

Abstract
The Pim protein kinases are frequently overexpressed in prostate cancer and certain forms of leukemia and lymphoma. 5-(3-Trifluoromethylbenzylidene)thiazolidine-2,4-dione (4a) was identified by screening to be a Pim-1 inhibitor and was found to attenuate the autophosphorylation of tagged Pim-1 in intact cells. Although 4a is a competitive inhibitor with respect to ATP, a screen of approximately 50 diverse protein kinases demonstrated that it has high selectivity for Pim kinases. Computational docking of 4a to Pim-1 provided a model for lead optimization, and a series of substituted thiazolidine-2,4-dione congeners was synthesized. The most potent new compounds exhibited IC50s of 13 nM for Pim-1 and 2.3 µM for Pim-2. Additional compounds in the series demonstrated selectivities of more than 2500-fold and 400-fold for Pim-1 or Pim-2, respectively, while other congeners were essentially equally potent toward the two isozymes. Overall, these compounds are new Pim kinase inhibitors that may provide leads to novel anticancer agents.
Introduction
Pim-1a and Pim-2 are serine/threonine protein kinases that were originally cloned as proviral insertions in murine (Pim) T cell lymphomas.1 The Pim-2 gene is 53% identical to Pim-1, with the greatest divergence occurring at the amino and carboxy termini of the encoded proteins. These kinases share the ability to transform lymphoma cells, although it is not clear that they have completely overlapping biochemical mechanisms of action. The Pim protein kinases were first implicated in the development of prostate cancer by DNA microarray analyses that showed that Pim-1 is overexpressed in human prostate cancer and that its expression correlates with clinical outcome.2 In humans, enhanced levels of nuclear Pim-2 in the tumor cells has been associated with a higher risk of prostate-specific antigen recurrence and with perineural invasion of the prostate gland.3 Consistently, overexpression of Pim-1 has been reported to be related to the grade of prostate cancer,4 with moderate-to-strong cytoplasmic staining of Pim-1 being seen in tumors of 68% of patients with a Gleason score of 7 or higher.5 Pim-1 also is overexpressed in prostate intraepithelial neoplasia, and Pim staining may be helpful in differentiating benign glands from intraepithelial neoplasia.6
Additionally, overexpression of Pim-1 and Pim-2 in human prostate cancer cells markedly enhances their growth as tumors in nude mice7 and enforced expression of Pim-1 prevents cell death from factor withdrawal-induced apoptosis.8 This result may be explained by the observation that Pim can phosphorylate the proapoptotic protein BAD, leading to sequestration by 14–3–3 proteins and inhibition of apoptosis.9–11 This phosphorylation event abrogates the ability of BAD to regulate the activity of the Bcl-xL protein. It has now been shown that Pim-1 plays an important role in the ability of the FLT-3 protein kinase to stimulate leukemic growth by phosphorylating and inactivating BAD and preventing cell death.12
In spite of the interest in Pim-mediated signaling, there are few known inhibitors of these enzymes. The potential for the development of Pim-selective inhibitors is enhanced by the crystal structure of Pim-1, which has been recently solved by multiple groups.13–15 Importantly, the hinge region that contains the ATP binding site has a novel architecture, containing an additional amino acid residue not found in other protein kinases. This residue, proline-123, is incapable of making hydrogen bonds with ATP because of lack of a key amide H-bond donor. This structural feature is a key determinant in the binding mode of Pim inhibitors,13 and several groups have reported structurally novel Pim kinase inhibitors. For example, the Meggers group identified ruthenium half-sandwich complexes, which mimic staurosporine and inhibit Pim-1 and other kinases.16,17 Also, imidazo[1,2-b]pyridazines that inhibit Pim and block the growth of leukemia cells in vitro have recently been described.18,19 A similar chemotype is being developed for Pim inhibition by the SuperGen company.20,21 In 2007, substituted pyridones22 and specific flavinoids23,24 were shown to be Pim inhibitors. However, it is well-known that staurosporine analogues and flavinoids inhibit a variety of protein kinases, leaving the therapeutic usefulness of such compounds unclear. Therefore, improved inhibitors of Pim-1 are required not only for basic research, but also as lead compounds for developing novel anticancer agents. Pierce, et al. recently reported the use of virtual screening for the identification of several Pim-1 inhibitors with a variety of structures.25 The pharmacologic properties of these agents remain to be described. The present report describes the identification of a new chemotype of isozyme-selective Pim inhibitors with high potency.
Results and Discussion
Identification and Characterization of the Novel Pim-1 Inhibitor, 5-(3-Trifluoromethylbenzylidene)thiazolidine-2,4-dione (4a)
Because of the relative lack of high-affinity inhibitors of Pim kinases, we conducted a high-throughput screen for novel inhibitors using recombinant human Pim-1 and S6 kinase/Rsk-2 peptide 2 as the substrate. Compounds from the DIVERSet collection of the ChemBridge Corporation were tested at a final concentration of 100 µM, and hits were defined as compounds that inhibited Pim-1 activity by at least 50%. The known Pim-1 inhibitor (S)-616,17 was used as a positive control (Figure 6). Nearly 50000 compounds were screened, and 10 compounds with IC50s of 20 µM or less were identified (0.034%). This low hit rate reflects the assay design that utilized a relatively high concentration of ATP to select for allosteric or high-affinity ATP-binding site-directed compounds. The four most-potent screening hits are shown in Table 1. Of the Pim-1 inhibitors with IC50s < 20 µM, four compounds contain a benzylidene-linked thiazolidine or imidazolidine nucleus substituted at the 2- and 4- positions by oxo- or thioxo-moieties.
Figure 6.

Structures and potencies of reported Pim-1 inhibitors.
Table 1.
Structures and Potencies of Pim-1 Inhibitors from the ChemBridge Library
| Compound number |
Name | Structure | IC50(µM) |
|---|---|---|---|
| 5721666 (4a) | 5-(3-Trifluoromethylbenzylidene)thiazolidine-2,4-dione | 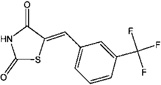 |
3 |
| 7642857 (F7) | 2-(4,6-Dimethylpyrimidin-2-ylsulfanyl)-1-(4-fluoro-3-methylphenyl)ethanone | 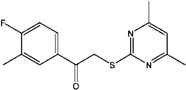 |
5 |
| 5378227 | 4-(2,4-Dioxothiazolidin-5-ylidenemethyl)benzoic acid methyl ester | 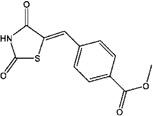 |
10 |
| 7644127 (H4) | 4-[3-(4-Phenylaminophenyl)thioureidomethyl]benzenesulfonamide | 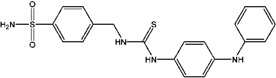 |
12 |
The most potent compound of this chemotype (4a), as well as two structurally dissimilar compounds (2-(4,6-dimethylpyrimidin-2-ylsulfanyl)-1-(4-fluoro-3-methylphenyl)ethanone (F7) and 4-[3-(4-phenylaminophenyl)thioureidomethyl]benzenesulfonamide (H4) (Table 1), were tested for their activities against Pim-1 in an independent in vitro assay and in a cellular assay. Pim-1 protein kinase when overexpressed in prostate cancer cells has been shown to increase the phosphorylation of 4E-BP1.7 On the basis of this in vivo activity, 4E-BP1 protein was shown to be a good substrate for Pim protein kinases in vitro as well (unpublished data). In the first assay, recombinant human Pim-1 protein was incubated with His-tagged 4E-BP1 protein and the test compounds to determine their effects toward a full-length substrate protein. As indicated in Figure 1, all three of the compounds inhibited Pim-1 activity toward 4E-BP1, with 4a and H4 being somewhat more potent than F7. As in the screening assays, (S)-6, a staurosporine-based Pim inhibitor,16,17 was used as a positive control, and this compound inhibited Pim-1 activity by approximately 50% at a concentration of 0.5 µM. Therefore, the inhibitory activities of the new compounds were confirmed by an independent assay.
Figure 1.

Inhibition of Pim-1 in a cell-free kinase assay. His-tagged 4E-BP1 protein was incubated with 0.1 µg Pim-1 protein kinase for 1 h at 30 °C along with [γ-32P]ATP and DMSO (solvent control, lane 1), 1.5 µM 4a (lane 2), 3 µM 4a (lane 3), 2.5 µM F7 (lane 4), 5 µM F7 (lane 5), 2.5 µM H4 (lane 6), 5 µM H4 (lane 7), or 0.5 µM (S)-6 (lane 8), respectively, as described in the Experimental Section. As a negative control, the sample in lane 9 lacked 4E-BP1. Radiolabeled 4E-BP1 was visualized by gel electrophoresis and autoradiography.
The potency of 4a for inhibition of Pim-1 kinase activity was determined using 4E-BP1 as the substrate. 4a caused a dose-dependent reduction in Pim-1-induced 4E-BP1 phosphorylation, with an IC50 of approximately 125 nM (data not shown). It is interesting to note that this is considerably more potent than was determined in the conditions of the screening assay, i.e., 3 µM. This difference in potency may arise from the higher catalytic efficiency of Pim-1 toward the full-length protein substrate in comparison with the S6K peptide used in the screening assay. Similarly, Pim-1 was found to be much more active using a substrate peptide that corresponds to Bad compared with the S6K peptide used in the initial studies. With the Bad peptide, the IC50s for 4a and most of the analogues discussed below were much lower than that for the S6K peptide, reflecting excellent inhibition of the enzyme operating under optimal conditions.
Importantly, the abilities of the compounds to inhibit Pim-1 activity in intact cells were also determined. In this assay, Pim-1 activity was measured as the extent of its autophosphorylation. 293T cells were transfected with Flag-Pim-1 and labeled with 32PO4 and then treated with the test compound for 1 h. As demonstrated in Figure 2, the amount of Flag-Pim-1 recovered from the cells did not change upon exposure to the test compounds. Pronounced autophosphorylation of Pim-1 was evident in all of the samples except those from cells treated with 4a, indicating that this compound is a very effective inhibitor of intracellular Pim-1 autophosphorylation. The lack of inhibition by the known Pim-1 inhibitor (S)-6 confirms that this compound is poorly delivered to the intracellular kinase, which is consistent with its known high-binding to serum proteins. As it is clearly advantageous that the Pim-1 inhibitor be cell permeable, subsequent studies were focused on 4a and its congeners.
Figure 2.

Inhibition of Pim-1 autophosphorylation in intact cells. cDNA-encoding Flag-Pim-1 was transfected into HEK-293T cells and cells were labeled with 32PO4 for 3 h. The following compounds (0.5 µM) were added and the incubations were continued for an additional 1 h: (S)-6 (lane 1), H4 (lane 2), 4a (lane 3), F7 (lane 4), or DMSO (solvent control, lane 5). Pim-1 was immunoprecipitated with Flag beads and 1/10 of the beads were analyzed by Western blotting (input), while the remaining material was analyzed by SDS-PAGE and autoradiography to visualize the autophosphorylation level of Pim-1 as described in the Experimental Section.
To test for competition with ATP, the effects of 4a at different ATP concentrations were determined. As indicated in Figure 3, 4a acts as a competitive inhibitor with respect to ATP, with a calculated Ki of 0.6 µM. To test the selectivity of 4a, it was sent to Caliper Life Sciences for profiling against a diverse panel of protein kinases. As shown in Table 2, 5 µM 4a inhibited Pim-1 and Pim-2 but did not significantly inhibit the other 47 serine/threonine- or tyrosine-kinases tested. Similar testing of a 4a analogue 16a indicated that this compound also is highly selective for Pim kinases, although the kinase DYRK1α was inhibited to a similar extent as Pim-1 and Pim-2. This finding is similar to the recent findings of Pagano et al.,26 who found Pim kinases and DYRK1α to be similarly sensitive to a series of 4,5,6,7-tetrabromon-1H-benzimidazoles. Together, the data indicate that this chemotype has excellent selectivity for Pim kinases.
Figure 3.

Kinetics of inhibition of Pim-1 by 4a. Pim-1 kinase activity was measured using the coupled assay in the presence of the indicated concentrations of ATP and 0 (●), 5 (■), or 10 (▲) µM 4a as described in the Experimental Section.
Table 2.
Selectivity of 4a and 16aa
| Kinase | 4a | 16a | Kinase | 4a | 16a |
|---|---|---|---|---|---|
| ABL | −4 | 6 | IRAK4 | 16 | 5 |
| AKT1 | −9 | 6 | JAK2 | 15 | 18 |
| AKT2 | 3 | −1 | JNK2 | 4 | 14 |
| AKT3 | 7 | 3 | KDR | 14 | −4 |
| AMPK | 2 | 7 | LCK | 2 | 3 |
| AurA | 6 | 4 | LYN | 3 | 8 |
| BMX | 8 | 10 | MAPKAPK2 | 0 | 5 |
| BTK | 10 | 7 | MARK1 | 5 | −6 |
| CAMK2 | −6 | −1 | MET | 3 | −6 |
| CAMK4 | 10 | −8 | MSK1 | −3 | −1 |
| CDK2 | 0 | −2 | MST2 | 2 | 1 |
| CHK1 | −10 | −20 | p38α | −2 | 0 |
| CHK2 | 1 | 4 | p70S6K | 10 | 8 |
| CK1δ | 3 | 7 | PAK2 | 3 | −1 |
| c-Raf | 9 | −1 | PDGFRα | 7 | 6 |
| c-TAK1 | 5 | 0 | PDK1 | 17 | 1 |
| DYRK1α | 20 | 68 | PIM1 | 73 | 36 |
| EGFR | −1 | −3 | PIM2 | 68 | 62 |
| Erk1 | 2 | 1 | PKA | 2 | 3 |
| Erk2 | 4 | −1 | PKCβ2 | −2 | −1 |
| FGFR1 | 3 | 1 | PKCζ | −2 | 3 |
| FLT1 | 18 | NT | PKD2 | −6 | −3 |
| FLT3 | −2 | 1 | PKGα | 1 | 0 |
| FLT3(D835Y) | 12 | 9 | PLK1 | 3 | 9 |
| FYN | 3 | 0 | PRAK | −3 | 4 |
| GSK3β | −1 | 3 | ROCK2 | 5 | 5 |
| HGK | −14 | 9 | RSK1 | 3 | 5 |
| IGF1R | 1 | −1 | SGK1 | 0 | 5 |
| Ikkβ | 12 | 10 | SRC | 4 | 4 |
| INSR | 5 | 4 | SYK | 5 | −3 |
The effects of 4a or 16a on the indicated protein kinases were determined through the Rapid Kinase Advisor profiling service from Caliper Life Sciences. Values represent the percent inhibition of each kinase at 5 µM of the test compound
Molecular Docking of 4a to Pim-1
The molecular structure for compound 4a, 5-[3-(trifluoromethyl)benzylidene]-1,3-thiazolidine-2,4-dione, was built and docked with the crystal structure of Pim-1 (PDB code: 1XWS) using the suite of programs within the DiscoveryStudio interface (Accelrys) (Figure 4). The ligand and protein structures were prepared for docking by the addition of hydrogens, followed by the assignment of atom types and charges within the CHARMm forcefield. This was followed by creation of the docking grids within a 6 Å radius of the native ligand and molecular mechanics/energy minimization docking of flexible 4a into the rigid binding site of Pim-1. After generation of 100 poses, plausible binding modes were generated through energetic ranking by CDocker nonbonded energies and similarity to known interactions for Pim-1 ligands within the RCSB protein databank. Evaluation with these criteria suggested a binding mode for 4a in which the thiazolidine NH group donates a H-bond to the carbonyl oxygen of Glu121 and the trifluorophenyl group makes numerous van der Waals contacts with the hydrophobic cleft. These results suggested that the thiazolidine ring in particular is a good candidate for modification, as according to the pharmacophore model shown in Figure 4 right, this ring system only contributes a hydrogen bond to the hinge region while making few other nonbonded contacts. While the thiazolidine ring lacks scope for diversity, the alternative imidazoline ring has potential for substitution on the N1 with hydrophobic groups as observed with the XWS ligand. The carbonyl groups could be replaced with hydrophobic functionality in order to exploit contacts with Pro123 and Leu44 among other residues. Appropriate modification of the aromatic ring also was indicated in initial modeling results to have potential for increasing potency of this series and an initial set of derivatives was proposed. Further refinement of the binding mode was completed subsequent to obtaining SAR information from the synthesized compounds.
Figure 4.

Computational docking of 4a to Pim-1. Left panel: docking of 5-[3-(trifluoromethyl)benzylidene]-1,3-thiazolidine-2,4-dione (4a) with the crystal structure of Pim-1 kinase (1XWS) showing interacting residues and pharmacophore centers near the ATP binding site. Right panel: 2-D pharmacophore for 4a bound to Pim-1.
Synthesis of 4a Analogues
As indicated above, the compound 4a and a few congeners were identified as Pim-1 inhibitors through screening a commercial library of compounds. Therefore, we synthesized a series of congeners to begin SAR studies of this chemotype, and the approach to the synthesis of 5-arylidene-2,4-thiazolidinediones (1a–4a, 8a–18a, and 20a–24a, as indicated in the Table 3) is shown in Scheme 1.
Table 3.
Biological effects of the new compoundsa
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| compound | geometric isomer |
R1 | R2 | R3 | R4 | IC50 for Pim-1 (µM) |
IC50 for Pim-2 (µM) |
IC50 for PC3 cells (µM) |
| 1a | Z | CH3 | H | H | H | 0.06 ± 0.02 | 4.4 ± 0.2 | > 100 |
| 2a | Z | H | CH3 | H | H | 0.16 ± 0.04 | 0.1 ± 0.1 | 97 ± 0 |
| 3a | Z | CF3 | H | H | H | 0.33 ± 0.13 | 0.08 ± 0.02 | 38 ± 31 |
| 4a | Z | H | CF3 | H | H | 0.024 ± 0.006 | 0.1 ± 0.3 | 17 ± 6 |
| 5a | Z | H | CF3 | H | CH2C6H5-4-OCF3 | 28 ± 22 | > 100 | > 100 |
| 6a | Z | H | CF3 | CH3OCH2O | H | 0.46 ± 0.32 | 1.5 ± 0.1 | 68 ± 18 |
| 7a | Z | H | CF3 | CH3O(CH2)2O | H | 0.28 ± 0.13 | 42 ± 12 | 80 ± 12 |
| 8a | Z | C2H5 | H | H | H | 0.6 ± 0.1 | 0.3 ± 0.1 | 65 ± 18 |
| 9a | Z | (CH3)2CH | H | H | H | 0.04 ± 0.03 | > 100 | 28 ± 13 |
| 10a | Z | CH3O | H | H | H | 5.1 ± 5.0 | 0.3 ± 0.1 | 11 ± 1 |
| 11a | Z | CF3O | H | H | H | 0.3 ± 0.2 | 0.5 ± 0.2 | 48 ± 27 |
| 12a | Z | H | CH3O | H | H | 0.65 ± 0.05 | 0.04 ± 0 | 73 ± 13 |
| 13a | Z | H | CF3O | H | H | 0.067 ± 0.061 | 0.9 ± 0.4 | 6.4 ± 2.4 |
| 14a | Z | C2H5O | H | H | H | 0.17 ± 0.04 | 43 ± 18 | 65 ± 17 |
| 15a | Z | H | CF2HCF2O | H | H | 0.073 ± 0.053 | 2.2 ± 1.1 | 3.2 ± 0.5 |
| 16a | Z | C3H7O | H | H | H | 0.15 ± 0.11 | 0.02 ± 0.01 | 48 ± 8 |
| 17a | Z | (CH3)2N | H | H | H | 6.0 ± 1.7 | 0.1 ± 0.1 | 66 ± 13 |
| 18a | Z | F | H | H | H | 0.013 ± 0.01 | 2.3 ± 0.1 | 93 ± 5 |
| 19a | Z | F | H | H | CH2C6H4-2-F | 45 ± 15 | > 100 | > 100 |
| 20a | Z | H | F | H | H | 0.13 ± 0.06 | 0.4 ± 0.2 | 83 ± 9 |
| 21a | Z | Cl | H | H | H | 0.04 ± 0.03 | 0.2 ± 0.1 | 49 ± 8 |
| 22a | Z | H | Cl | H | H | 0.60 ± 0.51 | 0.04 ± 0.15 | 63 ± 6 |
| 23a | Z | Br | H | H | H | 28 ± 23 | 0.09 ± 0.04 | 85 ± 12 |
| 24a | Z | H | Br | H | H | 45 ± 11 | 0.1 ± 0.02 | 58 ± 11 |
| 25b | CH2C6H4-2-F | 63 ± 3.0 | > 100 | 88 ± 10 | ||||
| 26c | E/Z (9/1) | Cl | H | H | CH2C6H5 | 69 ± 6.5 | > 100 | 67 ± 28 |
| 27c-1 | E | CH3O | H | H | CH2C6H5 | 38 ± 23 | > 100 | 74 ± 20 |
| 27c-2 | E/Z (83/17) | CH3O | H | H | CH2C6H5 | 75 ± 5.0 | > 100 | > 100 |
| 28c | E/Z (4/1) | C2H5O | H | H | CH2C6H5 | 73 ± 7.5 | > 100 | 18 ± 9.7 |
| 29c-1 | E | Br | H | H | CH2C6H5 | 58 ± 7.5 | > 100 | 68 ± 22 |
| 30c | E | (CH3)2N | H | H | CH2C6H5 | 78 ± 7.5 | > 100 | 57 ± 23 |
| 31c | E/Z (5/1) | H | H | CH3O | CH2C6H5 | 78 ± 2.5 | > 100 | 80 ± 15 |
Inhibition of Pim-1 and Pim-2 enzymatic activity and PC3 cell proliferation by arylidene-2,4-thiazolidinediones (1a–24a), 3-(2-fluorobenzyl)thiazolidine-2,4-dione (25b), and arylidenehydantoins (26c–31c) are shown. Data represents the mean ± SEM for at least 3 experiments.
Scheme 1.

General Synthesis of 5-Arylidene-2,4-thiazolidinediones
As described by Momose et al.27 and Bruno et al.,28 2,4-thiazolidinedione can undergo a Knoevenagel condensation with a variety of meta- and para-substituted benzaldehydes to produce 5-arylidene-2,4-thiazolidinones (Scheme 1). In this piperidine-catalyzed reaction, the exocyclic double bond is exclusively in the Z configuration because of the high degree of thermodynamic stability of this isomer. Thirty-three compounds within the benzylidene-thiazolidine-2,4-dione chemotype were synthesized and evaluated, demonstrating that these compounds can be made through a Knoevenagel condensation with a variety of meta- and para-substituted benzaldehydes with good-to-excellent yields.
Compounds 6a and 7a were suggested by the initial round of computational modeling based on the docking of the screening hit 4a. They were prepared with a three-step reaction applying ortho-lithiation strategies illustrated in Scheme 2. 2-Trifluoromethylphenol was treated with sodium hydride, followed by reaction with chloromethylmethyl ether or chloroethylmethyl ether to produce 2-methoxymethoxy-1-trifluoromethylbenzen (6a1) or 2-methoxyethoxy-1-trifluoromethylbenzene (7a1). Because methoxymethoxy and methoxyethoxy groups are strongly ortho-directing,29 further intermediates were ortho-metalated with nBuLi. The corresponding arylaldehydes (6a2 and 7a2) were obtained by addition of DMF to the metalated intermediates, followed by hydrolysis with water. The arylaldehydes were then condensed with 2,4-thiazolidinedione to afford the compounds 6a and 7a with quite low yields, probably due to the steric hindrance of the arylaldehydes by the 2-position substitution.
Scheme 2.

Synthesis of Compounds 6a and 7aa
a(a) NaH/DMF/Cl(CH2)nOCH3; (b) (1) nBuLi, −78 °C, Ar, (2) DMF, RT; (c) piperidine, reflux.
Also suggested by computational modeling, compounds 5a and 19a were synthesized via a three-component condensation in a one-pot reaction using the ionic solvent 1-n-butyl-3-methylimidazolium hexafluorophosphate as depicted in Scheme 3.30 However, an attempt to synthesize 3-(2-fluorobenzyl)-5-(4-methoxybenzylidene)thiazolidine-2,4-dione by this reaction was unsuccessful, producing only compound 25b, 3-(2-fluorobenzyl)thiazolidine-2,4-dione.
Scheme 3.

Synthesis of 3-Aryl-5-arylidene-2,4-thiazolidinediones
5-Arylidene-3-benzylhydantoins (26c–31c) were also suggested by computational modeling and were synthesized using the strategy as described in Scheme 4. Although this is similar to that used for the preparation of 5-arylidenethiazolidine-2,4-diones, the reactions did not always proceed smoothly and often resulted in a mixture of Z/E isomers, which were difficult to separate by routine silica gel chromatography. As expected, the E-isomer was the major compound in the Z/E-mixtures.31
Scheme 4.

Synthesis of 5-Arylidene-3-benzylhydantoins
Evaluation of 4a Analogues
Each of these compounds was tested for its ability to inhibit recombinant Pim-1 using the ATP-depletion assay described above, except that the ATP concentration was 0.1 µM and the Bad peptide was used as the phosphate acceptor. The results are summarized in Table 3. The data demonstrate that several new compounds are significantly more potent than the original screening hit, with IC50s for inhibition of Pim-1 ranging from 0.013 to 45 µM and for inhibition of Pim-2 ranging from 0.02 to >100 µM. Substitution of the thiazolidine nitrogen decreased the potency of the compounds by more than 1000-fold, e.g., 4a vs 5a. Benzylidines with Cl- or F-substitutions at the 4-position were approximately 10-fold more potent than the corresponding 3-substituted compounds, while 3-Br- and 4-Br-congeners had poor Pim-1 inhibitory activity but substantial Pim-2 inhibitory activity. Trifluoromethoxy-substituted compounds were 10- to 15-fold more potent than the corresponding methoxy compounds, e.g., 11a vs 10a. It is also interesting to note that certain compounds are significantly more potent toward either Pim-1 or -2 than the other isozyme. For example, 9a is more than 2500-fold selective for Pim-1, and 24a is more than 400-fold selective for Pim-2. This provides the opportunity to pharmacologically distinguish the roles of the kinase in prostate cancer biology. Compounds that inhibit both isozymes, e.g., 16a, may be more effective as therapeutic agents if there is redundancy in the functions of Pim-1 and Pim-2.
After the synthesis and evaluation of Pim inhibitors based on the initial computational design, retrospective analysis of the structure–activity relationship and further refinement of the 3-D pharmacophore was carried out. This analysis showed that a refined binding mode as shown in Figure 4 best explained the SAR and provided insights for the design of next generation compounds. The potency increase of the triflouromethoxy and the smaller halogen substitutions on the benzylidine can be attributed to more optimal interactions of these groups with Phe49 of the Pim-1 active site. The decreased binding affinity of analogues derivatized at R3 is explained by steric clashes of the ortho substitutions with Val52 or Ile185. This was not predicted in the initial binding hypothesis, which described a different conformation of the benzylidine group. Further optimization of activity and selectivity is possible through insights provided by the refined binding mode.
The cytotoxicity of each of the compounds toward PC3 prostate carcinoma cells was determined. The compounds exhibited IC50s that ranged from 3 to >100 µM, with the least effective Pim inhibitors generally having no toxicity at the highest dose tested. Because the IC50 for proliferation reflects both the affinity of the compound for the target and its ability to enter the cells, this series of compounds appears to have reasonably poor penetration. Alternately, inhibition of Pim alone may not be sufficient to drive these cells into apoptosis, and combinatorial studies with other signaling inhibitors and cytotoxic drugs will be conducted.
Finally, we have begun to characterize the in vivo toxicity and efficacy of a representative Pim inhibitor. Compound 16a was chosen because it has excellent potency for inhibition of both Pim-1 and Pim-2. Dose-escalation studies indicated that mice tolerate intraperitoneal doses of the compound of 50 mg/kg daily for 5 days, while 100 mg/kg is overtly toxic. Therefore, toxicity and antitumor studies were limited to doses of or below 50 mg/kg. As indicated in Table 4, subchronic dosing with 16a did not affect the levels of red or white blood cells, including lymphocytes, monocytes, and granulocytes, indicating that the compound does not have myelosuppressive effects. The blood chemistry profile also indicated that 16a does not have toxicity toward the liver as the albumin, alkaline phosphatase, and alanine aminotransferase levels were unchanged. Similarly, pancreatic function, monitored by amylase activity, and kidney function, monitored by electrolyte, blood urea nitrogen, and creatinine levels, were also unaltered in the 16a-treated mice. However, elevations in blood glucose levels were detected in mice treated with the highest dose of 16a, suggesting an alteration in metabolism, which is consistent with inhibition of signaling through mTOR, which is known to be regulated by Pim activity.32 To evaluate the antitumor activity of 16a, the compound was administered to Balb/c mice bearing tumors of JC murine mammary adenocarcinoma cells. As indicated in Figure 5, treatment of the animals with 16a for 5 days per week did not cause a loss of body weight, consistent with the toxicology studies described above. However, the compound reduced the growth of tumors by approximately 50%. Therefore, the Pim inhibitors of this chemotype have good potential for further optimization as anticancer agents.
Table 4.
Toxicologic Evaluation of 16aa
| parameter | units | control | 3 mg/kg 16a | 10 mg/kg 16a | 50 mg/kg 16a |
|---|---|---|---|---|---|
| white blood cells | 109/L | 3.55–9.83 | 3.73–7.99 | 4.95–7.03 | 3.65–7.1 |
| lymphocytes | 109/L | 3.44–8.88 | 3.61–6.78 | 3.94–5.84 | 3.5–5.62 |
| monocytes | 109/L | 0.04–0.36 | 0.11–0.34 | 0.08–0.28 | 0.04–0.35 |
| granulocytes | 109/L | 0.06–0.36 | 0.01–0.91 | 0.61–0.93 | 0.08–1.13 |
| red blood cells | 1012/L | 10.37–11.76 | 10.37–11.33 | 9.22–10.93 | 8.99–10.98 |
| hemoglobin | g/dL | 13.2–14 | 12.8–14 | 11.7–14.1 | 12.2–13.5 |
| albumin | g/dL | 3.2–3.4 | 3.3–3.4 | 2.9–3.4 | 2.6–3.1 |
| alkaline phosphatase | U/L | 87–134 | 85–127 | 85–113 | 80–86 |
| alanine aminotransferase | U/L | 47–87 | 40–134 | 44–46 | 37–52 |
| amylase | U/L | 926–981 | 878–1206 | 816–1054 | 760–967 |
| blood urea nitrogen | mg/dL | 15–19 | 17–19 | 11–16 | 14–22 |
| phosphate | mg/dL | 5.7–9.5 | 5.1–5.6 | 5.7–6.2 | 5.5–8.1 |
| creatinine | mMg/dL | 0.2–0.3 | 0.2–0.4 | 0.2–0.3 | 0.2–0.3 |
| Na+ | mmol/L | 150–155 | 150–155 | 144–151 | 145–153 |
| K+ | mmol/L | 7.7–7.9 | 7.3–7.5 | 7.2–8.2 | 6.3–6.7 |
| glucose | mg/dL | 70–94 | 80–90 | 87–129 | 136–163 |
The indicated doses of compound 16a were administered by intraperitoneal injection daily for 7 days and blood was collected after an additional 7 days of observation. The ranges of values for cell counts and blood chemistry are given.
Figure 5.

Antitumor activity of 16a. Female Balb/C mice were injected subcutaneously with JC cells (1 × 106) suspended in PBS. After palpable tumor growth, animals were treated five days per week by intraperitoneal injection of vehicle alone (▼) or 50 mg/kg of 16a (■). Values represent the mean ± standard error tumor volumes. n = 5 mice per group.
Conclusions
Pim protein kinases are receiving increasing attention because of their roles in prostate cancer and leukemias. Consequently, several types of Pim-1 inhibitors have recently been described (Figure 6), with several examples that take advantage of the unique structure of the ATP binding domain of these kinases. Substituted thiazolidine-2,4-diones provide a new chemotype that inhibits the Pim family of protein kinases with excellent selectivity in spite of their acting as ATP competitors. A binding mode that rationalizes this selectivity has been proposed by computational docking and provides a model for the further refinement of this chemotype. Interestingly, the current compounds include examples that selectively target Pim-1 or Pim-2, thereby providing new pharmacologic probes that may be useful for differentiating the biological roles of these Pim isozymes. Initial in vivo testing of a potent Pim-1 and Pim-2 inhibitor demonstrates the ability of these compounds to attenuate tumor growth in the absence of undue toxicity to the host. Therefore, the new Pim inhibitors have considerable promise for development as new antitumor and antileukemia agents.
Experimental Section
Chemistry
All chemical reagents were purchased from Sigma-Aldrich except ethanol (200 proof) that was from EMD Chemicals. Proton magnetic resonance and carbon magnetic resonance were recorded on Varian INOVA 400 MHz spectrometer. High resolution mass spectra were determined by the Department of Chemistry at the University of South Carolina. Radial chromatography was performed on a Harrison Research Chromatotron using EMD 60 PF254 containing gypsum. Thin layer chromatography was performed with Sigma-Aldrich silica gel aluminum-backed plate with UV visualization. The following synthetic conditions have not been optimized.
(Z)-5-(4-methylbenzylidene)thiazolidine-2,4-dione (1a)
2,4-Thiazolidinedione (118 mg, 1 mmol) and 4-tolualdehyde (119 µL, 121 mg, 1 mmol) were dissolved in ethanol (7 mL), followed by addition of piperidine (79 µL, 0.80 mmol). After refluxing for 24 h, the mixture was poured into water (~60 mL) and acidified with acetic acid to a pH of 3–4 and left at 4 °C overnight. After filtration and washing with methanol, a yellow solid was collected (150 mg, 0.68 mmol, 68% yield). 1H (DMSO) δ 7.76 (s, 1 H), 7.49 (d, J = 8.0 Hz, 2 H), 7.35 (d, J = 8.0 Hz, 2 H), 2.36 (s, 3 H). 13C NMR (DMSO) δ 168.1, 167.6, 140.9, 132.0, 130.5, 130.2 (2 C), 130.1 (2 C), 122.5, 21.2.
(Z)-5-(3-methylbenzylidene)thiazolidine-2,4-dione (2a)
2,4-Thiazolidinedione (117 mg, 1 mmol) and m-tolualdehyde (118 µL, 120 mg, 1 mmol) were dissolved in ethanol (7 mL), followed by addition of piperidine (79 µL, 0.8 mmol). After refluxing for 20 h, the yellow solution was poured into water (~60 mL) and acidified with acetic acid to a pH of 3–4, and left at 4 °C overnight. After filtration, the collected solid was carefully washed with methanol to afford an orange product (135 mg, 0.62 mmol, 62% yield). 1H (DMSO) δ 12.59 (bs, 1 H), 7.75 (s, 1 H), 7.37–7.45 (m, 3 H), 7.30 (d, J = 7.2 Hz, 1 H), 2.37 (s, 3 H). 13C NMR (DMSO) δ 168.1, 167.5, 138.8, 133.2, 132.0, 131.3, 130.7, 129.4, 127.2, 123.5, 21.1.
(Z)-5-(4-Trifluoromethylbenzylidene)thiazolidine-2,4-dione (3a)
2,4-Thiazolidinedione (119 mg, 1 mmol) and 4-trifluoromethylbenzaldehyde (136 µL, 177 mg, 1 mmol) were dissolved in ethanol (7 mL), followed by addition of piperidine (79 µL, 0.8 mmol). The mixture was refluxed for 24 h. The yellow solution was poured into water (~60 mL), and a yellow precipitate appeared immediately. The mixture was acidified with acetic acid to a pH of 3–4 and then left at 4 °C overnight. After filtration, the yellow solid was washed with methanol to afford the product (147 mg, 0.54 mmol, 54% yield). 1H (DMSO) δ 7.89 (d, J = 8.4 Hz, 2 H), 7.87 (s, 1 H), 7.81 (d, J = 8.4 H, 2 H). 13C NMR (DMSO) δ 167.7, 167.3, 137.2, 130.6 (2 C), 130.0, 129.7, 126.9, 126.2 (t, J = 3.8 Hz, 2 C), 120.4 (t, J = 271 Hz, 1 C).
(Z)-5-(3-Trifluoromethylbenzylidene)thiazolidine-2,4-dione (4a)
2,4-Thiazolidinedione (119 mg, 1 mmol) and 3-trifluoromethylbenzaldehyde (136 µL, 177 mg, 1 mmol) were dissolved in ethanol (7 mL), followed by addition of piperidine (79 µL, 0.8 mmol). After reflux for 20 h, the yellow solution was poured into water (~60 mL) and acidified with acetic acid to a pH of 3–4. After filtration, the solid was purified by radial chromatography (silica) eluted with 5% methanol in chloroform to afford an off-white white solid (79 mg, 0.29 mmol, 29% yield). Rf = 0.25, silica, 3% methanol in chloroform. 1H (CDCl3) δ 7.88 (s, 1 H), 7.75 (s, 1 H), 7.70 (d, J = 7.6 Hz, 1 H), 7.68 (d, J = 7.6 H, 1 H), 7.63 (J1 = J2 = 7.6 Hz, 1 H). 13C NMR (CDCl3) δ 166.5, 166.3, 133.8, 132.7, 132.5, 132.0 (t, J = 32.6 Hz, 1 C), 129.9, 127.1 (t, J = 3.8 Hz, 1 C), 127.0 (t, J = 3.8 Hz, 1 C), 124.6, 123.5 (t, J = 271 Hz, 1 C).
(Z)-3-(4-Trifluoromethoxybenzyl)-5-(3-trifluoromethylbenzylidene)thiazolidine-2,4-dione (5a)
2,4-Thiazolidinedione (59 mg, 0.5 mmol) and m-trifluorotolualdehyde (67 µL, 87 mg, 0.5 mmol) were dissolved in 1-butyl-3-methylimidazolium hexafluorophosphate, [bmim]PF6, (2 mL), followed by addition of Et3N (84 µL, 0.6 mmol) and 4-trifluoromethoxybenzyl bromide (0.6 mmol, 96 µL). The reaction was carried out at 60 °C for 17 h. After cooling down to room temperature, the mixture was extracted with ether (4 × 15 mL). After filtration, the concentrated residue was purified by radial chromatography (silica) eluted with 15–25% ethyl acetate in hexane to afford a semisolid (5 mg, 0.011 mmol, 2% yield). Rf = 0.28, 25% ethyl acetate in hexane. 1H (CDCl3) δ 7.92 (s, 1 H), 7.73 (s, 1 H), 7.59–7.70 (m, 3 H), 7.48 (d, J = 8.0 Hz, 2 H), 7.19 (d, J = 8.0 Hz, 2 H), 4.90 (s, 2 H). 13C NMR (CDCl3) δ 167.3, 165.9, 149.5, 134.2, 133.8, 132.8, 132.5, 132.1 (q, J = 33 Hz, 1 C), 130.8 (2 C), 130.1, 127.0–127.2 (m, 2 C), 123.8, 123.7 (q, J = 271 Hz, 1 C), 121.5 (2 C), 120.6 (q, J = 256 Hz, 1 C), 44.8. HR-MS (EI): calcd 447.0364 for C19H11F6NO3S, found 447.0360.
1-Methoxymethoxy-2-trifluoromethoxybenzene (6a1)
A suspension of 60% NaH (1.07 g, 27 mmol) in anhydrous DMF (15 mL) was chilled to 0 °C under argon, and 2-trifluoromethylphenol (3.72 g, 23 mmol) in DMF (5 mL) was added to it dropwise. The the mixture was allowed to warm to RT and stirred for 1 h, followed by the slow addition of chloromethyl methyl ether (2.97 g, 37 mmol) and the suspension was stirred overnight. Note: smoke was produced during this addition. Ice–water (30 mL) was then slowly added, and the suspension became clear solution. The mixture was extracted with ether (3 × 30 mL), and the collected ether was washed with 1 N NaOH (30 mL), 1 N HCl (30 mL), and 10% NaCl (30 mL) sequentially and then dried over sodium sulfate. After passing through a pad of silica eluted with ether, the solvent was removed with nitrogen flushing to afford a colorless oil (3.42 g, 72%). Rf = 0.44, 25% ethyl acetate in hexane. 1H (CDCl3) δ 7.57 (d, J = 7.6 Hz, 1 H), 7.46 (dd, J1 = 8.4 Hz, J2 = 7.6 Hz, 1 H), 7.22 (d, J = 8.4 Hz, 1 H), 7.04 (dd, J1 = J2 = 7.6 Hz, 1 H), 5.25 (s, 2 H), 3.49 (s, 3 H). 13C NMR (CDCl3) δ 155.0 (m, 1 C), 133.3, 127.1 (q, J = 5.3 Hz, 1 C), 123.8 (q, J = 270 Hz, 1 C), 121.2, 119.7 (q, J = 30.3 Hz, 1 C), 115.3, 94.3, 56.4.
Methoxymethoxy-3-trifluoromethylbenzaldehyde (6a2)
A dried (100 mL) three-necked flask was charged with freshly dried THF (20 mL) and chilled to <−70 °C under argon. N,N,N′,N′-tetramethylethylenediamine (480 µL, 3.25 mmol) and 2.5 M nBuLi in hexane (1.3 mL, 3.25 mmol) were then added and the system was recooled to <−70 °C. 1-Methoxymethoxy-2-trifluoromethylbenzene (432 mg, 2.1 mmol) in anhydrous THF (5 mL) was then added dropwise so that the temperature was kept below −70 °C and the mixture was stirred for 1 h. DMF (250 µL, 3.25 mmol) was then added to it, and the cooling bath was removed after 5 min. The temperature was gradually raised to room temperature and the color of the mixture changed to red from yellow and the reaction was continued for 20 h at room temperature. After dropwise addition of saturated NH4Cl (25 mL), the mixture was extracted with ethyl acetate (3 × 25 mL). The combined ethyl acetate was washed with 1 N HCl (25 mL), water (25 mL), and 10% NaCl (25 mL) sequentially and then dried over sodium sulfate. After passing through a pad of silica gel eluted with 25% ethyl acetate in hexane, the concentrated residue was purified by radial chromatography (silica) eluted with 5–15–25–50–100% ethyl acetate in hexane to afford a yellowish oil (Rf = 0.43, 25% ethyl acetate in hexane). NMR spectra indicated that the compound was not pure, so it was further purified by radial chromatography (silica) eluted with hexane–chloroform to afford the pure compound (100 mg, 20% yield). Rf = 0.19, chloroform. 1H (CDCl3) δ 10.32 (bd, J = 0.4 Hz, 1 H), 8.07 (dd, J1 = 7.6 Hz, J2 = 1.2 Hz, 1 H), 7.88 (dd, J1 = 7.6 Hz, J2 = 1.2 Hz, 1 H), 7.38 (dd, J1 = J2 = 7.6 Hz, 1 H), 5.13 (s, 2 H), 3.64 (s, 3 H). 13C NMR (CDCl3) δ 189.6, 158.7, 132.9 (q, J = 5.3 Hz, 1 C), 132.7, 131.7, 124.9, 123.2 (q, J = 171 Hz, 1 C), 102.7 (2 C), 58.2.
(Z)-5-(2-Methoxymethoxy-3-trifluoromethylbenzylidene)thiazolidine-2,4-dione (6a)
Piperidine (15 µL, 0.15 mmol) was added to a mixture of 2,4-thiazolidinedione (18 mg, 0.15 mmol) and 2-methoxymethoxy-3-trifluoromethyl-benzaldehyde (35 mg, 0.15 mmol) in ethanol (3 mL) and refluxed overnight. After cooling to room temperature, the mixture was poured into water (~30 mL) and acidified with acetic acid to a pH of 3–4. After filtration, the collected solid was dissolved in chloroform and passed through a pad of silica eluted with 5% methanol in chloroform. The concentrated residue was purified by radial chromatography (silica) eluted with 0–1–3% methanol in chloroform to afford the compound 6a (7 mg, 14% yield). Rf = 0.24, 3% methanol in chloroform. 1H (CDCl3) δ 8.20 (s, 1 H), 7.71 (d, J = 8.0 Hz, 1 H), 7.69 (d, J = 8.0 Hz, 1 H), 7.36 (dd, J1 = J2 = 8.0 Hz, 1 H), 5.05 (s, 2 H), 3.70 (s, 3 H). 13C NMR (DMSO) δ 167.0, 166.3, 156.2, 132.3, 129.5, 129.4 (q, J = 5.3 Hz, 1 C), 128.4, 125.8 (q, J = 30 Hz, 1 C), 125.0, 124.9, 123.2 (q, J = 271 Hz, 1 C), 102.5, 58.3. HR-MS (EI): clcd 333.0283 for C13H10F3NO4S, found 333.0283.
1-Methoxyethoxy-2-trifluoromethylbenzene (7a1)
A suspension of 60% NaH (1.05 g, 26 mmol) in DMF (15 mL) was chilled to 0 °C under argon, and 2-trifluoromethylphenol (3.38 g, 21 mmol) in DMF (5 mL) was added to it dropwise. The mixture was allowed to warm to RT and was stirred for 1 h, followed by the slow addition of bromoethyl methyl ether (3.59 g, 26 mmol). The reaction was continued for 18 h, and then ice–water (30 mL) was slowly added to it. The mixture was extracted with ether (3 × 30 mL), and the collected ether was washed with 1 N NaOH (30 mL), 1 N HCl (30 mL), and 10% NaCl (30 mL) sequentially and then dried over sodium sulfate. After filtration through a funnel with cotton, the solvent was removed with nitrogen flushing to afford a colorless oil (3.55 g, 77% yield). Rf = 0.62, ether. 1H (CDCl3) δ 7.55 (d, J = 7.6 Hz, 1 H), 7.46 (dd, J1 = J2 = 7.6 Hz, 1 H), 6.97–7.01 (m, 2 H), 4.18 (t, d = 4.8 Hz, 2 H), 3.78 (d, J = 4.8 Hz, 2 H), 3.45 (s, 3 H). 13C NMR (CDCl3) δ 156.9 (m, 1 C), 133.3, 127.1 (q, J = 5.3 Hz, 1 C), 123.8 (q, J = 270 Hz, 1 C), 120.4, 119.2 (q, J = 30.3 Hz, 1 C), 113.3, 71.0, 68.9, 59.6.
2-Methoxyethoxy-3-trifluoromethylbenzaldehyde (7a2)
A dried (100 mL) three-necked flask was charged with freshly dried THF (20 mL) and chilled to <−70 °C under argon. A solution of N,N,N′,N′-tetramethylethylenediamine (960 µL, 6.5 mmol) and 2.5 M nBuLi in hexane (2.6 mL, 6.5 mmol) was then added and the mixture was recooled to reached <−70 °C. 1-Methoxyethoxy-2-trifluoromethylbenzene (972 mg, 4.4 mmol) in THF (5 mL) was then added dropwise, keeping the temperature <−70 °C, and the mixture was stirred for 1 h. DMF (501 µL, 6.5 mmol) was then added and the cooling bath was removed after 5 min, allowing the temperature to gradually rise to RT for 20 h. After the dropwise addition of saturated NH4Cl (25 mL), the mixture was extracted with ethyl acetate (3 × 25 mL), and the combined ethyl acetate was washed with 1 N HCl (25 mL), water (25 mL), and 10% NaCl (25 mL) and dried over sodium sulfate. After passing through a pad of silica gel eluted with 25% ethyl acetate in hexane, the concentrated residue was purified by radial chromatography (silica) eluted with 5–15–25–50–100% ethyl acetate in hexane to afford an oil (Rf = 0.09, chloroform). A second cycle of radial chromatography (silica) eluted with hexane–chloroform was necessary to afford the product (32 mg, 0.13 mmol, 3% yield). Rf = 0.27, 1% methanol in chloroform. 1H (CDCl3) δ 10.5 (bd, J = 0.8 Hz, 1 H), 8.06 (dd, J1 = 7.6 Hz, J2 = 1.2 Hz, 1 H), 7.86 (dd, J1 = 7.6 Hz, J2 = 1.2 Hz, 1 H), 7.36 (dd, J1 = J2 = 7.6 Hz, 1 H), 4.24 (t, J = 4.4 Hz, 2 H), 3.79 (t, J = 4.4 Hz, 2 H), 3.45 (s, 3 H). 13C NMR (CDCl3) δ 189.3, 160.4, 133.0 (q, J = 5.2 Hz, 1 C), 132.9, 131.1, 125.5 (q, J = 31 Hz, 1 C), 124.7, 123.3 (q, J = 272 Hz, 1 C), 78.4, 71.6, 59.3.
(Z)-5-(2-Methoxyethoxy-3-trifluoromethyl-benzylidene)thiazolidine-2,4-dione (7a)
Piperidine (5.6 µL, 0.056 mmol) was added to a mixture of 2,4-thiazolidinedione (8 mg, 0.07 mmol) and 2-methoxyethoxy-3-trifluoromethyl-benzaldehyde (17 mg, 0.07 mmol) in ethanol (1.2 mL) and refluxed overnight, during which it became yellow. The mixture was poured into water (~12 mL) and acidified with acetic acid to a pH of 3–4 and then precipitated at 4 °C. After centrifugation and filtration, the collected yellow solid was dissolved in chloroform. After passing through a pad of silica eluted with 5% methanol in chloroform, the concentrated residue was purified by radial chromatography (silica) eluted with 0–1–3–5% methanol in chloroform to afford a white solid (11 mg, 45% yield). Rf = 0.24, 5% methanol in chloroform; Rf = 0.13, 3% methanol in chloroform. 1H (CDCl3) δ 8.88 (bs, 1 H), 8.30 (s, 1 H), 7.69 (d, J = 7.6 Hz, 1 H), 7.67 (d, J = 7.6 Hz, 1 H), 7.34 (dd, J1 = J2 = 7.6 Hz, 1 H), 4.08 (t, J = 4.8 Hz, 2 H), 3.80 (t, J = 4.8 Hz, 2 H), 3.51 (s, 3 H). 13C NMR (CDCl3) δ 166.8, 165.9, 157.3, 132.6, 129.3 (q, J = 4.6 Hz, 1 C), 129.2, 128.6, 125.6 (q, J = 30 Hz, 1 C), 125.5, 124.7, 123.3 (q, J = 272 Hz, 1 C), 76.6, 71.4, 59.4. HR-MS (EI): calcd 347.0439 for C14H12F3NO4S, found 347.0441.
(Z)-5-(4-ethylbenzylidene)thiazolidine-2,4-dione (8a)
2,4-Thiazolidinedione (119 mg, 1 mmol) and 4-ethoxybenzaldehyde (139 µL, 136 mg, 1 mmol) were dissolved in ethanol (7 mL), followed by the addition of piperidine (79 µL, 0.8 mmol). After refluxing for 20 h, the yellow solution was poured into water (~60 mL), acidified with acetic acid to a pH of 3–4, and incubated at 4 °C overnight. After filtration, the solid was washed with methanol to afford a yellow product (116 mg, 0.50 mmol, 50% yield). 1H (DMSO) δ 7.76 (s, 1 H), 7.51 (d, J = 8.4 Hz, 2 H), 7.38 (d, J = 8.4 Hz, 2 H), 2.66 (q, d = 7.6 Hz, 2 H), 1.20 (t, J = 7.6 Hz, 3 H). 13C NMR (DMSO) δ 168.1, 167.6, 147.0, 132.0, 130.7, 130.4 (2 C), 128.9 (2 C), 122.6, 28.3, 15.4.
(Z)-5-(4-Isopropylbenzylidene)thiazolidine-2,4-dione (9a)
2,4-Thiazolidinedione (117 mg, 1 mmol) and 4-methoxybenzaldehyde (148 mg, 151 µL, 1 mmol) were dissolved in ethanol (7 mL), followed by the addition of piperidine (79 µL, 0.8 mmol). After refluxing for 24 h, the yellow solution was poured into water (~60 mL), and acidified with acetic acid to a pH of 3–4. After filtration, it was subjected to radial chromatography (silica) eluted with 3–5% methanol in chloroform to afford the product (125 mg, 0.51 mmol, 51% yield). Rf = 0.31, 3% methanol in chloroform. 1H (CDCl3) δ 7.87 (s, 1 H), 7.44 (d, J = 8.0 Hz, 2 H), 7.34 (d, J = 8.0 Hz, 2 H), 2.96 (tt, J1 = J2 = 6.8 Hz, 1 H), 1.27 (d, J = 6.8 Hz, 6 H). 13C NMR (CDCl3) δ 167.8, 167.4, 152.6, 135.0, 130.8 (2 C), 130.7, 127.7 (2 C), 121.3, 34.4, 23.9 (2 C).
(Z)-5-(4-methoxybenzylidene)thiazolidine-2,4-dione (10a)
2,4-Thiazolidinedione (117 mg, 1 mmol) and 4-methoxybenzaldehyde (136 mg, 123 µL, 1 mmol) were dissolved in ethanol (7 mL), followed by the addition of piperidine (79 µL, 0.8 mmol). After refluxing for 20 h, the yellow solution was poured into water (~60 mL), acidified with acetic acid to a pH of 3–4, and incubated at 4 °C overnight. After filtration, the yellow solid was washed with methanol to afford the product (90 mg, 0.38 mmol, 38% yield). 1H (DMSO) δ 7.74 (s, 1 H), 7.56 (d, J = 8.8 Hz, 2 H), 7.10 (d, J = 8.8 Hz, 2 H), 3.83 (s, 3 H). 13C NMR (DMSO) δ 168.4, 168.1, 160.9, 132.0 (2 C), 131.6, 125.6, 114.9 (2 C), 55.5.
(Z)-5-(4-Trifluoromethoxybenzylidene)thiazolidine-2,4-dione (11a)
2,4-Thiazolidinedione (119 mg, 1 mmol) and 4-trifluoromethoxybenzaldehyde (144 µL, 192 mg, 1 mmol) were dissolved in ethanol (7 mL), followed by the addition of piperidine (79 µL, 0.8 mmol). After refluxing for 20 h, the solution was poured into water (~60 mL) and acidified with acetic acid to a pH of 3–4. After filtration, the solid dissolved in chloroform and was purified by radial chromatography (silica) eluted with 3–5% methanol in chloroform to afford the product (80 mg, 0.28 mmol, 28% yield). Rf = 0.28 (3% methanol in chloroform). 1H (CDCl3) δ 7.85 (s, 1 H), 7.55 (d, J = 8.4 Hz, 2 H), 7.32 (d, J = 8.4 H, 2 H). 13C NMR (CDCl3) δ 167.0, 166.8, 150.6 (t, J = 2.2 Hz, 1 H), 132.8, 131.9 (2 C), 131.4, 123.3, 121.4 (2 C), 120.4 (t, J = 257 Hz, 1 C). HR-MS (EI): calcd 289.0020 for C11H6F3NO3S, found 289.0014.
(Z)-5-(3-Methoxybenzylidene)thiazolidine-2,4-dione (12a)
2,4-Thiazolidinedione (117 mg, 1 mmol) was dissolved in ethanol (8 mL), followed by addition of m-anisaldehyde (122 µL, 1 mmol) and piperidine (79 µL, 0.8 mmol). After refluxing for 20 h, the yellow solution was poured into water (~60 mL), acidified with acetic acid to a pH of 3–4, and incubated at 4 °C overnight. After filtration, the yellow solid was washed with methanol to afford the product (115 mg, 0.49 mmol, 49% yield). 1H (DMSO) δ 7.77 (s, 1 H), 7.45 (dd, J1 = J2 = 8.0 Hz, 1 H), 7.16 (d, J = 8.0 Hz, 1 H), 7.15 (s,1 H), 7.07 (d, J = 8.0 Hz, 1 H), 3.81 (s, 3 H). 13C NMR (DMSO) δ 168.2, 167.7, 160.1, 134.9, 132.2, 130.9, 124.4, 122.3, 116.8, 115.8, 55.7.
(Z)-5-(3-Trifluoromethoxybenzylidene)thiazolidine-2,4-dione (13a)
2,4-Thiazolidinedione (118 mg, 1 mmol) and 4-tolualdehyde (143 µL, 190 mg, 1 mmol) were dissolved in ethanol (7 mL), followed by the addition of piperidine (79 µL, 0.80 mmol). After refluxing for 24 h, the mixture was poured into water (~60 mL) and acidified with acetic acid to a pH of 3–4. After filtration, the yellow solid was purified by radial chromatography (silica) eluted with 3% methanol in chloroform to afford a white solid product (78 mg, 0.27 mmol, 27% yield). Rf = 0.27, 3% methanol in chloroform. 1H (CDCl3) δ 7.88 (bs, 1 H), 7.83 (s, 1 H), 7.52 (dd, J1 = J2 = 8.0 Hz, 1 H), 7.43 (d, J = 8.0 Hz, 1 H), 7.34 (s, 1 H), 7.29 (d, J = 8.0 Hz, 1 H). 13C NMR (CDCl3) δ 167.5, 167.3, 150.0 (m, 1 C), 135.1, 132.8, 130.9, 128.4, 124.8, 123.0, 122.5, 120.6 (q, J = 257.3 Hz, 1 C).
(Z)-5-(4-Ethoxybenzylidene)thiazolidine-2,4-dione (14a)
2,4-Thiazolidinedione (117 mg, 1 mmol) and 4-ethoxybenzaldehyde (150 mg, 139 µL, 1 mmol) were dissolved in ethanol (7 mL), followed by the addition of piperidine (79 µL, 0.8 mmol). After refluxing for 20 h, the yellow solution was poured into water (~60 mL) and acidified with acetic acid to a pH of 3–4. After filtration, the solid was purified by precipitation from methanol–hexane to afford the yellow product (60 mg, 0.24 mmol, 24% yield). 1H (DMSO) δ 12.50 (bs, 1 H), 7.73 (s, 1 H), 7.54 (d, J = 8.6 Hz, 2 H), 7.08 (d, J = 8.6 Hz, 2 H), 4.10 (q, J = 6.8 Hz, 2 H), 1.35 (t, J = 6.8 Hz, 3 H). 13C NMR (DMSO) δ 168.5, 168.3, 160.4, 132.2 (2 C), 131.6, 125.6, 121.0, 115.4 (2 C), 63.7, 14.7.
(Z)-5-[3-(1,1,2,2-Tetrafluoroethoxy)benzylidene]thiazolidine-2,4-dione (15a)
2,4-Thiazolidinedione (117 mg, 1 mmol) and 3-(1,1,2,2-tetrafluoroethoxy)benzaldehyde (160 µL, 222 mg, 1 mmol) were dissolved in ethanol (7 mL), followed by the addition of piperidine (79 µL, 0.80 mmol). After refluxing for 20 h, the mixture was poured into water (~60 mL) and acidified with acetic acid to a pH of 3–4. After filtration, the solid dissolved in chloroform was purified by radial chromatography (silica) eluted with 3–5% methanol in chloroform to afford a solid product (101 mg, 0.31 mmol, 31% yield). Rf = 0.23, 3% methanol in chloroform. 1H (DMSO) δ 12.70 (bs, 1 H), 7.85 (s, 1 H), 7.65 (dd, J1 = J2 = 8.0 Hz, 1 H), 7.59 (d, J = 8.0 Hz, 1 H), 7.52 (s, 1 H), 7.40 (dd, J1 = 8.0 Hz, J2 = 1.2 Hz, 1 H), 6.84 (tt, J1 = 52 Hz, J2 = 3.2 Hz, 1 H). 13C NMR (DMSO) δ 167.6, 167.3, 148.7, 135.4, 131.3, 130.3, 128.1, 125.7, 123.3, 122.9, 116.6 (tt, J1 = 270 Hz, J2 = 28.1 Hz, 1 C), 107.9 (tt, J1 = 247 Hz, J2 = 40.2 Hz, 1 C). HR-MS (EI): calcd 321.0083 for C12H7F4NO3S, found 321.0080.
(Z)-5-(4-Propoxybenzylidene)thiazolidine-2,4-dione (16a)
2,4-Thiazolidinedione (117 mg, 1 mmol) and 4-propoxybenzaldehyde (164 mg, 158 µL, 1 mmol) were dissolved in ethanol (7 mL), followed by the addition of piperidine (79 µL, 0.8 mmol). After refluxing for 20 h, the yellow solution was poured into water (~60 mL), acidified with acetic acid to a pH of 3–4, and incubated at 4 °C overnight. After filtration, the solid was washed with methanol to afford the product (114 mg, 0.43 mmol, 43% yield). 1H (DMSO) δ 12.50 (bs, 1 H), 7.75 (s, 1 H), 7.54 (d, J = 8.8 Hz, 2 H), 7.09 (d, J = 8.8 Hz, 2 H), 4.01 (t, J = 6.8 Hz, 2 H), 1.75 (m, 2 H), 0.98 (t, J = 7.6 Hz, 3 H). 13C NMR (DMSO) δ 168.1, 167.6, 160.7, 132.3 (2 C), 132.1, 125.5, 120.3, 115.5 (2 C), 69.5, 22.1, 10.5. HR-MS (EI): calcd 263.0616 for C13H13NO3S, found 263.0618.
(Z)-5-(4-Dimethylaminobenzylidene)thiazolidine-2,4-dione (17a)
2,4-Thiazolidinedione (121 mg, 1 mmol) and 4-trifluoromethylbenzaldehyde (154 mg, 1 mmol) were dissolved in ethanol (7 mL), followed by the addition of piperidine (80 µL, 0.81 mmol). The mixture was refluxed for 3 h, and the resulting orange suspension was poured into water (~60 mL), acidified with acetic acid to a pH of 3–4, and incubated overnight at 4 °C. After filtration, the yellow solid was washed with methanol to afford the product (175 mg, 0.71 mmol, 71% yield). 1H (DMSO) δ 7.66 (s, 1 H), 7.44 (d, J = 8.8 Hz, 2 H), 6.82 (d, J = 8.8 Hz, 2 H), 3.32 (s, 6 H). 13C NMR (DMSO) δ 168.4, 167.8, 151.6, 133.1, 132.3 (2 C), 120.0, 115.9, 112.2 (2 C), 39.5 (2 C).
(Z)-5-(4-Fluorobenzylidene)thiazolidine-2,4-dione (18a)
2,4-Thiazolidinedione (119 mg, 1 mmol) and 4-trifluoromethylbenzaldehyde (107 µL, 126 mg, 1 mmol) were dissolved in ethanol (7 mL), followed by the addition of piperidine (79 µL, 0.80 mmol). After refluxing for 24 h, the mixture was poured into water (~60 mL) and acidified with acetic acid to a pH of 3–4. After filtration, the solid was purified by radial chromatography (silica) eluted with 3% methanol in chloroform to afford a yellow solid (91 mg, 0.41 mmol, 41% yield). Rf = 0.27, 3% methanol in chloroform. 1H (DMSO) δ 7.81 (s, 1 H), 7.67 (dd, J1 = 8.8 Hz, J2 = 5.6 Hz, 2 H), 7.39 (dd, J1 = J2 = 8.8 Hz, 2 H). 13C NMR (DMSO) δ 168.0, 167.6, 163.0 (d, J = 249 Hz, 1 C), 132.6 (d, J = 8.4 Hz, 2 C), 130.8, 129.9 (d, J = 3 Hz, 1 C), 123.6 (d, J = 2.2 Hz, 1 C), 116.7 (d, J = 22 Hz, 2 C).
(Z)-3-(2-Fluorobenzyl)-5-(4-fluorobenzylidene)thiazolidine-2,4-dione (19a)
2,4-Thiazolidinedione (117 mg, 1 mmol) and 2-fluorobenzbenzaldehyde (127 µL, 149 mg, 1.2 mmol) were dissolved in 1-butyl-3-methylimidazolium hexafluorophosphate, [bmim>]PF6, (2 mL), followed by the addition of Et3N (170 µL, 1.2 mmol) and 2-fluorobenzyl chloride (127 µL, 1.2 mmol). The mixture was stirred at 60 °C for 20 h. After cooling to room temperature, the mixture was extracted with ether (4 × 15 mL), passed through a pad of silica eluted with 25% ethyl acetate in hexane, and the concentrated residue was purified by radial chromatography (silica) eluted with 15–25% ethyl acetate in hexane to afford two products based on differences in their Rfs. One component is a semisolid identified as 3-(2-fluorobenzyl)thiazolidine-2,4-dione (25b). The other component was not pure and was purified again by radial chromatography (silica) eluted with hexane and chloroform to afford a white solid (5 mg, 0.02 mmol, 2% yield) (Rf = 0.33, silica, chloroform). 1H (CDCl3) δ 7.88 (s, 1 H), 7.50 (dd, J1 = 8.4 Hz, 2 H), 7.27–7.37 (m, 2 H), 7.17 (dd, J1 = J2 = 8.4 Hz, 2 H), 7.04–7.14 (m, 2 H), 5.00 (s, 2 H). 13C NMR (CDCl3) δ 167.4, 166.1, 163.9 (d, J = 253 Hz, 1 C), 160.9 (d, J = 247 Hz, 1 C), 133.2, 132.5 (d, J = 8.4 Hz, 2 C), 130.4 (d, J = 3.8 Hz, 1 C), 130.2 (d, J = 7.6 Hz, 1 C), 129.7 (d, J = 3.8 Hz, 1 C), 124.5 (d, J = 3.8 Hz, 1 C), 122.0 (d, J = 14.4 Hz, 1 C), 121.1 (d, J = 2.3 Hz, 1 C), 116.8 (d, J = 22.0 Hz, 2 C), 115.9 (d, J = 21.2 Hz, 1 C), 39.3 (d, J = 4.5 Hz, 1 C). HR-MS (EI): calcd 331.0479 for C17H11F2NO2S, found 331.0477.
(Z)-5-(3-Fluorobenzylidene)thiazolidine-2,4-dione (20a)
2,4-Thiazolidinedione (117 mg, 1 mmol) and 3-fluorobenzaldehyde (124 mg, 105 µL, 1 mmol) were dissolved in ethanol (7 mL), followed by the addition of piperidine (79 µL, 0.8 mmol). After refluxing for 20 h, the mixture was poured into water (~60 mL) and acidified with acetic acid to a pH of 3–4. After filtration, the solid dissolved in chloroform was purified by radial chromatography eluted with 5% methanol in chloroform to afford a yellowish product (100 mg, 0.45 mmol, 45% yield). Rf = 0.36, 5% methanol in chloroform. 1H (DMSO) δ 12.68 (bs, 1 H), 7.80 (s, 1 H), 7.59 (m, 1 H), 7.42–7.48 (m, 2 H), 7.34 (ddd, J1 = J2 = 8.0 Hz, J3 = 2 Hz, 1 H). 13C NMR (DMSO) δ 167.9, 167.6, 162.4 (d, J = 243.6 Hz, 1 C), 135.7 (d, J = 8.3 Hz, 1 C), 131.5 (d, J = 8.3 Hz, 1 C), 130.3, 125.7 (2 C), 117.3 (d, J = 21.2 Hz, 1 C), 116.8 (d, J = 22.8 Hz, 1 C).
(Z)-5-(4-Chlorobenzylidene)thiazolidine-2,4-dione (21a)
2,4-Thiazolidinedione (119 mg, 1 mmol) and 4-chlorobenzaldehyde (142 mg, 1 mmol) were dissolved in ethanol (8 mL), followed by the addition of piperidine (79 µL, 0.8 mmol). After refluxing for 20 h, the yellow solution was poured into water (~60 mL), acidified with acetic acid to a pH of 3–4 and incubated at 4 °C overnight. After filtration, the yellow solid was washed with methanol to afford the product (152 mg, 0.63 mmol, 63% yield). 1H (DMSO) δ 7.79 (s, 1 H), 7.61 (m, 4 H). 13C NMR (DMSO) δ 167.8, 167.4, 135.1, 132.1, 131.8 (2 C), 130.5, 129.5 (2 C), 124.6.
(Z)-5-(3-Chlorobenzylidene)thiazolidine-2,4-dione (22a)
2,4-Thiazolidinedione (119 mg, 1 mmol) and 3-chlorobenzaldehyde (118 µL, 1 mmol) were dissolved in ethanol (8 mL), followed by the addition of piperidine (79 µL, 0.8 mmol). After refluxing for 20 h, the yellow solution was poured into water (~60 mL), acidified with acetic acid to a pH of 3–4 and incubated at 4 °C overnight. After filtration, the yellow solid was washed with methanol to afford the product (113 mg, 0.47 mmol, 47% yield). 1H (DMSO) δ 7.79 (s, 1 H), 7.68 (s, 1 H), 7.52–7.60 (m, 3 H). 13C NMR (DMSO) δ 167.8, 167.4, 135.5, 134.1, 131.3, 130.3, 130.2, 130.1, 128.0, 125.7.
(Z)-5-(4-Bromobenzylidene)thiazolidine-2,4-dione (23a)
2,4-Thiazolidinedione (117 mg, 1 mmol) and 4-bromobenzaldehyde (185 mg, 1 mmol) were dissolved in ethanol (7 mL), followed by the addition of piperidine (79 µL, 0.8 mmol). After refluxing for 20 h, the yellow solution was poured into water (~60 mL), acidified with acetic acid to a pH of 3–4 and incubated at 4 °C overnight. After filtration, the yellow solid was washed with methanol to afford the product (220 mg, 0.77 mmol, 77% yield). 1H (DMSO) δ 7.76 (s, 1 H), 7.73 (d, J = 8.4 Hz, 2 H), 7.54 (d, J = 8.4 Hz, 2 H).C NMR (DMSO) δ 168.0, 167.8, 132.6, 132.5 (2 C), 131.9 (2 C), 131.4, 130.5, 124.0.
(Z)-5-(3-Bromobenzylidene)thiazolidine-2,4-dione (24a)
2,4-Thiazolidinedione (117 mg, 1 mmol) and m-bromobenzaldehyde (185 mg, 117 µL, 1 mmol) were dissolved in ethanol (7 mL), followed by the addition of piperidine (79 µL, 0.8 mmol). After refluxing for 20 h, the yellow solution was poured into water (~60 mL), acidified with acetic acid to a pH of 3–4 and incubated at 4 °C overnight. After filtration, the solid was suspended in chloroform, followed by centrifugation and discarding the supernatant to afford an off-white product (155 mg, 0.55 mmol, 55% yield). 1H (DMSO) δ 12.69 (bs, 1 H), 7.82 (dd, J1 = J2 = 2.0 Hz, 1 H), 7.78 (s, 1 H), 7.68 (ddd, J1 = 8.0 Hz, J2 = 2.0 Hz, J3 = 0.8 Hz, 1 H), 7.58 (d, J = 8.0 Hz, 1 H), 7.50 (dd, J1 = J2 = 8.0 Hz, 1 H). 13C NMR (DMSO) δ 167.7, 167.3, 135.7, 133.0, 132.9, 131.5, 130.2, 128.3, 125.6, 122.6.
3-(2-Fluorobenzyl)thiazolidine-2,4-dione (25b)
2,4-Thiazolidinedione (59 mg, 0.5 mmol) and p-methoxybenzaldehyde (61 µL, 68 mg, 0.5 mmol) were dissolved in 1-butyl-3-methylimidazolium hexafluorophosphate, [bmim]PF6, (2 mL), followed by the addition of Et3N (84 µL, 0.6 mmol) and 2-fluorobenzyl chloride (0.6 mmol, 72 µL). The reaction was carried out at 60 °C for 17 h, cooled to room temperature, and then extracted with ether (4 × 15 mL). TLC (25% ethyl acetate in hexane, silica) demonstrated the presence of two major UV spots: Rf1 = 0.26 (same Rf of p-methoxybeznaldehyde) and Rf2 = 0.19. After passing through a pad of silica eluted with 25% ethyl acetate in hexane, the concentrated residue was purified by radial chromatography (silica) eluted with 15–25% ethyl acetate in hexane to afford a yellowish semisolid (Rf2) (55 mg, 0.24 mmol, 24% yield). NMR spectra confirmed it was 3-(2-fluorobenzyl)thiazolidine-2,4-dione rather than 3-(2-fluorobenzyl)-5-(4-methoxybenzylidene)thiazolidine-2,4-dione. 1H (CDCl3) δ 7.24–7.32 (m, 2 H), 7.00–7.11 (m, 2 H), 4.85 (s, 2 H), 3.97 (s, 2 H). 13C NMR (CDCl3) δ 171.4, 171.1, 160.8 (d, J = 247 Hz, 1 C), 130.5 (d, J = 3.0 Hz, 1 C), 130.2 (d, J = 8.3 Hz, 1 C), 124.4 (d, J = 3.8 Hz, 1 C), 122.0 (d, J = 14 Hz, 1 C), 115.8 (d, J = 21 Hz, 1 C), 39.3 (d, J = 4.6 Hz, 1 C), 33.9.
(E/Z)-1-Benzyl-5-(4-chlorobenzylidene)imidazolidine-2,4-dione (26c)
1-Benzylhydantoin (190 mg, 1 mmol) and p-chlorobenzaldehyde (142 mg, 1 mmol) were dissolved in ethanol (7 mL), followed by the addition of piperidine (79 µL, 0.8 mmol). The mixture was refluxed for 24 h, and the yellowish solution was poured into water (~60 mL) and acidified with acetic acid to a pH of 5 and then was extracted with chloroform (3 × 30 mL). The collected organic solution was dried over sodium sulfate. After passing through a pad of silica column eluted with 25% ethyl acetate in hexane, the concentrated residue was purified by radial chromatography (silica) eluted with 15–50% ethyl acetate in hexane to afford three components: (A) Rf = 0.42 (25% ethyl acetate in hexane); (B) Rf = 0.14 (25% ethyl acetate in hexane); (C) 0.20 (50% ethyl acetate in hexane). Component (B) was the desired product (20 mg, 6% yield, E/Z ≈ 9/1). According to the NMR spectra, the (E)-isomer was the major component: 1H (DMSO) δ 8.87 (bs, 1 H), 7,27–7.40 (m, 7 H), 6.14 (s, 1 H), 4.91 (s, 2 H). 13C NMR (DMSO) δ 162.0, 153.4, 135.2, 135.0, 131.8 (2 C), 130.7, 129.3 (2 C), 128.8, 128.6 (2 C), 128.3, 127.2 (2 C), 117.9, 43.8. Z-isomer is the minor component: 1H (DMSO) δ 7,25–7.40 (m, 1 H), 7.24 (d, J = 8.0 Hz, 2 H), 7.14 (d, J = 8.0 Hz, 2 H), 6.97 (d, J = 8.0 Hz, 2 H), 6.80 (s, 1 H), 6.61 (dd, J1 = 8.0 Hz, J2 = 1.6 Hz, 2 H), 4.73 (s, 2 H). HR-MS (EI): calcd 312.0666 for C17H13ClN2O2, found 312.0663.
(E)-1-Benzyl-5-(4-methoxybenzylidene)imidazolidine-2,4-dione (27c-1) and (E/Z)-1-Benzyl-5-(4-methoxybenzylidene)imidazolidine-2,4-dione (27c-2)
1-Benzylhydantoin (190 mg, 1 mmol) and p-methoxybenzaldehyde (136 mg, 1 mmol) were dissolved in ethanol (7 mL), followed by the addition of piperidine (100 µL, 1 mmol). The mixture was refluxed for 22 h, and the yellowish solution was poured into water (~60 mL) and acidified with acetic acid to a pH of 3–4. After incubation at 4 °C for a few hours, the sample wash filtered and washed with cold water, producing an off-white solid, which quite dissolved in methanol or chloroform. TLC (25% ethyl acetate in hexane, silica) suggested two products, one was UV absorbent with an Rf = 0.06 and the other was fluorescent with an Rf = 0.02. When 50% ethyl acetate in hexane was used as the developing solution, TLC was showed only one UV tailing spot, Rf = 0.28 (no fluorescent spot). Similarly, when 1% or 3% methanol in chloroform was used as developing solution, TLC analyses indicated only one UV spot. Products were purified by radial chromatography (silica) eluted with 25% ethyl acetate in hexane (collection-1) and 3% methanol in chloroform (collection-2). NMR was shown that collection-1 was a mixture composed of 83% E-isomer and 17% Z-isomer, whereas collection-2 contained only the E-isomer (19 mg (collection-2) + 107 mg (collection-1) = 126 mg, 0.41 mmol, 41% yield). E-isomer: 1H (CDCl3) δ 7.79 (d, J = 8.8 Hz, 2 H), 7.27–7.39 (m, 5 H), 6.85 (d, J = 8.8 Hz, 2 H), 6.17 (s, 1 H), 4.91 (s, 2 H), 3.81 (s, 3 H). 13C NMR (CDCl3) δ 162.0, 160.7, 153.1, 135.4, 132.5 (2 C), 129.2 (2 C), 128.2, 127.2 (2 C), 126.8, 125.1, 119.9, 113.9 (2 C), 55.5, 43.8. HR-MS (EI): calcd 308.1161 for C18H16N2O3, found 308.1159.
(Z/E)-1-Benzyl-5-(4-ethoxybenzylidene)imidazolidine-2,4-dione (28c)
1-Benzylhydantoin (190 mg, 1 mmol) and p-ethoxybezyaldehyde (139 µL, 150 mg, 1 mmol) were dissolved in ethanol (7 mL), followed by the addition of piperidine (100 µL, 1 mmol). The mixture was refluxed for 24 h, and the yellowish solution was poured into water (~60 mL) and acidified with acetic acid. After filtration, the solid dissolved in chloroform was purified by radial chromatography (silica) eluted with 0–1–3% methanol in chloroform to afford a yellowish solid (136 mg, 0.50 mmol, 50% yield). Rf = 0.21, 3% methanol in chloroform. NMR analyses demonstrated a mixture of Z- and E-compounds (Z/E ≈ 1/4). The E-compound: 1H (CDCl3) δ 8.94 (bs, 1 H), 7.80 (d, J = 8.8 Hz, 2 H), 7.28–7.38 (m, 5 H), 6.84 (d, J = 8.8 Hz, 2 H), 6.17 (s, 1 H), 4.91 (s, 2 H), 4.04 (q, d = 6.8 Hz, 2 H), 1.40 (t, J = 6.8 Hz, 3 H). 13C NMR (CDCl3) δ 162.4, 160.1, 153.5, 135.4, 132.6 (2 C), 129.2 (2 C), 128.1, 127.2 (2 C), 126.6, 124.8, 120.1, 114.4 (2 C), 63.7, 43.7, 14.9. The Z-compound: 1H (CDCl3) δ 9.02 (bs, 1 H), 7.13 (d, J = 8.4 Hz, 2 H), 7.05 (d, J = 8.4 Hz, 2 H), 6.81–6.86 (m, 4 H), 6.67 (dd, J1 = 8.0 Hz, J2 = 2.0 Hz, 2 H), 4.79 (s, 2 H), 4.06 (q, J = 7.2 Hz, 2 H), 1.45 (t, J = 7.2 Hz, 3 H). 13C NMR (CDCl3) δ 162.3, 159.5, 155.7, 135.4, 131.2 (2 C), 128.6 (2 C), 127.9, 127.7 (2 C), 124.8, 114.7, 114.4 (2 C), 63.8, 45.2, 14.9. HR-MS (EI): calcd 322.1317 for C19H18N2O3, found 322.1309.
(E)-1-Benzyl-5-(4-bromobenzylidene)imidazolidine-2,4-dione (29c-1) and (E/Z)-1-Benzyl-5-(4-bromobenzylidene)imidazolidine-2,4-dione (29c-2)
1-Benzylhydantoin (190 mg, 1 mmol) and p-bromobenzaldehyde (185 mg, 1 mmol) were dissolved in ethanol (7 mL), followed by the addition of piperidine (100 µL, 1 mmol). The mixture was refluxed for 36 h, and the yellowish solution was poured into water (~60 mL) and acidified with acetic acid to a pH of 3 and kept at 4 °C overnight. After filtration and washing with methanol, a yellow solid was collected (9 mg, 0.025 mmol, 3% yield) and shown to be the E-isomer (29c-1). The remaining solution was passed through a pad of silica column and eluted with chloroform. The concentrated residue was purified by radial chromatography (silica) eluted with 0–1–3% methanol in chloroform to provide a solid which was a mixture of E/Z-isomer (37 mg, 0.1 mmol, 13% yield, E/Z ≈ 10/1). Rf = 0.45, 50% ethyl acetate in hexane; Rf = 0.31, 3% methanol in chloroform. The E-isomer: 1H (CDCl3) δ 7.60 (d, J = 8.4 Hz, 2 H), 7.44 (d, J = 8.4 Hz, 2 H), 7.28–7.40 (m, 5 H), 6.12 (s, 1 H), 4.91 (s, 2 H). 13C NMR (CDCl3) δ 161.5, 152.8, 135.0, 132.0 (2 C), 131.6 (2 C), 131.1, 129.3 (2 C), 128.9, 128.3, 127.2 (2 C), 123.6, 117.9, 43.8. HR-MS (EI): calcd 356.0160 for C17H13BrN2O2, found 356.0154.
(E)-1-Benzyl-5-(4-dimethylaminobenzylidene)imidazolidine-2,4-dione (30c)
1-Benzylhydantoin (190 mg, 1 mmol) and p-dimethylaminobenzaldehyde (149 mg, 1 mmol) were dissolved in ethanol (7 mL), followed by the addition of piperidine (100 µL). The mixture was refluxed for 6 h, resulting in a precipitate in a yellow solution. The reaction mixture was poured into water (~60 mL), acidified with acetic acid to a pH of 3, and kept at 4 °C overnight. After filtration and washing with methanol, a yellow solid (100 mg, 31% yield) was obtained. 1H (DMSO) δ 11.35 (bs, 1 H), 7.87 (d, J = 9.2 Hz, 2 H), 7.24–7.40 (m, 5 H), 7.65 (d, J = 9.2 Hz, 2 H), 6.28 (s, 1 H), 4.87 (s, 2 H), 2.94 (s, 6 H). 13C NMR (DMSO) δ 162.4, 152.9, 149.9, 136.1, 131.5 (2 C), 128.2 (2 C), 126.8, 126.5 (2 C), 124.2, 119.8, 118.3, 110.7 (2 C), 41.4, 39.2 (2 C). HR-MS (EI): calcd 321.1477 for C19H19N3O2, found 321.1472.
(E/Z)-1-Benzyl-5-(2-methoxybenzylidene)imidazolidine-2,4-dione (31c)
1-Benzylhydantoin (190 mg, 1 mmol) and o-methoxybenzaldehyde (122 µL, 136 mg, 1 mmol) were dissolved in ethanol (7 mL), followed by the addition of piperidine (100 µL), and the mixture was refluxed for 48 h. The solution was poured into water (~60 mL), acidified with acetic acid to a pH of 3, and kept at 4 °C overnight. After filtration and washing with water, a white solid was collected. The solid was dissolved in 50% ethyl acetate in hexane and passed a pad of silica column eluted with 50% ethyl acetate in hexane to afford a solid compound (145 mg, 0.47 mmol, 47% yield, E/Z ≈ 5/1). Rf = 0.37, 50% ethyl acetate in hexane; Rf = 0.24, 3% methanol in chloroform. The E-isomer: 1H (CDCl3) δ 9.15 (bs, 1 H), 7.62 (m, 1 H), 7.11–7.39 (m, 7 H), 6.85–6.88 (m, 1 H), 6.19 (s, 1 H), 4.91 (s, 2 H), 3.82 (s, 3 H). 13C NMR (CDCl3) δ 162.2, 159.5, 153.7, 135.1, 133.5, 129.3, 129.2 (2 C), 128.2, 127.2 (2 C), 126.9, 123.6, 119.6, 116.0, 114.9, 55.5, 43.7. HR-MS (EI): calcd 308.1161 for C18H16N2O3, found 308.1155.
Pim Kinase Assays
Pim protein kinase assays were conducted using multiple methods to ensure that the effects of the compounds were not due to any experimental artifacts. The primary screen and evaluation of the compounds shown in Table 3 was conducted using an ATP-depletion assay. Briefly, recombinant human Pim-1 (Upstate) was incubated with S6 kinase/Rsk-2 peptide 2 (KKRNRTLTK) (Upstate) as the substrate in the presence 100 µM of compounds from the screening library (DIVERSet collection of the ChemBridge Corporation), 1 µM ATP and 10 mM MgCl2 for 1 h. The Kinase-Glo luciferase kit (Promega) was used to measure residual ATP levels after the kinase reaction. For experiments that required higher ATP concentrations, Pim-1 kinase activity was monitored spectrophotometrically using a coupled assay in which ADP production is coupled to NADH oxidation catalyzed by pyruvate kinase and lactate dehydrogenase. Assays were carried out in 20 mM MOPS pH 7 containing 100 mM NaCl, 10 mM MgCl2, 2.5 mM phosphoenolpyruvate, 0.2 mM NADH, 30 µg/mL pyruvate kinase, 10 µg/mL lactate dehydrogenase, 2 mM dithiothreitol, 25 nM Pim-1, 100 µM S61 peptide (RRLSSLRA, American Peptide Company), and varying concentrations of ATP. Activity was measured by monitoring NADH oxidation as the decrease at 340 nm in a VersaMax microplate reader (Molecular Devices) at 25 °C. Reactions were initiated by the addition of ATP (typically 100 µM). Inhibitors (final 1% DMSO) were added just prior to the addition of ATP. In either case, IC50 values were determined using nonlinear regression with the program GraphPad Prism. In some experiments, Pim-1 kinase activity was determined using His-tagged 4E-BP1 as the substrate. The active Pim-1 protein (Upstate) was resuspended in kinase reaction buffer (10 mM MOPS, pH 7.4, 100 µM ATP, 15 mM MgCl2, 1 mM Na3VO4, 1 mM NaF, 1 mM DTT, and protease inhibitor cocktail). In each reaction (30 µL), 3 µg of His-4E-BP1 protein was used as substrate, and 10 μCi of [γ-32P] ATP were then added. Incubation was carried out at 30 °C for 30 min with agitation. The samples were then subjected to SDS-PAGE and 32P labeled 4E-BP1 was visualized by autoradiography. Finally, Pim-1 activity in intact cells was measured in some experiments. HEK-293T cells were transfected with Flag-Pim-1 for 24 h, and then were trypsined and divided into smaller dishes for overnight. Cells were washed once and incubated with phosphate-free media containing 10% phosphate-free FBS (Invitrogen, Carlsbad, CA) for 1 h. Cells were then incubated in medium containing 50 μCi/ml [32P]orthophosphate for 4 h, in which the test compounds were added for the final 1 h. To immunoprecipitate Pim-1, anti-Flag M2 agarose was added to the cell lysate and incubated for 3 h. A portion (10%) of the immunoprecipitates was used for Western blotting with anti-Flag antibodies (input). The other 90% of each sample was subjected to SDS-PAGE, and 32P-labeled Pim-1 was visualized by autoradiography.
Cytotoxicity Assays
Human prostate cancer PC3 cells were seeded in 96-well tissue culture dishes at approximately 10% confluency and allowed to attach and recover for 24 h. Varying concentrations of the test compounds are then added to each well, and the plates were incubated for an additional 48 h. The number of surviving cells was determined by the MTS assay (Promega). The percentage of cells killed was calculated as the percentage decrease in MTS metabolism compared with control cultures.
Toxicity Evaluations
Six week-old female Swiss Webster mice (approximately 25 g) were purchased from Charles River Laboratories. Groups of mice were injected intraperitoneally daily with vehicle (30% PEG-400, 5% Tween-80, 65% DMSO), or 3, 10, or 50 mg/kg 16a for a total of 7 days. Animals were monitored for an additional 7 days, and on the final day, blood was collected and analyzed for complete blood count and blood chemistry (Drug Metabolism and Clinical Pharmacology Core, MUSC).
Antitumor Assay
A syngeneic mouse tumor model that uses a transformed murine mammary adenocarcinoma cell line (JC, ATCC number CRL-2116) and Balb/C mice (Charles River) was performed as previously described.33 Animal care and procedures were in accordance with guidelines and regulations of the IACUC of the Medical University of South Carolina. Tumor cells (1 × 106) were implanted subcutaneously, and tumor volume was calculated using the equation: (L × W2)/2. Upon detection of tumors, mice were randomized into treatment groups. Treatment was then administered once per day, five days per week, thereafter consisting of intraperitoneal doses of 0 or 50 mg 16a/kg or vehicle (50% DMSO: 50% phosphate-buffered saline). Whole body weights and tumor volume measurements were performed three times per week.
Computational Modeling
The molecular structure for compound 4a was built and docked to the crystal structure of Pim-1 (PDB code: 1XWS) using the suite of programs within the DiscoveryStudio interface (Accelrys). The ligand and protein structures were prepared for docking by the addition of hydrogens, followed by the assignment of atom types and charges within the CHARMm forcefield. This was followed by creation of the docking grids within a 6 Å radius of the native ligand and molecular mechanics/energy minimization docking of flexible 4a into the rigid binding site of Pim-1.
Footnotes
Abbreviations: 4E-BP1, eukaryotic translation initiation factor 4E binding protein 1; mTOR, mammalian target of rapamycin; MTS, (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; MTS); Pim, proviral insertions in murine; S6K, S6 protein kinase.
References
- 1.Selten G, Cuypers HT, Berns A. Proviral activation of the putative oncogene Pim-1 in MuLV induced T-cell lymphomas. EMBO J. 1985;4(7):1793–1798. doi: 10.1002/j.1460-2075.1985.tb03852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, Pienta KJ, Rubin MA, Chinnaiyan AM. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001;412(6849):822–826. doi: 10.1038/35090585. [DOI] [PubMed] [Google Scholar]
- 3.Dai H, Li R, Wheeler T, Diaz de Vivar A, Frolov A, Tahir S, Agoulnik I, Thompson T, Rowley D, Ayala G. Pim-2 upregulation: biological implications associated with disease progression and perinueral invasion in prostate cancer. Prostate. 2005;65(3):276–286. doi: 10.1002/pros.20294. [DOI] [PubMed] [Google Scholar]
- 4.Xu Y, Zhang T, Tang H, Zhang S, Liu M, Ren D, Niu Y. Overexpression of PIM-1 is a potential biomarker in prostate carcinoma. J. Surg. Oncol. 2005;92(4):326–330. doi: 10.1002/jso.20325. [DOI] [PubMed] [Google Scholar]
- 5.Valdman A, Fang X, Pang ST, Ekman P, Egevad L. Pim-1 expression in prostatic intraepithelial neoplasia and human prostate cancer. Prostate. 2004;60(4):367–371. doi: 10.1002/pros.20064. [DOI] [PubMed] [Google Scholar]
- 6.Cibull TL, Jones TD, Li L, Eble JN, Ann Baldridge L, Malott SR, Luo Y, Cheng L. Overexpression of Pim-1 during progression of prostatic adenocarcinoma. J. Clin. Pathol. 2006;59(3):285–288. doi: 10.1136/jcp.2005.027672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen WW, Chan DC, Donald C, Lilly MB, Kraft AS. Pim family kinases enhance tumor growth of prostate cancer cells. Mol. Cancer Res. 2005;3(8):443–451. doi: 10.1158/1541-7786.MCR-05-0007. [DOI] [PubMed] [Google Scholar]
- 8.Lilly M, Kraft A. Enforced expression of the Mr 33,000 Pim-1 kinase enhances factor-independent survival and inhibits apoptosis in murine myeloid cells. Cancer Res. 1997;57(23):5348–5355. [PubMed] [Google Scholar]
- 9.Aho TL, Sandholm J, Peltola KJ, Mankonen HP, Lilly M, Koskinen PJ. Pim-1 kinase promotes inactivation of the pro-apoptotic Bad protein by phosphorylating it on the Ser112 gatekeeper site. FEBS Lett. 2004;571(1–3):43–49. doi: 10.1016/j.febslet.2004.06.050. [DOI] [PubMed] [Google Scholar]
- 10.Macdonald A, Campbell DG, Toth R, McLauchlan H, Hastie CJ, Arthur JS. Pim kinases phosphorylate multiple sites on Bad and promote 14-3-3 binding and dissociation from Bcl-XL. BMC Cell Biol. 2006;7:1. doi: 10.1186/1471-2121-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan B, Zemskova M, Holder S, Chin V, Kraft A, Koskinen PJ, Lilly M. The PIM-2 kinase phosphorylates BAD on serine 112 and reverses BAD-induced cell death. J. Biol. Chem. 2003;278(46):45358–45367. doi: 10.1074/jbc.M307933200. [DOI] [PubMed] [Google Scholar]
- 12.Kim KT, Levis M, Small D. Constitutively activated FLT3 phosphorylates BAD partially through pim-1. Br. J. Hamaetol. 2006;134(5):500–509. doi: 10.1111/j.1365-2141.2006.06225.x. [DOI] [PubMed] [Google Scholar]
- 13.Jacobs MD, Black J, Futer O, Swenson L, Hare B, Fleming M, Saxena K. Pim-1 ligand-bound structures reveal the mechanism of serine/threonine kinase inhibition by LY294002. J. Biol. Chem. 2005;280(14):13728–13734. doi: 10.1074/jbc.M413155200. [DOI] [PubMed] [Google Scholar]
- 14.Kumar A, Mandiyan V, Suzuki Y, Zhang C, Rice J, Tsai J, Artis DR, Ibrahim P, Bremer R. Crystal structures of proto-oncogene kinase Pim1: a target of aberrant somatic hypermutations in diffuse large cell lymphoma. J. Mol. Biol. 2005;348(1):183–193. doi: 10.1016/j.jmb.2005.02.039. [DOI] [PubMed] [Google Scholar]
- 15.Qian KC, Wang L, Hickey ER, Studts J, Barringer K, Peng C, Kronkaitis A, Li J, White A, Mische S, Farmer B. Structural basis of constitutive activity and a unique nucleotide binding mode of human Pim-1 kinase. J. Biol. Chem. 2005;280(7):6130–6137. doi: 10.1074/jbc.M409123200. [DOI] [PubMed] [Google Scholar]
- 16.Bregman H, Meggers E. Ruthenium half-sandwich complexes as protein kinase inhibitors: an N-succinimidyl ester for rapid derivatizations of the cyclopentadienyl moiety. Org. Lett. 2006;8(24):5465–5468. doi: 10.1021/ol0620646. [DOI] [PubMed] [Google Scholar]
- 17.Debreczeni JE, Bullock AN, Atilla GE, Williams DS, Bregman H, Knapp S, Meggers E. Ruthenium half-sandwich complexes bound to protein kinase Pim-1. Angew. Chem., Int. Ed. 2006;45(10):1580–1585. doi: 10.1002/anie.200503468. [DOI] [PubMed] [Google Scholar]
- 18.Pogacic V, Bullock AN, Fedorov O, Filippakopoulos P, Gasser C, Biondi A, Meyer-Monard S, Knapp S, Schwaller J. Structural analysis identifies imidazo[1,2-b]pyridazines as PIM kinase inhibitors with in vitro antileukemic activity. Cancer Res. 2007;67(14):6916–6924. doi: 10.1158/0008-5472.CAN-07-0320. [DOI] [PubMed] [Google Scholar]
- 19.Bullock AN, Debreczeni JE, Fedorov OY, Nelson A, Marsden BD, Knapp S. Structural basis of inhibitor specificity of the human protooncogene proviral insertion site in moloney murine leukemia virus (PIM-1) kinase. J. Med. Chem. 2005;48(24):7604–7614. doi: 10.1021/jm0504858. [DOI] [PubMed] [Google Scholar]
- 20.Bearss D, Liu X-H, Grand C, Gourley E, Lamb J, Lloyd M, Vankayalapati H. Discovery and characterization of a small molecule inhibitor for Pim-1 kinase; American Society of Hematology National Meeting, 2006; poster presentation. [Google Scholar]
- 21.Lamb J, Gourley E, Liu X-H, Grand C, Vankayalapati H, Warner S, Bearss D. A small molecule inhibitor of Pim-1 kinase with activity in both hematological and solid tumor malignancies. AACR-NCI-EOTRC International Conference on Molecular Targets and Cancer Therapeutics; San Francisco, CA. 2007. [Google Scholar]
- 22.Cheney IW, Yan S, Appleby T, Walker H, Vo T, Yao N, Hamatake R, Hong Z, Wu JZ. Identification and structure—activity relationships of substituted pyridones as inhibitors of Pim-1 kinase. Bioorg. Med. Chem. Lett. 2007;17(6):1679–1683. doi: 10.1016/j.bmcl.2006.12.086. [DOI] [PubMed] [Google Scholar]
- 23.Holder S, Lilly M, Brown ML. Comparative molecular field analysis of flavonoid inhibitors of the PIM-1 kinase. Bioorg. Med. Chem. 2007;15(19):6463–6473. doi: 10.1016/j.bmc.2007.06.025. [DOI] [PubMed] [Google Scholar]
- 24.Holder S, Zemskova M, Zhang C, Tabrizizad M, Bremer R, Neidigh JW, Lilly MB. Characterization of a potent and selective small-molecule inhibitor of the PIM1 kinase. Mol. Cancer Ther. 2007;6(1):163–172. doi: 10.1158/1535-7163.MCT-06-0397. [DOI] [PubMed] [Google Scholar]
- 25.Pierce AC, Jacobs M, Stuver-Moody C. Docking study yields four novel inhibitors of the protooncogene Pim-1 kinase. J. Med. Chem. 2008;51(6):1972–1975. doi: 10.1021/jm701248t. [DOI] [PubMed] [Google Scholar]
- 26.Pagano M, Bain J, Kazimierczuk Z, Sarno S, Ruzzene M, di Maira G, Elliott M, Orzeszko A, Cozza G, Meggio F, Pinna L. The selectivity of inhibitors of protein kinase CK2: an update. Biochem. J. 2008;415:353–365. doi: 10.1042/BJ20080309. [DOI] [PubMed] [Google Scholar]
- 27.Momose Y, Meguro K, Ikeda H, Hatanaka C, Oi S, Sohda T. Studies on antidiabetic agents. X. Synthesis and biological activities of pioglitazone and related compounds. Chem. Pharm. Bull. 1991;39(6):1440–1445. doi: 10.1248/cpb.39.1440. [DOI] [PubMed] [Google Scholar]
- 28.Bruno G, Costantino L, Curinga C, Maccari R, Monforte F, Nicolo F, Ottana R, Vigorita MG. Synthesis and aldose reductase inhibitory activity of 5-arylidene-2,4-thiazolidinediones. Bioorg. Med. Chem. 2002;10(4):1077–1084. doi: 10.1016/s0968-0896(01)00366-2. [DOI] [PubMed] [Google Scholar]
- 29.Wada A, Kanatomo S, Nagai S. ortho-Directed lithiation of (2-methoxy)ethoxy- and (2-dimethylamino)ethoxy-arenes. Chem. Pharm. Bull. 1985;33(3):1016–1022. [Google Scholar]
- 30.Yang D, Chen Z, Chen S, Zheng Q. Organic reactions in ionic liquids: ionic liquid-accelerated three-component reaction: a rapid, onepot synthesis of 3-alkyl-5-[(Z)-arylmethylidene]-1,3-thiazolidine-2,4-diones. Synthesis. 2003;12:1891–1894. [Google Scholar]
- 31.Tan S-F, Ang K-P, Fong Y-F. Z)- and (E)-5-Arylmethylene-hydantoins: spectroscopic properties and configuration assignment. J. Chem. Soc., Perkin Trans. 2. 1986:1941–1944. [Google Scholar]
- 32.Hammerman PS, Fox CJ, Birnbaum MJ, Thompson CB. Pim and Akt oncogenes are independent regulators of hematopoietic cell growth and survival. Blood. 2005;105(11):4477–4483. doi: 10.1182/blood-2004-09-3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee BD, French KJ, Zhuang Y, Smith CD. Development of a syngeneic in vivo tumor model and its use in evaluating a novel P-glycoprotein modulator, PGP-4008. Oncol. Res. 2003;14(1):49–60. doi: 10.3727/000000003108748603. [DOI] [PubMed] [Google Scholar]