

Abstract
The NRF2 pathway activates a cell survival response when cells are exposed to xenobiotics or are under oxidative stress. Therapeutic activation of NRF2 can also be used prior to insult as a means of disease prevention. However, prolonged expression of NRF2 has been shown to protect cancer cells by inducing the metabolism and efflux of chemotherapeutics, leading to both intrinsic and acquired chemoresistance to cancer drugs. This effect has been termed the “dark side” of NRF2. In an effort to combat this chemoresistance, our group discovered the first NRF2 inhibitor, the natural product brusatol, however the mechanism of inhibition was previously unknown. In this report, we show that brusatols mode of action is not through direct inhibition of the NRF2 pathway, but through the inhibition of both cap-dependent and cap-independent protein translation, which has an impact on many short-lived proteins, including NRF2. Therefore, there is still a need to develop a new generation of specific NRF2 inhibitors with limited toxicity and off-target effects that could be used as adjuvant therapies to sensitize cancers with high expression of NRF2.
Keywords: NRF2, brusatol, protein translation, chemoresistance, cancer
Introduction
Chemoresistance to current drug regimens for the treatment of cancer has become a major health concern leading to the usage of highly cytotoxic chemicals with many unwanted side effects. The molecular mechanisms that lead to chemoresistance are not well understood, resulting in the continued reliance on broad-spectrum, highly toxic chemotherapeutics. There are a number of potential avenues to combat chemoresistance. One is through personalized medicine, which would foster the development of highly-specific targeted therapies based on the molecular profile of each individual patient’s cancer. This means that the treatment would be more effective in a shorter amount of time due to the decreased likelihood that chemoresistance may occur. A second approach would be to develop a better molecular understanding of the mechanisms underlying chemoresistance and develop adjuvant therapies to increase the efficacy of chemotherapeutics [1]. In order to address this second option for targeted therapy, we developed the first inhibitor of the nuclear factor-erythroid factor 2-related 2 (NRF2) pathway based on the discovery that high levels of NRF2 are associated with resistance to chemotherapeutics [2].
NRF2 is a redox-sensitive transcription factor that maintains the crucial intracellular reductive/oxidative (redox) balance of the cell. To do so, it up-regulates genes that are involved in phase I and II drug metabolism, glutathione synthesis, and xenobiotic drug transport [3]. Because of the critical role played by NRF2 in cellular protection, timely activation by NRF2 is needed. As a result, NRF2 is constantly translated by the cell; however, under stress-free conditions, when NRF2 activation is not needed, its negative regulator KEAP1 constantly targets NRF2 for degradation [4,5]. KEAP1 is a substrate adaptor protein that is part of an E3 ubiquitin ligase complex that polyubiquitylates NRF2 and targets it for degradation by the 26S proteasome [4,5]. A critical cysteine residue in KEAP1, cysteine 151 (Cys151), can become oxidized or covalently modified by reactive oxygen species (ROS) or electrophiles, causing a conformational change in KEAP1, preventing polyubiquitylation and subsequent degradation of NRF2 [6]. Newly synthesized NRF2 then accumulates in the cytosol and translocates into the nucleus, where it forms a heterodimer with small MAF proteins, binds to an enhancer sequence, the antioxidant response element (ARE), in the promoter region of its target genes, and promotes the antioxidant response.
Early investigations determined that up-regulation of NRF2 with dietary phytochemicals (sulforaphane, cinnamaldehyde, etc.) could protect against cancer and other diseases, prompting the search for NRF2-inducing compounds [7,8]. However, the recognition that certain cancers overexpress NRF2, either through somatic mutations or epigenetic silencing of key negative regulators, and that chronic upregulation of NRF2 can lead to tissue damage and cancer progression, led to the concept of the “dark side” of NRF2 [2,3,9]. To overcome this “dark side”, our lab identified the first NRF2 inhibitor, brusatol, and proved that inhibition of NRF2 sensitized non-small cell lung cancer (NSCLC) cells to cisplatin treatment, making combination therapy with an NRF2 inhibitor a promising new initiative for the treatment of cancer with high levels of NRF2 [10]. While treatment with brusatol, a quassinoid compound extracted from Brucea javanica, was effective in the nanomolar range at inhibiting NRF2 signaling, the mode of action was unknown [10]. Despite this, brusatol was shown to be effective in enhancing the antitumor action of cisplatin in a mutant KRAS G12D-induced lung cancer model [11]. In the present study, the mode of action of brusatol as an NRF2 inhibitor was determined. Brusatol was found to be a general translation inhibitor, which causes a decline in the protein levels of short-lived proteins, including NRF2. While these findings do not negate previous studies showing inhibition of NRF2 by brusatol as an effective strategy to sensitize cancers to chemotherapies, they do argue for the development of new inhibitors with heightened specificity for inhibition of the NRF2 pathway.
Materials and Methods
Cell culture
A549 non-small cell lung carcinoma (NSCLC) cells were purchased from the American Type Culture Collection (ATCC) and were maintained with Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum at 37°C with 5% CO2.
RNA-seq profiling
A549 cells were plated at 80% confluence in D35 dishes and left to adhere overnight. Cells were treated (in duplicate) with 40 nM brusatol for 16 h and total RNA was extracted as previously described [12]. RNA-sequencing analysis was performed as previously described by The Genomics and Microarray Shared Resource at the University of Colorado [13]. A 5% false discovery rate (FDR) cut off was applied.
Fluorescent brusatol probe
The Immunoaffinity (IAF) tag was synthesized as previously described [14]. Briefly, a) brusatol (1) was treated with t-butyl-bromoacetate in basic dimethylformamide (DMF) and then b) deprotected in tri-fluoroacetic acid (TFA) to yield (2). c) Coupling with IAF tag (3) was carried out by treatment with HATU in EtNiPr2. A549 cells were treated with the brusatol-IAF probe (1 µM) for 4 h, which was followed by the addition of staining markers for the lysosome (LysoTracker red, Life Science Technology), endoplasmic reticulum (ER tracker, ThermoFisher Scientific), and Golgi (Golgi tracker, ThermoFisher Scientific) for 15 min. Live cell images were acquired with a deconvolution microscope.
Antibodies and western blot
Antibodies for NRF2, p53, p21, p97 (VCP), and GAPDH were purchased from Santa Cruz Biotechnology. The antibody used to detect GFP was purchased from GeneTex. For western blot experiments, A549 cells were seeded at 80% confluence and left to adhere overnight. After the indicated treatments, cells were harvested in 1× NuPAGE LDS Sample Buffer (Invitrogen) and subjected to SDS-polyacrylamide gel electrophoresis.
Dual reporters for translation inhibition
The FF-Ren and FF-EMCV IRES-Ren plasmids were a kind gift from Dr. Jerry Pelletier (McGill University). The mRFP-IRES-GFP plasmid was constructed from the pIRES-EGFP-Puro construct acquired from Addgene (Plasmid #45567). Briefly, mRFP from a previous construct was sub-cloned within the multiple cloning site using the Xho1 and Nhe1 digestion enzymes.
For dual luciferase experiments, A549 cells were seeded at 80% confluence and left to adhere overnight. Plasmid transfection for FF-Ren and FF-EMCV IRES-Ren was performed using the Lipofectamine 3000 transfection reagent according to the manufacturer’s instructions. After a 4 h transfection, the media/transfection reagent was removed and the cells were treated with indicated compounds in normal media for 16 h. Cells were lysed in a 1× passive lysis buffer (Promega) and a dual luciferase assay was performed with a luminometer (Turner BioSystems). The experiment was repeated 3 times, with triplicate wells, for acquisition of statistical significance as reported by the standard error of the mean.
For the dual fluorescence reporter (mRFP-IRES-GFP), A549 cells were seeded at 80% confluence in glass bottom D35 dishes and left to adhere overnight. Transfection of the mRFP-IRES-GFP plasmid was performed as mentioned previously for 4 h and treatment occurred for 16 h. After treatment, live cells images were acquired using the Zeiss Observer.Z1 microscope with the Slidebook 4.2.0.11 imaging program (Intelligent Imaging Innovations, Inc.).
Results
RNA-seq profiling reveals similar gene set enrichment patterns between brusatol and other translation inhibitors
The effects of brusatol on gene expression in A549 cells, a NSCLC cell line that has high, constitutive NRF2 expression, were analyzed by RNA-seq. These results were compared to publicly available gene enrichment data sets for a variety of different treatments, such as chemotherapeutics, hormones, cytokines, and other signaling factors, to try to infer a mechanism of action for brusatol. Brusatol treatment in A549 cells up-regulated the expression of 2,914 genes and down-regulated the expression of 2,991 genes. The top 10 up- and down-regulated genes are shown in Table 1. Interestingly, when comparing the gene expression modulation of brusatol with other compounds, brusatol shared a similar gene-set enrichment pattern with the translation inhibitor cycloheximide (Figure 1A). A more in-depth analysis revealed that brusatol and cycloheximide shared 1,339 up-regulated and 1,320 down-regulated genes (Figure 1B). As a follow-up, brusatol was compared to ricin and puromycin, two other translation inhibitors with distinct mechanisms of action. Brusatol also showed a similar gene-set enrichment pattern to both inhibitors (Figure 1C). Gene-sets that were shared between brusatol and ricin (176 genes up-regulated and 175 down-regulated) (Figure 1D) or brusatol and puromycin (136 up-regulated and 110 down-regulated) (Figure 1E) treatment were found to be equally significant, considering that ricin and puromycin regulated a smaller subset of genes compared to cycloheximide. These results indicated that brusatol might function as a protein translation inhibitor. Additionally, RNA-seq profiling after brusatol treatment revealed a 0.6 fold increase in NRF2 mRNA transcript levels. This phenomenon likely occurs as a compensation for the rapid loss of the NRF2 protein, and has been previously reported [15].
Table 1.
Top 10 gene expression changes associated with brusatol treatment in A549 cells.
| ID | Base Mean |
Fold Change |
log2 Fold Change |
p value | p adjusted | |
|---|---|---|---|---|---|---|
|
Up- regulated |
DUSP27 | 126.1137 | 506.8652 | 8.985458 | 0.002388 | 0.010455 |
| ATP10B | 78.41707 | 314.7887 | 8.29824 | 0.005938 | 0.022985 | |
| CYP1A1 | 75.48413 | 302.9777 | 8.243068 | 2.58E-14 | 6.45E-13 | |
| MAL2 | 259.7108 | 270.6116 | 8.08008 | 0.001155 | 0.005488 | |
| SEMA5A | 54.76455 | 219.5391 | 7.778334 | 1.65E-10 | 2.67E-09 | |
| ASCL2 | 42.4191 | 169.8235 | 7.407892 | 9.95E-08 | 1.08E-06 | |
| SPINK1 | 233.5567 | 155.7571 | 7.283154 | 0.000276 | 0.001527 | |
| PPP1R1B | 449.7914 | 143.133 | 7.161213 | 4.15E-05 | 0.000276 | |
| SLC5A1 | 85.09216 | 113.2232 | 6.823026 | 8.87E-05 | 0.000549 | |
| ACE2 | 51.0301 | 101.7502 | 6.668887 | 4.41E-09 | 5.84E-08 | |
|
Down- regulated |
NR2E3 | 29.39972 | 0.0068 | −7.20022 | 3.75E-06 | 3.08E-05 |
| GSTA2 | 158.6975 | 0.010111 | −6.62789 | 1.68E-05 | 0.000122 | |
| SNORD116-17 | 15.97673 | 0.015506 | −6.011 | 0.001941 | 0.008685 | |
| SNORD116-19 | 15.97673 | 0.015506 | −6.011 | 0.001941 | 0.008685 | |
| LOC93432 | 77.42563 | 0.031753 | −4.97695 | 0.002499 | 0.010873 | |
| SERPING1 | 15.76144 | 0.033605 | −4.89518 | 0.002523 | 0.010955 | |
| LDLRAD1 | 15.76144 | 0.03928 | −4.67005 | 0.009018 | 0.033027 | |
| PLCB2 | 18.31799 | 0.041617 | −4.5867 | 0.002526 | 0.010963 | |
| SEC14L3 | 36.5122 | 0.04227 | −4.56423 | 0.001689 | 0.007693 | |
| GSTM5 | 104.4568 | 0.046495 | −4.42679 | 1.24E-12 | 2.59E-11 | |
Figure 1.

RNA-seq profiling reveals similar gene set enrichment patterns between brusatol and other protein translation inhibitors. (A) Gene sets that were up-regulated (light gray) and down-regulated (dark gray) were acquired and compared to gene sets altered by brusatol treatment. The –log of the fold change of each treatment was plotted and compared to brusatol. (B) A Venn diagram representing the up- and down-regulated genes shared by both brusatol and cycloheximide. (C) Brusatol-modulated gene expression was compared to that of the translation inhibitors, ricin and puromycin, and the –log of the fold change caused by these treatments was also plotted and compared to brusatol. (D and E) Venn diagrams show the up- and down-regulated genes shared by the treatment of brusatol and ricin (D) and brusatol and puromycin (E).
Brusatol localizes to the endoplasmic reticulum
Given that the effects of brusatol treatment resembled that of other established protein translation inhibitors, we attempted to determine if brusatol co-localizes in the cell to a site of active translation. In order to do this, brusatol was chemically modified with a fluorescent immunoaffinity (IAF) tag to track its subcellular location using live cell fluorescent microscopy (Figure 2A). After a mixture of two distinct probes (henceforth brusatol-IAF) was successfully synthesized, A549 cells were treated with increasing concentrations of the brusatol-IAF mixture for 4 h to test if the tag affected the drug’s potency. As shown in Figure 2B, addition of the tag did not alter the native action of brusatol, since the inhibitory effect of 40 nM brusatol-IAF on NRF2 was similar to that of 40 nM brusatol. Moreover, there was a dose-dependent reduction of NRF2 protein levels by brusatol-IAF. Next, the localization of brusatol-IAF was tested. A549 cells were treated for 4 h with brusatol or brusatol-IAF (1 µM), followed by the addition of fluorescent probes that selectively stain the lysosome, ER, or Golgi. Live cell images were acquired via deconvolution microscopy and the merged images between brusatol-IAF and the different organelle tracker dyes indicated that brusatol-IAF localized to the endoplasmic reticulum (ER) (Figure 2C). Treatment with non-tagged brusatol was used as a negative control, and no autofluorescence was observed.
Figure 2.

Brusatol localizes to the endoplasmic reticulum. (A) Synthesis of the brusatol-IAF probe. Reagents and conditions: a) t-butyl-bromoacetate, K2CO3, DMF, rt, 65%; b) then TFA, CH2Cl2 to cleave t-butyl ester; c) tag (3) HATU, EtNiPr2, DMF, rt, 53%. Overall yield 34%. (B) A549 cells were treated with the indicated doses of brusatol-IAF for 4 h to determine if the probe maintained the native activity of brusatol, inhibition of NRF2 protein levels. (C) Images of live A549 cells obtained using deconvolution microscopy show brusatol-IAF localizes to the ER over the Golgi apparatus or lysosomes.
Inhibition of cap-dependent and cap-independent translation by brusatol
Since brusatol mimics the effects of other translation inhibitors and concentrates to the ER, the ability of brusatol to inhibit cap-dependent or cap-independent translation was measured. A549 cells were transfected with a plasmid that expresses mRFP under the control of a cytomegalovirus promoter (CMV, cap-dependent translation) and GFP under the control of an internal ribosomal entry site (IRES, cap-independent) (Figure 3A) and then treated with brusatol, cycloheximide (CHX, both cap-dependent and cap-independent inhibitor), and rapamycin (cap-dependent inhibitor). Brusatol and CHX abolished the expression of mRFP, and significantly reduced the GFP signal, as analyzed by live cell fluorescent microscopy (Figure 3B) and Western blot (Figure 3C). However, rapamycin only inhibited the expression of mRFP without effecting GFP expression (Figure 3B and C). In addition to GFP, the levels of the cap-dependent proteins NRF2 and p53, were shown to decrease with brusatol or CHX treatment (Figure 3C). Furthermore, the inhibitory effects of brusatol were quantified using the Firefly (FF) and Renilla (Ren) dual reporter constructs in A549 cells. In the FF-Ren construct, a T3 RNA polymerase promoter drives the expression of a FF-Ren fusion transcript that is translated in a cap-dependent manner. Conversely, in the FF-EMCV IRES-Ren construct, FF is translated in a cap-dependent manner (T3 promoter), but Ren is translated in a cap-independent manner due to the EMCV (Encephalomyocarditis virus) IRES upstream of it [16]. In accordance with the fluorescence reporter data, brusatol indeed inhibited the expression of both FF and Ren, as indicated by Relative Luciferase Activity (RLA), in both constructs in a dose-dependent manner, while the maximum inhibitory effect of CHX seemed to be reached at 12.5 µM, since 25 µM CHX did not further decrease FF and Ren expression (Figure 3D and E). Together, these results indicated that brusatol inhibits both cap-dependent and cap-independent translation.
Figure 3.
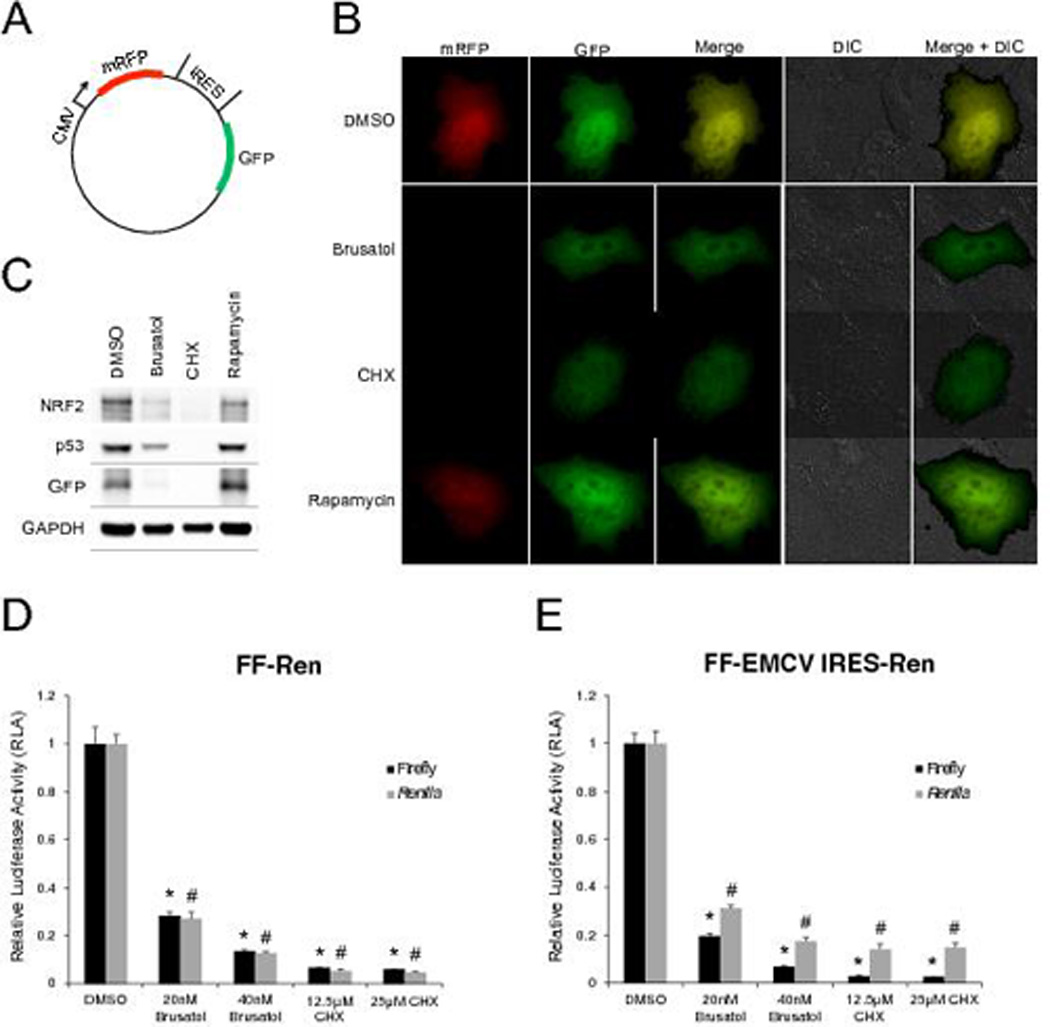
Inhibition of cap-dependent and cap-independent translation by brusatol. (A) Plasmid map of the dual fluorescent reporter constructed for live cell visualization (mRFP, cap-dependent translation; GFP, cap-independent translation). (B) Live-cell images of A549 cells after 16 h of indicated treatments. Differential interference contrast (DIC) images were also taken in conjunction with fluorescent images to define the cell body. (C) The experiment from (B) was performed under identical conditions, except cells were plated in D35 dishes and subjected to western blotting. (D and E) Brusatol and cycloheximide (CHX) inhibited the expression of both Firefly (FF) and Renilla (Ren) in a dose-dependent manner in both of the FF-Ren and FF-EMCV IRES-Ren constructs, indicating that brusatol inhibits cap-dependent and cap-independent translation. Data are shown as the mean ± SEM (n=3 independent groups) and the * (for Firefly) and # (for Renilla) symbols indicate p<0.05 vs control.
Brusatol inhibits the expression of short-lived proteins
Having identified that brusatol localized to the ER and inhibited both cap-dependent and cap-independent protein translation, it was predicted that brusatol would affect short-lived endogenous proteins or other endogenous proteins that are constantly translated. Therefore, the dose-dependent inhibitory effects of brusatol on NRF2, other short-lived proteins (p53 and p21), or long-lived proteins (the AAA+ chaperone p97 and GAPDH) were tested [10,17,18]. Bruceantin, another translation inhibitor with a chemical structure similar to that of brusatol was also tested. After a 4 h treatment, both brusatol and bruceantin reduced the protein levels of NRF2, p53, and p21 in a dose-dependent manner, while the expression of p97 and GAPDH were not affected (Figure 4A and B). Taken together, these results indicate that brusatol inhibits NRF2 and other short-lived, constantly translated proteins through inhibition of protein translation.
Figure 4.

Brusatol inhibits the expression of short-lived proteins. (A and B) Western blot analysis of short- (NRF2, p53, and p21) and long-lived (p97 and GAPDH) proteins in A549 cells after treatment with brusatol (A) and bruceantin (B) for 4 h.
Discussion
There is substantial evidence indicating that NRF2 up-regulation prevents cancer initiation, which has encouraged the development of NRF2 inducers, such as those found in fruits, vegetables, and other sources of natural products [7]. However, high constitutive levels of NRF2 can confer resistance to cancer cells, the dark-side of NRF2, which argues for the development of NRF2 inhibitors. After successfully identifying the first inhibitor, brusatol, we provided experimental evidence demonstrating that suppressing the NRF2 pathway leads to chemosensitization in a variety of cancer cell lines, and enhances the efficacy of cisplatin using the KRAS-G12D induced murine lung tumor model in vivo [11]. Given that it was the first of its class, we set out to elucidate the mechanism of action of brusatol in regulating NRF2 levels using A549 cells which have constitutively high NRF2 due to a mutation in KEAP1 [19].
In this study, we identified brusatol as a potent inhibitor of protein translation. RNA-seq profiling was initially used to assess changes to the transcriptome following brusatol treatment. Brusatol was shown to induce gene expression changes similar to those of cycloheximide, a known inhibitor of the translational elongation step during protein synthesis. Brusatol also showed a similar gene set enrichment pattern to ricin and puromycin, two other translation inhibitors, suggesting that brusatol may have a greater effect on the proteins with short half-lives.
In an effort to identify the target of brusatol, we modified brusatol with an immunoaffinity fluorescent tag (IAF), a method that has been previously reported to be effective for real time visualization of the subcellular localization of the tagged compound, and for identification of target proteins by immunoprecipitation using an antibody targeting the fluorescent probe [14]. Localization of brusatol-IAF to the ER, coupled with the gene set enrichment patterns shared between brusatol and other translation inhibitors, suggested that brusatol could be concentrating to ribosomes, since the majority of translation occurs at the ER. Consistent with the notion that brusatol may be an inhibitor of protein translation, early reports on brusatol and bruceantin claimed that these drugs inhibit the peptidyl transferase reaction in biochemical assays [20–23]. Furthermore, a crystal structure of a partial ribosome bound to bruceantin was reported in 2009, and molecular footprinting data suggested that bruceantin binds to specific nucleotides within the A-site of the ribosome, some of which are conserved between eukaryotes, prokaryotes, and archaea [24]. Most recently, using a mass spectrometry profiling approach, it was reported that brusatol is an inhibitor of proteins with short half-lives [25].
An interesting feature of brusatol is that its EC50, the effective concentration in reducing NRF2 protein levels to 50%, is 40 nM in most cancer cell lines tested. However, previous studies performed in rabbit reticulocyte lysate utilized brusatol in micromolar concentrations [20–23]. In order to address this finding, we also performed an in vitro transcription and translation assay and determined that brusatol inhibited translation in vitro with an EC50 of 1 µM, consistent with previous reports (data not shown). The large discrepancy between effective doses in vitro may be due to the fact that brusatol concentrates to the ER following cellular uptake (Figure 2C). Moreover, a previous structure-activity relationship study indicates that the hydrophobic side chain of related quassinoids is responsible for their cellular uptake and retention [20], supporting the hypothesis that brusatol and bruceantin concentrate to the ER. This allows for a lower effective concentration in live cells versus cellular lysate or biochemical assays, which are more dilute systems with a variable concentration of translational machinery compared to live cells. Additionally, brusatol blocks both cap-dependent and cap-independent translation, arguing against inhibition of PKR-like endoplasmic reticulum kinase (PERK), a membrane bound kinase in the ER that can inhibit cap-dependent translation through phosphorylation of eIF2α. It is now clear that brusatol is a global translation inhibitor that selectively targets short-lived proteins, including NRF2.
The unwanted side-effect of global protein translation inhibition makes the development of brusatol into a commonly used adjuvant for chemosensitization less desirable. Thus, there is once again a need to develop next generation inhibitors that specifically target the NRF2 pathway. There have been a growing number of reports of NRF2 inhibitors, however, many of these target upstream or unknown factors that may result in other off-target effects [26–28]. Encouragingly, a recently reported compound called ML385 was found to bind to the Neh1 domain of NRF2 and inhibit NRF2-MAFG heterodimerization, selectively interfering with NRF2 target gene expression and enhancing cytotoxicity in KEAP1 deficient NSCLC cells, compared to single agent treatment [29]. However, a reduction in NRF2 mRNA and protein levels was also reported using doses of 5 and 10µM, potentially indicating that there is either toxicity, or an effect on global protein translation associated with this compound, since inhibiting the NRF2-MAFG interaction should not affect NRF2 expression [29]. Specific NRF2 inhibitors will be highly effective for overcoming chemoresistance in tumors with high levels of NRF2, which is a major obstacle for cancer therapy, and these drugs will have a substantial impact on the future treatment of NSCLC and other cancers with high levels of NRF2.
Acknowledgments
The authors are grateful to Dr. Jerry Pelletier for kindly providing the dual Firefly/Renilla reporter constructs and to Dr. Matthew Dodson and Montserrat Rojo de la Vega for their critical review of this manuscript. This work was funded by the National Cancer Institute (RO1CA154377).
References
- 1.Chan BA, Hughes BG. Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Transl Lung Cancer Res. 2015;4(1):36–54. doi: 10.3978/j.issn.2218-6751.2014.05.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang XJ, Sun Z, Villeneuve NF, et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29(6):1235–1243. doi: 10.1093/carcin/bgn095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27(20):2179–2191. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kobayashi A, Kang MI, Okawa H, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24(16):7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24(24):10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23(22):8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3(10):768–780. doi: 10.1038/nrc1189. [DOI] [PubMed] [Google Scholar]
- 8.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 9.Hayes JD, McMahon M. The double-edged sword of Nrf2: subversion of redox homeostasis during the evolution of cancer. Mol Cell. 2006;21(6):732–734. doi: 10.1016/j.molcel.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Ren D, Villeneuve NF, Jiang T, et al. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc Natl Acad Sci U S A. 2011;108(4):1433–1438. doi: 10.1073/pnas.1014275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tao S, Wang S, Moghaddam SJ, et al. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014;74(24):7430–7441. doi: 10.1158/0008-5472.CAN-14-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen W, Sun Z, Wang XJ, et al. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol Cell. 2009;34(6):663–673. doi: 10.1016/j.molcel.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ooi A, Dykema K, Ansari A, et al. CUL3 and NRF2 mutations confer an NRF2 activation phenotype in a sporadic form of papillary renal cell carcinoma. Cancer Res. 2013;73(7):2044–2051. doi: 10.1158/0008-5472.CAN-12-3227. [DOI] [PubMed] [Google Scholar]
- 14.Yu WL, Guizzunti G, Foley TL, Burkart MD, La Clair JJ. An optimized immunoaffinity fluorescent method for natural product target elucidation. J Nat Prod. 2010;73(10):1659–1666. doi: 10.1021/np100371k. [DOI] [PubMed] [Google Scholar]
- 15.Olayanju A, Copple IM, Bryan HK, et al. Brusatol provokes a rapid and transient inhibition of Nrf2 signaling and sensitizes mammalian cells to chemical toxicity-implications for therapeutic targeting of Nrf2. Free Radic Biol Med. 2015;78:202–212. doi: 10.1016/j.freeradbiomed.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Novac O, Guenier AS, Pelletier J. Inhibitors of protein synthesis identified by a high throughput multiplexed translation screen. Nucleic Acids Res. 2004;32(3):902–915. doi: 10.1093/nar/gkh235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tecleab A, Zhang X, Sebti SM. Ral GTPase down-regulation stabilizes and reactivates p53 to inhibit malignant transformation. J Biol Chem. 2014;289(45):31296–31309. doi: 10.1074/jbc.M114.565796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee MS, Seo J, Choi DY, et al. Stabilization of p21 (Cip1/WAF1) following Tip60-dependent acetylation is required for p21-mediated DNA damage response. Cell Death Differ. 2013;20(4):620–629. doi: 10.1038/cdd.2012.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh A, Misra V, Thimmulappa RK, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3(10):e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liao LL, Kupchan SM, Horwitz SB. Mode of action of the antitumor compound bruceantin, an inhibitor of protein synthesis. Mol Pharmacol. 1976;12(1):167–176. [PubMed] [Google Scholar]
- 21.Fresno M, Gonzales A, Vazquez D, Jimenez A. Bruceantin, a novel inhibitor of peptide bond formation. Biochim Biophys Acta. 1978;518(1):104–112. doi: 10.1016/0005-2787(78)90120-x. [DOI] [PubMed] [Google Scholar]
- 22.Willingham W, Jr, Stafford EA, Reynolds SH, et al. Mechanism of eukaryotic protein synthesis inhibition by brusatol. Biochim Biophys Acta. 1981;654(2):169–174. doi: 10.1016/0005-2787(81)90168-4. [DOI] [PubMed] [Google Scholar]
- 23.Rodriguez-Fonseca C, Amils R, Garrett RA. Fine structure of the peptidyl transferase centre on 23 S-like rRNAs deduced from chemical probing of antibiotic-ribosome complexes. J Mol Biol. 1995;247(2):224–235. doi: 10.1006/jmbi.1994.0135. [DOI] [PubMed] [Google Scholar]
- 24.Gurel G, Blaha G, Moore PB, Steitz TA. U2504 determines the species specificity of the A-site cleft antibiotics: the structures of tiamulin, homoharringtonine, and bruceantin bound to the ribosome. J Mol Biol. 2009;389(1):146–156. doi: 10.1016/j.jmb.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vartanian S, Ma TP, Lee J, et al. Application of Mass Spectrometry Profiling to Establish Brusatol as an Inhibitor of Global Protein Synthesis. Mol Cell Proteomics. 2016;15(4):1220–1231. doi: 10.1074/mcp.M115.055509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang XJ, Hayes JD, Henderson CJ, Wolf CR. Identification of retinoic acid as an inhibitor of transcription factor Nrf2 through activation of retinoic acid receptor alpha. Proc Natl Acad Sci U S A. 2007;104(49):19589–19594. doi: 10.1073/pnas.0709483104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang X, Wang H, Fan L, et al. Luteolin inhibits Nrf2 leading to negative regulation of the Nrf2/ARE pathway and sensitization of human lung carcinoma A549 cells to therapeutic drugs. Free Radic Biol Med. 2011;50(11):1599–1609. doi: 10.1016/j.freeradbiomed.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 28.Bollong MJ, Yun H, Sherwood L, Woods AK, Lairson LL, Schultz PG. A Small Molecule Inhibits Deregulated NRF2 Transcriptional Activity in Cancer. ACS Chem Biol. 2015;10(10):2193–2198. doi: 10.1021/acschembio.5b00448. [DOI] [PubMed] [Google Scholar]
- 29.Singh A, Venkannagari S, Oh KH, et al. Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors. ACS Chem Biol. 2016 doi: 10.1021/acschembio.6b00651. [DOI] [PMC free article] [PubMed] [Google Scholar]