

ABSTRACT
Posttranslocational protein folding in the Gram-positive biofilm-forming actinobacterium Actinomyces oris is mediated by a membrane-bound thiol-disulfide oxidoreductase named MdbA, which catalyzes oxidative folding of nascent polypeptides transported by the Sec translocon. Reoxidation of MdbA involves a bacterial vitamin K epoxide reductase (VKOR)-like protein that contains four cysteine residues, C93/C101 and C175/C178, with the latter forming a canonical CXXC thioredoxin-like motif; however, the mechanism of VKOR-mediated reoxidation of MdbA is not known. We present here a topological view of the A. oris membrane-spanning protein VKOR with these four exoplasmic cysteine residues that participate in MdbA reoxidation. Like deletion of the VKOR gene, alanine replacement of individual cysteine residues abrogated polymicrobial interactions and biofilm formation, concomitant with the failure to form adhesive pili on the bacterial surface. Intriguingly, the mutation of the cysteine at position 101 to alanine (C101A mutation) resulted in a high-molecular-weight complex that was positive for MdbA and VKOR by immunoblotting and was absent in other alanine substitution mutants and the C93A C101A double mutation and after treatment with the reducing agent β-mercaptoethanol. Consistent with this observation, affinity purification followed by immunoblotting confirmed this MdbA-VKOR complex in the C101A mutant. Furthermore, ectopic expression of the Mycobacterium tuberculosis VKOR analog in the A. oris VKOR deletion (ΔVKOR) mutant rescued its defects, in contrast to the expression of M. tuberculosis VKOR variants known to be nonfunctional in the disulfide relay that mediates reoxidation of the disulfide bond-forming catalyst DsbA in Escherichia coli. Altogether, the results support a model of a disulfide relay, from its start with the pair C93/C101 to the C175-X-X-C178 motif, that is required for MdbA reoxidation and appears to be conserved in members of the class Actinobacteria.
IMPORTANCE It has recently been shown in the high-GC Gram-positive bacteria (or Actinobacteria) Actinomyces oris and Corynebacterium diphtheriae that oxidative folding of nascent polypeptides transported by the Sec machinery is catalyzed by a membrane-anchored oxidoreductase named MdbA. In A. oris, reoxidation of MdbA requires a bacterial VKOR-like protein, and yet, how VKOR mediates MdbA reoxidation is unknown. We show here that the A. oris membrane-spanning protein VKOR employs two pairs of exoplasmic cysteine residues, including the canonical CXXC thioredoxinlike motif, to oxidize MdbA via a disulfide relay mechanism. This mechanism of disulfide relay is essential for pilus assembly, polymicrobial interactions, and biofilm formation and appears to be conserved in members of the class Actinobacteria, including Mycobacterium tuberculosis.
KEYWORDS: Actinobacteria, disulfide bond formation, oxidative protein folding, pilus assembly, biofilm formation, oxidoreductases, polymicrobial interactions, sortase
INTRODUCTION
Oxidative protein folding via disulfide bond formation is a central process that is thought to occur in an oxidizing environment, such as the eukaryotic endoplasmic reticulum and the periplasm of Gram-negative bacteria (1). Protein-disulfide isomerase (PDI) is an example of disulfide bond-forming machines in the eukaryotes, whereas the thiol-disulfide oxidoreductase DsbA is an equivalent apparatus in Gram-negative bacteria (2). In the model organism Escherichia coli, DsbA catalyzes disulfide bond formation of nascent polypeptides exported by the Sec translocon (3). Subsequently, DsbA becomes reduced, and the oxidized state of DsbA is maintained by a membrane-bound oxidoreductase called DsbB (4, 5). Like PDIs, DsbA and DsbB contain a redox-active CXXC motif (6). In addition, DsbB possesses a second redox center with a pair of cysteine residues, one of which forms a mixed disulfide bond with DsbA during the process of electron transfer from this redox center to the CXXC motif (7). With the notorious exceptions of the Bacteroidetes, Fusobacteria, and Thermotogales genera and several anaerobes, the majority of Gram-negative bacteria have DsbA and DsbB homologs (8).
Intriguingly, although considered not to possess a periplasmic space, many Gram-positive bacteria, including Bacillus subtilis, Staphylococcus aureus, Mycobacterium tuberculosis, and Streptococcus gordonii, are known to harbor DsbA-like proteins (9–12). Recent findings (13, 14) of the thiol-disulfide oxidoreductase MdbA enzymes in Actinomyces oris and Corynebacterium diphtheriae cement the idea that disulfide bond formation is the major folding mechanism of nascent polypeptides exported by the Sec translocon in high-GC-content Gram-positive bacteria or members of the class Actinobacteria (15). In A. oris, an oral actinobacterium that utilizes fimbriae or pili for biofilm formation, polymicrobial interactions, and host cell adherence (16–18), MdbA is an essential enzyme that catalyzes posttranslocational folding of fimbrial proteins, as well as other protein precursors (13). The MdbA activity requires the cysteine residues within the catalytic CXXC motif, as their replacement with alanine abrogates pilus assembly, biofilm formation, and polymicrobial interactions (13). Since pilus assembly in A. oris is closely linked to polymicrobial interactions (18, 19), a Tn5 screen for mutants defective in coaggregation revealed that a bacterial vitamin K epoxide reductase (VKOR) analog is required to maintain the oxidized status of A. oris MdbA, as demonstrated by alkylation with methoxypolyethylene glycol-maleimide (Mal-PEG) (13). A. oris VKOR also harbors a catalytic CXXC motif; replacing the first cysteine of this motif with alanine (C175A) abrogates pilus assembly and bacterial coaggregation (13).
The first bacterial VKOR involved in disulfide bond formation was reported previously, i.e., M. tuberculosis VKOR; when expressed in an E. coli dsbB mutant, M. tuberculosis VKOR rescues the nonmotile defect of this mutant (8). M. tuberculosis VKOR appears to be a functional analog of DsbB, as the expression of this protein in the E. coli dsbB mutant restores the oxidized state of DsbA, which is otherwise in a reduced form (20). M. tuberculosis VKOR is a membrane protein with 4 catalytic cysteine residues forming two redox centers of disulfide bonds, one of which is in the redox catalytic CXXC motif; the substitution of alanine for any cysteine residues results in the nonmotile phenotype of E. coli (21). Interestingly, M. tuberculosis VKOR forms a mixed disulfide bond with E. coli DsbA via C57, the first cysteine residue of another redox center of VKOR (21), just as DsbB forms an intermediate with DsbA via C104 (22). While deletion of the VKOR gene causes a severe growth defect in M. tuberculosis, it remains unclear how the VKOR analog acts on the cognate disulfide bond-forming machine DsbA in this actinobacterium (20).
Here, we report the biochemical characterization of A. oris VKOR, which displays a topological arrangement similar to that of M. tuberculosis VKOR. A. oris VKOR harbors 4 redox-active cysteine residues, forming two pairs of redox centers (C93/C101 and C175/C178), with the latter constituting the redox-active CXXC motif. Similar to the phenotypes of the VKOR deletion mutant, in which MdbA is reduced (13), mutation of these Cys residues to alanine abrogates polymicrobial interactions, biofilm formation, and pilus assembly. Through this mutational analysis, we were able to isolate an MdbA-VKOR intermediate in which the C93 residue of VKOR is postulated to form a mixed disulfide bond with MdbA. Significantly, ectopic expression of M. tuberculosis VKOR in the A. oris VKOR mutant rescues the defects of this mutant, albeit not to the same level as A. oris VKOR. In contrast, the A. oris VKOR mutant expressing M. tuberculosis VKOR Cys-to-Ala variants defective in disulfide relay remains defective. We propose a model of VKOR-mediated reactivation of MdbA that appears to be conserved in the Actinobacteria.
RESULTS
Membrane topology of A. oris VKOR.
Based on various membrane topology prediction algorithms, including TMHMM (23), HMMTOP (24), TMPred (25), Predict Protein Open (26), CCTOP (27), and SPLIT (28), A. oris VKOR is expected to have five transmembrane (TM) domains with an N-in/C-out topology, in which the N terminus faces the cytoplasm and the C terminus faces toward the exoplasm (Fig. 1A). The first TM segment contains 2 Cys residues (C65 and C73), followed by 2 more Cys residues (C93 and C101) located within an exoplasmic loop connecting the first and second TM segments; in addition, the exoplasmic loop between the third and fourth TM segments harbors the catalytic CXXC motif (C175 and C178) (Fig. 1A).
FIG 1.
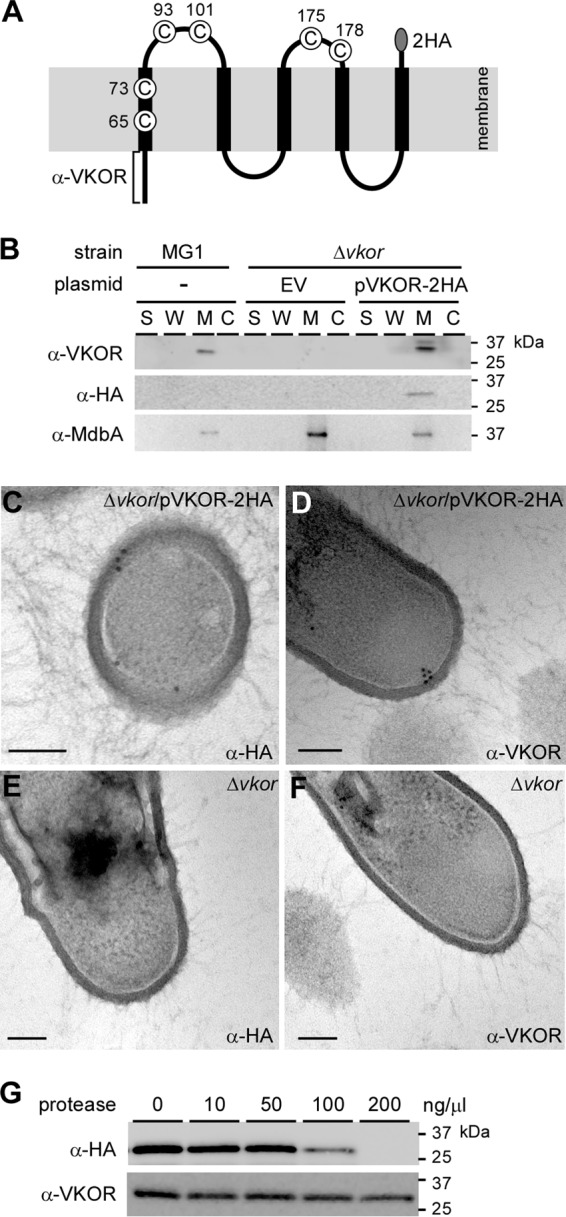
Membrane topology of A. oris VKOR. (A) Presented is a topological model of the membrane protein VKOR, which possesses 6 cysteine (C) residues. C175 and C178 are part of the catalytic CXXC motif. To detect VKOR, a recombinant VKOR protein harboring a C-terminal double HA tag (2HA) was generated; in addition, the N-terminal 56-amino-acid epitope was used to raise polyclonal antibodies (square bracket). (B) Cell fractionation was performed on the parental strain MG1 and its isogenic ΔVKOR deletion mutants containing an empty vector (EV) or the complementing plasmid pVKOR-2HA. Protein samples isolated from culture medium (S), cell wall (W), membrane (M), and cytoplasmic (C) fractions were blotted with antibodies against the first 56 amino acids of VKOR (α-VKOR) and the 2×HA tag (α-HA). Anti-MdbA Ab (α-MdbA) was used to detect the membrane-bound MdbA. (C to F) Ultrathin sections of cells of the indicated strains were subjected to immunoelectron microscopy, for which samples were treated with anti-HA (C and E) or anti-VKOR (D and F) Ab, followed by IgG-conjugated gold particles (12 nm). The samples were viewed using an electron microscope after staining with 1% uranyl acetate. Scale bars indicate 100 nm. (G) Protoplasts of the ΔVKOR mutant expressing HA-tagged VKOR (pVKOR-2HA) were treated with increasing concentrations of proteinase K. The protein samples obtained were immunoblotted with anti-VKOR and anti-HA Ab. Molecular mass markers are indicated.
To detect cellular localization of VKOR, we generated polyclonal antibodies (Ab) against the first 56 residues comprising the cytoplasmic loop of VKOR (i.e., anti-VKOR Ab). Additionally, we cloned a recombinant vector, pVKOR-2HA, expressing a VKOR protein with a double hemagglutinin (2×HA) tag at the C terminus (Fig. 1A). This vector was introduced into a VKOR deletion mutant (ΔVKOR mutant) (13). Log-phase cells of the parental MG1 strain and the ΔVKOR mutant, as well as this mutant carrying an empty vector (EV), were subjected to cell fractionation. Protein samples collected from the culture medium (S), cell wall (W), membrane (M), and cytoplasmic (C) fractions were analyzed by immunoblotting with specific antibodies against HA tags, VKOR, and MdbA, which is a membrane protein (13). As shown by the results in Fig. 1B, anti-VKOR Ab detected a band migrating between the 37- and 25-kDa markers in the membrane fraction of the MG1 strain (predicted VKOR molecular mass of 26.5 kDa). Deletion of VKOR eliminated membrane detection of VKOR, which was rescued by ectopic expression of VKOR with a 2×HA tag (Fig. 1B, last 8 lanes; note a slight upshift in the membrane fraction of strain pVKOR-2HA). When blotted with anti-HA antibodies, the HA-labeled VKOR signal was only detected in the membrane fraction of strain pVKOR-2HA (Fig. 1B). As a control, antibodies against the membrane-bound MdbA detected MdbA in the membrane fraction of all three strains. The results indicate that VKOR is indeed a membrane protein and the two antibodies are specific.
To verify the membrane localization of VKOR, cell pellets of the ΔVKOR mutant and the complementing strain with pVKOR-2HA were thin sectioned. Ultrathin sections on nickel grids were stained with anti-VKOR or anti-HA Ab, followed by IgG antibodies conjugated to 12-nm gold particles. The samples were viewed with an electron microscope after negative staining. In the ΔVKOR/pVKOR-2HA strain, HA-labeled gold particles were largely detected on the exoplasmic side of the cytoplasmic membrane (Fig. 1C), whereas VKOR-labeled gold particles were observed on the cytoplasmic side of the membrane (Fig. 1D). No gold particles were detected in the ΔVKOR mutant (Fig. 1E and F). The data suggest that the two epitopes for these antibodies are differentially located on either side of the membrane. To confirm this, we treated the protoplasts of the ΔVKOR/pVKOR-2HA strain with increased concentrations of protease. Protein samples from this treatment were analyzed by Western blotting. As expected, at a high concentration of protease, the HA-reactive signal representing the HA-tagged VKOR protein was not observed. In contrast, the VKOR signal was visible, although it was slightly reduced (Fig. 1C) Altogether, the findings support a topological model of A. oris VKOR that spans the membrane five times, with the N terminus facing the cytoplasm and the C terminus exoplasmic (Fig. 1A).
Requirement of the VKOR catalytic cysteine residues for polymicrobial interactions, biofilm formation, and pilus assembly.
Previously, cysteine residue C175 within the catalytic CXXC motif has been shown to be essential for VKOR's function (13). However, the functional role of other cysteines has not been investigated. To examine this, we generated VKOR variants with individual Cys residues mutated to Ala, and the resulting strains were analyzed for their ability to coaggregate with other oral bacteria, form biofilms, and assemble surface pili. For bacterial coaggregation, A. oris cells were mixed with Streptococcus oralis and coaggregation was scored visually (see Materials and Methods). As previously reported, the coaggregation of A. oris with S. oralis is dependent on FimA and VKOR (13), as the deletion of either gene abrogated coaggregation (Fig. 2A, first 4 wells). Ectopic expression of wild-type VKOR or VKOR variants with alanine substitutions for the TM-associated C65 or C73 rescued the coaggregation defect of the ΔVKOR mutant (Fig. 2A, 5th to 7th wells). However, like the C175A mutation, Ala substitutions for C93, C101, and C178 abrogated bacterial coaggregation (Fig. 2A, last 4 wells).
FIG 2.

Requirement of the VKOR exoplasmic cysteine residues for bacterial coaggregation, biofilm formation, and pilus polymerization. (A) Coaggregation of various A. oris strains with receptor polysaccharide (RPS)-positive S. oralis (So34) was performed in a 12-well flat-bottom plate and was imaged using an AlphaImager. (B) Biofilms produced by the indicated strains were quantified by a crystal violet retention method that measures absorbance at 580 nm using a microplate reader. The results are presented as the average values from three independent experiments performed in triplicate. Statistical analysis was performed using one-way ANOVA. ***, P ≤ 0.001. (C and D) Cell wall fractions of A. oris strains were immunoblotted with specific antibodies against FimA (α-FimA) (C) or FimP (α-FimP) (D). Monomeric (arrowheads) and polymeric (square brackets) forms of FimA and FimP and molecular mass markers are shown.
The same set of A. oris strains was subjected to biofilm formation assays, and biofilm production was quantified by a crystal violet retention method (13). Similar to the results for the coaggregation phenotype testing described above, strains carrying the C93A, 101A, C175A, or C178A mutation failed to form biofilms, whereas the C65A and C73A mutants displayed no apparent defects (Fig. 2B). Since bacterial coaggregation and biofilm formation in A. oris require surface pili (29), we then examined pilus polymerization in the cell envelope, whereby the cell wall fractions of the indicated strains were isolated by cell fractionation and analyzed by immunoblotting with antibodies against the fimbrial shaft proteins FimA and FimP. Consistent with the above-described results, compared to the results for the wild-type MG1 strain and the isogenic VKOR mutants that express VKOR variants with the C65A or C73A mutation, the C93A, C101A, C175A, and C178A mutants were not able to form pilus polymers, and this phenotype was accompanied by a high level of degradation of pilus monomers (Fig. 2C and D). Finally, surface display of pilus polymers was determined by electron microscopy (EM), in which A. oris cells were deposited on nickel grids and stained with 1% uranyl acetate. As shown by the results in Fig. 3, the pilus assembly defects of the C93A, C101A, C175A, and C178A mutants mirrored the defects of these strains observed in pilus polymerization assays, as shown by the results in Fig. 2C and D. Altogether, the results indicate that four Cys residues forming the two exoplasmic Cys pairs (C93/C101 and C175/C178) are required for the VKOR function. The findings also suggest that the two pairs are involved in catalytic reactions leading to MdbA reactivation.
FIG 3.

Requirement of the VKOR exoplasmic cysteine residues for surface assembly of pili. A. oris cells were immobilized on nickel grids and stained with 1% uranyl acetate. The samples were visualized by transmission electron microscopy. Scale bars represent 0.5 μm.
Formation of the MdbA-VKOR mixed disulfide complex requires C93.
Previous studies on the M. tuberculosis VKOR reactivation of DsbA in E. coli demonstrate that M. tuberculosis VKOR interacts with DsbA, forming a mixed disulfide intermediate using the first cysteine residue, C57, in the first redox center (21). To examine this possibility in A. oris, we prepared the membrane fractions of the ΔVKOR mutants harboring an empty vector or this vector expressing wild-type VKOR or its variants as described for the experiment whose results are shown in Fig. 2C. The samples were then boiled in sample buffer containing 1% SDS in the presence or absence of 10% β-mercaptoethanol (BME) prior to immunoblotting with anti-VKOR and anti-MdbA Ab. As shown by the results in Fig. 4A, in the samples without BME, we observed a high-molecular-mass complex that was reactive to anti-VKOR and anti-MdbA Ab and migrated between the 50- and 75-kDa markers in the strain expressing the VKOR variant with C101A but not in other strains. The signal of this MdbA-VKOR complex (predicted molecular mass of 62 kDa) was greatly diminished when the sample was treated with BME (Fig. 4B), leading to increased intensity of the VKOR and MdbA monomers (Fig. 4, lanes labeled pC101A). Furthermore, this complex was not found when both C93 and C101 were mutated to alanine (see Fig. S1 in the supplemental material). The results suggest that when C101 is mutated, VKOR forms a complex with MdbA via the VKOR C93 residue.
FIG 4.
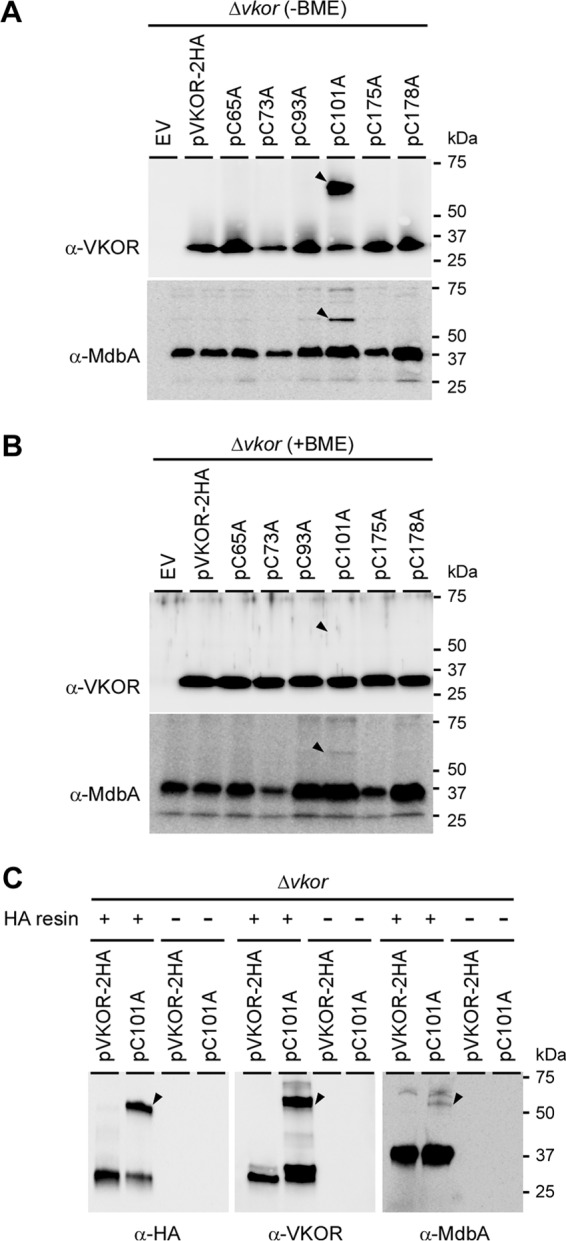
Identification of an MdbA-VKOR intermediate complex. (A and B) Protein samples obtained from the membrane fractions of A. oris strains were boiled in SDS-containing sample buffer in the presence (A) or absence (B) of 10% β-mercaptoethanol (BME) and subjected to SDS-PAGE, followed by immunoblotting with anti-VKOR or anti-MdbA Ab. Arrowheads indicate high-molecular-mass MdbA-VKOR complexes. (C) Clear lysates obtained from membrane fractions of the ΔVKOR mutant strains expressing wild-type VKOR and VKOR variants were incubated with anti-HA Ab–agarose beads, followed by immunoblotting with anti-VKOR or anti-MdbA Ab. The MdbA-VKOR complex (arrowheads) and molecular mass markers are indicated.
To confirm the MdbA-VKOR complex, we performed a pulldown assay for which the membrane lysates from the ΔVKOR mutant carrying pVKOR-2HA or pVKOR-C101A were incubated with anti-HA Ab–agarose beads or mock treated; the beads were collected by centrifugation and extensively washed, and bound proteins released from the beads were subjected to immunoblotting with anti-HA, anti-VKOR, and anti-MdbA Ab. Consistent with the results in Fig. 4A and B, the anti-HA Ab–agarose beads captured the MdbA-VKOR complex in the strain bearing pVKOR-C101A. The anti-HA Ab–agarose beads were reactive to all of the antibodies, i.e., the anti-HA, anti-VKOR and anti-MdbA Ab; this complex was not detected in the strain expressing wild-type VKOR and mock-treated samples (Fig. 4C).
Functionality of M. tuberculosis VKOR in A. oris.
As mentioned above, M. tuberculosis VKOR is a membrane protein, with 5 membrane-spanning domains arranged in an N-in/C-out configuration; the protein also possesses 4 catalytic cysteine residues (21). C57 and C65 are located within the first exoplasmic loop connecting the first and second transmembrane domains, whereas C139 and C142, as part of the catalytic CXXC motif, reside within the second exoplasmic loop (see Fig. 6A). These cysteine residues form two pairs of redox-reactive disulfide bonds involved in reoxidation of E. coli DsbA (21). The A. oris and M. tuberculosis VKOR proteins share 32% sequence identity; sequence alignment analysis of the two proteins by Clustal Omega (30) revealed many homologous segments with the two pairs of catalytic cysteines found within the conserved core domains (Fig. 5), suggesting that the two proteins are closely related.
FIG 6.

Functionality of M. tuberculosis VKOR in A. oris. (A) Presented is the membrane topology of M. tuberculosis VKOR. (B to K) The A. oris ΔVKOR mutant was transformed with plasmids expressing A. oris VKOR or M. tuberculosis VKOR or its Cys-to-Ala variants. The resulting strains were examined for their ability to aggregate with S. oralis (B), form biofilms (C), and assemble pili (D to K). Coaggregation and biofilm assays were performed as described in the legend to Fig. 2A and B. **, P ≤ 0.01. To examine pilus assembly, A. oris cells were subjected to EM as described in the legend to Fig. 3. Scale bars represent 0.5 μm. (L) The expression of the VKOR gene in A. oris ΔVKOR/pVKOR-2HA and A. oris ΔVKOR/pMtbVKOR was analyzed by quantitative RT-PCR. The results are presented as the average values from two independent experiments performed in triplicate. **, P ≤ 0.01. (M) Membrane fractions of the A. oris ΔVKOR mutant expressing M. tuberculosis VKOR or its variants, as well as A. oris VKOR-2HA or -C101A, were subjected to immunoblotting with anti-HA Ab in the absence of BME. Monomeric and complex forms of VKOR are marked by gray and black arrowheads, respectively.
FIG 5.

Sequence alignment of A. oris and M. tuberculosis VKOR proteins. The VKOR protein sequences of A. oris MG1 and M. tuberculosis Erdman were aligned using Clustal Omega (30). Conserved residues are highlighted, and the catalytic disulfide bonds are marked by square brackets.
To examine this, we cloned the M. tuberculosis VKOR gene of strain Erdman or its variants under the control of the A. oris VKOR promoter; the proteins expressed harbored an HA tag for Western blotting. The recombinant plasmids were introduced into the ΔVKOR mutant by electroporation. When the coaggregation of A. oris with S. oralis was examined, the strain expressing wild-type M. tuberculosis VKOR partially rescued the coaggregation defect of the A. oris ΔVKOR mutant compared to the results for the strain expressing A. oris VKOR (Fig. 6B). In contrast, strains expressing M. tuberculosis VKOR variants, i.e., C57A, C65A, C139A, and C142A mutants, known to be defective in disulfide relay (21), failed to aggregate with streptococci (Fig. 6B, last four plates).
The same set of strains described above was analyzed in the biofilm assay as described for the experiment whose results are shown in Fig. 2B. Compared to the ΔVKOR mutant harboring an empty vector, the strain expressing the wild-type M. tuberculosis VKOR protein had its biofilm formation enhanced by 50%, whereas strains expressing the catalytically inactive VKOR variants produced biofilms at the same level as the ΔVKOR mutant (Fig. 6C). Furthermore, these strains were subjected to electron microscopy using negative staining as described for the experiment whose results are shown in Fig. 3. Consistent with the above-described results, the ΔVKOR mutant expressing M. tuberculosis VKOR produced pili, albeit less abundantly than the wild-type MG1 strain and the ΔVKOR mutant expressing A. oris VKOR (Fig. 6D to G). In contrast, all the strains expressing M. tuberculosis VKOR mutants failed to assemble pili (Fig. 6H to K). Of note, the reduced coaggregation, biofilm formation, and pilus assembly phenotype of the ΔVKOR mutant expressing M. tuberculosis VKOR might be due to the reduced VKOR gene expression level in this strain, as detected by quantitative reverse transcription (qRT)-PCR (Fig. 6L).
Finally, to examine whether M. tuberculosis VKOR forms a complex with A. oris MdbA, membrane fractions of the A. oris ΔVKOR cells expressing M. tuberculosis wild-type VKOR and its cysteine mutant strains described above were subjected to Western blotting with anti-HA Ab. Intriguingly, similar to the A. oris ΔVKOR strain expressing A. oris VKOR-C101A, both monomeric and complex forms of M. tuberculosis VKOR were observed in all strains (Fig. 6M, first 5 lanes). Note that the mobility of these bands is faster than for A. oris VKOR species due to the smaller size of M. tuberculosis VKOR proteins. Altogether, the results indicate that M. tuberculosis VKOR is a functional analog of A. oris VKOR and suggest that both reductase enzymes catalyze reoxidation of a cognate oxidative folding machine by a mechanism that is conserved in the Actinobacteria.
DISCUSSION
While dsbA and dsbB are found in the majority of Gram-negative bacterial genomes, only a few bacterial lineages encode VKOR in place of dsbB; the list includes members of the class Actinobacteria, with the exception of C. diphtheriae, the Cyanobacteria, and some species of Spirochaetes and Deltaproteobacteria (8). Consistent with the view that bacterial VKOR is a functional homolog of DsbB, VKOR of the actinobacterium M. tuberculosis was shown to be capable of oxidizing DsbA in E. coli (20). This involves four exoplasmic catalytic cysteine residues, C57/C65 and C139/C142, of M. tuberculosis VKOR that participate in electron transfer during DsbA reoxidation (21). Nonetheless, how this disulfide relay mechanism takes place in M. tuberculosis remains unknown. Here, we present biochemical evidence of VKOR-mediated oxidation of the native thiol-disulfide oxidoreductase MdbA that involves four exoplasmic cysteine residues (C93/C101 and C175/C178) in A. oris.
Like M. tuberculosis VKOR, A. oris VKOR is a membrane protein spanning the membrane five times, with the N terminus facing the cytoplasm and the C terminus facing toward the exoplasm (Fig. 1A). This topological view is consistent with the models predicted by various membrane topology algorithms and validated by biochemical evidence and in situ electron microscopy, which demonstrate contrasting localization of the N and C termini (Fig. 1). Otherwise, an alternative model, i.e., an N-out/C-in arrangement, is not possible because the exoplasmic N terminus would not be protected by the cytoplasmic membrane from proteolytic cleavage (Fig. 1G). Thus, it is apparent that the M. tuberculosis and A. oris VKOR proteins share comparable topologies for the 4 exoplasmic cysteine residues, supporting the possibility that they function in a similar manner (see below), given the strong similarity of their primary protein sequences (Fig. 5).
Consistent with the results described above, individual alanine substitutions for the catalytic Cys residues, i.e., C93A, C101A, C175A, and C178A, abrogate polymicrobial interactions, biofilm formation, and pilus assembly (Fig. 2 and 3). These defective phenotypes are possibly due to the failure of the VKOR mutants to reoxidize the main thiol-disulfide oxidoreductase MdbA (13). Through this mutational analysis, combined with biochemical methods, we were able to trap an MdbA-VKOR intermediate only with the C101A mutation, supporting the direct interaction between the two oxidoreductases (Fig. 4). Since both C93 and C101 reside on the same exoplasmic loop of VKOR, we propose that these two cysteine residues form a redox center that allows electrons to be transferred from MdbA to C93 before passing through C101. During this process, an MdbA-VKOR intermediate is formed via a mixed disulfide bond between the MdbA C139 residue and the VKOR C93 residue. Subsequently, electron shuffling mediated by the second redox center, C175/C178, i.e., the CXXC motif, results in oxidized MdbA (Fig. 7). When C101 is mutated, the electron transfer between C93 and C101 is blocked, resulting in the formation of a stable mixed disulfide bond between MdbA and VKOR; otherwise, this transient intermediate complex would not be detected in wild-type cells under the current experimental conditions (Fig. 4). Such an intermediate has also been observed with M. tuberculosis VKOR and E. coli DsbA, where it is dependent on the first cysteine residue, i.e., C57, within the first exoplasmic loop of M. tuberculosis VKOR (Fig. 6A) (21).
FIG 7.

A model of VKOR-mediated reactivation of MdbA. VKOR and MdbA form a pair of thiol-disulfide oxidoreductases in A. oris. The cysteine residues C139 and C142 constitute the conserved catalytic CXXC motif of MdbA, whereas C175 and C178 in VKOR form the equivalent motif. After catalyzing oxidative folding of nascent polypeptides, MdbA is reduced. Reduced MdbA is proposed to be reoxidized by VKOR by a disulfide relay mechanism (see the text for details). During the electron transfer process, the C139 residue of MdbA forms a mixed disulfide bond with the C93 of VKOR, generating transient intermediates between MdbA and VKOR. VKOR becomes reduced, and reoxidation of the VKOR catalytic CXXC motif is proposed to be mediated by menaquinone (MQ). Intermediate forms of MdbA and VKOR are shown within square brackets. Dashed arrows represent possible multisteps. MQH2, menaquinol.
The findings described above suggest that M. tuberculosis VKOR and A. oris VKOR act similarly in the reoxidation of disulfide bond-forming machinery. Indeed, when expressed in the A. oris VKOR mutant, M. tuberculosis VKOR was able to rescue the coaggregation, biofilm formation, and pilus assembly defects of this mutant, albeit less efficiently; alanine substitution for M. tuberculosis VKOR C57, as well as other redox Cys residues, prevents the catalytic activities of the enzyme, although the formation of the M. tuberculosis VKOR-MdbA complex is not affected (Fig. 6), much like the M. tuberculosis VKOR complex with DsbA in E. coli (21). A possible explanation for this is that electron shuffling mediated by the two redox centers of M. tuberculosis VKOR in the heterologous systems E. coli and A. oris is less efficient than in the native mycobacteria; this is probably due to a less optimal interface between MdbA or DsbA and M. tuberculosis VKOR than in the native systems, like that of E. coli DsbA-DsbB (31). It is noteworthy that the N terminus of M. tuberculosis VKOR is shorter than that of A. oris VKOR. Whether or not this N terminus contributes to the reduced activity remains to be examined. As proposed by the model presented in Fig. 7, after reoxidation of MdbA, VKOR is reduced, and VKOR reoxidation may involve electron transfer from VKOR to a quinone, most likely menaquinone, since a bioinformatics search into the A. oris genome revealed a gene cluster for menaquinone biosynthesis (http://www.brop.org/). In E. coli, the cysteine pair within the catalytic CXXC motif of DsbB is the oxidation target of ubiquinone (32, 33). Thus, future experiments need to address whether menaquinone is an oxidizing reactant of A. oris VKOR through the catalytic cysteine pair C175/C178. Altogether, the results presented here suggest that A. oris VKOR utilizes an electron transfer mechanism of MdbA reoxidation that may be conserved in the Actinobacteria, including M. tuberculosis. In this regard, the MdbA-VKOR machine of A. oris provides an excellent experimental model, since genetic manipulations in A. oris are highly tractable (34, 35).
MATERIALS AND METHODS
Bacterial strains, primers, plasmids, and media.
The bacterial strains, primers, and plasmids used in this study are listed in Table 1. A. oris was grown in heart infusion (HI) broth or on HI agar plates at 37°C with 5% CO2. E. coli DH5α and BL21, used for cloning and protein purification, were grown in Luria-Bertani (LB) or Luria agar at 37οC. Kanamycin (Kan) or ampicillin (Amp) was added at 50 μg ml−1 or 100 μg ml−1, respectively. Streptococcus oralis So34 and OC1, used for coaggregation assays, were grown in HI broth or HI agar plates (in the presence of 1% glucose) in an anaerobic chamber at 37°C. All reagents and media were purchased from Sigma-Aldrich and Fisher Scientific unless otherwise indicated below.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Reference or source |
|---|---|---|
| Strains | ||
| A. oris MG1 | Parental strain | 19 |
| A. oris CW1 | ΔgalK; an isogenic derivative of MG1 | 19 |
| A. oris MR108 | VKOR deletion mutant; an isogenic derivative of CW1 | 13 |
| A. oris AR4 | ΔfimA; an isogenic derivative of CW1 | 18 |
| S. oralis So34 | Receptor polysaccharide (RPS) positive | 40 |
| S. oralis OC1 | RPS negative | 40 |
| Plasmids | ||
| pCWU10 | pCVD047 derivative with the kanamycin resistance gene from pJRD215 replacing its original ampicillin resistance gene; Kanr | 36 |
| pVKOR-2HA | pCWU10 derivative expressing wild-type VKOR from MG1 and carrying a double human influenza hemagglutinin (HA) tag at the C terminus | This study |
| pVKOR-C65A | Derivative of pVKOR-2HA harboring a C65A mutation | This study |
| pVKOR-C73A | Derivative of pVKOR-2HA harboring a C73A mutation | This study |
| pVKOR-C93A | Derivative of pVKOR-2HA harboring a C93A mutation | This study |
| pVKOR-C101A | Derivative of pVKOR-2HA harboring a C101A mutation | This study |
| pVKOR-C175A | Derivative of pVKOR-2HA harboring a C175A mutation | This study |
| pVKOR-C178A | Derivative of pVKOR-2HA harboring a C178A mutation | This study |
| pVKOR-C93AC101A | Derivative of pVKOR-2HA harboring C93A and C101A mutations | This study |
| pMtbVKOR | pCWU10 derivative expressing wild-type VKOR from M. tuberculosis Erdman and carrying the C-terminal HA tag | This study |
| pMtbVKOR-C57A | Derivative of pMtbVKOR harboring a C57A mutation | This study |
| pMtbVKOR-C65A | Derivative of pMtbVKOR harboring a C65A mutation | This study |
| pMtbVKOR-C139A | Derivative of pMtbVKOR harboring a C139A mutation | This study |
| pMtbVKOR-C142A | Derivative of pMtbVKOR harboring a C142A mutation | This study |
| pMCSG7 | Used for ligation-independent cloning for protein expression | 37 |
| pMCSG7-VKOR1–56 | Used for recombinant VKOR1–56 expression; VKOR1–56 is the sequence of 56 amino acids at the N-terminal end of VKOR from MG1 | This study |
Recombinant plasmids.
pVKOR-2HA is a recombinant plasmid that expresses A. oris VKOR with a double human influenza hemagglutinin (2×HA) tag at its C terminus and was constructed as follows. Primers VKOR-HindIII-F/VKOR-HA-dn and HA-up/HA-NdeI-R (see Table S1 in the supplemental material) were used to amplify the promoter and coding regions of the A. oris VKOR gene and the 2×HA fragment, respectively. Both PCR products were used as templates for overlapping PCR with primers VKOR-HindIII-F and HA-NdeI-R. The PCR product generated was gel purified and digested with HindIII and NdeI (New England BioLabs [NEB]) and subsequently ligated into pCWU10 (36). The resulting plasmid was introduced into DH5α, and plasmid DNA was purified for sequencing using primer pVKORseq (see Table S1) to confirm the insertion sequence. Finally, the vector was electroporated into the A. oris ΔVKOR mutant (13).
pMtbVKOR.
To construct a recombinant plasmid expressing Mycobacterium tuberculosis VKOR with a 2×HA tag, overlapping PCR was performed as mentioned above. Briefly, the primer set VKOR-HindIII-F/VKOR-Mtb-R (see Table S1 in the supplemental material) was used to amplify the A. oris VKOR gene promoter from A. oris MG1 chromosomal DNA, whereas the primer set VKOR-Mtb-F/Mtb-HA-R was used to amplify the M. tuberculosis VKOR coding sequence from M. tuberculosis Erdman chromosomal DNA. Of note, chromosomal DNA of the heat-killed M. tuberculosis strain Erdman (kindly provided by Jeffrey K. Actor and Shen-An Hwang, UTHealth) was isolated using a genomic DNA purification kit (Promega). The 2×HA fragment was obtained by PCR amplification using primers Mtb-HA-F and HA-NdeI-R (see Table S1). The three fragments were joined together by overlapping PCR using primers VKOR-HindIII-F and HA-NdeI-R. The PCR product generated was gel purified and digested with HindIII and NdeI, followed by ligation into pCWU10 to produce pMtbVKOR-2HA. The plasmid was introduced into the A. oris ΔVKOR mutant after its verification by DNA sequencing.
pMCSG7-VKOR1–56.
Primers LIC-VKOR-F and LIC-VKOR-R (see Table S1 in the supplemental material) were used to amplify the sequence coding the first 56 amino acids of A. oris VKOR. The PCR product generated was cloned into pMCSG7 using ligation-independent cloning (37), based on a published protocol (13). The resulting plasmid was introduced into E. coli BL21(DE3) after its verification by DNA sequencing.
Site-directed mutagenesis.
Cys-to-Ala mutations of VKOR were generated by a PCR-based method (36), using overlapping primers (see Table S1 in the supplemental material) and pVKOR-2HA or pMtbVKOR-2HA as the template. Prior to PCR amplification with Phusion DNA polymerase (NEB), all primers were phosphorylated at the 5′ end using T4 kinase (NEB). The PCR products were gel purified and cleaned by using DNA Clean & Concentrator (Zymo Research). The purified PCR products were ligated using Quick ligase (NEB) and circular plasmids and then transformed into E. coli DH5α for subsequent verification of mutations by DNA sequencing. The confirmed plasmids were electroporated into the A. oris ΔVKOR mutant.
Protein purification.
Protein purification was performed as previously reported (13). Briefly, E. coli cells harboring pMCSG7 grown in LB until reaching an optical density at 600 nm (OD600) of ∼0.6 were induced by 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG; Sigma) at 30°C for 3 h. Cell pellets were harvested by centrifugation and resuspended in EQ buffer (150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride [PMSF], 50 mM Tris-Cl, pH 7.5). The cell suspension was lysed in a French press, and clear lysates were obtained by centrifugation. The lysates were subjected to Ni-nitrilotriacetic acid (NTA) chromatography, and the purified proteins were desalted at 4°C using desalting columns (Merck), concentrated using Amicon ultra centrifugal filters (Merck), and stored at −20°C. A milligram of VKOR1–56 was used for antibody production (Cocalico Biologicals, Inc., PA).
Coaggregation and biofilm assays.
Coaggregation of A. oris and Streptococcus oralis was performed according to a previously published protocol with some modifications (13). Briefly, overnight cultures of A. oris and S. oralis were normalized to an OD600 of 2.0. Cells were harvested by centrifugation and washed twice in TBS (150 mM NaCl, 20 mM Tris-Cl, pH 7.4) plus 0.1 mM CaCl2 (TBSC). The washed cells were then suspended in 500 μl TBSC. Coaggregation was determined by mixing 750 μl of the A. oris suspension with 250 μl of the S. oralis suspension, followed by slight agitation. Images were recorded by a FluorChem Q (Alpha Innotech).
For biofilm formation, equivalent overnight cultures of A. oris strains were inoculated (1:100 dilution) into 24-well plates containing HI broth plus 1% sucrose. The plates were incubated at 37°C with 5% CO2 for 48 h. Biofilms were gently washed three times with 500 μl phosphate-buffered saline (PBS) and allowed to air dry. Biofilm production was quantified by using 1% crystal violet according to a published protocol (13). The results of three independent experiments performed in triplicate are reported.
Electron microscopy.
Log-phase cells of various A. oris strains were harvested and subjected to negative staining and thin-section electron microscopy as previously described (38). For immunogold labeling, ultrathin sections on grids were stained with anti-HA (1:10 dilution) or anti-VKOR1–56 (1:100) Ab, followed by staining with secondary antibody conjugated to 12-nm gold particles diluted 1:20 in PBS–1% bovine serum albumin (BSA). Finally, the grids were washed 5 times with distilled water and stained with 1% uranyl acetate for 1 min. The samples were viewed with a JEOL JEM-1400 electron microscope.
Cell fractionation and Western blotting.
Cell fractionation was performed as previously described (36). Briefly, log-phase cell cultures of various strains were obtained and normalized to an OD600 of 1.0 (36). Cells were fractionated into culture medium (S), cell wall (W), membrane (M), and cytoplasmic (C) fractions; proteins were precipitated using trichloroacetic acid (TCA) and washed with acetone. Protein concentrations were measured using a bicinchoninic acid (BCA) protein assay kit (Thermo Scientific) following the manufacturer's instructions. Protein samples were heated in loading buffer containing 1% SDS for 5 min prior to SDS-PAGE using 3-to-12% or 3-to-20% Tris-glycine gradient gels. Proteins were immunoblotted with specific antibodies (at dilutions of 1:5,000 for anti-MdbA Ab, 1:5,000 for anti-FimA Ab, 1:5,000 for anti-FimP Ab, 1:2,000 for anti-VKOR Ab, and 1:100 for anti-HA Ab).
Protease protection assays.
Protoplasts of log-phase A. oris cells were prepared as previously described (13, 36) and used in a protease protection assay (39). The protoplasts were treated with proteinase K (Sigma) in PBS at final concentrations of 0, 10, 50, 100, and 200 ng/μl at room temperature for 15 min. The reactions were quenched by the addition of 1 mM phenylmethylsulfonyl fluoride (PMSF; Sigma). After treatment, the protoplasts were collected by centrifugation and boiled in loading buffer containing 1% SDS for 5 min prior to immunoblotting.
HA pulldown assays.
Five milliliters of log-phase cell cultures of A. oris strains were harvested, and bacterial membrane fractions were obtained by cell fractionation as described above. The membrane fractions were suspended in 500 μl of EQ buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.5) plus 0.5% SDS, 100 μl of which was incubated with 50 μl of anti-HA Ab–agarose beads (Pierce) containing 1× protease inhibitors (GeneDepot). After overnight incubation at 4°C, the anti-HA Ab–agarose beads were collected by centrifugation at 580 × g and washed 5 times with 500 μl of EQ buffer containing 1 mM PMSF. Bound proteins were eluted from the beads by heating in 100 μl of 1× loading buffer without β-mercaptoethanol (BME) for 10 min at 80°C. Eluted proteins were subjected to SDS-PAGE with 12% Tris-glycine gels and immunoblotted with anti-HA, anti-VKOR, or anti-MdbA Ab.
qRT-PCR.
Total RNA was isolated from A. oris cells using the RNeasy minikit (Qiagen), following the manufacturer's instructions. Log-phase A. oris cells were harvested by centrifugation and lysed in 1 ml of RLT buffer (Qiagen) by using glass beads (0.1 mm; MP Biomedical), followed by centrifugation at 15,000 × g for 5 min. Seven hundred microliters of supernatants was transferred to DNase-RNase-free Eppendorf tubes and mixed well with 500 μl of chilled ethanol (200 Proof; Decon Laboratories, PA). The suspension was transferred onto columns prewashed with 500 μl of RW1 buffer (Qiagen) and subsequently washed twice with 500 μl of RPE buffer (Qiagen). The samples were eluted with 60 μl of nuclease-free water. After treatment with 5 μl of DNase (Qiagen) at 30°C for an hour, the samples were subjected to another purification with prewashed columns. The concentration of eluted RNA in 60 μl of nuclease-free water was determined by using a NanoDrop ND-1000 spectrophotometer.
Five hundred nanograms of RNA was used to make cDNA using random primers (Invitrogen) and Moloney murine leukemia virus (MMLV) reverse transcriptase (Invitrogen) as described by the manufacturer's protocol. Diluted cDNA samples (1:1,000) were used for quantitative PCR by using iTAQ SYBR green supermix (Bio-Rad) and the CFX96 Touch real-time PCR detection system (Bio-Rad). The primers used in quantitative PCRs are shown in Table S1 in the supplemental material. The data are presented as cycle threshold (ΔCT) values.
Statistical analysis.
Graphing of the results of biofilm assays, qRT-PCR, and statistical analysis was performed using GraphPad Prism 5, with significant differences calculated using one-way analysis of variance (ANOVA; Duncan's method, nonparametric). The results are presented as the average values from 3 independent experiments performed in triplicate ± standard deviations (SD). A nonparametric, two-tailed P value of ≤0.05, ≤0.01, or ≤0.001 was considered significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jeffrey K. Actor and Shen-An Hwang (UTHealth) for kindly providing heat-killed M. tuberculosis and our laboratory members for the critical review and discussion of the manuscript.
Research reported in this publication was supported by the National Institute of Dental and Craniofacial Research of the National Institutes of Health under award numbers DE017382 and DE025015 to H.T.-T.
We have no conflict of interest to declare.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00817-16.
REFERENCES
- 1.Kadokura H, Katzen F, Beckwith J. 2003. Protein disulfide bond formation in prokaryotes. Annu Rev Biochem 72:111–135. doi: 10.1146/annurev.biochem.72.121801.161459. [DOI] [PubMed] [Google Scholar]
- 2.Hatahet F, Boyd D, Beckwith J. 2014. Disulfide bond formation in prokaryotes: history, diversity and design. Biochim Biophys Acta 1844:1402–1414. doi: 10.1016/j.bbapap.2014.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bardwell JC, McGovern K, Beckwith J. 1991. Identification of a protein required for disulfide bond formation in vivo. Cell 67:581–589. doi: 10.1016/0092-8674(91)90532-4. [DOI] [PubMed] [Google Scholar]
- 4.Bardwell JC, Lee JO, Jander G, Martin N, Belin D, Beckwith J. 1993. A pathway for disulfide bond formation in vivo. Proc Natl Acad Sci U S A 90:1038–1042. doi: 10.1073/pnas.90.3.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Missiakas D, Georgopoulos C, Raina S. 1993. Identification and characterization of the Escherichia coli gene dsbB, whose product is involved in the formation of disulfide bonds in vivo. Proc Natl Acad Sci U S A 90:7084–7088. doi: 10.1073/pnas.90.15.7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chivers PT, Laboissiere MC, Raines RT. 1996. The CXXC motif: imperatives for the formation of native disulfide bonds in the cell. EMBO J 15:2659–2667. [PMC free article] [PubMed] [Google Scholar]
- 7.Kadokura H, Beckwith J. 2002. Four cysteines of the membrane protein DsbB act in concert to oxidize its substrate DsbA. EMBO J 21:2354–2363. doi: 10.1093/emboj/21.10.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dutton RJ, Boyd D, Berkmen M, Beckwith J. 2008. Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc Natl Acad Sci U S A 105:11933–11938. doi: 10.1073/pnas.0804621105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bolhuis A, Venema G, Quax WJ, Bron S, van Dijl JM. 1999. Functional analysis of paralogous thiol-disulfide oxidoreductases in Bacillus subtilis. J Biol Chem 274:24531–24538. doi: 10.1074/jbc.274.35.24531. [DOI] [PubMed] [Google Scholar]
- 10.Dumoulin A, Grauschopf U, Bischoff M, Thony-Meyer L, Berger-Bachi B. 2005. Staphylococcus aureus DsbA is a membrane-bound lipoprotein with thiol-disulfide oxidoreductase activity. Arch Microbiol 184:117–128. doi: 10.1007/s00203-005-0024-1. [DOI] [PubMed] [Google Scholar]
- 11.Davey L, Ng CK, Halperin SA, Lee SF. 2013. Functional analysis of paralogous thiol-disulfide oxidoreductases in Streptococcus gordonii. J Biol Chem 288:16416–16429. doi: 10.1074/jbc.M113.464578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Premkumar L, Heras B, Duprez W, Walden P, Halili M, Kurth F, Fairlie DP, Martin JL. 2013. Rv2969c, essential for optimal growth in Mycobacterium tuberculosis, is a DsbA-like enzyme that interacts with VKOR-derived peptides and has atypical features of DsbA-like disulfide oxidases. Acta Crystallogr D Biol Crystallogr 69:1981–1994. doi: 10.1107/S0907444913017800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reardon-Robinson ME, Osipiuk J, Chang C, Wu C, Jooya N, Joachimiak A, Das A, Ton-That H. 2015. A disulfide bond-forming machine is linked to the sortase-mediated pilus assembly pathway in the gram-positive bacterium Actinomyces oris. J Biol Chem 290:21393–21405. doi: 10.1074/jbc.M115.672253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reardon-Robinson ME, Osipiuk J, Jooya N, Chang C, Joachimiak A, Das A, Ton-That H. 2015. A thiol-disulfide oxidoreductase of the Gram-positive pathogen Corynebacterium diphtheriae is essential for viability, pilus assembly, toxin production and virulence. Mol Microbiol 98:1037–1050. doi: 10.1111/mmi.13172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reardon-Robinson ME, Ton-That H. 2016. Disulfide-bond-forming pathways in gram-positive bacteria. J Bacteriol 198:746–754. doi: 10.1128/JB.00769-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu C, Mishra A, Yang J, Cisar JO, Das A, Ton-That H. 2011. Dual function of a tip fimbrillin of Actinomyces in fimbrial assembly and receptor binding. J Bacteriol 193:3197–3206. doi: 10.1128/JB.00173-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mishra A, Devarajan B, Reardon ME, Dwivedi P, Krishnan V, Cisar JO, Das A, Narayana SV, Ton-That H. 2011. Two autonomous structural modules in the fimbrial shaft adhesin FimA mediate Actinomyces interactions with streptococci and host cells during oral biofilm development. Mol Microbiol 81:1205–1220. doi: 10.1111/j.1365-2958.2011.07745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mishra A, Wu C, Yang J, Cisar JO, Das A, Ton-That H. 2010. The Actinomyces oris type 2 fimbrial shaft FimA mediates co-aggregation with oral streptococci, adherence to red blood cells and biofilm development. Mol Microbiol 77:841–854. doi: 10.1111/j.1365-2958.2010.07252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mishra A, Das A, Cisar JO, Ton-That H. 2007. Sortase-catalyzed assembly of distinct heteromeric fimbriae in Actinomyces naeslundii. J Bacteriol 189:3156–3165. doi: 10.1128/JB.01952-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dutton RJ, Wayman A, Wei JR, Rubin EJ, Beckwith J, Boyd D. 2010. Inhibition of bacterial disulfide bond formation by the anticoagulant warfarin. Proc Natl Acad Sci U S A 107:297–301. doi: 10.1073/pnas.0912952107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Dutton RJ, Beckwith J, Boyd D. 2011. Membrane topology and mutational analysis of Mycobacterium tuberculosis VKOR, a protein involved in disulfide bond formation and a homologue of human vitamin K epoxide reductase. Antioxid Redox Signal 14:1413–1420. doi: 10.1089/ars.2010.3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guilhot C, Jander G, Martin NL, Beckwith J. 1995. Evidence that the pathway of disulfide bond formation in Escherichia coli involves interactions between the cysteines of DsbB and DsbA. Proc Natl Acad Sci U S A 92:9895–9899. doi: 10.1073/pnas.92.21.9895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 24.Tusnady GE, Simon I. 1998. Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J Mol Biol 283:489–506. doi: 10.1006/jmbi.1998.2107. [DOI] [PubMed] [Google Scholar]
- 25.Hofmann K, Stoffel W. 1993. TMBASE—a database of membrane spanning protein segments. Biol Chem Hoppe-Seyler 374:166. [Google Scholar]
- 26.Yachdav G, Kloppmann E, Kajan L, Hecht M, Goldberg T, Hamp T, Hönigschmid P, Schafferhans A, Roos M, Bernhofer M. 2014. PredictProtein—an open resource for online prediction of protein structural and functional features. Nucleic Acids Res 42:W337–W343. doi: 10.1093/nar/gku366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dobson L, Reményi I, Tusnády GE. 2015. CCTOP: a Consensus Constrained TOPology prediction web server. Nucleic Acids Res 43:W408–W412. doi: 10.1093/nar/gkv451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Juretic D, Zoranic L, Zucic D. 2002. Basic charge clusters and predictions of membrane protein topology. J Chem Inf Comput Sci 42:620–632. doi: 10.1021/ci010263s. [DOI] [PubMed] [Google Scholar]
- 29.Reardon-Robinson ME, Wu C, Mishra A, Chang C, Bier N, Das A, Ton-That H. 2014. Pilus hijacking by a bacterial coaggregation factor critical for oral biofilm development. Proc Natl Acad Sci U S A 111:3835–3840. doi: 10.1073/pnas.1321417111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inaba K, Murakami S, Suzuki M, Nakagawa A, Yamashita E, Okada K, Ito K. 2006. Crystal structure of the DsbB-DsbA complex reveals a mechanism of disulfide bond generation. Cell 127:789–801. doi: 10.1016/j.cell.2006.10.034. [DOI] [PubMed] [Google Scholar]
- 32.Kobayashi T, Kishigami S, Sone M, Inokuchi H, Mogi T, Ito K. 1997. Respiratory chain is required to maintain oxidized states of the DsbA-DsbB disulfide bond formation system in aerobically growing Escherichia coli cells. Proc Natl Acad Sci U S A 94:11857–11862. doi: 10.1073/pnas.94.22.11857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inaba K, Takahashi YH, Ito K, Hayashi S. 2006. Critical role of a thiolate-quinone charge transfer complex and its adduct form in de novo disulfide bond generation by DsbB. Proc Natl Acad Sci U S A 103:287–292. doi: 10.1073/pnas.0507570103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu C, Reardon-Robinson ME, Ton-That H. 2016. Genetics and cell morphology analyses of the Actinomyces oris srtA mutant. Methods Mol Biol 1440:109–122. doi: 10.1007/978-1-4939-3676-2_9. [DOI] [PubMed] [Google Scholar]
- 35.Wu C, Ton-That H. 2010. Allelic exchange in Actinomyces oris with mCherry fluorescence counterselection. Appl Environ Microbiol 76:5987–5989. doi: 10.1128/AEM.00811-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siegel SD, Wu C, Ton-That H. 2016. A type I signal peptidase is required for pilus assembly in the gram-positive, biofilm-forming bacterium Actinomyces oris. J Bacteriol 198:2064–2073. doi: 10.1128/JB.00353-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. 2002. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr Purif 25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 38.Wu C, Huang IH, Chang C, Reardon-Robinson ME, Das A, Ton-That H. 2014. Lethality of sortase depletion in Actinomyces oris caused by excessive membrane accumulation of a surface glycoprotein. Mol Microbiol 94:1227–1241. doi: 10.1111/mmi.12780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilson MJ, Carlson PE, Janes BK, Hanna PC. 2012. Membrane topology of the Bacillus anthracis GerH germinant receptor proteins. J Bacteriol 194:1369–1377. doi: 10.1128/JB.06538-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoshida Y, Ganguly S, Bush CA, Cisar JO. 2006. Molecular basis of L-rhamnose branch formation in streptococcal coaggregation receptor polysaccharides. J Bacteriol 188:4125–4130. doi: 10.1128/JB.01843-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.