

Abstract
Background and Purpose
CD163, a receptor for hemoglobin, is involved in hemoglobin clearance following intracerebral hemorrhage (ICH). In contrast to microglial/macrophage CD163, neuronal CD163 hemoglobin has not been well studied. The current study examined the expression of neuronal CD163 in a pig model of ICH as well as in vitro rat cortical neurons, and the impact of deferoxamine on that expression.
Methods
There were 2 parts to this study. In the in vivo part, piglets had injection of autologous blood into the right frontal lobe. The time course of CD163 expression and the effect of deferoxamine on the expression of CD163 after ICH were determined in the grey matter. In the in vitro part, the levels of CD163 and neuronal death, and the effect of deferoxamine were examined in rat cortical neurons culture treated with hemoglobin.
Results
CD163 positive cells were found and the CD163 protein levels were upregulated in the ipsilateral grey matter after ICH. The CD163 levels peaked at days 1 and 3. The CD163 positive cells were co-located with NeuN, HO-2 and TUNEL positive cells. Deferoxamine treatment attenuated ICH-induced CD163 upregulation, and significantly reduced both brain CD163 and hemoglobin levels at day 3. Treating neuronal cultures with hemoglobin for 24 hours resulted in CD163 upregulation and increased cell death. Deferoxamine significantly attenuated the hemoglobin-induced neuronal death and CD163 upregulation.
Conclusions
CD163 is expressed in neurons and upregulated after ICH. Deferoxamine reduced ICH-induced CD163 upregulation and brain cell death in vivo, and hemoglobin-induced CD163 upregulation and neuronal death in vitro.
Keywords: Cerebral hemorrhage, CD163 antigen, Deferoxamine, Swine, Hemoglobin
Subject Terms: Intracranial Hemorrhage
INTRODUCTION
Hemoglobin release from an intracerebral hemorrhage (ICH) has deleterious effects on perihematomal brain tissues, resulting in brain edema, neuronal death, and blood-brain barrier disruption1 .The haptoglobin-CD163-HO-1 pathway exerts as a first defense system in hemoglobin induced toxicity2, 3, and CD163 acts as a main hemoglobin receptor mediating the intracellular uptake of hemoglobin into macrophages4.
While macrophages play a central role in hemoglobin clearance, evidence indicates that other cell types may also be involved5. Hemoglobin mostly remains encapsulated within erythrocytes until they are phagocytosed and degraded by microglia and infiltrating macrophages6. However, hemoglobin can also be taken up by neural cells and metabolized locally7, 8. Neuronal exposure to hemoglobin for one day induces a concentration-dependent cell death9. While astrocytes and hippocampal neurons can take up hemoglobin10, neurons are more vulnerable to hemoglobin toxicity11, 12.
We and others have demonstrated that neurons can express the hemoglobin receptor CD16313, 14, but the trafficking of CD163 in neural cells has not been intensively investigated. The present study examined the expression of neuronal CD163 in vivo and in vitro. The effect of an iron chelator, deferoxamine, on CD163 expression was also examined.
METHODS
Animal preparation and intracerebral blood injection
Animal use protocols were approved by the University of Michigan Committee on the Use and Care of Animals and were conducted in accordance with United State Public Health Service’s Policy on Humane Care and Use of Laboratory Animals. A total of 78 male piglets (8–10 kg; Michigan State University, East Lansing) were used in this study. One piglet was excluded from this study because of brain infection at the second day after ICH. The ICH model was performed as previously described15–17. Piglets were sedated with ketamine (25 mg/kg, IM) and anesthetized with 2% isoflurane via nose cone. The isoflurane concentration was maintained at 1.0 to 1.5% during the surgical procedures. Body temperature was maintained at 37.5 ± 0.5 °C by a feedback-controlled heating pad (Gaymar, Orchard Park, NY). The right femoral artery was inserted with a polyethylene catheter (PE-160) to obtain blood for injection and to monitor arterial blood pressure, blood gases and glucose concentrations.
A cranial burr hole (1.5 mm) was then drilled 11 mm to the right of the sagittal and 11 mm anterior to the coronal suture. An 18-mm-long 20-gauge sterile plastic catheter was placed stereotaxically into the center of the right frontal cerebral white matter and cemented in place. Using an infusion pump, first 1 mL of autologous arterial blood (without anticoagulants) was infused over 15 minutes and another 1.5 mL of blood was injected over 15 minutes after a 5 minutes break. After injection, the needle was removed, the burr hole was filled with bone wax, and the skin incision closed with sutures. Sham animals underwent the same procedure without blood infusion.
Experimental groups
The in vivo study was divided into two parts. In the first part, piglets had 2.5 ml blood injected into the right frontal lobe. Brains were harvested at 4 hours (n=7), day 1 (n=7), day 3 (n=7) and day 7 (n=7). Sham piglets were euthanized at day 3 (n=7). Brains were used for histology and immunoblots. In the second part, piglets had blood injected into the frontal lobe and were treated with vehicle (saline) or deferoxamine (50 mg/kg, i.m. at 2 hours after ICH and then every 12 hours for 7 days). Brains were harvested at days 1, 3 and 7 (n=7 each group) for brain histological examines and Western blotting. The investigators were blinded for the treatment. YH preformed surgeries and RL and SC did all other measurements.
Brain in situ freezing and brain histology
Brains were frozen in situ by decanting liquid nitrogen into a 12 oz. bottomless foam cup adhered to the head with Dow-Corning high vacuum grease (Dow Corning, Midland, MI), as described previously18. The frozen head was then cut with a band saw into 5 mm thick coronal sections. Tissues were sampled from the regions of interest.
For brain histology, piglets were reanesthetized and the brains perfused in situ with 10% formalin. Brains were sectioned coronally. Paraffin embedded brain was cut into 10 μm thick sections.
Primary cortical neuronal cultures
Primary cortical neurons were obtained from embryonic day 17 Sprague-Dawley rats (Charles River Laboratories, MI). Cultures were prepared according to a previously described procedure with some modifications19, 20. Briefly, cortices were dissected, stripped of meninges, dissociated by a combination of 0.5% trypsin digestion and mechanical trituration, and plated in neurobasal medium with 2% B27, 0.5mM glutamine and 1% Antibiotic-Antimycotic in 6-well plates with a density of 2×106/cm2 and cultured in a humidified incubator at 37 °C with 5% CO2. Half of the culture media was changed every 3–4 days. The neurons were used at day-7.
Treatment of the neurons
The in vitro studies were divided into two parts. In the first part, primary cultured neurons were treated with either vehicle or hemoglobin at concentrations of 5 or 20μM (Cat No. 151234, MP Biomedicals, LLC). The hemoglobin concentrations used in this study were based on a prior study by Regan and Panter9. In the second part, neurons were treated with hemoglobin (5 or 20μM) with or without deferoxamine (5μM). Deferoxamine was applied 2 hours prior to hemoglobin administration. Cells were collected at 24 hours later and used for Western Blot and immunocytochemistry.
Neurotoxicity assay
Culture medium (50 μL) was taken after 24 hours treatment and neuronal death was quantified by lactate dehydrogenase (LDH) release assay (G1780, Promega Corporation, WI, USA) as previously described21. The mean basal LDH activity in sister cultures subjected to medium exchange (sham wash) alone was subtracted from all values to quantify the signal specific to the cytotoxic exposure, following the protocol of Koh and Choi22.
Western blot analysis
Western blot analysis was performed as previously described15, 17. Ipsi- and contralateral grey matter tissues were sampled. Protein concentration was determined by Bio-Rad protein assay kit (Hercules, CA). An equal amount of protein (50 μg) was suspended in loading buffer, denatured at 95°C for 5 minutes and loaded on an SDS-PAGE gel. After being electrophoresed and transferred onto Hybond-C pure nitrocellulose membrane (Amersham), the membrane was blocked with nonfat milk buffer for 1 hour and then incubated with the primary antibodies. The primary antibodies were polyclonal rabbit anti CD163 (Abcam, Ab87099, 1:1000), rabbit monoclonal anti hemoglobin subunit alpha (Abcam, Ab92492, 1:2000), monoclonal mouse anti-GAPDH (Fitzgerald, 10R-G109A, 1:1000000) and monoclonal mouse anti-β actin (Sigma-Aldrich, A3854, 1:100000). The membranes were then incubated with horseradish perixidase-conjugated secondary antibodies for 1 hour at room temperature. The second antibodies were HRP-conjugated goat anti-mouse IgG (1:2000, Bio-Rad) and HRP-conjugated goat anti-rabbit IgG (1:2000, Bio-Rad). Protein band densities were detected by Kodak X-OMAT film and quantified by NIH Image J. CD163 levels were expressed as the ratio of CD163/GAPDH or CD163/β actin. Hemoglobin level was expressed as the ratio of hemoglobin/GAPDH.
Immunonistochemistry and immunofluorescence staining
Immunohistochemical staining was performed using the avidin-biotin complex method as described previously15, 17. Paraffin-embedded brains were cut into 10-μm-thick sections. The sections were deparaffinized in xylene and rehydrated in a graded series of alcohol dilutions. Antigen retrieval was performed by the microwave method using citrate buffer (10mM, pH 6.4). Sections were permeabilized in 0.3% Triton X-100 (v/v) for 15 min and washed three times in PBS. Non-specific binding was blocked by 30 min treatment in 10% normal goat serum/PBS. The sections were incubated with primary antibody at 4 ℃ overnight. The primary antibody was polyclonal rabbit anti CD163 (Abcam, 87099, 1:500). After washing three-time in PBS, sections were incubated with biotinylated goat anti-rabbit IgG (1:500, Vector Laboratories) for 2 hours at room temperature, followed by 2 hours incubation in avidin-biotinylated peroxidase complex (1:500, Vectorstain Elite Kit) at room temperature. Diaminobenzidine (DAB; Invitrogen) was used as chromogen, with reactions sustained for 2–5 min at room temperature. For negative controls, the primary antibody was replaced with normal goat serum and no specific positive staining was detected. Stained sections were examined on an Olympus BX51 microscope. Images used for analysis were captured at the same contrast setting and exposure times.
For double labeling immunofluorescence staining, paraffin-embedded sections were treated with the same protocol as described above. The primary antibodies were monoclonal mouse anti-pig CD163 (AbD serotec, MCA2311, 1:500), monoclonal mouse anti-rat CD163 (AbD serotec, MCA342, 1:500), polyclonal rabbit anti CD163 (Abcam, ab87099, 1:500), polyclonal mouse anti-FOX3/NeuN (AbD serotec, MAB377, 1:1000), polyclonal rabbit anti HO-2 (Enzo Life Sciences, ADI-OSA-200, 1:500) and monoclonal mouse anti porcine GFAP (Novus Biologicals, NBP 1-22545, 1:500). Secondary antibodies were Alexa Fluor 488 donkey anti-mouse IgG (1:500, Invitrogen), and Alexa Fluor 594 donkey anti-rabbit IgG (1:500, Invitrogen). Fluoroshield™ with DAPI (F6057) was used for nuclear labeling.
Immunocytochemistry of isolated cells was performed by the indirect immunofluorescence method. The cultures were washed twice with PBS and fixed with 4% paraformaldehyde in PBS for 15 min, then washed twice more with PBS at room temperature. The staining protocol and the primary and second antibodies were the same as described above.
Terminal dUDP nick end labeling (TUNEL) staining and cell counting
TUNEL staining was performed to assess DNA fragmentation using an Apop Tag® Peroxidase In Situ Apoptosis Detection Kit (S7100, Millipore, Billerica, MA, USA) and Apop Tag® Fluorescein In Situ Apoptosis Detection Kit (S7110, Millipore, Billerica, MA, USA). Counting of TUNEL positive cells was performed on high-power images (×40 magnification) taken using a digital camera. Counts were performed on 3 areas in each brain section.
Statistical Analysis
Measurements were performed by personnel blinded to treatment groups. Data were expressed as mean ± standard deviation, and comparisons between groups were performed by Student t tests or one-way ANOVA as appropriate. Differences were considered significant at p < 0.05.
RESULTS
There were some CD163 positive cells in the ipsilateral grey matter, with fewer in contralateral grey matter at 4 hours after ICH. CD163 positive cells were located mostly in the perihematoma area and numbers increased at day 1, became abundant at day 3, and decreased at day 7 in the ipsilateral grey matter (Fig. 1A). CD163 protein levels were upregulated after ICH, it was peaked at day 3 (CD163/GAPDH: 0.57±0.34 vs. 0.03±0.03 at 4 hours, P >0.05, 0.79±0.43 at day 1, P <0.05 and 0.23±0.13 at day 7, p >0.05) in grey matter (Fig. 1B). Compared to sham-operated animals and contralateral hemisphere, ICH induced significantly higher CD163 protein levels in the ipsilateral grey matter at day 3 after ICH (CD163/GAPDH: 0.78±0.84 vs. 0.11±0.04 in sham, p<0.05, 0.01±0.01 in contralateral grey matter, p<0.01, Fig. 1C).
Figure 1.
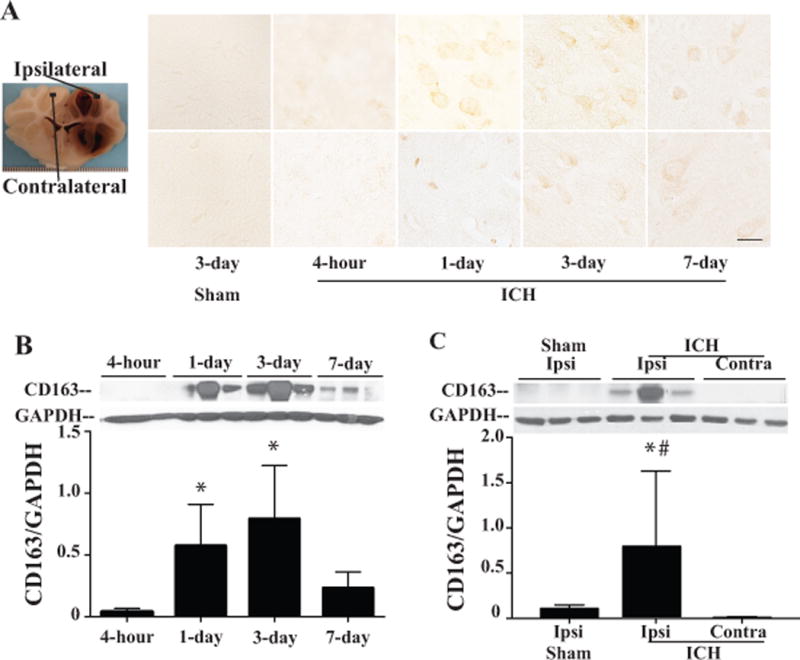
Time course of CD163 immunoreactivity (A) and protein levels (B) in the ipsilateral and contralateral grey matter after 2.5ml of autologous blood injected into the right frontal lobe. Scale bar=50μm. Values are mean ±SD, * p<0.05 vs. 4 hours. (C) Protein levels of CD163 in the ipsi- and contralateral grey matter after ICH, and in ipsilateral grey matter after sham operation at day 3. Values are mean ±SD, *p<0.05, vs. the ipsilateral side of sham, # p<0.01 vs. the contralateral side of ICH.
In order to determine whether CD163 expressed in neurons, double immunofluorescence staining was performed 24 hours after ICH. CD163 positive stained cells were co-localized with NeuN (a neuronal marker, Fig. 2), HO-2 (neuronal isoform of heme oxygenase, Fig. 2) and TUNEL (marker for double DNA strains damaged cells, Fig. 2) positive cells in the perihematoma area. Many neurons were also TUNEL positive. This indicated that CD163 expressed in the injured neurons after ICH. However, astrocytes did not express CD163 in this piglet ICH model (Fig. 2).
Figure 2.
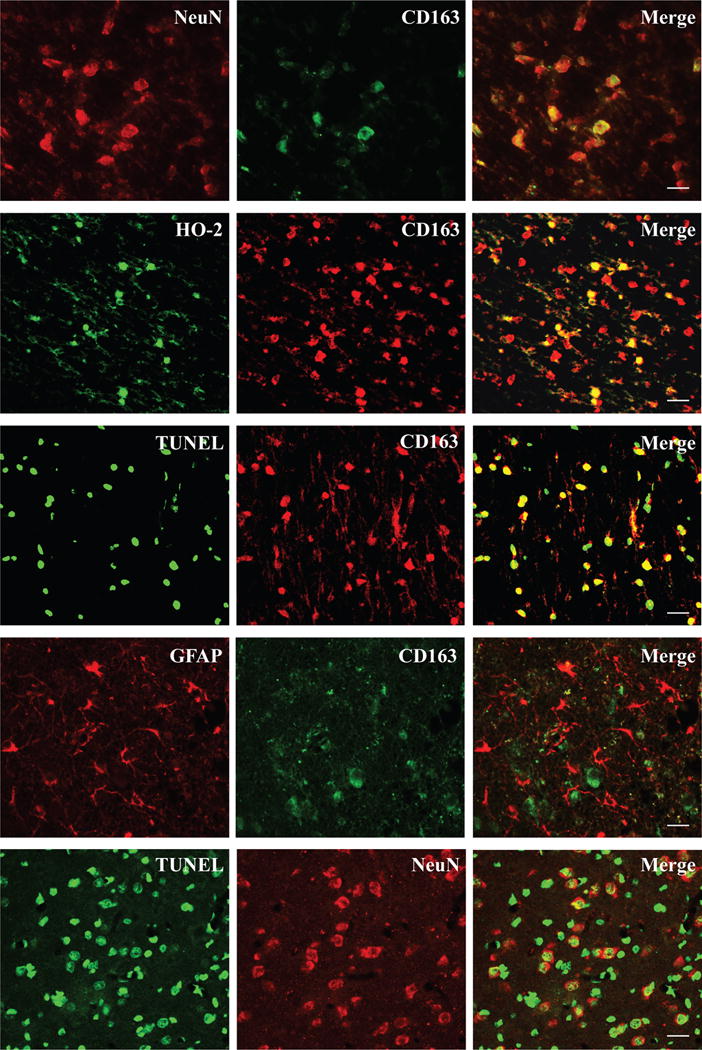
Double labeling of CD163 immunoreactivity with NeuN, HO-2, TUNEL, and GFAP positive cells in the perihematoma area at 24 hours after blood injection. Double labeling was also performed for TUNEL and NeuN. Scale bar= 50μm.
Systemic deferoxamine treatment significantly reduced CD163 immunoactivity and protein levels in the ipsilateral grey matter at day 1 (CD163/GAPDH: 0.04±0.03 vs. 0.34±0.14 in vehicle, p<0.01), day 3 (CD163/GAPDH: 0.09±0.03 vs. 0.70±0.48 in vehicle, p<0.05) and day 7 (CD163/GAPDH: 0.10±0.05 vs. 0.58±0.36 in vehicle, p<0.05) after ICH (Fig. 3A). Deferoxamine treatment reduced the numbers of the TUNEL positive cells in the peri-hemotoma region at day-1(TUNEL positive cells 495.30±22.82 vs. 258.50±63.39 in vehicle, p<0.01, Fig. 3B). And there was no TUNEL positive cell in the sham operation group. ICH induced significantly upregulation of hemoglobin in ipsilateral grey matter at day 3 (hemoglobin/GAPDH: 1.46±0.70 vs. 0.39±0.54 in sham, p<0.05, Fig. 3C), and that was blocked by deferoxamine treatment (hemoglobin/GAPDH: 0.77±0.27 vs. 1.46±0.70 in vehicle, p<0.05, Fig. 3C).
Figure 3.
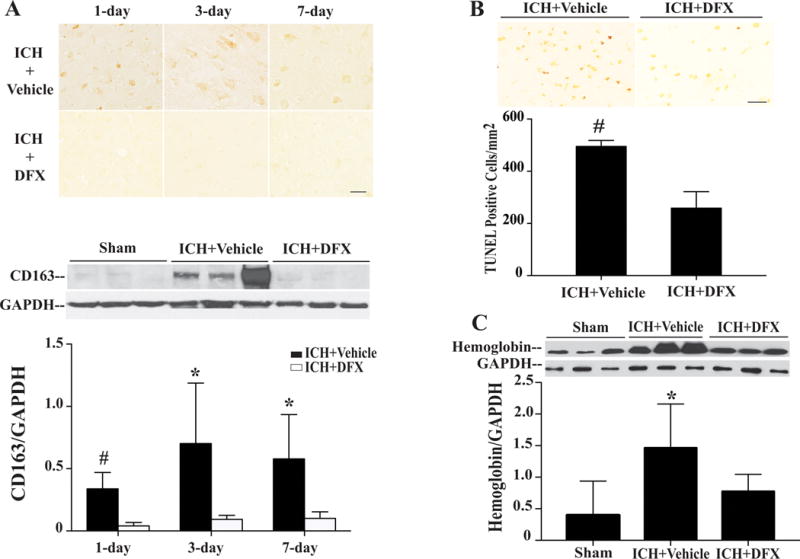
CD163 immunoreactivity and protein levels in the ipsilateral grey matter of piglets treated with deferoxamine or vehicle after ICH (A); numbers of TUNEL positive cells in the perihematoma cortex of piglets treated with deferoxamine or vehicle at day 1 after ICH (B); levels of hemoglobin in the ipsilateral grey matter of piglets at day 3 after ICH or sham operation treated with deferoxamine or vehicle (C). Scale bar= 50μm, values are mean ±SD, #p<0.01, *p<0.05 vs. the DFX-treated group.
To further confirm neuronal expression of CD163, primary cultured neurons were treated with hemoglobin (0, 5 or 20μM) for 24 hours. Immunofluorescence double labeling revealed co-localization of CD163 with NeuN, a neuronal marker (Fig. 4A). Treatment with 5μM hemoglobin caused a slight decrease in neuronal number and mild morphological changes. In contrast, treatment with 20μM hemoglobin caused marked decreases in neuronal numbers, cell body shrinkage and axonal disruption. Also, most degenerating neurons expressed weaker NeuN immunoreactivity (Fig. 4A). Neuronal CD163 levels were upregulated significantly after treatment with hemoglobin for 24 hours (0.69±0.03 in 5μM, p<0.05 and 0.75±0.12 in 20μM, p<0.01 vs. 0.39±0.11 in control, Fig. 4B).
Figure 4.
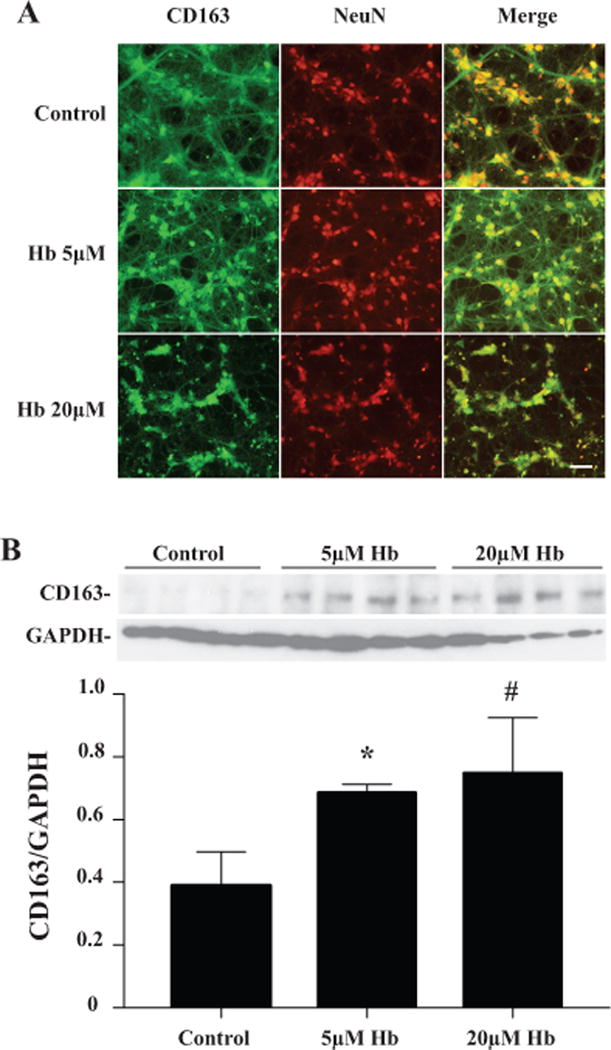
(A) Double labeling of CD163 and NeuN in primary cultured neuron treated with 0 (control), 5 or 20μM hemoglobin for 24 hours. Scale bar = 50μm. (B) Quantification of CD163 protein levels after hemoglobin exposure by Western blot. Values are mean ±SD, n=4, *p<0.05, #p<0.01 vs. the control.
To investigate the effect of deferoxamine on neuronal CD163 expression, cultured neurons were pretreated with deferoxamine (5μM) for 2 hours before hemoglobin (20μM) was administered. Immunocytochemistry revealed that axons were partially preserved in neurons that were pretreated with deferoxamine (Fig. 5A). Western blot analysis showed that CD163 levels were less in the deferoxamine-treated cells than in the vehicle-treated controls (0.39± 0.19 vs. 1.18±0.64, p<0.05; Fig. 5A). In addition, media LDH levels were significantly increased after treating neurons with 20μM of hemoglobin compared to controls (107±37 vs. 18 ± 4, p<0.01, Fig. 5B). This was attenuated by the pretreatment of deferoxamine (33 ±7 vs. 107 ±37 in vehicle, p<0.01, Fig. 5B).
Figure 5.
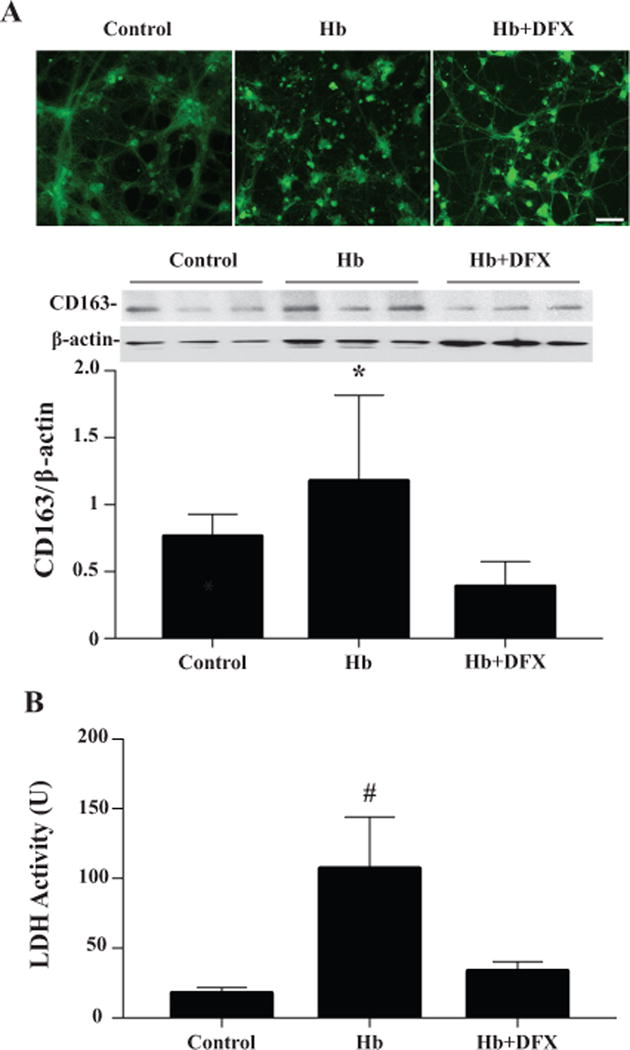
(A) CD163 immunoreactivity and protein levels in primary cultured neurons treated with hemoglobin (0 or 20μM) with or without deferoxamine (5μM) pretreatment. Scale bar= 50μm. (B) Media LDH levels 24 hours after the treatment of hemoglobin (0 or 20μM) with or without deferoxamine (5μM). Values are mean ± SD, *p<0.05, # p<0.01 vs. the other groups.
DISCUSSION
The major findings of this study are as follows: (1) CD163 is expressed in neurons in grey matter; (2) in the ipsilateral grey matter, CD163 protein levels increased at day 1 after ICH, peaked at day 3 and decreased at day 7; (3) hemoglobin induced CD163 upregulation in cultured neurons; and (4) deferoxamine reduced ICH-induced CD163 expression in vivo and hemoglobin-induced neuronal CD163 expression in vitro.
The present study using a piglet model indicated that neurons can express CD163. It is well known that CD163 is a hemoglobin receptor, predominantly expressed in microglia/ macrophages2. Our recent study showed that CD163 positive cells infiltrated into the hematoma from edge to center after ICH. A significant increase of CD163 positive cells in the hematoma was found at day 3 in the piglet model of ICH17. Recently, neuronal CD163 has been detected and may be involve in hemoglobin clearance after brain hemorrhage10, 13, 14. Neuronal express high levels of HO-2 levels and heme can be metabolized by HO-2 inside neurons23. Co-localization of HO-2 and CD163 revealed that both CD163 and HO-2 participate in hemoglobin degradation. Besides CD163-macrophage-HO-1 pathway, CD163-neuron-HO-2 pathway may also responsible for the degradation of hemoglobin (Fig. 6). A difference in CD163 expression, may explain why neurons are more vulnerable to hemoglobin toxicity than astrocytes11.
Figure 6.
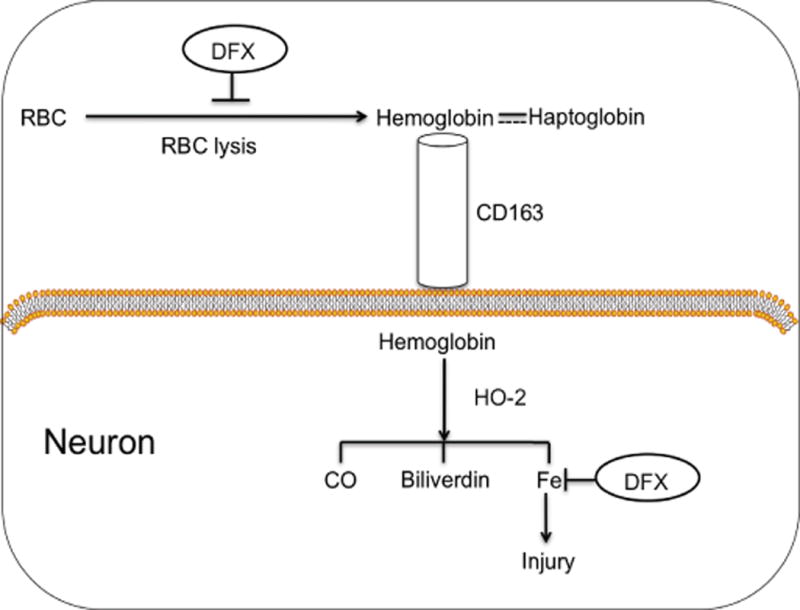
Model of the role of neuronal CD163 in relation to ICH and the effects of deferoxamine (DFX). Lysis or red blood cells (RBCs) in the clot leads to hemoglobin release into the extracellular space, which may be taken up into neurons via CD163 either alone or as part of a haptoglobin complex. Intracellularly, hemoglobin is degraded by heme oxygenase-2 (HO-2), producing iron, carbon monoxide and biliverdin. Deferoxamine may exert the neuroprotective effect by attenuating red blood cell lysis and iron chelation.
Our results indicate that hemoglobin upregulates CD163 expression in neurons. In the pig model, there was little CD163 expression in the brain of sham animals and in contralateral hemisphere after ICH. CD163 upregulation after ICH was time dependent. It was significantly upregulated at 24 hours and remained elevated at 3 days and then down regulated at 7 days. Neuron-like CD163 positive cells were also found in the contralateral grey matter, which may result from hemoglobin release from the clot. In the in vitro study, CD163 expression in cultured neurons was upregulated by hemoglobin in a dose-dependent manner. Therefore, ICH-induced increase of neuronal CD163 may result, at least in part, from hemoglobin release after ICH into perihematomal tissue as red blood cells lyse. DFX reduced the hemoglobin-induced increase of neuronal CD163 suggesting that iron may be involved in CD163 upregulation in neurons. In should be noted, however, that microglia also expressed CD163 in grey matter.
Hemoglobin is pivotal in the pathogenesis of ICH-induced brain damage24. Hemoglobin uptake into neurons is through CD163. In addition to uptake of the hemoglobin-haptoglobin complex, CD163 can also facilitate the uptake of free hemoglobin2. A recent study found that haptoglobin increases hemoglobin-induced neuronal death in vitro14. Some evidence also indicates that the neurotoxic effects of hemoglobin are HO-2 dependent. HO-2 is constitutively expressed by neurons and it accounts for most brain HO activity under normal conditions5. While HO-2 activity is not markedly increased over constitutive levels during exposure to hemoglobin25, deletion of HO-2 isoform not only reduced oxidative stress but also conferred resistance of neurons to hemoglobin12, 26, 27. Our current data showed many CD163 positive cells in the grey matter could be dying cells (TUNEL positive). In the current study, however, we did not establish a causal relationship between neuronal CD163 and neuronal death. Whether CD163 is involved in ICH-induced brain cell death should be determined in the future studies.
Deferoxamine is approved by the FDA for the treatment of acute iron intoxication and chronic iron overload in transfusion-dependent anemia. In the piglet ICH model, DFX treatment resulted in less hemoglobin accumulation, less ICH-induced upregulation of CD163 and less brain cell death in the grey matter. Our previous study demonstrated that deferoxamine can reduce ICH-induced neuronal death28. There is evidence that deferoxamine can inhibit hemin-induced erythrocyte lysis29 and lessen the rate of hematoma clearance in rats30. Therefore, reduced CD163 upregulation in the deferoxamine-treated group may be due to less erythrocyte lysis and less hemoglobin release.
The effect of hemoglobin on neuronal CD163 was also examined in neuronal culture. The nontoxic concentrations of deferoxamine are 0.6 to 5μM31, and the maximal nontoxic concentration of deferoxamine (5μM) was used in these experiments. The current in vitro study indicated that deferoxamine suppressed the expression of CD163 induced by hemoglobin in neurons and decreased the hemoglobin-induced neuronal death. There may be a role of iron in CD163 modulation.
A number of preclinical studies have shown the neuroprotective effects of deferoxamine after ICH28, 30, 32. Previous studies revealed that deferoxamine attenuates brain edema and reduces neuronal death after ICH32, but the underlying mechanisms are still not well understood. While the actions of deferoxamine may involve chelation of iron and reduced free radical production, it may also prevent neuronal death by stabilizing hypoxia-inducible factor-1 α (HIF-1α) protein in neurons33. In addition, in cultured neurons, some of the protective effects of deferoxamine were mediated by enhancing release of hemin from neurons rather than an effect on hemin intake34. The present study that shows deferoxamine can also modulate brain CD163 levels. Thus, it appears that deferoxamine has multiple and diverse neuroprotective properties in relation to hemorrhagic insults.
The involvement of neurons in the immediate response to hemoglobin might be responsible for some of ICH symptoms. For example, seizures may occur when the hemoglobin degradation is impaired10. Therapies targeting at neuronal CD163 may protect those cells by limiting hemoglobin uptake reducing the oxidative stress induced by hemoglobin/iron. This merits further study. Salvaging neurons by targeting hemoglobin-induced injury may also bring clinical benefits.
In summary, we found that neurons express CD163 and that brain levels are increased after ICH. Neuronal CD163 can be upregulated by hemoglobin and may be associated with hemoglobin-induced neuronal death (Fig. 6). Deferoxamine can modulate CD163 levels and cell death in vivo and in vitro.
Acknowledgments
Sources of Funding: This study was supported by grants NS-073959, NS-079157, NS-084049, NS-091545, NS-090925 and NS-096917 from the National Institutes of Health (NIH) and a University of Michigan and Peking University Joint Institute grant.
Footnotes
Conflict of Interests: the authors declared no conflict of interests.
References
- 1.Xi G, Keep RF, Hoff JT. Erythrocytes and delayed brain edema formation following intracerebral hemorrhage in rats. J Neurosurg. 1998;89:991–996. doi: 10.3171/jns.1998.89.6.0991. [DOI] [PubMed] [Google Scholar]
- 2.Thomsen JH, Etzerodt A, Svendsen P, Moestrup SK. The haptoglobin-cd163-heme oxygenase-1 pathway for hemoglobin scavenging. Oxid Med Cell Longev. 2013:523652. doi: 10.1155/2013/523652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao X, Song S, Sun G, Strong R, Zhang J, Grotta JC, et al. Neuroprotective role of haptoglobin after intracerebral hemorrhage. The Journal of Neuroscience. 2009;29:15819–15827. doi: 10.1523/JNEUROSCI.3776-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Madsen M, Graversen JH, Moestrup SK. Haptoglobin and cd163: Captor and receptor gating hemoglobin to macrophage lysosomes. Redox Rep. 2001;6:386–388. doi: 10.1179/135100001101536490. [DOI] [PubMed] [Google Scholar]
- 5.Chen-Roetling J, Lu X, Regan RF. Targeting heme oxygenase after intracerebral hemorrhage. Ther Targets Neurol Dis. 2015;2:e474. doi: 10.14800/ttnd.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schaer CA, Schoedon G, Imhof A, Kurrer MO, Schaer DJ. Constitutive endocytosis of cd163 mediates hemoglobin-heme uptake and determines the noninflammatory and protective transcriptional response of macrophages to hemoglobin. Circ Res. 2006;99:943–950. doi: 10.1161/01.RES.0000247067.34173.1b. [DOI] [PubMed] [Google Scholar]
- 7.Hua Y, Nakamura T, Keep RF, Wu J, Schallert T, Hoff JT, et al. Long-term effects of experimental intracerebral hemorrhage: The role of iron. J Neurosurg. 2006;104:305–312. doi: 10.3171/jns.2006.104.2.305. [DOI] [PubMed] [Google Scholar]
- 8.Wu J, Hua Y, Keep RF, Nakamura T, Hoff JT, Xi G. Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke. 2003;34:2964–2969. doi: 10.1161/01.STR.0000103140.52838.45. [DOI] [PubMed] [Google Scholar]
- 9.Regan RF, Panter SS. Neurotoxicity of hemoglobin in cortical cell culture. Neurosci Lett. 1993;153:219–222. doi: 10.1016/0304-3940(93)90326-g. [DOI] [PubMed] [Google Scholar]
- 10.Lara FA, Kahn SA, da Fonseca AC, Bahia CP, Pinho JP, Graca-Souza AV, et al. On the fate of extracellular hemoglobin and heme in brain. J Cereb Blood Flow Metab. 2009;29:1109–1120. doi: 10.1038/jcbfm.2009.34. [DOI] [PubMed] [Google Scholar]
- 11.Chen-Roetling J, Regan RF. Effect of heme oxygenase-1 on the vulnerability of astrocytes and neurons to hemoglobin. Biochem Biophys Res Commun. 2006;350:233–237. doi: 10.1016/j.bbrc.2006.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rogers B, Yakopson V, Teng ZP, Guo Y, Regan RF. Heme oxygenase-2 knockout neurons are less vulnerable to hemoglobin toxicity. Free Radic Biol Med. 2003;35:872–881. doi: 10.1016/s0891-5849(03)00431-3. [DOI] [PubMed] [Google Scholar]
- 13.Garton TP, He Y, Garton HJ, Keep RF, Xi G, Strahle JM. Hemoglobin-induced neuronal degeneration in the hippocampus after neonatal intraventricular hemorrhage. Brain Res. 2016;1635:86–94. doi: 10.1016/j.brainres.2015.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen-Roetling J, Regan RF. Haptoglobin increases the vulnerability of cd163-expressing neurons to hemoglobin. J Neurochem. 2016;139:586–595. doi: 10.1111/jnc.13720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou X, Xie Q, Xi G, Keep RF, Hua Y. Brain cd47 expression in a swine model of intracerebral hemorrhage. Brain Res. 2014;1574:70–76. doi: 10.1016/j.brainres.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xie Q, Gu Y, Hua Y, Liu W, Keep RF, Xi G. Deferoxamine attenuates white matter injury in a piglet intracerebral hemorrhage model. Stroke. 2014;45:290–292. doi: 10.1161/STROKEAHA.113.003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cao S, Zheng M, Hua Y, Chen G, Keep RF, Xi G. Hematoma changes during clot resolution after experimental intracerebral hemorrhage. Stroke. 2016;47:1626–1631. doi: 10.1161/STROKEAHA.116.013146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wagner KR, Xi G, Hua Y, Kleinholz M, de Courten-Myers GM, Myers RE. Early metabolic alterations in edematous perihematomal brain regions following experimental intracerebral hemorrhage. J Neurosurg. 1998;88:1058–1065. doi: 10.3171/jns.1998.88.6.1058. [DOI] [PubMed] [Google Scholar]
- 19.Jiang Y, Wu J, Hua Y, Keep RF, Xiang J, Hoff JT, et al. Thrombin-receptor activation and thrombin-induced brain tolerance. J Cereb Blood Flow Metab. 2002;22:404–410. doi: 10.1097/00004647-200204000-00004. [DOI] [PubMed] [Google Scholar]
- 20.He Y, Hua Y, Lee JY, Liu W, Keep RF, Wang MM, et al. Brain alpha- and beta-globin expression after intracerebral hemorrhage. Transl Stroke Res. 2010;1:48–56. doi: 10.1007/s12975-009-0004-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Regan RF, Rogers B. Delayed treatment of hemoglobin neurotoxicity. J Neurotrauma. 2003;20:111–120. doi: 10.1089/08977150360517236. [DOI] [PubMed] [Google Scholar]
- 22.Koh JY, Choi DW. Vulnerability of cultured cortical neurons to damage by excitotoxins: Differential susceptibility of neurons containing nadph-diaphorase. J Neurosci. 1988;8:2153–2163. doi: 10.1523/JNEUROSCI.08-06-02153.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robinson SR, Dang TN, Dringen R, Bishop GM. Hemin toxicity: A preventable source of brain damage following hemorrhagic stroke. Redox Rep. 2009;14:228–235. doi: 10.1179/135100009X12525712409931. [DOI] [PubMed] [Google Scholar]
- 24.Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral hemorrhage. Lancet Neurol. 2006;5:53–63. doi: 10.1016/S1474-4422(05)70283-0. [DOI] [PubMed] [Google Scholar]
- 25.Chen M, Regan RF. Time course of increased heme oxygenase activity and expression after experimental intracerebral hemorrhage: Correlation with oxidative injury. J Neurochem. 2007;103:2015–2021. doi: 10.1111/j.1471-4159.2007.04885.x. [DOI] [PubMed] [Google Scholar]
- 26.Chen-Roetling J, Cai Y, Regan RF. Neuroprotective effect of heme oxygenase-2 knockout in the blood injection model of intracerebral hemorrhage. BMC Res Notes. 2014;7:561. doi: 10.1186/1756-0500-7-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Regan RF, Chen J, Benvenisti-Zarom L. Heme oxygenase-2 gene deletion attenuates oxidative stress in neurons exposed to extracellular hemin. BMC Neurosci. 2004;5:34. doi: 10.1186/1471-2202-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu Y, Hua Y, Keep RF, Morgenstern LB, Xi G. Deferoxamine reduces intracerebral hematoma-induced iron accumulation and neuronal death in piglets. Stroke. 2009;40:2241–2243. doi: 10.1161/STROKEAHA.108.539536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sullivan SG, Baysal E, Stern A. Inhibition of hemin-induced hemolysis by desferrioxamine: Binding of hemin to red cell membranes and the effects of alteration of membrane sulfhydryl groups. Biochim Biophys Acta. 1992;1104:38–44. doi: 10.1016/0005-2736(92)90129-a. [DOI] [PubMed] [Google Scholar]
- 30.Hatakeyama T, Okauchi M, Hua Y, Keep RF, Xi G. Deferoxamine reduces neuronal death and hematoma lysis after intracerebral hemorrhage in aged rats. Transl Stroke Res. 2013;4:546–553. doi: 10.1007/s12975-013-0270-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guelman LR, Pagotto RM, Di Toro CG, Zieher LM. Deferoxamine antioxidant activity on cerebellar granule cells gamma-irradiated in vitro. Neurotoxicol Teratol. 2004;26:477–483. doi: 10.1016/j.ntt.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 32.Song S, Hua Y, Keep RF, Hoff JT, Xi G. A new hippocampal model for examining intracerebral hemorrhage-related neuronal death: Effects of deferoxamine on hemoglobin-induced neuronal death. Stroke. 2007;38:2861–2863. doi: 10.1161/STROKEAHA.107.488015. [DOI] [PubMed] [Google Scholar]
- 33.Karuppagounder SS, Alim I, Khim SJ, Bourassa MW, Sleiman SF, John R, et al. Therapeutic targeting of oxygen-sensing prolyl hydroxylases abrogates atf4-dependent neuronal death and improves outcomes after brain hemorrhage in several rodent models. Sci Transl Med. 2016;8:328ra329. doi: 10.1126/scitranslmed.aac6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen-Roetling J, Cai Y, Lu X, Regan RF. Hemin uptake and release by neurons and glia. Free Radic Res. 2014;48:200–205. doi: 10.3109/10715762.2013.859386. [DOI] [PMC free article] [PubMed] [Google Scholar]