

ABSTRACT
Despite the rising rates of resistance to dihydroartemisinin-piperaquine (DP), DP remains a first-line therapy for uncomplicated malaria in many parts of Cambodia. While DP is generally well tolerated as a 3-day DP (3DP) regimen, compressed 2-day DP (2DP) regimens were associated with treatment-limiting cardiac repolarization effects in a recent clinical trial. To better estimate the risks of piperaquine on QT interval prolongation, we pooled data from three randomized clinical trials conducted between 2010 and 2014 in northern Cambodia. A population pharmacokinetic model was developed to compare exposure-response relationships between the 2DP and 3DP regimens while accounting for differences in regimen and sample collection times between studies. A 2-compartment model with first-order absorption and elimination without covariates best fit the data. The linear slope-intercept model predicted a 0.05-ms QT prolongation per ng/ml of piperaquine (5 ms per 100 ng/ml) in this largely male population. Though the plasma half-life was similar in both regimens, peak and total piperaquine exposures were higher in those treated with the 2DP regimen. Furthermore, the correlation between the plasma piperaquine concentration and the QT interval prolongation was stronger in the population receiving the 2DP regimen. Neither the time since the previous meal nor the baseline serum magnesium or potassium levels had additive effects on QT interval prolongation. As electrocardiographic monitoring is often nonexistent in areas where malaria is endemic, 2DP regimens should be avoided and the 3DP regimen should be carefully considered in settings where viable alternative therapies exist. When DP is employed, the risk of cardiotoxicity can be mitigated by combining a 3-day regimen, enforcing a 3-h fast before and after administration, and avoiding the concomitant use of QT interval-prolonging medications. (This study used data from three clinical trials that are registered at ClinicalTrials.gov under identifiers NCT01280162, NCT01624337, and NCT01849640.)
KEYWORDS: cardiac safety, POPPK, QT prolongation, antimalarial agents, piperaquine
INTRODUCTION
Piperaquine, a bisquinoline antimalarial drug structurally similar to chloroquine (CQ) and other 4-aminoquilunes, was first synthesized in China and distributed as mass monotherapy as part of Chinese National Malaria Control Program campaigns starting in 1978. It was progressively abandoned in the late 1980s due to the emergence of Plasmodium falciparum resistance. It was rediscovered as a suitable partner compound for artemisinin combination therapies (ACTs) (1, 2). In recent years, dihydroartemisinin (DHA)-piperaquine (DP) has been widely adopted as a first-line treatment for uncomplicated malaria in areas of multidrug resistance, particularly in Southeast Asia.
While treatment with DP is generally a well-tolerated, low-cost, short-course regimen that results in a high cure rate, piperaquine, like chloroquine, is known to interfere with cardiac repolarization, prolonging the QT interval on surface electrocardiograms (EKGs). While the clinical significance remains poorly defined, piperaquine is known to inhibit a slow-rectifier potassium channel expressed by the human ether-a-go-go gene, known as IKr or the hERG channel. While piperaquine is a lipophilic drug with a large volume of distribution and long half-life, peak plasma levels decline rapidly and clinically significant QT interval prolongation typically occurs in the first 4 to 8 h after dosing, resolving over 24 h (3). As is the case with chloroquine, the risks of clinically significant events are thought to be relatively low, and DP is widely used in Cambodia and elsewhere, even though limited or no ability to monitor cardiac safety is available in those regions. A version of the drug (Eurartesim) manufactured under good manufacturing practices is licensed by the European Medicines Agency, and the associated labeling recommends a 3-h fast before and after dosing to reduce the effect of food on bioavailability.
The U.S. Army Medical Component of the Armed Forces Research Institute of Medical Sciences, in partnership with the National Malaria Control Program and Royal Cambodian Armed Forces, conducted three clinical trials of DP in Cambodia between 2010 and 2014 to evaluate the therapeutic and protective efficacy of DP. EKG monitoring was performed in all three studies to estimate the potential for clinically significant repolarization injury. In 2010 and 2011, a comparison of a 3-day DP (3DP) regimen and a compressed 2-day DP (2DP) regimen revealed that both had similar efficacies for the treatment of uncomplicated malaria in northern Cambodia. A mean prolongation of the QT interval corrected using Fridericia's formula (QTcF) of 20 to 30 ms between the predose QT interval and the QT interval at the time of the trough piperaquine levels at 24 h postdosing was measured (4). In 2012, a follow-on randomized, double-blind, placebo-controlled study evaluating a 2-day DP regimen as a monthly malaria prevention therapy was halted after 4 out of 69 volunteers met prespecified criteria to stop the study because of individual cardiac safety endpoints with a QTcF prolongation of >500 ms. An unblind review by the Data Safety Monitoring Board (DSMB) revealed a 46-ms mean QTcF prolongation over that achieved with placebo at the time of the maximum plasma concentration (Cmax) of piperaquine on day 2. A moderate, statistically significant correlation between the piperaquine concentration and the QTcF increase over the baseline was observed, and a strong correlation was observed for the four volunteers whose findings precipitated the cessation of the study (3). Lastly, in 2013 there was a moderate correlation between the piperaquine concentration and corrected QT interval (QTc) interval changes when DP was used as a 3-day regimen for the treatment of P. falciparum infection, though the effect was less pronounced than that found in the previous studies (5). Unfortunately, by then the efficacy of DP compared to that just 3 years earlier in the same area of Cambodia had declined dramatically.
The direct comparability of concentration-effect relationships is limited by the sampling intervals of the respective studies. The 2010 study omitted piperaquine Cmax values altogether, collecting only trough levels at 24 h postdosing. The 2012 study determined Cmax values at 4 and 28 h after the first and second doses, respectively, but collected no data during the terminal elimination phase. The 2013 study determined Cmax values after the first and third doses at 4 and 52 h, respectively. To better understand the relationship between plasma piperaquine levels and QT interval prolongation, we performed a population pharmacokinetic (POPPK) analysis with data from the 3 studies and incorporated these pharmacokinetic (PK) data in a PK-pharmacodynamic (PD) analysis of the effects of the piperaquine concentration on the QT interval.
RESULTS
There were 256 evaluable volunteers from the 3 DP studies. All subjects were of Khmer ethnicity, and nearly all were male. Baseline demographic data, the manually determined QTcF (QTcFm), serum electrolyte concentrations, and the time since the previous meal appear in Table 1. The latter two were not determined in the 2010 study. There were no significant differences among the 3 studies in the values of the parameters listed, with the exception of those observed between the numbers of subjects with moderate and severe QT interval prolongations determined on the basis of correction methods. Correction of the QT interval using Bazett's formula (QTcB) resulted in substantially fewer volunteers with both moderate (>30-ms) and severe (>60-ms) QT interval prolongations compared to the number of volunteers with moderate and severe QT interval prolongations when QTcF was used.
TABLE 1.
DP dosing regimens and characteristics of volunteers from three clinical trials conducted between 2010 and 2014 in Anlong Veng, Cambodia
| Characteristic | Value(s) for study conducted ina: |
||
|---|---|---|---|
| 2010 | 2012 | 2013 | |
| DP dosing regimenb | 2DP or 3DP | 2DP or placebo | 3DP |
| Patients studied | Patients infected with P. falciparum and P. vivax | Healthy subjects | Patients infected with P. falciparum or mixed P. falciparum and P. vivax and infections |
| Total no. of evaluable volunteersc | 80 (40 receiving 2DP regimen/40 receiving 3DP regimen) | 69 (47 receiving 2DP regimen/22 receiving placebo) | 107 |
| Median (IQR): | |||
| Age (yr) | 32 (27–49) | 26 (22–56) | 25 (21–34) |
| Wt (kg) | 56.2 (34–62) | 60.0 (55–65) | 56.0 (52–60) |
| Ht (cm) | 163 (160–167) | 165 (161–169) | 164 (160–168) |
| Body mass index (kg/m2) | 21.2 (20–22) | 22.0 (21–24) | 20.7 (19–23) |
| Baseline QTcFm (ms) | 400 (383–415) | 390 (378–403) | 393 (375–410) |
| Baseline QTcBm (ms) | 418 (413–425) | 396 (393–402) | 412 (411–421) |
| Maximum QTcFm prolongation (ms) | 431 (425–436) | 430 (425–440) | 430 (427–437) |
| Maximum QTcBm prolongation (ms) | 439 (435–447) | 436 (433–447) | 439 (436–445) |
| No. of volunteers with: | |||
| ΔQTcFm of >30 ms | 40 | 42 (39/3)d | 76 |
| ΔQTcBm of >30 ms | 40 | 26 (22/4)d | 43 |
| ΔQTcFm of >60 ms | 6 | 21 (21/0)d | 17 |
| ΔQTcBm of >6 0ms | 9 | 7 (5/2)d | 11 |
| Median (IQR): | |||
| Baseline serum K+ concne (mM) | Not assessed | 3.8 (3.7–4.1) | 3.6 (3.3–4.0) |
| Baseline serum Mg2+ concnf (mg/dl) | Not assessed | 1.9 (1.8–2.0) | 1.8 (1.6–2.0) |
| Time to previous meal prior to 1st dose (h) | Not assessed | 2.9 (1.9–4.5) | 2.7 (1.8–4.1) |
| Avg (IQR) time to previous meal prior to all doses (h) | Not assessed | 2.9 (2.0–3.3) | (3.6 (3–4.4) |
The total DHA-piperaquine phosphate (DP) dose was 360/2,880 mg.
All volunteers were male, except for 3 females in the study conducted in 2013.
Data in parentheses represent the number in each category receiving DP/placebo.
The normal K+ concentration range is 3.5 to 5.1 mM.
The normal Mg2+ concentration range is 1.8 to 2.4 mg/dl.
Observed plasma piperaquine concentrations and QT interval.
The median plasma piperaquine base (PIP) concentrations were significantly higher following the 2DP regimen than the 3DP regimen, with similar concentrations at 4 h postdosing being seen in the groups receiving the 2DP regimen in the trials conducted in 2010 and 2012 (Fig. 1). Figure 2 illustrates significant changes in the QTcFm over that at the baseline (ΔQTcFm) when the ΔQTcFm for the group receiving placebo in the study conducted in 2012 was compared to the ΔQTcFm values in all three studies, with the highest increase being seen in the group receiving the 2DP regimen in the study conducted in 2012. The mean ΔQTcFm determined from the QTcFm values at the times of the Cmax values at 4 and 28 h following the first and second doses of DP, respectively, was significantly higher in the group receiving the 2DP regimen in the study conducted in 2012 than the mean ΔQTcFm determined from the QTcFm values at the times of the Cmax values at 4 and 52 h following the first and third doses of DP, respectively, in the group receiving the 3DP regimen in the study conducted in 2013.
FIG 1.

Plots of observed maximum concentrations of piperaquine from three clinical studies evaluating DP (2010 to 2013). Blue, red, and green symbols, data from the 2010, 2012, and 2013 studies, respectively; circles, 2DP regimen; diamonds, 3DP regimen; horizontal bars, median and interquartile ranges; value above each column, the median piperaquine concentration; red bars above the columns, individual comparisons of statistical significance (**, significant differences [P < 0.01]; ****, very highly significant differences [P < 0.0001]).
FIG 2.

Plots of the medians and interquartile ranges of the observed QTcFm at 0, 24, 48, and 52 h after the first dose of DP or placebo (A) and the change in QTcFm over the baseline at 4, 24, 28, 48, and 52 h after the first dose (B) from the three clinical trials conducted between 2010 and 2013 in northern Cambodia. Light blue, orange, green, pink, dark blue, and red, data for times of 0, 4, 24, 28, 48, and 52 h after the first dose, respectively; horizontal bars, median and interquartile ranges; value above each column, median; red bars above the columns, individual comparisons of statistical significance (**, significant differences [P < 0.01]; ****, very highly significant differences [P < 0.0001]). Note that the volunteers in the 2010 and 2013 studies were treated for uncomplicated malaria, while those in the 2012 study were healthy volunteers administered DP as prophylaxis. The 2010 study collected trough drug levels and EKG results only at 24 and 48 h postdosing.
Population pharmacokinetics of piperaquine and its effect on the QT interval.
Preliminary findings relating the PIP concentration to effects on the QT interval led to the selection of a 2-compartment model over a 1-compartment model. Since a thorough analysis of covariates did not improve the fit, a covariate-free 2-compartment model with first-order absorption and elimination, interindividual random variability, and a log additive residual error was used as the final model. The data predicted by the model were in good agreement with the experimental data, as shown by the goodness of fit based on standard diagnostic plots (Fig. 3), particularly for individual predicted concentrations. Conditional weighted residuals were generally within 2 standard deviations of the mean. A visual predictive check of the model was also in good agreement with the experimental data (Fig. 4). Population pharmacokinetic parameter estimates for PIP from the clinical studies that evaluated the 3DP regimen are shown in Table 2. The final model was used to predict secondary parameters and found that the area under the concentration-time curve (AUC) and Cmax were significantly higher in the 2DP regimen than the 3DP regimen (Table 3). The compressed 2-day dosing regimen led to 2-fold higher levels of piperaquine exposure (AUC) and 4-fold higher Cmaxs, with little or no difference in half-lives being detected (Table 3).
FIG 3.

Basic goodness-of-fit plots for the final piperaquine population model. (A, B) Observed concentrations were plotted against population predicted natural logarithm-transformed concentrations (A) and against individual predicted concentrations (B) and compared to the line of identity (solid line). (C, D) Conditional weighted residuals were plotted against population predicted concentrations (C) and the time after dose administration (D).
FIG 4.
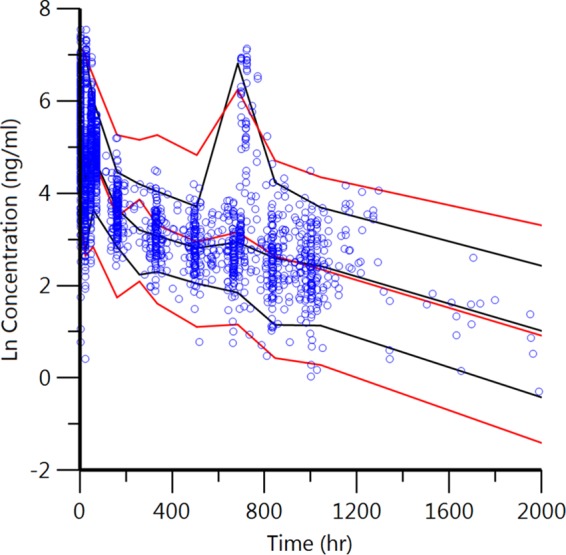
Visual predictive check of the final piperaquine model. Circles, observed data; red and black lines, 5th, 50th, and 95th percentiles of the observed and predicted data, respectively. The concentrations were transformed into their natural logarithms. The second peak at about 720 h (30 days) represents the second month of dosing from the DP prophylaxis study in 2012 in a small subset of subjects.
TABLE 2.
POPPK parameter estimates from the final model describing the PK of PIP in volunteers in three clinical trials of DP conducted from 2010 to 2013 in Cambodia
| Parametera | POPPK estimate (% RSE) | 95% CIb |
|---|---|---|
| Ka (1/h) | 0.06 (7.09) | 0.05–0.07 |
| V (liters) | 94.6 (16.5) | 64–125 |
| V2 (liters) | 19863 (3.8) | 18,392–21,334 |
| CL (liters/h) | 36.3 (3.3) | 34–39 |
| CL2 (liters/h) | 105 (6.1) | 92–117 |
| σ (SD) | 0.47 (2.0) | 0.45–0.49 |
| Beta t1/2 (h) | 512 (5.09) | 461–563 |
| t1/2 (days) | 21.3c |
Ka, absorption rate constant; V, volume of distribution; V2, volume of distribution of the peripheral compartment; CL, clearance; CL2, clearance of the peripheral compartment; σ, log of the additive residual error; beta t1/2, elimination-phase half-life; t1/2, half-life.
CI, confidence interval.
Calculated from the elimination-phase half-life.
TABLE 3.
Values of the secondary PK parameters in volunteers in three clinical trials of DP conducted from 2010 to 2013 in Cambodiaa
| Regimen | AUC (ng · h/ml)b | Beta_hl (h)b | t1/2 (days) | Tmax (h) | Cmax (ng/ml)b |
|---|---|---|---|---|---|
| 2DP | 27,460.5 (25,574–29,347) | 485 (427–543) | 20.0 (18–23) | 2.7 (1.9–3.5) | 562 (499–627) |
| 3DP | 11,875 (10,978–12,772) | 457 (407–506) | 19 (17–21) | 2.2 (1.5–3.0) | 138 (122–154) |
Data are presented as the mean (95% confidence interval). AUC, area under the concentration-time curve; Beta_h1, elimination phase half-life; t1/2, half-life; Tmax, time to maximum concentration of drug in plasma; Cmax, maximum concentration of drug in plasma.
The differences between the two regimens were statistically significant on the basis of an unpaired t test.
Plasma PIP concentrations were compared to the ΔQTcFm (Fig. 5). The strongest correlations were found in the group receiving the 2DP regimen in the 2012 chemoprophylaxis study (Fig. 5B). The correlations for the four volunteers whose findings precipitated the cessation of the study were strong on the basis of Spearman's ρ statistic, while the correlations for the other volunteers treated with the 2DP regimen in that study were moderate. In contrast, only weak correlations were observed in the 2010 and 2013 studies (Fig. 5A and C). Comparisons of plasma PIP concentrations with QTcBm revealed similar results, though overall the associations were weaker (see Fig. S2 in the supplemental material). Figure 5D describes the overall linear slope-intercept model of the effect of PIP exposure on ΔQTcFm. The additive residual error was sufficient to describe the QT interval prolongation following an oral dose of PIP, with the baseline intercept being 6.55 ms (relative standard error [RSE] = 10.01%) and the slope estimate being 0.05 ms per ng/ml, or a 5-ms increase for every 100 ng/ml of piperaquine (RSE = 5.26%).
FIG 5.

(A to C) Plots of the plasma piperaquine concentration versus ΔQTcFm over the QTcFm at the baseline. Pink, green, and yellow circles, results for volunteers receiving the normal 2DP regimen, volunteers receiving the normal 3DP regimen, and volunteers who were stopped from participating in the study, respectively. (D) Plot of the overall observed change in QTcFm over that at the baseline versus the plasma piperaquine concentration from linear slope-intercept modeling. Red circles, observed data; blue line, predicted values. The slope estimate was 0.05 ms per ng/ml of piperaquine (RSE = 5.26%) with a baseline intercept of 6.55 ms (RSE = 10.1%) and a standard deviation for the additive residual error of 22.4.
Somewhat surprisingly, a higher baseline serum potassium level was weakly associated with a higher ΔQTcFm (Fig. 6A). There was little effect of the baseline serum magnesium concentration or time since the previous meal on ΔQTcFm (Fig. 6B and C). There was a weak but statistically significant negative correlation between the maximum ΔQTcFm and the QTcFm at the baseline (Fig. 6D), suggesting that patients with longer baseline QTcFm values tended to experience less of a prolongation of the QTcFm over the QTcFm at the baseline. There were no effects of patient age or body temperature on the model.
FIG 6.

Effects of baseline serum potassium concentration (A), baseline serum magnesium concentration (B), time since the previous meal (C), and baseline QTcFm on the maximum change in the manually read QTcF (max ΔQTcFm) (D) for volunteers receiving dihydroartemisinin-piperaquine in northern Cambodia. ns, no significant difference.
DISCUSSION
By pooling the data sets from three clinical trials of the cardiac safety of dihydroartemisinin-piperaquine, a piperaquine population PK (POPPK) exposure-response model of the QT interval prolongation revealed that the risk of cardiotoxicity, manifested as a prolongation of the corrected QT interval, increased linearly with higher levels of piperaquine exposure. The compressed 2-day DP regimen resulted in a 2-fold higher level of exposure (AUC) and a 4-fold greater maximum piperaquine concentration than the more widely used 3-day DP regimen. Higher levels of exposure corresponded with a higher risk of a prolonged QT interval and greater changes in QTc over the baseline values.
Few covariates influenced the model. There were no effects of age or body temperature on the QT interval. The latter finding is perhaps unexpected, given the potential differences in the QT interval between healthy volunteers and malaria patients due to fever and tachycardia. In malaria patients, early measurements might be lengthened by fever and tachycardia, with the measurements declining as the patients defervesce clinically over 24 to 48 h. However, we did not see evidence for this phenomenon in our population. There were few differences in the QT interval parameters between the healthy and the infected groups (Fig. 2), and no effect of body temperature on the QT interval was found in our model. The baseline potassium and magnesium concentrations and the time since the previous meal had little or no additional influence on prolongation of the QT interval independently of the piperaquine concentration. Patients with longer baseline QT intervals tended to have less subsequent prolongations of the QT interval, a previously well-described phenomenon referred to as “regression to the mean.” As a result, there appear to be few, if any, measurable predictors of repolarization injury risk in settings where DP is likely to be used, as many facilities in areas where malaria is endemic lack the ability to monitor patients by the use of EKGs, and piperaquine levels are rarely, if ever, obtained outside of selected research settings. While QTcFm is the most widely reported method of correction of the QT interval, its reliance on manual reading and the possibility that it underestimates the true prolongation of the QT interval remain important caveats. Low levels of interreader variability and the observations in our data set indicating that QTcB appeared to underestimate rather than overestimate the prolongation of the QT interval compared to the values obtained by the use of QTcF provide reassurance in the overall accuracy of our results.
The overall pharmacokinetic properties of piperaquine defined in our POPPK model were consistent with those presented in previously published reports (1, 6). The results obtained with the POPPK model presented here are consistent with those of our previously reported individual noncompartmental analysis (NCA) of PIP indicating that the level of PIP exposure (Cmax and AUC) from the 2-day DP regimen was higher than that from the 3-day DP regimen (4). The maximum plasma concentrations of piperaquine achieved with oral dosing were reached at approximately 4 h postdosing, which corresponded to the time of the peak prolongation of the QT interval. The terminal half-life in our model was very long as a consequence of a significant distribution of piperaquine to the peripheral compartment. The resulting long half-life and large volume of distribution observed are characteristic of highly lipid-soluble drugs and are similar to the findings described in previous reports, in which large volumes of distribution and an extensive distribution in peripheral tissue were found (7–10). While analysis of covariates did not improve our model, previous reports have indicated that food has various effects on the bioavailability of piperaquine (10–12), with increased concentrations being achieved in the fed state. This finding supports the general recommendation for a 3-h fast before and after dosing.
The slope estimate (0.05 ms per ng/ml, or an increase of 5 ms for every 100 ng/ml piperaquine) of the piperaquine exposure-QT interval response from the pooled analysis is similar to that described in recent publications (3, 13). The QT-lengthening effects of piperaquine have been observed in clinical studies for more than a decade. DP therapy produced a mean prolongation of QTcF of 11 ms in a study of 62 patients with uncomplicated malaria in 2004 (14), though a less pronounced effect (2 to 4 ms) was seen in a Thai study of 56 patients with uncomplicated P. falciparum malaria (15). Electrocardiograms were performed at the time of peak drug levels only in the latter study. More recently, the cardiovascular effects of 3 days of DP (Euratesim) therapy for uncomplicated P. falciparum malaria determined in two pivotal studies were reviewed (16). Subjects receiving DP were more likely to have >60-ms QTcF prolongations on day 2 following the 3rd treatment dose than those taking artesunate-mefloquine in the Asian study or artemether-lumefantrine in an African study (16). A study of piperaquine alone and in combination with the novel antimalarial spiroindolone KAE609 in healthy volunteers showed that piperaquine increased the QTcF and was not influenced by KAE609 (17). Finally, nonlinear mixed-effects exposure-response modeling of the prolongation of the QT interval by single-dose combination regimens of piperaquine and compound OZ439 in 60 healthy volunteers revealed nearly identical changes in the QT interval from the average baseline QTcF, as was seen in the present study, with a mean slope increase of 0.047 ms per ng/ml piperaquine (13).
A number of antimalarials have known QT interval-prolonging effects as well as present a clinical risk for polymorphic ventricular tachycardia, known as torsades de pointes (TdP). Drugs in the amino alcohol class generally have the highest risk, with quinidine (QND), quinine (QN), and, particularly, halofantrine (HF) having significant dose-dependent cardiotoxicities (QND [tmt] QN ≈ HF) in in vitro studies (18, 19) and in vivo animal models (20). HF was also found to potentiate the QT interval prolongation induced by mefloquine, which is not known to be toxic on its own in an animal model (21). Clinically, both HF and QN had significant QT interval-prolonging effects compared to those of mefloquine, artemether-lumefantrine (22), and artesunate-amodiaquine (23) which did not.
While piperaquine has a clear QT interval-prolonging effect, its clinical significance remains less certain. Piperaquine, a 4-aminoquinoline drug, is essentially two covalently bonded chloroquine (CQ) molecules and as such would be expected to behave similarly to chloroquine with respect to its QT interval-prolonging effects. CQ is known to block the hERG channel at two aromatic residues (Tyr652 and Phe656) on the protein's S6 domain, increasing membrane depolarization in Xenopus oocytes (24). It is also known to block the sodium current (INa) and calcium current (ICa) channels in the right ventricular free wall of adult cats (25). Despite similarities to CQ, both the DHA-PIP combination and artemether-lumefantrine exhibited lower potentials to induce TdP than CQ did in the rabbit heart wedge model (26). While there have not been documented reports of clinically significant TdP events for either CQ or PIP to date, there are many potential limitations to accurate reporting from many of the austere settings where antimalarials are most heavily prescribed. We recently observed that DHA-PIP appeared to prolong the U wave, being governed by the slow rectifier potassium channel (Ik1) rather than the inward rectifier potassium channel (Ikr), and the clinical significance of U-wave prolongation remains poorly understood (3).
This study of pooled piperaquine concentrations attempted to estimate the effects of DP on the QT interval from three studies of DP with different treatment durations. The model improved the relative standard error (in percent) of the baseline and slope estimates of the piperaquine-corrected QT interval linear slope-intercept model to 10.1% and 5.26%, respectively. These values suggest that PK sampling and EKG monitoring at the time of Cmax after the first and last doses may have the greatest impact on the parameters estimated and limit interpretation of the concentration-QTc relationship from the 2010 study, where EKGs were not performed at the time of Cmax. This may explain why no significant differences in adverse cardiac events between the regimens were detected (4). The placebo-controlled 2012 study of a 2-day DP course demonstrated by far the strongest linear PK-PD relationships, despite the fact that the study itself was halted prematurely after four volunteers met prespecified rules to stop the study (QTcF > 500 ms).
Interpretation of the results obtained with the present model is further limited by the fact that nearly all volunteers were male: the study could have underestimated the risks in females, who may have higher levels of exposure and a greater risk for TdP (27). Nonpregnant females on the Thai-Myanmar border of comparable age and weight administered a standard course of DP had similar piperaquine half-lives but a Cmax nearly double that observed here in a population pharmacokinetic analysis (28). Another limitation common to most studies of the QT interval is the exclusion of those with QT intervals of greater than 450 ms. While this practice is necessary due to safety concerns, it has tended to systematically exclude those at the highest risk for prolongation of the QT interval. While prior reports have demonstrated that even a small amount of fat intake may increase the level of piperaquine exposure (8), our results did not provide additional support for this finding. The fasting time was deliberately limited to a minimum of 3 h, where practical, only in the 2013 study, though the mean fasting time was 1 to 3 h in the other two studies. This may have accounted for the lower peak concentrations and lower effects of PIP on the QT interval observed in 2013. Multiple piperaquine peak concentration increases postdosing have been observed in several studies and were possibly the result of erratic dissolution and/or absorption from gastric emptying, enterohepatic recirculation, and/or multisector intestinal absorption (7–10). Other factors, including plasma protein binding of >99% (16) and moderate concentrations in red blood cells (29), limit measurement of the total level of piperaquine exposure in the body, and thus, the actual levels in myocardial tissue are unlikely to be approximated. Pharmacogenomic factors increasing the risk of QT interval prolongation have recently been elucidated, though none have yet been identified for piperaquine. Despite this growing body of evidence, even where hereditary factors are known, the clinical significance of QT interval-prolonging effects remains undefined, given the variable risks for TdP independent of the degree of prolongation of the QT interval for some drugs (30).
The overall risk for QT interval prolongation with standard 3-day courses of DP appears to be transient and coincides with times of peak concentrations at 4 to 8 h after dosing. The clinical risks associated with the use of DP have yet to be clearly defined. There is a clear prolongation of the QT interval in a substantial proportion of the population and little or no monitoring capacity. Given that a prolonged QT interval is known to trigger episodes of TdP, further in vitro and in vivo studies of related mechanisms of action and the pharmacogenomics of the piperaquine-induced prolongation of the QT interval are warranted. For now, given the lack of otherwise identifiable or measurable risk factors in settings where DP is used, risk should be mitigated by avoiding the concomitant use of QT interval-prolonging medications or the treatment of those with long QT syndromes, enforcing a 3-h fast before and after administration, and using the standard 3-day course of DP rather than the compressed 2-day courses.
MATERIALS AND METHODS
Ethics statement.
This study pooled data from three clinical trials conducted in Cambodia between 2010 and 2014: WR1737 (ClinicalTrials.gov identifier NCT01280162), WR1849 (ClinicalTrials.gov identifier NCT01624337), and WR1877 (ClinicalTrials.gov identifier NCT01849640). All protocols were approved by the Cambodia National Ethics Committee for Health Research (NECHR) and the Walter Reed Army Institute of Research Institutional Review Board (WRAIR IRB). All study subjects provided written informed consent prior to participation, and all clinical trial protocols complied with the International Conference on Harmonization good clinical practice (ICH-GCP) guidelines.
Study design and participants.
Duo-cotecxin (Zhejiang Holley Nanhu Pharmaceutical, Zhenjiang, China), which contained 40 mg of dihydroartemisinin (DHA) and 360 mg of piperaquine base (PIP), was used in all three studies. Total cumulative doses of DHA-PIP of 360/2,880 mg were given to the participants orally as 2- or 3-day regimens (the 2DP and 3DP regimens, respectively). Those randomized to 2-day regimens received 4.5 tablets daily, with 3 tablets daily being given for the 3-day regimen. The study designs of and the participants in the three clinical trials have been described previously (3, 4, 31). Briefly, the 3 trials were (i) WR1737, which was an evaluation of the efficacy and safety of the 2DP regimen versus those of the 3DP regimen in Cambodian military personnel at risk for P. falciparum and P. vivax malaria conducted from September 2010 to March 2011 (referred to hereafter as the 2010 study); (ii) WR1849, which was performed to determine the protective efficacy of a monthly 2DP regimen in healthy volunteers (referred to hereafter as the 2012 study); and (iii) WR1877, which was an evaluation of the efficacy of the 3DP regimen with or without a single dose of 45 mg of primaquine for the treatment of uncomplicated P. falciparum or mixed P. falciparum-P. vivax malaria (referred to hereafter as the 2013 study).
Plasma piperaquine concentration measurement and electrocardiograms.
A total of 256 volunteers from the three studies for whom at least four plasma piperaquine levels were available and for whom at least one interpretable electrocardiogram was performed following the baseline study were considered evaluable for pharmacokinetic (PK)-pharmacodynamic (PD) modeling analysis. The plasma piperaquine concentration, reported as the piperaquine base (PIP) concentration, was analyzed as described previously by ultraperformance liquid chromatography coupled with mass spectrometry with a Xevo TQ-S mass spectrometer (Waters, Milford, MA, USA). The lower limit of quantification (LLOQ) was 0.54 ng/ml PIP (4). Blood sampling was performed on the basis of the individual study designs. The 2010 study collected samples for PIP concentration determination at 0 h (predosing), at 4, 24, 48, and 72 h postdosing, and then weekly until day 42. The 2012 study collected samples for PIP concentration determination at 0 h (predosing) and 4, 24, and 28 h postdosing for each initial monthly dose, while the 2013 study collected samples for PIP concentration determination at 0 h (predosing), at 4, 24, 48, 52, and 72 h postdosing, and then weekly for 6 weeks. In addition, blood sampling on the day of recurrence was performed for the 2010 and 2013 studies.
Resting 12-lead electrocardiograms were performed on each volunteer, who was in the supine position, at 0 h (predosing), at 24, 48, and 72 h postdosing, and then weekly after the first dose until day 42 in the 2010 study; at 0, 4, 24, and 28 h after the first dose each month for the 2012 study; and at 0, 4, 24, 48, and 52 h and weekly after the first dose for the 2013 study. In the 2010 study, a single EKG study was performed, and the results for all volunteers were read by a single reader, while in the 2012 and 2013 studies, three serial EKGs were recorded at least 5 min apart and the results were read by one of three assigned readers. A GE Mac 1200 electrocardiograph was used, and the results from all studies conducted in 2012 and 2013 were digitized. The average of the three QT and RR intervals was recorded manually using calipers. Details of the EKG measurements and the criteria used to halt the studies were reported previously (3, 4). Final decisions regarding subject disposition and the grading of adverse events were made by the principal investigators with concurrence from the DSMB on the basis of manual QTcF (QTcFm) measurements. The interrater variability for the three investigators responsible for measurement of the QT interval in the study was assessed by comparing independent readings of the same selection of 100 deidentified EKGs obtained in a blind manner. One-way analysis of variance with Tukey's posttest was used to compare the average manual QT interval, RR, and the resultant QTcF from three consecutive EKG complexes in lead II. A Bland-Altman plot was used to analyze the agreement between readers by plotting the percent difference from the mean values using GraphPad Prism software (v.6).
POPPK and concentration-versus-QT interval analysis.
The PIP population pharmacokinetic (POPPK) database included all evaluable data from subjects enrolled in the three clinical studies for whom at least one plasma piperaquine level was available. PK-PD relationships were evaluated using nonlinear mixed-effects modeling. Plasma PIP levels below the LLOQ and those obtained following repeated PIP dosing due to malaria recurrence were excluded from the model. The naive pooled data (NPD) approach and the first-order (FO) estimation method were used to produce the preliminary model. The final model was constructed using a first-order conditional estimate (FOCE). The covariates investigated in the POPPK analysis included age, body temperature at the time of measurement, weight, height, body mass index (BMI), and the time that the previous meal was taken before the first dose, and the covariates were investigated using covariate search stepwise hypothesis testing. Model discrimination was assessed by the use of objective function values (OFV), calculated as −2 log likelihood (−2LL). A change in the OFV of >3.84 was considered to be significant when P was <0.05 and when 1 degree of freedom (a difference in one parameter) was used. Diagnostic plots and the predictive check method were used to evaluate the model. An exposure-response model was evaluated to explore the relationship between plasma PIP exposure and the changes in QTcFm (ΔQTcFm; in milliseconds) over the QTcFm at the baseline. Nonparametric analysis was performed using the Spearman correlation with GraphPad Prism software (v.6; La Jolla, CA). Population pharmacokinetic modeling and linear slope-intercept models were developed using Phoenix NLME software (v.1.4; Certara USA, Princeton, NJ).
Supplementary Material
ACKNOWLEDGMENTS
We thank the volunteers as well as the clinical and laboratory staff who made the study possible.
The views expressed in this article are those of the authors and do not reflect the official policy of the U.S. Department of the Army, the U.S. Department of Defense, or the U.S. or Cambodian government.
Funding for the studies was provided by the U.S. Army Medical Material Development Activity, Fort Detrick, MD (WR1737 and WR1849), and the Armed Forces Health Surveillance Center's Global Emerging Infections Surveillance and Response System, U.S. Department of Defense (WR1877).
P. Vanachayangkul, C. Lon, M. Spring, M. Haigney, L. Cantilena, C. Lanteri, S. Chann, N. Buathong, S. Sok, M. Ittiverakul, W. Kuntawunginn, and D. Saunders designed the study. All authors were involved in data collection. P. Vanachayangkul, W. Ta-aksorn, C. Kodchakorn, M. So, M. Haigney, L. Cantilena, J. Manning, C. Lon, and D. Saunders analyzed and interpreted the data. All authors reviewed and approved the final manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02000-16.
REFERENCES
- 1.Davis TM, Hung TY, Sim IK, Karunajeewa HA, Ilett KF. 2005. Piperaquine: a resurgent antimalarial drug. Drugs 65:75–87. doi: 10.2165/00003495-200565010-00004. [DOI] [PubMed] [Google Scholar]
- 2.Gargano N, Cenci F, Bassat Q. 2011. Antimalarial efficacy of piperaquine-based antimalarial combination therapies: facts and uncertainties. Trop Med Int Health 16:1466–1473. doi: 10.1111/j.1365-3156.2011.02855.x. [DOI] [PubMed] [Google Scholar]
- 3.Manning J, Vanachayangkul P, Lon C, Spring M, So M, Sea D, Se Y, Somethy S, Phann ST, Chann S, Sriwichai S, Buathong N, Kuntawunginn W, Mitprasat M, Siripokasupkul R, Teja-Isavadharm P, Soh E, Timmermans A, Lanteri C, Kaewkungwal J, Auayporn M, Tang D, Chour CM, Prom S, Haigney M, Cantilena L, Saunders D. 2014. Randomized, double-blind, placebo-controlled clinical trial of a two-day regimen of dihydroartemisinin-piperaquine for malaria prevention halted for concern over prolonged corrected QT interval. Antimicrob Agents Chemother 58:6056–6067. doi: 10.1128/AAC.02667-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lon C, Manning JE, Vanachayangkul P, So M, Sea D, Se Y, Gosi P, Lanteri C, Chaorattanakawee S, Sriwichai S, Chann S, Kuntawunginn W, Buathong N, Nou S, Walsh DS, Tyner SD, Juliano JJ, Lin J, Spring M, Bethell D, Kaewkungwal J, Tang D, Chuor CM, Satharath P, Saunders D. 2014. Efficacy of two versus three-day regimens of dihydroartemisinin-piperaquine for uncomplicated malaria in military personnel in northern Cambodia: an open-label randomized trial. PLoS One 9:e93138. doi: 10.1371/journal.pone.0093138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saunders DL, Vanachayangkul P, Lon C, U.S. Army Military Malaria Research Program; National Center for Parasitology, Entomology, and Malaria Control (CNM); Royal Cambodian Armed Forces. 2014. Dihydroartemisinin-piperaquine failure in Cambodia. N Engl J Med 371:484–485. doi: 10.1056/NEJMc1403007. [DOI] [PubMed] [Google Scholar]
- 6.Tran TH, Dolecek C, Pham PM, Nguyen TD, Nguyen TT, Le HT, Dong TH, Tran TT, Stepniewska K, White NJ, Farrar J. 2004. Dihydroartemisinin-piperaquine against multidrug-resistant Plasmodium falciparum malaria in Vietnam: randomised clinical trial. Lancet 363:18–22. doi: 10.1016/S0140-6736(03)15163-X. [DOI] [PubMed] [Google Scholar]
- 7.Annerberg A, Lwin KM, Lindegardh N, Khrutsawadchai S, Ashley E, Day NP, Singhasivanon P, Tarning J, White NJ, Nosten F. 2011. A small amount of fat does not affect piperaquine exposure in patients with malaria. Antimicrob Agents Chemother 55:3971–3976. doi: 10.1128/AAC.00279-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hai TN, Hietala SF, Van Huong N, Ashton M. 2008. The influence of food on the pharmacokinetics of piperaquine in healthy Vietnamese volunteers. Acta Trop 107:145–149. doi: 10.1016/j.actatropica.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen TC, Nguyen NQ, Nguyen XT, Bui D, Travers T, Edstein MD. 2008. Pharmacokinetics of the antimalarial drug piperaquine in healthy Vietnamese subjects. Am J Trop Med Hyg 79:620–623. [PubMed] [Google Scholar]
- 10.Sim IK, Davis TM, Ilett KF. 2005. Effects of a high-fat meal on the relative oral bioavailability of piperaquine. Antimicrob Agents Chemother 49:2407–2411. doi: 10.1128/AAC.49.6.2407-2411.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tarning J, Lindegardh N, Lwin KM, Annerberg A, Kiricharoen L, Ashley E, White NJ, Nosten F, Day NP. 2014. Population pharmacokinetic assessment of the effect of food on piperaquine bioavailability in patients with uncomplicated malaria. Antimicrob Agents Chemother 58:2052–2058. doi: 10.1128/AAC.02318-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen DV, Nguyen QP, Nguyen ND, Le TT, Nguyen TD, Dinh DN, Nguyen TX, Bui D, Chavchich M, Edstein MD. 2009. Pharmacokinetics and ex vivo pharmacodynamic antimalarial activity of dihydroartemisinin-piperaquine in patients with uncomplicated falciparum malaria in Vietnam. Antimicrob Agents Chemother 53:3534–3537. doi: 10.1128/AAC.01717-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darpo B, Ferber G, Siegl P, Laurijssens B, Macintyre F, Toovey S, Duparc S. 2015. Evaluation of the QT effect of a combination of piperaquine and a novel anti-malarial drug candidate OZ439, for the treatment of uncomplicated malaria. Br J Clin Pharmacol 80:706–715. doi: 10.1111/bcp.12680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karunajeewa H, Lim C, Hung TY, Ilett KF, Denis MB, Socheat D, Davis TM. 2004. Safety evaluation of fixed combination piperaquine plus dihydroartemisinin (Artekin) in Cambodian children and adults with malaria. Br J Clin Pharmacol 57:93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mytton OT, Ashley EA, Peto L, Price RN, La Y, Hae R, Singhasivanon P, White NJ, Nosten F. 2007. Electrocardiographic safety evaluation of dihydroartemisinin piperaquine in the treatment of uncomplicated falciparum malaria. Am J Trop Med Hyg 77:447–450. [PubMed] [Google Scholar]
- 16.Keating GM. 2012. Dihydroartemisinin/piperaquine: a review of its use in the treatment of uncomplicated Plasmodium falciparum malaria. Drugs 72:937–961. doi: 10.2165/11203910-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 17.Stein DS, Jain JP, Kangas M, Lefevre G, Machineni S, Griffin P, Lickliter J. 2015. Open-label, single-dose, parallel-group study in healthy volunteers to determine the drug-drug interaction potential between KAE609 (cipargamin) and piperaquine. Antimicrob Agents Chemother 59:3493–3500. doi: 10.1128/AAC.00340-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kinoshita A, Yamada H, Kotaki H, Kimura M. 2010. Effects of anti-malarial drugs on the electrocardiographic QT interval modelled in the isolated perfused guinea pig heart system. Malar J 9:318. doi: 10.1186/1475-2875-9-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mbai M, Rajamani S, January CT. 2002. The anti-malarial drug halofantrine and its metabolite N-desbutylhalofantrine block HERG potassium channels. Cardiovasc Res 55:799–805. doi: 10.1016/S0008-6363(02)00448-0. [DOI] [PubMed] [Google Scholar]
- 20.Batey AJ, Lightbown ID, Lambert JP, Edwards G, Coker SJ. 1997. Comparison of the acute cardiotoxicity of the antimalarial drug halofantrine in vitro and in vivo in anaesthetized guinea-pigs. Br J Pharmacol 122:563–569. doi: 10.1038/sj.bjp.0701402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lightbown ID, Lambert JP, Edwards G, Coker SJ. 2001. Potentiation of halofantrine-induced QTc prolongation by mefloquine: correlation with blood concentrations of halofantrine. Br J Pharmacol 132:197–204. doi: 10.1038/sj.bjp.0703823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Touze JE, Heno P, Fourcade L, Deharo JC, Thomas G, Bohan S, Paule P, Riviere P, Kouassi E, Buguet A. 2002. The effects of antimalarial drugs on ventricular repolarization. Am J Trop Med Hyg 67:54–60. [DOI] [PubMed] [Google Scholar]
- 23.Adjei GO, Oduro-Boatey C, Rodrigues OP, Hoegberg LC, Alifrangis M, Kurtzhals JA, Goka BQ. 2012. Electrocardiographic study in Ghanaian children with uncomplicated malaria, treated with artesunate-amodiaquine or artemether-lumefantrine. Malar J 11:420. doi: 10.1186/1475-2875-11-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanchez-Chapula JA, Navarro-Polanco RA, Culberson C, Chen J, Sanguinetti MC. 2002. Molecular determinants of voltage-dependent human ether-a-go-go related gene (HERG) K+ channel block. J Biol Chem 277:23587–23595. doi: 10.1074/jbc.M200448200. [DOI] [PubMed] [Google Scholar]
- 25.Sanchez-Chapula JA, Salinas-Stefanon E, Torres-Jacome J, Benavides-Haro DE, Navarro-Polanco RA. 2001. Blockade of currents by the antimalarial drug chloroquine in feline ventricular myocytes. J Pharmacol Exp Ther 297:437–445. [PubMed] [Google Scholar]
- 26.Borsini F, Crumb W, Pace S, Ubben D, Wible B, Yan GX, Funck-Brentano C. 2012. In vitro cardiovascular effects of dihydroartemisinin-piperaquine combination compared with other antimalarials. Antimicrob Agents Chemother 56:3261–3270. doi: 10.1128/AAC.05688-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Khatib SM, LaPointe NMA, Kramer JM, Califf RM. 2003. What clinicians should know about the QT interval. JAMA 289:2120–2127. [DOI] [PubMed] [Google Scholar]
- 28.Tarning J, Rijken MJ, McGready R, Phyo AP, Hanpithakpong W, Day NP, White NJ, Nosten F, Lindegardh N. 2012. Population pharmacokinetics of dihydroartemisinin and piperaquine in pregnant and nonpregnant women with uncomplicated malaria. Antimicrob Agents Chemother 56:1997–2007. doi: 10.1128/AAC.05756-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hung TY, Davis TM, Ilett KF. 2003. Measurement of piperaquine in plasma by liquid chromatography with ultraviolet absorbance detection. J Chromatogr B Analyt Technol Biomed Life Sci 791:93–101. doi: 10.1016/S1570-0232(03)00209-5. [DOI] [PubMed] [Google Scholar]
- 30.van Noord C, Eijgelsheim M, Stricker BH. 2010. Drug- and non-drug-associated QT interval prolongation. Br J Clin Pharmacol 70:16–23. doi: 10.1111/j.1365-2125.2010.03660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spring MD, Lin JT, Manning JE, Vanachayangkul P, Somethy S, Bun R, Se Y, Chann S, Ittiverakul M, Sia-ngam P, Kuntawunginn W, Arsanok M, Buathong N, Chaorattanakawee S, Gosi P, Ta-aksorn W, Chanarat N, Sundrakes S, Kong N, Heng TK, Nou S, Teja-isavadharm P, Pichyangkul S, Phann ST, Balasubramanian S, Juliano JJ, Meshnick SR, Chour CM, Prom S, Lanteri CA, Lon C, Saunders DL. 2015. Dihydroartemisinin-piperaquine failure associated with a triple mutant including kelch13 C580Y in Cambodia: an observational cohort study. Lancet Infect Dis 15:683–691. doi: 10.1016/S1473-3099(15)70049-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.