

Summary
Cells exposed to hypoxia experience replication stress but do not accumulate DNA damage, suggesting sustained DNA replication. Ribonucleotide reductase (RNR) is the only enzyme capable of de novo synthesis of deoxyribonucleotide triphosphates (dNTPs). However, oxygen is an essential cofactor for mammalian RNR (RRM1/RRM2 and RRM1/RRM2B), leading us to question the source of dNTPs in hypoxia. Here, we show that the RRM1/RRM2B enzyme is capable of retaining activity in hypoxia and therefore is favored over RRM1/RRM2 in order to preserve ongoing replication and avoid the accumulation of DNA damage. We found two distinct mechanisms by which RRM2B maintains hypoxic activity and identified responsible residues in RRM2B. The importance of RRM2B in the response to tumor hypoxia is further illustrated by correlation of its expression with a hypoxic signature in patient samples and its roles in tumor growth and radioresistance. Our data provide mechanistic insight into RNR biology, highlighting RRM2B as a hypoxic-specific, anti-cancer therapeutic target.
Keywords: RNR, RRM2B, replication stress, hypoxia, nucleotides, DNA damage response, radiosensitivity, P53
Graphical Abstract
Highlights
-
•
RRM2B is induced in response to hypoxia in both cell models and patient datasets
-
•
RRM2B retains activity in hypoxic conditions and is the favored RNR subunit in hypoxia
-
•
Loss of RRM2B has detrimental consequences for cell fate, specifically in hypoxia
-
•
RRM2B depletion enhanced hypoxic-specific apoptosis and increased radiosensitivity
Foskolou et al. demonstrate that in hypoxic conditions the small subunit of the RNR enzyme is switched from RRM2 to RRM2B in order to facilitate nucleotide production and ongoing replication. They identify specific residues within RRM2B that are responsible for maintaining activity in hypoxia.
Introduction
Replication stress is a well-characterized tumor characteristic and has recently been considered as a potential new hallmark of cancer (Macheret and Halazonetis, 2015). Replication stress leads to the activation of the DNA damage response (DDR), which is associated with the early stages of cancer development (Bartkova et al., 2005, Gorgoulis et al., 2005). Hypoxia (low oxygen levels) is one of the most physiologically relevant driving forces of replication stress and is characterized by an increased number of stalled replication forks and significantly reduced replication rates. Importantly, hypoxia-induced replication stress occurs in the absence of DNA damage (Olcina et al., 2013). Regions of hypoxia are observed in preneoplastic lesions in the majority of tumors. Hypoxia-induced replication stress is one of the factors proposed to contribute to the selection pressure to lose key components of the DDR, including p53 (Gorgoulis et al., 2005, Hammond et al., 2007, Graeber et al., 1996). The degree of tumor hypoxia is an indicator of poor patient prognosis due to resistance to most current therapies and increased metastatic spread (Begg et al., 2011, Höckel and Vaupel, 2001).
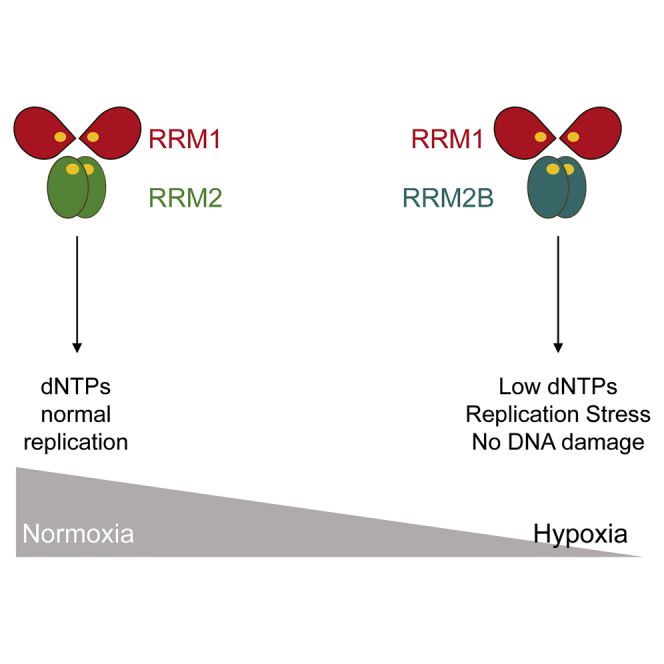
The importance of ribonucleotide reductase (RNR) lies in its unique ability to catalyze the rate-limiting step of the reduction of ribonucleotides (NDPs) to the corresponding deoxyribonucleotides (dNDPs), the precursors of DNA (Kolberg et al., 2004, Stubbe, 1998). Mammalian RNRs consist of two homodimeric subunits, which associate to form the holoenzyme. The large RRM1 dimer contains the catalytic site, whereas the smaller dimer (RRM2 or RRM2B) contains an oxygen-requiring di-iron tyrosyl-radical site, which is essential for catalysis (Kolberg et al., 2004, Uhlin and Eklund, 1994, Stubbe et al., 2003). RNR is highly regulated, as nucleotide imbalances can be detrimental to cell fate (Aye et al., 2015). Elevated deoxyribonucleotide triphosphate (dNTP) pools have been correlated with increased mutagenesis, while insufficient dNTPs cause replication stress and promote genomic instability (Bester et al., 2011, Burrell et al., 2013, Halazonetis et al., 2008, D’Angiolella et al., 2012).
Oxygen is essential for mammalian RNRs to oxidize the di-iron center found in the smaller subunit (RRM2 or RRM2B) (Huang et al., 2014). This generates the tyrosyl radical, which via a long-range pathway, transfers an electron to the catalytic site of RRM1, enabling the reduction of NDPs to dNDPs (Lundin et al., 2015, Kolberg et al., 2004, Stubbe et al., 2003). The mechanism by which iron is incorporated into the small subunit, and how oxygen activates the di-iron center for radical initiation, is still poorly understood and under intense investigation (Huang et al., 2014, Zhang et al., 2014). The oxygen dependency of the small RNR subunit has led to the logical assumption that RNR is inactive in severely hypoxic conditions, something that has been verified for the RRM1/RRM2 (R1/R2) version of the RNR enzyme (Brischwein et al., 1997, Chimploy et al., 2000, Thelander et al., 1983, Reichard, 1993, Probst et al., 1989, Nordlund and Reichard, 2006). In contrast, we demonstrate that RNR is able to maintain dNDP formation by switching subunits in hypoxic conditions (<0.1% O2). This switch is critical to preserve sufficient nucleotide levels for ongoing replication and avoid DNA damage accumulation and apoptosis in hypoxia. Interestingly, we found that the RRM1/RRM2B (R1/R2B) version of the RNR enzyme retains activity in hypoxia due to adaptation of RRM2B to these conditions through two distinct mechanisms. Specifically, we determined that most of the tyrosyl radical in RRM2B remained stable when exposed to <0.1% O2, a property that was diminished after disruption of the helix B open conformation (K37/K151) and the subsequent loss of the crosstalk between the two monomers. Furthermore, computational simulations suggest that Y164 plays a role in the fine-tuning of the oxygen-entering frequency of RRM2B, meaning it is likely to be a better oxygen-sequestering agent than RRM2. Collectively, our results reveal a critical role for the RNR (R1/R2B) enzyme in hypoxic conditions in maintaining sufficient dNTPs for ongoing replication and therefore preventing DNA damage, genomic instability, and subsequent loss of viability.
Results
The RRM2B Subunit of RNR Is Induced in Response to Hypoxia
Cells in hypoxic conditions (<0.1% O2) do not accumulate DNA damage, although they are characterized by the presence of replication stress, which leads to the activation of the DDR (Figures S1A and S1B) (Olcina et al., 2013). As predicted, hypoxia-induced replication stress leads to the accumulation of single-stranded DNA (ssDNA), which can be detected through the visualization of replication protein A (RPA) foci in S-phase cells (Pires et al., 2010). However, the percentage of cells in hypoxia with RPA foci decreases after prolonged exposure to these conditions (>6 hr) (Figure 1A). Therefore, our hypothesis is that although uncoupling of the helicases and polymerases occurs in hypoxia, replication can proceed. Using DNA fiber analysis, we confirmed that DNA replication is significantly slower in hypoxia (25% of normal rate) but is not completely abrogated (Figure 1B). dNTP incorporation assays were carried out in RKO cells exposed to hypoxia and demonstrated that, although the dNTP pools were disrupted (with the purines [dGTP, dATP] being more affected than the pyrimidines [dCTP, dTTP]), there was nucleotide availability in hypoxic conditions (Figures 1C and 1D). The differences observed between purine and pyrimidine pools are not fully understood. It has been suggested that pyrimidine levels persist due to compensatory activities of the pyrimidine salvage pathways or as a result of the deoxyuridine present in culture medium, which cells could uptake and phosphorylate, therefore contributing to pyrimidine levels, particularly dTTP (Eriksson et al., 1987, Bianchi et al., 1986). Interestingly, the pyrimidine salvage pathway has been reported to be more efficient than the purine pathway, suggesting an explanation for why purine levels are more sensitive to hydroxyurea (HU) and potentially hypoxia (Reichard, 1988). Our data demonstrate sufficient nucleotide availability in hypoxia to allow replication to proceed, albeit at a compromised rate; thus raising the possibility that RNR is able to retain some activity in hypoxia.
Figure 1.

Replication Is Compromised, but Not Abrogated, in Oxygen Levels below 0.1%
(A) RPA32 foci in RKO cells exposed to <0.1% O2 and representative images of RPA-negative (normoxia) and positive (<0.1% O2, 3 hr) cells. Scale bar, 10 μm.
(B) Replication rates by DNA fiber analysis in RKO cells exposed to <0.1% O2 and representative images of DNA fibers in normoxia and <0.1% O2 (5 hr). Scale bar, 10 μm.
(C and D) dNTP levels of purines (C) and pyrimidines (D) in RKO cells exposed to <0.1% O2 or the radical-scavenger hydroxyurea (HU) (2 mM, 6 hr). Data represent percentage of the control (normoxia).
(E) Immunoblots of RNR subunits in RKO cells exposed to <0.1% O2 for the times indicated.
(F) Representative images of RRM2B immunostaining in normoxia and < 0.1% O2 (18 hr) in RKO cells. Scale bar, 20 μm.
(G) mRNA levels of RNR subunits in RKO cells in <0.1% O2 normalized to 18S.
(H) Expression of RRM2B (log10 conversion) in a colorectal adenocarcinoma TCGA dataset is shown against the hypoxia-inducible signature (log10 conversion).
For all panels (except H), n = 3 (biological replicates); for (A)–(D) and (G), data show means ± SEM and one-way ANOVA analysis was applied; (ns) indicates a non-significant change. See also Figure S1.
Expression analysis of the RNR complex showed that the RRM2B protein is significantly induced (2- to 3.5-fold, depending on cell line) in response to hypoxia in cell lines, including colorectal, osteosarcoma, glioblastoma, and esophageal cancer cells, whereas the RRM2 small subunit decreased over time and the RRM1 remained unchanged or showed a slight decrease (Figures 1E, 1F, and S1C–S1F). As RKO cells showed the highest level of RRM2B expression, we chose it as a model line for the majority of our experiments (Figure S1G). Similar results were obtained when mRNA levels were assayed. RRM2B increased 4.6-fold after 24 hr in hypoxic conditions, whereas RRM2 and RRM1 mRNA levels decreased 12.3- and 2.5-fold, respectively (Figures 1G and S1H). Importantly, in silico TGCA (The Cancer Genome Atlas) analysis of colorectal adenocarcinoma patient cohorts demonstrated that RRM2B expression correlates significantly with the expression of a verified hypoxia signature (Figure 1H), suggesting that the oxygen-dependent overexpression of RRM2B also occurs in vivo (Li et al., 2014). In contrast, RRM1 and RRM2 expression did not correlate with the same hypoxic signature (Figures S1I and S1J). Interestingly, overexpression and genetic alterations in RRM2B correlated with worse overall and disease-free survival in colorectal cancer patients (Figures S1K–S1N).
The transcription factor HIF-1 (hypoxia-inducible factor 1) is known to mediate significant gene expression changes in response to hypoxia and has roles in DNA replication, DNA repair, and respiration (Hubbi et al., 2013, Fukuda et al., 2007, Crosby et al., 2009). Therefore, we investigated if the induction of RRM2B in hypoxia was dependent on HIF-1α by utilizing RKOHIF-1α +/+ and RKOHIF-1α −/− cells exposed to hypoxia (Figures 2A, S2A, and S2B). Interestingly, both the mRNA and the protein levels of RRM2B were induced in hypoxia irrespective of HIF-1α status, in contrast to the well-documented HIF-1α target GLUT1. Next, using RKOHIF-1α +/+ cells exposed to either 2% or <0.1% O2, we investigated the oxygen dependency of the induction of RRM2B protein. RRM2B was induced in response to the lower level of hypoxia (<0.1% O2), where a robust p53 induction was also observed but did not increase in response to 2% O2 despite HIF-1α stabilization (Figure 2B). This finding is in agreement with our previous studies demonstrating that the lower level of hypoxia (<0.1% O2) induces replication stress and that this is the signal that initiates the DDR (including p53 stabilization) (Hammond et al., 2002, Olcina et al., 2013).
Figure 2.

RRM2B Is Induced in Hypoxia
(A) mRNA levels of RRM2B and GLUT1 in RKOHIF-1α+/+ and RKOHIF-1α−/− cells in <0.1% O2 assayed by qPCR and normalized to 18S.
(B) Immunoblots of RKO cells exposed to 2% and <0.1% O2 for the times indicated. p53 and RRM2B protein induction are observed only in <0.1%O2.
(C and D) Immunoblots (C) and RRM2B mRNA levels (D) of HCT116p53+/+ and HCT116p53−/− exposed to <0.1% O2 for the times indicated.
(E) qPCR for p53 ChIP in RKO cells treated with Adriamycin (2 μM, 6 hr) or exposed to either normoxia or <0.1% O2 (6 hr).
(F) Immunoblots of RKO cells treated as in (E).
(G) Expression of RRM2B (log10 conversion) in the colorectal adenocarcinoma TCGA datasets is shown against hypoxia dependent p53-inducible group of genes (log10 conversion).
(H) Immunoblots of RNR subunits in H1299p53−/− (non-small cell lung carcinoma) cells exposed to <0.1% O2 for the times indicated.
For all panels (except G), n = 3 (biological replicates); data in (A) and (D) represent mean ± SEM; and (E) shows representative mean of technical triplicates ± RQmax/RQmin. HIF-1α status and p53 status was examined by two-way ANOVA analysis; two-tailed Student’s t test was applied in (E); (ns) indicates a non-significant change. See also Figure S2.
RRM2B was first characterized as a p53-regulated RNR subunit (p53R2) (Tanaka et al., 2000). Here, further analysis of the molecular pathways mediating RRM2B induction in hypoxia demonstrated that the hypoxic overexpression of RRM2B occurs in a p53-dependent manner (Figures 2C, 2D, and S2C–S2H). To rule out the possibility of an indirect mechanism of induction of RRM2B by p53, chromatin immunoprecipitation (ChIP) assays were carried out and demonstrated that p53 binds directly to the p53-response element at the RRM2B locus leading to transcriptional overexpression (Figures 2E, 2F, and S2I–S2K). Interestingly, although p53 expression was increased in response to the DNA damaging agent Adriamycin, this did not correlate with increased p53 binding to the p53-response element in RRM2B (Figures 2E and 2F). Most importantly, analysis of the TCGA colorectal adenocarcinoma patient cohorts showed that RRM2B expression significantly correlated with a recently identified group of hypoxia-inducible p53-dependent genes (Leszczynska et al., 2015), suggesting that hypoxia- and p53-dependent expression of RRM2B occurs in human cancers (Figure 2G). Interestingly, in p53 null cell lines (H1299p53−/−, HCT116p53−/−) a mild (1.3- to 1.7-fold) increase in RRM2B protein levels was also observed in hypoxia (Figures 2C and 2H). These findings suggest that additional post-translational p53-independent mechanisms exist for RRM2B stabilization and therefore the importance of RRM2B in hypoxic conditions.
RRM2B Replaces RRM2 in Hypoxia
In order to investigate the biological significance of hypoxia-induced RRM2B, we first verified that it forms a complex with the RRM1 subunit to reconstitute the R1/R2B holoenzyme in <0.1% O2. Immunoprecipitation assays demonstrated that increased levels (5.3-fold) of RRM2B protein were bound to the RRM1 subunit in hypoxia whereas the levels of RRM2 bound to RRM1 decreased by 1.8-fold (Figures 3A, S3A, and S3B). Next, we asked if the hypoxia-formed R1/R2B enzyme was functional. Small interfering RNA (siRNA)-mediated loss of RRM2B led to significantly lower intracellular dNTP levels in hypoxia (50%–55% less pyrimidines and 25%–30% less purines compared to the control [siCTL]) (Figures 3B and S3C). In contrast, the loss of RRM2B did not significantly affect the dNTP pools in normoxic conditions (Figure S3D). In addition, fluorescence-activated cell sorting (FACS) analysis demonstrated that S-phase U2OS cells lacking RRM2B incorporate 37.5% less BrdU than the control-treated cells in hypoxia (Figures 3C and S3E). These findings demonstrate that depletion of RRM2B in hypoxia leads to further disruption of the dNTP pools and indicate that ongoing replication is disrupted.
Figure 3.

Effects of RRM2B Depletion in Hypoxia
(A) Immunoprecipitation of RRM1 followed by immunoblotting for RRM2B and RRM2 in normoxia and <0.1% O2 (18 hr).
(B) dNTP levels in RKO cells treated with non-specific (siCTL) or siRRM2B and exposed to <0.1% O2 (16 hr).
(C) FACS analysis of U2OS cells treated with siCTL or siRRM2B and exposed to normoxia or <0.1% O2 (3 hr). Cells were pulsed with bromodeoxyuridine (BrdU) (20 μM) 30 min before collection.
(D) RPA32 foci in RKORRM2B+/+ and RKORRM2B−/− cells after exposure to <0.1% O2.
(E) 53BP1 foci in RKORRM2B+/+ and RKORRM2B−/− cells exposed to normoxia or <0.1% O2 (6 hr).
(F) Representative images of 53BP1 foci in RRM2B-negative RKO cells treated with siRRM2B and exposed to normoxia or <0.1% O2 (6 hr). Scale bar, 20 μm.
(G) Colony survival assay in RKO cells treated with siCTL or siRRM2B and exposed to normoxia or <0.1% O2 (24 hr).
(H) Apoptosis detected morphologically in RKO cells treated with siCTL or siRRM2B and exposed to normoxia or <0.1% O2 (19 hr).
(I) RKORRM2B+/+ and RKORRM2B−/− cells were grown as xenografts in mice (n = 4 mice per each group). Where indicated, irradiation (10 Gy) was given when tumors reached ∼100 mm3.
(J) Representative images of co-localization of cleaved caspase-3 (apoptosis) with PIMO (hypoxic areas) in RKORRM2B+/+ or RKORRM2B−/− xenografts. Scale bars, 50 μm.
(K and L) Tumors were removed on day 28 post-implantation (from Figure S5H), and the level of apoptosis was quantified in normoxic areas (PIMO negative) (K) and hypoxic areas (PIMO positive) (L). Images from three different tumors (n = 3) per group were counted.
For all panels, n = 3 (biological replicates) unless otherwise stated. Data show mean ± SEM and two-tailed Student’s t test was applied, except in (D), where one-way ANOVA analysis was applied, and (I), where two-way ANOVA analysis was applied. (ns) indicates a non-significant change. See also Figures S3 and S4.
To further investigate the hypoxic role of RRM2B, we used CRISPR/Cas9 technology to construct a RRM2B knockout cell line (RKORRM2B−/−) (Figures S3F–S3I). RRM2B-depleted RKO cells (both knockout and siRNA treated) showed a persistent formation of RPA foci during long (24 hr) exposures to hypoxia, suggesting an accumulation of ssDNA and failure to complete DNA replication (Figures 3D and S4A–S4C). Furthermore, cells with reduced levels of RRM2B formed 53BP1 foci indicating the presence of double-strand breaks (DSBs) specifically in hypoxia (Figures 3E, 3F, and S4D–S4F). No evidence of elevated DNA damage was observed in control cells or in cells lacking RRM2B in normoxia; this was attributed to the RRM2-dependent supply of dNTPs when oxygen is abundant. The increased DSBs observed in hypoxic cells with depleted RRM2B correlated with (1) a significant loss of viability measured by colony survival assay (77% fewer cells survived after 24 hr of hypoxia when RRM2B was silenced in comparison to siCTL) (Figure 3G) and (2) the induction of apoptosis as determined both morphologically and by induction of PARP cleavage (Figures 3H and S4G). Notably, xenograft tumors of RKORRM2B+/+ and RKORRM2B−/− cells showed delayed tumor growth and increased radiosensitivity when RRM2B was lacking (Figures 3I and S4H). Additionally, a significant increase in apoptosis specifically in the hypoxic regions of the RRM2B−/− tumors was observed (Figures 3J–3L) as determined by the presence of cleaved caspase-3 (apoptosis) in the pimonidazole (PIMO)-positive (hypoxic) regions. Increased apoptosis in the hypoxic fraction of the tumors offers a likely explanation for the increased radiosensitivity observed (Figure 3I).
The data presented so far suggest that the basal levels of dNTPs provided by R1/R2B (as opposed to R1/R2) are sufficient for ongoing replication in hypoxic cells, preventing the collapse of replication forks that would ultimately lead to DSBs and loss of viability. These data indicate that RRM2B contributes to the increased resistance of the most aggressive fraction of tumors; therefore, we investigated the hypoxic adaptation of RRM2B by determining the enzymatic properties of both forms of RNR enzyme (R1/R2 and R1/R2B) in normoxia and hypoxia.
RRM2B Retains Enzymatic Activity in Hypoxia
RRM1, RRM2, and RRM2B recombinant proteins were overexpressed and purified from E. coli (Figures S5A and S5B), without additional iron or reductants, to prevent the variability associated with in vitro assembly of the active cluster of the tyrosyl radical (Cotruvo and Stubbe, 2011). We quantified the iron bound to RRM2 and RRM2B using inductively coupled plasma (ICP) mass spectrometry and found comparable iron incorporation between the two recombinant proteins (Figure S5C). The level of iron incorporation was ∼10% of the level that has been reported in a fully loaded RRM2, which would in turn predict lower enzymatic activity (Aye et al., 2012b, Ochiai et al., 1990).
The abilities of the two forms of the RNR enzyme (R1/R2B and R1/R2) to reduce cytidine diphosphate (CDP) (substrate) to deoxycytidine diphosphate (dCDP) (product) were assayed in normoxic and hypoxic conditions (<0.1% O2). As hypothesized, the R1/R2B was able to sustain activity in both <0.1% O2 and normoxia with no statistically significant difference in the total amount of dCDP formed under either condition after 30 min (Figure 4A). In fact, the R1/R2B enzyme continued converting CDP to dCDP for over 2 hr in hypoxia (Figures 4B and S5D). In contrast, the R1/R2 enzyme did not sustain activity in hypoxia (Figure 4C) and stopped producing dCDPs after 15 min in <0.1% O2 (Figures 4D and S5D). Importantly, we verified that the R1/R2 enzyme did not stop dCDP formation in hypoxia due to lack of available CDP (Figure S5E). Collectively, these data suggest that the compromised R1/R2 activity observed at <0.1% O2 was a result of limited oxygen availability. It is important here to state that the oxygen does not affect the stability of the tyrosyl radical, and it is only required for tyrosyl radical formation (Stubbe, 2003). Once assembled, the tyrosyl radical can catalyze multiple turnovers until regeneration is necessary, with mammalian R1/R2 being stable for 15–25 min in vitro (Cotruvo and Stubbe, 2011). The key difference observed between R1/R2B and R1/R2 in hypoxia offers a mechanistic explanation for why the R1/R2B form of RNR is preferred in hypoxic conditions. It should be noted that, as previously described, the overall activity of the RRM2B-containing RNR enzyme is significantly lower than that of the RRM2-containing RNR (Shao et al., 2004) (Figures 4B and 4D). Therefore, although our data suggest that R1/R2B is the preferred RNR enzyme in hypoxia, the levels of available nucleotides are still reduced compared to normoxia, thus explaining the continued replication stress in these conditions (Figure 1A).
Figure 4.

RRM2B Retains Activity in Hypoxia
(A) Product formation (percentage of the maximum, where maximum is the dCDP levels at 30 min in normoxia) for R1/R2B enzyme in normoxia and <0.1% O2.
(B) dCDP (μM) in <0.1% O2 for R1/R2B for the times indicated. Activity of R1/R2B enzyme at 37°C at 5 min in <0.1% O2 was 19.57 nmol/min/mg RRM2B protein. Gray columns indicate the amount of dCDP formed up to 15 min in <0.1% O2, and red columns indicate the amount of dCDP formed after 15 min in <0.1% O2.
(C) Product formation (percentage of the maximum, where maximum is the dCDP levels at 30 min in normoxia) for R1/R2 enzyme in normoxia and <0.1% O2.
(D) dCDP (μM) in <0.1% O2 for R1/R2 for the times indicated. Activity of R1/R2 enzyme at 37°C at 5 min in <0.1% O2 was 97.74 nmol/min/mg RRM2 protein. Gray columns indicate the amount of dCDP formed up to 15 min in <0.1% O2, and red columns indicate the amount of dCDP formed after 15 min in <0.1% O2.
(E and F) Characterization of the oxygen tunnels (T1–T3) of RRM2B (E) and RRM2 (F).
(G and H) EPR spectra of the tyrosyl radical of RRM2B (G) and RRM2 (H) in normoxia and <0.1% O2, respectively.
(I) Quantification of (G) and (H). Data present electron spins per β subunit.
For all panels, n = 3 (biological replicates); for (A) and (C), data represent mean ± SEM and two-way ANOVA was applied; for (B) and (D), data represent mean ± SEM and two-tailed Student’s t test was applied; (ns) indicates non significant change. See also Figure S5.
Adaption of RRM2B to Hypoxic Environments
Given the differences in normoxic and hypoxic activity between RRM2B and RRM2, we compared the potential oxygen accessibility for the proteins using a modeling approach. Using the only available published structures of RRM2B (PDB: 3HF1) and RRM2 (PDB: 3VPN) (Smith et al., 2009), we investigated the theoretical differences and/or similarities in the dynamics of oxygen diffusion between the two proteins using classical molecular dynamics (MD) simulations. The intention of this study was to understand how molecular oxygen accesses the core of the protein and to probe whether any differences could be related to the activities of RRM2/RRM2B in normoxia versus hypoxia. Our analysis revealed three principal oxygen cavity tunnels (T1–T3) in both proteins, which could be employed as access points (Figures 4E, 4F, and S5F). Analysis of T1–T3 suggested that RRM2B could act as a better oxygen-sequestering agent than RRM2. Specifically, we found that the predicted oxygen-entering frequencies are greater for RRM2B (68% at monomer 1 and 22% at monomer 2) than for RRM2 (14% at monomer 1 and 0% at monomer 2) (Table 1), which indicate differential oxygen-turnover susceptibilities between RRM2B and RRM2. In addition, free-energy calculations of oxygen entry through tunnels T1, T2, and T3 (Figure S5G) suggest that the origin of the selectivity between RRM2B and RRM2 for oxygen turnover resided primarily in tunnels T2 and T3, meaning that the energy barriers for oxygen to cross T2 and T3 are higher for RRM2 than for RRM2B.
Table 1.
O2 Residence Times around the Fe Metallocenter for RRM2B and RRM2 Proteins
| System | Time (ns) | O2, Fe Residence Time (ns) | O2 Entering Events | Entering Frequency (%) | |
|---|---|---|---|---|---|
| RRM2B monomer 1 | 300 | O2, 79 | 25 | 25 | 68 |
| O2, 80 | 19 | ||||
| O2, 120 | 36 | ||||
| O2, 125 | 25 | ||||
| RRM2B monomer 2 | 300 | O2, 93 | 49 | 8 | 22 |
| O2, 107 | 9 | ||||
| O2, 126 | 38 | ||||
| O2, 175 | 103 | ||||
| RRM2 monomer 1 | 300 | O2, 19 | 3 | 5 | 14 |
| O2, 22 | 11 | ||||
| O2, 37 | 2 | ||||
| O2, 142 | 5 | ||||
| RRM2 monomer 2 | 300 | O2, 8 | 19 | 0 | 0 |
| O2, 65 | 20 | ||||
| O2, 88 | 14 | ||||
| O2, 122 | 9 | ||||
We then performed electron paramagnetic resonance (EPR) spectroscopy to monitor the stability of the tyrosyl radical of the two proteins in hypoxic conditions (Figures 4G and 4H). Strikingly, we observed that 66% ± 3.35% of the tyrosyl radical in RRM2B remained stable for 1 hr at 37°C in <0.1% O2, while in the same conditions, only 43% ± 4% remained stable in RRM2 (Figure 4I). Collectively, our results provide compelling evidence that RRM2B is able to retain activity in hypoxia while the activity of RRM2 is compromised and therefore highlight a specific role for RRM2B in the hypoxic stress response.
Hypoxic Activity of RRM2B Is Dependent on K37/K151 and Y164
Despite the fact that RRM2 and RRM2B share 83% sequence homology, there are distinct functional and structural differences (Shao et al., 2004, Smith et al., 2009, Zhou et al., 2010, Xue et al., 2006, Wang et al., 2009b). The switch of C202 in RRM2 to Y164 in RRM2B results in an open phenylalanine channel specifically in RRM2B (Smith et al., 2009), which is connected with oxygen T3 (Figures S5F and S5H). It has been proposed that F183 in RRM2B versus Y221 in RRM2 contributes to the differences in the enzymatic activity between the two proteins (Zhou et al., 2010). In addition, RRM2B has unique antioxidant capabilities, which are partially due to the Y241 and Y331 residues (H279 and Y369 in RRM2) (Xue et al., 2006). Finally, RRM2B through K37 and K151 (E76 and E190 in RRM2) exhibits greater crosstalk between its secondary structures, which stabilizes helix B in an open conformation (Smith et al., 2009). Based on these key differences between RRM2 and RRM2B (Figure S6A) and in order to determine the regions of RRM2B responsible for hypoxic adaptation, we constructed, overexpressed, and purified seven recombinant RRM2B mutants: Y164C, Y164F, F183Y, K37E/K151E, Y241H, Y331F, and Q127K (Figure S6B). The Q127K mutant was generated as a negative control, as it diminishes enzymatic activity due to disruption of the stability of the radical cluster (Zhou et al., 2010).
The seven purified mutants showed the same secondary structure, determined by circular dichroism, as the wild-type RRM2B recombinant protein (Figure S6C). The levels of iron incorporation were also similar for the mutant with the exception of Q127K, as expected (Figure S6D). Reduction of CDP to dCDP was assayed in hypoxia for these mutants. Sustained reduction of CDP to dCDP was observed for 2 hr for most mutants, except Y164C and K37E/K151E, which both appeared to stop reducing substrate after ∼30 min in hypoxia, reminiscent of R1/R2 (Figures S6E–S6K). For these two mutants, we then compared dCDP formation in normoxia and hypoxia and found that the ability to reduce CDP to dCDP in hypoxia was compromised in both the K37E/K151E and Y164C mutants (Figures 5A and 5B). These results indicate that these specific residues are critical to the role of RRM2B in hypoxia and that without them, RRM2B activity is not sustained, similar to RRM2.
Figure 5.

Critical Roles of K37/K151 and Y164 in RRM2B
(A and B) Product formation (percentage of the maximum, where maximum is the dCDP levels at 30 min in normoxia) for K37E/K151E (A) and Y164C (B) in normoxia and <0.1% O2.
(C) EPR spectra of the tyrosyl radical of Y164C, K37E/K151E, and Q127K (as a negative control) in normoxia and <0.1% O2.
(D) Quantification of (C). Data present electron spins per β subunit.
(E) The RRM2B phenylalanine network around Y164 and phenylalanine conformation in Y164C mutation. Distance plot reveals the effect of Y164C in F95-F197 distance. Color code: WT (black), Y164C (red).
(F and G) dATP (F) and dTTP (G) levels in RKORRM2B−/− cells transfected with CTL, WT, Y164C, or K37E/K151E and exposed to <0.1% O2 (16 hr).
(H) Immunoblot for PARP cleavage in RKORRM2B−/− cells treated as in (F) and (G) plus Q127K and exposed to <0.1% O2 (19 hr).
(I) Apoptosis detected morphologically in RKORRM2B−/− cells treated as in (H).
(J) Schematic representation of our proposed model. Hypoxia leads to severely compromised activity of RRM2, leading to replication stress. RRM2B is then induced through the DDR pathway to maintain ongoing replication. However, insufficient dNTPs are generated by R1/R2B, and replication stress is unresolved. The importance of RRM2B activity is that while it does not resolve replication stress, it does maintain replication fork integrity and prevents the accumulation of DNA damage and loss of genome stability.
For (A), n = 3; for (B), n = 4 (biological replicates) and two-way ANOVA was applied; for (C), n = 2 (biological replicates); for (F)–(I), n = 3 (biological replicates); data represent means ± SEM and two-tailed Student’s t test was applied. See also Figures S6 and S7.
To further probe the role of K37/K151 and Y164, we performed additional EPR analyses and MD simulations. Using EPR spectroscopy, we observed that while most of the RRM2B mutants behaved as the WT-RRM2B, which retains 60%–79% of its tyrosyl radical in hypoxia (Figure S7A), the K37E/K151E mutant retained only 28% of its tyrosyl radical in these conditions (Figures 5C and 5D). Since K37/K151 residues affect helix B of RRM2B monomer 2, the bending angle of the helices was estimated and showed that helix B in RRM2B is in an open conformation (20°–25° bending angle), whereas in RRM2, it is closed (8°–10°) (Figure S7B). Interestingly, MD simulations for the K37E/K151E variant showed a closure of helix B after 300 ns from 20° to 8° bending angle, therefore resembling the closed conformation of RRM2 (Figures S7B and S7C). Although these are theoretical studies that provide only indications of the protein conformation, the distortion of helix B in monomer 2 of the K37E/K151E mutant could suggest loss of the essential crosstalk between the two monomers, which may explain the loss of tyrosyl radical stability in hypoxia as demonstrated by our EPR analysis.
Additionally, MD simulations showed a 4.8-fold lower oxygen entering frequency for the Y164C mutant in comparison to WT-RRM2B, while we did not observe any changes in the oxygen entering frequency for the K37E/K151E mutant (Table 2). This could be explained by disruption of the Y164-F197 and Y164-F198 contacts in monomer 1 of RRM2B, which could cause side-chain rearrangement of F95 in the Y164C mutant. This rearrangement may result in F95 moving closer to F197, leading to closure of the RRM2B specific phenylalanine channel (Figures 5E and S5H), therefore explaining the lower oxygen-entering frequency in Y164C mutant.
Table 2.
O2 Residence Times around the Fe Metallocenter for RRM2B Mutants
| System | Time (ns) | O2, Fe Residence Time (ns) | O2 Entering Events | Entering Frequency (%) | |
|---|---|---|---|---|---|
| Y164C monomer 1 | 300 | O2, 57 | 30 | 8 | 14 |
| O2, 79 | 6 | ||||
| O2, 124 | 9 | ||||
| O2, 139 | 60 | ||||
| Y164C monomer 2 | 300 | O2, 1 | 9 | 17 | 29 |
| O2, 105 | 14 | ||||
| O2, 167 | 7 | ||||
| O2, 191 | 16 | ||||
| K37E/K151E monomer 1 | 300 | O2, 37 | 30 | 37 | 64 |
| O2, 40 | 32 | ||||
| O2, 47 | 47 | ||||
| O2, 27 | 27 | ||||
| K37E/K151E monomer 2 | 300 | O2, 9 | 9 | 6 | 16 |
| O2, 188 | 4 | ||||
| O2, 190 | 5 | ||||
| O2, 198 | 3 | ||||
Finally, in order to demonstrate that our findings are relevant in cells experiencing hypoxia and that the hypoxic adaptation of RRM2B depends on the identified residues (Y164 and K37/K151), we performed rescue experiments by transfecting our RKORRM2B−/− cells with the following constructs: pRRM2BWT, pRRM2BY164C, pRRM2BK37E/K151E, and pcDNA3.1 (CTL). As expected, when WT-RRM2B was reintroduced, a significant increase in the intracellular nucleotide levels was observed in hypoxia, whereas reintroduction of Y164C and K37E/K151E showed significantly lower dNTP levels compared to WT-RRM2B (Figures 5F, 5G, S7D, and S7E). Additionally, by monitoring apoptosis, we observed that WT-RRM2B rescued the null phenotype from apoptosis, whereas loss of the key residues (Y164 and K37/K151) abrogated this rescue (Figures 5H and 5I), therefore validating our in vitro experiments and theoretical studies. Overall, we demonstrate the critical importance of the hypoxic induction of RRM2B to mitigate replication stress and determine the molecular adaptations of RRM2B to hypoxia to support this function (Figure 5J).
Discussion
We identified that in response to the physiological stress of hypoxia, RNR responds by isoform switching favoring RRM2B over RRM2. In hypoxia the activity of RRM2 is severely compromised due to the lack of available oxygen, leading to replication stress. RRM2B is able to retain its activity in hypoxia and is therefore induced to compensate and facilitate ongoing replication. This property of RRM1/RRM2B, although not sufficient to resolve replication stress, does preserve replication fork integrity and prevent the accumulation of DNA damage in hypoxia. We verified that depletion of RRM2B results in lower dNTP levels in hypoxic cells and that this has detrimental consequences for cell fate (failure to complete DNA replication, DNA damage, and loss of viability). Importantly, loss of RRM2B in a xenograft model showed delayed tumor growth, increased radiosensitivity, and increased apoptosis specifically in hypoxic areas, further highlighting the biological importance of the hypoxic induction of RRM2B.
Our data suggest that RRM2B is capable of retaining activity in hypoxia through two mechanisms: (1) increased oxygen-entering frequency and (2) enhanced stability of the tyrosyl radical. Our MD analyses suggested that Y164 could increase the oxygen-entering frequency through oxygen tunnel T3 in monomer 1 by keeping F95 an optimum distance from F197. We also found that through the K37/K151 residues, the RRM2B protein retains 66%–70% of its tyrosyl radical in hypoxia. Interestingly, monomer 2 of RRM2B showed a higher oxygen-entering frequency than RRM2 (22% and 0%, respectively), suggesting that the two monomers of the small RNR subunits could react differently in the overall production of the tyrosyl radical. It is possible that monomer 2 contributes more to the tyrosyl radical in RRM2B than in RRM2, possibly due to its helix B open conformation, and that this could be more relevant in hypoxic conditions. In support of this hypothesis, EPR spectra of the double mutant, K37E/K151E, showed a marked reduction in the tyrosyl radical content in hypoxia, but not in normoxia. We propose that the tyrosyl radical of monomer 2 is better protected in RRM2B, therefore contributing to the prolonged activity of the enzyme in hypoxic environments. Our proposed model, where both monomers contribute to tyrosyl radical formation, is in agreement with the established finding that the tyrosyl radical is equally distributed between each small subunit, suggesting the possibility of generation of two tyrosyl radicals per dimer (Cotruvo and Stubbe, 2011). It is important to note that our MD simulations were carried out with the only currently available crystal structure of human RRM2B protein (PDB: 3HF1), which has limitations in its active site configuration and specifically in the iron center. However, we considered it unlikely that as a result of this, there would be large conformational changes in the global structure of the protein compared to its native state, at least to a degree that would influence our MD analysis. Indeed, this is supported by the results of our recombinant mutant analysis and, most importantly, our rescue experiments using our RRM2B knockout cell line. Collectively, this work highlights the importance of Y164, K37, and K151 for RRM2B activity in hypoxia. It is important to note that the recombinant proteins used in our biochemical assays were assembled without additional iron. The biochemistry of RNR, especially the reconstitution of the tyrosyl radical, is extremely complex and not fully understood. Additional studies that also consider the relevant factors for accurate assembly are required and will likely further illuminate the role of RRM2B in both normoxia and hypoxia.
To date, mammalian RRM2B has been primarily associated with DNA repair and mitochondrial DNA synthesis (Wang et al., 2009b, Pontarin et al., 2012). However, it is probable that RRM2B has been evolutionarily maintained in order to be used as the hypoxic-specific RNR small subunit, especially as variants of RRM2B protein have been found to be responsible for continued cell division in anoxia tolerant vertebrates (Sandvik et al., 2012). Here, we propose that one of the principal functions of RRM2B is to act as the hypoxic-specific RNR subunit in order to be able to react promptly when this physiologically relevant stress occurs. It is tempting to speculate that through antioxidant and catalase-like properties (Xue et al., 2006), RRM2B might even play a role in increasing immediate oxygen availability.
Interestingly, a number of previous reports have demonstrated that RRM2B is frequently affected by copy-number changes, typically showing gains, in a broad range of cancers (Chae et al., 2016, Jørgensen et al., 2013). This has led to the suggestion that RRM2B is a tumor promoter (Aye et al., 2015). Our study demonstrates that increased RRM2B expression in hypoxia maintains replication and prevents DNA damage, therefore providing a plausible explanation for why RRM2B is so often amplified in cancers.
Our data suggest that targeting R1/R2B enzyme specifically in hypoxic tumor cells might be an effective therapeutic strategy. RNR is a well-established therapeutic target, and a number of RNR inhibitors (such as gemcitabine, clofarabine, hydroxyurea, and triapine) are being used clinically (Manegold et al., 2000, Aye and Stubbe, 2011, Aye et al., 2012a, Levin, 1992, Sterkers et al., 1998, Hehlmann et al., 1993, Nutting et al., 2009). Interestingly, delivery of siRNA against RRM2 by phosphorothionate oligodeoxynucleotides (GTI-2040) (Lee et al., 2003) is in clinical trials for various solid tumors (Juhasz et al., 2006, Leighl et al., 2009, Oh and Park, 2009). It is tempting to speculate that if this approach was modified to target RRM2B and was employed alongside patient stratification to identify those with significant tumor hypoxia, it may be an effective way to target the most aggressive and treatment-resistant fraction of tumors.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat polyclonal anti-RRM1 (T-16) | Santa Cruz | Cat# sc-11733 |
| Goat polyclonal anti-RRM2 (N-18) | Santa Cruz | Cat# sc-10844 |
| Goat polyclonal anti-p53R2 (N-16) (RRM2B) | Santa Cruz | Cat# sc-10840 |
| Mouse monoclonal anti-Chk1 (G-4) | Santa Cruz | Cat# sc-8408 |
| Mouse monoclonal anti-p53 (DO-1) | Santa Cruz | Cat# sc-126 |
| Rabbit polyclonal anti-p53 (FL-393) | Santa Cruz | Cat# sc-6243 |
| Mouse monoclonal anti-β-Actin Antibody (AC-15) | Santa Cruz | Cat# sc-69879 |
| Mouse anti-Human HIF-1α Clone 54 | BD Transduction Laboratories | Cat# 610958 |
| Mouse anti-BrdU Clone B44 | BD Transduction Laboratories | Cat# 347580 |
| Rat monoclonal anti BrdU Clone BU1/75 (ICR1) | Bio-Rad | Cat# OBT0030CX |
| Mouse monoclonal anti-RRM2 Clone 1E1 | Bio-Rad | Cat# MCA3434Z |
| Rabbit polyclonal anti-KAP-1 | Bethyl / Universal Biologicals | Cat# A300-274 |
| Rabbit polyclonal anti-Phospho KAP-1 (S824) | Bethyl / Universal Biologicals | Cat# A300-767 |
| Rabbit polyclonal anti-Phospho KAP-1 (S473) | BioLegend | Cat# 644602 RRID:AB_2241094 |
| Rabbit polyclonal anti-53BP1 | Novus Biologicals | Cat# NB100-904 |
| Rabbit polyclonal anti-Phospho-p53 (Ser15) | Cell Signaling | Cat# 9284 |
| Rat monoclonal anti-RPA32/RPA2 (4E4) | Cell Signaling | Cat# 2208 |
| Rabbit polyclonal anti-Phospho-Chk1 (Ser296) | Cell Signaling | Cat# 2349 |
| Rabbit polyclonal anti-Phospho-Chk1 (Ser317) | Cell Signaling | Cat# 2344 |
| Rabbit polyclonal anti-Phospho-Chk1 (Ser345) | Cell Signaling | Cat# 2341 |
| Rabbit polyclonal anti-PARP | Cell Signaling | Cat# 9542 |
| Rabbit polyclonal anti-Cleaved Caspase-3 (Asp175) | Cell Signaling | Cat# 9661 |
| Anti-mouse IgG, HRP-linked | Cell Signaling | Cat# 7076 |
| Anti-rabbit IgG, HRP-linked | Cell Signaling | Cat# 7074 |
| Rabbit polyclonal anti-beta Tubulin antibody | Abcam | Cat# ab6046 |
| Mouse monoclonal anti-pimonidazole Clone 4.3.11.3) | Chemicon International | Cat# HP1-100 |
| Alexa Fluor 680 goat anti-mouse IgG (H+L) | Invitrogen | Cat# A21057 |
| Alexa Fluor 680 goat anti-rabbit IgG (H+L) | Invitrogen | Cat# A21076 |
| Alexa Fluor 680 donkey anti-goat IgG (H+L) | Invitrogen | Cat# A21084 |
| IRDye 800CW donkey anti-rabbit IgG | Li-Cor | Cat# 926-32213 |
| IRDye 800CW donkey anti-mouse IgG | Li-Cor | Cat# 926-32212 |
| Alexa Fluor 488-conjugated goat anti-rabbit IgG | Invitrogen | Cat# A11070 |
| Alexa Fluor 488-conjugated donkey anti-rabbit IgG | Invitrogen | Cat# A21206 |
| Alexa Fluor 488-conjugated goat anti-mouse IgG | Invitrogen | Cat# A11017 |
| Alexa Fluor 488-conjugated donkey anti-goat IgG | Invitrogen | Cat# A11055 |
| Alexa Fluor 594-conjugated goat anti-rabbit IgG | Invitrogen | Cat# A11072 |
| Alexa Fluor 594-conjugated goat anti-rat IgG | Invitrogen | Cat# A11007 |
| Alexa Fluor 594-conjugated goat anti-mouse IgG | Invitrogen | Cat# A11020 |
| Alexa Fluor 594-conjugated donkey anti-goat IgG | Invitrogen | Cat# A11058 |
| Alexa Fluor 555-conjugated goat anti-rat IgG | Invitrogen | Cat# A21434 |
| Alexa Fluor 488-conjugated goat anti-mouse F(ab’)2 fragment | Invitrogen | Cat# A11017 |
| Pierce Recombinant Protein A/G | Thermo Fisher Scientific | Cat# 21186 |
| Bacterial and Virus Strains | ||
| BL21-Gold(DE3) Competent Cells | Agilent Technologies | Cat# 230132 |
| One Shot TOP10 Chemically Competent E. coli | Thermo Fisher Scientific | Cat# C404010 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Hydroxyurea (HU) | Sigma | Cat# H8627 |
| Doxorubicin hydrochloride (Adriamycin) | Sigma | Cat# D1515 |
| 5-Bromo-2′-deoxyuridine (BrdU) | Sigma | Cat# B5002 |
| Propidium iodide (PI) | Sigma | Cat# S7109 |
| Protease Inhibitor Cocktail | Roche | Cat# 04693159001 |
| CldU (5-chloro-2′-deoxyuridine) | Sigma | Cat# C6891 |
| IdU (5-iodo-2′-deoxyuridine) | Sigma | Cat# I7125 |
| Cytidine 5′-diphosphocholine sodium salt dehydrate (CDP) | Sigma | Cat# C9755 |
| Protein G Sepharose, Fast Flow | Sigma | Cat# P3296 |
| Isopropyl β-D-1-thiogalactopyranoside (IPTG) | Sigma | Cat# I6758 |
| Deoxyadenosine 5′-Triphosphate, [8-3H(N)] (dATP-H3) | Perkin Elmer | Cat# NET268250UC |
| Deoxythymidine 5′-Triphosphate ((dTTP) Tetrasodium Salt) (dTTP-H3) | Perkin Elmer | Cat# NET520A250UC |
| TRIzol Reagent | Thermo Fisher Scientific | Cat# 15596018 |
| DharmaFECT 1 | Dharmacon | Cat# T-2001 |
| Lipofectamine Ltx | Thermo Fisher Scientific | Cat# 15338100 |
| ProLong Gold | Thermo Fisher Scientific | Cat# P36931 |
| Critical Commercial Assays | ||
| Hypoxyprobe-1 Kit for the Detection of Tissue Hypoxia | Chemicon International | Cat# HP1-100 |
| Real-Time and Dynamic Monitoring of Cell Proliferation and Viability for Adherent Cells | ACEA Biosciences Inc/ Cambridge Biosc | Cat# 00380601050 |
| SYBR Green PCR Master Mix | Applied Biosystems | Cat# 4309155 |
| Verso cDNA Synthesis Kit | Thermo Fisher Scientific | Cat# AB1453B |
| HisTrap HP columns | GE Healthcare | Cat# 17-5248-02 |
| QIAprep Spin Miniprep Kit | QIAGEN | Cat# 27106 |
| QuickChange II XL Site-Directed Mutagenesis Kit | Agilent Technologies | Cat# 200521 |
| QIAquick PCR Purification Kit | QIAGEN | Cat# 28106 |
| QIAquick Gel Extraction Kit | QIAGEN | Cat# 28704 |
| Deposited Data | ||
| Original imaging data | this study | http://dx.doi.org/10.17632/bp95v48kgm.1 |
| Experimental Models: Cell Lines | ||
| RKO | ATCC | CRL-2577 |
| HCT116 | ATCC | CCL-247 |
| H1299 | ATCC | CRL-5803 |
| U2OS | ATCC | HTB-96 |
| U87-MG | ATCC | HTB-14 |
| OE21 | ECACC | 96062201 |
| RKOHIF-1α+/+ and RKOHIF-1α−/− | Dang L.H. Laboratory | Dang et al., 2006 |
| HCT116p53+/+ and HCT116p53−/− | Vogelstein B. Laboratory | Bunz et al., 1998 |
| RKORRM2B−/− | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| MICE: Female BALB/c nude were used for xenograft experiments | Charles River, UK | |
| Oligonucleotides | ||
| siRNA-RRM2B (sequence: UGAGUUUGUAGCUGACAGAUU) | Sigma | Piao et al., 2009 |
| siRNA#2 - RRM2B (sequence: GGAACAAGCUUAAAGCAGA) | Ambion / Life Technologies | s224156 |
| siRNA-p53 (sequence: GUAAUCUACUGGGACGGAA) | Ambion / Life Technologies | Leszczynska et al., 2015 |
| AllStars Negative Control siRNA | QIAGEN | SI03650318 |
| Primers for RRM2B Forward-ChIP (sequence: CTTGCTGGGAAATCTTGACC) | Sigma | Tanaka et al., 2007 |
| Primers used for site-directed mutagenesis Table S1 | N/A | N/A |
| Primers used for dNTP incorporation assay Table S1 | N/A | N/A |
| Primers used for quantitative PCR (qPCR) Table S1 | N/A | N/A |
| Recombinant DNA | ||
| Plasmids used in this work are listed in Table S2 | N/A | N/A |
| Software and Algorithms | ||
| 7500 FAST Real-Time PCR thermocycler was used with v2.0.5 software | Applied Biosystems | http://www6.appliedbiosystems.com/support/software/7500/ |
| The software CAVER 3.0 was used for the analysis of the evolution of lateral fenestrations during the MD simulations | N/A | N/A |
| The particle mesh Ewald (PME) algorithm was used for evaluation of electrostatics interactions | Darden et al., 1993 | N/A |
| The multi time step algorithm Verlet-I/r-RESPA was used to integrate the equations of motion | Tuckerman et al., 1992, Verlet, 1967 | N/A |
| The POPS (Parameter OPtimsed Surfaces) algorithm was used for calculation of the Solvent-Accessible Surface Area (SASA) of both proteins RRM2B and RRM2 | Cavallo et al., 2003, Fraternali and Van Gunsteren, 1996 | N/A |
| The SETTLE algorithm was used for constrained the lengths of covalent bonds involving hydrogen atoms | Miyamoto and Kollman, 1992 | N/A |
| Other | ||
| For the analysis of RRM2B expression and genetic alterations in colorectal cancer datasets the cBioPortal and Prognoscan | http://www.cbioportal.org/; http://www.abren.net/PrognoScan/ | Gao et al., 2013, Cerami et al., 2012, Mizuno et al., 2009 |
| Gene Expression Omnibus; Smith et al., 2010 | https://www.ncbi.nlm.nih.gov/geo | accession number GSE17536 |
| ICP-MS was performed with ICP-MS Trace Element Analysis | https://www.earth.ox.ac.uk/research/services/geochemical-analysis/icpms/ | N/A |
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to, and will be fulfilled by, the lead contact, Dr. Ester M. Hammond (ester.hammond@oncology.ox.ac.uk).
Experimental Model and Subject Details
Source of cell lines used in the study is reported in the reagent and resource table.
Method Details
Cell culture and treatments
Source of cell lines used in the study is reported in the reagent and resource table. Cells were grown in DMEM with 10% FBS, in a standard humidified incubator at 37°C and 5% CO2. All cell lines were routinely mycoplasma tested and found to be negative. RKORRM2B−/− was constructed in this work using CRISPR-Cas9 technology as previously described (Ran et al., 2013). RRM2B gene has three isoforms and in order to construct a full knock-out cell line two 20-bp target sgRNA sequences were used targeting exon 1: TTCGGCGGAGTCTGCGCGAT (isoforms 1 and 3) and AAATGTTATTCGCCGCGGTC (isoform 2). Lipofectamine Ltx (Invitrogen) was used for plasmid transfections according to manufacturer’s recommendations (the plasmids used are listed in Table S2). Dharma-FECT 1 reagent (Thermo Fisher Scientific) was used for siRNA knockdown according to manufacturer’s instructions. RKO or U2OS cells were transfected with the siRNA sequences reported in the reagent and resource table in a final concentration of 50 nM. Drugs were purchased from Sigma unless otherwise stated. Hydroxyurea (HU) was used at a concentration of 2 mM for 6 hr. Adriamycin was used at a concentration of 2 μM for 6 hr. BrdU (5-Bromo-2′-deoxyuridine) was used at a concentration of 20 μM.
Hypoxia treatment
Hypoxia treatments were carried out in a Bactron II anaerobic chamber (Shell labs) or an In vivo2 400 (Baker Ruskinn) (for oxygen concentrations at 2%). For experiments at < 0.1% O2 cells were plated on glass dishes. Cells were harvested inside the chamber with equilibrated solutions. Oxygen concentrations were periodically verified using an Oxylite probe (Oxford Optronix, UK).
Immunobloting and Immonufluorescence
For immunoblots, cells were lysed in UTB (9 M urea, 75 mM Tris-HCl pH 7.5 and 0.15 M β-mercaptoethanol) and sonicated briefly. The Odyssey infrared imaging technology was used (LI-COR Biosciences) and the Odyssey analysis system was used for quantification of the immunoblots. In each case, experiments were carried out in triplicate and a representative blot is shown unless otherwise stated. The antibodies used in this study are listed in the reagent and resource table.
For immunofluorescence, cells were seeded on autoclaved cover glasses (Menzel-Glaser). At the end of each experiment cells were fixed with 4% fixation buffer (4% (w/v) paraformaldehyde in PBS), lysed with 1% PBS-Triton X-100 and then incubated with blocking buffer (2% (w/v) BSA in 0.1% PBS-Triton X-100). Incubation with the appropriate primary and then secondary antibody followed (diluted in 2% (w/v) BSA - 0.1% PBS-Triton X-100) (antibodies used are listed in the reagent and resource table). Cells were visualized using a LSM780 (Carl Zeiss Microscopy Ltd) confocal microscope. At least 100 cells were counted per condition. Due to the presence of 53BP1 foci in the nuclei of unstressed cells, induction of DNA damage was quantified by counting cells with more than six foci. Induction of ssDNA was quantified by counting cells with more than six RPA32 foci.
RT-qPCR
RNA was prepared using Trizol (Invitrogen/Life Technologies). For qPCR expression analysis cDNA was reverse transcribed from total RNA using Verso kit (Thermo Scientific). qPCR was performed using SYBR Green PCR Master Mix kit (Applied Biosystems) in a 7500 FAST Real-Time PCR thermocycler with v2.0.5 software (Applied Biosystems). mRNA fold change was calculated using a 2-ΔΔCt method in relation to the 18S reference gene. The qPCR graphs show the mean of three biological replicates ± s.e.m. The primers used for qPCR are listed in Table S1.
Flow cytometry (FACS)
FACS analysis was performed as previously described (Olcina et al., 2013). U2OS cells were treated with either RRM2B siRNA or negative control siRNA and were placed in normoxia or < 0.1% O2 (3 hr). Samples were pulsed with BrdU (20 μM) for 30 min before collection. FACS analysis was carried out using a Becton Dickinson FACSort. Samples were later analyzed using CellQuest Pro and ModFit LT software. siRNAs and antibodies used are listed in the reagent and resource table.
DNA Fiber analysis and dNTP pool determination
Fiber analysis was carried out as previously described (Foskolou et al., 2016). In brief, sub confluent cells (50%–60% confluency) were sequentially pulse labeled with 25 mM CldU and 250 mM IdU for 20 min each and left at 21% O2 in a 37°C incubator (normoxic samples). Cells were washed once with fresh warm media before the addition of the second (IdU) label. For hypoxic treated samples, cells were first placed for 2 hr in the hypoxic chamber (< 0.1% O2) and then treated with 25 mM CldU for the indicated times, followed by the addition of 250 mM IdU for as long as needed for all samples to be in hypoxia for 6 hr in total. At the end of the treatment, labeled cells were lysed and spread with spreading buffer (200 mM Tris-HCl, pH 7.4, 50 mM EDTA, 0.5% SDS) in a tilting the slide. DNA fibers were then fixed with methanol/acetic acid (3:1 ratio) and stained with rat anti-BrdU monoclonal antibody (for detection of CldU labeled tracts) and mouse anti-BrdU monoclonal antibody (for detection of IdU labeled tracts). Source of antibodies is reported in the reagent and resource table. Fibers were imaged using a Bio-Rad Radiance or LSM780 (Carl Zeiss Microscopy Ltd) confocal microscope and analyzed using ImageJ software (NIH). The length of the fiber tracts that had incorporated both labels (CldU and IdU) was measured and replication rates were calculated with the following formula (VCldU (kb/min) = [(x ∗ 0.132 μm) ∗ 2.59 kb / μm] / t (min), where x = length of CldU).
dNTP pool determination in whole-cell extracts was carried out as previously described (D’Angiolella et al., 2012). In brief, cells were seeded in glass dishes and treated either in < 0.1% O2 (for the indicated times) or with 2 mM HU (6 hr). The normoxic samples were processed immediately after the beginning of the hypoxic and/or HU treatment. Cells were collected and resuspended in 60% iced cold methanol. Samples were then boiled, centrifuged and the supernatant was dried by centrifugal evaporation and finally dissolved in H2O. For the preparation of each primer mix a different set of primers was used (listed in Table S1) supplemented with either [3H]dATP or [3H]dTTP (PerkinElmer). For dGTP and dCTP determination 1 U of Thermo Sequenase DNA Polymerase (GE Healthcare) was used; for dTTP determination 1.25 U Klenow Fragment (Thermo Fisher Scientific) and for dATP determination 0.625 U Klenow Fragment (Thermo Fisher Scientific) was used. The reactions were started by addition of the enzyme. Reactions were incubated for 1 hr either at 48°C (for dGTP and dCTP mixtures) or at room temperature (for dTTP and dATP mixtures). Following incubation, the reaction mixture were spotted onto Whatman DE81 paper (GE Healthcare) and let dry. The papers were then washed with 5% Na2HPO4, followed by rinsing with distilled H2O and 100% EtOH. After being dried, the radioactivity on the papers was measured in a LS 6500 Multi-Purpose Scintillation Counter (Beckman CoulterTM) using 3.5 mL Optiphase HiSafe 3 (PerkinElmer) counting fluid. Data present percentage of dNTP incorporation compared to the positive control of each experiment. In experiments where both siRNA and hypoxia treatment were used, a normalization to each normoxic control was preceded the final analysis.
Immunoprecipitation (IP) and Chromatin IP (ChIP)
IP was carried out as previously described (Zhou et al., 2003). Briefly, RKO cells were lysed in lysis buffer (100 mM NaCl, 20 mM Tris-HCl pH 7.4, 5 mM MgCl2, 0.5% NP-40) with freshly added phosphatase/protease cocktail inhibitors (Roche). The lysates were incubated with Protein G Sepharose beads (Sigma) and the antibody of interest RRM1 (Santa Cruz Biotechnology: sc-11733) or a control IgG antibody (Invitrogen: A10535) with agitation, at 4°C overnight. Lysates were washed with lysis buffer and solubilized in SDS-PAGE sample buffer. Antibodies used were: RRM1 (Santa Cruz Biotechnology: sc-11733), RRM2B (Santa Cruz Biotechnology: sc-10840), RRM2 (AbD Serotec, MCA3434Z) and for the detection of RRM2 the Pierce Recombinant Protein A/G (Thermo Fisher Scientific, 21186) secondary antibody was used.
ChIP in RKO cells treated as indicated in figure legend was carried out as previously described (Leszczynska et al., 2015). For each sample 2 μg of combined mouse p53 (DO-1; sc-126) and rabbit p53 (FL-393; sc-6243) (Santa Cruz) antibodies were used for immunoprecipitation. Combined non-specific mouse (7076) and rabbit (7074) (Cell Signaling) were used as control IgGs. A no antibody control sample was also included in each experiment. Bound fraction and input were analyzed by qPCR using specific primer set for the RRM2B locus: CTTGCTGGGAAATCTTGACC (Tanaka et al., 2007). Fold enrichment is expressed as a % of input and is normalized to total p53 in each sample. Graphs show the mean of technical triplicates ± RQmax and RQmin; n = 3.
Colony survival and apoptosis
RKO cells were treated either with RRM2B siRNA or negative control siRNA and exposed to normoxia or hypoxia (< 0.1% O2 for 24 hr). Subsequently all cells were placed in normoxic incubator and left for 9 days to form colonies, which were visualized by crystal violet staining. Graphs show the mean of three biological replicates ± s.e.m.
Apoptosis assay were performed as previously described (Leszczynska et al., 2015). In brief, both adherent and floating cells were collected and fixed with 4% paraformaldehyde. Samples were then washed and the nuclei were stained with ProLong Gold mounting medium with DAPI (Thermo Fisher Scientific). Apoptosis was plotted as the percentage of cells with fragmented DNA per field of view, with at least ten fields of view quantified per experiment. Each apoptosis experiment was performed at least 3 times.
xCELLigence proliferation assay
The xCELLigence Real-Time Cell Analyzer (ACEA Biosciences) DP Instrument equipped with an E-Plate was used for proliferation assays. Cells were seeded into 100 μL of media in 96X microplates. The attachment, spreading and proliferation of the cells were monitored every 15 min for 60 hr. For quantification, the cell index at indicated time points was averaged from three independent experiments. Values are presented as mean ± SEM of three independent experiments.
Xenografts and tissue IF staining
All animal procedures were performed in accordance with current UK legislation and were approved by the University of Oxford Biomedical Services Ethical Review Committee, Oxford, UK. For the tumor growth curves (xenografts), female BALB/c nude mice or female athymic nude mice (Charles River, UK) were randomized and injected subcutaneously with 3 × 106 RKORRM2B+/+ or RKORRM2B−/− cells in 25% (v/v) matrigel and serum-free DMEM. Tumor growth was monitored until they reached approximately 300-400 mm3 (volume = height x depth x width x π / 6). For the irradiation experiment, the tumors in half of the animals were irradiated with 10 Gy when tumor volumes reached 100 mm3. Tumors were measured regularly, and tumor growth was plotted as a mean of tumor volumes ± SEM. Mice were injected with 50 mg/kg of pimonidazole (PIMO) 2 hr before the end of the experiment. Tumors were split in half and either preserved in formalin for wax embedding or were snap frozen and embedded in optimal cutting temperature compound (OCT).
Tissue IF staining were performed as previously described (Leszczynska et al., 2015). In brief, tumors embedded in OCT were sectioned (5 μm) and froze down at −20°C. Sections were immediately fixed with 4% paraformaldehyde and then quenched with 50 mM NH4Cl. Blocking was performed with 1% BSA in TBS, followed by incubation with Mouse on Mouse (M.O.M.) Blocking Reagent (Vector Laboratories). The sections were stained with PIMO and cleaved caspase-3 primary antibodies, followed by secondary antibodies (listed in the reagent and resource table) and mounted with ProLong Gold mounting medium with DAPI (Invitrogen/Life technologies). The apoptotic cells were counted within and outside of hypoxic regions. Images were taken from at least 3 RKORRM2B+/+ and 3 RKORRM2B−/− tumors. Graphs show the percentage of apoptotic cells from all normoxic or hypoxic regions.
TCGA RNA-sequencing (RNA-seq) analysis and analysis of colorectal cancer datasets
Raw RNA-seq data for 382 colorectal adenocarcinoma tumors were downloaded from the TCGA project (accessed through cBioportal: http://www.cbioportal.org/). To examine tumor-associated HIF-activity (referred to as hypoxia signature), raw data for each sequenced gene were rescaled to set the median equal to 1, and HIF-activity was quantified by averaging the normalized expression of 44 target genes, associated with HIF activity (encoding ADM, GFBP3, EDN2, PFKFB4, FLT1, TFR2, BNIP3L, TGFA, BNIP3, PGK1, EGLN1, LDHA, EGLN3, CP, TGFB3, PFKFB3, HK1, TFRC, EDN1, CDKN1A, CA9, HMOX1, SERPINE1, LOX, NDRG1, CA12, PDK1, VEGFA, ERO1L, RORA, P4HA1, MXI1, SLC2A1(GLUT1), STC2, MIF, DDIT4, ENO1, CXCR4, PLOD1, P4HA2, GAPDH, PGAM1, TMEM45A and PIM1) (Li et al., 2014). Log10 conversion of the hypoxia signature was plotted against Log10 conversion of raw data for RRM1, RRM2 and RRM2B. Two-tailed p value shown on each graph for each Pearson and Spearman r (correlation coefficient).
To examine tumor-associated p53-activity (referred to as p53 group), raw data for each sequenced gene were rescaled to set the median equal to 1, and p53-activity was quantified by averaging the normalized expression of 6 p53 target genes, associated with hypoxia-induced p53 activity (encoding BTG2, CYFIP2, INPP5D, KANK3, PHLDA3 and SULF2) (Leszczynska et al., 2015, Leszczynska et al., 2016). Log10 conversion of the p53 signature was plotted against Log10 conversion of raw data for RRM2B (also rescaled to set the median equal to 1). Two-tailed p value shown on each graph for each Pearson and Spearman r (correlation coefficient).
For the analysis of RRM2B expression and genetic alterations in colorectal cancer datasets the cBioPortal (http://www.cbioportal.org/) and Prognoscan (http://www.abren.net/PrognoScan/) tools were used on the 5th Dec 2016 (Gao et al., 2013, Cerami et al., 2012, Mizuno et al., 2009). Using cBioPortal the TCGA (provisional) dataset was analyzed (RNA Seq V2 RSEM) for alterations, including mutations, putative copy-number alterations, mRNA expression, mutations and survival probability (629 cancer patients). For the Prognoscan analysis, RRM2B expression was checked in all available colorectal cancer datasets and Kaplan-Meier graphs were extracted only for the statistically significant (p < 0.05) dependence between RRM2B expression (probe 223342_at) and survival probability. This analysis used the publicly available dataset at Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo) with the accession number GSE17536 (Smith et al., 2010).
Protein overexpression and purification
The 6 × His-tagged hRRM1 (pET28a-RRM1) was kindly provided by Prof JoAnne Stubbe (Massachusetts Institute of Technology, Cambridge, USA), expressed in E.coli BL21-Gold (DE3) competent cells (Agilent Technologies) and as previously described (Ando et al., 2016). The 6 × His-tagged hRRM2 and hRRM2B (pET28a-RRM2, pET28a-RRM2B) were kindly provided by Dr. Yun Yen (Beckman Research Institute at City of Hope, Duarte, USA). The proteins were expressed in E.coli BL21-Gold (DE3) competent cells (Agilent Technologies) and purified as previously described (Shao et al., 2004) with minor modifications. Specifically, an overnight culture of the transformed bacteria was diluted 80-fold in 600 mL of 2 × YT medium containing 50 μg/ml kanamycin. Each culture was grown at 37°C until OD600nm = 0.7 - 0.9 and then induced by 1 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG) and incubated overnight at 18°C. Cells were harvested by centrifugation and the pellets were lysed with appropriate amount (5 ml/gr of pellet) Lysis Buffer (50 mM NaH2PO4 pH 7.0; 0.1% Triton X-100; 10 mM β-mercaptoethanol; freshly added EDTA-free Protease Inhibitors and DNase I) at 4°C with vigorous agitation until the lysate was homogeneous. The lysate was then sonicated (60% Amplitude 30 s ON / 30 s OFF) and clarified by centrifugation at 48,000 × g for 20 min at 4°C. Soluble lysate was passed through a His-Trap HP purification column (GE Healthcare), washed with at least 30-fold bed volume of Wash Buffer (50 mM NaH2PO4 pH 7.0; 800 mM NaCl; 50 mM imidazole; 0.1% Triton X-100; 10 mM β-mercaptoethanol) and finally eluted with Elution Buffer (50 mM NaH2PO4 pH 7.0; 300 mM NaCl; 125 mM imidazole). The protein then underwent buffer exchange into 25 mM Tris-HCl, pH 7.5 before being concentrated and stored at −80°C. The purified proteins showed approximately 90% purity. The apo-forms of RRM2 and RRM2B recombinant proteins used in ICP-MS experiments were overexpressed and purified as previously described (Wang et al., 2009a). Site-specific mutations in RRM2B (Y164C; Y164F; F183Y; K31E/K151E; Y241H; Y331F and Q127K) were generated by PCR, using the pET28a-RRM2B as a template and the QuickChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies). All constructs were verified by DNA sequencing. Expression and purification of the hRRM2B mutated proteins were performed in the same way as for hRRM2B.
dCDP formation assays
The activity of recombinant RNR was measured using the following reduction method. The enzyme concentration (R1/R2B or R1/R2) was the same in all cases and the limited factor was the presence or not of oxygen (normoxia versus hypoxia). Specifically, a final concentration of 1.25 μM of purified RRM1 was incubated with 2.5 μM of either RRM2 or RRM2B protein. The reaction mixture contained 0.1 mM CDP, 50 mM HEPES (pH 7.5), 6 mM DTT, 8 mM magnesium acetate, 2 mM ATP and 1 mM NADPH in a final volume of 400 μl. The RNR proteins purified were active without reassembling the iron center (Shao et al., 2004, Zhou et al., 2010). In hypoxic samples (< 0.1% O2) the reaction mixture was assembled and the assay was carried out within an anaerobic chamber (Bactron, Shel Labs). Reactions were initiated by the addition of CDP, incubated at 37°C and aliquots of 50 μl were quenched by the addition of 10 μl of 3% tricarboxylic acid over a time course of the reaction. Subsequent dCDP and CDP levels were analyzed by HPLC. Graphs present either percentage of dCDP formation when normoxia and hypoxia were compared (data were normalized against the normoxic samples at 37°C at 30 min where the dCDP formation was considered 100%) or in μM when the accumulation of dCDP formed in < 0.1% O2 at a 0-120 min time course was examined.
Determination of CDP/dCDP
CDP and dCDP levels following RNR activity assays were determined by ion-pairing HPLC with UV detection (270 nm). HPLC was performed using a Shimadzu system with a photodiode array detector. The column was an ACE C 18, 3 μm, 125 × 3 mm (Hichrom) maintained at 35°C. A gradient separation was achieved using 10% methanol, 10 mM KH2PO4, 10 mM TBAOH, pH 6.9 (A) and 30% methanol, 50 mM KH2PO4, 6 mM TBAOH, pH 7.0 (B), with a linear gradient of 25 – 47% B over 8 min, 47 – 80% B over 1 min, then returning to the started conditions 3 min. The flow rate was 0.6 ml/min and the running time was 16.5 min.
Circular dichroism spectrometry
Circular dichroism (CD) spectra (Chirascan CD/Fluorimeter spectrometer (Applied Photophysics, UK) of hRRM2B and the mutants (0.2 mg/mL) were recorded in the wavelength range 260 to 185 nm in 100 mmol/L potassium phosphate buffer (pH 7.5). All measurements were carried out at 37°C using a 0.1-cm path length quartz curette. The data pitch was 0.1 nm with a 1-nm bandwidth at a scan speed of 1.0 nm/s. Each spectrum shown represents the average of three. All CD data were expressed as the mean residue ellipticity, [θ], deg·cm2·dmol-1.
Inductively coupled plasma mass spectrometry (ICP-MS)
The Thermo Finnigan Element 2 ICP-MS was used for quantitation of iron content in the recombinant purified proteins (40 μg of protein was used per 50 μl of reaction). Calibrations were obtained using external standards (a series of standards of known Fe concentrations were prepared and analyzed to gain a calibration linear, prior to the measurement of the samples). An external standard was diluted and measured from a dilution of High Purity Standards 10 ppm ICP-MS-68 A standard. All blanks, standards and samples were also spiked with 1 ng/g Rh, so that any general instrument drift could be normalized. Dilutions were made using a 2% HNO3 solution, prepared using in-house distilled nitric acid and 18.2 Mohm DI water. All data results were first reported as elemental concentrations and then calculated in nM of Fe per μM of protein. Additional information can be found at (https://www.earth.ox.ac.uk/research/services/geochemical-analysis/icpms/).
Electron paramagnetic resonance (EPR)
CW-EPR spectra were collected in the Center for Advanced ESR (CAESR) in the Inorganic Chemistry Laboratory at the University of Oxford. X-band measurements utilized a Bruker-Biospin Micro EMXplus spectrometer equipped with a PremiumX microwave bridge, a cylindrical TE011-mode resonator (SHQE-W), an ESR-900 liquid helium cryostat, and an Oxford Instruments ITC-503 s temperature controller. Measurements were performed within the limit of resolution by temperature, 40K, and under non-saturating conditions.
The protein concentration for all EPR samples was 200 μM except for RRM2 protein, which was 70 μM. The protein samples (RRM2B, RRM2 or RRM2B mutants) were diluted in 50 mM HEPES, pH 7.5 and were incubated either at 37°C for 5 min with oxygen present (normoxia) or at 37°C for 60 min in a Bactron II anaerobic chamber (< 0.1% O2). At the end of each treatment 20% (v/v) glycerol was added for vitrification during the low-temperature recordings. Samples were then transferred to EPR tubes and quick-frozen by immersion in liquid nitrogen. Spin quantitation was performed using the Bruker spectrometer hardware spin calibration, given the input dimensions of the precision Wilmad PQ-706 tubes. The results were verified with a 1 mM CuII EDTA in 50 mM HEPES pH 7.5, 20% (v/v) glycerol that was measured under non-saturating conditions at 100 K with 50 μW microwave power. The results show spin equivalents per β-subunit.
Molecular Dynamics (MD) simulations
System setup: MD simulations of O2 transport on hRRM2B (PDB: 3HF1; resolution 2.60 Å) and hRRM2 (PDB: 3VPN; resolution 2.25 Å) were performed (Smith et al., 2009). The RRM2B structure comprises residues S29 to F311. The 3VPN X-ray structure is that of the E106D variant of native RRM2, and comprises residues M65 to M350. Four simulations were performed in which 200 randomly chosen water molecules outside the solvated protein were replaced by 200 O2 molecules (0.5 M) termed the 100% oxygen level simulation. The random distribution of oxygen molecules precludes any potential artifacts arising from initial distribution. An additional two sets of simulations were performed to control for concentration effects; the first set up has a tenfold reduction in oxygen concentration, termed 10% oxygen level simulation with effective concentration of 50 mM, for RRM2B and RRM2. The second set does not include any oxygen molecules, termed 0% oxygen level simulation. Each system was solvated in a box of dimensions (80, 80, 92) Å using the Solvate plug-in of VMD (Humphrey et al., 1996), with the total system size comprising ∼61,000 atoms. In accordance with the Uniprot sequence alignment two mutants of native RRM2B were prepared with the Mutate plugin in VMD (Humphrey et al., 1996), namely Y164C and K37E/K151E.
MD equilibration protocol
Calculations were done with NAMD 2.9 (Phillips et al., 2005), using the CHARMM 27 protein force-field (Brooks et al., 2009) with the CMAP correction (Buck et al., 2006), together with the TIP3P water model (Jorgensen et al., 1983). Oxygen Lennard-Jones parameters were taken from CHARMM (Cohen et al., 2005, Cohen et al., 2006). For O2 gas access in RRM2B and RRM2, the iron centers were modeled using CHARMM 27 force-field, with iron Lennard-Jones parameters εO = 0.00 kcal/mol and Rmin/2 = 0.65 Å. Oxygen (O2) was modeled using CHARMM 27 Lennard-Jones parameters from heme oxygen, εO = −0.12 kcal/mol and Rmin/2 = 1.7 Å, as well as bond spring constants and bond spring lengths for O2 as 1.23 Å and 600 kcal/mol/Å-1, respectively. RRM2B contains the same diiron/dityrosyl cofactor that RRM2 does, and both RRM2B and RRM2 are biologically active as homodimers. In RRM2B, the active site iron coordination environment in Monomer-1 is a mono-iron site, and Monomer-2 has a di-iron site. In RRM2B, the active site iron coordination environment consists in both Monomer-1 and 2 of mono-iron sites.
After 1,000 steps of Conjugate-Gradient minimization, four classical molecular dynamics (MD) simulations of the O2-containing solvated protein were carried out for 300 ns in the NPT ensemble. The high oxygen concentration was necessary to ensure sufficient sampling of protein cavities by O2 on the accessible timescale of current simulations. It was verified that no gas clustering occurred in the solvent at this concentrations. The particle mesh Ewald (PME) algorithm was used for evaluation of electrostatics interactions beyond 12 Å, with a PME grid spacing of 1 Å, and NAMD defaults for spline and κ values (Darden et al., 1993). A cut-off at 12 Å was applied to non-bonded forces, both electrostatics and Van der Waals forces were smoothly switched off between the cut-off distance of 12 Å, and the switching distance of 10 Å, using the default NAMD switching function. A Verlet neighbor list with pairlist distance of 14 Å was used to only evaluate non-bonded neighboring forces within the pairlist distance (Verlet, 1967). The lengths of covalent bonds involving hydrogen atoms were constrained by the SETTLE algorithm (Miyamoto and Kollman, 1992) in order to be able to use a 2-fs time-step. The multi time step algorithm Verlet-I/r-RESPA (Tuckerman et al., 1992, Verlet, 1967) was used to integrate the equations of motion. Non-bonded short-range forces were computed for each time step, while long-range electrostatic forces were updated every 2 time steps. The pressure was kept at 1 atm by the Nosé-Hoover Langevin piston (Langevin, 1908, Nosé, 1984a, Nosé, 1984b), with a damping time constant of 50 fs and a period of 100 fs. The temperature was maintained at 300 K by coupling the system to a Langevin thermostat, with a damping coefficient of 1 ps-1. A total of 3.0 μs of MD simulations were accrued.
Code availability
The standard MPI version 2.9 of the MD simulation code NAMD was employed (Phillips et al., 2005). A free license can be obtained at http://www.ks.uiuc.edu/Research/namd/. Version 1.9 of Visual Molecular Dynamics (VMD) was employed for analysis, with license obtained at http://www.ks.uiuc.edu/Research/vmd/.
MD simulation analysis
The Solvent-Accessible Surface Area (SASA) of both proteins RRM2B and RRM2 was calculated using the POPS (Parameter OPtimsed Surfaces) algorithm (Cavallo et al., 2003, Fraternali and Van Gunsteren, 1996). The bending angle of helices was estimated using Bendix in VMD (Dahl et al., 2012).
The Diffusion-Reaction Model (Wang et al., 2013) presented in Equation (1) was adopted to describe the diffusion of O2 into the RNR active site:
| (1) |
This is a kinetic two-step model consisting of an initial diffusion step of the gas molecule, followed by a metal-gas molecule chemical reaction step (Wang et al., 2011, Wang and Blumberger, 2012). Here, a classical force-field (FF) was employed, and only the diffusion and binding of O2 gas molecules to the RNR iron center were modeled:
| (2) |
The simulation of O2 diffusing in the solvated RNR enzymes yields diffusion statistics that enable the construction of a probability density map of the O2-distribution within RNR (RRM2B or RRM2). This distribution is then divided into a set of M discrete protein cluster states that correspond to the probability maxima. Two parameters are used to characterize the diffusion of O2. First, a distance criterion is established to determine when an oxygen molecule has entered into the active site of hRRM2B and hRRM2, defined as the difference between the center-of-mass (COM) of the iron ion and the O2 molecule. Second, cut-off criteria of 8 Å, 9 Å and 10 Å were employed to generate statistics of the O2 entering. An O2 molecule is counted as having entered the cluster if dCOM (O2-Fe) < c where c is 8 or 9 Å.
Free-energy simulations of tunnel entry
Oxygen entry tunnels were assigned and characterized from classical MD simulations. As previously described, O2 entry into WT RRM2B and RRM2 was identified to occur primarily through Tunnels T1, T2 and T3. Based on this assignment, equilibrium Adaptive-Biasing Force (ABF) MD simulations (Chipot and Pohorille, 2007, Darve and Pohorille, 2001, Darve et al., 2008, Van Gunsteren, 1989) were performed on oxygen entry into tunnels T1, T2 and T3 in Monomer-1 to determine relative differences in the thermodynamics of oxygen entry into RRM2B and RRM2. In total, six ABF simulations per RNR were performed, totalling ≈0.5 μs of MD simulation dynamics. Each ABF simulation was performed with a force bias on a single oxygen molecule meaning these simulations constitute single-molecule biased MD simulations, using the collective variable ξ center-of-mass (COM) distance between Fe(III) and O2, , with ξ ranging from 3 Å to 15Å, in bins of width 0.25 Å. Each ABF simulation obtains ergodic sampling by applying every 500 timesteps a biasing force (3) onto the Newtonian equations of motion, whose value is updated on-the-fly (Den Otter, 2000, Den Otter and Briels, 1998) From the result, free-energy gradient along ξ, the Potential of Mean Force (PMF) along ξ is obtained from numerical integration of the gradient.
| (3) |
| (4) |
Tunnel T1 in Monomer-1 extends for ∼30 Å and connects the two faces of the protein. It was simulated as two separate tunnels of length ∼15 Å in Monomer 1, denoted Tunnel T1 and T1’, and was compared to a single simulation of Tunnel T1 in Monomer 2. Two simulations of Tunnels T2 and T3 were performed in RRM2B and RRM2.
Analysis of fenestrations
The software CAVER 3.0 was used for the analysis of the evolution of lateral fenestrations during the MD simulations, using 100 evenly chosen snapshots from the MD simulations, which is every 2 ns. The outer surface of the protein is calculated by rolling a large spherical probe around the surface of the protein and then internal cavities are identified using a smaller spherical probe (a radius of 0.8 Å was used here, and 12 probe spheres used). All identified tunnels are grouped into clusters based on relative proximities and with an 8 Å cut off for each cluster node, meaning that tunnels differing from the node by more than 8 Å are excluded. The values for shell radius and depth (15 Å) influence the definition of the protein molecular surface.
Statistical analysis
The statistical significance of differences between datasets was determined assessed by using the following test. Student’s t test (two-tailed paired, except for dNTP incorporation assays, where a two-tailed unpaired test was applied) was used in all experiments where two means were compared. One-way analysis of variance (ANOVA) was used to compare several means and two-way ANOVA was used for determination in a response that is affected by two factors (one- or two-way ANOVA analysis is indicated at the figure legends). Statistical significance was assumed if p < 0.05 or lower and is noted in the figures. Error bars represent mean ± standard error of the mean (s.e.m.).
Data and Software Availability
Original imaging data have been deposited to Mendeley Data and are available at http://dx.doi.org/10.17632/bp95v48kgm.1
Author Contributions
I.P.F. conceived, designed, performed, and interpreted the majority of the experiments and wrote the manuscript. C.J. performed and interpreted the MD simulations. K.B.L. performed the xenografts, immunohistochemistry (IHC), some experiments, and TCGA analysis. M.M.O. advised on some experiments and carried out TCGA analysis. B.H. assisted in developing the CRISPR clone. V.D. advised on the dNTP assay. H.T. assisted with enzyme assays. W.K.M. performed and analyzed the EPR studies. C.D. supervised, designed, and interpreted the MD simulations. E.F. supervised, designed, and interpreted in vitro enzymatic assays. E.M.H. conceived the project; conceived, designed, and interpreted the majority of the experiments; and wrote the manuscript. All authors commented on the manuscript.
Acknowledgments
We thank Michael Stratford, Adam Hardy, Lisa Folkes, Mick Woodcock, Graham Brown, Philip Holdship, Ludovico Buti, Haoran Li, JoAnne Stubbe, and Yun Yen. CRUK Oxford Cancer Centre, CRUK Hartree Centre resources and the National Service were used for the Computational Chemistry Software. This work was supported by a Cancer Research UK (CR-UK) (H3RWGJ00.H307.1) grant (to E.M.H.), CR-UK Oxford Centre Prize DPhil Studentship C38302/A12981 (to I.P.F.), and a King’s College London GTA studentship (to C.J.).
Published: April 13, 2017
Footnotes
Supplemental Information includes seven figures and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2017.03.005.
Supporting Citations
The following references appear in the Supplemental Information: Hammond et al., 2006, Sherman and Fyfe, 1989, Wang et al., 2007.
Supplemental Information
References
- Ando N., Li H., Brignole E.J., Thompson S., McLaughlin M.I., Page J.E., Asturias F.J., Stubbe J., Drennan C.L. Allosteric inhibition of human ribonucleotide reductase by dATP entails the stabilization of a hexamer. Biochemistry. 2016;55:373–381. doi: 10.1021/acs.biochem.5b01207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aye Y., Stubbe J. Clofarabine 5′-di and -triphosphates inhibit human ribonucleotide reductase by altering the quaternary structure of its large subunit. Proc. Natl. Acad. Sci. USA. 2011;108:9815–9820. doi: 10.1073/pnas.1013274108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aye Y., Brignole E.J., Long M.J., Chittuluru J., Drennan C.L., Asturias F.J., Stubbe J. Clofarabine targets the large subunit (α) of human ribonucleotide reductase in live cells by assembly into persistent hexamers. Chem. Biol. 2012;19:799–805. doi: 10.1016/j.chembiol.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aye Y., Long M.J., Stubbe J. Mechanistic studies of semicarbazone triapine targeting human ribonucleotide reductase in vitro and in mammalian cells: tyrosyl radical quenching not involving reactive oxygen species. J. Biol. Chem. 2012;287:35768–35778. doi: 10.1074/jbc.M112.396911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aye Y., Li M., Long M.J., Weiss R.S. Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene. 2015;34:2011–2021. doi: 10.1038/onc.2014.155. [DOI] [PubMed] [Google Scholar]
- Bartkova J., Horejsí Z., Koed K., Krämer A., Tort F., Zieger K., Guldberg P., Sehested M., Nesland J.M., Lukas C. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- Begg A.C., Stewart F.A., Vens C. Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer. 2011;11:239–253. doi: 10.1038/nrc3007. [DOI] [PubMed] [Google Scholar]
- Bester A.C., Roniger M., Oren Y.S., Im M.M., Sarni D., Chaoat M., Bensimon A., Zamir G., Shewach D.S., Kerem B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–446. doi: 10.1016/j.cell.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi V., Pontis E., Reichard P. Changes of deoxyribonucleoside triphosphate pools induced by hydroxyurea and their relation to DNA synthesis. J. Biol. Chem. 1986;261:16037–16042. [PubMed] [Google Scholar]
- Brischwein K., Engelcke M., Riedinger H.J., Probst H. Role of ribonucleotide reductase and deoxynucleotide pools in the oxygen-dependent control of DNA replication in Ehrlich ascites cells. Eur. J. Biochem. 1997;244:286–293. doi: 10.1111/j.1432-1033.1997.00286.x. [DOI] [PubMed] [Google Scholar]
- Brooks B.R., Brooks C.L., 3rd, Mackerell A.D., Jr., Nilsson L., Petrella R.J., Roux B., Won Y., Archontis G., Bartels C., Boresch S. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck M., Bouguet-Bonnet S., Pastor R.W., MacKerell A.D., Jr. Importance of the CMAP correction to the CHARMM22 protein force field: dynamics of hen lysozyme. Biophys. J. 2006;90:L36–L38. doi: 10.1529/biophysj.105.078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunz F., Dutriaux A., Lengauer C., Waldman T., Zhou S., Brown J.P., Sedivy J.M., Kinzler K.W., Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Burrell R.A., McClelland S.E., Endesfelder D., Groth P., Weller M.C., Shaikh N., Domingo E., Kanu N., Dewhurst S.M., Gronroos E. Replication stress links structural and numerical cancer chromosomal instability. Nature. 2013;494:492–496. doi: 10.1038/nature11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallo L., Kleinjung J., Fraternali F. POPS: a fast algorithm for solvent accessible surface areas at atomic and residue level. Nucleic Acids Res. 2003;31:3364–3366. doi: 10.1093/nar/gkg601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae Y.K., Anker J.F., Carneiro B.A., Chandra S., Kaplan J., Kalyan A., Santa-Maria C.A., Platanias L.C., Giles F.J. Genomic landscape of DNA repair genes in cancer. Oncotarget. 2016;7:23312–23321. doi: 10.18632/oncotarget.8196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimploy K., Tassotto M.L., Mathews C.K. Ribonucleotide reductase, a possible agent in deoxyribonucleotide pool asymmetries induced by hypoxia. J. Biol. Chem. 2000;275:39267–39271. doi: 10.1074/jbc.M006233200. [DOI] [PubMed] [Google Scholar]
- Chipot C., Pohorille A. Springer; 2007. Free Energy Calculations. [Google Scholar]
- Cohen J., Kim K., Posewitz M., Ghirardi M.L., Schulten K., Seibert M., King P. Molecular dynamics and experimental investigation of H(2) and O(2) diffusion in [Fe]-hydrogenase. Biochem. Soc. Trans. 2005;33:80–82. doi: 10.1042/BST0330080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J., Arkhipov A., Braun R., Schulten K. Imaging the migration pathways for O2, CO, NO, and Xe inside myoglobin. Biophys. J. 2006;91:1844–1857. doi: 10.1529/biophysj.106.085746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotruvo J.A., Stubbe J. Class I ribonucleotide reductases: metallocofactor assembly and repair in vitro and in vivo. Annu. Rev. Biochem. 2011;80:733–767. doi: 10.1146/annurev-biochem-061408-095817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby M.E., Kulshreshtha R., Ivan M., Glazer P.M. MicroRNA regulation of DNA repair gene expression in hypoxic stress. Cancer Res. 2009;69:1221–1229. doi: 10.1158/0008-5472.CAN-08-2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angiolella V., Donato V., Forrester F.M., Jeong Y.T., Pellacani C., Kudo Y., Saraf A., Florens L., Washburn M.P., Pagano M. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell. 2012;149:1023–1034. doi: 10.1016/j.cell.2012.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl A.C.E., Chavent M., Sansom M.S.P. Bendix: intuitive helix geometry analysis and abstraction. Bioinformatics. 2012;28:2193–2194. doi: 10.1093/bioinformatics/bts357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang D.T., Chen F., Gardner L.B., Cummins J.M., Rago C., Bunz F., Kantsevoy S.V., Dang L.H. Hypoxia-inducible factor-1alpha promotes nonhypoxia-mediated proliferation in colon cancer cells and xenografts. Cancer Res. 2006;66:1684–1936. doi: 10.1158/0008-5472.CAN-05-2887. [DOI] [PubMed] [Google Scholar]
- Darden T., York D., Pedersen L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089. [Google Scholar]
- Darve E., Pohorille A. Calculating free energies using average force. J. Chem. Phys. 2001;115:9169. [Google Scholar]
- Darve E., Rodríguez-Gómez D., Pohorille A. Adaptive biasing force method for scalar and vector free energy calculations. J. Chem. Phys. 2008;128:144120. doi: 10.1063/1.2829861. [DOI] [PubMed] [Google Scholar]
- Den Otter W.K. Thermodynamic integration of the free energy along a reaction coordinate in Cartesian coordinates. J. Chem. Phys. 2000;112:7283. [Google Scholar]
- Den Otter W.K., Briels W.J. The calculation of free-energy differences by constrained molecular-dynamics simulations. J. Chem. Phys. 1998;109:4139. [Google Scholar]
- Eriksson S., Skog S., Tribukait B., Wallström B. Deoxyribonucleoside triphosphate metabolism and the mammalian cell cycle. Effects of hydroxyurea on mutant and wild-type mouse S49 T-lymphoma cells. Exp. Cell Res. 1987;168:79–88. doi: 10.1016/0014-4827(87)90417-4. [DOI] [PubMed] [Google Scholar]
- Foskolou I.P., Biasoli D., Olcina M.M., Hammond E.M. Measuring DNA Replication in Hypoxic Conditions. Adv. Exp. Med. Biol. 2016;899:11–25. doi: 10.1007/978-3-319-26666-4_2. [DOI] [PubMed] [Google Scholar]
- Fraternali F., Van Gunsteren W.F. An efficient mean solvation force model for use in molecular dynamics simulations of proteins in aqueous solution. J. Mol. Biol. 1996;256:939–948. doi: 10.1006/jmbi.1996.0139. [DOI] [PubMed] [Google Scholar]
- Fukuda R., Zhang H., Kim J.W., Shimoda L., Dang C.V., Semenza G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis V.G., Vassiliou L.V., Karakaidos P., Zacharatos P., Kotsinas A., Liloglou T., Venere M., Ditullio R.A., Jr., Kastrinakis N.G., Levy B. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- Graeber T.G., Osmanian C., Jacks T., Housman D.E., Koch C.J., Lowe S.W., Giaccia A.J. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. 1996;379:88–91. doi: 10.1038/379088a0. [DOI] [PubMed] [Google Scholar]
- Halazonetis T.D., Gorgoulis V.G., Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- Hammond E.M., Denko N.C., Dorie M.J., Abraham R.T., Giaccia A.J. Hypoxia links ATR and p53 through replication arrest. Mol. Cell. Biol. 2002;22:1834–1843. doi: 10.1128/MCB.22.6.1834-1843.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond E.M., Mandell D.J., Salim A., Krieg A.J., Johnson T.M., Shirazi H.A., Attardi L.D., Giaccia A.J. Genome-wide analysis of p53 under hypoxic conditions. Mol. Cell. Biol. 2006;26:3492–3504. doi: 10.1128/MCB.26.9.3492-3504.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond E.M., Kaufmann M.R., Giaccia A.J. Oxygen sensing and the DNA-damage response. Curr. Opin. Cell Biol. 2007;19:680–684. doi: 10.1016/j.ceb.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Hehlmann R., Heimpel H., Hasford J., Kolb H.J., Pralle H., Hossfeld D.K., Queisser W., Löffler H., Heinze B., Georgii A., The German CML Study Group Randomized comparison of busulfan and hydroxyurea in chronic myelogenous leukemia: prolongation of survival by hydroxyurea. Blood. 1993;82:398–407. [PubMed] [Google Scholar]
- Höckel M., Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J. Natl. Cancer Inst. 2001;93:266–276. doi: 10.1093/jnci/93.4.266. [DOI] [PubMed] [Google Scholar]
- Huang M., Parker M.J., Stubbe J. Choosing the right metal: case studies of class I ribonucleotide reductases. J. Biol. Chem. 2014;289:28104–28111. doi: 10.1074/jbc.R114.596684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbi M.E., Kshitiz, Gilkes D.M., Rey S., Wong C.C., Luo W., Kim D.H., Dang C.V., Levchenko A., Semenza G.L. A nontranscriptional role for HIF-1α as a direct inhibitor of DNA replication. Sci. Signal. 2013;6:ra10. doi: 10.1126/scisignal.2003417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926. [Google Scholar]
- Jørgensen C.L., Ejlertsen B., Bjerre K.D., Balslev E., Nielsen D.L., Nielsen K.V. Gene aberrations of RRM1 and RRM2B and outcome of advanced breast cancer after treatment with docetaxel with or without gemcitabine. BMC Cancer. 2013;13:541. doi: 10.1186/1471-2407-13-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhasz A., Vassilakos A., Chew H.K., Gandara D., Yen Y. Analysis of ribonucleotide reductase M2 mRNA levels in patient samples after GTI-2040 antisense drug treatment. Oncol. Rep. 2006;15:1299–1304. [PubMed] [Google Scholar]
- Kolberg M., Strand K.R., Graff P., Andersson K.K. Structure, function, and mechanism of ribonucleotide reductases. Biochim. Biophys. Acta. 2004;1699:1–34. doi: 10.1016/j.bbapap.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Langevin P. On the theory of Brownian motion. C.R. Acad Sci. 1908;146:530–533. [Google Scholar]
- Lee Y., Vassilakos A., Feng N., Lam V., Xie H., Wang M., Jin H., Xiong K., Liu C., Wright J., Young A. GTI-2040, an antisense agent targeting the small subunit component (R2) of human ribonucleotide reductase, shows potent antitumor activity against a variety of tumors. Cancer Res. 2003;63:2802–2811. [PubMed] [Google Scholar]
- Leighl N.B., Laurie S.A., Chen X.E., Ellis P., Shepherd F.A., Knox J.J., Goss G., Burkes R.L., Pond G.R., Dick C. A phase I/II study of GTI-2040 plus docetaxel as second-line treatment in advanced non-small cell lung cancer: a study of the PMH phase II consortium. J. Thorac. Oncol. 2009;4:1163–1169. doi: 10.1097/JTO.0b013e3181a949b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leszczynska K.B., Foskolou I.P., Abraham A.G., Anbalagan S., Tellier C., Haider S., Span P.N., O’Neill E.E., Buffa F.M., Hammond E.M. Hypoxia-induced p53 modulates both apoptosis and radiosensitivity via AKT. J. Clin. Invest. 2015;125:2385–2398. doi: 10.1172/JCI80402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leszczynska K.B., Göttgens E.L., Biasoli D., Olcina M.M., Ient J., Anbalagan S., Bernhardt S., Giaccia A.J., Hammond E.M. Mechanisms and consequences of ATMIN repression in hypoxic conditions: roles for p53 and HIF-1. Sci. Rep. 2016;6:21698. doi: 10.1038/srep21698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin V.A. The place of hydroxyurea in the treatment of primary brain tumors. Semin. Oncol. 1992;19(3 Suppl 9):34–39. [PubMed] [Google Scholar]
- Li B., Qiu B., Lee D.S., Walton Z.E., Ochocki J.D., Mathew L.K., Mancuso A., Gade T.P., Keith B., Nissim I., Simon M.C. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature. 2014;513:251–255. doi: 10.1038/nature13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin D., Berggren G., Logan D.T., Sjöberg B.M. The origin and evolution of ribonucleotide reduction. Life (Basel) 2015;5:604–636. doi: 10.3390/life5010604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheret M., Halazonetis T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015;10:425–448. doi: 10.1146/annurev-pathol-012414-040424. [DOI] [PubMed] [Google Scholar]
- Manegold C., Zatloukal P., Krejcy K., Blatter J. Gemcitabine in non-small cell lung cancer (NSCLC) Invest. New Drugs. 2000;18:29–42. doi: 10.1023/a:1006327729228. [DOI] [PubMed] [Google Scholar]
- Miyamoto S., Kollman P.A. SETTLE: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992;13:952–962. [Google Scholar]
- Mizuno H., Kitada K., Nakai K., Sarai A. PrognoScan: a new database for meta-analysis of the prognostic value of genes. BMC Med. Genomics. 2009;2:18. doi: 10.1186/1755-8794-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordlund P., Reichard P. Ribonucleotide reductases. Annu. Rev. Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- Nosé S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984;52:255–268. [Google Scholar]
- Nosé S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984;81:511–519. [Google Scholar]
- Nutting C.M., van Herpen C.M., Miah A.B., Bhide S.A., Machiels J.P., Buter J., Kelly C., de Raucourt D., Harrington K.J. Phase II study of 3-AP Triapine in patients with recurrent or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2009;20:1275–1279. doi: 10.1093/annonc/mdn775. [DOI] [PubMed] [Google Scholar]
- Ochiai E., Mann G.J., Gräslund A., Thelander L. Tyrosyl free radical formation in the small subunit of mouse ribonucleotide reductase. J. Biol. Chem. 1990;265:15758–15761. [PubMed] [Google Scholar]
- Oh Y.K., Park T.G. siRNA delivery systems for cancer treatment. Adv. Drug Deliv. Rev. 2009;61:850–862. doi: 10.1016/j.addr.2009.04.018. [DOI] [PubMed] [Google Scholar]
- Olcina M.M., Foskolou I.P., Anbalagan S., Senra J.M., Pires I.M., Jiang Y., Ryan A.J., Hammond E.M. Replication stress and chromatin context link ATM activation to a role in DNA replication. Mol. Cell. 2013;52:758–766. doi: 10.1016/j.molcel.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kalé L., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao C., Jin M., Kim H.B., Lee S.M., Amatya P.N., Hyun J.W., Chang I.Y., You H.J. Ribonucleotide reductase small subunit p53R2 suppresses MEK-ERK activity by binding to ERK kinase 2. Oncogene. 2009;28:2173–2184. doi: 10.1038/onc.2009.84. [DOI] [PubMed] [Google Scholar]
- Pires I.M., Bencokova Z., Milani M., Folkes L.K., Li J.L., Stratford M.R., Harris A.L., Hammond E.M. Effects of acute versus chronic hypoxia on DNA damage responses and genomic instability. Cancer Res. 2010;70:925–935. doi: 10.1158/0008-5472.CAN-09-2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontarin G., Ferraro P., Bee L., Reichard P., Bianchi V. Mammalian ribonucleotide reductase subunit p53R2 is required for mitochondrial DNA replication and DNA repair in quiescent cells. Proc. Natl. Acad. Sci. USA. 2012;109:13302–13307. doi: 10.1073/pnas.1211289109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst H., Schiffer H., Gekeler V., Scheffler K. Oxygen dependent regulation of mammalian ribonucleotide reductase in vivo and possible significance for replicon initiation. Biochem. Biophys. Res. Commun. 1989;163:334–340. doi: 10.1016/0006-291x(89)92140-2. [DOI] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichard P. Interactions between deoxyribonucleotide and DNA synthesis. Annu. Rev. Biochem. 1988;57:349–374. doi: 10.1146/annurev.bi.57.070188.002025. [DOI] [PubMed] [Google Scholar]
- Reichard P. From RNA to DNA, why so many ribonucleotide reductases? Science. 1993;260:1773–1777. doi: 10.1126/science.8511586. [DOI] [PubMed] [Google Scholar]
- Sandvik G.K., Tomter A.B., Bergan J., Zoppellaro G., Barra A.L., Røhr A.K., Kolberg M., Ellefsen S., Andersson K.K., Nilsson G.E. Studies of ribonucleotide reductase in crucian carp-an oxygen dependent enzyme in an anoxia tolerant vertebrate. PLoS ONE. 2012;7:e42784. doi: 10.1371/journal.pone.0042784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao J., Zhou B., Zhu L., Qiu W., Yuan Y.C., Xi B., Yen Y. In vitro characterization of enzymatic properties and inhibition of the p53R2 subunit of human ribonucleotide reductase. Cancer Res. 2004;64:1–6. doi: 10.1158/0008-5472.can-03-3048. [DOI] [PubMed] [Google Scholar]
- Sherman P.A., Fyfe J.A. Enzymatic assay for deoxyribonucleoside triphosphates using synthetic oligonucleotides as template primers. Anal. Biochem. 1989;180:222–226. doi: 10.1016/0003-2697(89)90420-x. [DOI] [PubMed] [Google Scholar]
- Smith P., Zhou B., Ho N., Yuan Y.C., Su L., Tsai S.C., Yen Y. 2.6 A X-ray crystal structure of human p53R2, a p53-inducible ribonucleotide reductase. Biochemistry. 2009;48:11134–11141. doi: 10.1021/bi9001425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.J., Deane N.G., Wu F., Merchant N.B., Zhang B., Jiang A., Lu P., Johnson J.C., Schmidt C., Bailey C.E. Experimentally derived metastasis gene expression profile predicts recurrence and death in patients with colon cancer. Gastroenterology. 2010;138:958–968. doi: 10.1053/j.gastro.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterkers Y., Preudhomme C., Laï J.L., Demory J.L., Caulier M.T., Wattel E., Bordessoule D., Bauters F., Fenaux P. Acute myeloid leukemia and myelodysplastic syndromes following essential thrombocythemia treated with hydroxyurea: high proportion of cases with 17p deletion. Blood. 1998;91:616–622. [PubMed] [Google Scholar]
- Stubbe J. Ribonucleotide reductases in the twenty-first century. Proc. Natl. Acad. Sci. USA. 1998;95:2723–2724. doi: 10.1073/pnas.95.6.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbe J. Di-iron-tyrosyl radical ribonucleotide reductases. Curr. Opin. Chem. Biol. 2003;7:183–188. doi: 10.1016/s1367-5931(03)00025-5. [DOI] [PubMed] [Google Scholar]
- Stubbe J., Nocera D.G., Yee C.S., Chang M.C. Radical initiation in the class I ribonucleotide reductase: long-range proton-coupled electron transfer? Chem. Rev. 2003;103:2167–2201. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- Tanaka H., Arakawa H., Yamaguchi T., Shiraishi K., Fukuda S., Matsui K., Takei Y., Nakamura Y. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature. 2000;404:42–49. doi: 10.1038/35003506. [DOI] [PubMed] [Google Scholar]
- Tanaka T., Ohkubo S., Tatsuno I., Prives C. hCAS/CSE1L associates with chromatin and regulates expression of select p53 target genes. Cell. 2007;130:638–650. doi: 10.1016/j.cell.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Thelander L., Gräslund A., Thelander M. Continual presence of oxygen and iron required for mammalian ribonucleotide reduction: possible regulation mechanism. Biochem. Biophys. Res. Commun. 1983;110:859–865. doi: 10.1016/0006-291x(83)91040-9. [DOI] [PubMed] [Google Scholar]
- Tuckerman M., Berne B.J., Martyna G.J. Reversible multiple time scale molecular dynamics. J. Chem. Phys. 1992;97:1990–2001. [Google Scholar]
- Uhlin U., Eklund H. Structure of ribonucleotide reductase protein R1. Nature. 1994;370:533–539. doi: 10.1038/370533a0. [DOI] [PubMed] [Google Scholar]
- Van Gunsteren W.F. Methods for calculation of free energies and binding constants: successes and problems. In: van Gunsteren W.F., Weiner P.K., editors. Computer Simulation of Biomolecular Systems: Theoretical and Experimental Applications. Escom; 1989. pp. 27–59. [Google Scholar]
- Verlet L. Computer “experiments” on classical fluids. I. Thermodynamical properties of Lennard-Jones molecules. Phys. Rev. 1967;159:98–103. [Google Scholar]
- Wang P.H., Blumberger J. Mechanistic insight into the blocking of CO diffusion in [NiFe]-hydrogenase mutants through multiscale simulation. Proc. Natl. Acad. Sci. USA. 2012;109:6399–6404. doi: 10.1073/pnas.1121176109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Lohman G.J., Stubbe J. Enhanced subunit interactions with gemcitabine-5′-diphosphate inhibit ribonucleotide reductases. Proc. Natl. Acad. Sci. USA. 2007;104:14324–14329. doi: 10.1073/pnas.0706803104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Lohman G.J., Stubbe J. Mechanism of inactivation of human ribonucleotide reductase with p53R2 by gemcitabine 5′-diphosphate. Biochemistry. 2009;48:11612–11621. doi: 10.1021/bi901588z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Zhenchuk A., Wiman K.G., Albertioni F. Regulation of p53R2 and its role as potential target for cancer therapy. Cancer Lett. 2009;276:1–7. doi: 10.1016/j.canlet.2008.07.019. [DOI] [PubMed] [Google Scholar]
- Wang P.-H., Best R.B., Blumberger J. A microscopic model for gas diffusion dynamics in a [NiFe]-hydrogenase. Phys. Chem. Chem. Phys. 2011;13:7708–7719. doi: 10.1039/c0cp02098b. [DOI] [PubMed] [Google Scholar]
- Wang P.-H., Bruschi M., De Gioia L., Blumberger J. Uncovering a dynamically formed substrate access tunnel in carbon monoxide dehydrogenase/acetyl-CoA synthase. J. Am. Chem. Soc. 2013;135:9493–9502. doi: 10.1021/ja403110s. [DOI] [PubMed] [Google Scholar]
- Xue L., Zhou B., Liu X., Wang T., Shih J., Qi C., Heung Y., Yen Y. Structurally dependent redox property of ribonucleotide reductase subunit p53R2. Cancer Res. 2006;66:1900–1905. doi: 10.1158/0008-5472.CAN-05-2656. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Li H., Zhang C., An X., Liu L., Stubbe J., Huang M. Conserved electron donor complex Dre2-Tah18 is required for ribonucleotide reductase metallocofactor assembly and DNA synthesis. Proc. Natl. Acad. Sci. USA. 2014;111:E1695–E1704. doi: 10.1073/pnas.1405204111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B., Liu X., Mo X., Xue L., Darwish D., Qiu W., Shih J., Hwu E.B., Luh F., Yen Y. The human ribonucleotide reductase subunit hRRM2 complements p53R2 in response to UV-induced DNA repair in cells with mutant p53. Cancer Res. 2003;63:6583–6594. [PubMed] [Google Scholar]
- Zhou B., Su L., Yuan Y.C., Un F., Wang N., Patel M., Xi B., Hu S., Yen Y. Structural basis on the dityrosyl-diiron radical cluster and the functional differences of human ribonucleotide reductase small subunits hp53R2 and hRRM2. Mol. Cancer Ther. 2010;9:1669–1679. doi: 10.1158/1535-7163.MCT-10-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.