (see Table S1 in the supplemental material), estimated using nonlinear kinetics, indicated that the transfected HEK-OATP1B1, HEK-OATP2B1, HEK-OATP1B3, HEK-OAT1, HEK-OAT3, HEK-OCT1, and HEK-OCT2 cells were suitable for use in the uptake experiments.
Screening for PAS uptake by HEK cells overexpressing the transporters.
We performed a preliminary screen for PAS transport via SLC transporters by using Xenopus laevis oocytes. This screen revealed that PAS is transported by OATP1B1, OAT1, OAT3, OCT1, and OCT2 in oocytes injected with cRNA for these transporters at levels 3-, 9-, 5-, 4-, and 6-fold higher, respectively, than those in control oocytes not injected with transporter cRNA (not shown). In contrast, the level of PAS uptake by oocytes overexpressing the transporters OATP2B1, OATP1B3, OCTN1, OCTN2, MATE1, and MATE2K was negligible (a less than 2-fold increase over that for the control). We found that PAS uptake by HEK293 cells overexpressing the SLC transporters was also marked higher than that by control (mock-transfected) cells, consistent with the findings of our previous screening for uptake by X. laevis oocytes. The level of uptake of PAS by HEK-1B1, HEK-OAT1, HEK-OAT3, HEK-OCT1, and HEK-OCT2 cells was 3-, 13-, 5-, 4-, and 6-fold higher than that by mock-transfected cells, respectively (Fig. 1). We further evaluated OATP1B1-, OAT1-, OAT3-, OCT1-, and OCT2-mediated PAS transport in the presence of representative inhibitors. As expected, a concentration gradient of rifampin, probenecid, and verapamil, representative inhibitors of these transporters, strongly inhibited PAS transport (Fig. 2 and S4). Next, we evaluated the net uptake kinetics of these transporters by using a range of PAS concentrations and found Km values of 50.0 ± 13.6, 78.0 ± 18.2, 100.6 ± 23.6, 20.3 ± 4.6, and 28 ± 6.8 μM for HEK-1B1, HEK-OAT1, HEK-OAT3, HEK-OCT1, and HEK-OCT2, respectively (Fig. 3 and Table 1).
FIG 1.
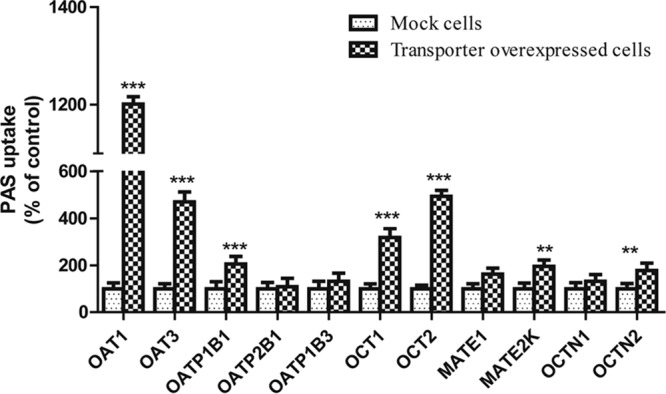
Screening for uptake of PAS by cells overexpressing SLC transporters. PAS (100 μM) uptake by stably transfected HEK cells overexpressing SLC transporters and mock-infected cells (control) was tested. Data are presented as the mean ± SDs from three or more independent experiments. Significant differences compared with the percent uptake for the control (mock-transfected cells) are indicated. **, P > 0.01; ***, P > 0.001.
FIG 2.
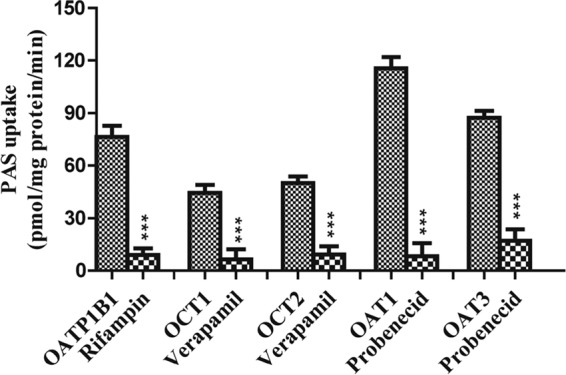
Effect of representative inhibitors on the uptake of PAS by stably transfected HEK293 cells. Positive-control inhibitors of PAS uptake were 20 μM rifampin for OATP1B1, 10 μM verapamil for OCT1, 40 μM verapamil for OCT2, and 30 μM probenecid for OAT1 and OAT3. Data are presented as the means ± SDs from three or more independent experiments. Significant differences compared with the percent uptake for the control (no inhibitor) are indicated. ***, P > 0.001.
FIG 3.

Kinetics of PAS uptake by stably transfected HEK-OATP1B1, HEK-OCT1, HEK-OCT2, HEK-OAT1, and HEK-OAT3 cells. The intracellular uptake kinetics of PAS via OATP1B1 (A), OCT1 (B), OCT2 (C), OAT1 (D), and OAT3 (D) were derived from an in vitro experiment with PAS at concentrations ranging from 1 to 400 μM, and the results are normalized to those for the control. Data are presented as the means ± SDs from three or more independent experiments.
TABLE 1.
Kinetic parameters for PAS uptake by stably transfected HEK-OATP1B1, HEK-OCT1, HEK-OCT2, HEK-OAT1, and HEK-OAT3 cellsa
| Transporter | Km (μM) | Vmax (pmol/min/mg protein) |
|---|---|---|
| OATP1B1 | 50.0 ± 13.6 | 129.7 ± 17.5 |
| OCT1 | 20.3 ± 4.6 | 48.7 ± 12.2 |
| OCT2 | 28.7 ± 6.8 | 70.7 ± 9.8 |
| OAT1 | 78 ± 18.2 | 1,898 ± 112 |
| OAT3 | 100.0 ± 23.6 | 1,572 ± 207 |
The intracellular kinetics of PAS uptake were derived from an in vitro experiment with PAS at concentrations ranging from 1 to 400 μM, and the data are normalized to those for the control. Data are presented as the means ± SDs from three or more independent experiments. Km, Michaelis-Menten constant; Vmax, maximum rate of uptake.
Transepithelial screening of PAS transport by ABC transporters.
We also investigated whether PAS is a substrate for the ABC transporters P-gp, BCRP, MRP1, and MRP2 using a bidirectional efflux assay. Cells constructed to overexpress the ABC transporters showed functional activity with the probe substrate (Fig. S2). The net flux ratios (NFRs) of the transporters from the apical-to-basal direction and the basal-to-apical direction were not markedly different (less than 2-fold) (Fig. 4). These data suggest that ABC transporters do not play a major role in PAS transport and that PAS is not a substrate of this ATP-dependent transport. Furthermore, the corresponding inhibitors did not increase the level of PAS accumulation in cells overexpressing P-gp, BCRP, MRP1, and MRP2 (Fig. S3).
FIG 4.
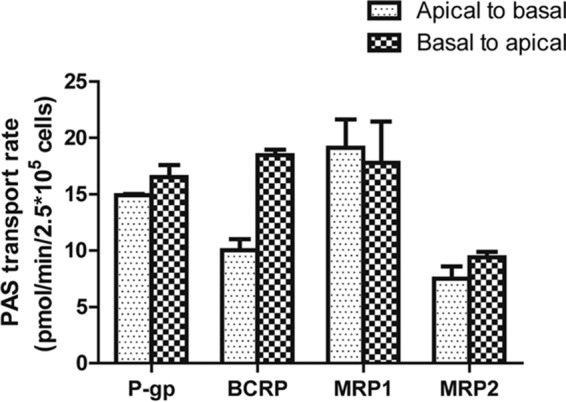
Screening of transepithelial transport of PAS via ABC transporters. The rate of transport of 10 μM PAS in the apical-to-basal and basal-to-apical directions in monolayers of LLCPK cells overexpressing P-gp, LLCPK cells overexpressing BCRP, MDCKII cells overexpressing MRP1, and MDCKII cells overexpressing MRP2 is shown. Each data point presents the mean ± SD from three or more independent experiments.
Effect of NSAIDs on PAS uptake via organic anion and cation transporters.
We evaluated the inhibitory potential of nonsteroidal anti-inflammatory drugs (NSAIDs) on PAS uptake via OAT1, OAT3, OCT1, and OCT2 in vitro using the stably transfected HEK293 cells. For this experiment, we tested different NSAIDS for strong inhibition of OAT- and OCT-mediated PAS uptake. The NSAIDs acetyl-salicylic acid, ibuprofen, diclofenac, naproxen, and indomethacin strongly inhibited (50 to 70%) PAS uptake by HEK-OAT1 and HEK-OAT3 cells compared to the level of uptake by control cells (Fig. 5). Among the NSAIDs tested, indomethacin and diclofenac showed the highest levels of inhibition of PAS uptake via OAT1 and OAT3 (Fig. 5). We measured the half-maximal (50%) inhibitory concentrations (IC50s) of probenecid, acetyl-salicylic acid, indomethacin, ibuprofen, diclofenac, and naproxen to be 16.3, 368, 1.91, 5.1, 1.8, and 4.1 μM, respectively, for OAT1-mediated PAS uptake inhibition and 24.6, 597, 2.18, 1.98, 2.0, and 4.6 μM, respectively, for OAT3-mediated PAS uptake inhibition. In the case of OCT1- and OCT2-mediated PAS uptake inhibition by the NSAIDs, no significant inhibitory effect was observed. Ibuprofen, indomethacin, and diclofenac showed mild inhibition of OCT1- and OCT2-mediated PAS uptake (Fig. 5).
FIG 5.

Inhibitory effects of NSAIDs and PPIs on OAT1-, OAT3-, OCT1-, and OCT2-mediated PAS uptake. The level of uptake of 20 μM PAS into stably transfected HEK-OAT1 and HEK-OAT3 cells (A) and HEK-OCT1 and HEK-OCT2 cells (B) in the presence or absence of NSAIDs and PPIs is shown. The levels of PAS uptake via OAT1, OAT3, OCT1, and OCT2 in the absence of any uptake inhibitor as a control and the inhibition of uptake by the NSAIDs and PPIs in HEK-OAT1, HEK-OAT3, HEK-OCT1, and HEK-OCT2 cells were also determined. Probenecid (30 μM), verapamil (30 μM), acetyl-salicylic acid (30 μM), ibuprofen (2 μM), indomethacin (5 μM), diclofenac (5 μM), naproxen (5 μM), and omeprazole were used for inhibition of OAT1- and OAT3-mediated (A) and OCT1- and OCT2-mediated (B) PAS uptake, and lansoprazole (1, 10, and 50 μM) was used for inhibition of OAT1- and OAT3-mediated (C) and OCT1- and OCT2-mediated (D) PAS uptake. Data are presented as the means ± SDs from three or more independent experiments. Significant differences compared with the percent uptake for the control (no inhibitor) are indicated: *, P > 0.05; **, P > 0.01; ***, P > 0.001.
Effect of PPIs on PAS uptake via organic anion and cation transporters.
Among the proton pump inhibitors (PPIs), omeprazole and lansoprazole also strongly inhibited PAS uptake via the OAT1 and OAT3 transporters (Fig. 5). The estimated IC50s of omeprazole and lansoprazole for inhibition of PAS uptake via OAT1 were 1.76 and 4.13 μM, respectively, and those for inhibition of PAS uptake via OAT3 were 5.1 and 2.18 μM, respectively (Fig. 6). Also, omeprazole and lansoprazole inhibited PAS uptake via OCTs (Fig. 5). The estimated IC50s of omeprazole and lansoprazole for inhibition of PAS uptake via OCT1 were 17.1 and 25.2 μM, respectively, and those for inhibition of PAS uptake via OCT2 were 5.5 and 13.0 μM, respectively. Additionally, the inhibitory effects of metformin and cimetidine on PAS transport via OCT1 and OCT2 were evaluated in vitro. Again, metformin (200 μM) and cimetidine (100 μM) strongly inhibited PAS uptake by HEK-OCT1 and HEK-OCT2 cells (Fig. 7). Metformin and cimetidine markedly decreased the levels of OCT1- and OCT2-mediated PAS uptake by approximately 50 and 56%, respectively. The inhibitory effects of metformin and cimetidine on PAS uptake were dose dependent, and their IC50s for inhibition of PAS uptake via OCT1 were 179.1 and 107.5 μM, respectively, and those for inhibition of PAS uptake via OCT2 were 125.6 and 116.5 μM, respectively (Fig. 6). We also evaluated the inhibitory effects of verapamil and quinidine on PAS uptake via OCT1 and OCT2. The estimated IC50s of verapamil and quinidine for inhibition of PAS uptake via OCT1 were 1.2 and 4.84 μM, respectively, and those for inhibition of PAS uptake via OCT2 were 21.4 and 10.7 μM, respectively.
FIG 6.
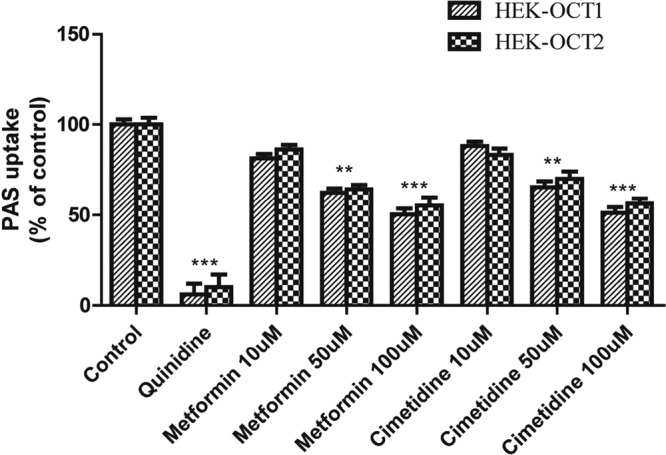
Inhibitory effects of metformin and cimetidine on OCT1- and OCT2-mediated PAS uptake. The level of uptake of 5 μM PAS by stably transfected HEK-OCT1 and HEK-OCT2 cells in the presence or absence of the inhibitor drugs is shown. The level of OCT1- and OCT2-mediated uptake in the absence of any uptake inhibitor as a control and the inhibition of uptake by the inhibitor drugs were determined in HEK-OCT1 and HEK-OCT2 cells. Quinidine (20 μM) and metformin and cimetidine (10, 50, 100 μM) were used for OCT1- and OCT2-mediated PAS uptake inhibition. Data are presented as the means ± SDs from three or more independent experiments. Significant differences compared with the percent uptake for the control (no inhibitor) are indicated. **, P > 0.01; ***, P > 0.001.
FIG 7.

Evaluation of the concentration-dependent inhibition of OAT- and OCT-mediated PAS uptake by NSAIDs, PPIs, metformin, and cimetidine. The kinetics of NSAIDs and PPIs on inhibition of 20 μM PAS uptake by stably transfected HEK-OAT1 (●) and HEK-OAT3 (○) cells and of cimetidine and metformin on 5 μM PAS uptake by stably transfected HEK-OCT1 (●) and HEK-OCT2 (○) cells were estimated. The NSAIDs, PPIs, metformin, and cimetidine showed potent inhibition of PAS uptake by HEK-OAT1, HEK-OAT3, HEK-OCT1, and HEK-OCT2 cells according to the concentration gradient. The estimated IC50 was calculated by nonlinear kinetics using WinNonlin software (version 5.1). Data are presented as the means ± SDs from three or more independent experiments and are the level of uptake as a percentage of that for the control (no inhibitor).
Drug-drug interaction index prediction.
To assess the clinical relevance of this study's outcomes, we estimated the DDI index for the interaction of PAS with inhibitors of each transporter, using clinical plasma concentrations (Table S2) in a static model-based approach. The DDI index revealed mild to moderate elevation of the DDI index values compared to the cutoff values in FDA guidelines (>1.25 for OAT1B1, >0.1 for OCT and OATs) for PAS uptake inhibition and possible in vivo DDIs. Among the drugs tested, rifampin showed the highest potential in vivo DDI, with a DDI index value of 6.6 for OATP1B1-mediated PAS uptake inhibition. The estimated DDI index values for the PAS interaction with probenecid, indomethacin, ibuprofen, diclofenac, and naproxen were 2.23, 0.02, 0.53, 0.02, and 0.65, respectively, for OAT1 and 1.48, 0.02, 1.35, 0.02, and 0.60, respectively, for OAT3 (Table 2). Similarly, the DDI index values for the PAS interaction with omeprazole and lansoprazole were 0.05 and 0.03, respectively, for OAT1 and 0.02 and 0.07, respectively, for OAT3. The DDI index values for the PAS interaction with verapamil, quinidine, cimetidine, and metformin were 2.85, 1.48, 71.2, and 0.06, respectively, for OCT1 and 0.002, 0.65, 68.2, and 0.08, respectively, for OCT2, suggesting a strong possibility of DDIs with PAS in vivo mediated by the OCT transporters (Table 3).
TABLE 2.
DDI index values estimated from the kinetics of OAT1- and OAT3-mediated PAS uptake inhibition by NSAIDs and PPIs in vitroa
| Inhibitor drug | OAT1 |
OAT3 |
||||
|---|---|---|---|---|---|---|
| IC50 (μM) | DDI index |
IC50 (μM) | DDI index |
|||
| [I] = [I]max | [I] = [I]max,u | [I] = [I]max | [I] = [I]max,u | |||
| Probenecid | 16.3 ± 3.6 | 14.9 ± 2.4 | 2.23 ± 0.96* | 24.6 ± 4.2 | 9.91 ± 1.21 | 1.48 ± 0.26* |
| Indomethacin | 1.91 ± 0.20 | 2.13 ± 0.2 | 0.02 ± 0.001 | 2.18 ± 0.25 | 1.87 ± 0.22 | 0.02 ± 0.002 |
| Ibuprofen | 5.1 ± 0.78 | 53.0 ± 8.9 | 0.53 ± 0.09* | 1.98 ± 0.24 | 135.6 ± 18 | 1.35 ± 0.18* |
| Diclofenac | 1.8 ± 0.12 | 5.4 ± 0.33 | 0.02 ± 0.001 | 2.0 ± 0.35 | 4.65 ± 0.82 | 0.02 ± 0.003 |
| Naproxen | 4.13 ± 0.25 | 65.4 ± 4.0 | 0.65 ± 0.04* | 4.6 ± 0.23 | 58.4 ± 2.91 | 0.60 ± 0.02* |
| Omeprazole | 1.76 ± 0.11 | 1.75 ± 0.11 | 0.05 ± 0.003 | 5.1 ± 0.78 | 0.62 ± 0.10 | 0.02 ± 0.003 |
| Lansoprazole | 4.13 ± 0.26 | 1.18 ± 0.07 | 0.03 ± 0.002 | 2.18 ± 0.25 | 2.25 ± 0.27 | 0.07 ± 0.002 |
The DDI index values were determined using the inhibition constant (IC50) with the maximum concentration in plasma (Cmax; bound plus unbound drug) ([I]max) and the maximum unbound concentration ([I]max,u) of the anti-TB drugs following the regulatory guidelines described in the text. DDI index values represent means ± SDs obtained from the inhibition constants from three or more independent experiments. *, the result is significant according to FDA guidance for prototypical substrates, in which the DDI index values are greater than the corresponding cutoff values recommended by the regulatory authorities for an [I]max,u value of ≥0.1. The cutoff value is expressed as the value according to the upper limit of the equivalence range suggested by FDA.
TABLE 3.
DDI index values estimated from the kinetics of OCT1- and OCT2-mediated PAS uptake inhibition by the inhibitor drugs in vitroa
| Inhibitor drug | OCT1 |
OCT2 |
||||
|---|---|---|---|---|---|---|
| IC50 (μM) | DDI index |
IC50 (μM) | DDI index |
|||
| [I] = [I]max | [I] = [I]max,u | [I] = [I]max | [I] = [I]max,u | |||
| Verapamil | 1.2 ± 0.35 | 0.28 ± 0.01 | 2.85 ± 0.1* | 21.4 ± 5.2 | 0.015 ± 0.001 | 0.002 ± 0.001 |
| Cimetidine | 107.5 ± 19.5 | 89.0 ± 17.5 | 71.2 ± 14* | 111.5 ± 17 | 85.2 ± 13.1 | 68.2 ± 10.5* |
| Metformin | 179.1 ± 33 | 0.06 ± 0.01 | 0.06 ± 0.01 | 125.6 ± 18 | 0.08 ± 0.01 | 0.08 ± 0.013 |
| Quinidine | 4.84 ± 1.4 | 2.97 ± 0.82 | 1.48 ± 0.41* | 10.7 ± 1.73 | 1.29 ± 0.20 | 0.65 ± 0.10* |
| Omeprazole | 17.1 ± 2.25 | 0.18 ± 0.02 | 0.005 ± 0.001 | 5.53 ± 1.31 | 0.58 ± 0.16 | 0.02 ± 0.001 |
| Lansoprazole | 25.2 ± 3.7 | 0.21 ± 0.02 | 0.005 ± 0.001 | 13.0 ± 3.51 | 0.41 ± 0.10 | 0.01 ± 0.003 |
The DDI index values were determined using the inhibition constant (IC50) with the maximum concentration in plasma (Cmax; bound plus unbound) ([I]max) and the maximum unbound concentration ([I]max,u) of the anti-TB drugs following the regulatory guidelines described in the text. DDI index values represent means ± SDs obtained from the inhibition constants from three or more independent experiments. *, the result is significant according to FDA guidance for prototypical substrates, in which the DDI index values are greater than the corresponding cutoff values recommended by the regulatory authorities for an [I]max,u value of ≥0.1. The cutoff value is expressed as the value according to the upper limit of the equivalence range suggested by FDA.
DISCUSSION
In this study, we comprehensively assessed the specificity of PAS as a substrate of clinically important SLC transporters, including OATP1B1, OATP2B1, OATP1B3, OAT1, OAT3, OCT1, OCT2, MATE1, MATE2K, OCTN1, and OCTN2, and the ABC transporters P-gp, BCRP, MRP1, and MRP2. Among the 11 SLC transporters, we found that the cellular accumulation of PAS was mediated by the OATP1B1, OAT1, OAT3, OCT1, and OCT2 transporters, as evidenced by 3-, 13-, 5-, 4-, and 6.0-fold increases in the level of PAS uptake by HEK293 cells overexpressing OATP1B1, OAT1, OAT3, OCT1, and OCT2, respectively, relative to the uptake by mock-transfected HEK293 cells. We further confirmed the specificity of PAS uptake by examining OATP1B1-, OAT1-, OAT3-, OCT1-, and OCT2-mediated PAS uptake in the presence of well-characterized inhibitors of these transporters. Rifampin, probenecid, and verapamil were selected as inhibitors of OATP1B1, OATs, and OCTs, respectively. In OATP1B1-overexpressing cells, 20 μM rifampin inhibited PAS uptake by 93.1%. Similarly, 10 and 40 μM verapamil inhibited PAS uptake by cells overexpressing OCT1 or OCT2 by 87.1% and 92%, respectively. Finally, 30 μM probenecid inhibited PAS uptake by OAT1- and OAT3-overexpressing cells by 95% and 91.1%, respectively (Fig. 2). The different potencies that we observed for these inhibitors correlated well with their different IC50s. Collectively, these results show that these transporters are involved in the PAS uptake process.
None of the ABC transporters that we evaluated played a significant role in PAS efflux. Our bidirectional efflux assay showed that PAS transport in the basal-to-apical direction was less than or equal to that in the apical-to-basal direction, with a net flux ratio (NFR) of <2. These data indicate that PAS is not a substrate of the ABC efflux transporters that we tested (Fig. 4). Despite the results of a previous study indicating that inhibition of P-gp efflux caused an increased PAS concentration in the brain (22, 23), our in vitro transport data found no involvement of P-gp in PAS transport. The half-life of orally administered PAS is reported to be 1.5 to 2 h, with the concentration remaining in plasma within 4 to 5 h after administration of a single conventional dose being negligible. Within 24 h, more than 80% is excreted in the urine. More than 50% of the excreted PAS is acetylated (24). Half of the PAS remaining after glomerular filtration is eliminated by tubular secretion (including very effective secretion of its metabolites), consistent with our results showing that the involvement of renal uptake transporters may play a major role in the elimination of PAS (4).
PAS resistance is becoming a frequent issue, limiting its efficacy, although the exact mechanism remains unknown. An early in vitro study reported that low PAS concentrations only delayed the emergence of PAS resistance, but higher concentrations suppressed the growth of resistant mutants (25). Our study revealed possible drug interactions with PAS that may cause clinical adverse effects in patients. The most common adverse events in patients administered PAS are gastrointestinal effects (anorexia, diarrhea, nausea, and vomiting) and hypothyroidism. The latter occurs more frequently when PAS is administered concomitantly with ethionamide, but thyroid function returns to normal when the drug is discontinued (26). Hepatitis occurs in 0.3 to 0.5% of cases, and allergic reactions (fever, rash, and pruritus), hemolytic anemia, agranulocytosis, leukopenia, thrombocytopenia, malabsorption syndrome, and increased thyroid volume are rare, as are cardiovascular adverse effects (pericarditis), neurological adverse effects (encephalopathy), respiratory adverse effects (eosinophilic pneumonia), and ocular adverse effects (optic neuritis). PAS should be used with caution in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency and in those who are allergic to aspirin (21).
We also tested for DDIs of PAS with commonly prescribed drugs, such as NSAIDs, PPIs, metformin, and cimetidine. Our data showed a strong potential DDI with probenecid, which may explain clinical reports of increased PAS concentrations and toxicity in patients also taking probenecid (19, 20). Among the NSAIDs, diclofenac also increased the PAS concentration in plasma in clinical studies. In agreement with the results of these studies, our in vitro data showed the potent inhibition of PAS uptake by diclofenac, although the DDI index value did not reach the cutoff value. The potent inhibition of PAS uptake by diclofenac probably happened owing to the lower unbound plasma concentration and was also probably affected by the tissue concentrations. With the exception of the DDI with diclofenac, the DDIs suggest that nonselective NSAIDS will increase the adverse effects of PAS.
PAS has been reported to increase the hypoglycemic effects of sulfonylurea and to increase the risk of bleeding when administered in conjunction with oral anticoagulants, thrombolytics, or salicylates (21). On the other hand, although NSAIDs showed mild inhibition of OCT-mediated uptake, the level of inhibition was minor. In contrast, extensive studies of omeprazole and lansoprazole have reported the inhibition of renal OATs and DDIs with methotrexate (a substrate of OATs) in vitro and in vivo (27, 28). We also suspected similar drug interactions based on our in vitro data, which showed that omeprazole and lansoprazole inhibited OAT1- and OAT3-mediated PAS uptake by more than 70%. Although it has to be considered that the amount of PAS is markedly lower than the amount of acetyl-PAS, the hepatic uptake transporters can play a major role compared to the role played by renal uptake transporters, as we found a higher DDI index value for rifampin. Perhaps those SLC transporters can play role in its level of expression and thus its tissue distribution. However, the DDI index value that we determined failed to reach the cutoff value, suggesting that the DDI between PAS and NSAIDs has a negligible effect. We have previously published data on the inhibitory effect of PAS on OAT and OCT transporters, and it seems that these transporters are strongly associated with its uptake (29).
Metformin is an inhibitor and substrate of OCTs and is extensively used to treat diabetes. It is also commonly coadministered with anti-TB drugs. In our in vitro study, metformin potently inhibited PAS uptake via OCT1 and OCT2 (Fig. 3), but its estimated DDI index value did not support the existence of a clinical DDI. This is in contrast to the findings described in a recently published study in which we reported a potential DDI between PAS and metformin and in which PAS inhibited metformin uptake via OCT1 and OCT2 in vitro (29). In this study, verapamil, cimetidine, and quinidine strongly inhibited PAS uptake via OCT1 and OCT2, and the DDI index values were much higher than the cutoff values (Table 2), indicating possible clinical DDIs.
PAS concentrations strongly correlate with NAT1 genetic polymorphisms. As was previously reported from a clinical study, NAT1 genetics and their relationship to serum PAS concentrations confirmed that the NAT1*14 allele is a loss-of-function allele. In contrast, there was no evidence that NAT1*10 is associated with decreased activity (30). These studies suggest that the influence of transporters and NAT must be considered to fully understand the disposition and pharmacokinetics (PK) of PAS.
As we have discovered that the OATP1B1, OAT1, OAT3, OCT1, and OCT2 transporters actively transport PAS, genetic polymorphisms in these transporters and their effects on PAS transport and PK may also be clinically relevant. We have published the findings of a recent study in which metformin PK were greatly affected by polymorphisms in OCT1 and OCT2 among Koreans (31, 32). In addition to the allelic effects of the metabolic enzyme (NAT1), interference with the disposition of PAS in the liver and kidney by drugs inhibiting OATPs, OATs, and OCTs provides a complementary mechanistic rationale for its DDIs. The static model is widely used for the prediction of DDIs because of its simplicity; however, it can result in false-negative DDI predictions; also, as described in the FDA draft guidance, the DDI index for determination of the disposition of a drug considers only a single route of administration, but clinical DDIs can be impacted by several routes of administration (33).
To our knowledge, this is the first comprehensive characterization of PAS transport, in which PAS was revealed to be a novel substrate of several SLC uptake transporters. These data suggest a mechanism by which PAS may potentially interact with several therapeutic classes of drugs. These in vitro data could be useful for further studies of PAS in vivo to understand its drug interactions in greater detail.
MATERIALS AND METHODS
The chemicals [3H]estrone-3-sulfate ([3H]ES; 1.66 TBq/mmol), [3H]-estradiol 17β-d-glucuronide ([3H]E2G; 1.27 TBq/mmol), [3H]para-aminohippurate ([3H]PAH; 0.166 TBq/mmol), and [3H]N-methyl-4-phenylpyridinium acetate ([3H]MPP+; 82.1 Ci/mmol) were purchased from PerkinElmer (Waltham, MA, USA). Probenecid, verapamil, cimetidine, para-aminosalicylic acid (PAS), acetyl-salicylic acid, indomethacin, diclofenac, ibuprofen, naproxen, omeprazole, lansoprazole, and rifampin were purchased from Sigma-Aldrich. The other reagents used in this study were purchased from commercial suppliers offering the highest purity.
Cell lines preparation and culture.
Human embryonic kidney 293 (HEK) cells were stably transfected according to a previously reported method (34), and we have recently published the findings of a study using such stably transfected cells (29, 31, 35). In brief, stably transfected HEK-OCT1, HEK-OCT2, HEK-OAT1, HEK-OAT3, HEK-OATP1B1, HEK-OATP2B1, and HEK-OATP1B3 cells were grown in tissue culture flasks in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% nonessential amino acids, 2 mM l-glutamine, and 100 U/ml penicillin-streptomycin (Invitrogen) at approximately 37°C in an atmosphere supplemented with 5% CO2. When the cells were at confluence, cells were harvested using trypsin-EDTA and resuspended in culture medium. This process was repeated as necessary to obtain sufficient numbers of cells for each experiment.
Synthesis of cRNA for the transport study using Xenopus laevis oocytes.
The cRNA synthesis and uptake experiments were performed as described previously (36). The capped OATP, OAT, and OCT cRNAs were synthesized in vitro using T7 RNA polymerase with linear plasmid DNA. X. laevis oocytes were digested in ORII solution, which contained 82.5 mM NaCl, 2 mM KCl, 1 mM MgCl2, 5 mM HEPES, pH 7.4, and 1.5 mg/ml collagenase, for 90 min at room temperature. After digestion, defolliculated oocytes were injected with 50 ng of the capped cRNA and incubated at 18°C in Barth's solution [88 mM NaCl, 1 mM KCl, 0.33 mM Ca(NO3)2, 0.4 mM CaCl2, 0.8 mM MgSO2, 2.4 mM NaHCO3, 10 mM HEPES, pH 7.4] that contained 50 ng/ml gentamicin and 2.5 mM pyruvate. After incubation for 2 days, uptake experiments were performed at room temperature in ND96 solution (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, pH 7.4). The uptake reaction was initiated by replacing the ND96 solution with ND96 solution containing PAS for both control oocytes and oocytes expressing uptake transporters (i.e., OATPs, OATs, and OCTs) and terminated by the addition of ice-cold ND96 solution after 30 min for OATPs and OATs and 60 min for OCTs. After the oocytes were washed three times, they were sonicated in 120 μl of 100% acetonitrile two times at 3°C for 5 s each time and centrifuged at 13,000 × g for 10 min at 4°C. Aliquots of the supernatant were injected into a liquid chromatography-tandem mass spectrometry (LC-MS/MS) system for detection of the anti-TB drugs.
Uptake of radiolabeled prototype substrate by transporter-overexpressing HEK cells.
Evaluation of the effect of transporter overexpression on the cellular uptake of the labeled transporter substrates was performed as described previously (37). To measure the cellular uptake of the radiolabeled substrates, HEK-OATP1B1, HEK-OATP2B1, HEK-OATP1B3, HEK-OCT1, HEK-OCT2, HEK-OAT1, HEK-OAT3 HEK-OCTN1, HEK-OCTN2, HEK-MATE1, and MATE2K cells were seeded in 24-well culture plates at a density of 2 × 105 cells/well and incubated at 37°C in an atmosphere supplemented with 5% CO2 for 1 day. After the cells reached 90% confluence, they were washed twice with phosphate-buffered saline (PBS) and incubated for 20 min in Dulbecco's modified Eagle medium lacking FBS or penicillin-streptomycin. The cells were then incubated in medium containing 45 nM [3H]ES, 59 nM [3H]E2G, 25 nM [3H]MPP+, 25 nM [3H]l-carnitine, 20 μM [14C]tetraethylammonium, or 89 nM [3H]PAH for 5 min at 37°C. This medium was then aspirated, and the cells were washed three times with ice-cold PBS and lysed in 1% Triton X-100 for 30 min at room temperature on a shaker. The uptake of substrate by the cells was quantitated by measuring the radioactivity in the lysate by using a liquid scintillation counter (PerkinElmer).
Characterization of PAS uptake by HEK cells overexpressing the transporters.
The uptake of PAS was quantitated by methods similar to those described above for the uptake of the prototypical transporter substrates. Cells were incubated in medium containing 100 μM PAS for 5 min at 37°C, and then the medium was immediately aspirated. The cells were washed three times with ice-cold PBS, and lysates were prepared for LC-MS/MS by adding 100 μl acetonitrile (70%) in a 1.5-ml tube, sonicating for 3 to 5 s, and centrifuging at 13,000 rpm for 10 min. An internal standard (IS) for LC-MS/MS was added to each supernatant. Supernatants were vortexed for 3 s and then transferred to vials for detection by LC-MS/MS. A 120-μl aliquot of each lysate was analyzed using LC-MS/MS to quantify uptake. In this experiment, positive-control substrates were [3H]MPP+ for OCT1 and OCT2, [3H]ES for OATP1B1 and OATP2B1, [3H]E2G for OATP1B3, 20 μM [14C]TEA for OCTN1, MATE1, and MATE2K, 25 nM [3H]l-carnitine for OCTN2, and [3H]PAH for OAT1 and OAT3.
Measurement of transepithelial transport of PAS.
For measurement of the transepithelial transport of PAS, we used LLC-PK1 cells overexpressing MDR1 and BCRP and MDCK cells overexpressing MRP1/2. The cells were grown on tissue culture flasks in DMEM supplemented with 10% fetal bovine serum, 1% nonessential amino acids, 2 mM l-glutamine, and 100 U/ml penicillin-streptomycin. The cells were incubated at 37°C in a humidified atmosphere with 5% CO2–95% air. The cells were seeded in 12-well Transwell membranes (pore size, 0.4 μm; filter, translucent; Greiner Bio-One, Alphen aan den Rijn, The Netherlands) at a density of 2 × 105 cells/insert. The cell monolayer was cultured, and the medium was refreshed two times over the 5-day period of culture. A vectorial transport study was performed in overexpressed cells after a transepithelial electrical resistance (TEER) value of the seeded cells of 200 to 250 Ω cm2 was reached. Transport experiments were initiated by washing the monolayer three times with transport medium (Dulbecco's modified phosphate-buffered saline [DPBS]; sodium bicarbonate, 0.35 g/liter; glucose, 1.95 g/liter, pH 7.4), followed by preincubation of the cell monolayer for 20 min. Studies of the flux in the apical-to-basal direction and basal-to-apical direction were performed by introducing 0.5 ml of the drug solution into the apical side and 1.5 ml into the basal side, respectively. For measurement of the transport of 10 μM PAS in the apical-to-basal direction, the insert was transferred to a well containing fresh transport medium every 15 min for 1 h. For measurement of transport in the basal-to-apical direction, the transport medium in the apical side was replaced with 0.35 ml of fresh incubation medium every 15 min for 1 h. A mixture consisting of 20 μl of transport medium with 90 μl of 100% acetonitrile containing rifabutin as an internal standard (10 μg/ml) was injected into an LC-MS/MS system for the determination of the PAS concentration. The apparent permeability coefficient (Papp; in centimeters per second) was calculated using the following equation: (ΔQ/Δt)/AC0, where ΔQ/Δt is the rate of appearance of the drug in the receiver chamber (in micrograms per minute), A is the cross-sectional area (in square centimeters) of the semipermeable membrane of the transwell, and C0 is the initial donor concentration in the apical side (in micrograms per milliliter).
Evaluation of PAS uptake inhibition and possible drug interactions.
To evaluate PAS uptake by specific transporters, we used rifampin to inhibit OATP1B1, verapamil and cimetidine to inhibit OCT1 and OCT2, and probenecid, proton pump inhibitors (PPIs), and nonsteroidal anti-inflammatory drugs (NSAIDs) to inhibit OAT1 and OAT3. Cells were incubated in medium containing 100 μM PAS for 5 min at 37°C in the presence of each transporter inhibitor. PAS uptake was then quantitated as described above. Studies of the kinetics of these inhibitors were performed using the same methods described above in the presence of inhibitors at concentration ranging from 1 to 200 μM.
Determination of kinetic parameters.
Analysis the of the kinetics of PAS uptake was performed using a substrate concentration range of 1 to 400 μM. Prior to performance of the experiments determining the kinetics of PAS uptake, the linearity of cellular uptake over time was determined for each cell line. The rates of cellular uptake of PAS were normalized by incubation time and the total protein content. Net uptake rates were calculated as the difference in the rate of uptake by transfected and wild-type cells for each concentration.
For each study drug for which results are reported, experiments were performed in triplicate under all conditions. The values obtained in each experiment are expressed as the mean ± standard deviation (SD). To ensure that inhibition of uptake occurred only with the selected transporters, we used mock-transformed cells and positive controls in all experiments. The kinetic parameters Km and Vmax were calculated using the Michaelis-Menten equation, as follows: V = (Vmax × S)/(Km + S), where V indicates the velocity of substrate uptake (in picomoles per minute per milligram of protein), S indicates the concentration of substrate added in the medium (in micromolar), Km is the Michaelis-Menten constant (in micromolar), and Vmax is the maximum rate of uptake. Calculations were performed using the Phoenix software package (WinNonlin, version 5.1; Pharsight Corp., Mountain View, CA, USA).
The half-maximal (50%) inhibitory concentration (IC50) of each inhibitor was determined by measuring the percent inhibition of OCT1-, OCT2-, OAT1-, and OAT3-mediated substrate uptake over a range of inhibitor concentrations. IC50s were determined using the inhibitory effect model: E = E0 · {1 − [C/(C + IC50)]}, where E is the effect, E0 is the baseline effect, IC50 is the concentration that produces a 50% inhibitory effect, and C is concentration. The values were determined by the use of WinNonlin software (version 5.1; Pharsight Corp., Mountain View, CA, USA).
The level of uptake of radiolabeled prototypical substrates in vitro was calculated as the amount of substrate associated with the cells (in disintegrations per minute per well or picomoles per well) divided by the initial concentration in the buffer (in disintegrations per minute per milliliter or picomoles per well) and the total amount of protein in each lysate (in milligrams of protein per well). The net uptake was determined by subtracting the total amount of uptake by control cells from the amount of uptake by cells overexpressing the transporters and was expressed as a percentage of the amount of uptake by control cells (referred to as percent of control). The kinetic parameter concentration-dependent uptake was evaluated by the Michaelis-Menten equation by a previously published method (38).
DDI index prediction using a static model.
To add clinical value to our study, we calculated the DDI indexes of the inhibitor drugs in our in vitro study according to FDA guidance (33) using the maximum plasma concentration (Cmax) and the maximum unbound plasma concentration (Cmax,u) (clinical pharmacokinetic data) (see Table S2 in the supplemental material) for each drug at its IC50. According to FDA guidelines, a different method was used for OATP1B1, OCTs, and OATs (33, 38, 39). Briefly, the method described by the FDA draft guidance and others is as follows: for OATP1B1, as a first step, we applied the equation R = 1 + Cmax/Ki, where R is the drug-drug interaction index and Cmax indicates the maximum concentration of the inhibitor (bound plus unbound) systemically present in the circulation. As a second step, FDA recommends the use of the following equation: R = 1+ [I]u,inlet,max/Ki, where [I]u,inlet,max indicates the maximum estimated concentration of unbound inhibitor entering the liver. The next equation is [I]u,inlet,max = fu · [Imax + (Ka · Fa · Fg · dose)/Qh)], where fu is the unbound fraction of an inhibitor in the systemic circulation, calculated by assuming the blood-to-plasma (B/P) ratio; Imax is the maximum concentration of an inhibitor present in blood; Ka is the absorption rate constant of the inhibitor; Fa is the absorbed fraction of the inhibitor; Fg is the fraction of the absorbed inhibitor dose from gut wall extraction; dose is the dose of the inhibitor; and Qh is the hepatic blood flow rate (97 liters/h) (for the European Union, from the Committee for Human Medicinal Products, 2012; for Japan, from the Ministry of Health, Labor, and Welfare, 2014). We determined the R value by using Ka equal to 0.1/min and an Fa × Fg value of 1 to avoid the risk of false-negative DDI predictions in our study, as described previously. For the OCTs and OATs, we simply calculated the drug-drug interaction index using the equation Cmax/IC50, where Cmax indicates the maximum concentration and Cmax,u indicates the maximum unbound concentration of the inhibitor present in the systemic circulation (27, 33, 39–41).
Statistical analysis.
Two-sided Student's t tests were used to determine the statistical significance of the differences between control and test data. Results are expressed as means ± SDs. P values of <0.05 are considered to indicate a statistically significant difference from the value for the control and were determined using GraphPad Prism software (version 6; San Diego, CA).
Supplementary Material
ACKNOWLEDGMENTS
We thank Lim Su Jeong, Cho Munju, Yeon Jung Yoon, and Dong Jun Lee for assisting with the transepithelial transport experiments and quantification by LC-MS/MS.
We declare no conflicts of interest or other relevant affiliations, financial involvement, or agreement/interest with any organization or governing body. In addition, no technical assistance was used in preparing the manuscript.
This research was supported by a grant of the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (grant number HI14C1063) and a grant of the Korean Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number HI15C1537).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02392-16.
REFERENCES
- 1.Lehmann J. 1964. Twenty years afterward historical notes on the discovery of the antituberculosis effect of paraaminosalicylic acid (PAS) and the first clinical trials. Am Rev Respir Dis 90:953–956. [DOI] [PubMed] [Google Scholar]
- 2.Rengarajan J, Sassetti CM, Naroditskaya V, Sloutsky A, Bloom BR, Rubin EJ. 2004. The folate pathway is a target for resistance to the drug para-aminosalicylic acid (PAS) in mycobacteria. Mol Microbiol 53:275–282. doi: 10.1111/j.1365-2958.2004.04120.x. [DOI] [PubMed] [Google Scholar]
- 3.European Medicines Agency. 2013. para-Aminosalicylic acid Lucane. European Medicines Agency, London, United Kingdom. [Google Scholar]
- 4.Peloquin CA. 2002. Therapeutic drug monitoring in the treatment of tuberculosis. Drugs 62:2169–2183. doi: 10.2165/00003495-200262150-00001. [DOI] [PubMed] [Google Scholar]
- 5.Wan SH, Pentikainen PJ, Azarnoff DL. 1974. Bioavailability of aminosalicylic acid and its various salts in humans. 3. Absorption from tablets. J Pharm Sci 63:708–711. [DOI] [PubMed] [Google Scholar]
- 6.Lehmann J. 1969. The role of the metabolism of p-aminosalicylic acid (PAS) in the treatment of tuberculosis. Interaction with the metabolism of isonicotinic acid hydrazide (INH) and the synthesis of cholesterol. Scand J Respir Dis 50:169–185. [PubMed] [Google Scholar]
- 7.Zheng J, Rubin EJ, Bifani P, Mathys V, Lim V, Au M, Jang J, Nam J, Dick T, Walker JR, Pethe K, Camacho LR. 2013. para-Aminosalicylic acid is a prodrug targeting dihydrofolate reductase in Mycobacterium tuberculosis. J Biol Chem 288:23447–23456. doi: 10.1074/jbc.M113.475798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.MacGregor AG, Somner AR. 1954. The anti-thyroid action of para-aminosalicylic acid. Lancet 267:931–936. [DOI] [PubMed] [Google Scholar]
- 9.Egelund EF. 1954. Progress report on therapeutic and toxic effects of combinations of isoniazid, streptomycin, and para-aminosalicylic acid; United States Public Health Service cooperative investigation of antimicrobial therapy of tuberculosis. Am Rev Tuberc 69:1–12. [PubMed] [Google Scholar]
- 10.Saifullah B, Hussein MZ, Hussein-Al-Ali SH, Arulselvan P, Fakurazi S. 2013. Sustained release formulation of an anti-tuberculosis drug based on para-amino salicylic acid-zinc layered hydroxide nanocomposite. Chem Cent J 7:72. doi: 10.1186/1752-153X-7-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joerger M, Huitema AD, van den Bongard HJ, Baas P, Schornagel JH, Schellens JH, Beijnen JH. 2006. Determinants of the elimination of methotrexate and 7-hydroxy-methotrexate following high-dose infusional therapy to cancer patients. Br J Clin Pharmacol 62:71–80. doi: 10.1111/j.1365-2125.2005.02513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walsky RL, Obach RS. 2004. Validated assays for human cytochrome P450 activities. Drug Metab Dispos 32:647–660. doi: 10.1124/dmd.32.6.647. [DOI] [PubMed] [Google Scholar]
- 13.Li AP. 2007. In vitro evaluation of metabolic drug-drug interactions: a descriptive and critical commentary. Curr Protoc Toxicol Chapter 4:Unit 4.25. [DOI] [PubMed] [Google Scholar]
- 14.World Health Organization. 2015. MDR-TB guidelines. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 15.Ho RH, Kim RB. 2005. Transporters and drug therapy: implications for drug disposition and disease. Clin Pharmacol Ther 78:260–277. doi: 10.1016/j.clpt.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 16.Fredriksson R, Nordstrom KJ, Stephansson O, Hagglund MG, Schioth HB. 2008. The solute carrier (SLC) complement of the human genome: phylogenetic classification reveals four major families. FEBS Lett 582:3811–3816. doi: 10.1016/j.febslet.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 17.International Transporter Consortium, Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, Hoffmaster KA, Ishikawa T, Keppler D, Kim RB, Lee CA, Niemi M, Polli JW, Sugiyama Y, Swaan PW, Ware JA, Wright SH, Yee SW, Zamek-Gliszczynski MJ, Zhang L. 2010. Membrane transporters in drug development. Nat Rev Drug Discov 9:215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hagenbuch B. 2010. Drug uptake systems in liver and kidney: a historic perspective. Clin Pharmacol Ther 87:39–47. doi: 10.1038/clpt.2009.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Breitenbucher RB, Amatuzio DS, Falk A. 1952. The effect of probenecid (benemid) in enhancing para-aminosalicylic acid concentrations in the blood. Am Rev Tuberc 66:228–232. [DOI] [PubMed] [Google Scholar]
- 20.Sirota JH, Yu TF, Gutman AB. 1952. Effect of benemid (p-[di-N-propylsulfamyl]-benzoic acid) on urate clearance and other discrete renal functions in gouty subjects. J Clin Invest 31:692–701. doi: 10.1172/JCI102651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arbex MA, Varella MDCL, Siqueira HR, Mello FA. 2010. Antituberculosis drugs: drug interactions, adverse effects, and use in special situations. Part 2. Second line drugs. J Bras Pneumol 36:641–656. [DOI] [PubMed] [Google Scholar]
- 22.Li XZ, Nikaido H. 2009. Efflux-mediated drug resistance in bacteria: an update. Drugs 69:1555–1623. doi: 10.2165/11317030-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong L, Xu C, O'Neal S, Bi H-C, Huang M, Zheng W, Zeng S. 2014. Roles of P-glycoprotein and multidrug resistance protein in transporting para-aminosalicylic acid and its N-acetylated metabolite in mice brain. Acta Pharmacol Sin 35:1577–1585. doi: 10.1038/aps.2014.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peloquin CA, Zhu M, Adam RD, Singleton MD, Nix DE. 2001. Pharmacokinetics of para-aminosalicylic acid granules under four dosing conditions. Ann Pharmacother 35:1332–1338. [DOI] [PubMed] [Google Scholar]
- 25.Singh B, Mitchison DA. 1955. Bactericidal activity of streptomycin and isoniazid in combination with p-aminosalicylic acid against Mycobacterium tuberculosis. J Gen Microbiol 12:76–84. doi: 10.1099/00221287-12-1-76. [DOI] [PubMed] [Google Scholar]
- 26.Andries A, Isaakidis P, Das M, Khan S, Paryani R, Desai C, Dalal A, Mansoor H, Verma R, Fernandes D, Sotgiu G, Migliori GB, Saranchuk P. 2013. High rate of hypothyroidism in multidrug-resistant tuberculosis patients co-infected with HIV in Mumbai, India. PLoS One 8:e78313. doi: 10.1371/journal.pone.0078313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chioukh R, Noel-Hudson MS, Ribes S, Fournier N, Becquemont L, Verstuyft C. 2014. Proton pump inhibitors inhibit methotrexate transport by renal basolateral organic anion transporter hOAT3. Drug Metab Dispos 42:2041–2048. doi: 10.1124/dmd.114.058529. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki K, Doki K, Homma M, Tamaki H, Hori S, Ohtani H, Sawada Y, Kohda Y. 2009. Co-administration of proton pump inhibitors delays elimination of plasma methotrexate in high-dose methotrexate therapy. Br J Clin Pharmacol 67:44–49. doi: 10.1111/j.1365-2125.2008.03303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parvez MM, Kaisar N, Shin HJ, Jung JA, Shin JG. 2016. Inhibitory interaction potential of 22 antituberculosis drugs on organic anion and cation transporters of the SLC22A family. Antimicrob Agents Chemother 60:6558–6567. doi: 10.1128/AAC.01151-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minchin RF, Hanna PE, Dupret JM, Wagner CR, Rodrigues-Lima F, Butcher NJ. 2007. Arylamine N-acetyltransferase I. Int J Biochem Cell Biol 39:1999–2005. doi: 10.1016/j.biocel.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 31.Kang HJ, Song IS, Shin HJ, Kim WY, Lee CH, Shim JC, Zhou HH, Lee SS, Shin JG. 2007. Identification and functional characterization of genetic variants of human organic cation transporters in a Korean population. Drug Metab Dispos 35:667–675. doi: 10.1124/dmd.106.013581. [DOI] [PubMed] [Google Scholar]
- 32.Song IS, Shin HJ, Shim EJ, Jung IS, Kim WY, Shon JH, Shin JG. 2008. Genetic variants of the organic cation transporter 2 influence the disposition of metformin. Clin Pharmacol Ther 84:559–562. doi: 10.1038/clpt.2008.61. [DOI] [PubMed] [Google Scholar]
- 33.Food and Drug Administration. 2012. Drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations. Food and Drug Administration, Silver Spring, MD. [Google Scholar]
- 34.Chen L, Takizawa M, Chen E, Schlessinger A, Segenthelar J, Choi JH, Sali A, Kubo M, Nakamura S, Iwamoto Y, Iwasaki N, Giacomini KM. 2010. Genetic polymorphisms in organic cation transporter 1 (OCT1) in Chinese and Japanese populations exhibit altered function. J Pharmacol Exp Ther 335:42–50. doi: 10.1124/jpet.110.170159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parvez MM, Jung JA, Shin HJ, Kim DH, Shin JG. 2016. Characterization of 22 antituberculosis drugs for inhibitory interaction potential on organic anionic transporter polypeptide (OATP)-mediated uptake. Antimicrob Agents Chemother 60:3096–3105. doi: 10.1128/AAC.02765-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kusuhara H, Sekine T, Utsunomiya-Tate N, Tsuda M, Kojima R, Cha SH, Sugiyama Y, Kanai Y, Endou H. 1999. Molecular cloning and characterization of a new multispecific organic anion transporter from rat brain. J Biol Chem 274:13675–13680. doi: 10.1074/jbc.274.19.13675. [DOI] [PubMed] [Google Scholar]
- 37.Hirano M, Maeda K, Shitara Y, Sugiyama Y. 2004. Contribution of OATP2 (OATP1B1) and OATP8 (OATP1B3) to the hepatic uptake of pitavastatin in humans. J Pharmacol Exp Ther 311:139–146. doi: 10.1124/jpet.104.068056. [DOI] [PubMed] [Google Scholar]
- 38.Izumi S, Nozaki Y, Maeda K, Komori T, Takenaka O, Kusuhara H, Sugiyama Y. 2015. Investigation of the impact of substrate selection on in vitro organic anion transporting polypeptide 1B1 inhibition profiles for the prediction of drug-drug interactions. Drug Metab Dispos 43:235–247. doi: 10.1124/dmd.114.059105. [DOI] [PubMed] [Google Scholar]
- 39.Ito K, Iwatsubo T, Kanamitsu S, Ueda K, Suzuki H, Sugiyama Y. 1998. Prediction of pharmacokinetic alterations caused by drug-drug interactions: metabolic interaction in the liver. Pharmacol Rev 50:387–412. [PubMed] [Google Scholar]
- 40.Lau YY, Huang Y, Frassetto L, Benet LZ. 2007. Effect of OATP1B transporter inhibition on the pharmacokinetics of atorvastatin in healthy volunteers. Clin Pharmacol Ther 81:194–204. doi: 10.1038/sj.clpt.6100038. [DOI] [PubMed] [Google Scholar]
- 41.Pan X, Wang L, Grundemann D, Sweet DH. 2013. Interaction of ethambutol with human organic cation transporters of the SLC22 family indicates potential for drug-drug interactions during antituberculosis therapy. Antimicrob Agents Chemother 57:5053–5059. doi: 10.1128/AAC.01255-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.