

Abstract
The complement system is a powerful effector arm of innate immunity that typically confers protection from microbial intruders and accumulating debris. In many clinical situations, however, the defensive functions of complement can turn against host cells and induce or exacerbate immune, inflammatory, and degenerative conditions. Although the value of inhibiting complement in a therapeutic context has long been recognized, bringing complement-targeted drugs into clinical use has proved challenging. This important milestone was finally reached a decade ago, yet the clinical availability of complement inhibitors has remained limited. Still, the positive long-term experience with complement drugs and their proven effectiveness in various diseases has reinvigorated interest and confidence in this approach. Indeed, a broad variety of clinical candidates that act at almost any level of the complement activation cascade are currently in clinical development, with several of them being evaluated in phase 2 and phase 3 trials. With antibody-related drugs dominating the panel of clinical candidates, the emergence of novel small-molecule, peptide, protein, and oligonucleotide-based inhibitors offers new options for drug targeting and administration. Whereas all the currently approved and many of the proposed indications for complement-targeted inhibitors belong to the rare disease spectrum, these drugs are increasingly being evaluated for more prevalent conditions. Fortunately, the growing experience from preclinical and clinical use of therapeutic complement inhibitors has enabled a more evidence-based assessment of suitable targets and rewarding indications as well as related technical and safety considerations. This review highlights recent concepts and developments in complement-targeted drug discovery, provides an overview of current and emerging treatment options, and discusses the new milestones ahead on the way to the next generation of clinically available complement therapeutics.
Keywords: Complement, Inflammation, Therapeutics, Immune modulation
1. Introduction
Therapeutic inhibition of the human complement system is far from a recent concept, and the use of complement inhibitors for the treatment of arthritic diseases or transplantation-related complications was already suggested almost 50 years ago [1, 2]. Yet despite several breakthroughs and tremendous progress in target characterization and inhibitor design, the translation of this appealing proposition into the clinic has taken way more time and effort than anticipated [3–5]. It has only been in the past decade that complement-targeted therapy has finally moved into the awareness of the broader research community, clinicians and the pharmaceutical industry alike. The introduction of the first complement-specific inhibitors to the clinic and the discovery of new diseases strongly associated with inappropriate complement activation have clearly contributed to this important milestone. Meanwhile, complement inhibitors are being successfully used in several diseases, numerous novel inhibitors have entered clinical development, and our growing clinical experience is finally allowing an evidence-based discussion about the potential and limitations of this approach [5–7]. Along the way, the field has seen a remarkable diversification in terms of targets, indications, and inhibitory concepts, suggesting an even broader application of complement inhibitors in the clinic.
The attractiveness and challenges of selecting the complement system as a target for therapeutic intervention are both founded in its intricate functional and molecular organization [8–10]. As a key part of the innate host defense machinery, complement contributes to the rapid recognition and elimination of particles, such as microbial intruders or apoptotic cells, that impose a potential threat. The response has to be rapid and comprehensive to prevent risk to the host, but selective enough to avoid damage to healthy cells. Complement typically achieves this delicate balance by employing a cascade-type network of close to 50 proteins, including activators, regulators, and receptors (see below and Fig. 1), and through extensive crosstalk with other defense systems ranging from innate and adaptive immune pathways and the cytokine system to coagulation [8–10].
Figure 1. Targets and inhibitors for pharmacological intervention in the complement cascade.
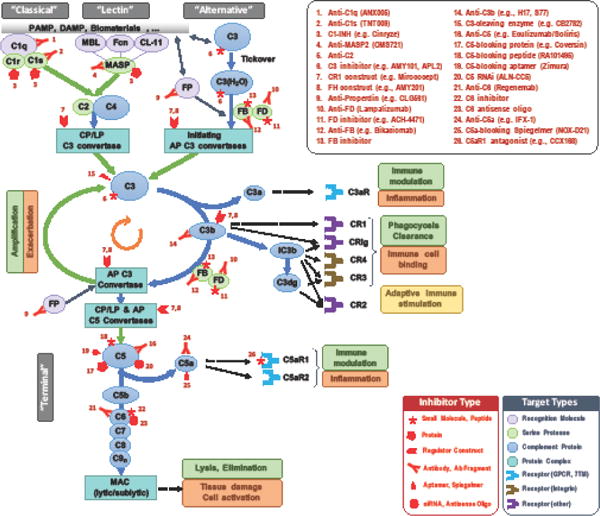
Simplified version of the complement cascade showing initiation on foreign/altered surfaces via the classical (CP), lectin (LP) and alternative (AP) pathways, generation of AP and CP/LP C3 convertases, amplification via the AP, and effector generation mainly via the AP and the terminal pathway. Some components, particularly the natural regulators/inhibitors, are not shown. Major effector functions in both physiological and pathophysiological contexts are listed in green and orange boxes, respectively (a yellow box signifies ambivalent function). Blue arrows represent conversion and/or assembly, green arrows enzymatic cleavage, and black dotted arrows signaling events. The various complement components are colored to signify major target classes as relevant for drug development (see legend). Inhibitors currently listed in the pipelines of pharmaceutical/biotech companies or known to be in clinical development are shown at their respective target, with red symbols to mark the inhibitor type (see legend); numbers refer to the inhibitor list in the upper right corner. Abbreviations: CL-11, collectin 11; CR, complement receptor; CRIg, CR of the immunoglobulin family; FB, factor B; Fcn, ficolins; FD, factor D; FP, properdin (factor P); MAC, membrane attack complex; MASP, MBL-associated serine protease; MBL, mannose-binding lectin.
However, the sheer number of interactions and processes involved in this immune triage also renders complement prone to error, with potentially devastating clinical consequences [11, 12]. For example, transplants and biomaterials are often recognized as foreign intruders that induce an “appropriate” complement response against an inappropriate target. Massive confrontation with infection- or damage-related triggers, such as during sepsis of trauma, can lead to an excessive complement-driven inflammatory reaction that can cause more damage than the underlying insult. An inability to efficiently clear immune complexes or accumulating debris can contribute to autoimmune, age-related, and neurodegenerative disorders. Also, in many cases, dysregulation of the complement network as a result of deficiencies, gain- or loss-of-function mutations, and other genetic alterations, will exacerbate tissue damage and inflammation initiated by various causes.
The unique position of complement as an early danger sensor, acting directly on the triggering cell or material surface, and as an orchestrator of downstream cellular and humoral immune responses makes complement an interesting pharmacological target [6, 7]. Inhibiting or reshaping the complement response can prevent much of the disease-driven damage before it propagates further and may be more efficient than blocking individual cytokines or other later-stage mediators. Yet the complexity and diversity of the complement reaction and crosstalk also impose challenges, and it is unlikely that a single therapeutic approach will be effective on all complement-related disorders. Moreover, some clinical conditions may be associated but not dominated by complement activity, and may therefore not benefit significantly from complement-targeted intervention. The identification of promising indications, the selection of the appropriate complement target, and the choice of the ideal inhibitors are therefore critical for arriving at a successful therapeutic strategy.
2. Spoiled for choice: points of intervention in the complement cascade
2.1. The complement system in health, disease and therapy
In order to achieve selectivity toward foreign and altered cells while allowing rapid reactivity, complement relies on a tiered and closely regulated cascade system (Fig. 1) [8, 10]. Circulating recognition molecules detect damage- or pathogen-associated molecular patterns on target surfaces and induce distinct complement activation routes. The classical pathway (CP) is primarily triggered by the binding of C1q to antibody-antigen complexes, whereas initiation of the lectin pathway (LP) typically involves the recognition of carbohydrate structures by mannose-binding lectin (MBL), ficolins, or certain collectins. These recognition events lead to the activation of the plasma proteins C4 and C2 by complex-associated serine proteases and the formation of C3 convertases on the activating surface. Binding of the abundant plasma component C3 to these convertases induces its cleavage, with covalent deposition of the opsonin C3b. In addition, continuous “tick-over” activation of C3 in solution and/or on surfaces via the alternative pathway (AP) also leads to C3b deposition.
Whereas healthy host cells express and recruit a panel of regulators to keep activation in check, the complement response is quickly amplified on non- or insufficiently protected surfaces, culminating in the generation of potent effectors. Enabled by two serine proteases (factors B and D), surface-deposited C3b can form additional C3 convertases to transform more C3 into C3b, thereby generating an amplification loop that is fueled by the AP and often dominates the overall response. An increasing density of C3b gradually leads to a shift in convertase reactivity toward complement component C5, the cleavage of which results in the generation of C5b and formation of lytic or sublytic membrane attack complexes (MAC).
The activation of C3 and C5 also leads to the release of the anaphylatoxins C3a and C5a, respectively, which act as potent immune modulators. C5a, in particular, has strong chemotactic and pro-inflammatory activities that, among other effects, recruit immune cells to the site of activation. The opsonins C4b and C3b and their degradation fragments (e.g., iC3b, C3dg) bind to various complement receptors (CR) and mediate adherence and immune complex removal (via CR1), phagocytosis (mostly via CR3, CR4, and CRIg), or stimulation of B-cell responses (via CR2). It is the differential involvement of recognition molecules and regulators that ultimately decides whether the final result will be a strong defense response toward an intruder, the “silent” homeostatic removal of cellular waste, or an exaggerated reaction with clinical consequences.
The intricate interplay between complement proteins not only defines their physiological and pathophysiological involvement but also offers a broad range of pharmacological targets that allow intervention at various stages of the complement cascade (Fig. 1). Indeed, clinical development programs are currently reported for inhibitors against more than a dozen distinct complement targets, covering every level from initiation pathways and the amplification loop to effector generation and complement-mediated signaling (Table 1) [6, 7]. While the recent renaissance of complement-targeted drug discovery has been spearheaded by biotech companies, interest in these drugs has also reached big pharma, with several key players working on complement programs. Moreover, various inhibitors have been granted orphan status because of unmet clinical needs in rare diseases, providing important incentives and likely expediting clinical development [13].
Table 1.
Complement therapeutics with known active development programs
| Compound (Company) | Main Target | Class | Clinical Phase (Trial No.)1 | Indications2 |
|---|---|---|---|---|
| Initiation Pathways | ||||
| Cinryze (Shire) | C1r/s, MASP | Protein | Clinic P3 (NCT02547220) | HAE Transplantation |
| Berinert (CSL Behring) | C1r/s, MASP | Protein | Clinic P1/2 (NCT02134314) | HAE Transplantation |
| Cetor (Sanquin) | C1r/s, MASP | Protein | Clinic | HAE |
| Ruconest (Pharming) | C1r/s, MASP | Protein | Clinic | HAE |
| ANX005 (Annexon) | C1q | Antibody | PC | Neurodegenerative |
| TNT009 (True North) | C1s | Antibody | P1 (NCT02502903) | CAD & others |
| N/A (Prothix) | C2 | Antibody | PC | N/A |
| OMS721 (Omeros) | MASP-2 | Antibody | P2 (NCT02222545) P2 (NCT02682407) |
TMA Glomerulopathies |
| OMS906 (Omeros) | MASP-3 | Antibody | PC | PNH & others |
| CLG561 (Novartis) | Properdin | Antibody | P2 (NCT02515942) | AMD |
| NM9401 (Novelmed) | Properdin | Antibody | PC | N/A |
| Activation & Amplification | ||||
| AMY-101 (Amyndas) | C3 | Peptide | PC | Transplantation,C3G, Periodontitis, PNH |
| APL-1 (Apellis) | C3 | Peptide | P1 | COPD |
| APL-2 (Apellis) | C3 | Peptide (PEGylated) | P1 (NCT02588833) P2 (NCT02503332) |
PNH AMD |
| CB 2782 (Catalyst) | C3 | Enzyme | PC | IRI |
| AMY-201 (Amyndas) | C3b,convertases | Protein | PC | PNH, AMD |
| Mirococept (MRC) | C3b,convertases | Protein | P2 (EMPIRIKAL) | Transplantation |
| Bikaciomab (Novelmed) | FB | Antibody | PC | AMD |
| N/A (Novartis) | FB | Small Molecule | ||
| Lampalizumab (Genentech) | FD | Antibody | P3 (NCT02247531) P3 (NCT02247479) |
AMD AMD |
| ACH-4471 (Achillion) | FD | Small Molecule | P1 (ACTRN12616000082404p) | PNH |
| ‘Compound 6’ (Novartis) | FD | Small Molecule | PC | AMD |
| Terminal Pathway & Effectors | ||||
| Soliris (Alexion) | C5 | Antibody | Clinic P2-P3 |
PNH, aHUS Various3 |
| ALXN1210 (Alexion) | C5 | Antibody | P2 (NCT02605993) | PNH |
| ALXN5500 (Alexion) | C5 | Antibody | P1 | N/A |
| LFG316 (Novartis) | C5 | Antibody | P2 (NCT02763644) P2 (NCT02515942) |
TMA AMD |
| Coversin (Akari) | C5 | Protein | P2 (NCT02591862) | PNH |
| RA101495 (Ra Pharma) | C5 | Peptide | P1 (ACTRN12615001143516) | PNH |
| Zimura (Ophthotech) | C5 | Aptamer | P2/3 (NCT02686658) | AMD |
| ALN-CC5 (Alnylam) | C5 | RNAi | P1/2 (NCT02352493) | PNH |
| Regenemab(Regenesance) | C6 | Antibody | PC | PNH, ALS, others |
| IFX-1 (InflaRx) | C5a | Antibody | P2 (NCT02246595) | Sepsis |
| ALXN-1007 (Alexion) | C5a | Antibody | P2 (NCT02245412) P2 (NCT02128269) |
GVHD APS |
| NOX-D21 (Noxxon) | C5a | Spiegelmer | PC | N/A |
| CCX168 (Chemocentryx) | C5aR1 | Small Molecule | P2 (NCT02222155) P2 (NCT02464891) |
ANCA Vasculitis aHUS |
Only major/select clinical trials shown; PC, preclinical, P1-3, clinical phase 1–3; ClinicalTrials.gov ID shown in parentheses (anzctr.org.au for ACH-4471 and RA101495).
Abbeviations: aHUS, atypical hemolytic uremic syndrome; ALS, amyotrophic lateral sclerosis; AMD, age-related macular degeneration; APS, antiphospholipid syndrome; C3G, C3 glomerulopathy; CAD, cold agglutinin disease; COPD, chronic obstructive pulmonary disease; GVHD, graft versus host disease; HAE, hereditary angioedema; IRI, ischemia-reperfusion injury; PNH, paroxysmal nocturnal hemoglobinuria; TMA, thrombotic microangiopathies. 3For a recent list of trials we refer to Ref. [6].
Despite these promising efforts, the current clinical arsenal is still limited to two complement drugs: the therapeutic anti-C5 antibody eculizumab (Soliris; Alexion Pharmaceuticals) and preparations of the physiological regulator C1 esterase inhibitor (C1-INH; various manufacturers). Furthermore, the clinical availability of those drugs is restricted in many markets because of pricing and other considerations. Where they are available, however, the use of these drugs has dramatically changed the management of several rare diseases, providing important first evidence for the effectiveness and safety of long-term complement-targeted intervention. Still, both drug classes act at rather peripheral points of the cascade and therefore may not be suitable for treating certain disorders. The exploration and clinical evaluation of alternative points of intervention is therefore highly important for broadening the use of complement inhibitors.
2.2. Controlling complement initiation
Blocking any of the initiation pathways promises to control unwanted complement activation at an upstream stage, while potentially allowing for physiological activity through the remaining pathways. However, such a tailored approach typically presupposes the dominant involvement of one specific initiation route in the pathology. Among the examples that largely fulfill this condition are autoimmune hemolytic anemias, in which binding of auto-reactive IgG and/or IgM to circulating erythrocytes triggers the CP and leads to complement-mediated lysis, without much involvement of the LP or AP. Although several diseases with strong association with the LP have been described in animal models in recent years, the value of specifically targeting LP activation remains to be established in most cases.
Until now, plasma-purified (Cinryze, Shire; Berinert, CSL Behring; Cetor, Sanquin) or recombinant (Ruconest, Pharming) preparations of C1-INH have been the only clinical compounds to exert control of the initiation pathways. Importantly, the plasma protein C1-INH acts as a broad serine protease inhibitor that blocks initiating proteases of both the CP (i.e., C1r, C1s) and the LP (i.e., MASP-2, MASP-x), but also non-complement proteases of the coagulation and contact systems (e.g., kallikrein) [14]. C1-INH is currently approved for the treatment of hereditary angioedema, a rare inherited blood disorder caused by a deficiency in functional C1-INH; although shifts in complement levels are often observed, the clinical manifestations (episodic swellings of the face, extremities, or other organs) appear to be mainly caused by the bradykinin system. Meanwhile, C1-INH preparations have been evaluated in other disorders, such as sepsis and ischemia-reperfusion injury (including myocardial infarction) [6], and Cinryze has recently received fast-track designation from the FDA for an investigational use in antibody-mediated rejection (AMR) during kidney transplantation [15].
Although the broad activity of C1-INH may have advantages in such complex conditions involving both complement and coagulation, other diseases would benefit from true pathway-specific approaches. A C1s-specific mAb (TNT009; True North) is currently in Phase 1 trials for various antibody-mediated indications, including autoimmune hemolytic anemia (for which it received orphan drug designation [16]), cold agglutinin disease, AMR, and bullous pemphigoid. Moreover, Annexon has developed an mAb (ANX005) acting at the C1q level for neurodegenerative and autoimmune disorders. Omeros has established a MASP program with an mAb against MASP-2 (OMS721) as a clinical candidate for the treatment of atypical hemolytic uremic syndrome (aHUS) and other thrombotic microangiopathies (TMA); this company recently reported positive results from a phase 2 trial in aHUS with a limited number of patients treated, and has announced plans for phase 3 trials [17]. In addition, Omeros is also developing an anti-MASP-3 mAb (OMS906) at a preclinical stage. Finally, complement activation via the CP and LP may be interrupted by targeting the CP/LP C3 convertase, an approach that appears to be followed by Prothix using an anti-C2 mAb [18]; this strategy may mimic the dual pathway activity of C1-INH without exerting its complement-independent effects.
2.3. Interfering with terminal pathway effector generation
As compared to C1-INH, eculizumab acts at the opposite end of the complement cascade by controlling the terminal pathway of complement activation. This humanized antibody binds to a site on C5 that prevents its activation by C5 convertases,[19] thereby impairing the release of C5a and the formation of the MAC while leaving opsonic pathways intact [20]. This approach is considered to be most successful in disorders that are largely driven by the lytic or cell-damaging action of the MAC and/or the pro-inflammatory activity of C5a. Indeed, eculizumab was initially approved in 2007 for the treatment of paroxysmal nocturnal hemoglobinuria (PNH), an ultra-rare disease in which a lack of complement regulators on clonal populations of blood cells leads to MAC-mediated lysis of erythrocytes, with resulting anemia and thrombotic complications [20, 21]. The approval for aHUS followed 4 years later; although aHUS shares some symptoms with PNH (i.e., hemolysis, thrombosis), the disease is largely defined by insufficient complement regulation on endothelial cells, resulting in tissue damage and inflammation that primarily affects the kidney [22].
The introduction of eculizumab has dramatically improved the therapeutic options for both PNH and aHUS, but the high treatment cost (which can surpass $500,000 per year) restricts its availability and has initiated debates among health care professionals and politicians [23]. Moreover, not all patients benefit sufficiently from eculizumab treatment, with a few individuals being non-responders as a result of point mutations in the epitope on their C5 [24]. Still, the overall clinical success of this drug has encouraged evaluation in other disorders, and eculizumab has been or is currently being assessed in more than 20 clinical trials for conditions ranging from transplant-rejection to asthma [6]. It has also spurred the development of a whole range of alternative C5-targeted strategies, from small molecules to small interfering RNAs (siRNAs), some of which are discussed in the next section. Meanwhile, Alexion itself has announced next-generation versions of eculizumab. ALXN1210 is a longer-acting anti-C5 antibody that promises monthly instead of bi-weekly dosing and is currently being evaluated for use in PNH (ClinicalTrials.gov identifiers NCT02598583 and NCT02605993) [25]. Another version with an improved half-life (ALXN5500) is being developed in partnership with Xencor [26].
Whereas most C5-targeted drugs impair both MAC- and C5a-mediated effector functions, other approaches aim to suppress each path individually. The rationale behind these strategies is to only eliminate the disease-driving effector without interfering with other functions. Conversely, such approaches require that the dominant effector pathway is known and that secondary effectors do not become relevant during inhibition. Although MAC formation involves five proteins (C5b, C6, C7, C8, C9) that can serve as potential targets, and it is controlled by regulators (CD59, clusterin, vitronectin) that can serve as templates, little clinical development has been reported in this area. An exception is the work of Regenesance, a company that lists three C6-targeted approaches, including a humanized mAb (Regenemab), antisense nucleotides, and small molecules in preclinical stages, with PNH and neurological diseases as potential indications.
Pharmacological interference with C5a signaling has been the more common strategy in recent years and can be achieved by blocking either C5a or its major receptor, C5aR1 (CD88). The anti-C5a antibody IFX-1 (InflaRx) has recently been positively evaluated in a phase 2 study (NCT02246595) in patients suffering from early septic organ dysfunction [27]. Similarly, an anti-C5a aptamer (Spiegelmer NOX-D21) is in preclinical development, and a precursor molecule has been used in models of acute inflammation, including sepsis [28]. Finally, Alexion is evaluating an anti-C5a antibody in phase 2 trials of graft-versus-host disease (NCT02245412) and antiphospholipid syndrome (NCT02128269).
The use of small-molecule C5aR1 antagonists to block C5a-mediated inflammatory signaling has been spearheaded by the orally bioavailable cyclic peptidomimetic PMX53, which had been evaluated in various disorders and has undergone several company transitions (Promics, Peptech, Arana, Cephalon) [6, 7, 29]. Meanwhile, a derivative (PMX205, Alsonex) with improved efficacy and blood-brain-barrier penetrance has received orphan designation from the FDA for amyotrophic lateral sclerosis (ALS), but no clinical development plans have been announced. Another C5aR1 antagonist (CCX168) has been evaluated in two phase 2 trials (CLEAR, NCT02222155) for ANCA-associated vasculitis, a rare inflammatory autoimmune disease caused by anti-neutrophil cytoplasmic antibodies (ANCA), with strong involvement of C5a-mediated priming. CCX168 showed positive results in the CLEAR study, and ChemoCentryx has announced plans for phase 3 [30]. Other phase 2 trials in aHUS (NCT02464891) and IgA nephropathy (NCT02384317) have been initiated.
2.4. Central intervention in the amplification loop
Despite the potential advantages of peripheral complement inhibition, these approaches may not be sufficient in certain disorders. This limitation is particularly applicable to diseases with complex complement involvement, those driven by the activity of the amplification loop, and cases in which opsonins act as major effectors. An emerging example is C3 glomerulopathy (C3G), a group of related conditions in which dysregulation of the alternative pathway resulting from genetic alterations and/or autoantibodies leads to consumption of C3 in circulation and massive deposition of C3 fragments on the glomeruli of the kidneys [31]. In this and other instances, central complement inhibition at the level of C3 and the C3 convertases may be considered [32].
C3 activation is the point of convergence of all three complement initiation pathways and acts as a central platform for the generation of nearly all effectors; the deposition of C3b fuels the amplification of the complement response and is a prerequisite for the formation of C5 convertases [33]. Although there are currently no C3-targeted inhibitors on the market, several candidates in clinical development either act on C3 itself or control the activity of the C3 convertase [32].
While compounds acting on the initiation pathways (see above) prevent the formation of the CP/LP convertases, there are several approaches that aim to control the AP convertase that fuels amplification. Antibodies against C3b (H17, Elusys; S77, Genentech) and factor B (Bikaciomab, Novelmed) prevent the initial assembly of the C3 proconvertase (i.e., C3bB complex) [34, 35]; however, no clinical trials have yet been reported for these inhibitors. The subsequent step in convertase formation, i.e. the conversion of C3bB into the final AP C3 convertase by factor D (FD), has gained more attention recently. Genentech has developed an anti-FD antibody (lampalizumab) for the treatment of geographic atrophy in age-related macular degeneration (AMD; see below) after intravitreal injection. Lampalizumab has shown encouraging results in phase 2 trials in a subset of AMD patients with certain genetic predispositions, and two phase 3 trials have been initiated (NCT02247531, NCT02247479).
In parallel, small-molecule FD inhibitors are being developed by Achillion and Novartis (another Novartis patent also describes small FB inhibitors [36]) [37, 38]. Achillion recently announced the initiation of phase 1 trials of its clinically developed molecule (ACH-4471) in healthy volunteers (ACTRN12616000082404p), with a potential application in PNH [39]; it will be particularly interesting to see the PK properties of this drug candidate in order to determine whether therapeutic drug levels can be maintained after oral administration. In addition, Achillion is developing ophthalmic and inhalational FD inhibitors for AMD and chronic obstructive pulmonary disease (COPD), respectively. Novartis’ FD and FB inhibitors have been evaluated in experimental disease models,[40, 41] but no official clinical development plans have been announced.
Another potential target for convertase formation is properdin, a modulator that is known to stabilize the AP C3 convertase complex and may also be involved in its initiation. A fully-human anti-properdin Fab (CLG561) is developed by Novartis for use in AMD [42]; it is currently evaluated as monotherapy or in combination with the anti-C5 mAb LFG316 (see below) in a phase 2 trial for geographic atrophy (NCT02515942). Moreover, an antibody (NM9401) and small molecules for properdin inhibition appear to being developed by Novelmed [6]. The natural complement regulators are powerful modulators of convertase activity, either accelerating the decay of the convertase complex or degrading C3b, and have long inspired the design of convertase inhibitors (see below) [32, 43, 44]. Factor H (FH) and CR1 (CD35) are currently at the center of regulator-based development efforts, with a truncated, membrane-targeted version of CR1 (Mirococept) currently being tested in clinical trials for kidney transplantation (UKCRN 16181; see below). In addition to the use of regulators as cofactors for C3b degradation, the converting serine protease factor I (FI) itself has also been suggested as a therapeutic entity [45].
The other major approach to controlling C3b deposition involves targeting native C3 itself. One option is to deplete C3 in circulation before it can be activated by surface-bound convertases. This strategy has been spearheaded by the use of cobra venom factor (CVF), a C3 homolog that forms stable convertases in solution; a humanized form of CVF has been clinically developed, but no further plans have been reported [46]. Another depleting protease (CB 2782), derived from human membrane-type serine protease 1 (MTSP-1), is being developed by Catalyst for ischemia-reperfusion injury, with other candidates being considered for AMD. Finally, C3 may also be protected from being activated by any of the convertases, an approach that is employed by the compstatin family of C3 inhibitors [47]. These cyclic peptides bind to a site on C3 that blocks binding to the convertases, thereby comprehensively preventing C3b formation by all three major initiation routes. An early version of compstatin (APL-1) is currently being developed by Apellis for inhalation treatment of COPD and, in a long-acting PEGylated form (APL-2) has entered clinical trials for wet AMD (Phase 1; NCT02461771), dry AMD (Phase 2; NCT02503332), and PNH (Phase 1; NCT02588833, NCT02264639). Meanwhile, a next-generation candidate (AMY-101), based on the compstatin analog Cp40 [48], with improved activity and pharmacokinetic properties, is being developed by Amyndas for several indications; it has received orphan designation from EMA and FDA for both PNH and C3G [13, 49].
3. A new diversity in complement inhibition approaches
3.1. From small molecules to antibodies and other biologics
In many ways, complement-targeted drug discovery illustrates and reflects several trends that have been transforming the pharmaceutical industry. Small-molecule approaches have been at the center of development efforts since the early days and, thanks to their druggability, the serine proteases of the cascade have stood in the spotlight. Indeed, several attempts have been made to develop inhibitors for complement proteases such as C1s and FD, yet limitations concerning target specificity and/or pharmacokinetic properties had provided challenges [50]. The advent of biologics, and in particular therapeutic antibodies, has profoundly changed the field and led to a surge of novel inhibitors [51]. Perhaps even more than in other therapeutic areas, antibodies offer a critical advantage; many functions of the complement cascade are driven by protein-protein interactions (PPI) that are difficult to inhibit with small molecules, making large and site-specific proteins such as antibodies attractive alternatives. By targeting exosites involved in substrate binding rather than the catalytic center, antibodies can also circumvent some of the specificity problems of traditional protease inhibitors, as in the case of the anti-FD antibody fragment lampalizumab [52]. As evident from this and the previous section, at least 15 antibody-based candidates are currently in various stages of clinical development, with one antibody on the market (eculizumab) and another in phase 3 trials (lampalizumab).
While this area continues to grow, meanwhile, other PPI inhibitors have been emerging (Fig. 2); these inhibitors include therapeutic proteins based on complement regulators (e.g., TT30; see below), natural immune evasion mediators (e.g., OmCI) or unrelated protein scaffolds (e.g. SOBI002), oligonucleotide-based ligands such as aptamers (e.g., Zimura) or Spiegelmers (e.g., NOX-D21), or peptides (e.g., compstatin analogs). The last example also illustrates an intriguing renaissance of lower molecular weight inhibitors in the complement field. Whereas C5aR1 antagonists dominated this class for a long time, PPI inhibitors against C3, C5, and other targets are now in clinical development, and even the aforementioned serine protease inhibitor category has seen new attractive candidates that appear to overcome some of the limitations of the initial compounds. As compared to traditional biologics, such molecules may offer advantages regarding production cost and oral bioavailability, and it will be interesting to follow them through clinical assessment. In any case, the complement inhibitor field has become more diverse than ever and currently offers a spectrum of distinct approaches (Fig. 2).
Figure 2. Sources of complement inhibitors.
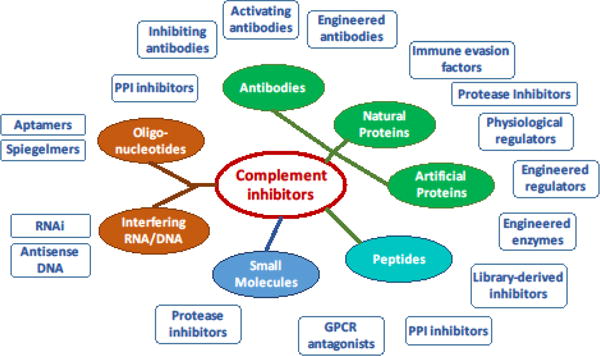
Complement-modulating entities are currently derived from or synthesized as a broad spectrum of chemical classes, ranging from proteins and peptides (green, cyan) to oligonucleotides (orange) and small synthetic molecules (blue). The boxes provide typical examples of inhibitor classes resulting from each type of molecule. Abbreviations: PPI, protein-protein interaction; RNAi, RNA interference.
3.2. C5 inhibitors as example for diversification of therapeutic approaches
This expansion and diversification in inhibitor development has been most obvious in the case of C5-targeted therapeutics, a situation that is hardly surprising in view of the therapeutic and economic success of eculizumab. Biosimilars of eculizumab (from Epirus, BioXpress and potentially other manufacturers) have already been announced with a marketing timeframe of 2020, when the patent protection on eculizumab expires. Meanwhile, Novartis has developed a distinct, fully human anti-C5 antibody (LFG316) based on phage-display technology that blocks terminal pathway activation in humans and NHP [53]. Novartis has previously evaluated LFG316 in wet AMD and is currently conducting clinical trials in dry AMD, uveitis, and PNH (NCT02515942, NCT01526889, NCT02534909, respectively). A recombinant anti-C5 minibody (Mubodina, Adienne) and an RGD-tagged derivative (Ergidina) had been in development, but no further plans for either have been reported.
Although the name would suggest a molecular relationship to antibodies, the C5-binding “affibody” SOBI002 (Swedish Orphan Biovitrum) is based on a small protein A scaffold fused to an albumin-binding domain to improve plasma residence. After a phase 1 trial with transient adverse effects (NCT02083666), the company announced that they would terminate the development of SOBI002 and focus on other candidates [54]. Another protein-based therapeutic with C5-inhibitory potency is Coversin (Akari), a drug candidate corresponding to the tick-derived inhibitor OmCI. This evasion protein of the lipocalin family harbors binding sites for both C5 and leukotriene B4, and potently inhibits convertase-mediated cleavage of C5 across several species [19, 55]. Coversin has been evaluated in phase 1 studies, and a phase 2 trial in PNH for patients with resistance to eculizumab (due to C5 polymorphism [24]) is imminent (NCT02591862). While these protein inhibitors are already much smaller in size (<20 kDa) than antibodies, Ra Pharma is aiming to decrease the size of C5 inhibitors even further. Their macrocyclic peptide-like inhibitor RA101495 shows high affinity for C5 and prevents its activation, and it is positioned for use in PNH via subcutaneous (self-)administration; Ra announced the initiation of phase 1 trials in volunteers (ACTRN12615001143516) [56]. While the above-mentioned inhibitors are all amino acid-based (antibodies, other proteins, and peptides), Zimura (Ophthotech) is a chemically synthesized, PEGylated aptamer (oligonucleotide) with C5-inhibiting activity; it is currently being tested in combination with anti-VEGF therapy in phase 2 for idiopathic polypoidal choroidal vasculopathy (NCT02397954) and in a phase 2/3 trial as a monotherapy for dry AMD (NCT02686658).
A completely different, nucleotide-based approach is the principle behind the RNAi therapeutic candidate ALN-CC5 (Alnylam), which consists of an siRNA with a GalNAc3 tag for hepatocyte targeting; rather than inhibiting the circulating C5 protein, subcutaneously administered ALN-CC5 is designed to suppress the synthesis of C5 in the liver. A phase 1/2 study in health volunteers and PNH patients is ongoing (NCT02352493), and interim results have shown a reduction in plasma C5 of up to 99% with a maximum of 86% serum hemolysis inhibition at the highest dose; Alnylam announced to focus on combination therapy of ALN-CC5 with eculizumab in poor responders of the drug in upcoming phase 2 trials [57, 58]. This latest example illustrates an increasingly explored therapeutic option that involves interference with complement on the genetic level and is slowly finding its way into complement-targeted treatment. In addition to ALN-CC5 by Alnylam and the above-mentioned C6-suppressing locked-nucleic acid (LNA) program by Regenesance, Ionis has shown initial preclinical data for antisense oligonucleotides against FB and FD [59, 60]. The primary target for these approaches is the expression of complement proteins by the liver, and such genetic antisense approaches may provide a comprehensive inhibition of systemic secretion. However, given the growing awareness of local complement production by various tissues and immune cells [61], it will be interesting to see which disorders will benefit most from this strategy.
3.3. Tapping natural templates for the design of new inhibitors
Throughout the development of complement-targeted drugs, the use of natural templates has always had great significance; their importance is particularly obvious in the case of the regulator-based protein inhibitors for controlling convertase activity [43, 44]. Members of the regulator of complement activation (RCA) family are all composed of complement control protein (CCP) domains and confer convertase decay activity and/or act as cofactors for the degradation of C3b or C4b by FI. Among them, CR1 (CD35) has the broadest spectrum of activity and had early been considered for therapeutic development. The extracellular part of CR1, consisting of 30 CCP domains, was originally developed as TP10 by Avant for use in cardiopulmonary bypass surgery, among other indications, and more recently by Celldex (as CDX-1135). Although CDX-1135 showed promising results in a limited trial in a C3G patient [62], Celldex has discontinued the program, partially because of enrollment issues [63]. Similarly, use of the major soluble AP regulator factor H (FH; 20 CCP domains) has thus far proven difficult. Attempts have therefore been made to render these templates more drug-like and to facilitate their production by reducing their size. In the case of CR1, this modification has resulted in Mirococept (see above), which contains only three CCP domains as well as a special lipopeptide tether that allows for painting of cell surfaces, such as the kidney endothelium or Langerhans islets in transplantation [64, 65].
In contrast to membrane-bound CR1, FH is a plasma protein that controls AP activation in the circulation via its N-terminal domains; however, the C-terminus of FH has important pattern recognition capabilities that allow the regulator to bind to host cells and control convertase activity on their surfaces. In recent years, engineered forms of FH have been introduced that directly link the regulatory and surface-recognition domains; interestingly, these mini-FH molecules (e.g., AMY-201, Amyndas) appear to have additional therapeutic advantages over FH, since they bind better to late-stage opsonins (i.e., iC3b, C3dg) that accumulate on host cells under complement attack [66–68]. This opsonin-targeting approach has been spearheaded by TT30 (Taligen, Alexion) and similar molecules; in these engineered proteins, the regulatory part of FH is fused to the C3dg-binding CCP domains of CR2 (CD21) [44, 69]. This class of therapeutics has provided important proof-of-concept about the value of directing complement inhibitors directly to the surface on which the complement attack occurs [44]. TT30 has been clinically developed for treatment of PNH and found safe in phase 1 trials [70], but no further development steps have been announced. Meanwhile, the targeted regulation strategy has been adapted to different regulatory units and/or targeting entities (e.g., TT32/CR1-CR2 [71], CD59-CRIg [72]) or dual-pathway activity (e.g., FH-MAP1 [73]). Finally, a targeting moiety can also be attached to the surface as a protective coating in order to recruit regulators from circulation; for example, the FH-binding peptide 5C6 has been shown to adsorb FH on biomaterial and cell surfaces and protect them from AP activation [74, 75].
Recruitment of FH to surfaces is not only an interesting targeting approach but also showcases the use of microbial/parasitic immune evasion strategies for complement inhibition [33]. Every organism that comes into contact with blood risks an attack by complement, and many pathogens have therefore developed intricate strategies for evading this defense system [76]. Among the most popular tactics is the binding of FH to the microbial surface to confer protection, an approach that can be mimicked by the 5C6 peptide (see above) [75]. Although aspects such as immunogenicity may need to be considered, some evasion molecules can also be directly used as therapeutic inhibitors. The tick-derived C5 inhibitor coversin (see above) is currently the most developed example in this category, and a new class of unrelated C5 inhibitors secreted by ticks (referred to as RaCI) have recently been reported [19]. Other exogenous natural inhibitors such as CVF (in humanized form), the staphylococcal C5aR1 antagonist CHIPS, or the vaccinia virus-derived convertase regulator VCP have been considered for therapeutic purposes [5–7, 43]. Finally, pharmacological targeting of microbial immune evasion proteins may hold promise in antiinfective and/or vaccination strategies [77, 78].
4. New frontiers: rare, re-emerging, and unexpected indications
Unbalanced complement activation has, meanwhile, been linked to numerous diseases, and genome-wide association studies continue to identify complement genes that can contribute to clinical conditions [6, 11, 79]. Yet despite the long list of complement-related disorders, finding the right indication has proven to be unexpectedly challenging. The reasons can be diverse and range from poor translation from the animal model to selection of non-ideal patient cohorts, and technical issues such as study design, inhibitor selection, and drug administration, among others. In this section, we explore the potential of therapeutic complement inhibition in major disease areas and highlight emerging indications. Within the scope of this review, no in-depth insight into complement’s pathological involvement can be given for each disease, and we refer to specialized articles for this purpose [11, 12].
4.1. Exploring the rare disease market: focus on blood and kidney disorders
Thus far, the rare/orphan disease field has benefitted most from the availability of complement inhibitors [13], largely as a result of the presence of several rare disorders with a well-defined, complement-focused disease mechanism and limited treatment options. This is particularly evident in PNH, in which complement-mediated hemolysis is the defining symptom; the success of eculizumab has made this ultra-rare condition a highly common indication for complement-targeted drugs. Although C5 remains a target of major interest, intervention at upstream levels is being increasingly explored, since the ongoing opsonization of PNH erythrocytes with C3 fragments can contribute to the insufficient response of a considerable fraction of PNH patients to anti-C5 treatment [21]. Inhibitors acting at the level of C1s/MASP (e.g., C1-INH [80]), C3 (e.g., compstatin [81]), the C3 convertase (e.g., mini-FH [67]), or FD (e.g., ACH-4471 [38]) have shown efficacy in ex vivo models, and some are currently being evaluated in clinical trials.
Similarly, alternative strategies are being investigated in the case of aHUS, with anti-MASP2 (OMS721) due to enter phase 3 trials for this rare indication [17]. Although the clinical impact of eculizumab as a treatment option for HUS during a recent outbreak of Shiga toxin-producing E. coli is being debated, typical forms of HUS and other TMA remain an area of interest [82, 83]. As an apparent susceptibility hot spot for endogenous complement attack, kidney-related disorders have steadily gained attention in recent years. In addition to aHUS, another prominent example is C3G, with dense deposit disease and C3 glomerulonephritis. Despite the highly diverse involvement of genetic alteration and autoantibodies in these rare and difficult-to-treat disorders, excessive activation and consumption of C3 is the common factor in all of them [31]. Consequently, C3-targeted intervention is considered a favorable path, with soluble CR1 showing clinical promise in a limited trial, and compstatin Cp40 being active in in vitro models [62, 84]. ANCA-associated vasculitis, on the other hand, appears to benefit considerably from selective inhibition of C5aR1 signaling, as the successful phase 2 trial with CCX168 has shown [30]. Notwithstanding this promising progress in orphan diseases, the search for alternative indications continues and has revealed challenges as well as novel therapeutic avenues.
4.2. Back to the roots: Considerations in arthritis, transplantation and hemodialysis
In some cases, the complexity and diversity of the underlying disease mechanism is a complicating factor. As mentioned above, rheumatoid arthritis (RA) and transplantation were identified as “obvious” candidates for complement-directed therapy very early on, but progress has been slow. In RA, numerous animal models had suggested that complement inhibition would have promising effects, yet clinical trials with anti-C5 antibodies and C5aR1 antagonists remained discouraging [85, 86]. It appears that the fully established disease is largely cytokine-driven, explaining the recent success of anti-TNF therapy and the fact that complement inhibition seems to be insufficient to break the inflammatory cycle at that stage. However, newer studies have pointed to a significant, yet complex, role in initial disease development and have opened the door for future complement-targeted strategies for preventing and treating the early stages of RA [87].
Although significant progress has been achieved in defining the role of complement in transplantation-related complications, several aspects remain elusive and appear to be context-specific [11, 88, 89]. It now seems clear that complement is involved in numerous adverse events, ranging from donor death-induced “conditioning” and ischemia-reperfusion injury (IRI) during transplantation to cellular and antibody-mediated rejection, making the system a prime therapeutic target. The clinical availability of C1-INH and eculizumab has allowed the use of complement drugs in transplantation settings, and the first clinical trials have been performed [90]. The results look promising but indicate that treatment success may not be homogeneous. It will therefore be interesting to follow and compare the ongoing and planned clinical studies with complement inhibitors acting at various levels (e.g., C1-INH, mirococept, eculizumab) in order to identify suitable complement targets and treatment strategies.
Among the most intriguing aspects of treatment with these complement inhibitors is that in some primate models, a state of accommodation can be achieved within 2–3 weeks of complement inhibition, with sustained prevention of complement attack thereafter [91]; however, neither the conditions for this behavior or its mechanism is fully understood as yet. Meanwhile, in related research, the potential for using complement drugs to enable transplantation across ABO and HLA incompatibility barriers has moved into the spotlight [92], and the development of transgenic pigs expressing human complement regulators (with the αGal epitope removed) has rekindled interest in xenotransplantation [93]. All in all, transplantation remains a focal area and a promising indication for complement therapy.
In other cases, the medical need has changed, or has appeared to change as the result of improvement in medical equipment or treatment options. In hemodialysis (HD), for example, early cellulose-based filters caused massive complement activation, whereas synthetic polymer alternatives have been thought to be more biocompatible [94, 95]. However, several studies have meanwhile shown that even modern filters induce significant complement activation; given the high frequency of HD treatment, “chronic acute” inflammatory triggers may occur and potentially contribute to HD-related complications such as anemia, cardiovascular disease, and decreased quality of life [94]. Complement-targeted treatment of HD-induced inflammation has therefore experienced a new surge of interest in recent years, and recent NHP studies with compstatin (Cp40) suggest that a single dose of the inhibitor prior to the HD session can alleviate complement activation throughout the treatment [96]. Cp40 may be similarly effective for other biomaterial-triggered conditions, such as during procedures involving extracorporeal circuits and surgical implants or complement activation-related pseudoallergic (CARPA) reactions to drug vehicles (e.g. liposomes) [95, 97]. In all these cases, a time-restricted intervention has the potential to prevent unwanted inflammatory triggers. Indeed, control of complement activation during cardiopulmonary bypass surgery was among the first indications during clinical trials of complement inhibitors such as soluble CR1 (i.e., TP10) and may see a revival with these new therapeutic options [95].
4.3. The promise and challenge of modulating complement in degenerative diseases
The challenges of translating the disease association of complement into a treatment strategy are perhaps best illustrated in the case of AMD. After polymorphisms in the FH gene were identified in 2005 as a strong risk factor for the development of this major cause of blindness in the elderly [98], many companies focused their effort on this common disease and potentially lucrative market. Several lines of evidence have also confirmed an important, yet complex, role for a dysregulated complement system in disease progression [99], but initial clinical trials have thus far largely produced results below expectations and have dampened initial enthusiasms. Even in the case of the successful anti-FD (lampalizumab) trial in geographic atrophy, an advanced form of dry AMD, substantial treatment effects appear to have been observed mainly in a subgroup of patients that carried polymorphic variants of both FH and FI [100]. This result suggests that the underlying disease mechanisms, including those relating to complement activation, may be more diverse than expected, with likely implications for the selection of patient cohorts and treatment strategies. In addition to the entrance of anti-FD (lampalizumab) into phase 3 trials, inhibitors acting on C3 (e.g., the compstatin analog APL-2) and C5 (e.g., Zimura) are currently being evaluated, and the results are eagerly awaited.
In a broader context, AMD is representative of a more generalized pathological involvement of complement in age-related and degenerative diseases, which are often driven by accumulating debris. An emerging hypothesis points toward a dual role for complement in the progression of such diseases; whereas complement’s waste disposal functions may exert a protective effect in early stages, this involvement may become adverse or even deleterious if the debris cannot be efficiently removed and excessive complement activation is triggered, with inflammatory and cell-damaging consequences. The “fitness” of the cascade, largely defined by the complotype of polymorphisms/mutations in complement genes [101], is likely of high importance in these chronic, slowly developing disorders.
Whereas an age-related contribution has, for example, been reported in atherosclerosis [102], there is a particular interest in Alzheimer’s disease (AD) and other neurodegenerative/neurological diseases [90]. Animal models support the two-edged effect of complement in AD, in which C1q-mediated triggering of the CP and subsequent generation of terminal pathway effectors fuel an inflammatory milieu that contribute to disease progression. In an AD model, experimental treatment with PMX205 showed positive effect on disease parameters [103]. Moreover, genome-wide association studies identified polymorphisms in CR1 as a potential risk factor for AD [104], thereby suggesting other potential complement targets in this disorder. Meanwhile, disease association with complement, with potential translational value for complement therapy, has been described for other neurological and neurodenerative diseases including Parkinson’s and Huntington’s disease, multiple sclerosis, amyotrophic lateral sclerosis, and schizophrenia, and we refer to specialized reviews for information concerning the often complex and incompletely explored pathological involvement of complement [90, 105]. As compared to the vascular space and many other tissues, our understanding of complement function in the brain and CNS is still limited, and the applicability of the current inhibitor arsenal largely remains to be explored. However, a wealth of studies indicate that neurological disorders constitute an exciting new frontier for complement therapy [105].
4.4. Dampening excessive complement activation during injury and infection
Whereas most of the hitherto-discussed indications involve chronic or frequently episodic conditions, therapeutic intervention may also be considered during acute situations (Fig. 3). Complement has been shown to be strongly involved in the systemic inflammatory response syndrome (SIRS) observed during sepsis, burns, or trauma, and these conditions have all been considered for therapeutic intervention [106]. In sepsis, a massive infection can trigger an inflammatory host response so severe that it leads to multi-organ failure, septic shock, and death. Although complement inhibition in connection with infection may sound counterintuitive, de-escalation of the vicious inflammatory reaction is paramount, even more because the pathogen may already have been cleared or can be controlled by antibiotic treatment. As a key adverse mediator in sepsis, the anaphylatoxin C5a has been an obvious target, and C5a-scavenging entities such the NOX-D21 Spiegelmer and the anti-C5a antibody IFX-1 have shown great promise in preclinical and clinical studies, respectively. Moreover, compstatin analogs potently prevent organ damage, lung fibrosis, and improve other clinical parameters in NHP models of E. coli-induced sepsis [107, 108], and several studies have suggested that simultaneous inhibition of both complement and TLR can confer additional therapeutic benefit [106, 109]. Although sepsis remains a highly challenging indication with translational hurdles, it would be good to see a treatment for this severe condition moving forward.
Figure 3. Considerations concerning the site and time-frame of clinical manifestations in complement-targeted therapy.
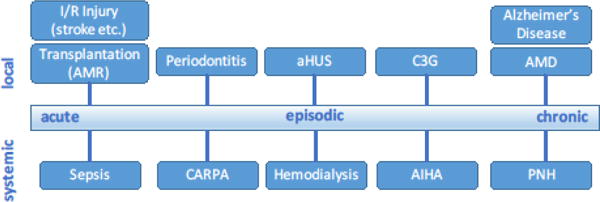
Major examples of complement-related conditions are shown in the context of whether they primarily manifest systemically or locally (e.g., in a specific tissue or organ), and whether they occur as an acute, episodic, or chronic event. Abbreviations: aHUS, atypical hemolytic uremic syndrome; AIHA, autoimmune hemolytic anemia; AMD, age-related macular degeneration; AMR, antibody-mediated rejection; C3G, C3 glomerulopathy; CARPA, complement activation-related pseudoallergy; I/R, ischemia/reperfusion; PNH, paroxysmal nocturnal hemoglobinuria.
Similar to sepsis, the sudden release of DAMPs during trauma can trigger massive complement activation, with C5a acting as a strong inflammatory agent, the control of which may produce a beneficial outcome [110]. Of note, however, there may be differences in the influence of complement in the initial acute phase and during later stages, and there is an increasing body of evidence that also shows a role for complement in fracture and wound healing, among other processes [110, 111]. Complement’s involvement in many of these regenerative mechanisms are still insufficiently understood and may likely be complex and context-specific. The onset and duration of the treatment and the target of intervention may therefore need to be more carefully considered under such circumstances, a caution that is underscored by a recent study using a spinal cord injury model, in which blockage of C5aR1 signaling had different outcomes in the acute and chronic phases of the injury [112].
The interplay between infection, inflammation, and complement is even more intriguing in the case of periodontitis, in which the keystone pathogen Porphyromonas gingivalis creates an inflammatory milieu (e.g., by directly cleaving C5 to release C5a) that favors its survival, leading to dysbiosis, and drives gum disease and bone loss [113, 114]. Mouse models of ligature-induced periodontitis have shown that the genetic absence of either C5aR1 or C3 strongly impairs disease progression and bone loss, although C3 deficiency had a more profound effect on the sustenance of dysbiosis [115, 116]. In NHP models, the compstatin analog Cp40 not only reduced inflammation and bone loss in a ligature-induced disease setting but, more recently, was also shown to exert strong inhibitory effects on markers of inflammation and bone resorption during treatment of the naturally occurring disease [115, 117]. These studies point to complement as a surprising target in an inflammatory disease with a strong infectious component and may open new avenues in the treatment of periodontal disease. They also raise the question of whether similar effects might be achieved in other conditions influenced by microbial dysbiosis. Although studies concerning the interplay between complement and the microbiome are only slowly beginning to emerge, there are early indications that complement inhibition may, for example, influence the skin microbiome and, conversely, that the commensal microbiome affects the expression of complement components [118].
4.5. Turing the tables in cancer therapy
Finally, whereas treatment of the conditions discussed above relies on inhibition or “re-balancing” of complement, the cytotoxic effects of the cascade can be harvested for therapeutic purposes via targeted activation on malignant cells. Complement-dependent cytotoxicity (CDC) is a key mechanism undergirding the use of therapeutic antibodies employed in the treatment of cancer and autoimmune diseases [119, 120]. These antibodies bind to antigens on the target cell (e.g., CD20 on B-cells in the case of rituximab) and facilitate the killing and removal of the cells by engaging complement via the CP, with the goal of achieving direct cell damage through MAC formation. Moreover, opsonization may help clear circulating target cells via phagocytosis, and the release of anaphylatoxins recruits immune cells and influences the expression of Fc receptors that support antibody-dependent cytotoxicity.
As simple and elegant as this approach sounds, there are caveats regarding its capacity and efficacy. For one thing, many cancer cells overexpress complement regulators, increasing the difficulty in achieving activation thresholds. The right dosing also appears to be critical, since excessive amounts of activating antibodies can exhaust the plasma levels of some complement components [119, 120]. Measures to improve CDC, such as the use of regulator-blocking inhibitors or siRNAs or the supplementation of fresh-frozen plasma to prevent complement exhaustion, are therefore being considered. In addition, antibody engineering allows for the optimization of cytotoxic properties to produce next-generation drugs with improved efficacy. Interestingly, as documented by an increasing body of literature, the role of complement in cancer appears to be highly complex and context-dependent [120, 121]. In addition to complement activation and regulation on the cancer cell itself, immune crosstalk and cell recruiting (e.g., mediated via C5a) have been shown to significantly influence the tumor microenvironment and affect proliferation. In some models, selective inhibition of complement effectors has been shown to have a profound effect on reducing tumor growth and/or metastasis [120–123]. Although more translational research still needs to be performed, these examples illustrate the intricate and diverse involvement of complement in disease processes and the need for careful evaluation to identify ideal treatment strategies.
5. Modulating an immune modulator: reconsidering the challenges
5.1. Reassessing safety considerations
Therapeutic interference with a host defense system such as complement naturally raises questions about safety and feasibility, and such concerns certainly have had an impact on progress in this target area. The more clinical experience we gain with complement-targeted drugs, however, the stronger grows our confidence in this approach. Most importantly, the data currently collected from the indicated and off-label use of existing inhibitors, the results from an increasing number of clinical trials, and insights gained from biomedical research on complement are enabling us to better discuss and reassess some of the long-standing reservations about therapeutic complement inhibition.
Indeed, the various roles of complement in health and disease appear to be a conundrum: In addition to the century-old appreciation of complement as an anti-microbial defense system, the past few decades have revealed much broader functions for this system in immune surveillance, waste disposal, tissue development and repair, metabolic processes, and coordination of immune responses [9, 10]. Although this involvement of complement in host protection may be seen as warning flag when pharmacological intervention is being considered, this protective role needs to be viewed in context. In many of the processes mentioned above, complement is an important contributor but is not the only system to maintain a certain task, since there is often considerable redundancy and coordination in physiological responses. Even more importantly, the significance of complement can wane in some cases, as is likely true for its basic defense functions. As adaptive immunity matures and grows more forceful and versatile, the influence of innate defense, including complement, becomes less prominent. This hypothesis agrees with observations in individuals with primary deficiencies in complement proteins, some of whom experience recurrent infection during childhood and adolescence but decreasing incidence of infection when approaching adulthood [124]. Moreover, evolutionary pressure has led many pathogens to develop complement-counteracting strategies [76, 125]. Finally, even after MAC formation is blocked, some bacteria are still efficiently killed in serum by other factors [126]. Only a restricted set of pyogenic bacteria, typically including Neisseria meningitidis, Streptococcus pneumoniae, and Haemophilus influenza, appear to be prominent in patients with complement deficiencies [124]. Other clinical symptoms are less common or uniform, and vary with the type of deficiency; for example, a lack of early components (e.g., C1q) is linked to a higher frequency of systemic lupus erythematosus (SLE) and SLE-like disorders [127]. However, the exact influence of complement in autoimmune conditions is still being investigated, with C1q and C3 likely exerting diverse roles (i.e., C3 deficiency may have a protective effect) [128]. Finally, in knockout mice, the genetic absence of individual complement components is often clinically uneventful, at least in the absence of specific triggers. This finding does not disallow the importance of complement as a host defense system but rather suggests that its impact may depend on the exact insult and the individual’s immune status, and a functional impartment may be at least partially compensated by other systems. After all, complement is designed as a “silent system” that should only become active under certain conditions, and the body’s intricate immune crosstalk is primarily a means of translating the sensing of an insult into downstream effector signals. The inadvertent or excessive triggering of complement, and a lack of proper regulation, are often more likely to cause clinical complication than is a lack of activity.
Still, pharmacological manipulation of complement is not, and should not, be taken lightly, and the clinical development of complement-targeted drugs requires careful oversight. As in other therapeutic areas, the disease burden and potential risk for the patient have to be weighed and considered individually. Chronic diseases being treated or proposed to be treat with complement-related drugs, such as PNH, aHUS, and C3G, are life-threatening conditions with very limited treatment options at present. Meanwhile, we have also gained real clinical experience with extended use of complement inhibitors. C1-INH preparations have been used for the treatment of HAE for many years and are considered safe and effective therapeutic options [129]. Similarly, the long-term experience with eculizumab in PNH has been highly positive thus far, showing strong efficacy in most patients, with negligible risk for developing immunogenicity (i.e., human antihuman antibodies) or severe infection when the recommended measures are followed concerning prophylactic meningococcal vaccination and antibiotic treatment upon signs of infection [130–133]. At least initially, such precautionary measures may likely be included for other complement drugs to further minimize risk of infection; in some cases, they may be extended to include vaccines for other complement-susceptible bacteria such as pneumococci or H. influenza (see above).
Many of the patients considered for long-term, systemic complement therapy are already under tight supervision, which facilitates early recognition of adverse events and adjustments of the treatment protocol. Unlike primary deficiencies, pharmacological complement inhibition can be interrupted and is expected to allow recovery of residual complement activity within hours in many cases; treatment with fresh-frozen plasma can even accelerate this process. Even residual complement activity is expected to confer significant antimicrobial protection, given that recurrent infection is not apparent in patients with hypocomplementemia (e.g., due to C3G). It is worth noting that even broad inhibitors such as compstatin do not block complement activity completely, but instead leave some aspects intact, such as tickover, conformational activation of C3 on surfaces, C3 cleavage by coagulation proteases, and/or deposition of the C4b opsonin by initiating pattern recognition complexes [47, 134, 135].
Careful patient monitoring will remain critical for the clinical development of complement drugs, but we currently have an increasingly evidence-based safety assessment. Most importantly, safety aspects also need to be viewed in the right context and evaluated for each indication, target, and treatment option. Whereas the considerations mentioned above mainly reflect the situation in chronic diseases with long-term systemic application of complement-targeted inhibitors, the assessment is typically even more favorable in cases of acute clinical situations requiring short-term treatment or treatment at intervals. During hemodialysis, for example, a single bolus injection of Cp40 is sufficient to suppress filter-induced complement activation during the 4-hour procedure, with the drug being washed out shortly after treatment [96]. In acute systemic inflammation, such as after trauma or during sepsis, the complement-inhibitory treatment is not only time-restricted, but patients typically are already under antimicrobial control. Although clinical experience still needs to be gained, the use of targeted inhibitors such as TT30 or mini-FH may also contribute to a more directed inhibition of the complement response at the site of activation, while conferring higher residual vascular activity [68, 87]. Finally, the application type and site need to be considered (Fig. 3); in conditions for which local application of drugs is appropriate, such as AMD or periodontal disease, the impact on systemic complement activity is considered negligible [117, 136].
5.2. Overcoming technical challenges
Another hurdle that has been affecting early drug development efforts in the complement therapeutics field are the plasma concentrations and/or turnover rates of some complement targets; C3, for example, can circulate at baseline concentrations of up to ~2 mg/ml [6, 32]. Thanks to improvements in inhibitor design (e.g., the use of antibodies and long-acting drug derivatives) and drug administration (e.g., local, subcutaneous), these limitations can often be overcome and improved. In typical treatment protocols, eculizumab is currently administered every other week, with improved next-generation variants being reported to significantly extend the interval between treatments [25]. Other approaches such as the RNAi-mediated knock-down of C5 secretion by the liver using ALN-CC5 may even allow quarterly subcutaneous administration [57]. In this context, it has to be considered that most complement proteins are not only synthesized in the liver but also locally secreted in individual tissues and by immune cells [61, 137], which may affect the efficacy of local and/or systemic complement inhibition. Moreover, potential roles of intracellular complement activation are increasingly discussed and might introduce new aspects and opportunities for focused complement intervention [61].
In many cases, treatment regimens can be tuned in a context-specific manner. Whereas addition of PEG or albumin-binding moieties to compstatin derivatives has generally been shown to increase their plasma residence [81, 138], C3-saturating drug concentrations can even be achieved via subcutaneous administration of the untagged compstatin analog Cp40 [81]. Subcutaneous injection is currently being considered for many biological complement inhibitors and may allow for patient self-administration in chronic diseases such as PNH. The site of injection may also considerably influence the pharmacokinetic profile, as has been seen for local (i.e., intravitreal) versus systemic administration of lampalizumab [136]. The revival of small-molecule inhibitors and antagonists (e.g., ACH-4471, CCX168) makes the advent of the first orally bioavailable complement drugs more likely, and gene-suppressing approaches such as RNAi or antisense oligonucleotides can further change the treatment landscape. Most of these options are still in clinical evaluation, and the results are eagerly awaited, but in any case, the diversity and creativity in the current inhibitory strategies should pave the way to improved treatment options for many complement-mediated disorders.
6. Conclusion and Outlook
Complement-targeted therapy has developed into a very lively area over the past few years. Potent complement inhibitors are now on the market and are increasingly used in approved and off-label indications that demonstrate the potential of controlling complement. It is encouraging to see that several new drug candidates covering a broad spectrum of targets have meanwhile reached phase 2 or even phase 3 trials, and efforts from both academia and biotechnological/pharmaceutical industry suggest that there are other promising concepts and products yet to come. This innovation is crucial, given that our increasing knowledge about complement’s role in highly diverse clinical conditions suggests that there will not be a single target or inhibitor that suits all needs. Hence, the availability of complement-targeted drugs interfering at various levels of the cascade would enable better tailoring of therapeutic strategies and make this approach accessible to a broader spectrum of indications. The experience with AMD or C3G, in which often only a subgroup of patients responded favorably in clinical trials, illustrates that careful stratification of indications and patient cohorts will be critical for avoiding discouraging outcomes and for identifying the patients who may benefit most from complement-targeted therapies. The availability of genetic and/or diagnostic tools will be of high importance in this context, as will continuous elucidation of disease mechanisms. The identification of new and unexpected indications, such as for periodontal disease or neurological disorders, demonstrates the impact of collaborative efforts between academic, clinical, and pharmaceutical science and may open new markets. The molecular, functional, and conceptual diversity of the currently developed clinical candidates suggests broad options concerning selective inhibition, targeting, and drug administration, with potential implications for accessible indications, treatment options, cost, and clinical availability. The results from the ongoing clinical trials are therefore eagerly awaited, with the hope of a soon-to-be extended arsenal of potent complement therapeutics.
Highlights.
Therapeutic complement inhibition offers a promising approach for controlling inflammatory, immune, and degenerative diseases
Complement inhibitors currently in the clinic show high efficacy and good long-term safety but are not applicable in all indications
Several new candidate drugs covering various targets within the complement cascade are in clinical development
Use of complement inhibitors in disease models and in the clinic has revealed several promising indications
Acknowledgments
We thank Dr. Deborah McClellan for her excellent editorial assistance. This work was supported by grants for the National Institutes of Health (AI068730, AI030040) and the National Science Foundation (1423304) and by funding from the European Community’s Seventh Framework Programme, under grant agreement number 602699 (DIREKT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baker BR, Erickson EH. Irreversible enzyme inhibitors. CLII. Proteolytic enzymes. X. Inhibition of guinea pig complement by substituted benzamidines. J Med Chem. 1969;12:408–414. doi: 10.1021/jm00303a016. [DOI] [PubMed] [Google Scholar]
- 2.Muller-Eberhard HJ. Chemistry and reaction mechanisms of complement. Adv Immunol. 1968;8:1–80. doi: 10.1016/s0065-2776(08)60464-2. [DOI] [PubMed] [Google Scholar]
- 3.Lachmann PJ, Smith RA. Taking complement to the clinic–has the time finally come? Scand. J Immunol. 2009;69:471–478. doi: 10.1111/j.1365-3083.2009.02258.x. [DOI] [PubMed] [Google Scholar]
- 4.Sahu A, Lambris JD. Complement inhibitors: a resurgent concept in anti-inflammatory therapeutics. Immunopharmacology. 2000;49:133–148. doi: 10.1016/s0162-3109(00)80299-4. [DOI] [PubMed] [Google Scholar]
- 5.Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol. 2007;25:1265–1275. doi: 10.1038/nbt1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan BP, Harris CL. Complement, a target for therapy in inflammatory and degenerative diseases. Nat Rev Drug Discov. 2015;14:857–877. doi: 10.1038/nrd4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: therapeutic interventions. J Immunol. 2013;190:3839–3847. doi: 10.4049/jimmunol.1203200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement System Part I – Molecular Mechanisms of Activation and Regulation. Front Immunol. 2015;6:262. doi: 10.3389/fimmu.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement System Part II: Role in Immunity. Front Immunol. 2015;6:257. doi: 10.3389/fimmu.2015.00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: pathophysiological mechanisms. J Immunol. 2013;190:3831–3838. doi: 10.4049/jimmunol.1203487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turing offensive. Nature Reviews: Nephrology. 2016 doi: 10.1038/nrneph.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reis ES, Mastellos DC, Yancopoulou D, Risitano AM, Ricklin D, Lambris JD. Applying complement therapeutics to rare diseases. Clin Immunol. 2015;161:225–240. doi: 10.1016/j.clim.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeerleder S. C1-inhibitor: more than a serine protease inhibitor. Semin Thromb Hemost. 2011;37:362–374. doi: 10.1055/s-0031-1276585. [DOI] [PubMed] [Google Scholar]
- 15.Shire’s CINRYZE® (C1 esterase inhibitor [human]) Receives FDA Fast Track Designation for Investigation in the Treatment of Antibody Mediated Rejection (AMR) in Patients Receiving Kidney Transplants. Shire; Oct 13, 2015. Press Release. https://www.shire.com/newsroom/2015/october/shires-cinryze (date accessed: 16 February 2016) [Google Scholar]
- 16.True North Therapeutics Receives Orphan Drug Designation in the European Union for TNT009 for the Treatment of Autoimmune Hemolytic Anemia, including Cold Agglutinin Disease (CAD) True North Therapeutics; Feb 25, 2016. Press Release. http://www.truenorthrx.com/news-events/press-releases/true-north-therapeutics-receives-orphan-drug-designation-european-union-tnt009-treatment-autoimmune-hemolytic-anemia-including-cold-agglutinin-disease-cad/ (date accessed: 28 February 2016) [Google Scholar]
- 17.Omeros Announces Additional Positive Data in OMS721 Phase 2 Clinical Trial. Omeros; Aug 18, 2015. Press Release. http://investor.omeros.com/phoenix.zhtml?c=219263&p=irol-newsArticle_Print&ID=2080177 (date accessed: 16 February 2016) [Google Scholar]
- 18.Boross P, Yildiz C, Simons PJ, Boon L, Hack CE. Altant Conference 2016: Innate Host Defence Utrecht; The Netherlands. A monoclonal antibody against complement C2, as a novel complement inhibitor; 2016. [Google Scholar]
- 19.Jore MM, Johnson S, Sheppard D, Barber NM, Li YI, Nunn MA, et al. Structural basis for therapeutic inhibition of complement C5. Nat Struct Mol Biol. 2016;23:378–386. doi: 10.1038/nsmb.3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol. 2007;25:1256–1264. doi: 10.1038/nbt1344. [DOI] [PubMed] [Google Scholar]
- 21.Mastellos DC, Ricklin D, Yancopoulou D, Risitano A, Lambris JD. Complement in paroxysmal nocturnal hemoglobinuria: exploiting our current knowledge to improve the treatment landscape. Expert Rev Hematol. 2014;7:583–598. doi: 10.1586/17474086.2014.953926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship TH, et al. Atypical aHUS: State of the art. Mol Immunol. 2015;67:31–42. doi: 10.1016/j.molimm.2015.03.246. [DOI] [PubMed] [Google Scholar]
- 23.Coyle D, Cheung MC, Evans GA. Opportunity cost of funding drugs for rare diseases: the cost-effectiveness of eculizumab in paroxysmal nocturnal hemoglobinuria. Med Decis Making. 2014;34:1016–1029. doi: 10.1177/0272989X14539731. [DOI] [PubMed] [Google Scholar]
- 24.Nishimura J, Yamamoto M, Hayashi S, Ohyashiki K, Ando K, Brodsky AL, et al. Genetic variants in C5 and poor response to eculizumab. N Engl J Med. 2014;370:632–639. doi: 10.1056/NEJMoa1311084. [DOI] [PubMed] [Google Scholar]
- 25.Sahelijo L, Mujeebuddin A, Mitchell D, Larouche R, Yu Z-X, Zhang Y, et al. First in Human Single-Ascending Dose Study: Safety, Biomarker, Pharmacokinetics and Exposure-Response Relationships of ALXN1210, a Humanized Monoclonal Antibody to C5, with Marked Half-Life Extension and Potential for Significantly Longer Dosing Intervals. Blood. 2015;126:4777–4777. [Google Scholar]
- 26.Adis Insight. Drug Profile: ALXN 5500. 2015 (Date accessed: 28 February 2016). http://adisinsight.springer.com/drugs/800042004.
- 27.InflaRx announces positive phase IIa top-line results from the SCIENS trial investigating IFX-1, a first-in-class anti-complement C5a antibody. InflaRx; Jan 28, 2016. Press Release. http://www.inflarx.de/dam/Inflarx/News/20160128-press-release-IFX-1_PIIa-SCIENS_ENG_final/20160128_press release_IFX-1_PIIa_SCIENS_ENG_final.pdf (date accessed: 28 February 2016) [Google Scholar]
- 28.Hoehlig K, Maasch C, Shushakova N, Buchner K, Huber-Lang M, Purschke WG, et al. A novel C5a-neutralizing mirror-image (l-)aptamer prevents organ failure and improves survival in experimental sepsis. Mol Ther. 2013;21:2236–2246. doi: 10.1038/mt.2013.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woodruff TM, Nandakumar KS, Tedesco F. Inhibiting the C5-C5a receptor axis. Mol Immunol. 2011;48:1631–1642. doi: 10.1016/j.molimm.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 30.ChemoCentryx Announces Positive Results in Phase II ANCA-Associated Vasculitis CLEAR Trial of Orally Administered Complement 5a Receptor Inhibitor CCX168. ChemoCentryx; Jan 6, 2016. Press Release. http://ir.chemocentryx.com/releasedetail.cfm?releaseid=948998 (date accessed: 26 February 2016) [Google Scholar]
- 31.Zipfel PF, Skerka C, Chen Q, Wiech T, Goodship T, Johnson S, et al. The role of complement in C3 glomerulopathy. Mol Immunol. 2015;67:21–30. doi: 10.1016/j.molimm.2015.03.012. [DOI] [PubMed] [Google Scholar]
- 32.Ricklin D, Lambris JD. Therapeutic control of complement activation at the level of the central component C3. Immunobiology. 2016;221:740–746. doi: 10.1016/j.imbio.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ricklin D. Manipulating the mediator: modulation of the alternative complement pathway C3 convertase in health, disease and therapy. Immunobiology. 2012;217:1057–1066. doi: 10.1016/j.imbio.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paixao-Cavalcante D, Torreira E, Lindorfer MA, Rodriguez de Cordoba S, Morgan BP, Taylor RP, et al. A humanized antibody that regulates the alternative pathway convertase: potential for therapy of renal disease associated with nephritic factors. J Immunol. 2014;192:4844–4851. doi: 10.4049/jimmunol.1303131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katschke KJ, Jr, Stawicki S, Yin J, Steffek M, Xi H, Sturgeon L, et al. Structural and functional analysis of a C3b-specific antibody that selectively inhibits the alternative pathway of complement. J Biol Chem. 2009;284:10473–10479. doi: 10.1074/jbc.M809106200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flohr S, Hommel U, Lorthiois ELJ, Maibaum JK, Ostermann N, Randl SA, et al. Pyrrolidine derivatives and their use as complement pathway modulators. (Patent Application: WO2014002052).Novartis AG. 2014 [Google Scholar]
- 37.Abdel-Magid AF. Factor D Inhibitors for the Treatment of AMD: Patent Highlight. ACS Med Chem Lett. 2012;3:781–782. doi: 10.1021/ml300233n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gavriilaki E, Thanassi JA, Yang G, Yuan X, Huang M, Brodsky RA. Small Molecule Factor D Inhibitors Block Complement Activation in Paroxysmal Nocturnal Hemoglobinuria and Atypical Hemolytic Uremic Syndrome [Abstract] Blood. 2015;126:275–275. doi: 10.3324/haematol.2016.153312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Achillion Initiates Phase 1 Study of ACH-4471, First Orally-Administered Small Molecule Complement Factor D Inhibitor. 2016 Feb 11; Press Release. http://ir.achillion.com/releasedetail.cfm?ReleaseID=954439 (date accessed: 28 February 2016)
- 40.Anderson K, Liao S-M, Maibaum J, Cumin F, Delgado O, Erkenez AD, et al. A novel oral small molecule Factor D inhibitor blocks complement activation in the blood and ocular tissues. ARVO Annual Meeting; Seattle, WA. 2016. [Google Scholar]
- 41.Crowley M, Delgado O, Anderson K, Zheng W, Sellner H, Mainol N, et al. Ocular complement activation and inhibition in rodent models of endotoxin-induced uveitis. ARVO 2016 Annual Meeting; Seattle, WA. 2016. [Google Scholar]
- 42.Johnson L, Splawski I, Baker L, Carrion A, Nguyen A, Twarog M, et al. Generation and characterization of CLG561: a fully-human, anti-properdin Fab for the treatment of age-related macular degeneration. ARVO 2016 Annual Meeting; Seattle, WA. 2016. [Google Scholar]
- 43.Ricklin D, Lambris JD. Progress and trends in complement therapeutics. Adv Exp Med Biol. 2013;735:1–22. doi: 10.1007/978-1-4614-4118-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holers VM, Rohrer B, Tomlinson S. CR2-mediated targeting of complement inhibitors: bench-to-bedside using a novel strategy for site-specific complement modulation. Adv Exp Med Biol. 2013;735:137–154. doi: 10.1007/978-1-4614-4118-2_9. [DOI] [PubMed] [Google Scholar]
- 45.Lachmann PJ, Lay E, Seilly DJ, Buchberger A, Schwaeble W, Khadake J. Further studies of the down-regulation by Factor I of the C3b feedback cycle using endotoxin as a soluble activator and red cells as a source of CR1 on sera of different complotype. Clin Exp Immunol. 2016;183:150–156. doi: 10.1111/cei.12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vogel CW, Finnegan PW, Fritzinger DC. Humanized cobra venom factor: structure, activity, and therapeutic efficacy in preclinical disease models. Mol Immunol. 2014;61:191–203. doi: 10.1016/j.molimm.2014.06.035. [DOI] [PubMed] [Google Scholar]
- 47.Mastellos DC, Yancopoulou D, Kokkinos P, Huber-Lang M, Hajishengallis G, Biglarnia AR, et al. Compstatin: a C3-targeted complement inhibitor reaching its prime for bedside intervention. Eur J Clin Invest. 2015;45:423–440. doi: 10.1111/eci.12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qu H, Ricklin D, Bai H, Chen H, Reis ES, Maciejewski M, et al. New analogs of the clinical complement inhibitor compstatin with subnanomolar affinity and enhanced pharmacokinetic properties. Immunobiology. 2013;218:496–505. doi: 10.1016/j.imbio.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Amyndas’ lead candidate AMY-101 receives orphan drug status from the FDA and the EMA for the treatment of C3 glomerulopathy. Amyndas Pharmaceuticals; Feb 29, 2016. Press Release. http://www.fiercepharma.com/press-releases/amyndas-lead-candidate-amy-101-receives-orphan-drug-status-fda-and-ema-trea (date accessed: 2 March 2016) [Google Scholar]
- 50.Qu H, Ricklin D, Lambris JD. Recent developments in low molecular weight complement inhibitors. Mol Immunol. 2009;47:185–195. doi: 10.1016/j.molimm.2009.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Melis JP, Strumane K, Ruuls SR, Beurskens FJ, Schuurman J, Parren PW. Complement in therapy and disease: Regulating the complement system with antibody-based therapeutics. Mol Immunol. 2015;67:117–130. doi: 10.1016/j.molimm.2015.01.028. [DOI] [PubMed] [Google Scholar]
- 52.Katschke KJ, Jr, Wu P, Ganesan R, Kelley RF, Mathieu MA, Hass PE, et al. Inhibiting alternative pathway complement activation by targeting the factor D exosite. J Biol Chem. 2012;287:12886–12892. doi: 10.1074/jbc.M112.345082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roguska M, Splawski I, Diefenbach-Streiber B, Dolan E, Etemad-Gilbertson B, Rondeau J-M, et al. Generation and Characterization of LFG316, A Fully-Human Anti-C5 Antibody for the Treatment of Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci. 2014;55:3433–3433. [Google Scholar]
- 54.Sobi report for the second quarter 2015. Sobi; Jul 16, 2015. Press Release. http://hugin.info/134557/R/1939169/699553.pdf (date accessed: 28 February 2016) [Google Scholar]
- 55.Hepburn NJ, Williams AS, Nunn MA, Chamberlain-Banoub JC, Hamer J, Morgan BP, et al. In vivo characterization and therapeutic efficacy of a C5-specific inhibitor from the soft tick Ornithodoros moubata. J Biol Chem. 2007;282:8292–8299. doi: 10.1074/jbc.M609858200. [DOI] [PubMed] [Google Scholar]
- 56.Pharmaceuticals Announces Ra. Initiation of First-in-Human Clinical Trial of Novel Complement C5 Inhibitor. Ra Pharmaceuticals; Dec 2, 2015. Press Release. http://www.rapharma.com/global/downloads/Ra-Pharma-PR-120115-Phase-1-ASH-FINAL.pdf (date accessed: 2 March 2016) [Google Scholar]
- 57.Najaan N, Hill A, Taubel J, Bush J, Borodovsky A, Kawahata N, et al. A Subcutaneously Administered Investigational RNAi Therapeutic (ALN-CC5) Targeting Complement C5 For Treatment Of PHN And Complement-Mediated Diseases: Interim Phase 1 Study Results. 53rd ERA-EDTA Congress; Vienna, Austria. 2016. [Google Scholar]
- 58.Updated Healthy Volunteer Data from Phase 1/2 Clinical Study with ALN-CC5 for the Treatment of Complement-Mediated Diseases. Alnylam Pharmaceuticals; May 22, 2016. Press Release. http://www.alnylam.com/capella/presentations/updated-healthy-volunteer-data-from-aln-cc5/ (date accessed: 24 May 2016) [Google Scholar]
- 59.Grossman TR, Hettrick LA, Johnson RB, Hung G, Peralta R, Watt A, et al. Inhibition of the alternative complement pathway by antisense oligonucleotides targeting complement factor B improves lupus nephritis in mice. Immunobiology. 2015 doi: 10.1016/j.imbio.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 60.Grossman TR. Complement: driver of inflammation and therapeutic target in diverse diseases. Chicheley Hall, UK: Royal Society; 2015. Antisense therapies targeting complement in renal disease. [Google Scholar]
- 61.Kolev M, Le Friec G, Kemper C. Complement–tapping into new sites and effector systems. Nat Rev Immunol. 2014;14:811–820. doi: 10.1038/nri3761. [DOI] [PubMed] [Google Scholar]
- 62.Zhang Y, Nester CM, Holanda DG, Marsh HC, Hammond RA, Thomas LJ, et al. Soluble CR1 therapy improves complement regulation in C3 glomerulopathy. J Am Soc Nephrol. 2013;24:1820–1829. doi: 10.1681/ASN.2013010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Celldex Reports Fiscal 2013 Business/Financial Results and Outlines 2014 Strategy. Celldex Therapeutics; Mar 3, 2014. Press Release. http://files.shareholder.com/downloads/ABEA-39HH7S/1545539254x0x730319/9BAC77CA-DAAD-4F55-8231-77E4BF9C6140/CLDX_News_2014_3_3_General_Releases.pdf (date accessed: 16 February 2016) [Google Scholar]
- 64.Xiao F, Ma L, Zhao M, Smith RA, Huang G, Jones PM, et al. APT070 (mirococept), a membrane-localizing C3 convertase inhibitor, attenuates early human islet allograft damage in vitro and in vivo in a humanized mouse model. Br J Pharmacol. 2016;173:575–587. doi: 10.1111/bph.13388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Patel H, Smith RA, Sacks SH, Zhou W. Therapeutic strategy with a membrane-localizing complement regulator to increase the number of usable donor organs after prolonged cold storage. J Am Soc Nephrol. 2006;17:1102–1111. doi: 10.1681/ASN.2005101116. [DOI] [PubMed] [Google Scholar]
- 66.Hebecker M, Alba-Dominguez M, Roumenina LT, Reuter S, Hyvarinen S, Dragon-Durey MA, et al. An engineered construct combining complement regulatory and surface-recognition domains represents a minimal-size functional factor H. J Immunol. 2013;191:912–921. doi: 10.4049/jimmunol.1300269. [DOI] [PubMed] [Google Scholar]
- 67.Schmidt CQ, Bai H, Lin Z, Risitano AM, Barlow PN, Ricklin D, et al. Rational engineering of a minimized immune inhibitor with unique triple-targeting properties. J Immunol. 2013;190:5712–5721. doi: 10.4049/jimmunol.1203548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schmidt CQ, Harder MJ, Nichols EM, Hebecker M, Anliker M, Hochsmann B, et al. Selectivity of C3-opsonin targeted complement inhibitors: A distinct advantage in the protection of erythrocytes from paroxysmal nocturnal hemoglobinuria patients. Immunobiology. 2016;221:503–511. doi: 10.1016/j.imbio.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fridkis-Hareli M, Storek M, Mazsaroff I, Risitano AM, Lundberg AS, Horvath CJ, et al. Design and development of TT30, a novel C3d-targeted C3/C5 convertase inhibitor for treatment of human complement alternative pathway-mediated diseases. Blood. 2011;118:4705–4713. doi: 10.1182/blood-2011-06-359646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Risitano AM, Storek M, Sahelijo L, Doyle M, Dai Y, Weitz IC, et al. Safety and Pharmacokinetics of the Complement Inhibitor TT30 in a Phase I Trial for Untreated PNH Patients [Abstract]. ASH 57th Annual Meeting & Exposition; Orlando, FL. 2015. [Google Scholar]
- 71.Holers M, Banda N, Mehta G, Fridkis-Hareli M, Or E, Storek M, et al. The human complement receptor type 2 (CR2)/CR1 fusion protein TT32, a targeted inhibitor of the classical and alternative pathway C3 convertases, prevents arthritis in active immunization and passive transfer models and acts by CR2-dependent targeting of CR1 regulatory activity. Immunobiology. 2012;217:1210–1210. [Google Scholar]
- 72.Ruseva MM, Ramaglia V, Morgan BP, Harris CL. An anticomplement agent that homes to the damaged brain and promotes recovery after traumatic brain injury in mice. Proc Natl Acad Sci U S A. 2015;112:14319–14324. doi: 10.1073/pnas.1513698112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nordmaj MA, Munthe-Fog L, Hein E, Skjoedt MO, Garred P. Genetically engineered fusion of MAP-1 and factor H domains 1–5 generates a potent dual upstream inhibitor of both the lectin and alternative complement pathways. FASEB J. 2015;29:4945–4955. doi: 10.1096/fj.15-277103. [DOI] [PubMed] [Google Scholar]
- 74.Nilsson PH, Ekdahl KN, Magnusson PU, Qu H, Iwata H, Ricklin D, et al. Autoregulation of thromboinflammation on biomaterial surfaces by a multicomponent therapeutic coating. Biomaterials. 2013;34:985–994. doi: 10.1016/j.biomaterials.2012.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu YQ, Qu H, Sfyroera G, Tzekou A, Kay BK, Nilsson B, et al. Protection of nonself surfaces from complement attack by factor H-binding peptides: implications for therapeutic medicine. J Immunol. 2011;186:4269–4277. doi: 10.4049/jimmunol.1003802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol. 2008;6:132–142. doi: 10.1038/nrmicro1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Georgoutsou-Spyridonos M, Ricklin D, Pratsinis H, Perivolioti E, Pirmettis I, Garcia BL, et al. Attenuation of Staphylococcus aureus-Induced Bacteremia by Human Mini-Antibodies Targeting the Complement Inhibitory Protein Efb. J Immunol. 2015;195:3946–3958. doi: 10.4049/jimmunol.1500966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.van der Veen S, Johnson S, Jongerius I, Malik T, Genovese A, Santini L, et al. Nonfunctional variant 3 factor H binding proteins as meningococcal vaccine candidates. Infect Immun. 2014;82:1157–1163. doi: 10.1128/IAI.01183-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.de Cordoba SR, Tortajada A, Harris CL, Morgan BP. Complement dysregulation and disease: from genes and proteins to diagnostics and drugs. Immunobiology. 2012;217:1034–1046. doi: 10.1016/j.imbio.2012.07.021. [DOI] [PubMed] [Google Scholar]
- 80.DeZern AE, Uknis M, Yuan X, Mukhina GL, Varela J, Saye J, et al. Complement blockade with a C1 esterase inhibitor in paroxysmal nocturnal hemoglobinuria. Exp Hematol. 2014;42:857–861 e851. doi: 10.1016/j.exphem.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Risitano AM, Ricklin D, Huang Y, Reis ES, Chen H, Ricci P, et al. Peptide inhibitors of C3 activation as a novel strategy of complement inhibition for the treatment of paroxysmal nocturnal hemoglobinuria. Blood. 2014;123:2094–2101. doi: 10.1182/blood-2013-11-536573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nester CM, Brophy PD. Eculizumab in the treatment of atypical haemolytic uraemic syndrome and other complement-mediated renal diseases. Curr Opin Pediatr. 2013;25:225–231. doi: 10.1097/MOP.0b013e32835df4a3. [DOI] [PubMed] [Google Scholar]
- 83.Noris M, Mescia F, Remuzzi G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol. 2012;8:622–633. doi: 10.1038/nrneph.2012.195. [DOI] [PubMed] [Google Scholar]
- 84.Zhang Y, Shao D, Ricklin D, Hilkin BM, Nester CM, Lambris JD, et al. Compstatin analog Cp40 inhibits complement dysregulation in vitro in C3 glomerulopathy. Immunobiology. 2015;220:993–998. doi: 10.1016/j.imbio.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mojcik CF, Kremer J, Bingham C, Burch F, Vitiello S, McCroskery E, et al. Results of a phase 2B study of the humanized anti-C5 antibody eculizumab in patients with rheumatoid arthritis. Ann Rheum Dis. 2004;63:301–301. [Google Scholar]
- 86.Vergunst CE, Gerlag DM, Dinant H, Schulz L, Vinkenoog M, Smeets TJ, et al. Blocking the receptor for C5a in patients with rheumatoid arthritis does not reduce synovial inflammation. Rheumatology (Oxford) 2007;46:1773–1778. doi: 10.1093/rheumatology/kem222. [DOI] [PubMed] [Google Scholar]
- 87.Holers VM. Targeting mechanisms at sites of complement activation for imaging and therapy. Immunobiology. 2015 doi: 10.1016/j.imbio.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sacks SH, Zhou W. The role of complement in the early immune response to transplantation. Nat Rev Immunol. 2012;12:431–442. doi: 10.1038/nri3225. [DOI] [PubMed] [Google Scholar]
- 89.Fremeaux-Bacchi V, Legendre CM. The emerging role of complement inhibitors in transplantation. Kidney Int. 2015;88:967–973. doi: 10.1038/ki.2015.253. [DOI] [PubMed] [Google Scholar]
- 90.Morgan BP. The role of complement in neurological and neuropsychiatric diseases. Expert Rev Clin Immunol. 2015;11:1109–1119. doi: 10.1586/1744666X.2015.1074039. [DOI] [PubMed] [Google Scholar]
- 91.Chen Song S, Zhong S, Xiang Y, Li JH, Guo H, Wang WY, et al. Complement inhibition enables renal allograft accommodation and long-term engraftment in presensitized nonhuman primates. Am J Transplant. 2011;11:2057–2066. doi: 10.1111/j.1600-6143.2011.03646.x. [DOI] [PubMed] [Google Scholar]
- 92.Biglarnia AR, Ekdahl KN, Nilsson B. Complement Interception Across Humoral Incompatibility in Solid Organ Transplantation: A Clinical Perspective. Adv Exp Med Biol. 2015;865:211–233. doi: 10.1007/978-3-319-18603-0_13. [DOI] [PubMed] [Google Scholar]
- 93.Cooper DK, Ekser B, Tector AJ. Immunobiological barriers to xenotransplantation. Int J Surg. 2015;23:211–216. doi: 10.1016/j.ijsu.2015.06.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.DeAngelis RA, Reis ES, Ricklin D, Lambris JD. Targeted complement inhibition as a promising strategy for preventing inflammatory complications in hemodialysis. Immunobiology. 2012;217:1097–1105. doi: 10.1016/j.imbio.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ekdahl KN, Lambris JD, Elwing H, Ricklin D, Nilsson PH, Teramura Y, et al. Innate immunity activation on biomaterial surfaces: a mechanistic model and coping strategies. Adv Drug Deliv Rev. 2011;63:1042–1050. doi: 10.1016/j.addr.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Reis ES, DeAngelis RA, Chen H, Resuello RR, Ricklin D, Lambris JD. Therapeutic C3 inhibitor Cp40 abrogates complement activation induced by modern hemodialysis filters. Immunobiology. 2015;220:476–482. doi: 10.1016/j.imbio.2014.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Szebeni J. Complement activation-related pseudoallergy: a stress reaction in blood triggered by nanomedicines and biologicals. Mol Immunol. 2014;61:163–173. doi: 10.1016/j.molimm.2014.06.038. [DOI] [PubMed] [Google Scholar]
- 98.Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.van Lookeren Campagne M, Strauss EC, Yaspan BL. Age-related macular degeneration: Complement in action. Immunobiology. 2015 doi: 10.1016/j.imbio.2015.11.007. [DOI] [PubMed] [Google Scholar]
- 100.Regillo CD, Yaspan BL, Li Z, Dressen A, van Lookeren Campagne M, Graham R, et al. On behalf of the MAHALO Study Investigators Lampalizumab (Anti-factor D) in Patients with Geographic Atrophy: the MAHALO Phase II Results. 117th Annual AAO meeting; New Orleans, LA. 2013. [Google Scholar]
- 101.Harris CL, Heurich M, Rodriguez de Cordoba S, Morgan BP. The complotype: dictating risk for inflammation and infection. Trends Immunol. 2012;33:513–521. doi: 10.1016/j.it.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hovland A, Jonasson L, Garred P, Yndestad A, Aukrust P, Lappegard KT, et al. The complement system and toll-like receptors as integrated players in the pathophysiology of atherosclerosis. Atherosclerosis. 2015;241:480–494. doi: 10.1016/j.atherosclerosis.2015.05.038. [DOI] [PubMed] [Google Scholar]
- 103.Fonseca MI, Ager RR, Chu SH, Yazan O, Sanderson SD, LaFerla FM, et al. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J Immunol. 2009;183:1375–1383. doi: 10.4049/jimmunol.0901005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 105.Brennan FH, Lee JD, Ruitenberg MJ, Woodruff TM. Therapeutic targeting of complement to modify disease course and improve outcomes in neurological conditions. Semin Immunol. 2016 doi: 10.1016/j.smim.2016.03.015. [DOI] [PubMed] [Google Scholar]
- 106.Barratt-Due A, Pischke SE, Brekke OL, Thorgersen EB, Nielsen EW, Espevik T, et al. Bride and groom in systemic inflammation–the bells ring for complement and Toll in cooperation. Immunobiology. 2012;217:1047–1056. doi: 10.1016/j.imbio.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 107.Silasi-Mansat R, Zhu H, Georgescu C, Popescu N, Keshari RS, Peer G, et al. Complement inhibition decreases early fibrogenic events in the lung of septic baboons. J Cell Mol Med. 2015 doi: 10.1111/jcmm.12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Silasi-Mansat R, Zhu H, Popescu NI, Peer G, Sfyroera G, Magotti P, et al. Complement inhibition decreases the procoagulant response and confers organ protection in a baboon model of Escherichia coli sepsis. Blood. 2010;116:1002–1010. doi: 10.1182/blood-2010-02-269746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Barratt-Due A, Thorgersen EB, Egge K, Pischke S, Sokolov A, Hellerud BC, et al. Combined inhibition of complement C5 and CD14 markedly attenuates inflammation, thrombogenicity, and hemodynamic changes in porcine sepsis. J Immunol. 2013;191:819–827. doi: 10.4049/jimmunol.1201909. [DOI] [PubMed] [Google Scholar]
- 110.Huber-Lang M, Kovtun A, Ignatius A. The role of complement in trauma and fracture healing. Semin Immunol. 2013;25:73–78. doi: 10.1016/j.smim.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 111.Rafail S, Kourtzelis I, Foukas PG, Markiewski MM, DeAngelis RA, Guariento M, et al. Complement deficiency promotes cutaneous wound healing in mice. J Immunol. 2015;194:1285–1291. doi: 10.4049/jimmunol.1402354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Brennan FH, Gordon R, Lao HW, Biggins PJ, Taylor SM, Franklin RJ, et al. The Complement Receptor C5aR Controls Acute Inflammation and Astrogliosis following Spinal Cord Injury. J Neurosci. 2015;35:6517–6531. doi: 10.1523/JNEUROSCI.5218-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hajishengallis G, Abe T, Maekawa T, Hajishengallis E, Lambris JD. Role of complement in host-microbe homeostasis of the periodontium. Semin Immunol. 2013;25:65–72. doi: 10.1016/j.smim.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hajishengallis G, Maekawa T, Abe T, Hajishengallis E, Lambris JD. Complement Involvement in Periodontitis: Molecular Mechanisms and Rational Therapeutic Approaches. Adv Exp Med Biol. 2015;865:57–74. doi: 10.1007/978-3-319-18603-0_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Maekawa T, Abe T, Hajishengallis E, Hosur KB, DeAngelis RA, Ricklin D, et al. Genetic and intervention studies implicating complement C3 as a major target for the treatment of periodontitis. J Immunol. 2014;192:6020–6027. doi: 10.4049/jimmunol.1400569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Abe T, Hosur KB, Hajishengallis E, Reis ES, Ricklin D, Lambris JD, et al. Local complement-targeted intervention in periodontitis: proof-of-concept using a C5a receptor (CD88) antagonist. J Immunol. 2012;189:5442–5448. doi: 10.4049/jimmunol.1202339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Maekawa T, Briones RA, Resuello RR, Tuplano JV, Hajishengallis E, Kajikawa T, et al. Inhibition of pre-existing natural periodontitis in non-human primates by a locally administered peptide inhibitor of complement C3. J Clin Periodontol. 2016 doi: 10.1111/jcpe.12507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chehoud C, Rafail S, Tyldsley AS, Seykora JT, Lambris JD, Grice EA. Complement modulates the cutaneous microbiome and inflammatory milieu. Proc Natl Acad Sci U S A. 2013;110:15061–15066. doi: 10.1073/pnas.1307855110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Taylor RP, Lindorfer MA. The role of complement in mAb-based therapies of cancer. Methods. 2014;65:18–27. doi: 10.1016/j.ymeth.2013.07.027. [DOI] [PubMed] [Google Scholar]
- 120.Mamidi S, Hone S, Kirschfink M. The complement system in cancer: Ambivalence between tumour destruction and promotion. Immunobiology. 2015 doi: 10.1016/j.imbio.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 121.Pio R, Ajona D, Lambris JD. Complement inhibition in cancer therapy. Semin Immunol. 2013;25:54–64. doi: 10.1016/j.smim.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, et al. Modulation of the antitumor immune response by complement. Nat Immunol. 2008;9:1225–1235. doi: 10.1038/ni.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vadrevu SK, Chintala NK, Sharma SK, Sharma P, Cleveland C, Riediger L, et al. Complement c5a receptor facilitates cancer metastasis by altering T-cell responses in the metastatic niche. Cancer Res. 2014;74:3454–3465. doi: 10.1158/0008-5472.CAN-14-0157. [DOI] [PubMed] [Google Scholar]
- 124.R ES, Falcao DA, Isaac L. Clinical aspects and molecular basis of primary deficiencies of complement component C3 and its regulatory proteins factor I and factor H. Scand J Immunol. 2006;63:155–168. doi: 10.1111/j.1365-3083.2006.01729.x. [DOI] [PubMed] [Google Scholar]
- 125.Cagliani R, Forni D, Filippi G, Mozzi A, De Gioia L, Pontremoli C, et al. The mammalian complement system as an epitome of host-pathogen genetic conflicts. Mol Ecol. 2016 doi: 10.1111/mec.13558. [DOI] [PubMed] [Google Scholar]
- 126.Berends ET, Mohan S, Miellet WR, Ruyken M, Rooijakkers SH. Contribution of the complement Membrane Attack Complex to the bactericidal activity of human serum. Mol Immunol. 2015;65:328–335. doi: 10.1016/j.molimm.2015.01.020. [DOI] [PubMed] [Google Scholar]
- 127.Macedo AC, Isaac L. Systemic Lupus Erythematosus and Deficiencies of Early Components of the Complement Classical Pathway. Front Immunol. 2016;7:55. doi: 10.3389/fimmu.2016.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Scott D, Botto M. The paradoxical roles of C1q and C3 in autoimmunity. Immunobiology. 2015 doi: 10.1016/j.imbio.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 129.Wu MA, Zanichelli A, Mansi M, Cicardi M. Current treatment options for hereditary angioedema due to C1 inhibitor deficiency. Expert Opin Pharmacother. 2016;17:27–40. doi: 10.1517/14656566.2016.1104300. [DOI] [PubMed] [Google Scholar]
- 130.Risitano AM. Paroxysmal Nocturnal Hemoglobinuria in the era of complement inhibition. Am J Hematol. 2016 doi: 10.1002/ajh.24323. [DOI] [PubMed] [Google Scholar]
- 131.Loschi M, Porcher R, Barraco F, Terriou L, Mohty M, de Guibert S, et al. Impact of eculizumab treatment on paroxysmal nocturnal hemoglobinuria: A treatment versus no-treatment study. Am J Hematol. 2015 doi: 10.1002/ajh.24278. [DOI] [PubMed] [Google Scholar]
- 132.Hillmen P, Muus P, Szer J, Hill A, Hochsmann B, Kulasekararaj A, et al. Assessment of human antihuman antibodies to eculizumab after long-term treatment in patients with paroxysmal nocturnal hemoglobinuria. Am J Hematol. 2016;91:E16–17. doi: 10.1002/ajh.24280. [DOI] [PubMed] [Google Scholar]
- 133.Hillmen P, Muus P, Roth A, Elebute MO, Risitano AM, Schrezenmeier H, et al. Long-term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2013;162:62–73. doi: 10.1111/bjh.12347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang J, Wang L, Xiang Y, Ricklin D, Lambris JD, Chen G. Using an in vitro xenoantibody-mediated complement-dependent cytotoxicity model to evaluate the complement inhibitory activity of the peptidic C3 inhibitor Cp40. Clin Immunol. 2016;162:37–44. doi: 10.1016/j.clim.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hamad OA, Mitroulis I, Fromell K, Kozarcanin H, Chavakis T, Ricklin D, et al. Contact activation of C3 enables tethering between activated platelets and polymorphonuclear leukocytes via CD11b/CD18. Thromb Haemost. 2015;114:1207–1217. doi: 10.1160/TH15-02-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Loyet KM, Good J, Davancaze T, Sturgeon L, Wang X, Yang J, et al. Complement inhibition in cynomolgus monkeys by anti-factor d antigen-binding fragment for the treatment of an advanced form of dry age-related macular degeneration. J Pharmacol Exp Ther. 2014;351:527–537. doi: 10.1124/jpet.114.215921. [DOI] [PubMed] [Google Scholar]
- 137.Marsh JE, Zhou W, Sacks SH. Local tissue complement synthesis–fine tuning a blunt instrument. Arch Immunol Ther Exp (Warsz. 2001;49(Suppl 1):S41–46. [PubMed] [Google Scholar]
- 138.Huang Y, Reis ES, Knerr PJ, van der Donk WA, Ricklin D, Lambris JD. Conjugation to albumin-binding molecule tags as a strategy to improve both efficacy and pharmacokinetic properties of the complement inhibitor compstatin. ChemMedChem. 2014;9:2223–2226. doi: 10.1002/cmdc.201402212. [DOI] [PMC free article] [PubMed] [Google Scholar]