

Abstract
Approximately 90% of all cancer deaths arise from the metastatic dissemination of primary tumors. Metastasis is the most lethal attribute of colorectal cancer. New data regarding the molecules contributing to the metastatic phenotype, the pathways they control and the genes they regulate are very important for understanding the processes of metastasis prognosis and prevention in the clinic. The purpose of this study was to investigate the role of T-LAK cell-originated protein kinase (TOPK) in the promotion of colorectal cancer metastasis. TOPK is highly expressed in human metastatic colorectal cancer tissue compared with malignant adenocarcinoma. We identified p53-related protein kinase (PRPK) as a new substrate of TOPK. TOPK binds with and phosphorylates PRPK at Ser250 in vitro and ex vivo. This site plays a critical role in the function of PRPK. Cell lines stably expressing mutant PRPK (S250A), knockdown TOPK, knockdown PRPK or knockdown of both TOPK and PRPK significantly inhibited liver metastasis of human HCT116 colon cancer cells in a xenograft mouse model. Therefore, we conclude that TOPK directly promotes metastasis of colorectal cancer by modulating PRPK. Thus, these findings may assist in the prediction of prognosis or development of new therapeutic strategies against colon cancer.
Abbreviations: MAPKK, mitogen-activated protein kinase; MAPK, mitogen-activated protein kinase; MEK, mitogen-activated protein kinase; ERK1/2, extracellular signal-regulated kinase; RSK2, ribosomal S6 kinase 2; MSK1, mitogen and stress activated kinase 1; JNK1/2, c-Jun N-terminal kinase 1/2; Cdk1/cyclin B, cyclin dependent kinase 1/cyclin B1; SOS, Son of Sevenless; EGF, epidermal growth factor; CRC, colorectal cancer
Keywords: Kinase, Phosphorylation, TOPK, PRPK, Metastasis
Highlights
-
•
Knockdown of TOPK or PRPK significantly inhibits liver metastasis of human colon cancer cells in vivo.
-
•
PRPK is a newly identified substrate of TOPK and TOPK, but not Akt, phosphorylates PRPK at Ser250.
-
•
Phosphorylation Ser250 is important for the function of PRPK in promoting liver metastasis of colon cancer cells in vivo.
Colorectal cancer (CRC) is one of the most prevalent cancers worldwide. Identifying new mechanisms contributing to metastasis of colon cancer cells to the liver is important for finding novel targets for chemotherapy. T-LAK-cell-originated protein kinase (TOPK) is a valuable prognostic marker in patients with sporadic CRC and 30–40% of CRC patients may benefit from the inhibition of TOPK. We identified p53-related protein kinase (PRPK) as a novel substrate for TOPK. The TOPK/PRPK signaling axis is directly involved in colon metastasis to the liver and its inhibition could lead to effective anti-metastatic therapies in patients with colon cancer.
1. Introduction
Metastasis is defined as the formation of secondary tumor foci in one or more organs at a distance from the primary lesion (Nguyen and Massague, 2007). Although the precise molecular events leading to the acquisition of the metastatic phenotype remain largely unclear, the coordination and cooperation of several signal transduction pathways in metastasis has been suggested. Examples of these pathways include the mitogen-activated protein kinase (MAPK) pathway (Krueger et al., 2001), focal adhesion kinase (FAK) pathway (Sieg et al., 2000), Src pathway (Yeatman, 2004) and the Akt pathway (Irie et al., 2005). The T-LAK-cell-originated protein kinase (TOPK) is a serine-threonine kinase member of the MAPKK family. We have studied TOPK function over the last few years and have shown that TOPK is involved in many cellular processes, including tumor development, cell growth, apoptosis and inflammation (Zykova et al., 2006, Oh et al., 2007, Zhu et al., 2007, Zykova et al., 2010, Li et al., 2011). We first discovered that TOPK serves as an oncogenic MEK that exerts positive feedback on ERK2 to promote colorectal cancer formation in vitro and in vivo (Zhu et al., 2007). Interest in the function of TOPK as an oncogene and in the development of new inhibitors of TOPK has dramatically increased (Vishchuk et al., 2016, Xiao et al., 2016, Zeng et al., 2016). However, a clear mechanism explaining how TOPK regulates the process of colon cancer metastasis to the liver has not yet been elucidated. In this study, we investigated the role of TOPK in colon cancer metastasis to the liver and identified the p53-related protein kinase (PRPK) as a novel substrate of TOPK.
PRPK was first cloned from an interleukin-2-activated cytotoxic T-cell subtraction library and was shown to up-regulate the transcriptional activity of p53 when transfected into COS-7 cells. Thus the protein was named “p53-related protein kinase” and the authors suggested that PRPK might play an important role in cell cycle or apoptosis (Abe et al., 2001). Later these same authors concluded that they could not rule out the possibility that PRPK did not directly phosphorylate p53 due to the fact that binding and phosphorylation p53 at Ser15 was shown in the presence of an activating COS-7 cell lysate, suggesting that the phosphorylation status of p53 is regulated not only by PRPK, but also by other kinases (Abe et al., 2006). The p53 protein also remains phosphorylated on Ser15 even after depletion of PRPK, suggesting that this is not the major role of PRPK in proliferating cells (Peterson et al., 2010). Human PRPK is a homolog to the yeast kinase piD261/Bud32 (Bud32) and PRPK can partially complement Bud32 deficiency (Facchin et al., 2003). PRPK can be activated and provides a functional link between this kinase and the Akt signaling pathway (Facchin et al., 2007). However, the biological function of PRPK remains elusive. Herein we showed that TOPK is involved in colorectal cancer metastasis to the liver through its phosphorylation of PRPK at Ser250.
2. Materials and Methods
2.1. Cell Culture
Human HCT116, HT29, HCT15, DLD1, WiDr colon cancer cells or CCD-18Co normal colon cells were from America Type Culture Collection (ATCC, Manassas, VA). The Lim1215 human colorectal cancer cell line was a gift from Dr. Robert H. Whitehead (Vanderbilt University, Nashville, TN) (Whitehead et al., 1985). Сells were purchased from ATCC between years 2009 and 2015. ATCC tests these cells by isoenzyme analysis to confirm human origin, DNA fingerprinting analysis of cell line-specific polymorphic markers, growth curve analysis to check doubling times, microscope-based morphology check and mycoplasma detection. All cell lines were matched with their identities and mycoplasma-free. Cells were maintained according to the ATCC instructions before being frozen. Each vial of frozen cells was thawed and maintained for a maximum of 8 weeks. HCT116 cells were cultured in McCoy's 5A medium. HT29 and HCT15 cells were cultured in DMEM/high glucose and DLD1 cells were cultured in RPMII-1649 medium. WiDr and CCD-18Co cells were cultured in MEM. All media were from Thermo Scientific Hyclone Laboratories, Inc. (Logan, UT) with 10% fetal bovine serum (FBS), 2 mM l-glutamine, and 25 μM/ml gentamicin. The medium for culturing Lim1215 cells contained HEPES (25 mM), insulin (0.6 μg/ml), hydrocortisone (1 μg/ml) and 1-thioglycerol (10 μM). Cells were grown in monolayers at 37 °C in a 5% CO2 incubator.
2.2. Antibodies and Reagents
The PBK/TOPK (Cat: 4942) and phosphor-PBK/TOPK (Thr9) (Cat# 4941) antibodies were from Cell Signaling Technology, Inc. (Beverly, MA). Antibodies to detect PRPK (F-9) (Cat# sc-100350), HA (F7) (Cat# sc-7392) and β-actin (C4) (Cat# sc-47778) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-V5 (Cat# R960-25) was from Invitrogen (Carlsbad, CA) and the GST-PRPK full-length recombinant protein (Cat# H00112858-P01) was from Novus Biologicals (Littleton, CO). Anti-Flag (Cat# F3165) was from Sigma (St Louis, MO). The Ki67 antibody (Clone SP-6) (Cat# RM-9106) and Mitomycin C (Cat# 32-581-0) were from Thermo Fisher Scientific (Waltham, MA) and the synthesized PRPK peptides were from Peptide 2.0 (Chantilly, VA). The active kinases ERK1 (Cat# 14-439), ERK2 (Cat# 14-550), RSK2 (Cat# 14-480), MEK1 (Cat# 14-429), JNK1 (Cat# 14-327), JNK2 (Cat# 14-329), MSK1 (Cat# 14-548), Akt1 (Cat# 14-276) or Akt2 (Cat# 14-339), and the H2B recombinant protein (Cat# 14-491) were from Millipore (Billerica, MA, USA) and active TOPK (Cat# T14-10G) was from SignalChem (Richmond, BC, Canada). The pQE-81L-PRPK plasmid for purification of the His-PRPK protein was a gift from Lorenzo A. Pinna (Facchin et al., 2003) and the TOPKT9A plasmid for purification of His-TOPK mutant was a gift from Dr. Y. Abe (Matsumoto et al., 2004). The epidermal growth factor (EGF) (Cat# 354001) was from BD Biosciences (Bedford, MA). The protein A/G plus-agarose immunoprecipitation reagent was from Santa Cruz Biotechnology, Inc. (Cat# sc-2003) and the JetPEI transfection reagent (Cat# GDSP10110) was from (Qbiogene, Inc., Montreal, Quebec, Canada).
2.3. Construction of Plasmids
To construct the PRPK overexpressing plasmids, the PRPK coding region was amplified by PCR using two primers (5′-GACGACGACAAGATGGCGGCGGCCAGAGCTACTACG-3′ as the sense primer and 5′-GAGGAGAAGCCCCCCAACCATGGACCTCTTTCTTCC-3′ as the anti-sense primer) for the pCMV-Sport6-PRPK plasmid (Thermo Scientific, Inc., Huntsville, AL) followed by their introduction into the pET46Ek/LIC and pcDNA3/V5-HisA vectors, respectively. The topk-T9A gene (a gift from Dr. Y. Abe, Department of Pathology, and Division of Molecular Pathology, Ehime University School of Medicine, Ehime, Japan) was amplified by PCR and then cloned into pET-46 using a pET-46 Ek/LIC kit (Novagen, USA). The PRPKS250A or TOPKT9A mutant was generated using the QuikChange Lightning Site-directed Mutagenesis kit (Stratagene, La Jolla, CA). All recombinant plasmids that were constructed were confirmed by restriction mapping and DNA sequencing.
2.4. Lentiviral Infection
Two pLKO.1-shTOPK vectors from Addgene Inc. (Cambridge, MA) were used for preparing HCT116 cells stably expressing knockdown TOPK (shTOPK/HCT116) as previously reported (Kim et al., 2012). Cells were examined for TOPK expression. To prepare viral particles, the pLKO.1 viral vector (for shMock cells) or pLKO.1-shTOPK viral vectors (for shTOPK cells) or pLKO.1-shPRPK (for shPRPK cells) with packaging vectors pMD2.0G and psPAX from Addgene Inc. (Cambridge, MA) were transfected into HEK293T cells (7 × 105/60-mm dish) using JetPEI transfection reagent following the manufacturer's suggested protocols. The transfection mix in 10% FBS/DMEM without antibiotics was incubated with cells for 12 h. Then the medium was replaced with 5 ml of fresh 10% FBS/DMEM with antibiotics (penicillin/streptomycin). The viral particles from cells were harvested after 24 or 48 h (10 ml total volume each) and were kept at − 80 °C. Viral particles (1 ml) and normal medium (9 ml) with 8 μg/ml of polybrane (Millipore, Billerica, MA) were mixed and infected into 60% confluent HCT116 cells overnight. The cell culture medium was replaced with fresh complete growth medium and incubated 24 h and then cells were selected with 1.5 μg/ml puromycin (A. G. Scientific, Inc., San Diego, CA) for 36 h. The selected cells were used for examination of PRPK expression. To prepare TOPK and PRPK double knockdown stable HCT116 cells (shPRPK/shTOPK/HCT116), PRPK-viral particles for pLKO.1-shPRPK were infected into 60% confluent shTOPK/HCT116 cells overnight. The cell culture medium was replaced with fresh complete growth medium and cells were used for experiments without selection with puromycin.
2.5. Bacterial Expression and Purification of Fusion Proteins
His-tagged proteins, including pQE81L-PRPK, pET46-WtPRPK, pET46-MutPRPK (S250A), were expressed in BL21 (DE3) bacteria (Novagen, USA) and purified as described for purification of peroxiredoxin 1 (Prx1) (Zykova et al., 2010). The His-TOPKT9A (pET46-TOPKT9A) fusion protein was expressed in BL21 (DE3) bacteria (Novagen, USA). Bacteria were grown at 37 °C to an absorbance of 0.9–1.0 at 600 nm and induced with 0.5 mM IPTG overnight at 15 °C and harvested by centrifugation. Cell pellets were suspended in 50 mM Tris (pH 8.0) lysis buffer containing 200 mM NaCl and 10 mM imidazole. After sonication and centrifugation, the supernatant fraction was incubated with Ni-NTA-agarose beads (Qiagen, Germany) overnight at 4 °C. Beads were washed with lysis buffer and PBS and His-proteins eluted with 100 mM imidazole.
2.6. In Vitro Kinase Assay
A purified fusion pQE81L-PRPK (His) protein was used as a substrate for in vitro kinase assays with active kinases (ERK1, ERK2, RSK2, MEK1, MSK1, JNK1, JNK2, TOPK, Akt1 or Akt2) or with the pET46TOPKT9A (His) protein as the inactive TOPK. The synthesized PRPK peptides, pET46-WtPRPK, and pET46-MutPRPK were used as substrates for active TOPK. After SDS-PAGE, samples with or without [γ-32P] ATP were visualized by autoradiography or Western blotting with a specific antibody, respectively.
2.7. Phosphorylated PRPK (Ser250) Antibody Preparation
The phosphor-PRPK (Ser250) antibody (anti-rabbit) was prepared by BioSynthesis, Inc. (Lewisville, TX). The antiserum was made using a synthesized 16-mer phosphopeptide consisting of an N-terminal cysteine (for coupling) and the sequence CLDEVRLRGRKR[pS]MVG.
2.8. Western Blotting and Immunoprecipitation
Western blotting and immunoprecipitation methods with HEK293 cells or HCT116 cells were conducted as reported previously (Zhu et al., 2012).
2.9. IHC Analyses for Tissue Array and Samples of Livers From Athymic Nude Mice
The colon cancer tissue array (C0702) with malignant adenocarcinoma (30 cases, 43%), metastatic adenocarcinoma (30 cases, 43%) and adjacent normal tissues (10 cases, 14%) including TNM and pathology grade, were from US Biomax Inc. (Rockville, MD). Livers from mice were extracted and fixed for 24 h in 10% (v/v) neutral-buffered formalin and then transferred into 70% ethanol for H&E and IHC staining. The fixed tissues were dehydrated in ascending grades of ethanol and xylene, and then embedded in paraffin wax. Sections (4 μm) were cut with a microtome and mounted on Superfrost®/plus microscope slides.
IHC was performed on the tissue array using antibodies to detect TOPK (1:100) and samples of livers from athymic nude mice for H&E staining and stained with a Ki67 antibody (1:150) as a proliferation marker. The biotinylated secondary antibody was rabbit anti-mouse IgG (1:200, in 10% normal rabbit serum; Vector Laboratories). The slides were developed in diaminobenzidine (DAB) and counter stained with hematoxylin. The stained slides were dehydrated and mounted in permount. Images were captured and analyzed using the Image-Pro-Plus 6.2 software program.
2.10. Migration Assay and Cell Invasion
For the wound closure assay, cells were plated in 35-mm culture dishes and grown to 80% confluence and treated with mitomycin C (10 μg/ml) for 2 h to inhibit proliferation (Rahman-Roblick et al., 2007) and then used for a wound-healing assay in normal medium. A single scratch was made by pulling a plastic tip across the cells. Photographs were taken immediately after scratching and 18 h later for WtPRPK and MutPRPK or 4 days later for knockdown cells using a microscope and a Casio Digital Camera (EX-Z1000 with 10.1 megapixels). For the Matrigel invasion assay, a MatrigelTM Invasion Chamber (6-well plate) with an 8-micron pore size membrane was used (BD Biosciences, Bedford, MA) following the manufacturer's suggested protocol. Cells that migrated through the membrane were fixed with 100% cold methanol. The membrane was mounted on a glass slide using Fluoro-Gel II with DAPI (Electron Microscopy Sciences, Hatfield, PA) (Mehrotra et al., 2010) and visualized by confocal microscope (NIKON C1si Confocal Spectral Imaging System; magnification 60 ×). Invading cells were counted from five random fields in two membranes.
2.11. Liver Metastasis Model
All animal experiments were performed using a protocol approved by the University of Minnesota Institutional Animal Care and Use Committee (IACUC). Athymic nude mice (5–6 weeks old; female and male) for a xenograft mouse model were from Harlan TekLAD (Madison, WI). Mice were anesthetized using an i.p. injection of a ketamine hydrochloride and xylazine hydrochloride solution (Sigma). Under aseptic conditions, a small longitudinal incision was made in the left upper flank to visualize the spleen, and cells (0.5 × 106 each) expressing shMock, shTOPK, shPRPK, shPRPK/shTOPK, WtPRPK or MutPRPK in 25 μl of PBS were injected under the spleen capsule using a 27-gauge needle. Animals injected with shMock, shTOPK, shPRPK and shPRPK/shTOPK cells were sacrificed at 10 days or at 7 days for mice injected with Mock, WtPRPK and MutPRPK cells. Livers were isolated at that time. After photographs were taken, livers were fixed in 10% buffered formalin and paraffin-embedded. Tumors were enumerated by visual inspection and liver sections were stained with H&E. Livers were also subjected to IHC using an antibody to detect Ki67, a proliferation marker.
2.12. Statistical Analysis
All quantitative results are expressed as mean values ± S.D. Statistically significant differences were obtained using the Student's t-test or one-way ANOVA. A p value of < 0.05 was considered to be statistically significant.
3. Results
3.1. TOPK Binds With and Phosphorylates PRPK at Ser250 In Vitro
We used His-tagged PRPK as a substrate and screened several common kinases to identify a kinase that could phosphorylate PRPK. The phosphorylation was visualized by autoradiography in the presence of [γ-32P] ATP and results indicated that TOPK phosphorylates PRPK (Fig. 1a, left). TOPK is phosphorylated at Thr9 by Cdk1/cyclin B (Abe et al., 2007, Matsumoto et al., 2004) and phosphorylation is required for its kinase activity (Gaudet et al., 2000). We purified a TOPKT9A fusion protein and an in vitro kinase assay indicated that PRPK was phosphorylated by active TOPK (Fig. 1a, right). Akt has been reported to phosphorylate PRPK at Ser250 (Facchin et al., 2007). We examined the phosphorylation of PRPK by TOPK or Akt (Fig. 1b). Unexpectedly, the in vitro kinase assay results indicated that neither Akt1 nor Akt2 could phosphorylate PRPK in vitro. Phosphorylation of histone H2B was used as a positive control for Akt1 and Akt2. Next, we co-transfected the pcDNA3-HA-TOPK and pcDNA3.1-V5-PRPK plasmids into HEK293 cells and immunoprecipitated the tagged proteins with anti-V5 and then probed with anti-HA. The results indicated that TOPK co-immunoprecipitated with PRPK after overexpression in HEK293 cells (Fig. 1c). Conversely, endogenous TOPK was immunoprecipitated from HCT116 cells and PRPK was detected by Western blot with a specific antibody. This result also indicated that TOPK could co-immunoprecipitate with PRPK in HCT116 cells (Fig. 1d).
Fig. 1.

TOPK binds with and phosphorylates PRPK at Ser250 in vitro. (a) The pQE-81L-PRPK (His) fusion protein was used as a substrate for an in vitro kinase assay with active protein kinases as shown (left panel) and for His-TOPKT9A fusion protein as inactive compare with TOPK, active (right panel). (b) Phosphorylation of PRPK by TOPK or Akt1/2 was compared using in vitro kinase assays and histone H2B was used as positive control for Akt1/2. (c) TOPK binds with PRPK in HEK293 cells after transient transfection. The pcDNA3-HA-TOPK and pcDNA3-V5-PRPK plasmids were co-transfected into HEK 293 cells, immunoprecipitated with anti-V5 and then probed with anti-HA. (d) TOPK binds with PRPK in HCT116 colon cancer cells. Endogenous TOPK was immunoprecipitated from HCT116 cells and then probed with anti-PRPK. (e) Five PRPK peptides were designed for an in vitro kinase assay with active TOPK. (f) For in vitro kinase assays, a wildtype His-PRPK (Wt) or His-PRPK-S250A mutant (S250A) protein was used as a substrate for active TOPK. For (a), (b), (e) or (f, upper panel), reactive products were resolved by SDS-PAGE and visualized by autoradiography. Reactive products for (f, lower panel) were resolved by SDS-PAGE and visualized by Western blot with specific antibodies.
Because TOPK is a serine/threonine kinase, we examined the potential phosphorylation of serine and threonine residues in PRPK using NetPhos 2.0 (Diella et al., 2004) and then synthesized 5 peptides (Fig. 1e, bottom) with the highest predicted phosphorylation scores (Supplementary Table 1). The results of the in vitro kinase assays using P1-P5 peptides with active TOPK and [γ-32P] ATP showed that Ser250 was consistently and strongly phosphorylated by TOPK (Fig. 1e, P3). To confirm the results of the peptide mapping, we conducted an in vitro kinase assay using wildtype His-PRPK (Wt) and a His-PRPK-S250A mutant (S250A) in the presence of active TOPK and [γ-32P] ATP. The results indicated that mutation of Ser250 to alanine abrogated the phosphorylation of PRPK by TOPK (Fig. 1f, upper). This result confirmed that TOPK phosphorylates PRPK at Ser250 only. The antibody to detect phosphorylated PRPK (Ser250) was prepared as described in Materials and Methods. Western blot analysis with this antibody confirmed that TOPK phosphorylated PRPK at Ser250 (Fig. 1e, bottom). Overall, these results indicated that TOPK binds with PRPK and phosphorylates PRPK at Ser250.
3.2. TOPK is Overexpressed in Metastatic Colorectal Tissue and Promotes Migration and Invasion of Cancer Cells
Our earlier study showed that TOPK is highly expressed in human colorectal cancer tissues and colon cancer cells and plays an important role in neoplastic transformation (Zhu et al., 2007). TOPK is highly expressed in human patient metastatic colorectal cancer tissues (Zlobec et al., 2010) and in stage I lung adenocarcinoma with metastatic capability (Wei et al., 2012). Using tissue array analysis, we compared the abundance of TOPK in metastatic colon adenocarcinoma tissues, malignant colon adenocarcinoma tissues and normal colorectal tissue. Results indicated that the abundance of TOPK (Fig. 2a) was significantly higher in metastatic colon adenocarcinoma tissue compared to malignant colon adenocarcinoma tissues (p < 0.02). In contrast, neither protein was highly expressed in normal colorectal tissue.
Fig. 2.

TOPK is overexpressed in metastatic colorectal cancer tissue and human colon cancer cells and promotes migration and invasion of HCT116 colon cancer cells. (a) IHC examination of the expression of TOPK in malignant human colorectal cancer tissue (30 cases, 43%) or metastatic adenocarcinoma tissues (30 cases, 43%) and matching normal colorectal tissue (10 cases, 14%). Photos from one representative case are shown. Data are shown as means ± S.D. The asterisk (*, p < 0.04) indicates a significant increase in TOPK protein expression in malignant colon cancer tissues compared with normal tissues. The asterisks (***, p < 0.02) indicate a significant increase in TOPK protein expression in metastatic cancer tissues compared with malignant cancer tissues. (b) Expression of TOPK, p-TOPK and p-PRPK in different colon cancer cell lines and the CCD-18Co normal colon cell line. (c) Efficiency of TOPK knockdown (shTOPK) in HCT116 colon cancer cells compared with shMock. (d) ShMock/HCT116 cells are compared with shTOPK/HCT116 cells in a wound closure assay. A single scratch was made with a plastic tip across the cells. Photographs were taken immediately after the scratch and 4 days later using a microscope and a Casio Digital Camera (EX-Z1000 with 10.1 megapixels). Experiments were repeated twice. (e) Cell migration was compared in shMock/HCT116 cells or shTOPK/HCT116 cells (1.5 × 105) using a Matrigel invasion chamber. Cells that migrated through the membrane were stained with DAPI and visualized by the NIKON C1si Confocal Spectral Imaging System (magnification 60 ×). Invading cells were counted from five random fields in two membranes. Experiments were repeated at least twice. Asterisks (**) indicate a significant (p < 0.001) decreased migration of shTOPK expressing cells compared with shMock cells.
We examined the abundance of TOPK or p-PRPK in different human colon cancer cell lines and the CCD-18C0 normal colon cell line (Fig. 2b). Results indicated that colon cancer cells with high expression of TOPK show increased PRPK phosphorylation. Based on this result, we chose the HCT116 colon cancer cell line to study the function and relationship of TOPK and PRPK.
To continue investigating the role of TOPK in tumor metastasis, we silenced the expression of TOPK (Fig. 2c) in HCT116 colon cancer cells. We used a migration or wound closure assay to compare cells expressing shTOPK, sequence 1 or 2 (shTOPK#1 or shTOPK#2) with cells expressing shMock. Results of this assay indicated that shTOPK (Fig. 2d) dramatically inhibited migration compared with cells expressing shMock. Cells were also stained with DAPI in a Matrigel invasion assay system and examined after 4 days. These results also showed that cells expressing shTOPK (Fig. 2e) exhibited attenuated invasion through Matrigel inserts compared with shMock cells. Overall, these findings indicated that TOPK is highly expressed in human patient metastatic colorectal cancer tissues and promotes the migration and invasion of HCT116 cancer cells.
3.3. The Phosphorylation of PRPK at Ser250 is Important for the Function of PRPK In Vivo
To investigate the function of the phosphorylation of PRPK at Ser250, we generated HCT116 cells stably overexpressing vector (Mock), wildtype PRPK (WtPRPK) or mutant PRPK-S250A (MutPRPK; Fig. 3a, upper panels). Phosphorylation of PRPK in MutPRPK stable cells was substantially lower than that observed in WtPRPK cells after treatment with EGF (Fig. 3a, upper, right). The findings obtained from a wound closure assay indicated that WtPRPK, but not the MutPRPK, promotes migration of cells after 18 h (Fig. 3a, bottom panels). A Matrigel invasion assay with DAPI staining confirmed that MutPRPK cells exhibited less invasion through the Matrigel inserts compared with WtPRPK cells (Fig. 3b). Overall, these results indicated that overexpression of PRPK promotes migration and invasion of HCT116 colon cancer cells.
Fig. 3.

Mutant PRPK (Ser250Ala) decreases cell migration and invasion of HCT116 colon cancer cells and the Ser250 site of PRPK are important for the function of PRPK in a human colon cancer HCT116 cell xenograft metastasis model. (a, upper left panel) HCT116 cells were transfected with pcDNA3-V5-vector (Mock), pcDNA3-V5-PRPK (WtPRPK) or pcDNA3-V5-PRPK-S250A mutant (MutPRPK) and selected with G418 (200 μM/ml) for 5 days. Expression of PRPK in Mock, WtPRPK, or MutPRPK stable cell lines was detected with an antibody against the V5 tag and β-actin was used to verify equal protein loading. (a, upper right panel) Phosphorylation of PRPK was detected in WtPRPK or MutPRPK stable cell lines after treatment by EGF (20 μg/ml, 15 min). (a, lower panel) Cells stably expressing Mock, Wt-PRPK or Mut-PRPK were subjected to a wound closure assay. Photographs were taken immediately after scratching and 18 h later using a microscope and a Casio Digital Camera (EX-Z1000 with 10.1 megapixels). Experiments were repeated twice. (b) Migration of cells stably expressing Mock, WtPRPK or MutPRPK after 18 h was determined by staining with DAPI and visualized by confocal microscope (NIKON C1si Confocal Spectral Imaging System; magnification 60 ×). Invading cells were counted from five random fields in two membranes and experiments were repeated twice. Asterisks (**) indicate a significant (p < 0.002) decreased migration of MutPRPK cells compared with WtPRPK cells. (c) Quantification of the area of liver metastasis using the ImagePro Plus 6.2 software program. The area of metastasis in livers of mice (6 mice/group) injected with WtPRPK HCT116 cells is significantly increased (***, p = 0.036) compared to livers from mice injected with Mock HCT116 cells. The metastasis (arrows) in the liver and tumor in the spleen (circles) are shown (one representative mouse per group) at 7 days after injection of cells. (d) The area of metastasis in livers from mice injected with MutPRPK HCT116 cells is significantly decreased (***, p = 0.011) compared with the area of metastasis in livers of mice injected with WtPRPK HCT116 cells. Metastasis (arrows) in the liver is shown (one representative mouse per group) at 7 days after injection of cells. (e and f) H&E staining of non-metastatic and metastatic liver tissue (4 μm sections) is shown (100 × magnification).
The liver is the primary site for colorectal carcinoma metastasis and therefore we chose a liver metastasis model to further study the role of PRPK phosphorylation at Ser250 in promoting metastasis. We conducted two sets of experiments for comparison of metastasis from the spleen to the liver. In the first experimental set, we injected Mock (as a negative control) or WtPRPK-transfected HCT116 colorectal cancer cells into the spleen to examine metastasis (Fig. 3c). In the second experimental set, we injected WtPRPK (as a positive control) or MutPRPK HCT116 colorectal cancer cells into the spleen to investigate metastasis (Fig. 3d). After 7 days, livers were isolated and metastasis was evaluated. Livers from mice injected with WtPRPK cells showed a significant amount of metastatic tumor growth compared with livers from mice injected with Mock cells (Fig. 3c). Livers from mice injected with MutPRPK cells showed significantly reduced metastasis compared with livers from mice injected with WtPRPK cells (Fig. 3d). Liver metastases (arrows/circles) are shown (one representative mouse per group). H&E staining was conducted in non-metastatic and metastatic liver tissue (Fig. 3e, f). Overall, these findings further indicate that the Ser250 residue of PRPK plays a critical role in promoting liver metastasis of HCT116 colon cancer cells (see Supplementary Fig. 1).
3.4. TOPK is Required for EGF-induced PRPK Phosphorylation
The phosphorylation of TOPK induced by epidermal growth factor (EGF) facilitates the function of TOPK (Zhu et al., 2007). Therefore the next series of experiments were designed to examine a potential correlation between TOPK and PRPK corresponding with increased TOPK activity. HCT116 colon cancer cells expressing shMock or shTOPK were treated with EGF and phosphorylation of PRPK was detected. The data show that PRPK phosphorylation was induced in a time-dependent manner following EGF treatment and the phosphorylation of PRPK was decreased in shTOPK knockdown cells (Fig. 4a). These data further supported the idea that TOPK is required for EGF-induced PRPK phosphorylation.
Fig. 4.
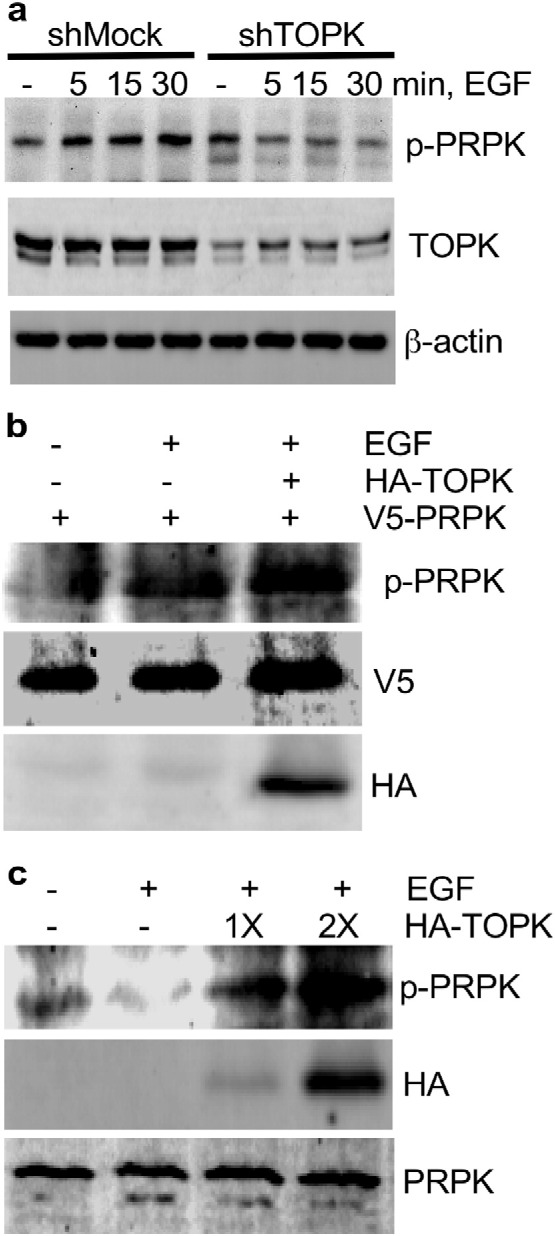
TOPK is required for EGF-induced PRPK phosphorylation. (a) EGF induces a time-dependent phosphorylation of PRPK and phosphorylation of PRPK decreases with depletion of TOPK in HCT116 cells. Total TOPK was used as an internal control for confirmation of TOPK deficiency (shTOPK). β-Actin was used to verify equal protein loading. (b and c) TOPK promotes phosphorylation of PRPK in HEK293 cells after treatment with EGF. The same amount of pcDNA3-HA-TOPK and pcDNA-V5-PRPK (b) and increasing amounts of pcDNA3-HA-TOPK (c) were transiently transfected into HEK293 cells and phosphorylation of PRPK was detected by Western blot analysis with specific antibodies after EGF (20 ng/ml) treatment for 30 min.
The pcDNA3-HA-TOPK plasmid was co-transfected with the pcDNA3.1-V5-PRPK plasmid into HEK293 cells and cells were then stimulated with EGF. Phosphorylation of PRPK was detected by Western blot (Fig. 4b). In addition, when increasing amounts of pcDNA3-HA-TOPK were transfected into HEK293 cells, phosphorylation of PRPK increased dose-dependently (Fig. 4c). All these results demonstrated positive relationships between TOPK and PRPK in cells after EGF treatment.
3.5. Knocking Down TOPK, PRPK or Both Proteins Inhibits Metastasis of Human Colon Cancer In Vivo
We examined the effect of knocking down expression of TOPK, PRPK or both proteins on metastasis of HCT116 cells in a xenograft mouse model. HCT116 colon cancer cells stably expressing shMock, shTOPK, shPRPK or both shTOPK/shPRPK were established by lentiviral infection (Fig. 5a, insert). All 4 cell types were used to further examine the role of TOPK and PRPK in colon cancer cell metastasis in vivo. HCT116 colon cancer cells expressing shMock, shTOPK, shPRPK or shPRPK/shTOPK were injected into the spleen of athymic nude mice. After 10 days, mice were euthanized and livers extracted for evaluation of metastasis. The area of liver metastasis found in mice expressing shTOPK, shPRPK and especially shPRPK/shTOPK was significantly decreased compared to livers from mice expressing shMock (Fig. 5a, top panel). Representative liver metastases (arrows/circles; bottom panel Fig. 5a) are shown (see Supplementary Fig. 2). Liver sections were also stained with H&E (Fig. 5b) and IHC was performed to detect Ki67, a proliferation maker (Fig. 5c). These data confirmed that blocking TOPK or PRPK protein expression significantly reduces liver metastasis of HCT116 colon cancer cells in vivo. Overall, our results clearly demonstrate that the TOPK/PRPK pathway is important for the promotion of metastasis of HCT116 colon cancer cells to the liver.
Fig. 5.

Knockdown of TOPK, PRPK or both inhibits metastasis of human colon cancer HCT116 cells in a xenograft mouse model. (a, insert) Knockdown efficiency of TOPK (shTOPK), PRPK (shPRPK) or both (shPRPK/shTOPK) in HCT116 colon cancer cells before intrasplenic inoculation. (a) Quantification and standardization of the area of liver metastasis was analyzed using the ImagePro Plus 6.2 software program. The area of liver metastasis in mice (5 mice/group) injected with shTOPK, shPRPK, or shPRPK/shTOPK HCT116 cells is significantly decreased compared to mice injected with cells expressing vector (shMock). Tumors in the spleen (circles) and liver metastasis (arrows or circles) are shown (one representative mouse per group) at 10 days after injection of cells. Asterisks (***) indicate a significant (p < 0.002) decreased area of liver metastasis in mice injected with shTOPK or shPRPK cells compared with shMock. The asterisks (****) indicate a significant (p < 0.0005) decreased area of liver metastasis in mice injected with shPRPK/shTOPK cells compared with shMock cells. (b) H&E staining and (c) Ki67 staining as a proliferation maker of non-metastatic and metastatic liver tissue (4 μm section; 100 × magnification).
Supplementary Fig. 2.

Suppressing TOPK and/or PRPK expression decreases metastasis of colon cancer cells to the liver. Cells (0.5 × 106) in 25 μl of PBS were injected using a 27-gauge needle into the spleen capsule of each mouse. Animals injected with HCT116 cells expressing shMock, shTOPK, shPRPK or shTOPK/shPRPK were sacrificed at day 10 (n = 5 mice per group). Metastasis in the liver (circles or arrows) and tumors in the spleen (circles) are shown.
4. Discussion
The metastatic process comprises a complex cascade in which many growth factors are involved. In order to metastasize, a cancer cell must first escape from its contacts with neighboring cells and the extracellular matrix (ECM) (Frisch and Screaton, 2001). They then must enter the circulation, arrest in the microcirculation, invade a different tissue compartment and finally form a new colony (Weigelt and Peterse, 2005). Many growth factors, including EGF (Kim and Muller, 1999), hepatocyte growth factor (Gao and Vande Woude, 2005), and transforming growth factor beta (Bierie and Moses, 2006), are derived from mesenchymal, stromal, and tumor cells. These growth factors activate the MAPK, FAK, Src and Akt signaling pathways, and are implicated in triggering tumor cell migration or invasion (Irie et al., 2005, Long et al., 2010). Activation of the MAPK pathway is a frequent event in tumorigenesis and MAPKs have been implicated in cell adhesion, migration and invasion (Reddy et al., 2003). Among the MAPKs, MAPK kinase (MEK) and extracellular signal-regulated kinases (ERKs) are unique because no substrates for MEK1/2 have been identified other than the ERKs. We identified the T-cell-originated protein kinase (TOPK) as another ERK1/2 kinase (Zhu et al., 2007). MEK1/2 is considered to be excellent targets for the development of inhibitors against the MAPK pathway (Sebolt-Leopold and Herrera, 2004). However, current MEK inhibitors have only been moderately effective (Sebolt-Leopold and Herrera, 2004), possibly because of the existence of a negative feedback loop between ERK2 and MEK1/2 (Eblen et al., 2004) and ERK2 with Ras and SOS, which hinders the inhibitory process (Langlois et al., 1995). Earlier, we reported that TOPK functions as an active form of MEK in cancer tissues and a positive feedback loop existing between TOPK and ERK2 promotes cell transformation (Zhu et al., 2007). Based on the ubiquitous expression of MEK1/2 in normal and tumor tissues and the existing negative feedback between ERK2 and MEK, we propose that TOPK might be a key kinase that regulates metastasis in the MAPK pathway. TOPK promotes cell migration and corresponds with poor prognosis in lung cancer (Shih et al., 2011). Previously reported data revealed that TOPK expression was correlated with clinical outcome in human CRC. The prognostic and predictive value of TOPK in CRC has been reported (Zlobec et al., 2010). TOPK was overexpressed in tumors with a median of 90% positive cell staining compared with 5% staining in normal tissue (p < 0.001). Moreover, TOPK seems to be a valuable prognostic factor because patients with diffuse TOPK expression treated with cetuximab or panitumumab exhibited poor outcome. However, a detailed mechanism explaining how TOPK regulates colon metastasis liver has not been elucidated.
Our gene expression analysis revealed a higher expression of TOPK in metastatic colon adenocarcinomas compared with malignant adenocarcinomas. In this study, we provide evidence showing that TOPK promotes metastasis of colorectal carcinoma, which is mediated through its phosphorylation of PRPK at Ser250. PRPK is also reported to be phosphorylated by Akt at Ser250 (Facchin et al., 2007). Our in vitro kinase assay results clearly showed that PRPK can be phosphorylated by TOPK at Ser250 but not by Akt1 or Akt2. Our results suggest that phosphorylation of PRPK might occur independently of Akt through TOPK. This is the first report to show that PRPK is a substrate of TOPK that has an important role in colon cancer metastasis. TOPK and PRPK promoted liver metastasis of HCT116 human colon cancer cells in a xenograft mouse model. Blocking expression of TOPK, PRPK or both in HCT116 colon cancer cells dramatically reduced the migration and invasion of these cancer cells and metastatic properties in vitro and in vivo. Phosphorylation of PRPK at Ser250 plays an essential role in promoting metastasis. The mutation of Ser250 of PRPK to alanine attenuated PRPK's promotion of metastasis of HCT116 human colon cancer cells to the liver in a xenograft mouse model.
Overall, our results indicated that the TOPK/PRPK pathway promotes metastasis of HCT116 colon cancer cells to the liver of mice. Therefore, TOPK and PRPK are directly involved in metastasis and their inhibition could provide effective anti-metastatic therapies in patients with colon cancer.
The following are the supplementary data related to this article.
Identification of potential phosphorylation sites on PRPK.
Supplementary Fig. 1.

PRPK (Ser250) phosphorylation is important for the function of PRPK. HCT116 colorectal cancer cells transfected with Mock, wildtype (Wt) PRPK (Ser250) or mutant (Mut) PRPK (Ser250Ala) were injected into the spleen of each mouse (n = 6 mice per group). After 7 days, livers and spleens were isolated and metastasis in the liver (circle or arrows) and tumors in the spleen (circle) were evaluated. Livers from mice injected with WtPRPK-transfected cells (middle panel) showed a substantial amount of metastatic tumor growth compared with livers from mice injected with Mock-transfected cells (upper panel). Livers from mice injected with MutPRPK-transfected cells (lower panel) showed markedly reduced metastasis compared with livers from mice injected with WtPRPK cells.
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
ZD designed research; TAZ, FZ, LW, HL, RB, DYL, KY carried out research experiments; TAZ, FZ, LW, RB analyzed data; TAZ and AMB wrote the manuscript.
Acknowledgments
This work was supported by The Hormel Foundation and National Institutes of Health grants CA187027, CA027502, CA196639, and CA16601. We thank Dr. Robert H. Whitehead (Vanderbilt University, Nashville, TN) for gift of the Lim1215 human colorectal cancer cell line, Dr. Lorenzo A. Pinna (Universita di Padova, Padova, Italy) for gift of the pQE-81L-PRPK plasmid for purification of the His-PRPK protein and Dr. Yasuhito Abe (Department of Pathology, and Division of Molecular Pathology, Ehime University School of Medicine, Ehime, Japan) for the gift of the TOPK-T9A plasmid for purification of the His-PRPKT9A mutant protein.
References
- Abe Y., Matsumoto S., Wei S., Nezu K., Miyoshi A., Kito K., Ueda N., Shigemoto K., Hitsumoto Y., Nikawa J., Enomoto Y. Cloning and characterization of a p53-related protein kinase expressed in interleukin-2-activated cytotoxic T-cells, epithelial tumor cell lines, and the testes. J. Biol. Chem. 2001;276:44003–44011. doi: 10.1074/jbc.M105669200. [DOI] [PubMed] [Google Scholar]
- Abe Y., Takeuchi T., Imai Y., Murase R., Kamei Y., Fujibuchi T., Matsumoto S., Ueda N., Ogasawara M., Shigemoto K., Kito K. A Small Ras-like protein Ray/Rab1c modulates the p53-regulating activity of PRPK. Biochem. Biophys. Res. Commun. 2006;344:377–385. doi: 10.1016/j.bbrc.2006.03.071. [DOI] [PubMed] [Google Scholar]
- Abe Y., Takeuchi T., Kagawa-MIKI L., Ueda N., Shigemoto K., Yasukawa M., Kito K. A mitotic kinase TOPK enhances Cdk1/cyclin B1-dependent phosphorylation of PRC1 and promotes cytokinesis. J. Mol. Biol. 2007;370:231–245. doi: 10.1016/j.jmb.2007.04.067. [DOI] [PubMed] [Google Scholar]
- Bierie B., Moses H.L. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer. 2006;6:506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- Diella F., Cameron S., Gemund C., Linding R., Via A., Kuster B., Sicheritz-Ponten T., Blom N., Gibson T.J. Phospho.ELM: a database of experimentally verified phosphorylation sites in eukaryotic proteins. BMC Bioinforma. 2004;5:79. doi: 10.1186/1471-2105-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eblen S.T., Slack-Davis J.K., Tarcsafalvi A., Parsons J.T., Weber M.J., Catling A.D. Mitogen-activated protein kinase feedback phosphorylation regulates MEK1 complex formation and activation during cellular adhesion. Mol. Cell. Biol. 2004;24:2308–2317. doi: 10.1128/MCB.24.6.2308-2317.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facchin S., Lopreiato R., Ruzzene M., Marin O., Sartori G., Gotz C., Montenarh M., Carignani G., Pinna L.A. Functional homology between yeast piD261/Bud32 and human PRPK: both phosphorylate p53 and PRPK partially complements piD261/Bud32 deficiency. FEBS Lett. 2003;549:63–66. doi: 10.1016/s0014-5793(03)00770-1. [DOI] [PubMed] [Google Scholar]
- Facchin S., Ruzzene M., Peggion C., Sartori G., Carignani G., Marin O., Brustolon F., Lopreiato R., Pinna L.A. Phosphorylation and activation of the atypical kinase p53-related protein kinase (PRPK) by Akt/PKB. Cell. Mol. Life Sci. 2007;64:2680–2689. doi: 10.1007/s00018-007-7179-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch S.M., Screaton R.A. Anoikis mechanisms. Curr. Opin. Cell Biol. 2001;13:555–562. doi: 10.1016/s0955-0674(00)00251-9. [DOI] [PubMed] [Google Scholar]
- Gao C.F., Vande Woude G.F. HGF/SF-Met signaling in tumor progression. Cell Res. 2005;15:49–51. doi: 10.1038/sj.cr.7290264. [DOI] [PubMed] [Google Scholar]
- Gaudet S., Branton D., Lue R.A. Characterization of PDZ-binding kinase, a mitotic kinase. Proc. Natl. Acad. Sci. U. S. A. 2000;97:5167–5172. doi: 10.1073/pnas.090102397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie H.Y., Pearline R.V., Grueneberg D., Hsia M., Ravichandran P., Kothari N., Natesan S., Brugge J.S. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J. Cell Biol. 2005;171:1023–1034. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.J., Li Y., Reddy K., Lee M.H., Kim M.O., Cho Y.Y., Lee S.Y., Kim J.E., Bode A.M., Dong Z. Novel TOPK inhibitor HI-TOPK-032 effectively suppresses colon cancer growth. Cancer Res. 2012;72:3060–3068. doi: 10.1158/0008-5472.CAN-11-3851. [DOI] [PubMed] [Google Scholar]
- Kim H., Muller W.J. The role of the epidermal growth factor receptor family in mammary tumorigenesis and metastasis. Exp. Cell Res. 1999;253:78–87. doi: 10.1006/excr.1999.4706. [DOI] [PubMed] [Google Scholar]
- Krueger J.S., Keshamouni V.G., Atanaskova N., Reddy K.B. Temporal and quantitative regulation of mitogen-activated protein kinase (MAPK) modulates cell motility and invasion. Oncogene. 2001;20:4209–4218. doi: 10.1038/sj.onc.1204541. [DOI] [PubMed] [Google Scholar]
- Langlois W.J., Sasaoka T., Saltiel A.R., Olefsky J.M. Negative feedback regulation and desensitization of insulin- and epidermal growth factor-stimulated p21ras activation. J. Biol. Chem. 1995;270:25320–25323. doi: 10.1074/jbc.270.43.25320. [DOI] [PubMed] [Google Scholar]
- Li S., Zhu F., Zykova T., Kim M.O., Cho Y.Y., Bode A.M., Peng C., Ma W., Carper A., Langfald A., Dong Z. T-LAK cell-originated protein kinase (TOPK) phosphorylation of MKP1 protein prevents solar ultraviolet light-induced inflammation through inhibition of the p38 protein signaling pathway. J. Biol. Chem. 2011;286:29601–29609. doi: 10.1074/jbc.M111.225813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long W., Yi P., Amazit L., Lamarca H.L., Ashcroft F., Kumar R., Mancini M.A., Tsai S.Y., Tsai M.J., O'malley B.W. SRC-3Delta4 mediates the interaction of EGFR with FAK to promote cell migration. Mol. Cell. 2010;37:321–332. doi: 10.1016/j.molcel.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto S., Abe Y., Fujibuchi T., Takeuchi T., Kito K., Ueda N., Shigemoto K., Gyo K. Characterization of a MAPKK-like protein kinase TOPK. Biochem. Biophys. Res. Commun. 2004;325:997–1004. doi: 10.1016/j.bbrc.2004.10.133. [DOI] [PubMed] [Google Scholar]
- Mehrotra S., Languino L.R., Raskett C.M., Mercurio A.M., Dohi T., Altieri D.C. IAP regulation of metastasis. Cancer Cell. 2010;17:53–64. doi: 10.1016/j.ccr.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D.X., Massague J. Genetic determinants of cancer metastasis. Nat. Rev. Genet. 2007;8:341–352. doi: 10.1038/nrg2101. [DOI] [PubMed] [Google Scholar]
- Oh S.M., Zhu F., Cho Y.Y., Lee K.W., Kang B.S., Kim H.G., Zykova T., Bode A.M., Dong Z. T-lymphokine-activated killer cell-originated protein kinase functions as a positive regulator of c-Jun-NH2-kinase 1 signaling and H-Ras-induced cell transformation. Cancer Res. 2007;67:5186–5194. doi: 10.1158/0008-5472.CAN-06-4506. [DOI] [PubMed] [Google Scholar]
- Peterson D., Lee J., Lei X.C., Forrest W.F., Davis D.P., Jackson P.K., Belmont L.D. A chemosensitization screen identifies TP53RK, a kinase that restrains apoptosis after mitotic stress. Cancer Res. 2010;70:6325–6335. doi: 10.1158/0008-5472.CAN-10-0015. [DOI] [PubMed] [Google Scholar]
- Rahman-Roblick R., Roblick U.J., Hellman U., Conrotto P., Liu T., Becker S., Hirschberg D., Jornvall H., Auer G., Wiman K.G. p53 targets identified by protein expression profiling. Proc. Natl. Acad. Sci. U. S. A. 2007;104:5401–5406. doi: 10.1073/pnas.0700794104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy K.B., Nabha S.M., Atanaskova N. Role of MAP kinase in tumor progression and invasion. Cancer Metastasis Rev. 2003;22:395–403. doi: 10.1023/a:1023781114568. [DOI] [PubMed] [Google Scholar]
- Sebolt-Leopold J.S., Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat. Rev. Cancer. 2004;4:937–947. doi: 10.1038/nrc1503. [DOI] [PubMed] [Google Scholar]
- Shih M.C., Chen J.Y., Wu Y.C., Jan Y.H., Yang B.M., Lu P.J., Cheng H.C., Huang M.S., Yang C.J., Hsiao M., Lai J.M. TOPK/PBK promotes cell migration via modulation of the PI3K/PTEN/AKT pathway and is associated with poor prognosis in lung cancer. Oncogene. 2011;31:2389–2400. doi: 10.1038/onc.2011.419. [DOI] [PubMed] [Google Scholar]
- Sieg D.J., Hauck C.R., Ilic D., Klingbeil C.K., Schaefer E., Damsky C.H., Schlaepfer D.D. FAK integrates growth-factor and integrin signals to promote cell migration. Nat. Cell Biol. 2000;2:249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- Vishchuk O.S., Sun H., Wang Z., Ermakova S.P., Xiao J., Lu T., Xue P., Zvyagintseva T.N., Xiong H., Shao C., Yan W., Duan Q., Zhu F. PDZ-binding kinase/T-LAK cell-originated protein kinase is a target of the fucoidan from brown alga Fucus evanescens in the prevention of EGF-induced neoplastic cell transformation and colon cancer growth. Oncotarget. 2016;7:18763–18773. doi: 10.18632/oncotarget.7708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei D.C., Yeh Y.C., Hung J.J., Chou T.Y., Wu Y.C., Lu P.J., Cheng H.C., Hsu Y.L., Kuo Y.L., Chen K.Y., Lai J.M. Overexpression of T-LAK cell-originated protein kinase predicts poor prognosis in patients with stage I lung adenocarcinoma. Cancer Sci. 2012;103:1–8. doi: 10.1111/j.1349-7006.2011.02197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelt B., Peterse J.L., van't Veer L.J. Breast cancer metastasis: markers and models. Nat. Rev. Cancer. 2005;5:591–602. doi: 10.1038/nrc1670. [DOI] [PubMed] [Google Scholar]
- Whitehead R.H., Macrae F.A., St John D.J., Ma J. A colon cancer cell line (LIM1215) derived from a patient with inherited nonpolyposis colorectal cancer. J. Natl. Cancer Inst. 1985;74:759–765. [PubMed] [Google Scholar]
- Xiao J., Duan Q., Wang Z., Yan W., Sun H., Xue P., Fan X., Zeng X., Chen J., Shao C., Zhu F. Phosphorylation of TOPK at Y74, Y272 by Src increases the stability of TOPK and promotes tumorigenesis of colon. Oncotarget. 2016;7:24483–24494. doi: 10.18632/oncotarget.8231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeatman T.J. A renaissance for SRC. Nat. Rev. Cancer. 2004;4:470–480. doi: 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- Zeng X., Liu L., Zheng M., Sun H., Xiao J., Lu T., Huang G., Chen P., Zhang J., Zhu F., Li H., Duan Q. Pantoprazole, an FDA-approved proton-pump inhibitor, suppresses colorectal cancer growth by targeting T-cell-originated protein kinase. Oncotarget. 2016;7:22460–22473. doi: 10.18632/oncotarget.7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F., Zykova T.A., Kang B.S., Wang Z., Ebeling M.C., Abe Y., Ma W.Y., Bode A.M., Dong Z. Bidirectional signals transduced by TOPK-ERK interaction increase tumorigenesis of HCT116 colorectal cancer cells. Gastroenterology. 2007;133:219–231. doi: 10.1053/j.gastro.2007.04.048. [DOI] [PubMed] [Google Scholar]
- Zhu F., Zykova T.A., Peng C., Zhang J., Cho Y.Y., Zheng D., Yao K., Ma W.Y., Lau A.T., Bode A.M., Dong Z. Phosphorylation of H2AX at Ser139 and a new phosphorylation site Ser16 by RSK2 decreases H2AX ubiquitination and inhibits cell transformation. Cancer Res. 2012;71:393–403. doi: 10.1158/0008-5472.CAN-10-2012. [DOI] [PubMed] [Google Scholar]
- Zlobec I., Molinari F., Kovac M., Bihl M.P., Altermatt H.J., Diebold J., Frick H., Germer M., Horcic M., Montani M., Singer G., Yurtsever H., Zettl A., Terracciano L., Mazzucchelli L., Saletti P., Frattini M., Heinimann K., Lugli A. Prognostic and predictive value of TOPK stratified by KRAS and BRAF gene alterations in sporadic, hereditary and metastatic colorectal cancer patients. Br. J. Cancer. 2010;102:151–161. doi: 10.1038/sj.bjc.6605452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zykova T.A., Zhu F., Lu C., Higgins L., Tatsumi Y., Abe Y., Bode A.M., Dong Z. Lymphokine-activated killer T-cell-originated protein kinase phosphorylation of histone H2AX prevents arsenite-induced apoptosis in RPMI7951 melanoma cells. Clin. Cancer Res. 2006;12:6884–6893. doi: 10.1158/1078-0432.CCR-06-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zykova T.A., Zhu F., Vakorina T.I., Zhang J., Higgins L.A., Urusova D.V., Bode A.M., Dong Z. T-LAK cell-originated protein kinase (TOPK) phosphorylation of Prx1 at Ser-32 prevents UVB-induced apoptosis in RPMI7951 melanoma cells through the regulation of Prx1 peroxidase activity. J. Biol. Chem. 2010;285:29138–29146. doi: 10.1074/jbc.M110.135905. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Identification of potential phosphorylation sites on PRPK.