

Summary
The protein α-synuclein has a central role in the pathogenesis of Parkinson’s disease (PD). Similar to other proteins that accumulate in neurodegenerative disease, however, the function of α-synuclein remains unknown. Localization to the nerve terminal suggests a role in neurotransmitter release and over-expression inhibits regulated exocytosis, but previous work has failed to identify a clear physiological defect in mice lacking all three synuclein isoforms. Using adrenal chromaffin cells and neurons, we now find that both over-expressed and endogenous synuclein serve to accelerate the kinetics of individual exocytotic events, promoting cargo discharge and reducing pore closure (‘kiss-and-run’). Thus, synuclein exerts dose-dependent effects on dilation of the exocytotic fusion pore. Remarkably, mutations that cause PD abrogate this property of α-synuclein without impairing its ability to inhibit exocytosis when over-expressed, indicating a selective defect in normal function.
Introduction
Despite the established role of multiple proteins in the pathogenesis of neurodegenerative disease, we know remarkably little about their function. In Parkinson’s disease (PD) as well as in the related conditions Dementia with Lewy Bodies (DLB) and Multiple System Atrophy, the peripheral membrane protein α-synuclein accumulates in characteristic inclusions1. Mutations in α-synuclein also produce a dominantly inherited form of PD2–7, demonstrating that the protein has a causative role. Indeed, α-synuclein gene duplication and particularly triplication produce a severe form of familial PD8, implicating the wild type (wt) protein in disease. Synuclein thus has a central role in PD. However, the normal function of α-synuclein remains poorly understood.
α-Synuclein normally localizes to the nerve terminal, suggesting a role in neurotransmitter release9. Consistent with this, modest over-expression (insufficient to produce inclusions or overt toxicity) inhibits the regulated exocytosis of large dense core vesicles (LDCVs) and synaptic vesicles10–12. However, the loss of synuclein has less effect, with minimal or no increase in glutamate release reported in triple knockout (TKO) mice lacking α-synuclein as well as closely related β- and γ- isoforms13, 14. Knockout mice lacking α- and γ-synuclein show an increase in evoked dopamine release15 but the physiological change responsible remains unknown. Although over-expression inhibits regulated exocytosis, the role of endogenous α-synuclein has thus remained unknown.
α-Synuclein binds specifically to anionic membranes with high curvature16–18, but can also deform the lipid bilayer. Synuclein aggregates membranes in yeast19, 20, tubulates artificial membranes in vitro21 and when over-expressed in mammalian cells, can produce mitochondrial fragmentation22, 23. However, membrane deformation is generally considered to have an important role in endocytosis rather than exocytosis. The effect of over-expressed synuclein on exocytosis has thus been difficult to explain on the basis of membrane curvature-sensing or -promoting properties. Alternatively, synuclein has been suggested to serve as chaperone for the SNARE complex, but without apparent effect on transmitter release14.
How might membrane deformation by synuclein influence regulated exocytosis? In the course of exocytosis, synaptic vesicles form a fusion pore that dilates before full collapse into the plasma membrane. However, the pore can also close as part of a ‘kiss-and-run’ mechanism that immediately regenerates the vesicle24. Regulation of membrane curvature might thus affect behavior of the fusion pore. Since classical transmitters such as glutamate escape rapidly, postsynaptic recording might not detect a change in fusion pore kinetics. We have therefore used imaging to monitor directly individual exocytotic events. Single synaptic vesicle fusion events are difficult to detect by imaging, so we have focused on peptidergic large dense core vesicles (LDCVs) due to their size (70–200 nm diameter) and relatively slow release. Adrenal chromaffin cells have been used extensively to study the process of regulated exocytosis, including release by kiss-and-run25, and previous work has indeed demonstrated the inhibition of LDCV exocytosis by synuclein over-expression in chromaffin cells10. α-Synuclein+ Lewy pathology also occurs at high frequency in the adrenal gland of patients with PD and DLB26, with effects on catecholamine release into the circulation27.
Results
Synuclein over-expression accelerates the kinetics of individual exocytotic events in chromaffin cells
To understand how synuclein influences release, we first infected primary cultures from the postnatal mouse adrenal medulla with a lentivirus encoding human α-synuclein. Double staining for human α-synuclein and the LDCV protein secretogranin II (SgII) confirmed expression of the human protein within chromaffin cells (Supplementary Fig. 1a). We also assessed the expression of endogenous α-synuclein using an antibody that recognizes the protein from multiple species. Comparison of chromaffin cells from wt and synuclein TKO mice lacking all synuclein isoforms shows that chromaffin cells express endogenous α-synuclein, and the lentivirus confers modest over-expression (Supplementary Fig. 1b,c).
To study individual exocytotic events, we used a fusion of brain derived neurotrophic factor (BDNF) to the ecliptic pHluorin, a modified form of the green fluorescent protein with enhanced pH sensitivity28. Quenched at the low pH of LDCVs, BDNF-pHluorin fluorescence increases on exposure to the external medium by exocytosis29. We monitored the behavior of BDNF-pHluorin by TIRF microscopy during depolarization with 45 mM K+. Since kiss-and-run may occur more frequently at high external Ca++30, we also used 5 mM external Ca++ to sample a wider variety of exocytotic events. α-Synuclein over-expression reduces the number of exocytotic events detected using BDNF-pHluorin (Fig. 1a), as suggested previously by amperometry10. A change in Ca++ entry cannot account for this because Ca++ entry does not significantly differ from wt or TKO (Supplementary Fig. 2).
Figure 1. α-Synuclein modulates the kinetics of peptide release.
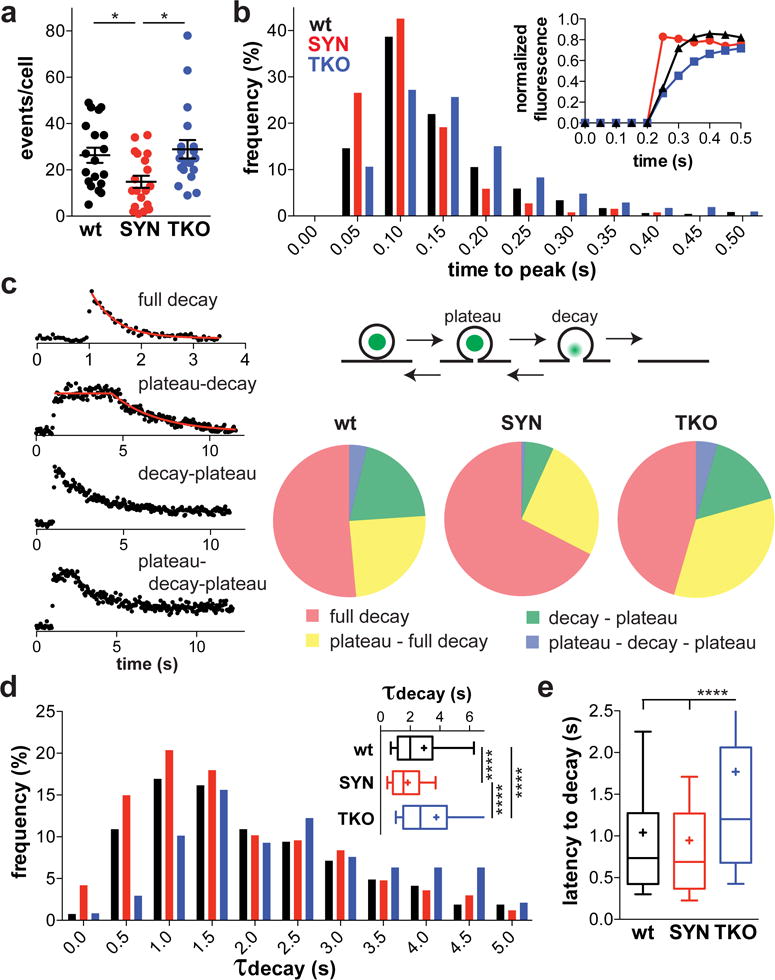
(a) Chromaffin cells from wt mice were transduced with lentiviruses encoding BDNF-pHluorin and either human α-synuclein (SYN) or empty vector (wt). Over-expression of α-synuclein reduces the number of exocytotic events evoked over 50 s by depolarization with 45 mM K+. Cells from the synuclein TKO show no difference from wt cells. *, p = 0.01 by one-way ANOVA (F(2, 54) = 4.991). n = 19 cells for each group from 3 independent cultures (b) Synuclein affects the rise time of exocytotic events. For each exocytotic event, the time to reach 90% maximum fluorescence was determined. Inset shows the average rise time of a single representative cell from each group (wt, n = 46 events; SYN, n = 34 events; TKO, n = 30 events). The histogram represents the frequency of events with rise time in the 50 ms bin indicated (p < 0.0001 by Kolmogorov-Smirnov test). wt, n = 473 events; SYN, n = 256 events; TKO, n = 518 events
(c) Exocytotic events belong to four distinct classes (left). In full decay, the fluorescence immediately decays to baseline. In plateau-decay, the fluorescence decay begins after a variable latency. In decay-closure, the fluorescence decays with no latency but the decay arrests before return to baseline. Plateau-decay-closure involves both a latency before decay and incomplete decay. The diagram (upper right) illustrates our interpretation of the traces. The proportion of event types differed among all three groups (p < 0.0001 by Chi-square for pair-wise as well as the comparison of all three groups). (d) Synuclein influences the rate of BDNF release. For all full decay events, the time constant of fluorescence decay (τdecay) was determined by fitting to a single exponential. The histogram represents the distribution of events with different τdecay (p < 0.0001 for WT versus SYN and TKO vs SYN; p < 0.001 for WT vs TKO by Kolmogorov-Smirnov test). wt, n = 266 events; SYN, n = 167 events; TKO, n = 237 events
(e) For all events with non-zero latency to decay, the time from reaching 90% maximal fluorescence to the onset of decay was determined (wt, n = 134 events; SYN, n = 66 events; TKO, n = 218 events). ****, p < 0.0001 by Kruskal-Wallis one-way ANOVA with Dunn’s post-hoc test; H = 55.22 (d) and 39.45 (e)
Individual exocytotic events show a series of characteristic changes due to the over-expression of α-synuclein. First, synuclein over-expression increases the rate of fluorescence rise. Although BDNF-pHluorin unquenching occurs rapidly at exocytosis, the fluorescence of many events increases over more than a single frame31 and this reflects buffering as well as dissolution of the LDCV core32. Alkalinization or approach to the plasma membrane might produce similar behavior, but loss of a preloaded dye33 accompanies or precedes events with slow rise times (Supplementary Fig. 4a,b). Thus, synuclein over-expression increases the rate of H+ loss at exocytosis (Fig. 1b and Supplementary Fig. 3a–c). Second, after peak fluorescence, the rate of fluorescence decay (due to peptide release) also increases even when analyzed as means per cell rather than aggregated, individual exocytotic events (Supplementary Fig. 3d). These results suggest acceleration of the release event by α-synuclein. However, the fluorescence events fall into at least four distinct classes (Fig. 1c and Supplementary Fig. 3e). Many events decay immediately to baseline (full decay) whereas others remain at maximum fluorescence for an interval before full decay (plateau-decay). In still others, decay is interrupted, suggesting constriction if not closure of the fusion pore, with or without a plateau preceding the decay (decay-plateau or plateau-decay-plateau). To determine whether the interruption of fluorescence decay reflects full pore closure, we quenched residual events using external solution adjusted to pH 5.5 with the impermeant buffer MES (Supplementary Fig. 4c). All events in the process of decay show quenching by the acidic buffer (Supplementary Fig. 4d), demonstrating exocytosis and either full collapse or persistence of a dilated fusion pore rather than movement away from the plasma membrane. Indeed, the H+-ATPase inhibitor bafilomycin does not influence the time course of events in any genotype (Supplementary Fig. 4e), confirming that loss of fluorescence indicates the release of peptide, not reacidification. Most of the stable events (plateau) also show quenching by low external pH, but a fraction do not (Supplementary Fig. 4c,d). Thus, complete closure of the fusion pore occurs only in stable events, but incomplete closure can still limit the loss of peptide. Consistent with the acceleration of release, synuclein over-expression increases the proportion of events that undergo full decay (Fig. 1c). Within the group undergoing full decay, however, synuclein over-expression also increases the rate of decay (Fig. 1d, Supplementary Fig. 3f). Thus, synuclein accelerates release independent of effects on pore closure, suggesting a role early in exocytosis to promote peptide release. Over-expression of synuclein also has no effect on the number of docked vesicles or their lumenal pH (Supplementary Fig. 5), arguing against a general disturbance of LDCVs as cause for the change in exocytosis.
Loss of synuclein prolongs the kinetics of release
The difficulty detecting clear effects on transmitter release in knockout mice11, 13, 14 suggests that α-synuclein over-expression may simply produce toxicity that secondarily affects release. It was therefore of great interest to determine how the loss of synuclein influences release kinetics. Consistent with previous work in neurons11, 13, 14, chromaffin cells from synuclein TKO mice show no clear change in event number relative to wt (Fig. 1a). However, analysis of event distribution reveals an increase in the time to peak fluorescence (Fig. 1b and Supplementary Fig. 3a). Loss of the synucleins modestly reduces the proportion of events with full decay (Fig. 1c) and also redistributes the decay time constants to longer values (Fig. 1d). In addition, TKO cells show greatly increased latency to decay among those events that do not decay immediately (Fig. 1e). (Over-expression does not affect this parameter presumably because it converts those with a short latency to full decay.) Thus, loss of synuclein prolongs release, suggesting a similar role for the endogenous and over-expressed protein in exocytosis. Similar to over-expression, the synuclein TKO also has no effect on LDCV docking or pH (Supplementary Fig. 5).
Although we used 5 mM external Ca++ for these experiments because it may promote kiss-and-run, we also examined BDNF-pHluorin events at the more physiological 2 mM Ca++. In this condition, as at 5 mM Ca++, over-expression of human α-synuclein both inhibits the exocytosis of chromaffin granules and accelerates the loss of BDNF-pHluorin (Supplementary Fig. 6a,b). The proportion of events again shifts to those with full decay, at the expense of those with interrupted release, and the time constant for fluorescence decay shortens (Supplementary Fig. 6c,d). The TKO also shows prolonged decay relative to wt (Supplementary Fig. 6d). Thus, synuclein has similar effects on release at 2 and 5 mM Ca++.
Synuclein inhibits closure of the fusion pore (‘kiss-and-run’)
The effect of synuclein on release could reflect changes in the fusion pore or in the solubility of dense core vesicle cargo. Indeed, the same LDCVs can release different substances at different rates34, 35, indicating that the properties of the aggregated peptide can influence the rate of release. It seems unlikely that a cytoplasmic protein such as synuclein would influence lumenal contents, but to distinguish further between effects on the fusion pore and on peptide solubility, we used a construct with the pHluorin inserted into a lumenal loop of the vesicular monoamine transporter VMAT236, a polytopic membrane protein that localizes to LDCVs. Stimulation of endocrine cells expressing the fusion produces discrete exocytotic events consistent with LDCVs, and we used bafilomycin for these experiments to prevent vesicle reacidification. In chromaffin cells, α-synuclein over-expression also inhibits the exocytosis of VMAT2-pHluorin (Fig. 2b). As a membrane protein, VMAT2-pHluorin cannot undergo release, and its decay therefore reflects spread within the plasma membrane and endocytosis, processes limited by the fusion pore37. Indeed, we observe events that spread and others that do not, as well as variation in the time course of fluorescence decay (Fig. 2a). Although the proportion of events with a latency to decay does not differ (46% for synuclein over-expression, 44% for control), the latency to decay shortens with over-expression of α-synuclein (Fig. 2b,c). The long duration of VMAT2-pHluorin events also enables us to determine how many remain accessible to the external solution. We find that a substantial fraction of the persistent events in control chromaffin cells are protected from quenching by MES-buffered solution at pH 5.5 (Fig. 2d,e), indicating kiss-and-run. In cells over-expressing human α-synuclein, this fraction declines substantially (Fig. 2e), suggesting inhibition of pore closure. However, synuclein over-expression also shortens the time constant of fluorescence decay for VMAT2-pHluorin (Fig. 2f), again supporting an independent effect on the rate of release, and a skew in the distribution of events per cell does not account for the effect on latency to decay or rate of decay (Supplementary Fig. 7). The analysis of membrane protein exocytosis thus supports an effect of synuclein on peptide release that cannot be explained by changes in cargo solubility.
Figure 2. The analysis of VMAT2-pHluorin reveals effects of α-synuclein on the fusion pore.
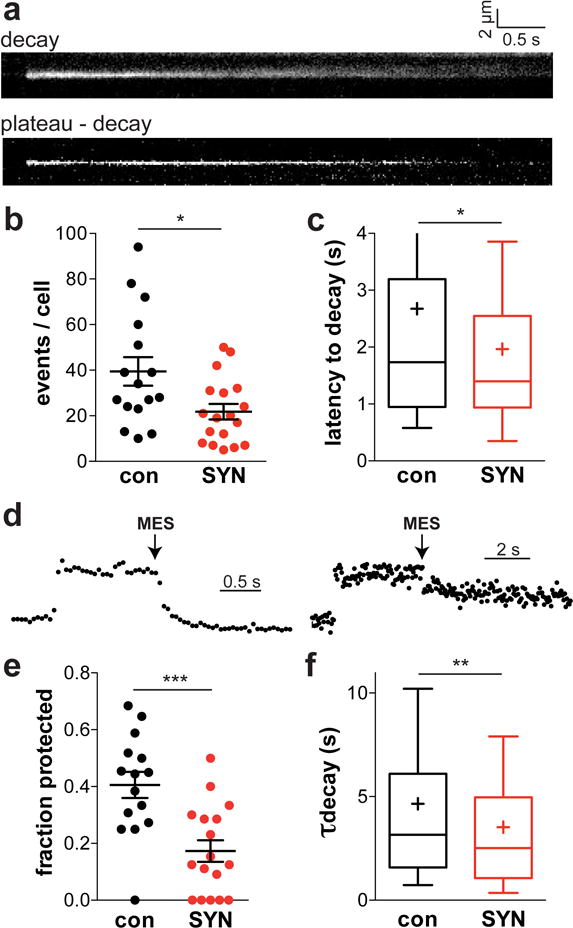
(a) Wild type chromaffin cells were transduced with lentiviruses encoding VMAT2-pHluorin and either human α-synuclein or empty vector and depolarized 3–5 days later with 45 mM K+ in the presence of H+ pump inhibitor bafilomycin to inhibit vesicle reacidification. The kymographs of two exocytotic events illustrate the observed variation in fluorescence time course and spread. after depolarization with 45 mM K+. Bar indicates 0.5 s. (b) α-Synuclein over-expression reduces the number of VMAT2-pHluorin exocytotic events (p = 0.0154 by unpaired, two-tailed t test; t(32) = 2.560). con, n = 16 cells; SYN, n = 18 cells from 3 independent cultures (c) Synuclein over-expression also reduces the latency to fluorescence decay (p = 0.0347 by Mann-Whitney; U = 42.00). con, n = 153 events; SYN, n = 128 events (d) Representative traces showing a VMAT2-pHluorin event quenched (left) and not quenched (right) by pH 5.5. (e) After depolarization for 30 s in 45 mM K+, the chromaffin cells were challenged at pH 5.5. Over-expression of α-synuclein reduced the proportion of events protected from quenching at low pH (p = 0.0005 by unpaired t-test; t(30) = 3.863). con, n = 222 events from 15 cells; SYN, n = 133 events from 17 cells (f) The time constant of fluorescence decay shortens with α-synuclein over-expression (p = 0.0012 by Mann-Whitney; U = 44.00). con, n = 348 events; SYN, n = 267 events. *, p < 0.05; **, p < 0.01; ***, p < 0.001
Endogenous and over-expressed synuclein promote fusion pore dilation in neurons
Does synuclein also affect the properties of individual exocytotic events in neurons? To address this, we first expressed human α-synuclein in primary rat hippocampal neurons, imaging cotransfected BDNF-pHluorin. Quantitative western analysis using an antibody that recognizes both human and rodent α-synuclein shows 4–5-fold over-expression (Supplementary Fig. 8). As previously reported, stimulation at 50 Hz evokes discrete exocytotic events primarily in axons38, 39 (Fig. 3a). The average number of events per coverslip does not significantly change with synuclein over-expression (Fig. 3c), but quenching at low pH reveals a dramatic effect of synuclein on the proportion of events accessible to external solution (p < 0.0001 by Chi-square) (Fig. 3d). With over-expression, more events decay before the addition of low pH solution (Fig. 3a,d), indicating that as in chromaffin cells over-expressing synuclein, the events decay more rapidly. Even among those events that remain, however, synuclein over-expression increases the proportion externally quenchable by low pH (Fig. 3d), demonstrating that α-synuclein has an effect on the fusion pore independent of release rate.
Figure 3. α-Synuclein influences fusion pore closure in neurons.

(a) Rodent hippocampal neurons were transfected with BDNF-pHluorin and imaged 14–20 days later, stimulating at 50 Hz for 5 s followed immediately by quenching of the cell surface fluorescence at pH 5.5. The arrow indicates an event quenchable at low pH, and the arrowheads events resistant to quenching. Scale bar, 5 μm. (b) Sample BDNF-pHluorin traces show sensitivity to quenching by pH 5.5 applied between the dashed lines (left) and resistance to quenching (right). (c) Average event frequency per coverslip (mean ± SEM) for BDNF-pHluorin expressing rat hippocampal neurons co-transfected with α-synuclein (SYN) or empty vector (con) and stimulated as in (a) (above) (p = 0.13 by unpaired, two-tailed t test; t(16) = 1.582). n = 9 cells from 2 independent cultures (d) Classifying events as either already decayed at the time of acid exposure or if not, unquenched or quenched by low pH, α-synuclein over-expression reduces the proportion of unquenched events (p < 0.0001 by Chi-square test) (left panel). n = 281 events (con), 158 events (α-syn). Synuclein over-expression also increases the proportion of quenchable events per coverslip independent of those already decayed (right panel). *, p < 0.05 by unpaired t-test (t(15) = 2.145) (e) Mouse neurons transfected with BDNF-pHluorin were stimulated at 50 Hz for 5 s and superfused with rapidly oscillating (1.33 Hz) Tyrode’s solutions at pH 7.8 and 6.4. Top trace shows an exocytotic event with oscillation that persists until fluorescence decay, indicating that the fusion pore remains open until the peptide is released. Middle trace shows an event that does not decay completely but shows oscillation throughout, indicating that the fusion pore does not close. Bottom trace shows an event where the oscillation stops (arrow) before full peptide release, indicating pore closure. (f) The proportion of event types differs in wt and synuclein TKO neurons (p = 0.01 by Chi-square). n = 100 events from 7 (wt) and 9 (TKO) coverslips (g) Among events with pore closure, the cumulative frequency distribution shows no significant difference between wt and synuclein TKO neurons in time to pore closure (p = 0.63 by Kolmogorov-Smirnov). Inset shows mean ± SEM (n.s., not significant). n = 44 events for wt and 63 events for TKO
Neurons express high levels of multiple synuclein isoforms, suggesting that the effect of the TKO might be greater than in chromaffin cells, which express modest levels. We were thus surprised that the synuclein TKO had little effect on the proportion of events already decayed, unquenchable or quenchable by low pH (Supplementary Fig. 9). However, BDNF-pHluorin events in neurons persist for many seconds (Fig. 3b), making it difficult to detect any further prolongation due to the loss of synuclein, and the acid quench limits characterization of the fusion pore to a single time point. We therefore turned to reporter NPY-pHluorin due to its more rapid release from LDCVs34, 39. To determine how much of the decay in fluorescence reflects peptide release or reacidification, we again used the H+-ATPase inhibitor bafilomycin. A small subset of NPY-pHluorin events decay slowly, with a time constant more than 5 s, and their proportion is reduced by bafilomycin (Supplementary Fig. 10a). At the same time, bafilomycin increases the proportion of events with no decay by a similar amount. Thus, a small fraction of events exhibit pore closure and reacidify slowly after endocytosis. However, the vast majority of events are rapid, with a time constant of decay considerably less than 5 s, and bafilomycin does not significantly change their proportion (Supplementary Fig. 10a). Bafilomycin also fails to alter the decay kinetics of most events (Supplementary Fig. 10b). Thus, fast NPY-pHluorin events reflect peptide release and only the longest events undergo pore closure.
Very similar to the results in chromaffin cells, over-expression of α-synuclein in neurons shortens both the latency of NPY-pHluorin events to decay and the time constant for fluorescence decay once this begins (Fig. 4a–c). Conversely, the loss of all three synucleins dramatically increases the latency of NPY-pHluorin to decay, and the time constant of fluorescence decay (Fig. 4d,e). The TKO thus exhibits a more pronounced effect on the kinetics of LDCV exocytosis in neurons than in chromaffin cells, presumably because neurons express much higher levels of endogenous synuclein, and the difference cannot be attributed to a skewed distribution of events per cell (Supplementary Fig. 11). It is interesting to note that the effect of synuclein on NPY-pHluorin fluorescence decay appears to involve only the more slowly decaying exocytotic events (Fig. 4c,e), which the experiment with bafilomycin suggests undergo pore closure (Supplementary Fig. 10). Presumably, it is difficult to detect acceleration of more rapidly decaying events because they are already very rapid. However, we also find that the latency from event appearance to the onset of decay shortens with over-expression of synuclein and lengthens in the TKO, but only for the events that decay rapidly (tau < 1 s), which are more common (Supplementary Fig. 12).
Figure 4. Over-expressed and endogenous synuclein exert similar effects on peptide release.
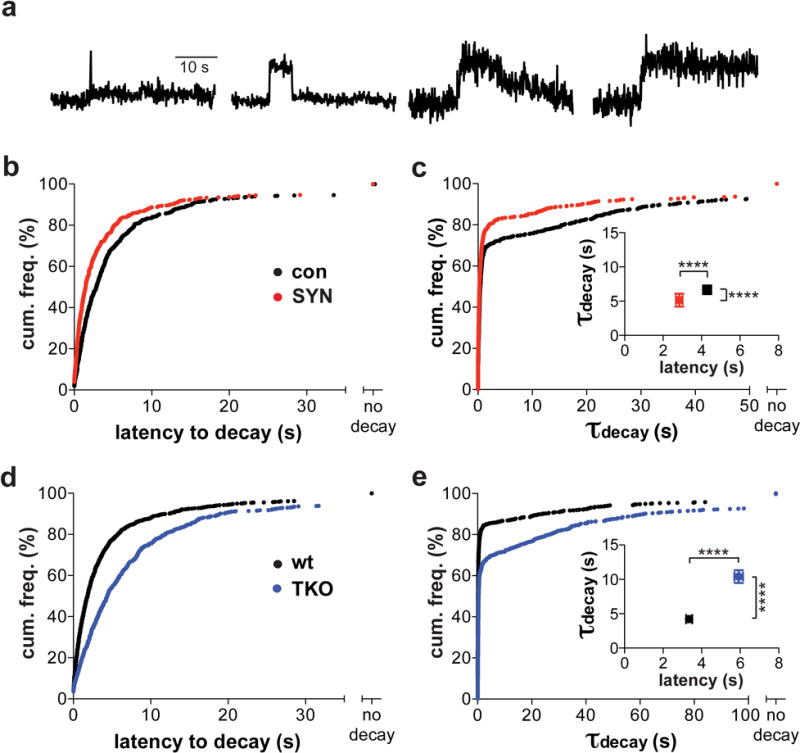
(a) Sample fluorescence traces of NPY-pHluorin in cultured rodent neurons stimulated at 50 Hz for 5 s. Individual traces were fit to a plateau with single exponential decay. Events that failed to exhibit fluorescence decay (right) were scored as no decay. (b,d) The latency to decay was combined with non-parametric “no decay” data and the cumulative frequency distribution plotted. (b) Overexpression of α-synuclein (SYN) in rat neurons significantly decreased latency to decay compared to controls transfected with empty vector (con) (p < 0.0001 by Kolmogorov-Smirnov). control, n = 652 events / 5 coverslips; α-syn, n = 586 events / 8 coverslips / 2 cultures (d) Latency to decay of NPY-pHluorin events increased in neurons from synuclein TKO mice relative to wt controls (p < 0.0001). wt, n = 928 events / 10 coverslips; TKO, n = 769 events / 11 coverslips / 3 cultures (c,e) Cumulative frequency histograms for the time constants of fluorescence decay (τdecay) by NPY-pHluorin, including events with no decay. (c) Overexpression of α-synuclein in rat neurons reduced latency to decay and τdecay (p < 0.001 by Kolmogorov-Smirnov). (e) Loss of synuclein in neurons from TKO mice increases NPY-pHluorin τdecay relative to neurons from wild type animals (p < 0.0001). Insets in (c) and (e) indicate mean ± SEM for the latency to decay and τdecay for decaying events in rat (c) and mouse (e) neurons. ****, p < 0.0001 by Mann-Whitney; U = 130046 (latency) and 149764 (tau) in (c); U = 221111 (latency) and 227995 (tau) in (e)
In chromaffin cells and neurons, synuclein over-expression affects the accessibility to quenching by external H+, as well as the kinetics of peptide release, indicating changes in behavior of the fusion pore. To monitor fusion pore closure with higher temporal resolution, we oscillated the pH of neuronal cultures between 6.4 and 7.8. If the fusion pore is open, the changes in pH will affect fluorescence of the peptide-pHluorin. When the oscillation in fluorescence stops, this indicates pore closure. We used BDNF-pHluorin for this experiment because the events last longer and can thus provide information about pore closure relatively long after exocytosis begins. Figure 3e shows sample traces for events where the pore remains open until peptide release, where closure does not occur during the period imaged, and for a closure event. Inactivation of all three synuclein genes substantially increases the proportion of events with pore closure, at the expense of those without closure (p = 0.01 by Chi-square) (Fig. 3f). However, loss of synuclein does not affect the time to pore closure (Fig. 3g). Taken together with the NPY-pHluorin data, these results suggest that synuclein prevents interruption of release by pore closure in two ways, first by increasing the rate of release and second, by preventing pore closure.
Synuclein localizes to dense core vesicles
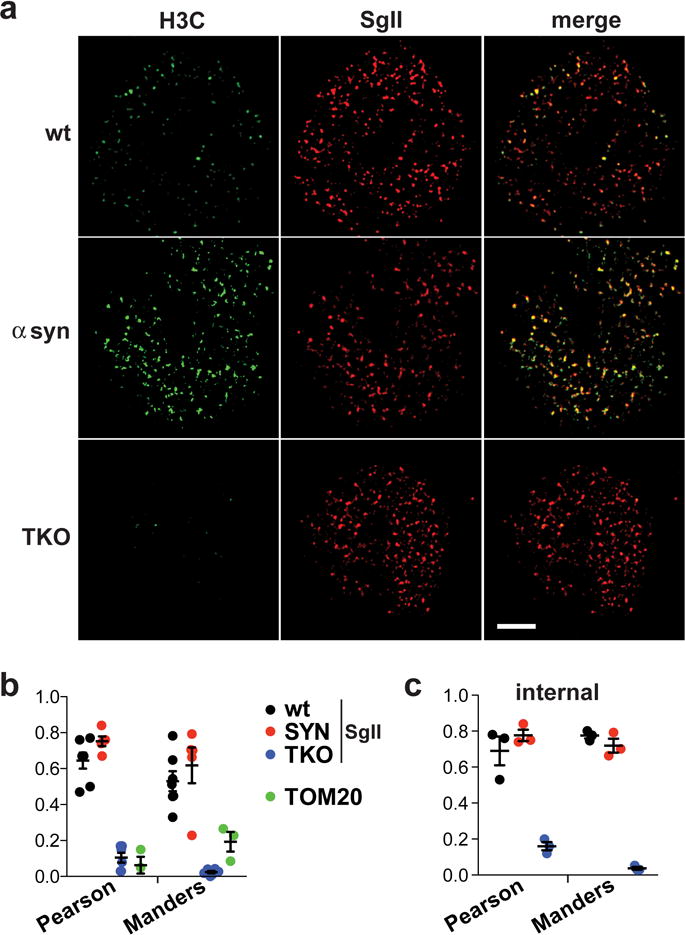
Does synuclein affect behavior of the fusion pore directly or indirectly? Despite its presynaptic location, its original identification in a preparation of synaptic vesicles and its preference for artificial membranes with high curvature17, α-synuclein exhibits only weak association with synaptic vesicles by gradient fractionation and photobleaching40, 41. We also failed to detect any specific localization of either over-expressed or endogenous synuclein with SgII in chromaffin cells using a number of commercially available antibodies. As shown in Supplementary Fig. 1, these antibodies produce diffuse labeling. We were therefore surprised to find that an antibody to the homologous canary protein synelfin specifically labels LDCVs in these cells. Using this antibody, which recognizes both α- and β-synuclein42, over-expressed human α-synuclein colocalizes extensively with LDCV protein SgII by structured illumination microscopy (Fig. 5). The same antibody detected endogenous synuclein on secretory granules in bovine chromaffin cells (personal communication, M.A. Bittner and R.W. Holz). Endogenous synuclein also colocalizes with SgII, and the immunoreactivity for synuclein is specific because the TKO shows very low background staining (Fig. 5 and Supplementary Fig. S13). Supporting the specificity for LDCVs, endogenous synuclein shows little colocalization with mitochondria (Fig. 5b and Supplementary Fig. 14). Further, the colocalization persists on LDCVs away from as well as at the plasma membrane, indicating that synuclein associates with LDCVs before docking. Due perhaps to conformational specificity for the membrane-bound or multimeric protein43–45, this antibody thus provides what may be the first direct histological evidence for the specific localization of synuclein to neurosecretory vesicles, indicating the potential for a direct effect on release.
Figure 5. Over-expressed and endogenous synuclein localizes to secretory granules in adrenal chromaffin cells.
(a) Chromaffin cells from wt or synuclein TKO mice were transduced with lentivirus encoding either human α-synuclein (SYN) or empty vector, cultured for 72 h and immunostained for α-synuclein (H3C, green) as well as the dense core vesicle protein secretogranin II (SgII, red) The images were obtained using structured illumination and shown here as reconstructions of a 120 nm-thick slice located within 0.5 μm of the cell-coverglass interface. Size bar, 2.5 μm. (b) The extent of SgII colocalization with synuclein was quantified using Pearson’s correlation coefficient (R) and Manders overlap coefficient (M1). The extent of wt synuclein colocalization with the mitochondrial protein TOM20 is shown in green. n = 7 cells for wt, 5 cells for SYN, 6 cells for TKO and 3 cells for TOM20 (c) Similar colocalization measures for a slice located 0.5–1.0 μm deeper inside the cell shows that the localization of synuclein to secretory vesicles is not limited to the docked pool; n = 3 cells. Values in b and c indicate mean ± SEM
The effect on fusion pore dilation is conserved among other synuclein isoforms but selectively inhibited by the mutations associated with Parkinson’s disease
The N-terminus of α-synuclein contains seven 11 amino acid repeats that form an amphipathic α-helix on interaction with membranes containing acidic phospholipid headgroups16. In addition to membrane association, the repeats may contribute to functional effects, such as on the fusion pore. Since the three synuclein isoforms show strong sequence conservation in the repeats and diverge at the more hydrophilic C-terminus, we tested the role of the N-terminus by examining the effects of β- and γ-synuclein. Transduced into wild type mouse chromaffin cells along with BDNF-pHluorin, human β- and γ-synuclein both reduce the number of exocytotic events (Fig. 6a), similar to the effect of α-synuclein in chromaffin cells and the effect of multiple synuclein isoforms on synaptic vesicle exocytosis11. In the events that remain, β- and γ-synuclein also accelerate peptide release (Fig. 6b), again similar to α-synuclein. As the only conserved domain, the N-terminal repeats thus appear responsible for both the effects on event number and kinetics.
Figure 6. Fusion pore dilation is a conserved function of the synucleins that is impaired by PD-associated point mutations.

Chromaffin cells from wt mice were transduced with lentiviruses encoding BDNF-pHluorin and either mutant human α-synuclein (A30P, A53T), human β-synuclein (β-syn), human γ-synuclein (γ-syn) or empty vector (con). (a) Relative to control, over-expression of mutant α-, β- or γ-synuclein all caused a reduction in the number of exocytotic events evoked over 50 s by depolarization with 45 mM K+ (**, p = 0.0046 by one-way ANOVA with Tukey’s post hoc test; F(4, 60) = 4.027). con, n = 17 cells; A30P, n = 14 cells; A53T, n = 10 cells; β-syn, n = 11 cells; γ-syn, n = 13 cells from 3 independent cultures (b) β- and γ-Synuclein both accelerate the kinetics of individual BDNF-pHluorin release events. However, the two PD-associated mutants do not affect release kinetics. The cumulative frequency distribution includes the decay constants for all BDNF-pHluorin events that decayed to baseline. Expression of either β- or γ- but not mutant α-synuclein, shifted the decay constants to shorter values relative to control (p < 0.0001 by Kolgomorov-Smirnov test for con vs. β-syn and con vs. γ-syn). The inset shows a 10–90 percentile box and whisker plot of the decay constants (mean represented as “+”). ****, p < 0.0001 by Kruskal-Wallis one-way ANOVA with Dunn’s post hoc test (H = 76.76), con vs. β-syn and con vs. γ-syn; n = 610 events for control, 296 for A30P, 172 for A53T, 210 for β-synuclein and 265 for γ-synuclein
(c,d) Chromaffin cells from wild type mice infected with either empty vector or lentivirus encoding A30P or A53T human α-synuclein were immunostained for α-synuclein (H3C, green) as well as SgII (red) and visualized by TIRF microscopy (c). Size bar, 2.5 μm. (d) The extent of SgII colocalization with synuclein was assessed using the Manders coefficient. n.s., not significant (p = 0.6147 by one-way ANOVA; F(4, 21) = 0.6781); n = 3 cells for TKO, 7 for control, 5 for A30P, 6 for A53T, 4 for TKO-A30P and 4 for TKO-A53T (e) The expression of wild type and mutant human α-synuclein was assessed by immunofluorescence with the syn-1 antibody (left) and human α-, β- and γ-synuclein with a pan-synuclein antibody (right). n = 6 cells for control, 8 for A30P, 7 for A53T using the syn-1 antibody, and 7 for control, 7 for β-synuclein and 7 for γ-synuclein using the pan-synuclein antibody
In previous work, we found that the mutations associated with PD do not impair the ability of α-synuclein to inhibit regulated exocytosis10, 11. The A30P mutant has less effect on synaptic vesicles because it does not concentrate at presynaptic boutons46, but the A53T mutant behaves very similarly to wild type. In chromaffin cells, both of these mutants reduce the frequency of exocytotic events (Fig. 6a), presumably because the small size of the cell eliminates the requirement for accumulation in a distant process10. Thus, we anticipated that the mutations would have similarly little effect on dilation of the fusion pore by synuclein. However, the mutations associated with PD all occur within a restricted region at the N-terminus, at the end of the second repeat (A30P) and within the fourth (E46K, H50Q, G51D, A53T)2–7, suggesting interference with a specific function. Remarkably, we observe that A30P and A53T α-synuclein both fail to accelerate peptide release (Fig. 6b). This is particularly surprising because the wild type rodent α-synuclein gene contains the pathogenic threonine residue at this position. Other sequence differences between the species must therefore account for the pathogenicity of this residue. Independent of mechanism, the PD-associated mutations affect one function of synuclein (pore dilation) but not another (inhibition of exocytosis).
The preserved ability to inhibit exocytosis suggests that the PD-associated mutants localize normally to LDCVs. Indeed, immunofluorescence using the H3C antibody shows unimpaired colocalization of both mutants with SgII by TIRF microscopy (Fig. 6c). Since this antibody does not distinguish between endogenous and introduced synuclein, we also examined the mutants in chromaffin cells from synuclein TKO mice, and again observe no difference from endogenous synuclein in colocalization with SgII (Fig. 6c and Supplementary Fig. 15).
Discussion
The results show that synuclein influences behavior of the exocytotic fusion pore. Over-expression accelerates the release event, reducing the time to peak fluorescence (pore dilation), speeding fluorescence decay (peptide release) and preventing pore closure. The loss of synuclein produces opposite effects, increasing the time to peak fluorescence, prolonging decay and increasing the likelihood of pore closure. Thus, both over-expressed and endogenous synuclein promote dilation of the fusion pore. Effects on pore closure are to some extent secondary to changes in the detection of undischarged cargo, but synuclein also appears to prevent the closure of persistent events. Nonetheless, the effect on pore opening suggests that synuclein acts early in exocytosis.
Changes in the fusion pore suggest an explanation for the inconsistent effects of synuclein on transmitter release reported in the literature. Fusion pore dilation would be expected to limit the release of neuromodulators such as monoamines and peptides that dissociate slowly from a lumenal matrix, rather than classical transmitters such as glutamate that escape rapidly through even a small pore. Indeed, synuclein over-expression and loss both affect the release of dopamine10, 15. In contrast, loss of synuclein has little effect on glutamate release13, 14. Despite this apparent difference, we anticipate the same effects on fusion of any vesicle to which synuclein binds.
We also provide evidence for the specific association of endogenous as well as over-expressed synuclein with neurosecretory vesicles in cells, indicating the potential for direct effects on the fusion pore. Since synuclein associates with LDCVs before docking at the plasma membrane, it is possible that synuclein acts before fusion to influence its properties. Indeed, previous work showing that synuclein increases the number of SNARE complexes14 suggests one mechanism for the effects of synuclein reported here. Increased SNARE complex formation might be expected to increase the force that drives fusion pore dilation and hence promote cargo release47. However, over-expression of synuclein also inhibits the extent of synaptic vesicle exocytosis11, and the results presented here confirm that this effect extends to LDCVs10. It is difficult to reconcile the observed inhibition of release with a role for synuclein as chaperone for the SNARE complex14. Indeed, synuclein inhibits the fusion of membranes in vitro as well as in vivo through direct effects on the lipid bilayer22, 48–50.
Alternatively, synuclein may promote SNARE complex accumulation by inhibiting exocytosis, thereby preventing the disassembly of complexes present on vesicles primed for fusion. In either case, synuclein appears to have a dual role. Physiologically, synuclein promotes dilation of the fusion pore, a dose-dependent effect shared by endogenous and over-expressed protein. On the other hand, inhibition of exocytosis may be restricted to synuclein over-expression13, 15, suggesting a pathological role. Nonetheless, the inhibition of exocytosis appears dose-dependent11, very similar to the effect on fusion pore kinetics.
The effect of mutations associated with PD shows that the two activities of synuclein, to promote pore dilation and to inhibit exocytosis, are distinguishable. A30P and A53T mutations do not impair the inhibition of exocytosis by synuclein over-expression. However, they both eliminate the effect of synuclein on fusion pore dilation. Since the loss of synuclein affects fusion pore dilation but not the number of exocytotic events, the PD mutations appear to produce a selective loss in the normal function of the protein. The preserved ability of over-expressed mutant and wild type synuclein to inhibit exocytosis may be required to produce degeneration.
Methods
Rodent strains
Synuclein triple knockout (TKO) mice were produced by crossing α/β-synuclein double KO mice (Jackson Laboratory, stock # 006390) to a γ-synuclein KO line51 generously provided by L. Lustig. TKO mice were maintained as homozygotes and C57Bl/6 animals used as wild type (wt) controls since this strain contributed ~90% of the genetic background of the synuclein TKO line (K. Nakamura, personal communication). All rodent procedures were performed according to guidelines established by the UCSF IACUC.
Antibodies
The rat monoclonal antibody to human α-synuclein (15G7) was obtained from Alexis Biochemicals52, the mouse monoclonal antibody to rodent α-synuclein (Syn-1) from BD Biosciences53, the the goat polyclonal antibody to TOM20 (C-20) from Santa Cruz Biotechnology54, the guinea pig polyclonal antibody to vesicular glutamate transporter 1 (VGLUT1) from EMD Millipore55 and the rabbit polyclonal antibodies to secretogranin II (K55101R) from Meridian Life Science56 and to actin (A2066) from Sigma57. The H3C antibody to synuclein developed by J. George was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of NIH and maintained at the University of Iowa, Department of Biology, Iowa City, IA 52242. The anti-rat antibody conjugated to Alexa Fluor 488, anti-mouse antibody conjugated to Alexa Fluor 488, anti-goat antibody conjugated to Alexa Fluor 594 and anti-rabbit antibody conjugated to Alexa Fluor 594 were all obtained from Thermo Fisher Scientific. The anti-mouse antibody conjugated to Cy5 and anti-guinea pig antibody conjugated to Cy3 were obtained from Jackson ImmunoResearch. The anti-mouse antibody conjugated to IRDye800 and anti-rabbit antibody conjugated to IRDye680 were obtained from Rockland Immunochemicals.
Chromaffin cell culture
Adrenal chromaffin cells were isolated as previously described58. Briefly, whole adrenal glands were harvested from 4–6 week old mice and placed in ice cold Ca++ -, Mg++ - free (CMF) Hank’s balanced salt solution (HBSS) supplemented with penicillin and streptomycin (pen/strep). Adrenal medullae were isolated and digested in the same solution containing collagenase type I (2.6 mg/ml, Worthington Labs), BSA (3 mg/ml), DNase I (0.15 mg/ml, Sigma) and hyaluronidase I (0.15 mg/ml, Sigma) at 37° C for 30 min, shaking at 800 rpm in a thermomixer (Eppendorf). Digested medullae were resuspended completely and the enzymatic reaction quenched with 10 volumes cold CMF-HBSS. Cells were pelleted at 300 g for 10 min at 4°C and resuspended in Dulbecco’s Modified Eagle’s medium (DME-H21) supplemented with 10% FBS (Hyclone) and pen/strep. Cell suspensions were plated drop-wise on glass chambers (LabTek) coated with poly-L-lysine and allowed to adhere for 45 min at 37° C/5% CO2, followed by addition of pre-warmed lentiviral supernatant. Lentiviral transduction was performed overnight, and media replaced the next morning.
Neuronal culture and transfection
Primary neuronal cultures were prepared from P0 Sprague-Dawley rat pups and transfected by electroporation as previously described (Amaxa)11. Briefly, 0.8 μg pCAGGS vectors containing either NPY-pHluorin or BDNF-pHluorin were cotransfected with 0.1 μg synaptophysin-mCherry and either 0.1 μg pCAGGS or 0.1 μg pCAGGS-α-synuclein per 4 × 105 cells. Cultures were maintained in Minimum Essential Media (MEM) containing 21 mM glucose, 5% FBS, 2% B27 (Gibco), 1% Glutamax (Gibco) and Mito+ serum extender (BD Biosciences). 5-FU and uridine were added on day 3 in vitro (DIV3) to inhibit glial growth.
Mouse primary neuronal cultures were prepared from P0 pups, electroporated and plated as described above, except cysteine-activated papain (Worthington) was used to dissociate hippocampi before trituration. On DIV1, 75% of the MEM media was replaced with Neurobasal medium (Gibco) supplemented with 2% B27 and 1.5% Glutamax. 5-FU and uridine were added on DIV8 to inhibit glial growth.
Lentivirus production
Low passage HEK293T cells were seeded onto 6-well plates and transfected overnight with a mixture of the third generation lentiviral vector pJHUMCS encoding the gene of interest, as well as accessory plasmids pREV, pVSVG and pPRE, using Fugene HD transfection reagent (Promega) and the manufacturer’s instructions. Cells were switched into chromaffin cell culture medium the next morning and 24 h later the culture medium was collected and cell debris sedimented at 1000 g. The viral supernatant was either used immediately or aliquoted and frozen at −80° C.
Immunofluorescence
Chromaffin cells were fixed by adding an equal volume of 4% formaldehyde in CMF-PBS to the culture medium and incubating for 20 min at room temperature. Cells were blocked and permeabilized in CMF-PBS containing 2% BSA, 1% fish skin gelatin and 0.02% saponin (blocking buffer). Primary antibodies were either diluted 1:13 (H3C), 1:500 (15G7, Syn-1, C-20) or 1:1000 (K55101R) in blocking buffer and incubated overnight at 4° C. Fluorescent dye-conjugated secondary antibodies were also diluted 1:500 in blocking buffer. For epifluorescence, cell staining was visualized using an upright fluorescence microscope (Axioskop; Zeiss) with 63× 1.25 N.A. oil objective (Zeiss) and a Coolsnap HQ CCD camera (Photometrics). Images were acquired using Metamorph software and analyzed in ImageJ (NIH).
Cultured hippocampal neurons were fixed in PBS containing 4% formaldehyde and 4% sucrose for 20 minutes at room temperature, blocked and permeabilized in PBS containing 5% calf serum and 0.05 % saponin. The fixed neurons were incubated in the same solution with monoclonal H3C anti-synuclein (1:13) and guinea pig anti-VGLUT1 antibodies (1:5000), followed by secondary detection using Cy3- or Cy5-conjugated antibodies. Images of fluorescent cell staining were acquired by confocal laser scanning microscopy using a Zeiss LSM510 microscope.
Structured illumination microscopy (SIM)
Isolated chromaffin cells were plated onto high precision coverslips (Zeiss), fixed 3–5 days later and immunostained as described above. Samples were imaged on a Nikon Ti microscope equipped with a 100× 1.49 NA Apo TIRF objective and an Andor Xyla sCMOS camera. Structured illumination images were acquired as a 15-slice z-stack with 120 nm step size and reconstructed using Nikon Elements software outfitted with the NIS-A N-SIM analysis module. Colocalization was quantified with the Coloc2 module in ImageJ.
Total internal reflection fluorescence (TIRF) microscopy
Wild type (wt) or synuclein TKO chromaffin cells were plated onto glass chamber slides (Lab-Tek) coated with poly-L-lysine, immediately transduced with lentivirus and imaged live 3–5 days later. Images were acquired at 20 Hz using an inverted TIRF microscope (Ti-E; Nikon) equipped with 50 mW Agilent MLC400B 488 nm laser, quad N-STORM TIRF filter set (405/488/561/647), 525/50 emission filter, 100× Plan Apo 1.49 N.A. oil objective (Nikon) and Andor iXon Ultra 897 high speed EMCCD camera (Oxford Instruments). Cells were imaged in modified Tyrode’s solution containing (in mM) 140 NaCl, 10 HEPES-NaOH, pH 7.4, 10 glucose, 4.5 KCl, 5 CaCl2, 1 MgCl2) and exocytosis stimulated by adding an equal volume of high KCl solution (osmotically balanced with NaCl), for a final K+ concentration of 45 mM. Individual exocytotic events were identified manually in ImageJ software by placing 5×5 pixel regions of interest (ROIs) over the center of events and the average intensity profiles extracted using the Time Series Analyzer plugin. An automated algorithm identified exactly the same events as well as several others that did not rise significantly (>20%) above the background, or that reflected the mobility of vesicles with unquenched residual fluorescence and were therefore excluded from the analysis (data not shown). The mean ROI intensity of the 20 preceding frames was used to define event onset, and subtracted as background. The onset of event decay was defined as the first frame dropping below an established higher level. Curve fitting, analysis of event parameters and all subsequent statistical analyses were performed using Graphpad Prism Version 6.05. The representative traces shown in Figures 1c and 2d were normalized to peak ΔF.
To quantify the rate of fluorescence decay (full decay events), the decay was fit to a single exponential. To quantify the latency to decay, the traces were fit to a plateau followed by single exponential decay and overlapping events or any latency shorter than the temporal resolution of the experiment (1 frame, 50 ms) were excluded from the analysis. To determine whether reacidification contributes to the decay of BDNF-pHluorin fluorescence, chromaffin cells were incubated in imaging buffer with Bafilomycin A1 (0.6 μM, EMD Millipore) for 2 min and stimulated with high K+ in imaging buffer that also contains 0.6 μM bafilomycin.
To assess the state of the fusion pore, imaging buffer at pH 5.5 was applied 30 s after stimulation with high K+. To determine the fraction of events protected from quenching, all stable exocytotic events visible immediately before the addition of low pH solution were selected using 5×5 pixel ROIs and the background subtracted. Since the spread of VMAT2-pHluorin into the plasma membrane affects local background fluorescence, classification as protected required (i) fluorescence intensity above the local background before event onset and (ii) punctate fluorescence despite the acid challenge.
For dual color TIRF imaging experiments, adrenal chromaffin cells transduced with lentivirus encoding BDNF-pHluorin were incubated in basal imaging buffer containing 20 μM FFN20633 for 1 h in 5% CO2 at 37° C, washed twice, stimulated and imaged immediately as described above. Dual color movies were acquired by alternating 30 ms excitation with 405 nm and 488 nm laser lines in triggered acquisition mode (NIS-Elements software, Nikon).
To compare the extent of H3C colocalization with SgII in cells expressing α-synuclein point mutants as well as β- and γ- isoforms, images were acquired on an inverted TIRF microscope (Ti-E; Nikon) using a 100× Plan Apo 1.49 N.A. oil objective (Nikon) and an Andor iXon Ultra 897 high speed EMCCD camera (Oxford Instruments). The Coloc2 plugin (ImageJ) was then used to determine the Manders overlap coefficient for SgII-positive punctae that colabeled with H3C.
Fluo-5F Imaging
Fluo-5F AM was reconstituted using anhydrous DMSO to a stock concentration of 3 mM. Immediately before imaging, the stock was diluted to a final concentration of 6 μM in imaging buffer. Wild type and TKO chromaffin cells transduced with lentivirus encoding either empty vector or human α–synuclein were incubated in Fluo-5F for 15 min at room temperature and washed three times with imaging buffer before imaging. Calcium influx was assessed by imaging Fluo-5F fluorescence at an acquisition rate of 20 Hz during depolarization with high K+ using the same laser and filter settings described above for pHluorin imaging. F0 values were determined by tracing the outline of the entire cell footprint and measuring the average pixel intensity during 20 frames immediately before stimulation. The maximum average intensity across the footprint, typically achieved within the first 1–2 s of stimulation, was used to calculate peak ΔF/F0.
Live cell imaging: neurons
Transfected neurons were imaged at 14–20 DIV at room temperature (24° C) as previously described11, 59. Neurons were imaged in standard Tyrode’s solution (in mM, 119 NaCl, 2.5 KCl, 2 MgCl2, 2 CaCl2, 30 glucose and 25 HEPES, pH 7.4). In low pH Tyrode’s (pH 5.5 and 6.4), HEPES was replaced with 25 mM MES. All buffers contained the glutamate receptor antagonists 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 μM) and D,L-2-amino-5-phosphonovaleric acid (APV, 50 μM). To induce LDCV exocytosis, cells were stimulated at 50 Hz for 5 s. The ecliptic pHluorin was imaged at 470/40 nm excitation and 525/50 nm emission, and mCherry at 572/35 nm excitation and 632/60 nm emission. Images were acquired in streaming mode (10 Hz) using a QuantEM:512SC EMCCD camera (Photometrics) and a 63× 1.2 N.A. water objective. The representative traces shown in Figures 3b, 3e and 4a were normalized to peak ΔF.
To assess the fusion pore by quenching of BDNF-pHluorin fluorescence at low pH, a 5 s pulse of pH 5.5 Tyrode’s solution was applied immediately after the 50 Hz stimulus, followed by washout in standard Tyrode’s solution. Exocytic events were identified manually in the image time series (10 Hz acquisition frequency) recorded from rat (control or +α-synuclein-overexpressing) or mouse (wt or synuclein TKO) neuronal cultures. Stationary events exhibiting rapid and single-step pHluorin unquenching (exocytosis) were scored for sensitivity to (“quenched”) or protection from (“unquenched”) quenching by acidic Tyrode’s solution. Fluorescence events that decayed before application of the low-pH buffer were scored as “already decayed”.
To probe further the state of the LDCV fusion pore, mouse neurons expressing BDNF-pHluorin were exposed to rapid pH oscillation as previously described60. Neurons were imaged in a chamber containing standard Tyrode’s solution (pH 7.4) and stimulated at 50 Hz for 5 s. High pH (7.8) and low pH (6.4) Tyrode’s solutions were delivered alternately to the imaging field via a theta pipette (~100 μm tip diameter) every 375 ms and images acquired continuously at 13.3 Hz. To reduce noise, a Kalman filter (Image J) was applied to the image stacks. Exocytotic events were identified manually as for BDNF-pHluorin in the experiment with quenching at pH 5.5, and 4×4 pixel regions of interest (ROIs) used to extract the fluorescence with the Time Series Analyzer plugin of ImageJ. Traces in which the pH-induced oscillation of pHluorin fluorescence stopped abruptly were scored as “pore closure” events, and the time from event onset to termination of oscillation determined. Events that showed oscillation throughout imaging were scored as “no closure”. Events that decayed to baseline but exhibited fluorescence oscillation throughout their lifetime were scored as “open until decay”. When required for scoring and quantitation, background fluorescence from areas adjacent to ROIs was subtracted. A total of 100 events each from wild type (7 coverslips) and synuclein TKO (9 coverslips) were scored. Groups were compared by the Chi-square test, and time-to-closure data displayed as cumulative frequency histograms.
For the analysis of NPY-pHluorin kinetics, exocytotic events imaged at 10 Hz were identified manually as for BDNF-pHluorin, and 4×4 pixel ROIs used to extract the fluorescence. For each event, the kinetics were fit to a plateau (of x seconds) and single exponential decay (F= IF(x<x0, F0, Plateau + Fspan * e-K*(x-x0))) by nonlinear regression (GraphPad Prism). Latency to decay (x) and decay time constant (τ) values for individual events were pooled across 3 independent experiments (10–11 coverslips total for each condition). Events that failed to decay were scored as “no decay” and included in the analysis. To inhibit reacidification, bafilomycin (0.6 μM) was added to the neurons immediately before imaging. The data, including non-parametric “no decay” events, were displayed as cumulative probability histograms, and groups compared by Kolmogorov-Smirnov. Latency and τ of decaying events were compared by Mann-Whitney. For Bafilomycin A1 experiments, events were grouped by decay kinetics (τ < 5s, τ > 5s or no decay) and compared by the Chi-square test.
Quantitative immunoblotting
Primary rat hippocampal neurons were transfected by electroporation with either pCAGGS or pCAGGS-α-synuclein, plus BDNF-pHluorin and synaptophysin-mCherry as described above. After culturing for 14 days, cells were solubilized in PBS containing 1% Triton X-100, 1 mM EGTA, 1 mM MgCl2 and protease inhibitors (Roche Complete). Lysates were sedimented at 1300 g to remove nuclei, and 6 μg protein was separated by polyacrylamide gel electrophoresis. After transfer to nitrocellulose, membranes were immunoblotted for α-synuclein (Syn-1; 1:500) and actin as a loading control (A2066; 1:500) followed by detection by IRDye-conjugated secondary antibodies. Immunoreactivity was detected by fluorescence scanner (LI-COR Biosciences) and quantified in ImageJ.
Statistical analysis
No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications11, 29, 31. At least two independent cultures were used for each experiment. Cumulative frequency distributions were compared using the Kolmogorov-Smirnov test and normally distributed data by two-tailed t-test for paired data and one-way ANOVA with Tukey’s post hoc test for multiple comparisons. For data not normally distributed, the Mann-Whitney U test was used for paired comparison and the Kruskal-Wallis one-way ANOVA with Dunn’s post hoc comparison for multiple comparisons. In box and whisker plots, the boxes represent the middle two quartiles, the whiskers 10% and 90% of the events, the line the median and + the mean. Event proportions were compared by Chi-square test. The organization of the experiments was not randomized. Data collection and analysis were not performed blind to the conditions of the experiments, except for the NPY-pHluorin data from neurons, of which >80% was analyzed blind to genotype.
Supplementary Material
Acknowledgments
We thank members of the Edwards lab for discussion, D. Jullié for help with the pH oscillation experiment, A. Bertholet (UCSF) for the TOM20 antibody and B. Calagui and S. Batarni for technical assistance. We also thank K. Bohannon, M. Bittner and R. Holz for sharing data and providing suggestions. This work was supported by grants from NINDS (NS062715), NIDA (DA10154) and the Weill Institute for Neurosciences (to R.H.E.), the John and Helen Cahill Family Endowment for Research on Parkinson’s Disease (to R.H.E.), a fellowship from NINDS (to T.L.) and a fellowship from the A.P. Giannini Foundation (to J.B.).
Footnotes
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Author contributions
R.H.E., T.L. and J.B. designed the research and wrote the manuscript. T.L. and J.B. performed the experiments and analyzed the data, with assistance from C.T. K.T. provided essential technical assistance with the chromaffin cell imaging experiments.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol. 2013;9:13–24. doi: 10.1038/nrneurol.2012.242. [DOI] [PubMed] [Google Scholar]
- 2.Polymeropoulos MH, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 3.Kruger R, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Gen. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 4.Zarranz JJ, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 5.Appel-Cresswell S, et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Movement Dis. 2013;28:811–813. doi: 10.1002/mds.25421. [DOI] [PubMed] [Google Scholar]
- 6.Lesage S, et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol. 2013;23:459–471. doi: 10.1002/ana.23894. [DOI] [PubMed] [Google Scholar]
- 7.Proukakis C, et al. A novel alpha-synuclein missense mutation in Parkinson disease. Neurology. 2013;80:1062–1064. doi: 10.1212/WNL.0b013e31828727ba. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singleton AB, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 9.Bendor JT, Logan TP, Edwards RH. The function of alpha-synuclein. Neuron. 2013;79:1044–1066. doi: 10.1016/j.neuron.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larsen KE, et al. Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci. 2006;26:11915–11922. doi: 10.1523/JNEUROSCI.3821-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nemani VM, et al. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott DA, et al. A pathologic cascade leading to synaptic dysfunction in alpha-synuclein-induced neurodegeneration. J Neurosci. 2010;30:8083–8095. doi: 10.1523/JNEUROSCI.1091-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greten-Harrison B, et al. alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci U S A. 2010;107:19573–19578. doi: 10.1073/pnas.1005005107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burre J, et al. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Senior SL, et al. Increased striatal dopamine release and hyperdopaminergic-like behaviour in mice lacking both alpha-synuclein and gamma-synuclein. Eur J Neurosci. 2008;27:947–957. doi: 10.1111/j.1460-9568.2008.06055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 17.Pranke IM, et al. alpha-Synuclein and ALPS motifs are membrane curvature sensors whose contrasting chemistry mediates selective vesicle binding. J Cell Biol. 2011;194:89–103. doi: 10.1083/jcb.201011118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jensen MB, et al. Membrane curvature sensing by amphipathic helices: A single liposome study using alpha-synuclein and annexin B12. J Biol Chem. 2011;286:42603–42614. doi: 10.1074/jbc.M111.271130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cooper AA, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soper JH, et al. {alpha}-synuclein-induced aggregation of cytoplasmic vesicles in Saccharomyces cerevisiae. Mol Biol Cell. 2008;19:1093–1103. doi: 10.1091/mbc.E07-08-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varkey J, et al. Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J Biol Chem. 2010;285:32486–32493. doi: 10.1074/jbc.M110.139576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kamp F, et al. Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010;29:3571–3589. doi: 10.1038/emboj.2010.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakamura K, et al. Direct membrane association drives mitochondrial fission by the Parkinson Disease-associated protein {alpha}-synuclein. J Biol Chem. 2011;286:20710–20726. doi: 10.1074/jbc.M110.213538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alabi AA, Tsien RW. Perspectives on kiss-and-run: role in exocytosis, endocytosis, and neurotransmission. Ann Rev Physiol. 2013;75:393–422. doi: 10.1146/annurev-physiol-020911-153305. [DOI] [PubMed] [Google Scholar]
- 25.Taraska JW, Perrais D, Ohara-Imaizumi M, Nagamatsu S, Almers W. Secretory granules are recaptured largely intact after stimulated exocytosis in cultured endocrine cells. Proc Natl Acad Sci U S A. 2003;100:2070–2075. doi: 10.1073/pnas.0337526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fumimura Y, et al. Analysis of the adrenal gland is useful for evaluating pathology of the peripheral autonomic nervous system in lewy body disease. J Neuropathol Exp Neurol. 2007;66:354–362. doi: 10.1097/nen.0b013e3180517454. [DOI] [PubMed] [Google Scholar]
- 27.Turkka JT, Juujarvi KK, Lapinlampi TO, Myllyla VV. Serum noradrenaline response to standing up in patients with Parkinson’s disease. Eur Neurol. 1986;25:355–361. doi: 10.1159/000116034. [DOI] [PubMed] [Google Scholar]
- 28.Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- 29.Dean C, et al. Synaptotagmin-IV modulates synaptic function and long-term potentiation by regulating BDNF release. Nat Neurosci. 2009;12:767–776. doi: 10.1038/nn.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ales E, et al. High calcium concentrations shift the mode of exocytosis to the kiss-and-run mechanism. Nat Cell Biol. 1999;1:40–44. doi: 10.1038/9012. [DOI] [PubMed] [Google Scholar]
- 31.Matsuda N, et al. Differential activity-dependent secretion of brain-derived neurotrophic factor from axon and dendrite. J Neurosci. 2009;29:14185–14198. doi: 10.1523/JNEUROSCI.1863-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holz RW. Evidence that catecholamine transport into chromaffin vesicles is coupled to vesicle membrane potential. Proc Natl Acad Sci U S A. 1978;75:5190–5194. doi: 10.1073/pnas.75.10.5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu G, et al. New fluorescent substrate enables quantitative and high-throughput examination of vesicular monoamine transporter 2 (VMAT2) ACS Chem Biol. 2013;8:1947–1954. doi: 10.1021/cb400259n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perrais D, Kleppe IC, Taraska JW, Almers W. Recapture after exocytosis causes differential retention of protein in granules of bovine chromaffin cells. J Physiol. 2004;560:413–428. doi: 10.1113/jphysiol.2004.064410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fulop T, Radabaugh S, Smith C. Activity-dependent differential transmitter release in mouse adrenal chromaffin cells. J Neurosci. 2005;25:7324–7332. doi: 10.1523/JNEUROSCI.2042-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Onoa B, Li H, Gagnon-Bartsch JA, Elias LA, Edwards RH. Vesicular monoamine and glutamate transporters select distinct synaptic vesicle recycling pathways. J Neurosci. 2010;30:7917–7927. doi: 10.1523/JNEUROSCI.5298-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiang HC, et al. Post-fusion structural changes and their roles in exocytosis and endocytosis of dense-core vesicles. Nat Comm. 2014;5:3356. doi: 10.1038/ncomms4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van de Bospoort R, et al. Munc13 controls the location and efficiency of dense-core vesicle release in neurons. J Cell Biol. 2012;199:883–891. doi: 10.1083/jcb.201208024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Asensio CS, et al. Self-assembly of VPS41 promotes sorting required for biogenesis of the regulated secretory pathway. Dev Cell. 2013;27:425–437. doi: 10.1016/j.devcel.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fortin DL, et al. Neural activity controls the synaptic accumulation of alpha-synuclein. J Neurosci. 2005;25:10913–10921. doi: 10.1523/JNEUROSCI.2922-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Unni VK, et al. In vivo imaging of alpha-synuclein in mouse cortex demonstrates stable expression and differential subcellular compartment mobility. PLoS One. 2010;5:e10589. doi: 10.1371/journal.pone.0010589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.George JM, Jin H, Woods WS, Clayton DF. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995;15:361–372. doi: 10.1016/0896-6273(95)90040-3. [DOI] [PubMed] [Google Scholar]
- 43.Bartels T, Choi JG, Selkoe DJ. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burre J, Sharma M, Sudhof TC. alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci U S A. 2014;111:E4274–4283. doi: 10.1073/pnas.1416598111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang L, et al. alpha-synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr Biol. 2014;24:2319–2326. doi: 10.1016/j.cub.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fortin DL, et al. Lipid rafts mediate the synaptic localization of alpha-synuclein. J Neurosci. 2004;24:6715–6723. doi: 10.1523/JNEUROSCI.1594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shi L, et al. SNARE proteins: one to fuse and three to keep the nascent fusion pore open. Science. 2012;335:1355–1359. doi: 10.1126/science.1214984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Braun AR, Sachs JN. alpha-Synuclein reduces tension and increases undulations in simulations of small unilamellar vesicles. Biophys J. 2015;108:1848–1851. doi: 10.1016/j.bpj.2015.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nuscher B, et al. Alpha-synuclein has a high affinity for packing defects in a bilayer membrane: a thermodynamics study. J Biol Chem. 2004;279:21966–21975. doi: 10.1074/jbc.M401076200. [DOI] [PubMed] [Google Scholar]
- 50.DeWitt DC, Rhoades E. alpha-Synuclein can inhibit SNARE-mediated vesicle fusion through direct interactions with lipid bilayers. Biochemistry. 2013;52:2385–2387. doi: 10.1021/bi4002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ninkina N, et al. Neurons expressing the highest levels of gamma-synuclein are unaffected by targeted inactivation of the gene. Mol Cell Biol. 2003;23:8233–8245. doi: 10.1128/MCB.23.22.8233-8245.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fellner L, et al. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia. 2013;61:349–360. doi: 10.1002/glia.22437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baksi S, Tripathi AK, Singh N. Alpha-synuclein modulates retinal iron homeostasis by facilitating the uptake of transferrin-bound iron: Implications for visual manifestations of Parkinson’s disease. Free Rad Biol Med. 2016;97:292–306. doi: 10.1016/j.freeradbiomed.2016.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Langone F, et al. Metformin protects skeletal muscle from cardiotoxin induced degeneration. PLoS One. 2014;9:e114018. doi: 10.1371/journal.pone.0114018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Turner TN, et al. Loss of delta-catenin function in severe autism. Nature. 2015;520:51–56. doi: 10.1038/nature14186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hao Z, et al. Impaired maturation of large dense-core vesicles in muted-deficient adrenal chromaffin cells. J Cell Sci. 2015;128:1365–1374. doi: 10.1242/jcs.161414. [DOI] [PubMed] [Google Scholar]
- 57.Zhang S, Wang P, Ren L, Hu C, Bi J. Protective effect of melatonin on soluble Abeta1-42-induced memory impairment, astrogliosis, and synaptic dysfunction via the Musashi1/Notch1/Hes1 signaling pathway in the rat hippocampus. Alzheimers Res Ther. 2016;8:40. doi: 10.1186/s13195-016-0206-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sirkis DW, Edwards RH, Asensio CS. Widespread dysregulation of peptide hormone release in mice lacking adaptor protein AP-3. PLoS Genet. 2013;9:e1003812. doi: 10.1371/journal.pgen.1003812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hua Z, et al. v-SNARE composition distinguishes synaptic vesicle pools. Neuron. 2011;71:474–487. doi: 10.1016/j.neuron.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jullie D, Choquet D, Perrais D. Recycling endosomes undergo rapid closure of a fusion pore on exocytosis in neuronal dendrites. J Neurosci. 2014;34:11106–11118. doi: 10.1523/JNEUROSCI.0799-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.