

Abstract
Background
Excessive alcohol (EtOH) consumption causes imbalances in protein metabolism. EtOH impairs protein synthesis in C2C12 myoblasts via a FoxO1-AMPK-TSC2-mTORC1 pathway and also induces degradation. As the underlying regulatory signaling cascades for these processes are currently poorly defined, we tested the hypothesis that alcohol-induced autophagy is mediated via activation of the PIK3C3 complex that is regulated by FoxO1-AMPK.
Methods
C2C12 myoblasts were incubated with EtOH for various periods of time and autophagy pathway-related proteins were assessed by Western blotting and immunoprecipitation. Expression of targeted genes was suppressed using electroporation of specific siRNAs and chemical inhibitors.
Results
Incubation of C2C12 myoblasts with 100 mM EtOH increased the autophagy markers LC3B-II and ATG7, whereas levels of SQSTM1/p62 decreased. The lysosomal inhibitor bafilomycinA1 caused a similar response, although there was no additive effect when combined with ETOH. EtOH altered ULK1 S555 and S757 phosphorylation in a time- and AMPK-dependent manner. The activation of AMPK and ULK1 was associated with increased BECN1 (S93, S14) and PIK3C3/VPS34 (S164) phosphorylation as well as increased total ATG14 and PIK3C3. These changes promoted formation of the ATG14-AMBRA1-BECN1-PIK3C3 pro-autophagy complex that is important in autophagosome formation. EtOH-induced changes were not associated with increased production of PtdIns3P, which may be due to enhanced PIK3C3 complex binding with 14-3-3ϴ. Reduction of AMPK using siRNA suppressed the stimulatory effect of EtOH on BECN1 S93, S14 and PIK3C3 S164 phosphorylation in a time-dependent manner. Likewise, knockdown of AMPK or chemical inhibition of FoxO1 attenuated phosphorylation of ULK1 at both residues. Knockdown of ULK1 or BECN1 antagonized the effect of EtOH on LC3B-II, SQSTM1and ATG7 protein expression.
Conclusions
EtOH-induced autophagy is mediated through changes in phosphorylation and interaction of various PIK3C3 complex components. This, in turn, is regulated either directly via FoxO1-AMPK, or indirectly via the FoxO1-AMPK-ULK1 signaling cascade in a mTORC1-independent or-dependent manner.
Keywords: Class III phosphatidylinositol 3-Kinase, FoxO1-AMPK, autophagy, alcohol, skeletal muscle
INTRODUCTION
Excessive alcohol (ethanol; EtOH) consumption causes diverse metabolic and functional changes in skeletal and cardiac muscle. A hallmark of prolonged alcohol abuse is the loss of lean body mass arising from an impairment in protein balance as evidenced by the decrease in protein synthesis and/or increase in protein degradation (Steiner and Lang, 2015). Previously, EtOH was shown to decrease protein synthesis in C2C12 mouse myoblasts, whereas its effect on global protein degradation in these cells was equivocal (Hong-Brown et al., 2007, Hong-Brown et al., 2012). However, recent data suggest that EtOH can increase muscle protein breakdown at least in part by stimulating lysosomal autophagy (Guo and Ren, 2012, Thapaliya et al., 2014), but the underlying signaling mechanisms have not been fully defined.
Macroautophagy (autophagy) is a catabolic system that delivers cytoplasmic content (protein, lipid, organelles) to the lysosome for degradation. Activation of autophagy in response to various stress conditions is an essential mechanism maintaining cell viability. As such, autophagy is a highly regulated process and is central in maintaining cellular protein homeostasis (Meijer and Codogno, 2004). Autophagy also plays a central role in development, tissue remodeling, and is altered in a number of human diseases of striated muscle (Piano and Phillips, 2014, Yamada et al., 2012, Guo et al., 2012). Multistep processes coordinate the action of various autophagy-related (ATG) proteins on Unc-51 like kinase-1 (ULK1) and vacuolar protein sorting 34 (VPS34/PIK3C3) complexes. These proteins, in turn, function as important regulators of autophagy initiation and progression.
PIK3C3 complex is a class III phosphatidylinositol 3-Kinase (PtdIns3K) implicated as a critical regulator for a number of cellular events including endocytic trafficking and autophagy (Backer, 2008, Jaber et al., 2012, Funderburk et al., 2010, Tassa et al., 2003). This protein is also involved in mammalian target of rapamycin (mTOR) signaling in response to nutrient starvation (Nobukuni et al., 2005, Yuan et al., 2013, Chang et al., 2009). PIK3C3 phosphorylates the 3-posititon of phosphatidylinositol thereby producing phosphatidylinositol 3-phosphate (PtdIns3P), a key membrane marker for both intracellular trafficking and autophagosome formation (Vieira et al., 2001, Zhong et al., 2009, Schu et al., 1993). The mammalian PIK3C3 protein resides in multiple complexes that are responsible for different cellular functions. Core components of various PIK3C3 complexes include PIK3C3, BECN1 and PIK3R4/p150 (vps15). BECN1 participates in PIK3C3 complex formation and recruits additional proteins including ATG14/Barkor (BECN1-associated autophagy-related key regulator), ultraviolet irradiation resistance-associated gene (UVRAG) and the activating molecule in BECN1-regulated autophagy (AMBRA-1). These ATG14, UVRAG or AMBRA1 containing complexes each function as a positive regulator of BECN1-PIK3C3 complexes and have important roles in autophagy (Matsunaga et al., 2009, Sun et al., 2008, Itakura et al., 2008, Zhong et al., 2009, Liang et al., 2006, Cianfanelli et al., 2015). Additionally, Bif1, VMP1 and Rubicon have also been reported as components of the PIK3C3 complex, suggesting that many different PIK3C3 complexes exist that can potentially impact cellular function (Shi et al., 2014, Takahashi et al., 2007).
The PIK3C3 complex is regulated via phosphorylation events that ultimately affect its interaction with various accessory proteins. Thus, in response to stress, the composition of the PIK3C3 complex depends upon which components are phosphorylated. For example, with glucose deprivation, phosphorylation of BECN1 at serine (S) 91/S94 activates the pro-autophagic PIK3C3 complex containing ATG14 or UVRAG (Kim et al., 2013). These complexes mediate autophagosome formation and maturation, respectively (Zhong et al., 2009, Itakura et al., 2008, Matsunaga et al., 2009, Sun et al., 2008). Conversely, phosphorylation of PIK3C3 at S164 inhibits the non-autophagic PIK3C3 complex, thereby suppressing PtdIns3P production (Kim et al., 2013). Similar to situations involving energy stress, amino acid deprivation differentially regulates a PIK3C3 complex that activates autophagy, while coordinately inhibiting the non-autophagic complex. This induction does not require BECN1 phosphorylation at S93, suggesting an additional mechanism is involved in PIK3C3 regulation. In contrast, amino acid starvation enhances S14-phosphorylation of BECN1 (Russell et al., 2013). At present, the precise role that PIK3C3 complexes play in EtOH-induced autophagy remains undefined.
AMP-activated protein kinase (AMPK) is a major sensor of nutrient and cellular energy status that is activated under stress conditions (e.g., hypoxia, glucose withdrawal, EtOH) (Gwinn et al., 2008, Hong-Brown et al., 2010). Signaling from AMPK regulates the autophagy pathway (Russell et al., 2014, Meijer and Codogno, 2007) and data from independent studies indicate AMPK regulates nutrient- and alcohol-induced autophagy via phosphorylation of ULK1 (Kim et al., 2011, Lee et al., 2010, Shang et al., 2011, Kandadi et al., 2013). Furthermore, while AMPK regulates multiple PIK3C3 complexes following glucose deprivation (Kim et al., 2013), the role of AMPK in regulating PIK3C3 complexes as a mechanism underlying EtOH-induced autophagy in muscle is unknown. Previously, we reported EtOH enhances AMPK phosphorylation and activity, while concurrently inhibiting mTORC1 function and protein synthesis (Hong-Brown et al., 2010; Hong-Brown et al., 2015). Although recent studies showed that forkhead box 3a (FoxO3a) is a key factor in regulating EtOH and stress-induced autophagy in cultured hepatocytes and other tissues (Ni et al., 2013, Sanchez et al., 2012, Nepal and Park, 2013), it is unclear whether this signaling cascade regulates the process in muscle.
In this study, we tested the hypothesis that alcohol-induced autophagy in myoblasts is mediated via activation of the PIK3C3 complex that is regulated by the activation of AMPK. Our data reveal that EtOH activates the pro-autophagy ATG14-PIK3C3 complex by increasing BECN1 phosphorylation (S14 and S93) and ATG14 levels. In parallel, EtOH inhibits the non-autophagy PIK3C3 complex by enhancing total PIK3C3 and PIK3C3 (S164) phosphorylation. EtOH-induced phosphorylation and dephosphorylation of ULK1 at S555 and S757 alters the interaction between AMPK and ULK1, and increases the association of ULK1 with BECN1, ATG14 and PIK3C3. Stimulation of autophagy by EtOH is regulated by FoxO1-AMPK signaling which mediates its effects via activation of ULK1 and ATG14-containing PIK3C3 complexes.
MATERIALS AND METHODS
Materials
EtOH was purchased from Fisher Scientific Co. (Springfield, NJ). Antibodies against phosphorylated (p)-ULK1 (S555), p-ULK1 (S757) and p-BECN1 (S93) were obtained from Cell Signaling Technology (Beverly, MA). LC3B, LC3A, ATG7, ATG12-5, SQSTM1/p62, total PIK3C3 and β-actin were from the same source with the latter serving as a control for equal protein loading of samples. The p-BECN1 (S15) antibody was from Abbiotec (San Diego, CA). The mouse (m) AMPKα1/2, BECN1 (m), ULK1 (m) siRNA, scrambled siRNA as well as antibodies against 14-3-3ϴ and AMBRA1 were purchased from Santa Cruz Biotechnology (Dallas, Texas). Antibody to ATG14 was from Medical & Biological Laboratories Co., LTD (Woburn, MA) and PIK3C3-p-34 (S164) was purchased from Biorbyt (San Francisco, CA). The AMPK inhibitor Compound C and FoxO1 inhibitor AS1842856 were from EMD Millipore Corporation (Billerica, MA). Phenylmethanesulfonyl fluoride (PMSF), bafilomycin A1, protease, and phosphatase I and II inhibitor cocktails were from Sigma Aldrich (St. Louis, MO). Cell culture media and fetal bovine serum (FBS) were from Mediatech, Inc (Manassas, VA).
Cell cultures and transfection
C2C12 mouse myoblasts (American Type Culture Collection; Manassas, VA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, penicillin (100 U/ml), streptomycin (100 μg/ml) and amphotericin (25 μg/ml) at 37° C in 5% CO2. Cells were plated on 6-well or 10 cm dishes and grown to confluence. All studies were conducted using cells at the early passage (P3–P7) of the myoblast stage. Upon confluence, cells were treated with 100 mM EtOH as previously described and harvested after 1 h unless otherwise indicated (Hong-Brown et al., 2015). A concentration of 100 mM EtOH was used because it produces an optimal effect on protein synthesis without being cytotoxic to myoblasts. During the incubation, plates were sealed with parafilm to minimize the evaporation of EtOH. Myoblasts were used because of the difficulty in effectively transfecting fully differentiated myotubes which was necessary for pursuing mechanistic studies.
For transient expression, the siRNA target or scrambled sequences were transfected into C2C12 myoblasts by electroporation (Amaxa Biosystems nucleofector II, Germany) and the Amaxa cell line nucleofector kit V (Lonza Walkersville Inc., Walkersville, MD). All transfections were performed following the manufactures’ protocol. Experiments were carried out 48 h post-transfection and cells were harvested thereafter for immunoblot analysis.
Drug treatment
For studies using the FoxO1 or lysosomal inhibitors, myoblasts were pre-treated as noted in the figure legends. The concentration and incubation times for reagents were chosen based on dose- and time-response curves derived from our preliminary studies and the literature (Tanaka et al., 2010). Total protein was determined using the bicinchoninic acid reagent (BCA) protein assay kit with bovine serum albumin (BSA) as a standard (Pierce, Rockford, IL). The data were normalized and expressed as a percentage of the appropriate time-matched control value.
PtdIns3K activity assay
PIK3C3 complex activity was determined using a PtdIns3K Elisa kit from Echelon Biosciences (Salt Lake City, UT) according to the manufacturer’s protocol. Briefly, PtdIns3K was immunoprecipitated from control and EtOH-treated cell lysates utilizing anti-PIK3C3 or ATG14 antibodies. In these experiments phosphatidylinositol was used as substrate and the activity was monitored for the detection of PtdIns3P production.
Autophagic detection
Autophagic activity in myoblasts was measured using a Cyto-ID autophagy detection kit (Enzo Life Sciences, Farmingdale, NY) according to the manufacturer’s protocol. Cells expressing a GFP-LC3 fusion protein were visually monitored using a Leica SP8 scanning confocal microscope (Heidelberg, Germany), thereby allowing for the observation of fluorescence in vesicles produced during autophagy.
Immunoprecipitation and immunoblot analysis
C2C12 myoblasts, cultured in either 10 cm or 6-well plates, were incubated in the presence of EtOH and/or various reagents (bafilomycin A1 or FoxO1 inhibitor). Thereafter, cells were rinsed once with PBS and lysed in ice-cold 0.3% CHAPS (3-[(3-cholamindopropyl) dimethylammonio]-1-propanesulfonate) buffer containing PMSF and a cocktail of protease and phosphatase inhibitors. Soluble fractions of cell extracts were isolated by centrifugation at 14,000 rpm for 5 min at 4° C. For immunoprecipitation (IP), a 1:75 dilution of primary antibody (Ab) was added to equal amounts of protein (300–500 μg) from cell lysates and rotated overnight at 4° C. A total of 50 μl of a 50% slurry of protein A sepharose was then added for an additional 1 h. Immunoprecipitates were washed 3 times with lysis buffer and the precipitated proteins were denatured by the addition of 2× Laemmli sample buffer (LSB). Equal amounts of protein from cell lysates were electrophoresed on denaturing polyacrylamide gels and transferred to nitrocellulose or PVDF membranes. The resulting blots were blocked with 5% nonfat dry milk and incubated with the antibodies of interest (1:500–1000). Unbound primary antibody was removed by washing with TBS containing 0.05% Tween-20 (ICI Americas, Inc, Wilmington, DE) and blots were incubated with anti-rabbit immunoglobulin (1:3000) conjugated with horseradish peroxidase (HRP). On occasions where the denatured and reduced rabbit IgG light or heavy chains obscured protein bands of similar molecular weights, mouse anti-rabbit IgG (conformation specific L27A9) mAb (Cell Signaling) was added as a bridging antibody prior to incubation with an anti-mouse IgG, HRP-linked secondary antibody (Cell Signaling). Blots were incubated with an enhanced chemiluminescent detection system (GE Healthcare, Buckinghamshire, UK) and exposed to X-ray blue film (Cole Parmer, Vernon Hills, IL) or the FluroChem M multifluor system was used for visualization (Proteinsimple, Santa Clara, CA) and quantified with Image J (NIH, Bethesda, MD).
Statistical analysis
For experimental protocols with more than two groups, statistical significance was determined using one- or two-way ANOVA followed by the Dunnett’s post-hoc test. For experiments with only two groups, an unpaired Student’s t-test was performed. Data are presented as mean ± SEM of multiple (3–5 times) repeated experiments with 3–4 samples per experiment. A value of P < 0.05 was considered statistically significant.
RESULTS
Autophagy markers
Although EtOH increases autophagy in various cell types and tissues, the signaling cascades that regulate EtOH-induced autophagic responses in muscle cells have not been fully defined. In initial experiments, we verified the effects of EtOH on protein levels of known autophagy markers. Incubation of myoblasts with 100 mM EtOH for various times increased LC3B lipidation (LC3B-II) by ~ 60% as compared to time-matched controls (Fig. 1A). Likewise, an enhanced fluorescent signal of LC3B-II was observed following EtOH exposure using confocal fluorescent microscopy (Fig. 1, B and C). EtOH increased the amount of both LC3A-II (an isoform of LC3B) and ATG7, while ATG12-5 protein expression did not differ between control and EtOH-treated cells. Conversely, EtOH decreased SQSMT1 (sequestosome 1)/p62 protein relative to control values (Fig. 1, D and E).
Fig. 1.
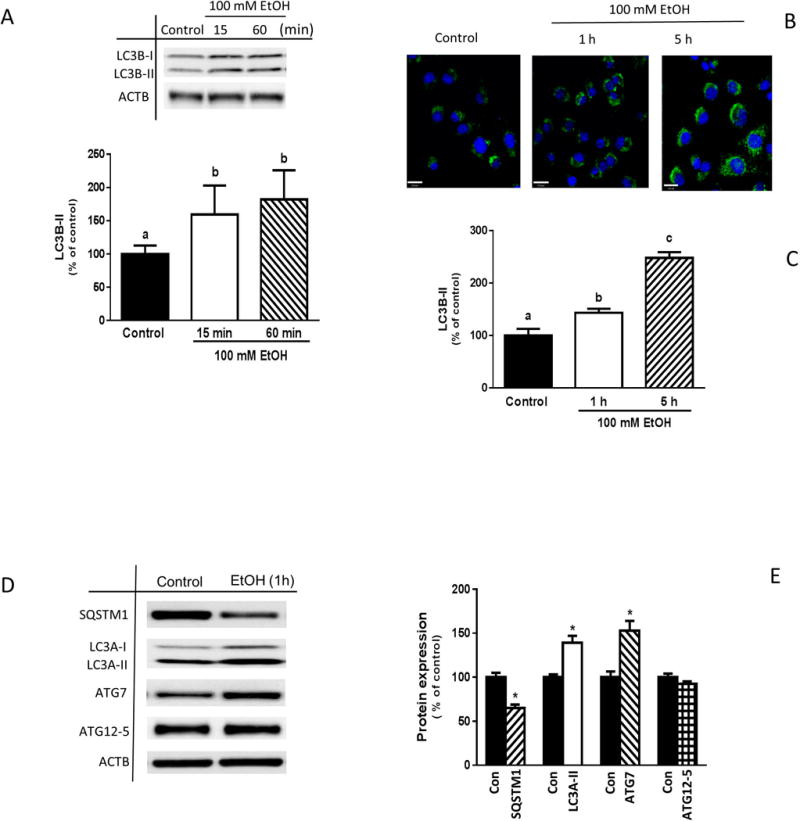
Effect of EtOH on the protein expression of autophagy markers. (A) C2C12 myoblasts were incubated in the presence or absence of 100 mM EtOH for the indicated times. Equal amounts of cell extracts were collected and analyzed via Western blotting using antibody against LC3B. (B) LC3B fluorescence levels in control and EtOH-treated myoblasts were examined utilizing scanning confocal fluorescence microscopy and a Cyto-ID autophagy detection kit as described under “Materials and Methods.” (C) The intensity of the fluorescence was quantified and presented as a bar graph. (D) The amount of LC3A, ATG7, SQSTM1/p62 and ATG12-5 was determined for myoblasts treated with EtOH for 1 h. (E) The protein levels were quantified and presented as a bar graph. Each bar represents the mean ± SEM of 4 independent experiments consisting of 4 replicate samples per experiment. Results were normalized to total protein and expressed as a percentage of basal control levels. Groups with different letters are significantly different from one another (P< 0.05). Group with the same letters are not significantly different. *P< 0.05 versus control values.
AMPK and FoxO1 mediate EtOH-induced changes in LC3B and SQSMT1
AMPK and the transcription factor FoxO3a are involved in regulating autophagy under starvation conditions as well as following EtOH exposure (Ni et al., 2013, Sanchez et al., 2012, Nepal and Park, 2013). Previously, we reported that AMPK and FoxO1 mediate the effects of EtOH on various signal transduction pathways (Hong-Brown et al., 2015). As such, EtOH increases both AMPK and FoxO1 activity towards a number of downstream targets that regulate protein synthesis and extracellular matrix components. To determine whether EtOH-induced changes in LC3B-II and SQSMT1 levels are regulated by AMPK, myoblasts were transfected with scrambled siRNA or a AMPKα1/2 siRNA. The selection of AMPKα1/2 siRNA was based on results from previous studies where we immune-isolated AMPKα1/2 for in vitro kinase assays. Knockdown of AMPK with siRNA resulted in a ≈60% decline in the amount of AMPK, compared to scrambled control values (Fig. 2A). While partial silencing of AMPK did not alter basal LC3B-II levels, the EtOH-induced increase in LC3B-II was prevented and the relative expression of LC3B-II was lower than scrambled control values (Fig. 2, B and C). Similar results were obtained when myoblasts were treated with the AMPK inhibitor compound C in the presence or absence of EtOH (data not shown). In addition, knockdown of AMPK prevented the EtOH-induced decrease in SQSMT1 (Fig. 2, B and D).
Fig. 2.
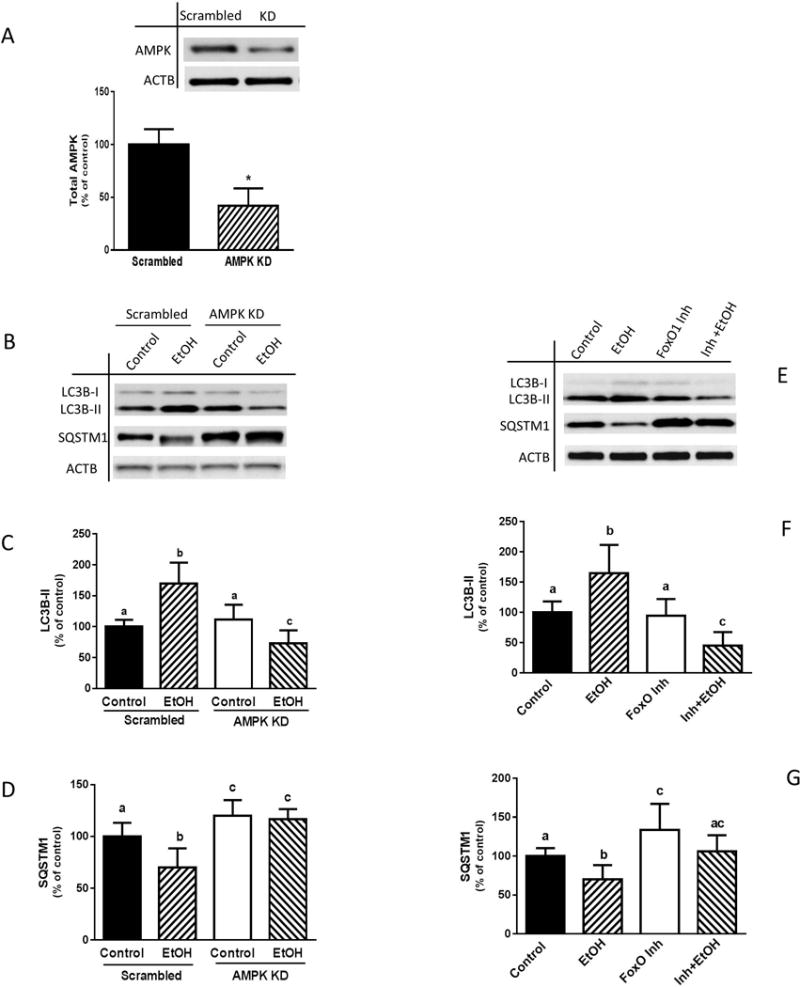
AMPK knockdown or inhibition of FoxO1 blocks EtOH-induced autophagy. (A) C2C12 myoblasts were transfected with scrambled siRNA or AMPKα1/2 siRNA. Cells were collected 48 h after transfection and analyzed by Western blotting using AMPK antibody. (B) Scrambled control or AMPK knockdown cells were incubated in the presence or absence of 100 mM EtOH for 1 h and equal amounts of cell extracts were analyzed via Western blotting using antibodies against LC3B or SQSTM1. (C–D) The protein levels were quantified on a bar graph. (E) Myoblasts were pretreated with 0.3 μM FoxO1 inhibitor for 1 h and then exposed to EtOH for an additional hour. Cells were collected and analyzed via immunoblotting using antibodies against LC3B or SQSTM1. (F–G) The protein levels were quantified on a bar graph. Each bar represents the mean ± SEM of 3 independent experiments consisting of 4 replicate samples per experiment. Results were normalized to total protein and expressed as a percentage of control levels. Groups with different letters are significantly different from one another (P< 0.05). Group with the same letters are not significantly different. *P< 0.05 versus scramble control values.
Next, we examined whether FoxO1 is involved in the regulation of EtOH-induced autophagy, as determined using our two autophagy markers. Incubation of myoblasts with a FoxO1 inhibitor did not alter the amount of basal LC3B-II (Fig. 2, E and F). However, this agent completely suppressed the stimulatory effect of EtOH, with LC3B-II levels remaining below control values. Likewise, this drug blocked the inhibitory effect of EtOH on SQSMT1 (Fig. 2, E and G). Collectively, these data demonstrate that both AMPK and FoxO1 are involved in the regulation of EtOH-induced autophagy in C2C12 myoblasts.
PIK3C3 and BECN1 phosphorylation
Phosphorylation events affect the formation of distinct PI3K complexes that either promote or inhibit autophagy. Following energy starvation, phosphorylation of BECN1 at S91/94 promotes the ATG14-containing pro-autophagy PIK3C3 complex, whereas phosphorylation of PIK3C3 at S164 blocks formation of the non-autophagy complex (Kim et al., 2013). Conversely, phosphorylation of S91/94 is not necessary for the induction of pro-autophagy PIK3C3 complexes in response to amino acid deprivation (Russell et al., 2013). Hence, we examined whether the EtOH-induced autophagy is associated with changes in the phosphorylation of BECN1 and PIK3C3. Incubation of myoblasts with EtOH rapidly increased BECN1 phosphorylation on S93 and S14 compared to control values (Fig. 3A). The increase in BECN1 S93 phosphorylation was transient, whereas S14 phosphorylation remained elevated up to 1 h. EtOH also increased phosphorylation of PIK3C3 at S164 in myoblasts for at least 1 h (Fig. 3B). The observed phosphorylation changes were independent of alterations in total BECN1 levels, although EtOH did increase the total amount of both ATG14 and PIK3C3 (Fig. 3C).
Fig. 3.
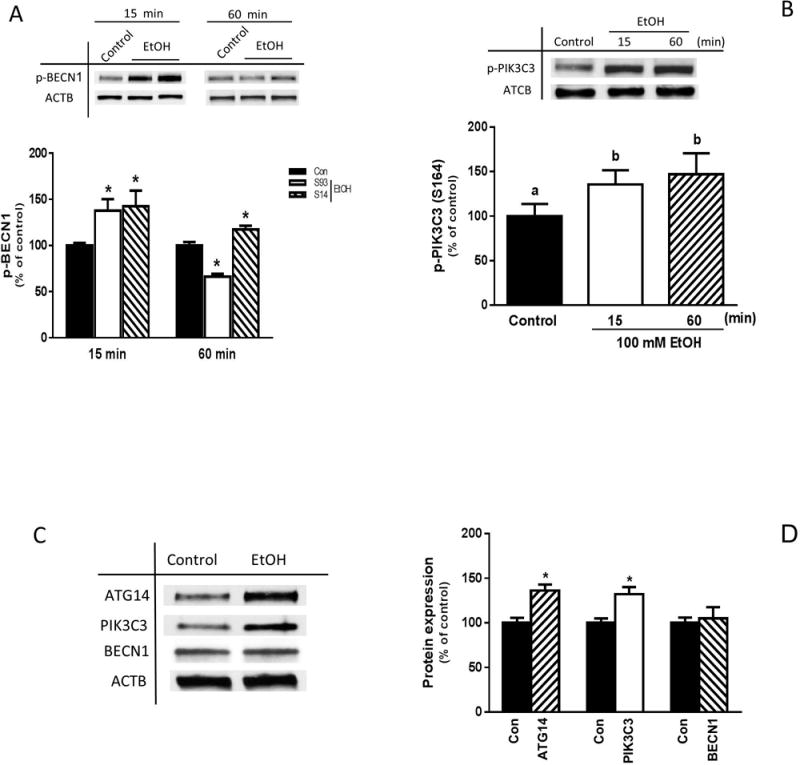
Time-dependent effects of EtOH on BECN1 and PIK3C3 phosphorylation. C2C12 myoblasts were incubated in the presence or absence of 100 mM EtOH for the indicated times. Cell lysates were subjected to immunoblotting utilizing antibodies against phosphorylated (p) BECN1 at serine (S) 93 and S14 (A), as well as p-PIK3C3 (S164) (B). (C) Cell lysates from myoblasts treated with EtOH for 15 min were analyzed via Western blotting using antibodies against the indicated proteins. (D) Protein levels were quantified and presented as a bar graph. Each bar represents the mean ± SEM of 4 independent experiments consisting of 4 replicate samples per experiment. Results were normalized to total protein and expressed as a percentage of time matched controls. Groups with different letters are significantly different from one another (P< 0.05). Group with the same letters are not significantly different. *P< 0.05 versus control values.
PIK3C3 interaction and binding with AMPK
Next, we determined whether EtOH-induced changes in PIK3C3 and BECN1 phosphorylation were associated with alterations in protein-protein interactions known to mediate downstream signaling events. Incubation of myoblasts with EtOH increased the association of PIK3C3 with BECN1, ATG14 and the negative regulator 14-3-3 θ, as compared to untreated control cells (Fig. 4, A and B). Enhanced binding of ATG14 with PIK3C3 and 14-3-3θ was also observed (Fig. 4, C and D). No interaction between BECN1 and UVRAG was detected (data not shown) because the presence of ATG14 and UVRAG in the same complex is mutually exclusive. EtOH increased the interaction of AMBRA1 with BECN1 and ATG14, while it decreased the binding between AMBRA1 and PIK3C3 (Fig. 4, E and F). Next, as assessed by an in vitro kinase assay, EtOH attenuated the PIK3C3 lipid kinase activity of immunoprecipitates isolated with antibodies against either ATG14 or PIK3C3 (Fig. 4G). Finally, we examined the interaction of the PIK3C3 complex with its upstream regulator AMPK. EtOH increased the interaction of AMPK with ATG14 and AMBRA1. In contrast, EtOH decreased the association between PIK3C3 and AMPK (Fig. 4, H and I). Together, these data suggest EtOH regulates the composition and activity of the PIK3C3 complex, changes mediated via the phosphorylation state of BECN1 and PIK3C3.
Fig. 4.
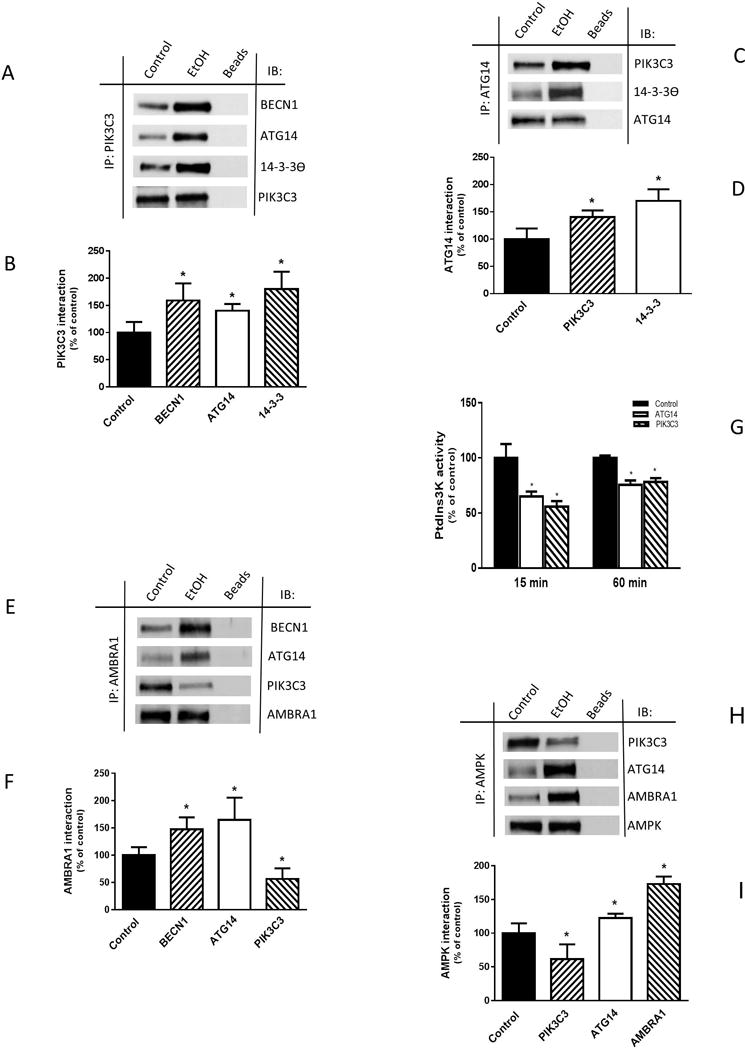
EtOH alters PIK3C3 complex component interaction and binding with AMPK. (A) C2C12 myoblasts were incubated in the presence or absence of 100 mM EtOH for 15 min and equal amounts of proteins from cell extracts (300–450 μg) were immunoprecipitated with antibody against PIK3C3 and then immunoblotted with BECN1, ATG14, or 14-3-3 antibodies. (B) The protein levels were quantified as bar graph and normalized with immunoprecipitated PIK3C3 which was assessed by Western blotting. (C) ATG14 was immunoprecipitated from equal amounts of cell lysate and immunoblotted with PIK3C3 or 14-3-3 antibodies. (D) Protein levels were quantified and normalized with immunoprecipitated ATG14, which was assessed by Western blotting. (E) AMBRA1 was immunoprecipitated from equal amounts of cell lysate and immunoblotted with BECN1, ATG14 and PIK3C3 (F) Protein levels were quantified and normalized with immunoprecipitated AMBRA1, which was assessed by Western blotting. (G) For PIK3C3 complex activity, ATG14 or PIK3C3 was immunoprecipitated from 250 μg of cell lysate, and the activity was determined using a PIK3C3 Elisa kit as described in “Material and Methods.” (H) AMPK was immunoprecipitated from equal amounts of cell lysate and immunoblotted with PIK3C3, ATG14 or AMBRA1 antibodies. (I) The protein levels were quantified as bar graph and normalized with immunoprecipitated AMPK which was assessed by Western blotting. Data are mean ± SEM of 3 independent experiments consisting of 3 replicate samples per experiment. *P< 0.05 versus the control values.
BECN1 mediates EtOH effect on LC3B, ATG7 and SQSMT1
To further characterize the role that BECN1 plays in EtOH-induced autophagy, myoblasts were transfected with scrambled or BECN1 specific siRNA. BECN1 was decreased by 90% as compared to scrambled controls (Fig. 5A). This knockdown of BECN1 blunted the stimulatory effect of EtOH on LC3B lipidation, with the amount of LC3B-II remaining below control values (Fig. 5, B and C). Knockdown of BECN1 also prevented the EtOH-induced decrease in SQSMT1 (Fig. 5, B and D). Finally, the reduction in BECN1 antagonized the changes in ATG7 observed in response to EtOH, when compared to scrambled controls (Fig. 5, B and E). These results suggest a role for BECN1 and the PIK3C3 complex as upstream regulators of the ethanol-induced autophagy markers LC3B and SQSMT1.
Fig. 5.
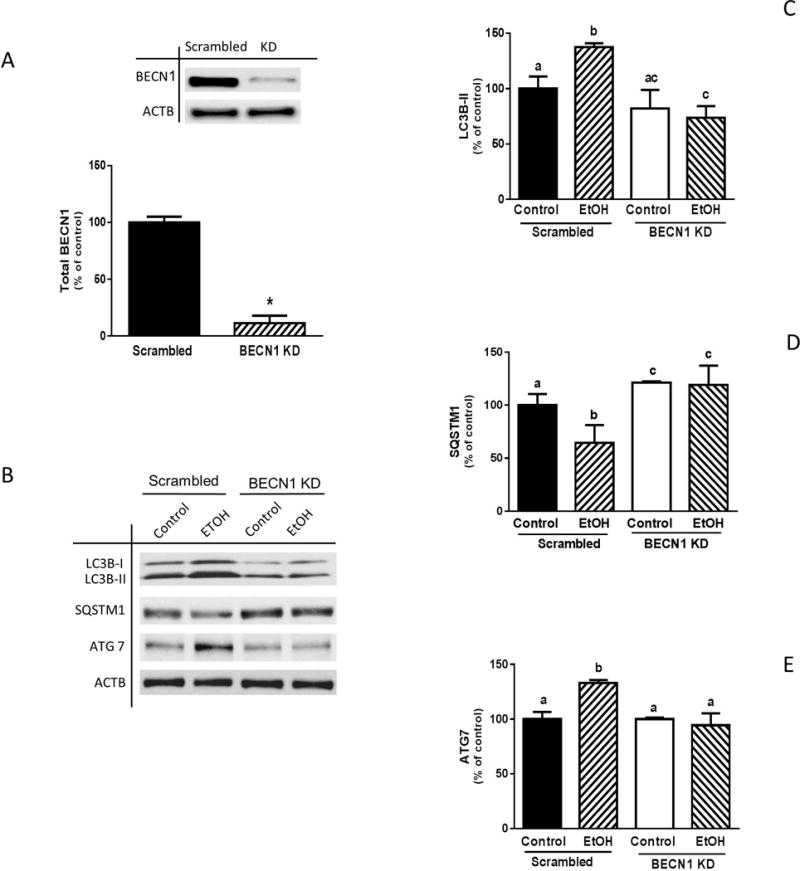
BECN1 knockdown alters EtOH-induced changes in LC3B, ATG7 and SQSTM1. (A) C2C12 myoblasts were transfected with scrambled siRNA or BECN1 siRNA. Cells were collected 48 post-transfection and analyzed by Western blotting using BECN1 antibody. (B) Scrambled and BECN1 knockdown cells were treated with EtOH for 1 h and equal amounts of cell lysates were analyzed via Western blotting using antibodies against LC3B (B–C), SQSTM1 (B & D) and ATG7 (B & E). Each bar represents the mean ± SEM of 3 independent experiments consisting of 3 replicate samples per experiment. Results were normalized to total protein and expressed as a percentage of scramble control levels. Groups with different letters are significantly different from one another (P< 0.05). Group with the same letters are not significantly different. *P< 0.05 versus control values.
ULK1 phosphorylation and its interaction with PIK3C3 complex and AMPK
ULK1 contains multiple phosphorylation sites and is differentially regulated by upstream kinases acting on various residues in response to stress conditions (Kim et al., 2011, Shang et al., 2011, Guo and Ren, 2012). To determine whether EtOH-induced autophagy in myoblasts is mediated by ULK1 phosphorylation, we assessed the dynamics of phosphorylation at S757 and S555, sites that are involved in “cytoplasmic” autophagy and mitophagy, respectively (Egan et al., 2011, Kim et al., 2011). Incubation of cells with EtOH for 15 min increased ULK1 phosphorylation at S555, compared to control values (Fig. 6A). However, phosphorylation was suppressed when cells were subjected to extended treatment (1 h), and remained below control levels for up to 4 h (data not shown). In contrast, short-term treatment with EtOH decreased ULK1 phosphorylation at S757 (Fig. 6B), while increased phosphorylation at this residue was detected after 1 h and remained elevated for up to 3 h (data not shown). The total amount of ULK1 was not altered at the various time points. These changes in ULK1 phosphorylation were associated with alterations in protein-protein interaction, as EtOH increased the binding of ULK1 with BECN1, ATG14 and PIK3C3 when compared to untreated control cells (Fig. 6, C and D). The association of ULK1 with its upstream regulator AMPK was also modulated in a time-dependent manner (Fig. 6, E and F).
Fig. 6.
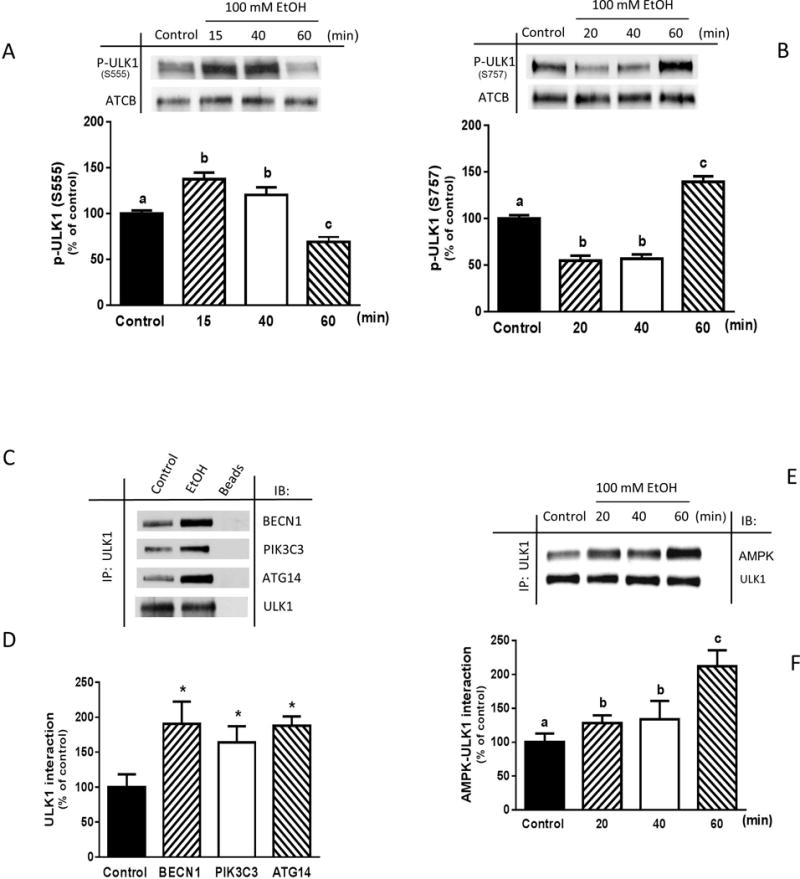
EtOH differentially phosphorylates ULK1 at S555 and S757, while increasing ULK 1 binding with the PIK3C3 complex and AMPK. C2C12 myoblasts were incubated in the presence or absence of 100 mM EtOH for the indicated times. Cell lysate was subjected to immunoblotting utilizing antibodies against p-ULK1 at S555 (A) and S757 (B). Results were normalized to total protein and expressed as a percentage of time matched control levels. (C) ULK1 was immunoprecipitated from equal amounts of cell extracts (300–450 μg) and then immunoblotted with BECN1, PIK3C3 or ATG14 antibodies. (D) The protein levels were quantified and normalized with immunoprecipitate ULK1 which was assessed by Western blotting. (E) ULK1was immunoprecipitated from equal amounts of cell extracts (300–450 μg) and then immunoblotted with AMPK. (F) Protein levels were quantified and normalized with immunoprecipitated ULK1 as assessed by Western blotting. Data are mean ± SEM of 3 independent experiments consisting of 4 replicate samples per experiment. Groups with different letters are significantly different from one another (P< 0.05). Group with the same letters are not significantly different. *P< 0.05 versus control values.
ULK1 knockdown attenuates EtOH effect on PIK3C3 and BECN1 phosphorylation
Because BECN1 is a direct target of ULK1 in cells subjected to stress conditions, we examined whether EtOH-induced changes in BECN1 and PIK3C3 phosphorylation were regulated by ULK1. Myoblasts were transfected with scrambled or ULK1-specific siRNA and cell extracts were collected. Knockdown of ULK1 with siRNA resulted in ~60% decrease in total ULK1, compared to scrambled control values (Fig. 7A). Furthermore, silencing of ULK1 attenuated the EtOH-induced increase in BECN1 phosphorylation at both S14 and S93 (Fig. 7, B, C and D), as well as the increase in PIK3C3 phosphorylation (Fig. 7B and E). These data suggest that ULK1 is an upstream regulator of the PIK3C3 complex in C2C12 myoblasts exposed to EtOH.
Fig. 7.
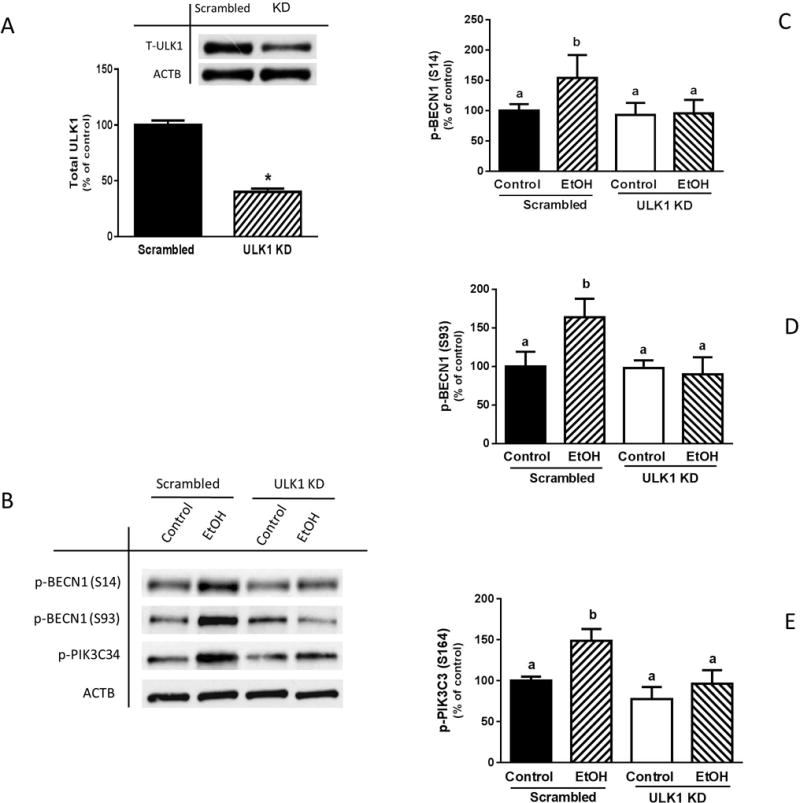
ULK1 knockdown attenuates EtOH-induced increase in BECN1 and PIK3C3 phosphorylation. (A) C2C12 myoblasts were transfected with scrambled siRNA or ULK1 siRNA (0.4 μg) for 48 h. Cells were collected thereafter and analyzed by Western blotting using ULK1 antibody. (B) Myoblasts were incubated with EtOH for 15 min and equal amount of cell lysates from scrambled control or ULK1 knockdown cells were analyzed via Western blotting using antibodies against p-BECN1 at S14 (B–C), S93 (B & D) and p-PIK3C3 S164 (B & E). Each bar represents the mean ± SEM of 3 independent experiments consisting of 4 replicate samples per experiment. Results were normalized to total protein and expressed as a percentage of scrambled control levels. Groups with different letters are significantly different from one another (P< 0.05). Group with the same letters are not significantly different. *P< 0.05 versus scrambled control values.
ULK1 mediates EtOH effect on ATG7 and LC3B
To study the link between ULK1 and EtOH-induced autophagy, myoblasts were transfected with scrambled or ULK1-specific siRNA. Knockdown of ULK1 blocked the increase in LC3B-II (Fig. 8, A and B) and ATG7 (Fig. 8, A and C) in EtOH-treated cells, compared to scrambled control values. In contrast, reduction of ULK1 by siRNA did not alter the EtOH-induced decrease of SQSTM1 (Fig. 8, A and D).
Fig. 8.
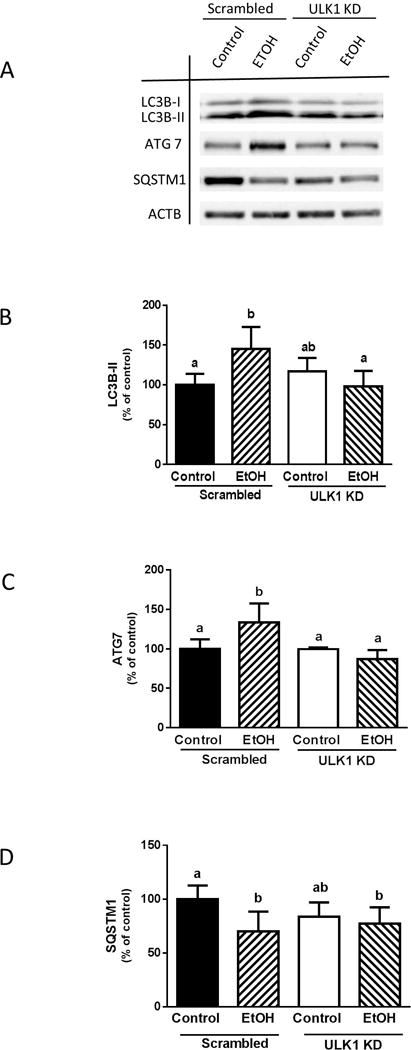
ULK1 knockdown suppresses the EtOH-induced increase in LC3B and ATG7. (A) Scrambled and ULK1 knockdown cells were incubated with EtOH as described above and the amount of LC3B-II (A–B), ATG7 (A & C), and SQSTM1 (A & D) were analyzed via Western blotting using indicated antibodies. Each bar represents the mean ± SEM of 3 independent experiments consisting of 3 replicate samples per experiment. Results were normalized to total protein and expressed as a percentage of scramble control levels. Groups with different letters are significantly different from one another (P< 0.05). Group with the same letters are not significantly different.
AMPK regulates BECN1, PIK3C3 and ULK1phosphorylation
To investigate whether the phosphorylation of BECN1, PIK3C3 and ULK1 was AMPK-dependent, myoblasts were transfected with scrambled or AMPKα1/2 siRNA. The knockdown of AMPK blunted the EtOH-induced increase of BECN1 (S93) phosphorylation after 15 min (Fig. 9, A and B). Likewise, partial silencing of AMPK prevented the EtOH-induced decrease at S93 BECN1 phosphorylation otherwise observed at 1 h (data not shown). In contrast, whereas AMPK knockdown did not affect the EtOH-induced increase in BECN1 (S14) phosphorylation at 15 min (Fig. 9, A and C), it did prevent the increase observed at the 1 h time point (Fig. 9, A and D). Reduction of AMPK by siRNA blocked EtOH-induced increases in PIK3C3 phosphorylation (Fig. 9, A and E). Comparable data were obtained when myoblasts were treated with the AMPK inhibitor Compound C (data not shown). These results suggest that phosphorylation of BECN1 S93 and PIK3C3 are directly regulated by AMPK at both time points, whereas phosphorylation of BECN1 at S14 was regulated at a later time (1 h) point. Together, these data indicate that AMPK regulates its downstream targets in a time-dependent manner.
Fig. 9.
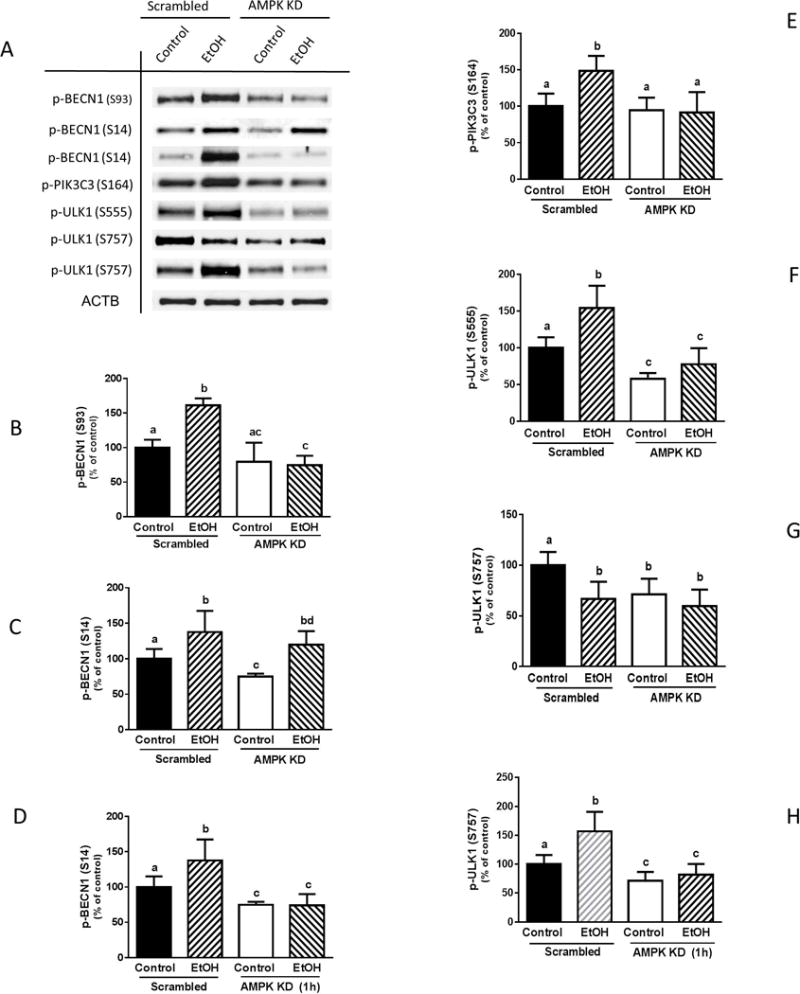
AMPKα1/2 knockdown inhibits the EtOH-induced phosphorylation of BECN1 (S93, S14) and PIK3C3 (S164), as well as ULK1 (S555 and S757). (A) Scrambled siRNA or AMPKα1/2 knockdown myoblasts were incubated with EtOH for 15 min or as indicated, and equal amounts of cell extracts were analyzed via Western blotting using antibodies against p-BECN1 at S93 (A–B), S14 (A & C–D), p-PIK3C3 S164 (A & E), p-ULK1 at S555 (A & F) and S757 (A & G–H). Each bar represents the mean ± SEM of 3 independent experiments consisting of 4 replicate samples per experiment. Results were normalized to total protein and expressed as a percentage of scramble control levels. Groups with different letters are significantly different from one another (P< 0.05). Group with the same letters are not significantly different.
Finally, AMPK knockdown also prevented the EtOH-induced increase in ULK1 phosphorylation observed at S555, with levels remaining below those of scrambled controls (Fig. 9, A and F). The suppressive effect of EtOH on S757 at early time points was not affected by AMPK silencing (Fig. 9, A and G). However, knockdown of AMPK did block the increased S757 phosphorylation otherwise observed when myoblasts were incubated with EtOH for 1 h (Fig. 9, A and H). These data suggest AMPK regulates both the early (S555) and late (S757) EtOH-mediated phosphorylation of ULK1.
DISCUSSION
We have previously reported that the FoxO1-sestrin3-AMPK signaling cascade mediates the ability of EtOH to inhibit mTOR function and protein synthesis in C2C12 myoblasts (Hong-Brown et al., 2015). Likewise, several studies have demonstrated a connection between AMPK and FoxO3a in the induction of autophagy, either under stress situations or following EtOH exposure (Nepal and Park, 2013, Ni et al., 2013, Sanchez et al., 2012). In the current study, we identified a role for AMPK and FoxO1 signaling in regulating autophagy in myoblasts. EtOH decreased SQSTM1, a selective autophagy adapter protein that recognizes and degrades a number of cargoes including organelles (Jin et al., 2013). For example, a reduction of SQSTM1 correlates with the induction of mitophagy in murine liver and in primary hepatocytes after alcohol (Ding et al., 2010). Our results are in agreement with previous studies using a variety of cell types subjected to starvation, hypoxia or EtOH (Pursiheimo et al., 2009, Wang et al., 2013, Kuma et al., 2004). In contradistinction, other studies report that EtOH increases SQSTM1 levels in mouse liver, HepG2 cells, and heart muscle cells (Thomes et al., 2015, Guo and Ren, 2012). These contrasting results are not unexpected because we and others have demonstrated differential effects of EtOH on AMPK-mTORC1 mediated signaling pathways in liver and muscle cells (Noh et al., 2011, (Hong-Brown et al., 2012, Garcia-Villafranca et al., 2008). The observed changes in LC3B-II and SQSTM1 were abolished in myoblasts in the presence of a FoxO1 inhibitor or when AMPK was decreased by siRNA, suggesting that both AMPK and FoxO1 are required to regulate EtOH-induced autophagy. Mechanistically, AMPK appears to mediate this effect via distinct kinase cascades, including ULK1 and the PIK3C3 complex.
ULK1 is a key initiator of starvation-induced autophagy and contains several phosphorylation sites that can differentially affect cellular targets. For example, enhanced ULK1 phosphorylation at S555 increases mitophagy under energy stress conditions (Egan et al., 2011). The increased phosphorylation at this site, coupled with a reduction in SQSTM1 led us to speculate that EtOH enhances autophagy in myoblasts by specifically targeting organelles, such as mitochondria, as reported in other cell types (Bonet-Ponce et al., 2015, Flores-Bellver et al., 2014, Ding et al., 2010). Furthermore, decreased ULK1 phosphorylation at S757 has been correlated with an induction of nonspecific autophagy as reported in studies during acute nutrient deprivation (Shang et al., 2011).
ULK1 phosphorylation appears to be regulated by AMPK, as there was an EtOH-induced increase in the interaction of AMPK with ULK1. Furthermore, knockdown of AMPK or inhibition of FoxO1 prevented the stimulatory effect of EtOH on ULK1 phosphorylation. ULK1 can be regulated by either AMPK- or mTOR-dependent pathways, depending on which residues are phosphorylated. For example, the ability of EtOH to increase S555 phosphorylation appears to be regulated by AMPK, since ULK1 (S555) is a direct substrate of AMPK (Egan et al., 2011). Conversely, enhanced phosphorylation of ULK1 at S757 by cellular stress can be controlled by mTOR as part of the AMPK-TSC2-mTORC1 signaling cascade (Kim et al., 2011). In this regard, we previously reported that EtOH increases AMPK and decreases mTORC1 activity via the same pathway (Hong-Brown et al., 2012). Hence, the EtOH-mediated decrease in ULK1 S757 may be due to the coordinate decrease in mTOR activity via the AMPK-TSC2-mTORC1 dependent pathway. Together, our data suggest that changes in the phosphorylation state of ULK1 at S555 and S757 in response to EtOH are regulated by AMPK and FoxO1, and this may involve both mTORC1-dependent and-independent pathways.
Phosphorylation of ULK1 by AMPK is important for the subsequent function of ULK1 in regulating downstream substrates including the PIK3C3 complex. Accumulating evidence indicates that alterations in phosphorylation and/or interaction of BECN1 and PIK3C3 are required for stress-induced autophagy (Kim et al., 2013). In the present study, EtOH-induced changes in BECN1 phosphorylation were associated with elevations in autophagy, as indicated by increased LC3B-II (Russel et al., 2013) and decreased SQSTM1 (Ding et al., 2010, Pursiheimo et al., 2009, Wang et al., 2013). The important role of BECN1 in EtOH-induced autophagy is further supported by the observation that knockdown of BECN1 blocked the EtOH-induced changes in LC3B, and SQSTM1. The EtOH-induced increase in BECN1 and PIK3C3 phosphorylation appears regulated by ULK1, as the interaction of this kinase with BECN1, ATG14 and PIK3C3 was increased. Likewise, siRNA knockdown of ULK1 prevented the effect of EtOH on BECN1 (S93, S14) and PIK3C3 (S164) phosphorylation. Moreover, decreasing AMPK by siRNA also prevented the phosphorylation of BECN1 (S93) phosphorylation and PIK3C3, consistent with reports whereby AMPK directly regulated BECN1 S93 upon glucose depletion (Kim and Guan, 2013, Kim et al., 2013). In contrast, knockdown of AMPK did not alter the short-term EtOH-induced increase in BECN1 S14 phosphorylation, a site regulated by ULK1 during amino acid starvation (Russell et al., 2013). Hence, EtOH-induced changes in the PIK3C3 complex are mediated either by AMPK or indirectly via ULK1 activation of different phosphorylated residues.
Increased BECN1 phosphorylation functions to recruit PIK3C3-specific subunits, such as ATG14, UVRAG and AMBRA1, and forming a pro-autophagy PIK3C3 complex. The central role played by ATG14 and AMBRA1 is supported by data indicating that siRNA silencing of ATG14 or AMBRA1 reduces autophagosome nucleation (Itakura et al., 2008, Matsunaga et al., 2009, Fimia et al., 2007). Likewise, down-regulation of AMBRA1 decreases the capacity of BECN1 to associate with PIK3C3 and reduces autophagy induction (Fiema et al., 2007). Conversely, overexpression of ATG14/Barkor or AMBRA1 activates autophagy and increases the number of autophagosomes (Sun et al., 2008). Hence, the ability of EtOH to increase association of the pro-autophagic proteins ATG14 and AMBRA1 with BECN1-PIK3C3 complexes may be essential for autophagosome formation and autophagy initiation (Matsunaga et al., 2009, Zhong et al., 2009). On the other hand, the lysosomal inhibitor bafilomycin A1 did not modulate the effect of EtOH on LC3B-II or SQSTM1 (data not shown). While this result is in agreement with a report examining HepG2 cells and liver from chronic alcohol-fed mice (Guo et al., 2015), it is in contrast to previous studies in myocytes where there was an additive effect of EtOH and lysosomal inhibitors (Thapaliya et al., 2014, Luo, 2014). Although there is no clear explanation for this discrepancy, it may be due to differences in cell types and/or the inhibitors used in the respective studies. Therefore, it is possible that EtOH affects the key components of the early, rather than later, stages of the autophagy pathway. Collectively, our data indicate that enhanced phosphorylation and interaction of BECN1 with accessory proteins within the PIK3C3 complex can be regulated directly by AMPK or indirectly via ULK1.
Activation of the PIK3C3 complex and synthesis of its product PtdIns3P are required for autophagosome formation and/or maturation during autophagy (Vieira et al., 2001, Zhong et al., 2009). Herein we demonstrated that EtOH decreased the amount of PtdIns3P, a result comparable to the starvation response (Kim et al., 2013). Hence, the reduction in PtdIns3P by EtOH may represent an important stress response involved in cell viability, whereby cells decrease metabolic activities associated with growth and differentiation. In this regard, the attenuation in PtdIns3P production may be due to an enhanced binding of the PIK3C3 complex with 14-3-3, a protein that negatively regulates the activity of the lipid kinase (Pozuelo-Rubio, 2011). This increased association appears regulated by an EtOH-induced decrease in the activity of protein phosphatase 2A towards AMPK (Hong-Brown et al., 2007), that increases AMPK-dependent phosphorylation of PIK3C3 thereby enhancing its binding with 14-3-3. Overall, these changes can have important consequences related to the survival of cells, some of which are associated with the suppression of apoptosis.
We propose the following model for EtOH-induced autophagy in myoblasts based on data from the current study and the published literature (Fig. 10). EtOH-induced changes in LC3B lipidation, SQSTM1 and ATG7 are regulated by FoxO1-AMPK either directly by affecting the PIK3C3 complex, or indirectly via ULK1 and PIK3C3 complex cascades. This, in turn, is mediated by both mTORC1-independent and-dependent pathways. AMPK increases ULK1 function through direct phosphorylation of the S555 residue. Alternatively, AMPK can regulate ULK1 by inactivating mTORC1 through phosphorylation of TSC2 and raptor. Inhibition of mTORC1 is correlated with a decreased ULK1 S757 phosphorylation and increased activity. Regardless, AMPK enhances the activity of ULK1 to regulate its downstream targets BECN1 and PIK3C3. Phosphorylation of BECN1 serves to recruit ATG14 and AMBRA1 for the formation of the pro-autophagy complex that may play an important role in autophagosome formation. Phosphorylation of PIK3C3 appears to be involved in inhibition of the non-autophagy complex. Hence, our data provide greater insight into the molecular mechanisms by which EtOH regulates muscle protein homeostasis.
Fig. 10.
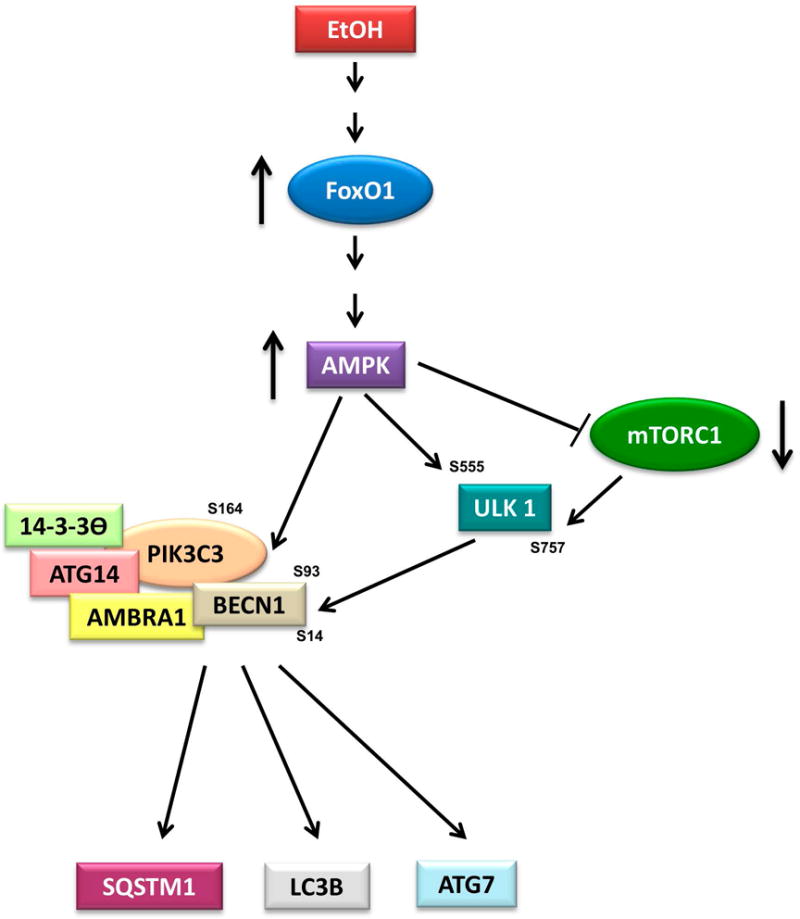
Proposed model for FoxO1 and AMPK regulation of EtOH-induced autophagy. EtOH increases LC3B lipidation and decreases SQSTM1, with this process being mediated via multiple signaling cascades. EtOH stimulates FoxO1 activity, which increases the phosphorylation and function of AMPK. Upon activation, the PIK3C3 complex can be directly regulated by AMPK through phosphorylation of BECN1 (S93, S14) and PIK3C3 (S164). Alternatively, AMPK can affect ULK1 function to regulate its downstream target PIK3C3 complex through phosphorylation of BECN1 and PIK3C3 via a mTORC1-independent and dependent pathway. The activated PIK3C3 complex increases LC3B-II and ATG7 levels, as well as decreases SQSTM1.
Acknowledgments
We thank Drs. Catherine Coleman, Kristen T. Crowell, and Abid A. Kazi for their technical support. Confocal images were generated using LeicaSP8 (S10OD010756-01A1) local in Microscopy Image Core Facility, Penn State, College of Medicine. This study was supported by National Institute of Health grant R37 AA-011290.
Abbreviations
- AMPK
AMP-activated protein kinase
- AMBRA1
activating molecule in BECN1-regulated autophagy
- ATG
autophagy-related
- Barkor
BECN1-associated autophagy-related key regulator
- BECN1
BECN1
- EtOH
ethanol
- FoxO1
forkhead transcription factor box1
- LC3
microtubule-associated protein light chain 3
- mTOR
mammalian target of rapamycin
- PIK3C3/VPS34
vacuolar protein sorting 34
- PtdIns3K
class III phosphatidylinositol 3-Kinase
- PtdIns3P
phosphatidylinositol 3-phosphate
- Rubicon
RUN domain and cysteine-rich domain containing, BECN1-interacting protein
- p62/SQSTM1
sequestosome 1
- ULK1
Unc-51 like kinase-1
- UVRAG
ultraviolet irradiation resistance-associated gene
Footnotes
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
References
- Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. The Biochemical journal. 2008;410:1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- Bonet-Ponce L, Saez-Atienzar S, da Casa C, Flores-Bellver M, Barcia JM, Sancho-Pelluz J, Romero FJ, Jordan J, Galindo MF. On the mechanism underlying ethanol-induced mitochondrial dynamic disruption and autophagy response. Biochimica et biophysica acta. 2015;1852:1400–1409. doi: 10.1016/j.bbadis.2015.03.006. [DOI] [PubMed] [Google Scholar]
- Chang YY, Juhasz G, Goraksha-Hicks P, Arsham AM, Mallin DR, Muller LK, Neufeld TP. Nutrient-dependent regulation of autophagy through the target of rapamycin pathway. Biochemical Society transactions. 2009;37:232–236. doi: 10.1042/BST0370232. [DOI] [PubMed] [Google Scholar]
- Cianfanelli V, De Zio D, Di Bartolomeo S, Nazio F, Strappazzon F, Cecconi F. Ambra1 at a glance. Journal of cell science. 2015;128:2003–2008. doi: 10.1242/jcs.168153. [DOI] [PubMed] [Google Scholar]
- Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, Lu B, Stolz DB, Clemens DL, Yin XM. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–1752. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–1125. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- Flores-Bellver M, Bonet-Ponce L, Barcia JM, Garcia-Verdugo JM, Martinez-Gil N, Saez-Atienzar S, Sancho-Pelluz J, Jordan J, Galindo MF, Romero FJ. Autophagy and mitochondrial alterations in human retinal pigment epithelial cells induced by ethanol: implications of 4-hydroxy-nonenal. Cell death & disease. 2014;5:e1328. doi: 10.1038/cddis.2014.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex–at the crossroads of autophagy and beyond. Trends in cell biology. 2010;20:355–362. doi: 10.1016/j.tcb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Villafranca J, Guillen A, Castro J. Ethanol consumption impairs regulation of fatty acid metabolism by decreasing the activity of AMP-activated protein kinase in rat liver. Biochimie. 2008;90:460–466. doi: 10.1016/j.biochi.2007.09.019. [DOI] [PubMed] [Google Scholar]
- Guo R, Hu N, Kandadi MR, Ren J. Facilitated ethanol metabolism promotes cardiomyocyte contractile dysfunction through autophagy in murine hearts. Autophagy. 2012;8:593–608. doi: 10.4161/auto.18997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R, Ren J. Deficiency in AMPK attenuates ethanol-induced cardiac contractile dysfunction through inhibition of autophagosome formation. Cardiovascular research. 2012;94:480–491. doi: 10.1093/cvr/cvs127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R, Xu X, Babcock SA, Zhang Y, Ren J. Aldehyde dedydrogenase-2 plays a beneficial role in ameliorating chronic alcohol-induced hepatic steatosis and inflammation through regulation of autophagy. Journal of hepatology. 2015;62:647–656. doi: 10.1016/j.jhep.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Molecular cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong-Brown LQ, Brown CR, Huber DS, Lang CH. Alcohol regulates eukaryotic elongation factor 2 phosphorylation via an AMP-activated protein kinase-dependent mechanism in C2C12 skeletal myocytes. The Journal of biological chemistry. 2007;282:3702–3712. doi: 10.1074/jbc.M606593200. [DOI] [PubMed] [Google Scholar]
- Hong-Brown LQ, Brown CR, Kazi AA, Huber DS, Pruznak AM, Lang CH. Alcohol and PRAS40 knockdown decrease mTOR activity and protein synthesis via AMPK signaling and changes in mTORC1 interaction. Journal of cellular biochemistry. 2010;109:1172–1184. doi: 10.1002/jcb.22496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong-Brown LQ, Brown CR, Kazi AA, Navaratnarajah M, Lang CH. Rag GTPases and AMPK/TSC2/Rheb mediate the differential regulation of mTORC1 signaling in response to alcohol and leucine. American journal of physiology. Cell physiology. 2012;302:C1557–1565. doi: 10.1152/ajpcell.00407.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong-Brown LQ, Brown CR, Navaratnarajah M, Lang CH. Adamts1 mediates ethanol-induced alterations in collagen and elastin via a FoxO1-sestrin3-AMPK signaling cascade in myocytes. Journal of cellular biochemistry. 2015;116:91–101. doi: 10.1002/jcb.24945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Molecular biology of the cell. 2008;19:5360–5372. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaber N, Dou Z, Lin RZ, Zhang J, Zong WX. Mammalian PIK3C3/VPS34: the key to autophagic processing in liver and heart. Autophagy. 2012;8:707–708. doi: 10.4161/auto.19627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Liu X, Klionsky DJ. SnapShot: Selective autophagy. Cell. 2013;152:368–368 e362. doi: 10.1016/j.cell.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandadi MR, Hu N, Ren J. ULK1 plays a critical role in AMPK-mediated myocardial autophagy and contractile dysfunction following acute alcohol challenge. Current pharmaceutical design. 2013;19:4874–4887. doi: 10.2174/1381612811319270010. [DOI] [PubMed] [Google Scholar]
- Kim J, Guan KL. AMPK connects energy stress to PIK3C3/VPS34 regulation. Autophagy. 2013;9:1110–1111. doi: 10.4161/auto.24877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152:290–303. doi: 10.1016/j.cell.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature cell biology. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. PloS one. 2010;5:e15394. doi: 10.1371/journal.pone.0015394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nature cell biology. 2006;8:688–699. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- Luo J. Autophagy and ethanol neurotoxicity. Autophagy. 2014;10:2099–2108. doi: 10.4161/15548627.2014.981916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, Akira S, Noda T, Yoshimori T. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nature cell biology. 2009;11:385–396. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. The international journal of biochemistry & cell biology. 2004;36:2445–2462. doi: 10.1016/j.biocel.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Meijer AJ, Codogno P. AMP-activated protein kinase and autophagy. Autophagy. 2007;3:238–240. doi: 10.4161/auto.3710. [DOI] [PubMed] [Google Scholar]
- Nepal S, Park PH. Activation of autophagy by globular adiponectin attenuates ethanol-induced apoptosis in HepG2 cells: involvement of AMPK/FoxO3A axis. Biochimica et biophysica acta. 2013;1833:2111–2125. doi: 10.1016/j.bbamcr.2013.05.013. [DOI] [PubMed] [Google Scholar]
- Ni HM, Du K, You M, Ding WX. Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. The American journal of pathology. 2013;183:1815–1825. doi: 10.1016/j.ajpath.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobukuni T, Joaquin M, Roccio M, Dann SG, Kim SY, Gulati P, Byfield MP, Backer JM, Natt F, Bos JL, Zwartkruis FJ, Thomas G. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:14238–14243. doi: 10.1073/pnas.0506925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh BK, Lee JK, Jun HJ, Lee JH, Jia Y, Hoang MH, Kim JW, Park KH, Lee SJ. Restoration of autophagy by puerarin in ethanol-treated hepatocytes via the activation of AMP-activated protein kinase. Biochemical and biophysical research communications. 2011;414:361–366. doi: 10.1016/j.bbrc.2011.09.077. [DOI] [PubMed] [Google Scholar]
- Piano MR, Phillips SA. Alcoholic cardiomyopathy: pathophysiologic insights. Cardiovascular toxicology. 2014;14:291–308. doi: 10.1007/s12012-014-9252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pursiheimo JP, Rantanen K, Heikkinen PT, Johansen T, Jaakkola PM. Hypoxia-activated autophagy accelerates degradation of SQSTM1/p62. Oncogene. 2009;28:334–344. doi: 10.1038/onc.2008.392. [DOI] [PubMed] [Google Scholar]
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nature cell biology. 2013;15:741–750. doi: 10.1038/ncb2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell research. 2014;24:42–57. doi: 10.1038/cr.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez AM, Csibi A, Raibon A, Cornille K, Gay S, Bernardi H, Candau R. AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. Journal of cellular biochemistry. 2012;113:695–710. doi: 10.1002/jcb.23399. [DOI] [PubMed] [Google Scholar]
- Schu PV, Takegawa K, Fry MJ, Stack JH, Waterfield MD, Emr SD. Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science. 1993;260:88–91. doi: 10.1126/science.8385367. [DOI] [PubMed] [Google Scholar]
- Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:4788–4793. doi: 10.1073/pnas.1100844108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Wu J, Fu M, Zhang B, Wang J, Yang X, Chi Y. Ambra1 modulates starvation-induced autophagy through AMPK signaling pathway in cardiomyocytes. Biochemical and biophysical research communications. 2014;452:308–314. doi: 10.1016/j.bbrc.2014.08.017. [DOI] [PubMed] [Google Scholar]
- Steiner JL, Lang CH. Dysregulation of skeletal muscle protein metabolism by alcohol. American journal of physiology. Endocrinology and metabolism. 2015;308:E699–712. doi: 10.1152/ajpendo.00006.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Fan W, Chen K, Ding X, Chen S, Zhong Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:19211–19216. doi: 10.1073/pnas.0810452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mule JJ, Pledger WJ, Wang HG. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nature cell biology. 2007;9:1142–1151. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Nagashima T, Shimaya A, Urano Y, Shimokawa T, Shibasaki M. Effects of the novel Foxo1 inhibitor AS1708727 on plasma glucose and triglyceride levels in diabetic db/db mice. European journal of pharmacology. 2010;645:185–191. doi: 10.1016/j.ejphar.2010.07.018. [DOI] [PubMed] [Google Scholar]
- Tassa A, Roux MP, Attaix D, Bechet DM. Class III phosphoinositide 3-kinase–Beclin1 complex mediates the amino acid-dependent regulation of autophagy in C2C12 myotubes. The Biochemical journal. 2003;376:577–586. doi: 10.1042/BJ20030826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapaliya S, Runkana A, McMullen MR, Nagy LE, McDonald C, Naga Prasad SV, Dasarathy S. Alcohol-induced autophagy contributes to loss in skeletal muscle mass. Autophagy. 2014;10:677–690. doi: 10.4161/auto.27918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomes PG, Trambly CS, Fox HS, Tuma DJ, Donohue TM., Jr Acute and Chronic Ethanol Administration Differentially Modulate Hepatic Autophagy and Transcription Factor EB. Alcoholism, clinical and experimental research. 2015;39:2354–2363. doi: 10.1111/acer.12904. [DOI] [PubMed] [Google Scholar]
- Vieira OV, Botelho RJ, Rameh L, Brachmann SM, Matsuo T, Davidson HW, Schreiber A, Backer JM, Cantley LC, Grinstein S. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. The Journal of cell biology. 2001;155:19–25. doi: 10.1083/jcb.200107069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Bower KA, Frank JA, Xu M, Luo J. Hypoxic preconditioning alleviates ethanol neurotoxicity: the involvement of autophagy. Neurotoxicity research. 2013;24:472–477. doi: 10.1007/s12640-013-9390-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada E, Bastie CC, Koga H, Wang Y, Cuervo AM, Pessin JE. Mouse skeletal muscle fiber-type-specific macroautophagy and muscle wasting are regulated by a Fyn/STAT3/Vps34 signaling pathway. Cell reports. 2012;1:557–569. doi: 10.1016/j.celrep.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan HX, Russell RC, Guan KL. Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy. 2013;9:1983–1995. doi: 10.4161/auto.26058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nature cell biology. 2009;11:468–476. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]