

ABSTRACT
Pathogenicity islands (PAIs) are mobile integrated genetic elements that contain a diverse range of virulence factors. PAIs integrate into the host chromosome at a tRNA locus that contains their specific bacterial attachment site, attB, via integrase-mediated site-specific recombination generating attL and attR sites. We identified conserved recombination modules (integrases and att sites) previously described in choleragenic Vibrio cholerae PAIs but with novel cargo genes. Clustered regularly interspaced short palindromic repeat (CRISPR)-associated proteins (Cas proteins) and a type VI secretion system (T6SS) gene cluster were identified at the Vibrio pathogenicity island 1 (VPI-1) insertion site in 19 V. cholerae strains and contained the same recombination module. Two divergent type I-F CRISPR-Cas systems were identified, which differed in Cas protein homology and content. The CRISPR repeat sequence was identical among all V. cholerae strains, but the CRISPR spacer sequences and the number of spacers varied. In silico analysis suggests that the CRISPR-Cas systems were active against phages and plasmids. A type III secretion system (T3SS) was present in 12 V. cholerae strains on a 68-kb island inserted at the same tRNA-serine insertion site as VPI-2 and contained the same recombination module. Bioinformatics analysis showed that two divergent T3SSs exist among the strains examined. Both the CRISPR and T3SS islands excised site specifically from the bacterial chromosome as complete units, and the cognate integrases were essential for this excision. These data demonstrated that identical recombination modules that catalyze integration and excision from the chromosome can acquire diverse cargo genes, signifying a novel method of acquisition for both CRISPR-Cas systems and T3SSs.
IMPORTANCE This work demonstrated the presence of CRISPR-Cas systems and T3SSs on PAIs. Our work showed that similar recombination modules can associate with different cargo genes and catalyze their incorporation into bacterial chromosomes, which could convert a strain into a pathogen with very different disease pathologies. Each island had the ability to excise from the chromosome as distinct, complete units for possible transfer. Evolutionary analysis of these regions indicates that they were acquired by horizontal transfer and that PAIs are the units of transfer. Similar to the case for phage evolution, PAIs have a modular structure where different functional regions are acquired by identical recombination modules.
KEYWORDS: CRISPR-Cas, type III secretion, Vibrio cholerae, pathogenicity islands
INTRODUCTION
Vibrio cholerae is the causative agent of the severe diarrheal disease cholera. The factors necessary for manifestation of this disease were acquired by horizontal gene transfer. The cholera toxin (CT), a potent enterotoxin whose effects are responsible for the copious rice-water diarrhea associated with the disease, is encoded on the lysogenic filamentous phage CTXΦ (1, 2). The toxin-coregulated pilus (TCP), a type IV pilus necessary for colonization of the small intestine, is found on a 41-kb pathogenicity island (PAI) named Vibrio pathogenicity island 1 (VPI-1). VPI-1 is inserted at the transfer-messenger RNA (tmRNA) locus, which contains its specific bacterial attachment site (attB), and encompasses open reading frames (ORFs) VC0817 to VC0847 on chromosome I of the biotype El Tor strain N16961 (3–6). Another PAI present in pathogenic isolates of V. cholerae is VPI-2, a 57-kb region inserted at a tRNA-serine locus, which contains its bacterial attB site between ORFs VC1757 and VC1810 on the genome of N16961. VPI-2 contains the gene VC1784, which encodes a sialidase that cleaves the amino-sugar sialic acid from terminal glycoproteins to expose the cell surface receptor for CT (7). ORF VC1784 is clustered next to the genes that encode a sialic acid catabolism and transport cluster (VC1773 to VC1783) (7). Thus, VPI-2 offers the bacterium a novel nutrient source in vivo that has been shown to give the bacterium a competitive advantage in the intestinal environment (8, 9). The pathogenic O1 and O139 serogroups of V. cholerae, which are responsible for epidemic cholera, contain CTXΦ, VPI-1, and VPI-2, all of which are absent from noncholeragenic strains. Thus, the acquisition of these genetic elements has allowed V. cholerae to become one of the most successful human pathogens, affecting millions of people each year.
Both VPI-1 and VPI-2 were shown to excise as complete units from the bacterial chromosome (10–13). The mechanisms of excision for both islands have recently been determined (13). The recombination modules of VPI-1 and VPI-2 consist of a cognate integrase from the tyrosine recombinase (TR) family, IntV1 (VC0847) and IntV2 (VC1758), respectively, and each island contains flanking attL and attR attachment sites marking the site of chromosomal integration. IntV1 and IntV2 share less than 50% amino acid identity and can catalyze both integrative and excisive recombination. VPI-2 contains two excisionases (also known as recombination directionality factors [RDFs]), VefA and VefB, while VPI-1 contains none (13). The RDFs were shown to be essential for VPI-2 excision and could induce VPI-1 excision. The cognate TR integrases for both VPI-1 and VPI-2 were shown to be essential for efficient excision of these islands from the bacterial chromosome (13). Various combinations of the components (integrases, RDFs, and att sites) of the VPI-1 and VPI-2 recombination modules were found in additional strains of V. cholerae and other Vibrio species, but they contained diverse cargo genes (11, 13–16). Of particular note were two island regions, one that contained a clustered regularly interspaced short palindromic repeat (CRISPR) and CRISPR-associated protein (Cas protein) system and a type VI secretion system (T6SS) gene cluster and a second island that carried genes encoding a type III secretion system (T3SS). These islands contained recombination modules (integrase and attL and attR attachments sites) described in VPI-1 and VPI-2 (11, 13, 14).
The CRISPR-Cas system is a bacterial immunity defense mechanism against invading foreign DNA such as bacteriophages and plasmids (17–22). CRISPR-Cas systems are widespread among prokaryotes, being found in 84% of archaeal and 47% of bacterial genomes (23, 24). The CRISPR repetitive region features direct repeats, which vary in size from 21 bp to 37 bp and can occur twice to over 100 times, depending on the species and the strain. These repeats are separated by nonrepetitive DNA sequences of similar size called spacers (25). The CRISPR array spacer sequences were shown to be acquired from previously infecting phage and serve as memory to protect against future infection (26; reviewed in reference 27). CRISPR-Cas systems are classified based on Cas protein content and arrangements in CRISPR-Cas loci, with two classes and six types defined (22, 28). However, aside from Cas1 and Cas3 proteins, Cas proteins in general are highly divergent, evolve rapidly, and exist in diverse numbers, making classification challenging (22, 28). All CRISPR-Cas systems identified to date contain Cas1 and Cas2 proteins (28). Type I CRISPR-Cas systems, specifically type I-E, have been described in V. cholerae strains, and type I-F has been described in bacteriophages that infect V. cholerae isolates (29–31). A signature of type I CRISPR-Cas systems is the presence of a Cas3 protein, which has a helicase domain and, in many Cas3 proteins, an HD family endonuclease domain. In type I-E and I-F systems, the cas4 gene is absent, and in type I-F systems, the cas2 and cas3 genes are fused (28).
The T3SS is a contact-dependent bacterial secretion system, an apparatus that spans the inner and outer bacterial membranes. When a bacterial cell containing a T3SS is in contact with a eukaryotic cell, it can pierce the eukaryotic membrane to inject bacterial effector proteins directly into the cell. These effector proteins can act on eukaryotic cell structures and signaling pathways, which significantly affects cell physiology (32). T3SSs have been described in V. cholerae isolates, mainly those that lack both CT and TCP (11, 33, 34). The T6SS is also a contact-dependent secretion system that transfers effector proteins to either a eukaryotic or prokaryotic cell, being functionally similar to the T3SS but evolutionarily unrelated. The T6SS was first described in V. cholerae as a novel secretion system by Putkatzki and colleagues and has since been identified in numerous species (35; reviewed in reference 36).
In this study, we determined the distribution of the CRISPR-Cas and T3SS islands, which we named VPI-6 and VPI-3, respectively, among V. cholerae strains. We identified a CRISPR-Cas island inserted at the tmRNA locus in three V. cholerae strains, RC385, RC586, and TMA11079-80. The CRISPR-Cas system in all three strains contained identical Cas1 proteins and type I-F CRISPR repeats; however, the number of spacers and their sequences varied among the strains. In V. cholerae HC-36A1 and 15 additional strains isolated in Haiti, a CRISPR-Cas system that differed from the one in RC385 was present. In these strains, the CRISPR-Cas system contained divergent Cas3 and Cas6 proteins and 3 additional Cas proteins, Csy1, Csy2, and Csy3. In all 16 Haiti strains, the type I-F CRISPR repeat and spacers were identical. The CRISPR-Cas islands in all strains contained a highly homologous recombination module and T6SS components Hcp1 and VgrG. Data from in silico analysis of spacer sequences suggest that these CRISPR systems are active against phages and plasmids. We identified a T3SS inserted at the tRNA-serine locus in 12 V. cholerae strains whose whole genome sequences are available. Bioinformatics and phylogenetic analyses showed that two divergent T3SSs were acquired separately in V. cholerae. The CRISPR and T3SS islands excised site specifically from the bacterial chromosome as complete units, and the cognate integrases IntV1 and IntV2 were required for excision. Overall, the data suggest that acquisition of CRISPR-Cas and secretion systems on islands may be common and suggest a mechanism by which different combinations of systems can arise in different isolates of the same species.
RESULTS AND DISCUSSION
Recombination module and cargo genes of VPI-6 in V. cholerae RC385.
Strain RC385 is a V. cholerae serogroup O135 strain isolated in 1998 from plankton in the Chesapeake Bay, USA (14). This strain contained a 29-kb region that was absent from pandemic strains and was variably present among V. cholerae strains (14). This region contains a clustered regularly interspaced short palindromic repeat–CRISPR-associated protein (CRISPR-Cas) cluster and several T6SS genes. The genomic region is inserted at the tmRNA locus and contains an integrase gene and attL and attR sites, indicating that it is a genomic island, which we named VPI-6 (Fig. 1). The recombination module (integrase and att sites) is homologous to the VPI-1 module that inserts at the same tmRNA locus in V. cholerae N16961. The CRISPR-Cas region in RC385 contains five ORFs, encoding three annotated Cas proteins (Cas1, Cas3, and Cas6) and two hypothetical proteins. The Cas3 protein shows the two signature domains of this protein, a helicase domain and an endonuclease domain. The CRISPR-Cas region contains a 4,528-bp CRISPR sequence with the 28-bp direct repeat GTTCACTGCCGCACAGGCAGCTTAGAAA and 75 unique spacer sequences of 32 bp (spacer 23 is 33 bp) (Table 1). The CRISPRMap program (37) determined the direct repeat sequence to be a type I-F system repeat. Using the CRISPRTarget program to identify spacer homology in the plasmid and phage databases, we found that 44 of the 75 spacers had hits to Vibrio phages (Fig. 2). Of these 44 spacers, 40 matched to regions within the same sequences of phages X29 (accession number KJ572845) and phi2 (accession number KJ545483). We examined the distribution of the 40 hits to phage X29 and found that the distribution was skewed to matches within structural proteins of this phage (Fig. 2, top). Three spacers each matched to the same sequences within the Kappa, K139, and PV95 phages and one spacer to phage VP16T (Fig. 2, bottom). These data confirm a role for this CRISPR-Cas system in phage immunity and suggest that this CRISPR-Cas system is active.
FIG 1.

Gene maps and comparisons of V. cholerae pathogenicity islands. VPI-1 and VPI-2 from N16961, VPI-3 from NRT36S, and VPI-6 from RC385 were compared. An entire secretion system is present on VPI-3, and a complete CRISPR-Cas system and a T6SS gene cluster are present on VPI-6. ORFs are designated by arrows, and the directions of arrows indicate directions of transcription. The conserved integrases of the islands, IntV2 from VPI-2 and VPI-3 and IntV1 from VPI-1 and VPI-6, are shown as shaded arrows and are located adjacent to a tRNA-serine and a tmRNA locus, respectively. Brackets connected by lines represent regions of identity between islands.
TABLE 1.
Direct repeats and CRISPR types from species examined
| Species and strain or phage | Type | Direct repeata |
|---|---|---|
| V. cholerae | ||
| O395 | I-E | GTCTTCCCCACGCAGGTGGGGGTGTTTC |
| Phage ICP1 | I-F | GTTAGCAGCCGCATAGGCTGCTTAAAGA |
| RC385 | I-F | GTTCACTGCCGCACAGGCAGCTTAGAAA |
| RC586 | I-F | GTTCACTGCCGCACAGGCAGCTTAGAAA |
| TM11079-80 | I-F | GTTCACTGCCGCACAGGCAGCTTAGAAAT |
| HC-36A1 | I-F | GTTCACTGCCGCACAGGCAGCTTAGAAA |
| V. metschnikovii CIP 69.14 | I-F | GTTCACTGCCGCACAGGCAGCTTAGAAA |
| I-C | GTCGCGTCTCCCGCAGGCGCGTGGATTGAAAC | |
| V. navarrensis | ||
| 2232 | I-F | GTTCACTGCCGCACAGGCAGCTTAGAAA |
| 0053-83 | I-F | GTTCACTGCCGTATAGGCAGCTTAGAAA |
| ATCC 51183 | I-F | GTTCACTGCCGCACAGGCAGCTTAGAAA |
| I-C | GTCGCGCCTCCCGCAGGCGCGTGGATTGAAAC | |
| V. fischeri 5F7 | I-F | GTTCACTGCCGTACAGGCAGCTTAGAAA |
| P. profundum SS9 | I-F | GTTCACTGCCGCACAGGCAGCTTAGAAA |
| P. aquimaris GCSL-P86 | I-F | GTTCACTGCCGCACAGGCAGCTTAGAAA |
Underlining indicates polymorphism relative to the RC385 repeat.
FIG 2.

RC385 CRISPR spacers that hit to Vibrio phage X29. (Top) Genome organization of phage X29 (41,569 bp). Horizontal arrows indicate ORFs, and vertical arrows indicate locations within the X29 genome to which spacers hit. (Bottom) List of spacers and their score and hits to phage X29.
The CRISPR sequence is located between the integrase IntV1 (VCRC385_RS05055) and ORF VCRC385_RS05060, which is annotated as a type I-F CRISPR-associated endoribonuclease, Cas6/Csy4 (Fig. 1). Traditional BLAST searches of VCRC385_RS05065 and VCRC385_RS05070 yielded no significant homology to known proteins in the database. An HHPred search (38) found that VCRC385_RS05070 was homologous to Cas7; this protein is followed by Cas3 (VCRC385_RS05075) and Cas1 (VCRC385_RS05080) homologues, annotated as type I subtype I-F. VCRC385_RS05085, which marks the end of the CRISPR-Cas region, is annotated as a hypothetical protein that contains a WYL domain and a helix-turn-helix domain, suggesting that it is a transcriptional regulator (Table 2). The prototype I-F system consists of six genes in a single operon, cas1, cas2-cas3, cas8f/cys1, cas5/csy2, cas7/csy3, and cas6f (28). Thus, the CRISPR-Cas system described here may be a novel variant of type I-F.
TABLE 2.
ORFs in V. cholerae RC385 VPI-6 and predicted homology to a CRISPR-Cas system and T6SS
| RC385 locus tag | ORF | Size (bp) | Gene product | V. cholerae strain | Homologue accession no. | Identity (%) |
|---|---|---|---|---|---|---|
| VCRC385_RS05055 | 1 | 412 | Integrase (IntV1) | N16961 | NP_230494 | 97 |
| VCRC385_RS05060 | 2 | 183 | Cas6/Csy4 | CAG76582 | 38 | |
| VCRC385_RS05065 | 3 | 351 | Hypothetical protein | |||
| VCRC385_RS05070 | 4 | 315 | Cas7 | Q97Y91 | 14 | |
| VCRC385_RS05075 | 5 | 978 | Cas3/Cas2 | CAG76578 | 37 | |
| VCRC385_RS05080 | 6 | 329 | Cas1 | CAG76577 | 58 | |
| VCRC385_RS05085 | 7 | 264 | Regulator | N16961 | NP_231407 | 38 |
| VCRC385_RS05090 | 8 | 172 | Hcp1 (T6SS) | N16961 | NP_232418 | 97 |
| VCRC385_RS05095 | 9 | 658 | VgrG (T6SS) | N16961 | NP_232419 | 69 |
| VCRC385_RS05100 | 10 | 262 | Hypothetical protein | |||
| VCRC385_RS05105 | 11 | 536 | Hypothetical protein | |||
| VCRC385_RS0318635 | 12 | 262 | Glycosyl hydrolase | |||
| VCRC385_RS05110 | 13 | 99 | Hypothetical protein (VCA0105) | N16961 | NP_232506 | 53 |
| VCRC385_RS0318630 | 14 | 262 | Glycosyl hydrolase | |||
| VCRC385_RS05115 | 15 | 209 | DUF2628 Domain | |||
| VCRC385_RS05120 | 16 | 179 | Hypothetical protein | |||
| VCRC385_RS05125 | 17 | 58 | VefA (VC1785) | N16961 | NP_231420 | 91 |
| VCRC385_RS05130 | 18 | 158 | RadC (VC1786) | N16961 | NP_231421 | 99 |
| VCRC385_RS05135 | 19 | 142 | Hypothetical protein (VC1804) | N16961 | NP_231439 | 91 |
| VCRC385_RS05140 | 20 | 136 | Hypothetical protein (VC1805) | N16961 | NP_231440 | 98 |
| VCRC385_RS05145 | 21 | 328 | GTPase (VC1806) | N16961 | NP_231441 | 97 |
| VCRC385_RS05150 | 22 | 213 | Hypothetical protein (VC0494) | N16961 | NP_230148 | 31 |
| VCRC385_RS05155 | 23 | 344 | Hypothetical protein (VC1808) | N16961 | NP_231442 | 30 |
| VCRC385_RS05160 | 24 | 66 | VefB (VC1809) | N16961 | NP_231443 | 97 |
In RC385, downstream of the CRISPR-Cas region is a gene that encodes Hcp1 (VCRC385_RS05090), a T6SS effector that shows 97% similarity to Hcp1 present in V. cholerae N16961. The next ORF, VCRC385_RS05095, also encodes a homologue of a T6SS protein, VgrG, that shares 69% similarity with VgrG from V. cholerae N16961. VgrG is followed by two ORFs annotated as encoding hypothetical proteins, followed by two ORFs (VCRC385_RS0318635 and VCRC385_RS0318630) encoding glycosyl hydrolases (Table 2). Two additional ORFs annotated as encoding hypothetical proteins were present before a VefA homologue. VefA (VCRC385_RS05125) marks the beginning of the region that shows intermittent homology with the 3′ region of VPI-2, and a VefB homologue marks the end of the island (Fig. 1 and Table 2). Thus, the CRISPR-Cas island contains the VPI-1 integrase and is flanked by att sites homologous to the att sites of VPI-1, which suggests that the region can excise from the chromosome as a complete unit.
The presence of CRISPR-Cas systems on mobile genetic elements has been shown in previous studies (14, 29, 30, 39). A CRISPR-Cas system was first identified in the classical biotype V. cholerae O395 strain, within a 17-kb genomic island that contained an integrase and was inserted between the homologues of ORFs VC0289-VC0290 in strain N16961 (14, 29). The CRISPR-Cas system in strain O395 consisted of eight Cas proteins, a 2,407-bp CRISPR sequence comprised of a 28-bp repeat (GTCTTCCCCACGCAGGTGGGGGTGTTTC), and 39 unique spacer sequences (Table 1). This CRISPR-Cas system was shown to be a functional type I-E system that was confined to classical strains of V. cholerae (31). This system showed no homology to the CRISPR-Cas system identified here in RC385, and the integrase shared <27% (E value, 2e−32) amino acid identity with IntV1 from strain N16961, indicating they are unrelated.
CRISPR-Cas systems also were described in bacteriophages from V. cholerae O1 serogroup strains, named ICP1 phages, isolated from cholera stool samples (30). These CRISPR-Cas systems consisted of six Cas proteins, Cas6f, Csy3, Csy2, Csy1, Cas2-Cas3, and Cas1, and a 28-bp repeat designated a type I-F (30). This repeat was similar to the repeat identified in RC385; however, there were seven nucleotide polymorphisms between RC385 and the ICP1 CRISPR-Cas system repeat (Table 1). The Cas1 protein of RC385 shared 38% amino acid identity with 96% query coverage with the Cas1 identified in ICP1_2006_E; all other ICP1_2006_E Cas proteins showed no homology with the Cas proteins in RC385. Outside of V. cholerae, a recent survey of Pseudomonas aeruginosa isolates identified a type I-C CRISPR-Cas system within an integrative and conjugative element (ICE) which is related to the conjugative plasmid pKLC102 (39). ICEs are mobile elements that contain both recombination modules (integrase and att sites) for site-specific integration and conjugative genes for transfer. This study found an ICE-containing CRISPR-Cas system present in multiple distinct lineages of the >600 isolates of P. aeruginosa examined and showed that the region was integrated at a tRNA-serine locus (39). In Legionella pneumophila, an intracellular pathogen, multiple CRISPR systems can be present within an isolate, and a noncanonical role in intracellular survival for these systems has been described (40). A recent study has shown that several L. pneumophila isolates contained a type I-C CRISPR-Cas system on the chromosome and a type I-F system on a 114-kb plasmid, and that study also demonstrated a role for these systems in immunity (41). Overall, it appears that many diverse CRISPR-Cas systems are associated with many different types of mobile genetic elements, indicating their ability to be horizontally transferred among isolates.
Comparative and phylogenetic analyses of the CRISPR-Cas system.
A CRISPR-Cas island similar to the island in RC385 was identified in two additional strains, Vibrio sp. strain RC586 and V. cholerae TM11079-80 at the same tmRNA locus. In RC586, the CRISPR-Cas island shared significant similarity with RC385; both of these strains were isolated in the late 1990s from the Chesapeake Bay, USA (Fig. 3A) (14–16). The RC586 putative cas1, cas3, cas7, and cas6 genes showed near identity with those present in RC385, as did the 3′ region of the island (Fig. 3A). In strain RC586, the CRISPR is a 2,007-bp sequence with the same 28-bp direct repeat as in RC385, but it contained 33 spacers (Table 1). Of the 33 spacers present in RC586, 24 showed identity to spacers in RC385 and matched to the same putative target sequences. RC385 spacer 7 (SP7) to SP11, SP12 to SP17, and SP21 to SP33 showed identity to spacers SP15 to SP19, SP57 to SP62, and SP63 to SP75 in RC586. Five additional RC586 spacers matched to X29 and phi2 phage sequences. The near identity in the CRISPR-Cas regions between the two strains but the difference in spacer numbers and sequences suggest that these were dynamic functional systems actively recruiting new spacer sequences.
FIG 3.

Comparison of the V. cholerae CRISPR-Cas islands among four V. cholerae strains. (A) The Artemis comparison tool (ACT) was used to examine nucleotide sequence differences, and the figure was generated using the genome comparison visualizer (50). Dark blocks represent forward regions of homology between the regions, and white blocks indicate no matches. (B) The evolutionary history of Cas1, Cas3, and Cas6 from 6 species was inferred using the neighbor-joining method, with bootstrap values (1,000 replicates) shown next to branches (57). The tree is drawn to scale, with branch lengths indicated. The evolutionary distances were computed using the p-distance method (58). Evolutionary analyses were conducted in MEGA6 (45).
Vibrio cholerae strain TM11079-80 is an environmental strain isolated in Brazil from sewage in 1980 (14). Sequence alignments of the CRISPR-Cas island with RC385 revealed significant homology with the IntV1, Cas1, and Cas6 proteins. The Hcp and VgrG ORFs were also highly conserved between the two strains, and this homology included an additional five ORFs (Fig. 3A). After the T6SS region, a transposase is present, followed by a protein from the ankyrin repeat (Ank) family, which marked the end of homology with the rest of the island present in RC385. Ankyrin repeat proteins in bacteria have been characterized as type IV secretion system effector proteins in Legionella pneumophila and Coxiella burnetii (42). The CRISPR in TM11079-80 is a 3,450-bp sequence and contained a 29-bp repeat with 57 spacers. The repeat shared identity with RC385 except for an additional nucleotide at the end of the sequence (Table 1). TM11079-80 had a transposase between Cas1 and Cas3, and a gap in homology exists in the hypothetical protein between Cas6 and Cas7 in RC385 (Fig. 3A). Of the 57 spacers identified, 16 spacers showed hits to Vibrio phages and plasmids (Table 3). Three spacers hit to the same sequence in phages fs2 and VFJ, and seven spacers hit to the same seven regions within phages Kappa, K139/VPUSM, and 8/919TP/VcP032/VcP032. Two spacers hit to plasmid p380 and one spacer to plasmid pYJ016 (Table 3). These data indicate that this CRISPR is active against both phages and plasmids.
TABLE 3.
CRISPR spacer sequence hits from TM11079-80
| Spacer no. | Spacer sequence | Score | Hit | Hit position |
|---|---|---|---|---|
| 2 | TGACTATAAACTTTGGACGGGTCGCCACTCTC | 31 | fs2/VFJ | 374–405 |
| 17 | TGTTACATCGAGCACTGGTAAACTTTTGGCAA | 31 | pYJ016 | 42119–42088 |
| 20 | GTTGTACTCAGTTTCATCGTCCGTAAACATGT | 28 | Plasmid p380 | 25520–25551 |
| 23 | GATCTTAAACATTCTGCGACCAGCTTTGTCTT | 28 | Plasmid p380 | 25451–25482 |
| 24 | TTTTCTAACGAGTCCATGCGATAAGCAAAAAA | 32 | KSF-1phi | 2964–2933 |
| 29 | ACGCTATTTGGCGAATTCGTCCACGTAAAAAC | 28 | Kappaa | 20778–20808 |
| 30 | TTCGCTTAGTTGAGTTTGAGCAAGATAAACAA | 30 | Kappa | 17314–17345 |
| 37 | ATTTGAGCGCCGAGTTTAAACGATAGTTCATA | 32 | X29/phi2 | 13260–13229 |
| 40 | TTCAAATAGAGGGCTCGGGCGGCTTGTCGGAT | 30 | Kappa | 15030–15061 |
| 43 | ATTCGAACTGCGGCATTCAGCATGGTTTTACC | 30 | Kappa | 24233–24264 |
| 44 | AGTAACAAAAAACTCGCCAAACGCATGGCGGA | 32 | Kappa | 5813–5844 |
| 45 | TCTACGTTTACTTCAAAGCAGTATCTTGCTTT | 32 | Kappa | 1125–1094 |
| 46 | TGTAAATAATATTTCTGCCAAAGTGTAGAGCG | 32 | fs2/VFJ | 4765–4734 |
| 47 | TGAACGAGTTGGACAAACTCAGGCTCAGGCGA | 26 | fs2/VFJ | 6040–6009 |
| 56 | AAACGATGAGATCTGTAAAAATGGGGTTTTGA | 32 | Kappa | 17487–17456 |
| 57 | ATTGTGTAAGGGTCGTTGTGCTTAAAGCGCTG | 22 | Phage pYD21 | 17672–17641 |
The spacer that hit to Kappa also hit to identical sequences in phages K139/VPUSM and 8/919TP/VcP032/VcP032.
Vibrio cholerae strain HC-36A1 is a non-O1 clinical strain isolated in Haiti in 2010 (43). It contained a CRISPR-Cas island at the same tmRNA locus as RC385, RC586, and TM11079-80. Strain HC-36A1 contained a Cas1 protein identical to that found in RC385; however, its Cas3 and Cas6 proteins diverged significantly from those present in RC385. In addition, HC-36A1 also contained homologues of Cas proteins, Csy1, Csy,2, and Cys3, which are components of type I-F systems (Fig. 3A). The 3′ region of the island shared homology with TM11079-80. The CRISPR in this strain consisted of a 2,367-bp sequence with 39 spacers and the same 28-bp direct repeat as in RC385 (Table 1). Of the 39 spacers identified, only five spacers gave hits with scores of >20; four spacers, 25, 26, 32, and 39, hit to phages X29/phi2, Kappa, fs2, and pYD21, respectively. Spacer 8 was interesting as it hit to two regions within the cholera toxin-containing phage CTX, within ig1 and ig2 intergenic regions with high scores of 28 and 30, respectively. A CRISPR-Cas system identical to the one present in HC-36A1 is also present in 15 additional V. cholerae strains, HC-50A1, HC-51A1, HC-52A1, HC-55A1, HC-56A1, HC-57A1, HC-59A1, HC-60A1, HC-78A1, HC-1A2, HC-55C2, HC-61A2, HC-02C1, HC-55B2, and HC-59B1.
Analysis of the distribution of the three proteins present in RC385, Cas1, Cas3, and Cas6 showed that they were all present in five additional species within the family Vibrionaceae: V. navarrensis strains 2232 (accession number JMCH01000043), 0053-83 (JMCF01000024), and ATCC 51183 (JMCG01000001.1/JMCG01000002.1); V. metschnikovii CIP 69.14 (ACZO01000006/ACZO01000007); V. fischeri 5F7 (MCGJ01000054); Photobacterium profundum SS9 (CR378681); and Photobacterium aquimaris GCSL-P86 (LZEZ01000003.1) (Fig. 4).
FIG 4.

CRISPR-Cas gene arrangement and content among the Vibrio species examined. Arrows indicate ORFs, and vertical lines indicate CRISPR arrays with repeats and spacers. Black arrows indicate a core chromosomal gene, and similarly shaded arrows indicate homologous ORFs with a 35% amino acid identity cut off with greater than 60% query coverage. Double hatched lines indicate CRISPR arrays located at different locations on the chromosomes in strains CIP69.14 and ATCC 51183.
Vibrio navarrensis is a recently described species that is closely related to V. vulnificus and is associated with infections in humans (44). In V. navarrensis strain 2232, the CRISPR-Cas system is homologous to that present in RC385. The repeat region marked the end of the contig, and therefore no IntV1 homologue was identified. The 28-bp direct repeat is identical to that in RC385, and the CRISPR was 1,107 bp with 18 spacers. In V. navarrensis strain 0053-83, the CRISPR-Cas system was homologous to the one present in HC-36A1, and the region contained an integrase that shows 83% identity to IntV1 (Fig. 4). This integrase was not in the same genomic location as intV1 in RC385 but occurred after the putative transcription regulator at the opposite end of the CRISPR region (Fig. 4). The system consists of six Cas proteins that show >96% sequence homology to Cas1, Cas3, Csy1, Csy2, Csy3, and Csy4/Cas6 in HC-36A1. The CRISPR in this strain is a 2,729-bp sequence with a 28-bp direct repeat and 45 spacers. Also the direct repeat in this V. navarrensis strain has two nucleotide polymorphisms in the middle of the repeat compared to RC385 (Table 1 and Fig. 4).
In V. navarrensis strain ATCC 51183, two CRISPR-Cas systems, a type I-F and a type I-C, and four CRISPR arrays were identified. On chromosome I, three CRISPR arrays are present, a type I-F system that is highly similar to the one present in V. navarrensis strain 0053-83 with six Cas proteins, Cas1, Cas3, Csy1/Cas8, Csy2/Cas5, Csy3/Cas7, and Csy4/Cas6. However, the 28-bp repeat is identical to that in V. cholerae HC-36A1. CRISPR1 is a 2,610-bp sequence with 43 spacers and is located at positions 12211 to 9601. The second type I-F CRISPR2 array with no associated CAS proteins is located at positions 3238102 to 3239750, with 27 spacers and the same direct repeat as CRISPR1. A third type I-F CRISPR3 array with no associated Cas proteins is present on chromosome II at positions 13861 to 12033, with 30 spacers and a repeat identical to that in CRISPR1. The type I-C CRISPR-Cas system consists of Cas3, Cas5, Cas8c, Cas7, Cas4, Cas1, and Cas2 and a 32-bp repeat that is classified as a type I-C with 28 spacers that varied in size from 33 bp to 35 bp, located at positions 1238174 to 1240043 on the chromosome (Table 1).
In V. metschnikovii CIP 69.14, two CRISPR-Cas systems were also identified, a type I-F and a type I-C, and six CRISPR arrays. The type I-F CRISPR-Cas system resembled that present in RC385 in gene arrangement, but no integrase or transposase genes were associated with the region. The CRISPR1 array, adjacent to the type I-F Cas proteins, was 1,227 bp with a 28-bp repeat identical to that in RC385 and contained 20 spacers. Four additional type I-F CRISPR arrays were identified, containing 5, 6, 7, and 10 spacers found elsewhere on the genome. The type I-C system had seven Cas proteins, Cas3, Cas5, Cas8c, Cas7, Cas4, Cas1, and Cas2, and a 32-bp repeat with one nucleotide mismatch with the I-C repeat from V. navarrensis strain ATCC 51183 and 20 unique spacers. Of the total of 65 spacers identified in this strain, two were redundant. In V. fischeri 5F7, the type I-F CRISPR-Cas system resembles HC-36A1 in the Cas proteins content and contains a 28-bp repeat with 1 nucleotide polymorphism with the RC385 repeat (Table 1). The CRISPR is a 3,508-bp sequence with 58 spacers (Fig. 4). The CRISPR-Cas systems identified in P. profundum and P. aquimaris were similar in Cas protein content to RC385 and had an identical 28-bp direct repeat and 1,588-bp and a 147-bp CRISPRs with 26 and 2 spacers, respectively.
The evolutionary histories of the type I-F CRISPR-Cas regions were inferred using the neighbor-joining method based on evolutionary distances computed using the p-distance method in MEGA6 (45). Phylogenetic analysis of the Cas1, Cas3, and Cas6 proteins showed that the CRISPR-Cas systems present in V. cholerae strains RC385 and RC586 were highly similar to each other and to a CRISPR-Cas system in V. navarrensis 2232, which suggests recent horizontal transfer between these species (Fig. 3B). The Cas1, Cas3, and Cas6 trees showed that V. metschnikovii CIP 69.14 was the strain with the CRISPR-Cas system next most closely related to that in RC385. Only in the Cas1 tree did all four V. cholerae strains cluster together and with the two Cas1 proteins from V. navarrensis 2232 and 0053-83. In the Cas3 tree, three V. cholerae strains clustered together, and strain HC-36A1 had the most divergent Cas3 protein examined, which clustered with Cas3 from V. navarrensis 0053-83. The Cas6 proteins from strains HC-36A1 and 0053-83 also branched divergently from the rest of the Cas6 proteins examined (Fig. 3B). In evolutionary terms, these data demonstrate that there is ongoing recombination and acquisition of CRISPR-Cas systems among closely related isolates and horizontal transfer among distantly related species. In support of this, a recent study showed that a type I-E CRISPR-Cas system could be transferred by natural transformation between V. cholerae isolates (31). A dynamic system would be expected in a region that is required for adaptive immunity to protect against constant attack from phages.
Recombination module and cargo genes of VPI-3 in V. cholerae NRT36S.
Vibrio cholerae NRT36S is a serogroup O31 strain isolated from a Japanese patient with travelers' diarrhea and contains a T3SS but not the genes that encode CT and TCP (34). The T3SS is found within a 68-kb island, which we named VPI-3, that is integrated into chromosome I between homologues of open reading frames (ORFs) VC1757 and VC1810 in strain N16961, at the same tRNA-serine locus integration site as VPI-2. Comparative analysis of VPI-3 from V. cholerae NRT36S and VPI-2 from N16961 showed significant homology in many regions (Fig. 1). The attL and attR attachment sites of VPI-3 share 100% homology with the att sites of VPI-2, and the integrase shares 98% nucleotide identity with the VPI-2 integrase. These data indicate that the recombination module of VPI-3 is highly homologous to that of VPI-2 and should function similarly. Downstream of the integrase gene in VPI-3 are the genes encoding a T3SS. The T3SS is at the same location as the type I restriction modification genes present in the 5′ region of VPI-2. The T3SS genes were followed by genes encoding sialic acid scavenging, transport, and catabolism proteins. This region shares 97% nucleotide sequence homology with the same region in VPI-2 (Fig. 1). Two excisionase genes are also present on VPI-3, vefA and vefB, and share 96% homology with those present on VPI-2. The last five genes in the 3′ end of the VPI-3 region also share homology with the same genes in VPI-2 and include vefB, which marks the end of the island (Fig. 1).
Comparative and phylogenetic analyses of VPI-3.
Using the ATPase from VPI-3 of NRT36S as a seed, we identified 11 additional T3SS-containing strains of V. cholerae whose whole genome sequences were available (Fig. 5A). In these 11 strains, the T3SS was present at the same tRNA-serine locus and contained identical recombination modules. The sialic acid catabolism region was also present in all isolates. The majority of the T3SS region was highly conserved among nine strains, but two strains, 1587 and 623-39, showed little homology to the other nine. Of the 49 ORFs within the T3SS region, 36 ORFs were highly divergent between strains 1587 and 623-39 and the other nine strains, which suggests that the T3SS regions were acquired independently in two separate events (Fig. 5A).
FIG 5.

Comparison of the V. cholerae T3SS island among 12 V. cholerae strains. (A) The Artemis comparison tool (ACT) was used to examine nucleotide sequence differences among VPI-3s. Dark blocks represent forward regions of homology between the island regions, and white regions indicate no homology. (B) The evolutionary history of IntV2, ATPase, and NanA among 12 V. cholerae strains was inferred using the neighbor-joining method, with bootstrap values (1,000 replicates) shown next to branches (57). The tree is drawn to scale, with branch lengths indicated. The evolutionary distances were computed using the p-distance method. Evolutionary analyses were conducted in MEGA6 (45).
To infer the evolutionary history of the T3SS islands and demonstrate further the different evolutionary histories of the regions, we constructed phylogenetic trees based on the integrase IntV2, the T3SS ATPase, and the NanA proteins of the 12 V. cholerae strains examined here (Fig. 5B). Analysis of the IntV2 proteins from the 12 V. cholerae strains showed that they were highly homologous to one another, as indicated by the small genetic distance. Similarly, the phylogenetic analysis of the NanA proteins showed that they were highly related to one another, with small branch lengths indicating limited divergence. The T3SS ATPases in strains NRT36S, AM-19226, CP1110, EM-1676A, V51, TMA21, 12129, VC35, HE-25, and P-18785 were all highly homologous, whereas the ATPases from strains 1587 and 623-39 clustered together but divergently from the other ATPases (Fig. 5B). These data suggest that the recombination module and the sialic acid region are core to the island, while the T3SS was acquired in two separate events within this core island. The near identity of VPI-6 among strains that are otherwise divergent from one another suggests, in evolutionary terms, recent horizontal transfer between strains.
To further determine the evolutionary history of the T3SS, we examined the phylogenetic relationships between the V. cholerae T3SS ATPases and those from other species (Fig. 6). These data showed that both T3SSs from V. cholerae strains were closely related to the T3SS-2 region on chromosome 2 of clinical strains of V. parahaemolyticus. In this species, there are two T3SS-2 variants described, designated T3SS-2α and T3SS-2β depending on the strain examined (46). The T3SS ATPases from the nine V. cholerae strains were highly related to T3SS-2α from V. parahaemolyticus RIMD2210633, an O3:K6 pandemic strain. The ATPase was also highly related to a T3SS ATPase present in V. mimicus and Grimontia hollisae strains. PCR detection assays were used to examine V. mimicus isolates for the presence of T3SS genes, and these studies suggested that the T3SS in these isolates was present on an island similar to VPI-3 (47). The T3SS region from G. hollisae ATCC 33564, whose genome sequence is available (accession number CP014056), is inserted at a tRNA-serine locus. The ATPases from strains 1587 and 623-39 clustered with the T3SS-2β ATPase from prepandemic V. parahaemolyticus strains (Fig. 6). Both T3SS-2 types in V. parahaemolyticus are present on chromosome 2 and are present only in clinical isolates. The presence of highly related ATPases in otherwise highly divergent species suggests horizontal gene transfer and by extension transfer of the entire region. All V. parahaemolyticus isolates contain a second, unrelated T3SS, which is present on chromosome 1 and clusters on a divergent branch of the ATPase tree with the related species V. alginolyticus, strains of which all contain a T3SS-1. In general, when T3SS-1 is present in a species, it is present in all isolates of that species, which suggests that T3SS-1 is the ancestral system. Taken together, these data further confirm that T3SS-2 was acquired by horizontal transfer. In support of this, a study has shown that a T3SS can be transferred between V. cholerae strains via natural transformation (48).
FIG 6.

Evolutionary analysis of ATPases from T3SS in Vibrionaceae and enteric species. The evolutionary history was inferred using the neighbor-joining method (57). The optimal tree with the sum of branch lengths of 1.99 is shown. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) is shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the p-distance method and are in the units of the number of amino acid differences per site. The analysis involved 48 amino acid sequences. All ambiguous positions were removed for each sequence pair. There were a total of 447 positions in the final data set. Evolutionary analyses were conducted in MEGA6 (45). The asterisk indicates species with two T3SSs.
The T3SS island can excise from the V. cholerae NRT36S bacterial genome.
It was previously demonstrated that VPI-2 of V. cholerae N16961 can excise from the chromosome (11–13). The excision of a T3SS-containing island has not yet been established. Thus, we sought to determine if VPI-3 could excise as a complete unit from the chromosome of NRT36S. VPI-3 excision was examined through PCR detection of attP and attB attachment sites (Fig. 7A). The attP site is created when the island excises and forms a circular intermediate and the attB site marks the empty chromosomal site after excision (Fig. 7A). In order to detect the attP site, an inverse PCR primer pair was designed within intV2 (the first gene within VPI-3) and vefB (the last gene within VPI-3) such that a PCR product will be obtained only if VPI-3 excises and forms a circular intermediate (CI) (Fig. 7A and B). Inverse PCR assays were performed on DNAs from V. cholerae NRT36S (VPI-3) and N16961 (VPI-2), and no product was detected in this first round of PCR (Fig. 7B). A second PCR assay was performed using the product of the first PCR as the template, with primers designed within an internal fragment of the inverse PCR product (Fig. 7B). A 497-bp PCR product was detected in NRT36S as well as in the control strain N16961, in which excision was previously shown (Fig. 7B). A two-stage nested PCR was also performed to detect the empty attB site left in the V. cholerae chromosome following excision of VPI-3. An attB PCR product was observed in the second round of the attB assay for V. cholerae strain NRT36S and the positive-control strain N16961 (Fig. 7C). V. cholerae strain SG-7 was used as a negative control, as it does not contain an island at the tRNA-serine locus. In strain SG-7, no excision was detected in the attP assay, and the empty attB site was detected in the first round of the attB PCR assay (Fig. 7B and C). The attB sequences were confirmed by sequencing. These data indicate that VPI-3 is capable of excision from the genome of V. cholerae NRT36S as a single unit for possible transfer.
FIG 7.
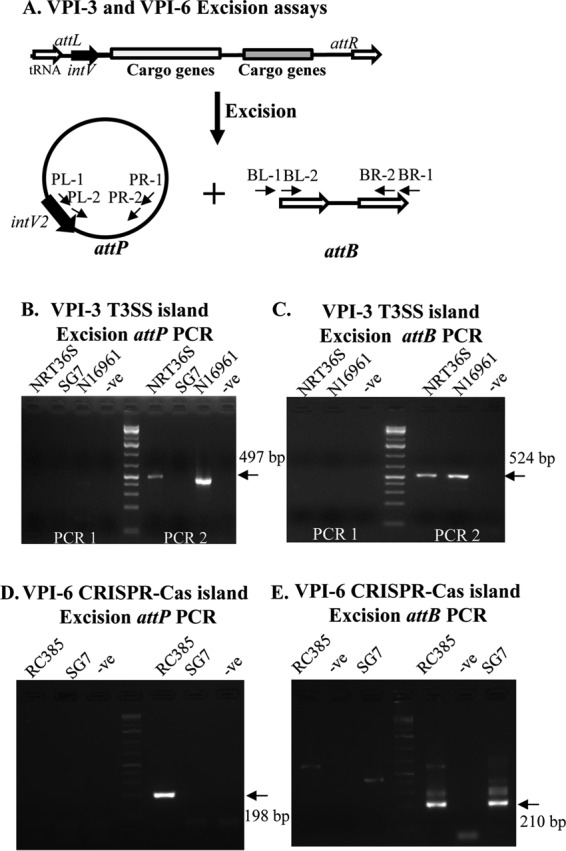
The T3SS and the CRISPR-Cas islands excise site specifically from the chromosome. (A) Primer design for PCR amplification of the attP and attB sites of V. cholerae strains. (B and C) PCR assays to detect attP and attB of VPI-3 in V. cholerae NRT36S and N16961. Strain SG7 was included as a negative control. (D and E) PCR assays to detect attP and attB of VPI-6 in V. cholerae RC385. Strain SG7 was included as a negative control.
VPI-2 excision was dependent upon the presence of the cognate integrase IntV2 (11–13). In order to examine whether the cognate integrase of NRT36S VPI-3 (IntV2) is also necessary for the excision, we constructed an in-frame deletion of intV2 to create strain MRCIntV2. We performed the attB excision assay on this strain to determine whether excision occurs (Fig. 8A). No PCR product was observed in either round of the attB assay for strain MRCIntV2, indicating that the cognate VPI-3 integrase is necessary for excision of this island (Fig. 8A).
FIG 8.
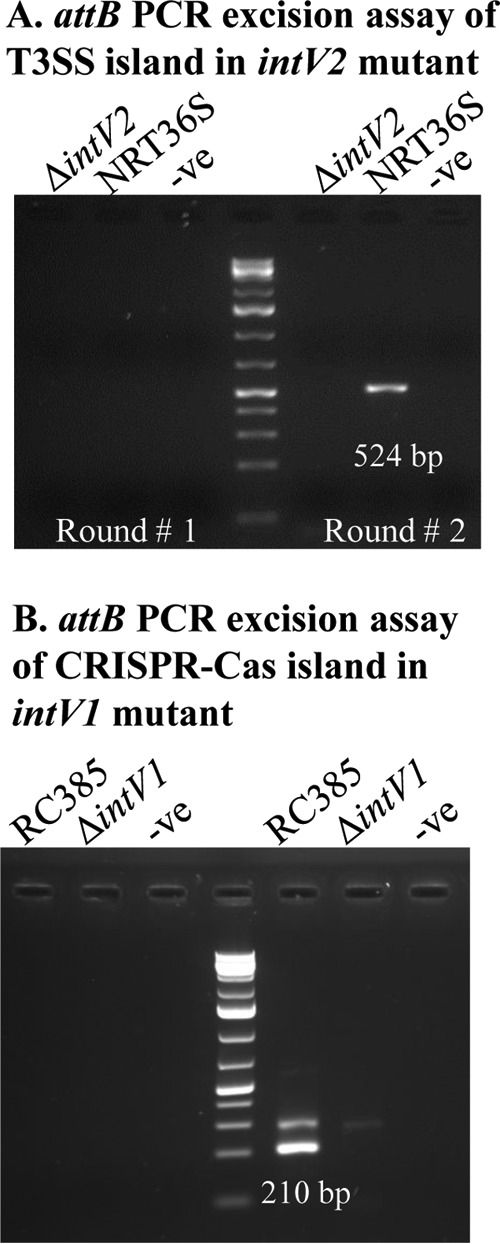
Integrases are essential for T3SS and CRISPR island excision. The two-stage attB PCR amplification assay was used to detect excision of VPI-3 in ΔintV2 and wild-type NRT36S (A) and of VPI-6 in ΔintV1 and wild-type RC385 (B). No PCR product was detected in the first round of PCR and wild-type attB excision products of 524 bp and 210 bp were detected in the second round of PCR for NRT36S and RC385, respectively, and no product was detected for NRT36S ΔintV2 or RC385 ΔintV1.
The CRISPR-Cas island can excise from the RC385 V. cholerae genome.
Next, we sought to determine whether the CRISPR-Cas island excised as a unit from the chromosome of V. cholerae RC385 (Fig. 7). Primers were designed to amplify the attP and attB sites of VPI-6 (Fig. 7D and E). In the attP assay, an expected 198-bp attP PCR product was detected in round two of PCR in V. cholerae RC385. In the attB PCR assay, a PCR product of 210 bp was detected in round two of PCR and was confirmed by sequencing (Fig. 7E). Strain SG-7 was used as a negative control, as it does not contain an island at the tmRNA locus, and as expected, no excision product was observed in the attP assay and an empty attB site was detected (Fig. 7D and E). These data indicated that VPI-6 can excise from the chromosome as a complete unit. A similar CRISPR-Cas island region in a V. cholerae strain isolated in Australia was also shown to excise from the genome, presumably using a similar mechanism (49).
To examine whether the cognate integrase of RC385 VPI-6 (IntV1) is also necessary for the excision, we constructed an in-frame deletion of intV1 to create mutant strain JDBIntV1. We performed the attB excision assay on wild-type and JDBIntV1 strains to determine whether excision occurs (Fig. 8B). No PCR product was observed in either round of the attB PCR assay for strain JDBIntV1, whereas a band of the expected size was present for the wild type, indicating that the cognate VPI-3 integrase is necessary for excision of this island (Fig. 8B).
These data confirm that conserved recombination modules that are widespread among isolates are also functionally conserved. These recombination modules can acquire new cargo genes and new combinations of cargo genes, suggesting a mechanism for the horizontal transfer and acquisition of T3SS and CRISPR-Cas systems as complete units.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
All bacterial strains and plasmids used in this study are listed in Table 4. Bacterial strains were grown in lysogeny broth (LB) (Fisher Scientific, Fair Lawn, NJ) overnight at 37°C aerobically at 225 rpm unless otherwise noted. The diaminopimelic acid (DAP) auxotroph Escherichia coli β2155λpir was supplemented with 0.3 mM DAP. When appropriate, medium was supplemented with the following antibiotics: 200 μg/ml streptomycin (Sm), 25 μg/ml chloramphenicol (Cm), and 100 μg/ml ampicillin (Amp). LB agar was supplemented with 10% sucrose for double cross screening.
TABLE 4.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Reference(s) or source |
|---|---|---|
| V. cholerae | ||
| N16961 | O1, El Tor strain, Bangladesh, clinical, 1975, VPI-1, VPI-2, VSP-I, VSP-II | 59 |
| SG7 | Non-O1, nonclinical, VPI negative | Lab collection |
| JDBIntV1 | RC385, intV1 deletion | This study |
| MRCIntV2 | NRT36S, intV2 deletion | This study |
| VPI-6 CRISPR strains | ||
| RC385 | O135, Chesapeake Bay, USA, plankton, 1998 | 14, 60 |
| RC586 | O133, Chesapeake Bay, USA, water, 1999 | 15, 16 |
| TM11079-80 | O1, El Tor, Brazil, sewage, 1980 | 14, 16 |
| HC-36A1 | −, Haiti, clinical, 2010 | 43 |
| VPI-3 T3SS strains | ||
| NRT36S | O31, Japan, clinical | 34 |
| AM-19926 | O39, Bangladesh, clinical, 2001 | 16 |
| TMA-21 | −, Brazil, sewage, 1980 | 16 |
| VC12129 | O1, El Tor, Australia, water, 1985 | 16 |
| EM1676A | O1, Bangladesh, water, 2011 | |
| NHCC008d | −, Bangladesh, clinical, 2010 | 43 |
| VC35 | −, Malaysia, clinical, 2004 | 43 |
| HE-25 | −, Haiti, water, 2010 | |
| P-18785 | −, Russia, clinical, 2005 | |
| CP1115 | O75, Florida, USA, clinical, 2010 | |
| 1587 | O12, Peru, clinical, 1994 | 16 |
| 623-39 | −, Bangladesh, water, 2002 | 16 |
| E. coli | ||
| DH5αλpir | Lab collection | |
| β2155λpir | Donor for bacterial conjugation, DAP auxotroph | Lab collection |
| Plasmids | ||
| pJET1.2 | Cloning vector, Ampr | |
| pJET1.2ΔintV1 | pJET1.2 harboring truncated intV1 gene | Fermentas |
| pJET1.2ΔintV2 | pJET1.2 harboring truncated intV2 gene | Fermentas |
| pDS132 | Suicide vector, SacB, Cmr | Lab collection |
| pDS132ΔintV1 | pDS132 harboring truncated intV1 gene | This study |
| pDS132ΔintV2 | pDS132 harboring truncated intV2 gene | This study |
Comparative genomics and phylogenetic analyses.
The FASTA sequence of the CRISPR-Cas island for each of the V. cholerae strains examined (RC385, RC586, and TMA11079-80) was downloaded from the NCBI genome database using the accession numbers NZ_GG774559.1, NZ_ADBD01000007.1, and NZ_ACHW01000035.1. The FASTA sequence for HC-36A1 (NZ_AXDR01000008.1) along with 15 additional strains with HC-XXXX strain numbers were downloaded from the NCBI database. The nucleotide sequence for each strain was compared to that of RC385 using the Artemis comparison tool (50). The VPI-3 region of V. cholerae NRT36S was assembled from raw whole-genome sequence data generated as previously described and obtained from Colin Stine's group (34). The NRT36S contigs were aligned to the T3SS region present in V. cholerae strain AM-19226 (GCA_000153785.1). The FASTA sequence for each of the strains examined was downloaded from the databases using the NCBI accession numbers GCA_000153785.1, GCA_000174295.1, GCA_000279265.1, GCA_000348345.1, GCA_000174115.1, GCA_000299495.1, GCA_000348425.1, GCA_000168895.1, GCA_000387605.1, GCA_000338215.1, and GCF_000152465.1. The nucleotide sequence for each strain was compared to that of NRT36S using the Artemis comparison tool (50). Using the T3SS ATPase from strain NRT36S as a seed, the NCBI genome database was examined for homologues (51). Protein sequences of the NanA and the integrase (IntV2) of VPI-3 from V. cholerae strains were obtained from NCBI database and uploaded into MEGA, where they were aligned using the ClustalW algorithm (45, 52, 53). After alignment, MEGA6 was used to generate neighbor-joining trees (45). Similarly, evolutionary analysis using MEGA6 was performed on the Cas1, Cas,3, and Cas6 proteins from species within the family Vibrionaceae that contained all three proteins (45).
CRISPR-Cas analysis.
The FASTA files for strains RC385, RC586, TMA11079-80, and HC-36A1 were used to identify CRISPR direct repeats and spacers in each sequence using the CRISPRFinder and CRISPRDetect programs (23, 24, 54). The CRISPRtionary program was used to determine how unique each spacer was to each strain (23, 24). We used the CRISPRMap program to assign type and subtype to each of the CRISPR-Cas regions (37). The spacer dictionaries generated by the CRISPRFinder and CRISPRDetect programs were used in the CRISPRTarget program to identify the most likely targets of CRISPR RNA spacers in the NCBI GenBank-phage and RefSeq-plasmid databases for all V. cholerae strains examined (55). Putative spacer target sequences were identified following a BLASTn search of the plasmid and bacteriophage databases (55). A cutoff score of 20 (the default value) was used for the analysis to filter out low-scoring hits from the output. Only scores above 22, which show greater than 80% sequence identity with targets, gave hits that consistently contained the Protospacer adjacent motif (PAM) sequence, a marker for a bona fide target.
VPI-3 and VPI-6 excision assays.
Genomic DNA was isolated from cells grown overnight in LB broth using the GNOME kit (MP Biomedicals, Santa Ana, CA) following the manufacturer's procedure and was used as the template in the attB assays. Plasmid DNA was isolated from cells grown overnight in LB broth using the Qiagen plasmid isolation kit (Qiagen, Valencia, CA) per the manufacturer's instructions and used as the template for the attP assay. Two-stage nested PCR assays were used to detect att sites present after excision of each island. In the chromosome following excision of either VPI-3 or VPI-6, an attB site is present that can be detected using the primer pairs listed in Table 5. Excision results in the formation of a circular intermediate that contains an attP site that can be detected by PCR using the primer pairs listed in Table 5. Since the excision event is rare under normal laboratory conditions, two-stage nested PCR assays are performed to detect both the attB and attP products. For PCR round one, 10 ng of V. cholerae genomic DNA was used as the template for the attB as previously described (13). One microliter of round one PCR cocktail was used as the template for round two of PCR, and products were analyzed on a 2% agarose gel. Vibrio cholerae SG7 was used as a negative control as it does not contain any island regions at the VPI-1 or VPI-2 insertion sites, and V. cholerae N16961 was used as a positive control as VPI-2 is inserted at the same tRNA-serine locus and was previously detected (13). The attP and attB PCR products were confirmed by sequencing. At least three biological replicates of each strain were examined.
TABLE 5.
Primers used in this study
| Category and primer | Sequence (5′→3′)a | Product size (bp) |
|---|---|---|
| Mutant primers | ||
| VC1758FF | AAGCAAACGCACTCAATGCG | |
| NRTintV2FR | GGATTTCTGCCTACTACCGT | 2,269 |
| VC1785A | tctagaGATTCGGTGAGTTGTCCGAG | |
| NRTintV2B | CATGAGCGAGAATTACTTGG | 523 |
| NRTintV2C | CCAAGTAATTCTCGCTCATGAAGCTACAGTGTCGCTGGTG | |
| NRTintV2D | gagctcGCCTGTGGCGAGATAGAGAC | 678 |
| RC385intSOEA | gtagcatgcTAGCCATTCGTTAGCGTGTC | 556 |
| RC385intSOEB | ATCGTCTTGTGGATCGATGC | |
| RC385intSOEC | GCATCGATCCACAAGACGATGCCCACGTTGACGATAACCAAG | 548 |
| RC385intSOED | tatgagctcGCCGCACAGGCAGCTTAGTT | |
| RC385intSOEFF | CTAGCTTCCGCTTGTAAGAC | 2,287 |
| RC385intSOEFR | GGGTTTAGACTTGGTATCAG | |
| Excision assay primers | ||
| VPI-3 primers | ||
| VPI-2attF | AGAGTGAAAGTCGCCAAAG | |
| VPI-2attR | GGTGCAATTTCGCATGTTGC | 524 |
| VPI-2 InvVC1808F | AGCTAGACAGATTAGCTAACC | |
| CnVC1758B | TTGCCATGAGCGAGAATTGC | 1,352 |
| NestVC1758comR | AGAATTGCTTGGACGTACGC | |
| NestVC1809comF | GCGTTAACTGAGAAAGTGTG | 461 |
| VPI-6 primers | ||
| VPI-1 attBR1 | TGTAAGACGGGGAAATCAGG | |
| RC385attBF1 | TTTGTTGATGAGCAGGATGG | 427 |
| VPI-1 attBR2 | ATTCGTTAGCGTGTCGG | |
| RC385attBF2 | AGTGAATCTTGATGAGACGC | 210 |
| NestVC0847F3 | TTTCTCTCTAGGTTTGGAGG | |
| RC385attPR1 | CTACTGCTTTATCAGGACCC | 413 |
| RC385attPF2 | GCTGCTATGGAATCTTGTGG | |
| RC385attPR2 | GGTACACTAAAAGGTACACC | 198 |
Lowercase and italics indicate restriction sites and complementary sequence tags, respectively.
Integrase mutant construction.
Integrase deletion mutants of V. cholerae NRT36S and RC385 were constructed using splicing by overlap extension (SOE) and homologous recombination (56). Primers were designed to create an in-frame deletion of the VPI-3 intV2 gene in strain NRT36S and the VPI-6 intV1 gene in strain RC385. These mutants were named MRCIntV2 and JDBIntV1, respectively, and are listed in Table 5. Briefly, for intV2 deletion, V. cholerae NRT36S genomic DNA was used as the template for PCR with VC1758A/NRTintv2B and NRTintv2C/NRTintv2D primer pairs to generate AB and CD products. Purified AB and CD products were used as the template for PCR with the primer pair VC1758A/NRTintv2D to create the truncated intV2 gene, utilizing the overlapping sequences found on NRTintv2B and NRTintv2C (Table 5). This intV2 truncated PCR product was blunt-end cloned into pJET1.2 (Fisher Scientific) and transformed into Escherichia coli DH5α. Plasmid DNA (pJET1.2ΔintV2) was isolated using the Qiagen plasmid minikit and digested with SacI and XbaI restriction enzymes (Fisher Scientific). The digested ΔintV2 fragment was cloned into the suicide vector pDS132, forming pDS132ΔintV2, and transformed into the DAP auxotroph E. coli β2155. Escherichia coli β2155 pDS132ΔintV2 served as the donor strain in conjugation with V. cholerae NRT36S. Transconjugants were selected for on LB agar supplemented with Sm and Cm, and colony PCR with primer pair VC1758A/NRTintv2D was used to detect the single-crossover event. Single-cross colonies were grown in the presence of 10% sucrose without Cm to detect homologous recombination and replacement of the full intV2 gene with the truncated intV2 in V. cholerae NRT36S. A similar strategy was used to construct an in-frame deletion of intV1 in RC385. The double-crossover events were screened with AD and flanking primer sets and confirmed by sequencing.
ACKNOWLEDGMENTS
We thank Nathan McDonald, Abish Regmi, Gwen Gregory, and Aoife Boyd for reviewing the manuscript and helpful discussions and three anonymous reviewers for constructive and invaluable feedback. We thank John Heidelberg, Christopher Grim, and Anwar Huq for help with locating V. cholerae strains.
M.R.C. was funded by a University of Delaware graduate fellowship award.
REFERENCES
- 1.De SN. 1959. Enterotoxicity of bacteria-free culture-filtrate of Vibrio cholerae. Nature 183:1533–1534. doi: 10.1038/1831533a0. [DOI] [PubMed] [Google Scholar]
- 2.Waldor MK, Mekalanos JJ. 1996. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 272:1910–1914. doi: 10.1126/science.272.5270.1910. [DOI] [PubMed] [Google Scholar]
- 3.Taylor RK, Miller VL, Furlong DB, Mekalanos JJ. 1987. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc Natl Acad Sci U S A 84:2833–2837. doi: 10.1073/pnas.84.9.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herrington DA, Hall RH, Losonsky G, Mekalanos JJ, Taylor RK, Levine MM. 1988. Toxin, toxin-coregulated pili, and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J Exp Med 168:1487–1492. doi: 10.1084/jem.168.4.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karaolis DK, Johnson JA, Bailey CC, Boedeker EC, Kaper JB, Reeves PR. 1998. A Vibrio cholerae pathogenicity island associated with epidemic and pandemic strains. Proc Natl Acad Sci U S A 95:3134–3139. doi: 10.1073/pnas.95.6.3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Umayam L, Gill SR, Nelson KE, Read TD, Tettelin H, Richardson D, Ermolaeva MD, Vamathevan J, Bass S, Qin H, Dragoi I, Sellers P, McDonald L, Utterback T, Fleishmann RD, Nierman WC, White O. 2000. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406:477–483. doi: 10.1038/35020000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jermyn WS, Boyd EF. 2002. Characterization of a novel Vibrio pathogenicity island (VPI-2) encoding neuraminidase (nanH) among toxigenic Vibrio cholerae isolates. Microbiology 148:3681–3693. doi: 10.1099/00221287-148-11-3681. [DOI] [PubMed] [Google Scholar]
- 8.Almagro-Moreno S, Boyd EF. 2009. Sialic acid catabolism confers a competitive advantage to pathogenic Vibrio cholerae in the mouse intestine. Infect Immun 77:3807–3816. doi: 10.1128/IAI.00279-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McDonald ND, Lubin JB, Chowdhury N, Boyd EF. 2016. Host-derived sialic acids are an important nutrient source that is required for optimal bacterial fitness in vivo. mBio 7:e02237-15. doi: 10.1128/mBio.02237-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rajanna C, Wang J, Zhang D, Xu Z, Ali A, Hou YM, Karaolis DK. 2003. The Vibrio pathogenicity island of epidemic Vibrio cholerae forms precise extrachromosomal circular excision products. J Bacteriol 185:6893–6901. doi: 10.1128/JB.185.23.6893-6901.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murphy RA, Boyd EF. 2008. Three pathogenicity islands of Vibrio cholerae can excise from the chromosome and form circular intermediates. J Bacteriol 190:636–647. doi: 10.1128/JB.00562-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Almagro-Moreno S, Napolitano MG, Boyd EF. 2010. Excision dynamics of Vibrio pathogenicity island-2 from Vibrio cholerae: role of a recombination directionality factor VefA. BMC Microbiol 10:306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carpenter MR, Rozovsky S, Boyd EF. 2016. Pathogenicity island cross talk mediated by recombination directionality factors facilitates excision from the chromosome. J Bacteriol 198:766–776. doi: 10.1128/JB.00704-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chun J, Grim CJ, Hasan NA, Lee JH, Choi SY, Haley BJ, Taviani E, Jeon YS, Kim DW, Brettin TS, Bruce DC, Challacombe JF, Detter JC, Han CS, Munk AC, Chertkov O, Meincke L, Saunders E, Walters RA, Huq A, Nair GB, Colwell RR. 2009. Comparative genomics reveals mechanism for short-term and long-term clonal transitions in pandemic Vibrio cholerae. Proc Natl Acad Sci U S A 106:15442–15447. doi: 10.1073/pnas.0907787106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haley BJ, Grim CJ, Hasan NA, Choi SY, Chun J, Brettin TS, Bruce DC, Challacombe JF, Detter JC, Han CS, Huq A, Colwell RR. 2010. Comparative genomic analysis reveals evidence of two novel Vibrio species closely related to V. cholerae. BMC Microbiol 10:154. doi: 10.1186/1471-2180-10-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haley BJ, Choi SY, Grim CJ, Onifade TJ, Cinar HN, Tall BD, Taviani E, Hasan NA, Abdullah AH, Carter L, Sahu SN, Kothary MH, Chen A, Baker R, Hutchinson R, Blackmore C, Cebula TA, Huq A, Colwell RR. 2014. Genomic and phenotypic characterization of Vibrio cholerae non-O1 isolates from a US Gulf Coast cholera outbreak. PLoS One 9:e86264. doi: 10.1371/journal.pone.0086264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mojica F, Díez-Villaseñor C, García-Martínez J, Soria E. 2005. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60:174–182. doi: 10.1007/s00239-004-0046-3. [DOI] [PubMed] [Google Scholar]
- 18.Mojica FJ, Diez-Villasenor C, Soria E, Juez G. 2000. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol Microbiol 36:244–246. doi: 10.1046/j.1365-2958.2000.01838.x. [DOI] [PubMed] [Google Scholar]
- 19.Mojica FJ, Ferrer C, Juez G, Rodriguez-Valera F. 1995. Long stretches of short tandem repeats are present in the largest replicons of the Archaea Haloferax mediterranei and Haloferax volcanii and could be involved in replicon partitioning. Mol Microbiol 17:85–93. doi: 10.1111/j.1365-2958.1995.mmi_17010085.x. [DOI] [PubMed] [Google Scholar]
- 20.Horvath P, Barrangou R. 2010. CRISPR/Cas, the immune system of bacteria and archaea. Science 327:167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 21.Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, van der Oost J, Koonin EV. 2011. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol 9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Makarova KS, Koonin EV. 2015. Annotation and classification of CRISPR-Cas systems. Methods Mol Biol 1311:47–75. doi: 10.1007/978-1-4939-2687-9_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grissa I, Vergnaud G, Pourcel C. 2007. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res 35:W52–W57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grissa I, Vergnaud G, Pourcel C. 2007. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8:172. doi: 10.1186/1471-2105-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jansen R, Embden JD, Gaastra W, Schouls LM. 2002. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43:1565–1575. doi: 10.1046/j.1365-2958.2002.02839.x. [DOI] [PubMed] [Google Scholar]
- 26.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 27.Hille F, Charpentier E. 26 September 2016. CRISPR-Cas: biology, mechanisms and relevance. Philos Trans R Soc Lond B Biol Sci doi: 10.1098/rstb.2015.0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Makarova K, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E, Haft DH, Horvath P, Moineau S, Mojica FJ, Terns RM, Terns MP, White MF, Yakunin AF, Garrett RA, van der Oost J, Backofen R, Koonin E. 2015. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol 13:722–736. doi: 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chakraborty S, Waise TM, Hassan F, Kabir Y, Smith MA, Arif M. 2009. Assessment of the evolutionary origin and possibility of CRISPR-Cas (CASS) mediated RNA interference pathway in Vibrio cholerae O395. In Silico Biol 9:245–254. [PubMed] [Google Scholar]
- 30.Seed K, Lazinski DW, Calderwood SB, Camilli A. 2013. A bacteriophage encodes its own CRISPR/Cas adaptive response to evade host innate immunity. Nature 494:489–491. doi: 10.1038/nature11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Box AM, McGuffie MJ, O'Hara BJ, Seed KD. 2016. Functional analysis of bacteriophage immunity through a type I-E CRISPR-Cas system in Vibrio cholerae and its application in bacteriophage genome engineering. J Bacteriol 198:578–590. doi: 10.1128/JB.00747-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hueck CJ. 1998. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev 62:379–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dziejman M, Serruto D, Tam VC, Sturtevant D, Diraphat P, Faruque SM, Rahman MH, Heidelberg JF, Decker J, Li L, Montgomery KT, Grills G, Kucherlapati R, Mekalanos JJ. 2005. Genomic characterization of non-O1, non-O139 Vibrio cholerae reveals genes for a type III secretion system. Proc Natl Acad Sci U S A 102:3465–3470. doi: 10.1073/pnas.0409918102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Y, Johnson JA, Pusch GD, Morris JG Jr, Stine OC. 2007. The genome of non-O1 Vibrio cholerae NRT36S demonstrates the presence of pathogenic mechanisms that are distinct from O1 Vibrio cholerae. Infect Immun 75:2645–2647. doi: 10.1128/IAI.01317-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, Nelson WC, Heidelberg JF, Mekalanos JJ. 2006. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci U S A 103:1528–1533. doi: 10.1073/pnas.0510322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Basler M. 2015. Type VI secretion system: secretion by a contractile nanomachine. Philos Trans R Soc Lond B Biol Sci 370:1679. doi: 10.1098/rstb.2015.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lange SJ, Alkhnbashi OS, Rose D, Will S, Backofen R. 2013. CRISPRmap: an automated classification of repeat conservation in prokaryotic adaptive immune systems. Nucleic Acids Res 41:8034–8044. doi: 10.1093/nar/gkt606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33:W244–248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Belkum A, Soriaga LB, LaFave MC, Akella S, Veyrieras JB, Barbu EM, Shortridge D, Blanc B, Hannum G, Zambardi G, Miller K, Enright MC, Mugnier N, Brami D, Schicklin S, Felderman M, Schwartz AS, Richardson TH, Peterson TC, Hubby B, Cady KC. 2015. Phylogenetic distribution of CRISPR-Cas systems in antibiotic-resistant Pseudomonas aeruginosa. mBio 6:e01796-15. doi: 10.1128/mBio.01796-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gunderson FF, Cianciotto NP. 2013. The CRISPR-associated gene cas2 of Legionella pneumophila is required for intracellular infection of amoebae. mBio 4:e00074-13. doi: 10.1128/mBio.00074-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rao C, Guyard C, Pelaz C, Wasserscheid J, Bondy-Denomy J, Dewar K, Ensminger AW. 2016. Active and adaptive Legionella CRISPR-Cas reveals a recurrent challenge to the pathogen. Cell Microbiol 18:1319–1338. doi: 10.1111/cmi.12586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pan X, Lührmann A, Satoh A, Laskowski-Arce MA, Roy C. 2008. Ankyrin repeat proteins comprise a diverse family of bacterial type IV effectors. Science 320:1651–1654. doi: 10.1126/science.1158160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Azarian T, Ali A, Johnson JA, Mohr D, Prosperi M, Veras NM, Jubair M, Strickland SL, Rashid MH, Alam MT, Weppelmann TA, Katz LS, Tarr CL, Colwell RR, Morris JG Jr, Salemi M. 2014. Phylodynamic analysis of clinical and environmental Vibrio cholerae isolates from Haiti reveals diversification driven by positive selection. mBio 5:e01824-14. doi: 10.1128/mBio.01824-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gladney L, Katz LS, Knipe KM, Rowe LA, Conley AB, Rishishwar L, Mariño-Ramírez L, Jordan IK, Tarr C. 2014. Genome sequences of Vibrio navarrensis, a potential human pathogen. Genome Announc 2:e01188-14. doi: 10.1128/genomeA.01188-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Okada N, Iida T, Park KS, Goto N, Yasunaga T, Hiyoshi H, Matsuda S, Kodama T, Honda T. 2009. Identification and characterization of a novel type III secretion system in trh-positive Vibrio parahaemolyticus strain TH3996 reveal genetic lineage and diversity of pathogenic machinery beyond the species level. Infect Immun 77:904–913. doi: 10.1128/IAI.01184-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okada N, Matsuda S, Matsuyama J, Park KS, de los Reyes C, Kogure K, Honda T, Iida T. 2010. Presence of genes for type III secretion system 2 in Vibrio mimicus strains. BMC Microbiol 10:302. doi: 10.1186/1471-2180-10-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morita M, Yamamoto S, Hiyoshi H, Kodama T, Okura M, Arakawa E, Alam M, Ohnishi M, Izumiya H, Watanabe H. 2013. Horizontal gene transfer of a genetic island encoding a type III secretion system distributed in Vibrio cholerae. Microbiol Immunol 57:334–339. doi: 10.1111/1348-0421.12039. [DOI] [PubMed] [Google Scholar]
- 49.Labbate M, Orata FD, Petty NK, Jayatilleke ND, King WL, Kirchberger PC, Allen C, Mann G, Mutreja A, Thomson NR, Boucher Y, Charles IG. 2016. A genomic island in Vibrio cholerae with VPI-1 site-specific recombination characteristics contains CRISPR-Cas and type VI secretion modules. Sci Rep 6:36891. doi: 10.1038/srep36891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 51.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Higgins DG, Thompson JD, Gibson TJ. 1996. Using CLUSTAL for multiple sequence alignments. Methods Enzymol 266:383–402. doi: 10.1016/S0076-6879(96)66024-8. [DOI] [PubMed] [Google Scholar]
- 53.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Biswas A, Staals RH, Morales SE, Fineran PC, Brown CM. 2016. CRISPRDetect: a flexible algorithm to define CRISPR arrays. BMC Genomics 17:356. doi: 10.1186/s12864-016-2627-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Biswas A, Gagnon JN, Brouns SJ, Fineran PC, Brown CM. 2013. CRISPRTarget: bioinformatic prediction and analysis of crRNA targets. RNA Biol 10:817–827. doi: 10.4161/rna.24046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 57.Saitou N, Nei M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425. [DOI] [PubMed] [Google Scholar]
- 58.Nei M, Jin L. 1989. Variances of the average numbers of nucleotide substitutions within and between populations. Mol Biol Evol 6:290–300. [DOI] [PubMed] [Google Scholar]
- 59.Kaper JB, Lockman H, Baldini MM, Levine MM. 1984. Recombinant nontoxinogenic Vibrio cholerae strains as attenuated cholera vaccine candidates. Nature 308:655–658. doi: 10.1038/308655a0. [DOI] [PubMed] [Google Scholar]
- 60.Taviani E, Grim CJ, Choi J, Chun J, Haley B, Hasan NA, Huq A, Colwell RR. 2010. Discovery of novel Vibrio cholerae VSP-II genomic islands using comparative genomic analysis. FEMS Microbiol Lett 308:130–137. doi: 10.1111/j.1574-6968.2010.02008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]