

Abstract
BACKGROUND
Familial combined hypolipidemia, a Mendelian condition characterized by substantial reductions in all 3 major lipid fractions, is caused by mutations that inactivate the gene angiopoietin-like 3 (ANGPTL3). Whether ANGPTL3 deficiency reduces risk of coronary artery disease (CAD) is unknown.
OBJECTIVES
The study goal was to leverage 3 distinct lines of evidence – a family that included individuals with complete (compound heterozygote) ANGPTL3 deficiency, a population based-study of humans with partial (heterozygote) ANGPTL3 deficiency, and biomarker levels in myocardial infarction (MI) patients – to test if ANGPTL3 deficiency is associated with lower risk for CAD.
METHODS
We assessed coronary atherosclerotic burden in 3 individuals with complete ANGPTL3 deficiency and 3 wild-type first-degree relatives using computed tomography angiography. In the population, ANGPTL3 loss-of-function (LOF) mutations were ascertained in up to 21,980 individuals with CAD and 158,200 controls. LOF mutations were defined as nonsense, frameshift, and splice-site variants, along with missense variants resulting in <25% of wild-type ANGPTL3 activity in a mouse model. In a biomarker study, circulating ANGPTL3 concentration was measured in 1,493 individuals presenting with MI and 3,232 controls.
RESULTS
The 3 individuals with complete ANGPTL3 deficiency showed no evidence of coronary atherosclerotic plaque. ANGPTL3 gene sequencing demonstrated that approximately 1 in 309 individuals was a heterozygous carrier for an LOF mutation. Compared to those without mutation, heterozygous carriers of ANGPTL3 LOF mutations demonstrated a 17% reduction in circulating triglycerides and a 12% reduction in low-density lipoprotein cholesterol. Carrier status was associated with a 34% reduction in odds of CAD (odds ratio: 0.66; 95% confidence interval: 0.44 to 0.98; p = 0.04). Individuals in the lowest tertile of circulating ANGPTL3 concentrations, compared with the highest, had reduced odds of MI (adjusted odds ratio: 0.65; 95% confidence interval: 0.55 to 0.77; p < 0.001).
CONCLUSIONS
ANGPTL3 deficiency is associated with protection from CAD.
Keywords: human genetics, loss of function mutations, myocardial infarction
Loss-of-function (LOF) mutations leading to complete deficiency of angiopoietin-like 3 (ANGPTL3) cause familial combined hypolipidemia, a Mendelian disorder characterized by low circulating concentrations of low-density lipoprotein (LDL) cholesterol, high-density lipoprotein (HDL) cholesterol, and triglycerides (TG) (1). ANGPTL3 is a hepatically-secreted protein first identified via positional cloning of a hypolipidemic mouse strain (2). ANGPTL3 acts as a potent inhibitor of lipoprotein lipase (LPL), the primary mechanism by which triglyceride-rich lipoproteins are cleared from the circulation (3). In addition, ANGPTL3 is an endogenous inhibitor of endothelial lipase (EL) (4). Loss of ANGPTL3 function appears to decrease triglyceride-rich lipoprotein and HDL cholesterol concentrations through loss of LPL and EL inhibition, respectively. The mechanism by which ANGPTL3 regulates LDL cholesterol metabolism remains unclear (5). The seemingly favorable implications of ANGPTL3 deficiency in reducing TG concentrations and circulating LDL cholesterol catalyzed drug development programs aiming to inhibit ANGPTL3 using either a monoclonal antibody (5) or an antisense oligonucleotide (6).
Although decreased atherosclerotic burden was observed in Angptl3 knockout mice (7), the relationship of ANGPTL3 deficiency to coronary artery disease (CAD) in humans remains uncertain. Individuals who carry LOF mutations in ANGPTL3 have lifelong reductions of circulating ANGPTL3 (8); as such, the clinical phenotypes of these individuals may inform the potential therapeutic efficacy of pharmacological ANGPTL3 inhibition.
Here, we tested the hypothesis that ANGPTL3 deficiency reduces risk of CAD in humans. We compared coronary atherosclerotic plaque burden in individuals who had complete ANGPTL3 deficiency (caused by compound heterozygous LOF mutations in ANGPTL3) with wild-type first-degree relatives. Next, we examined the coding regions of ANGPTL3 in up to 180,180 individuals, identified those who carried LOF mutations in this gene with the aid of mouse models, and determined whether those mutations were associated with a lower risk of CAD. Finally, we measured circulating ANGPTL3 concentrations in individuals presenting with a first-ever myocardial infarction (MI) and compared them to concentrations in individuals without MI.
METHODS
The original kindred used to map ANGPTL3 as a cause of familial combined hypolipidemia was recruited in the Lipid Research Clinic at the Washington University School of Medicine between 1994 and 1997. We recontacted all individuals from the kindred who inherited compound heterozygous LOF mutations in ANGPTL3 and invited them to participate in the current study. Three of the 4 compound heterozygous carriers (individuals II-1, II-2, and II-4 in Online Figure 1) were available to participate and were matched to 3 first-degree relatives who did not carry any ANGPTL3 LOF mutation (individuals II-8, II-7, and II-10 in Online Figure 1, respectively). Fasting laboratory values, including plasma lipids, were measured in all participants using standard clinical assays. Coronary computed tomography angiography (CCTA) was used to quantify coronary artery calcification and atherosclerotic plaque burden. Details of the image acquisition and post-acquisition processing are included in the Online Appendix. The Washington University School of Medicine Institutional Review Board approved all study protocols.
ASCERTAINMENT OF LOF MUTATIONS IN ANGPTL3
We identified carriers of an LOF mutation in ANGPTL3 using previously generated exome sequencing data from 9 case-control studies of the Myocardial Infarction Genetics Consortium. These included the ATVB (Italian Atherosclerosis Thrombosis and Vascular Biology) study (9), the ESP-EOMI (Exome Sequencing Project Early-Onset Myocardial Infarction) study (10), the South German Myocardial Infarction study (11), OHS (Ottawa Heart Study) (12), PROCARDIS (Precocious Coronary Artery Disease Study) (13), PROMIS (Pakistan Risk of Myocardial Infarction Study) (14), the Registre Gironi del COR (Gerona Heart Registry or REGICOR) study (15), the BHF-FHS (British Heart Foundation Family Heart Study) (16), and the Lubeck Myocardial Infarction study (16). Furthermore, we extracted ANGPTL3 sequence data from exome sequencing performed in the Jackson Heart Study (17), the BioImage study (18), and the ARIC (Atherosclerosis Risk in Communities) population-based cohort study (19) in addition to targeted sequencing in the Duke CATHGEN case-control study (20). Loss-of-function mutations included those leading to truncation via a premature stop codon (nonsense), insertions or deletions that scramble protein translation beyond the variant site (frameshift), or point mutations at sites of pre-messenger ribonucleic acid splicing that alter the splicing process (splice-site). Additional data for rs372257803, an intronic splice region variant in ANGPTL3 previously linked to significantly reduced circulating triglyceride levels (21), was obtained using high-quality genotype imputation in the United Kingdom Biobank (22), the PennCath study (23), and the Wellcome Trust Case Control Consortium Coronary Artery Disease study (24). All variant positions were based on the ANGPTL3 canonical transcript (ENST00000371129). Additional details on gene sequencing, the imputation of rs372257803, and study cohorts are included in the Online Appendix.
FUNCTIONAL VALIDATION OF MISSENSE VARIANTS IN ANGPTL3 LEADING TO LOF
Beyond the mutations that lead to LOF due to nonsense, frameshift, or splice-site disruption, studies based on evolutionary conservation suggested that about 20% of all missense mutations lead to severe decrements in protein function (25,26). We sought to experimentally define such variants in ANGPTL3 using a mouse model. Rare (minor allele frequency <1%) missense variants were prioritized if they were: 1) predicted to be damaging or possibly damaging by each of 5 in silico prediction algorithms (LRT score, MutationTaster, PolyPhen-2 HumDiv, PolyPhen-2 HumVar, and Sorting Intolerant From Tolerant); and 2) present in at least 2 sequenced individuals of the Myocardial Infarction Genetics Consortium cohorts.
For the set of ANGPTL3 missense variants identified previously, the functional significance of each variant was determined using adenoviral vectors developed to reconstitute the expression of the human ANGPTL3 ortholog in the livers of Angptl3 knockout mice. Vectors were engineered to contain either the wild-type ANGPTL3 gene or the missense variant of interest. Missense variants were annotated as LOF if they conferred <25% of wild-type activity as assessed by percent change in circulating TG levels and percent change in circulating cholesterol levels induced by expression. Additional details are described in the Online Appendix.
ANGPTL3 PLASMA CONCENTRATION
Using a previously validated enzyme-linked immunosorbent assay (Biovendor, Prague, Czech Republic) (27), plasma ANGPTL3 concentrations were measured in individuals from PROMIS (14), a study that included cases presenting with a first-ever MI and controls free of MI.
STATISTICAL ANALYSES
For changes in circulating triglyceride and total cholesterol levels in the mouse models, normality of data was assessed with the Shapiro-Wilk test and equality of variance was assessed using the F-test; p values were calculated using 2-sided paired samples t tests. The association of ANGPTL3 LOF mutations, analyzed in aggregate, with total cholesterol, LDL cholesterol, HDL cholesterol, and log-transformed TGs was assessed using linear regression with adjustment for the covariates of age, age squared, sex, study cohort, CAD status, and the first 5 principal components of ancestry. We accounted for the effect of lipid-lowering therapy in participants reporting such use at the time of lipid measurement by dividing the measured total cholesterol and LDL cholesterol by 0.8 and 0.7, respectively (28,29); HDL cholesterol and triglyceride values were not adjusted. The association of ANGPTL3 mutations with risk of CAD was determined via meta-analysis utilizing Cochran–Mantel–Haenszel statistics for stratified 2-by-2 tables as implemented previously (30). In calculating the study-specific odds ratio of disease, an adjustment of 0.5 was added to all counts in studies with zero mutation carriers in cases or controls. The association of circulating plasma ANGPTL3 concentration with MI was determined using multivariable logistic regression after stratification of the population into tertiles of ANGPTL3 concentration. Statistical analyses were performed using R version 3.2.2 software (The R Project for Statistical Computing, Vienna, Austria).
RESULTS
From the original kindred used to map this gene as a cause of familial combined hypolipidemia (1), we studied 3 individuals with complete ANGPTL3 deficiency due to compound heterozygous LOF mutations in ANGPTL3 and 3 matched first-degree relatives without an LOF ANGPTL3 mutation. As shown in Online Table 1, participants with complete ANGPTL3 deficiency continued to exhibit very low plasma lipid concentrations nearly 20 years after the initial report. An updated medical history was obtained. One participant with complete ANGPTL3 deficiency reported a history of type 2 diabetes mellitus, hypertension, and past tobacco use. Other characteristics and laboratory values are listed in Online Table 1. We performed CCTA in all 6 individuals. The coronary calcium score was 0 Agatston units (AU) for all participants with complete ANGPTL3 deficiency (Figure 1A). By contrast, 2 of the 3 matched controls had positive coronary calcium scores (6 AU for individual II-8 and 610 AU for individual II-7) (Figure 1B).
FIGURE 1. Coronary CT Angiography in Humans With and Without Complete ANGPTL3 Deficiency.



Representative sagittal computed tomography (CT) angiogram images show the right coronary artery (arrow) in (A) an individual with complete angiopoietin-like 3 (ANGPTL3) deficiency and (B) a matched first-degree relative without an ANGPTL3 mutation demonstrating calcified (open triangle) and noncalcified (closed triangle) plaque. (C) Total plaque burden representing the percentage of the coronary system affected by atherosclerosis is plotted by ANGPTL3 deficiency status.
We next calculated total plaque burden (a combination of both calcified and noncalcified plaque) for each participant. The total plaque burden was lower in the participants with complete ANGPTL3 deficiency (mean = 0%) compared to controls (mean = 39%) (Figure 1C, Online Table 1, and Online Figure 2). The small number of phenotyped individuals precluded robust statistical comparisons between groups.
ASCERTAINMENT OF ANGPTL3 LOF MUTATIONS
We next sought to characterize the clinical effects of ANGPTL3 LOF mutations in the population. Sequence data for ANGPTL3 were available in 13,914 individuals with CAD and 26,198 controls free of CAD. From these data, 21 LOF variants were identified, including 7 variants leading to premature stop codons, 2 variants predicted to disrupt splicing, and 12 frameshift indels (Online Table 2). Eleven rare missense variants underwent functional validation in a mouse model, of which 2 – p.Asp42Asn and p.Thr383Ser – were additionally included as validated LOF variants (Figure 2).
FIGURE 2. Mouse Model to Functionally Annotate ANGPTL3 Missense Variants.
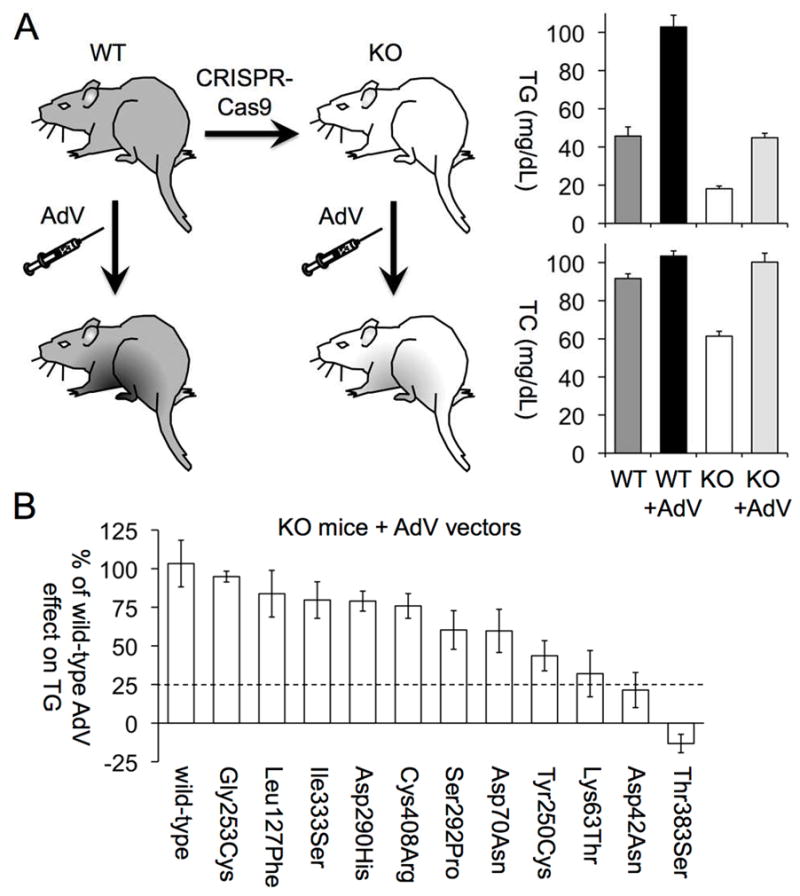
(A) This schematic illustrates the design of the mouse study and circulating triglyceride (TG) and total cholesterol (TC) levels of wild-type (WT) and Angptl3 knockout (KO) mice generated with CRISPR-Cas9, before and after treatment with adenovirus (AdV) expressing WT human ANGPTL3 (11 WT mice, 18 KO mice). (B) The effects of mutant ANGPTL3 alleles expressed from AdV in KO mice (5 to 6 in each group) on TG levels are shown as percentages of the effect of AdV expressing the WT ANGPTL3 allele on TG. Mutant ANGPTL3 alleles that had <25% of the WT allele effect on TG levels were annotated as loss-of-function alleles. (For additional details, including TC levels of mice before and after AdV injection, see Online Figure 3.) Abbreviations as in Figure 1.
In aggregate across all sequencing studies, an ANGPTL3 LOF mutation was identified in 130 of 40,112 participants (0.32%; 95% confidence interval [CI]: 0.27% to 0.39%). One homozygote was identified with a Gln192ArgfsTer5 frameshift mutation – a 56-year-old woman of African ancestry free of clinical CAD with LDL cholesterol of 112 mg/dl, HDL cholesterol of 44 mg/dl, and TGs of 56 mg/dl.
Among sequenced individuals of European ancestry, the most frequently observed inactivating variant was the intronic splice region variant rs372257803 (minor allele frequency = 0.17%). This variant was imputed in an additional 8,066 CAD cases and 140,068 controls, identifying an additional 68 heterozygous carriers of an ANGPTL3 LOF mutation.
ASSOCIATION OF ANGPTL3 LOF MUTATIONS WITH CIRCULATING LIPID LEVELS AND CAD RISK
Plasma lipid levels were available in up to 20,092 individuals of the Myocardial Infarction Genetics Consortium studies, including 60 heterozygous carriers of an ANGPTL3 LOF mutation. We found that individuals carrying an LOF ANGPTL3 mutation, when compared with noncarriers, had 11% lower total cholesterol (p = 0.0008), 12% lower LDL cholesterol (p = 0.04), and 17% lower TGs (p = 0.01) (Table 1). HDL cholesterol was not significantly different between the groups (p = 0.17).
TABLE 1.
Association of LOF Mutations in ANGPTL3 with Plasma Lipid Concentrations*
| Lipid fraction | Number of Participants | Percent Difference† | 95% CI | p Value |
|---|---|---|---|---|
| Total cholesterol | 19,783 | −10.9% | −17.3% to −4.5% | 0.0008 |
| LDL cholesterol | 18,231 | −11.8% | −21.5% to −2.1% | 0.04 |
| HDL cholesterol | 20,092 | −5.2% | −12.8% to 2.3% | 0.17 |
| Triglycerides | 19,282 | −17.3% | −31.1% to −3.4% | 0.01 |
In 20,092 individuals of the Myocardial Infarction Genetics Consortium.
Linear regression was used to test for an association between loss-of-function (LOF) angiopoietin-like 3 (ANGPTL3) mutations and plasma lipid concentrations using age, age squared, sex, study cohort, coronary artery disease, and the first 5 principal components of ancestry as covariates. Triglyceride concentrations were natural log-transformed prior to analysis.
CI = confidence interval; HDL = high-density lipoprotein; LDL = low-density lipoprotein.
A cohort-based meta-analysis stratified by ancestry was performed to determine the relationship between LOF mutations in ANGPTL3 and risk of CAD (Figure 3 and Online Table 2). We observed a 34% reduced risk of CAD among carriers of an ANGPTL3 LOF mutation as compared to noncarriers (odds ratio [OR] of disease for carriers = 0.66; 95% CI: 0.44 to 0.98; p = 0.04). This effect estimate was similar in a sensitivity analysis restricted to individuals in whom complete gene sequencing (as opposed to rs372257803 imputation) was performed (OR: 0.70; 95% CI: 0.46 to 1.06; p = 0.09).
FIGURE 3. Association of ANGPTL3 Loss-of-Function Mutations with Risk of CAD.

The association of ANGPTL3 mutations with risk of coronary artery disease (CAD) was determined via meta-analysis utilizing Cochran–Mantel–Haenszel statistics for ethnicity-specific stratified 2-by-2 tables. A reduced risk of CAD was noted among carriers of an ANGPTL3 loss-of-function variant (odds ratio of disease for carriers: 0.66; 95% confidence interval: 0.44 to 0.98; p = 0.04). AA = African ancestry; ARIC = Atherosclerosis Risk in Communities; ATVB = Italian Atherosclerosis Thrombosis and Vascular Biology; EA =European ancestry; ESP-EOMI = Exome Sequencing Project Early-Onset Myocardial Infarction; JHS = Jackson Heart Study; MI = myocardial infarction; OHS = Ottawa Heart Study; PROCARDIS = Precocious Coronary Artery Disease; PROMIS = Pakistan Risk of Myocardial Infarction Study; REGICOR = Registre Gironi del COR (Gerona Heart Registry); WTCCC = Wellcome Trust Case Control Consortium; other abbreviations as in Figure 1.
CIRCULATING PLASMA ANGPTL3 AND RISK OF MI
Protection from CAD among carriers of a rare, LOF mutation in ANGPTL3 led to the hypothesis that individuals with lower levels of circulating ANGPTL3 protein might similarly have reduced coronary risk. Plasma ANGPTL3 concentrations were measured in 1,493 cases presenting with a first-ever MI and 3,231 controls free of CAD from the PROMIS study. Consistent with our expectations, individuals in the lowest tertile of ANGPTL3 concentrations had significantly reduced risk of MI compared to those in the highest tertile (adjusted OR: 0.65; p = 2.2 x 10−7) (Table 2). This finding was modestly attenuated after additional adjustment for observed plasma LDL cholesterol and TGs (adjusted OR: 0.71; p = 0.0001).
TABLE 2.
Association of ANGPTL3 Concentration with MI Risk in PROMIS
| ANGPTL3 Concentration (ng/ml) | Adjusted Odds Ratio for MI (95% CI) | ||
|---|---|---|---|
|
| |||
| Model 1* | Model 2† | ||
| Tertile 1 (n = 1,575)‡ | 379–1,375 | 1.00 | 1.00 |
| Tertile 2 (n = 1,574) | 272–378 | 0.75 (0.64–0.88) | 0.79 (0.66–0.93) |
| Tertile 3 (n = 1,575) | 18–271 | 0.65 (0.55–0.77) | 0.71 (0.60–0.85) |
| p Value for trend | 2.2 x 10−7 | 0.0001 | |
Sex, current smoking, diabetes, and hypertension as covariates.
Model 1 plus LDL cholesterol and log-transformed triglycerides as covariates.
Individuals with the highest tertile of ANGPTL3 concentration served as the reference group for this analysis.
MI = myocardial infarction; other abbreviations as in Table 1.
DISCUSSION
We provided multiple lines of evidence suggesting that ANGPTL3 deficiency is associated with protection from CAD (Central Illustration). Detailed atherosclerotic phenotyping demonstrated an absence of coronary atherosclerotic plaque in individuals with complete ANGPTL3 deficiency. Genomic analysis of ANGPTL3 LOF variants, including functionally validated missense variants in up to 180,180 individuals, showed a 34% reduction in risk of CAD among heterozygous carriers. Finally, circulating ANGPTL3 concentrations were lower in healthy controls as compared to those presenting with MI.
CENTRAL ILLUSTRATION. ANGPTL3 Deficiency and Protection From Coronary Artery Disease.
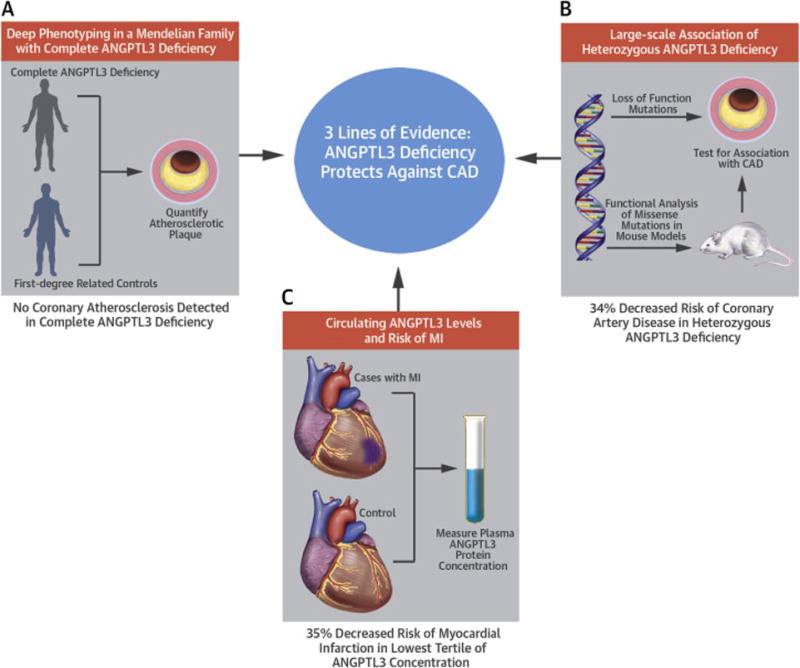
Multiple lines of evidence suggesting that angiopoietin-like 3 (ANGPTL3) deficiency is associated with protection from coronary artery disease (CAD). (A) A genotype-guided callback study of human “knockouts” for ANGPTL3, which used detailed atherosclerotic phenotyping, demonstrated an absence of coronary atherosclerotic plaque in individuals with complete ANGPTL3 deficiency. (B) Genomic analysis of ANGPTL3 loss-of-function variants, including missense variants that were experimentally found to disrupt ANGPTL3 function, found in up to 180,180 individuals showed a 34% reduction in risk of CAD among loss-of-function variant carriers. (C) Circulating ANGPTL3 protein concentrations were lower in healthy controls as compared to those presenting with a myocardial infarction.
These results permitted several conclusions. First, identifying families with extreme phenotypes of interest could facilitate both gene discovery as well as hypothesis-based phenotyping. Multiple independent groups have confirmed the impact of inactivating mutations in ANGPTL3 on decreasing lipid levels using family-based study designs (31–34). Here, we extended these observations by demonstrating that individuals with complete deficiency due to 2 inactivating mutations in the gene – effectively ‘human knockouts’ for ANGPTL3 – tended to have less coronary atherosclerosis as assessed by CCTA. This apparent protection from coronary atherosclerosis extended to a middle-aged participant (Online Table 1, individual II-1) with significant cardiovascular risk factors of type 2 diabetes mellitus, hypertension, and a history of cigarette smoking. Although suggestive, these results were based on a small number of family members. Large-scale gene sequencing in the population as presented here was used to confirm this observation.
Additionally, these findings lent support to ongoing drug development efforts focused on ANGPTL3 inhibition as a therapeutic strategy. Beyond a significant reduction in plasma LDL cholesterol and triglyceride concentrations, heterozygous ANGPTL3 LOF mutation carriers had a 34% decreased risk of CAD. Similar results were noted in a preliminary report from Dewey and colleagues (35). We also found that individuals with circulating plasma ANGPTL3 concentrations in the lowest tertile of the population (in a sense, mimicking the effect of pharmacological inhibition of ANGPTL3) had a 35% reduced risk of MI. This study adds ANGPTL3 to the list of therapeutic targets for coronary disease, which includes ANGPTL4 (16,36), APOC3 (11,37), LPA (38), NPC1L1 (30), and PCSK9 (39), that have been validated by finding LOF mutations that associate with protection from disease, highlighting the promise and potential of human genetic studies in identifying such targets (40).
Furthermore, these data added to a growing body of human genetics evidence linking regulation of lipoprotein lipase activity – the major mechanism by which circulating triglyceride-rich lipoproteins are hydrolyzed – with atherosclerosis. Studies in both mice and humans have demonstrated ANGPTL3 to be a potent inhibitor of LPL, particularly in the post-prandial state (41). Consistent with effects of rare mutations in 3 additional endogenous regulators of LPL, APOA5 (10), APOC3 (11,37), and ANGPTL4 (16,36), loss of ANGPTL3 function appears to result in gain of LPL activity, reduced triglyceride-rich lipoproteins, and protection from coronary disease. Beyond effects on TGs and LDL cholesterol, ANGPTL3 might affect glucose homeostasis as well as remodeling of HDL cholesterol particles by EL (4,42). The relative contributions of each of these mechanisms, as they relate to risk of CAD, warrant additional investigation.
Finally, we provided proof-of-concept for rare ANGPTL3 missense variant prioritization using a combination of bioinformatics tools and experimental characterization in vivo. Any given ANGPTL3 missense variant might perturb protein function via numerous potential mechanisms, including decreased expression, impaired hepatic secretion, or inability to bind and inhibit LPL. We developed an Angptl3 knockout mouse that exhibited a phenotype of very low triglycerides. We attempted to rescue this phenotype using adenoviral vectors producing either wild-type ANGPTL3 or a protein including the missense variant of interest. This proved useful in determining that only 2 of 11 screened missense variants led to near complete loss of ANGPTL3 protein function (i.e., <25% of wild-type activity). The ability to confidently annotate the functional consequences of rare missense variants in a gene of interest remains one of the biggest hurdles to rare variant gene discovery efforts (43). We present one approach to address this challenge.
STUDY LIMITATIONS
Important limitations to the present study should be recognized. First, CCTA was performed in only a subset of the original family used to identify ANGPTL3 as the cause of familial combined hypolipidemia. Second, the functional characterization of missense variants was performed in only 11 variants and alternative thresholds for defining LOF are possible. Third, genotype imputation was used to identify carriers of an ANGPTL3 splice-site imputation in some cohorts. However, a sensitivity analysis restricted to those studies in which complete sequencing of ANGPTL3 yielded similar results.
CONCLUSIONS
Deep phenotyping in a family, gene sequencing in the population, and biomarker analysis in cases and controls showed ANGPTL3 deficiency to be associated with a reduced risk of CAD. Whether pharmacological inhibition of ANGPTL3 function will prove useful in the treatment or prevention of CAD remains to be determined.
Supplementary Material
PERSPECTIVES.
COMPETENCY IN MEDICAL KNOWLEDGE
Hereditary combined hypolipidemia is caused by mutations that inactivate the gene angiopoietin-like 3 (ANGPTL3) and is characterized by low blood levels of all 3 major lipid fractions. Individuals with a loss-of-function ANGPTL3 mutation have reduced odds of CAD, suggesting that ANGPTL3 deficiency protects against CAD.
TRANSLATIONAL OUTLOOK
Interventions targeting ANGPTL3 should be considered for potential clinical application.
Acknowledgments
We are very grateful to the family presented here for participating in this study and for their ongoing contributions to furthering biomedical research. The authors also thank Ms. Teresa Roediger for coordinating the clinical imaging portion of the study. This study was supported by grants from the National Heart, Lung, and Blood Institute (R01HL131961 to NOS; K08HL114642 to NOS; R01 HL118744 to KM; R01 HL127564 to SK; R21 HL120781 to SK); the National Human Genome Research Institute (U54HG003067 to SG/EL, UM1HG008895 to SK/SG/EL, and UM1HG008853 to NOS); the Barnes-Jewish Hospital Foundation (NOS); the Fannie Cox Prize for Excellence in Science Teaching, Harvard University (KM); MGH Research Scholar Award (SK). AVK is supported by an ACCF/Merck Cardiovascular Research Fellowship, a John S. Ladue Memorial Fellowship at Harvard Medical School, and a KL2/Catalyst Medical Research Investigator Training award from Harvard Catalyst funded by the National Institutes of Health (NIH) (TR001100). DK is supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health (T32 HL007734). NJS is supported by the BHF and is a NIHR Senior Investigator. PROMIS fieldwork has been supported through grants awarded to DS, JD and PMF. Biomarker assays in PROMIS have been funded through grants awarded by NHLBI, NIH (RC2HL101834 and RC1TW008485) and the Fogarty International (RC1TW008485).
ABBREVIATIONS AND ACRONYMS
- ANGPTL3
angiopoietin-like 3
- EL
endothelial lipase
- HDL
high-density lipoprotein
- LDL
low-density lipoprotein
- LOF
loss of function
- LPL
lipoprotein lipase
- TC
total cholesterol
- TG
triglyceride
Footnotes
Financial Disclosures: NOS has received a research grant from AstraZeneca and consulting fees from Aegerion Pharmaceuticals. AVK has received consulting fees from Merck and Amarin. DJR has received consulting fees from Aegerion Pharmaceuticals, Alnylam Pharmaceuticals, Eli Lilly and Company, Pfizer, Sanofi, and Novartis; is an inventor on a patent related to lomitapide that is owned by the University of Pennsylvania and licensed to Aegerion Pharmaceuticals; and is a cofounder of Vascular Strategies and Staten Biotechnology. DS has received grants from Pfizer, Regeneron, Eli Lilly and Genenetech. SK has received grants from Bayer Healthcare, Aegerion Pharmaceuticals, and Regeneron Pharmaceuticals; and consulting fees from Merck, Novartis, Sanofi, AstraZeneca, Alnylam Pharmaceuticals, Leerink Partners, Noble Insights, Quest Diagnostics, Genomics PLC, and Eli Lilly and Company; and holds equity in San Therapeutics and Catabasis Pharmaceuticals. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose. This work was funded by the National Institutes of Health which had no involvement in the design and conduct of the study, in the collection, analysis, and interpretation of the data, or in the preparation, review, and approval of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Musunuru K, Pirruccello JP, Do R, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med. 2010;363:2220–7. doi: 10.1056/NEJMoa1002926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koishi R, Ando Y, Ono M, et al. Angptl3 regulates lipid metabolism in mice. Nat Genet. 2002;30:151–7. doi: 10.1038/ng814. [DOI] [PubMed] [Google Scholar]
- 3.Shimizugawa T, Ono M, Shimamura M, et al. ANGPTL3 decreases very low density lipoprotein triglyceride clearance by inhibition of lipoprotein lipase. J Biol Chem. 2002;277:33742–8. doi: 10.1074/jbc.M203215200. [DOI] [PubMed] [Google Scholar]
- 4.Shimamura M, Matsuda M, Yasumo H, et al. Angiopoietin-like protein3 regulates plasma HDL cholesterol through suppression of endothelial lipase. Arterioscler Thromb Vasc Biol. 2007;27:366–72. doi: 10.1161/01.ATV.0000252827.51626.89. [DOI] [PubMed] [Google Scholar]
- 5.Gusarova V, Alexa CA, Wang Y, et al. ANGPTL3 blockade with a human monoclonal antibody reduces plasma lipids in dyslipidemic mice and monkeys. J Lipid Res. 2015;56:1308–17. doi: 10.1194/jlr.M054890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sehgal A, Vaishnaw A, Fitzgerald K. Liver as a target for oligonucleotide therapeutics. J Hepatol. 2013;59:1354–9. doi: 10.1016/j.jhep.2013.05.045. [DOI] [PubMed] [Google Scholar]
- 7.Ando Y, Shimizugawa T, Takeshita S, et al. A decreased expression of angiopoietin-like 3 is protective against atherosclerosis in apoE-deficient mice. J Lipid Res. 2003;44:1216–23. doi: 10.1194/jlr.M300031-JLR200. [DOI] [PubMed] [Google Scholar]
- 8.Minicocci I, Santini S, Cantisani V, et al. Clinical characteristics and plasma lipids in subjects with familial combined hypolipidemia: a pooled analysis. J Lipid Res. 2013;54:3481–90. doi: 10.1194/jlr.P039875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atherosclerosis, Thrombosis, and Vascular Biology Italian Study Group. No evidence of association between prothrombotic gene polymorphisms and the development of acute myocardial infarction at a young age. Circulation. 2003;107:1117–22. doi: 10.1161/01.cir.0000051465.94572.d0. [No authors listed] [DOI] [PubMed] [Google Scholar]
- 10.Do R, Stitziel NO, Won HH, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518:102–6. doi: 10.1038/nature13917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crosby J, Peloso GM, Auer PL, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371:22–31. doi: 10.1056/NEJMoa1307095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McPherson R, Pertsemlidis A, Kavaslar N, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–91. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clarke R, Peden JF, Hopewell JC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361:2518–28. doi: 10.1056/NEJMoa0902604. [DOI] [PubMed] [Google Scholar]
- 14.Saleheen D, Zaidi M, Rasheed A, et al. The Pakistan Risk of Myocardial Infarction Study: a resource for the study of genetic, lifestyle and other determinants of myocardial infarction in South Asia. Eur J Epidemiol. 2009;24:329–38. doi: 10.1007/s10654-009-9334-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Senti M, Tomas M, Marrugat J, Elosua R REGICOR Investigators. Paraoxonase1-192 polymorphism modulates the nonfatal myocardial infarction risk associated with decreased HDLs. Arterioscler Thromb Vasc Biol. 2001;21:415–20. doi: 10.1161/01.atv.21.3.415. [DOI] [PubMed] [Google Scholar]
- 16.Stitziel NO, Stirrups KE, Masca NG, et al. Coding Variation in ANGPTL4, LPL, and SVEP1 and the Risk of Coronary Disease. N Engl J Med. 2016;374:1134–44. doi: 10.1056/NEJMoa1507652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peloso GM, Lange LA, Varga TV, et al. Association of Exome Sequences With Cardiovascular Traits Among Blacks in the Jackson Heart Study. Circ Cardiovasc Genet. 2016;9:368–74. doi: 10.1161/CIRCGENETICS.116.001410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baber U, Mehran R, Sartori S, et al. Prevalence, impact, and predictive value of detecting subclinical coronary and carotid atherosclerosis in asymptomatic adults: the BioImage study. J Am Coll Cardiol. 2015;65:1065–74. doi: 10.1016/j.jacc.2015.01.017. [DOI] [PubMed] [Google Scholar]
- 19.Li AH, Morrison AC, Kovar C, et al. Analysis of loss-of-function variants and 20 risk factor phenotypes in 8,554 individuals identifies loci influencing chronic disease. Nat Genet. 2015;47:640–2. doi: 10.1038/ng.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davies RW, Wells GA, Stewart AF, et al. A genome-wide association study for coronary artery disease identifies a novel susceptibility locus in the major histocompatibility complex. Circ Cardiovasc Genet. 2012;5:217–25. doi: 10.1161/CIRCGENETICS.111.961243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Helgadottir A, Gretarsdottir S, Thorleifsson G, et al. Variants with large effects on blood lipids and the role of cholesterol and triglycerides in coronary disease. Nat Genet. 2016;48:634–9. doi: 10.1038/ng.3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sudlow C, Gallacher J, Allen N, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12:e1001779. doi: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reilly MP, Li M, He J, et al. Identification of ADAMTS7 as a novel locus for coronary atherosclerosis and association of ABO with myocardial infarction in the presence of coronary atherosclerosis: two genome-wide association studies. Lancet. 2011;377:383–92. doi: 10.1016/S0140-6736(10)61996-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samani NJ, Erdmann J, Hall AS, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–53. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boyko AR, Williamson SH, Indap AR, et al. Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genet. 2008;4:e1000083. doi: 10.1371/journal.pgen.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yampolsky LY, Kondrashov FA, Kondrashov AS. Distribution of the strength of selection against amino acid replacements in human proteins. Hum Mol Genet. 2005;14:3191–201. doi: 10.1093/hmg/ddi350. [DOI] [PubMed] [Google Scholar]
- 27.Mehta N, Qamar A, Qu L, et al. Differential association of plasma angiopoietin-like proteins 3 and 4 with lipid and metabolic traits. Arterioscler Thromb Vasc Biol. 2014;34:1057–63. doi: 10.1161/ATVBAHA.113.302802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–78. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 29.Tobin MD, Sheehan NA, Scurrah KJ, Burton PR. Adjusting for treatment effects in studies of quantitative traits: antihypertensive therapy and systolic blood pressure. Stat Med. 2005;24:2911–35. doi: 10.1002/sim.2165. [DOI] [PubMed] [Google Scholar]
- 30.Stitziel NO, Won HH, Morrison AC, et al. Inactivating mutations in NPC1L1 and protection from coronary heart disease. N Engl J Med. 2014;371:2072–82. doi: 10.1056/NEJMoa1405386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin-Campos JM, Roig R, Mayoral C, et al. Identification of a novel mutation in the ANGPTL3 gene in two families diagnosed of familial hypobetalipoproteinemia without APOB mutation. Clin Chim Acta. 2012;413:552–5. doi: 10.1016/j.cca.2011.11.020. [DOI] [PubMed] [Google Scholar]
- 32.Minicocci I, Montali A, Robciuc MR, et al. Mutations in the ANGPTL3 gene and familial combined hypolipidemia: a clinical and biochemical characterization. J Clin Endocrinol Metab. 2012;97:E1266–75. doi: 10.1210/jc.2012-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pisciotta L, Favari E, Magnolo L, et al. Characterization of three kindreds with familial combined hypolipidemia caused by loss-of-function mutations of ANGPTL3. Circ Cardiovasc Genet. 2012;5:42–50. doi: 10.1161/CIRCGENETICS.111.960674. [DOI] [PubMed] [Google Scholar]
- 34.Noto D, Cefalu AB, Valenti V, et al. Prevalence of ANGPTL3 and APOB gene mutations in subjects with combined hypolipidemia. Arterioscler Thromb Vasc Biol. 2012;32:805–9. doi: 10.1161/ATVBAHA.111.238766. [DOI] [PubMed] [Google Scholar]
- 35.Dewey FE, Gusarova V, O’Dushlaine C, et al. Genetic and Pharmacological Inactivation of ANGPTL3 is Associated With Reduced Atherosclerotic Cardiovascular Disease. Circulation. 2016;134:A16563. [Google Scholar]
- 36.Dewey FE, Gusarova V, O’Dushlaine C, et al. Inactivating Variants in ANGPTL4 and Risk of Coronary Artery Disease. N Engl J Med. 2016;374:1123–33. doi: 10.1056/NEJMoa1510926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjaerg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. 2014;371:32–41. doi: 10.1056/NEJMoa1308027. [DOI] [PubMed] [Google Scholar]
- 38.Lim ET, Wurtz P, Havulinna AS, et al. Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet. 2014;10:e1004494. doi: 10.1371/journal.pgen.1004494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cohen JC, Boerwinkle E, Mosley TH, Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 40.Stitziel NO, Kathiresan S. Leveraging human genetics to guide drug target discovery. Trends Cardiovasc Med. 2016 Aug 26; doi: 10.1016/j.tcm.2016.08.008. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Minicocci I, Tikka A, Poggiogalle E, et al. Effects of angiopoietin-like protein 3 deficiency on postprandial lipid and lipoprotein metabolism. J Lipid Res. 2016;57:1097–107. doi: 10.1194/jlr.P066183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robciuc MR, Maranghi M, Lahikainen A, et al. Angptl3 deficiency is associated with increased insulin sensitivity, lipoprotein lipase activity, and decreased serum free fatty acids. Arterioscler Thromb Vasc Biol. 2013;33:1706–13. doi: 10.1161/ATVBAHA.113.301397. [DOI] [PubMed] [Google Scholar]
- 43.Zuk O, Schaffner SF, Samocha K, et al. Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci U S A. 2014;111:E455–64. doi: 10.1073/pnas.1322563111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.