

ABSTRACT
Hepatitis E virus (HEV) has emerged as a cause of chronic hepatitis among immunocompromised patients. Molecular assays have become important tools for the diagnosis and management of these chronically infected patients. A real-time reverse transcription-quantitative PCR (RT-qPCR) assay utilizing Pleiades probe chemistry and an RNA internal control for the simultaneous detection and quantification of HEV RNA in human serum was developed based on an adaptation of a previously described and broadly reactive primer set targeting the overlapping open reading frame 2/3 (ORF2/3) nucleotide sequence of HEV. A chimeric bovine viral diarrhea virus construct containing an HEV RNA insert (SynTura HEV) was developed, value assigned with the first World Health Organization (WHO) international standard for HEV RNA (code 6329/10), and used to prepare working assay calibrators and controls, which supported an assay quantification range of 100 to 5,000,000 IU/ml. The analytical sensitivity (95% detection rate) of this assay was 25.2 IU/ml (95% confidence interval [CI], 19.2 to 44.1 IU/ml). The assay successfully amplified 16 different HEV sequences with significant nucleotide mismatching in primer/probe binding regions, while evaluation of a WHO international reference panel for HEV genotypes (code 8578/13) showed viral load results falling within the result ranges generated by WHO collaborative study participants for all panel members (genotypes 1 to 4). Broadly reactive RT-qPCR primers targeting HEV ORF2/3 were successfully adapted for use in an assay based on Pleiades probe chemistry. The availability of secondary standards calibrated to the WHO HEV international standard can improve the standardization and performance of assays for the detection and quantification of HEV RNA.
KEYWORDS: HEV RNA, hepatitis E virus, RT-qPCR, quantification, viral load
INTRODUCTION
Hepatitis E virus (HEV) is a small, nonenveloped, positive-stranded RNA virus that is approximately 7.2 kb in length. The virus is a member of the Hepeviridae family and contains short 5′ and 3′ untranslated regions (UTRs) and 3 partially overlapping open reading frames (ORFs) (1). Currently, there are 4 major genotypes of HEV that have been implicated in human disease. HEV genotypes 1 and 2 appear to be restricted to human infection and have been strongly associated with waterborne, epidemic outbreaks of disease in subtropical regions of the developing world (2, 3). In contrast, autochthonous HEV infections occurring in industrialized countries and involving genotypes 3 and 4 have been shown to be zoonotic in origin and have been associated with direct animal exposure, ingestion of raw or undercooked meats, and transmission via contaminated blood products (4–6). These autochthonous HEV infections have occurred primarily in immunocompromised or immunosuppressed individuals (4, 7). More recently, chronic infection due to a camelid HEV strain (genotype 7) was reported in a liver transplant recipient (8).
In the setting of solid-organ transplantation (SOT), acute HEV infection can lead to chronic hepatitis E, with a potential for rapid progression to cirrhosis (9–12). Therefore, diagnostic testing for HEV is of particular importance among such patients exhibiting signs and symptoms of viral hepatitis, particularly in those cases that cannot be confirmed by routine virological testing. Additionally, due to the effects of immunosuppression and/or poor assay sensitivity, anti-HEV antibodies can remain undetected or be inconsistently detected in some of these patients, further complicating the diagnosis of hepatitis E and delaying appropriate medical intervention (13, 14).
Given these potential limitations of serological testing among immunocompromised patients, the diagnosis and management of both acute and chronic HEV infections require a combination of serological and nucleic acid amplification techniques (NATs) (15, 16). Molecular methods capable of accurately quantifying HEV viral load (VL) have also become increasingly important for monitoring changes in VL among chronically infected patients undergoing antiviral treatment. Significant decreases in serum or plasma VL or viral clearance during therapy may be important predictors of the virologic response (17–23).
Previous studies have shown that the performances of laboratory-developed tests (LDTs) for the detection and quantification of HEV RNA can vary greatly as a result of HEV diversity and assay design (24, 25). The assays shown to have the best overall performance characteristics are real-time reverse transcription-quantitative PCR (RT-qPCR) assays that target the overlapping region of ORF2 and ORF3 (24–27). Despite their important role in patient care, there are currently no commercial molecular-based assays for the detection of HEV RNA in human serum or plasma widely available in the United States.
In response to these important and yet largely unmet clinical needs, we developed and determined the performance characteristics of an RT-qPCR assay (HEV RT-qPCR) for the simultaneous detection and quantification of HEV RNA in human serum. This assay, based on Pleiades probe chemistry (28) and calibrated to the first World Health Organization (WHO) international standard for HEV RNA (code 6329/10) (29), is an adaptation of a previously described and broadly reactive primer/probe set targeting a conserved nucleotide sequence in the ORF2/3 region of HEV (30).
RESULTS
Analytical specificity of RT-qPCR.
Primer and probe sequences, based on an adaptation of a previously described and broadly reactive qPCR primer/probe set (30), were modified through the use of primers with 5′ flaps to enhance amplification efficiency (31), Pleiades probe chemistry (28), and the incorporation of modified bases to optimize probe performance (32). The specificity of these primer and probe sequences was initially assessed by performing BLASTn searches with each of these sequences. Results generated from sera of 50 healthy blood donors further confirmed the assay specificity, with minimal background fluorescence being observed between qPCR cycles 15 to 50 (ΔRn mean = 0.016; standard deviation [SD] = 0.006) and during the dissociation curve analysis (−ΔRn/ΔT mean = 0.002; SD = 0.001). DNA oligonucleotide constructs corresponding to 16 HEV sequences exhibiting high levels of nucleotide mismatching within the HEV ORF2/3 primer and probe binding regions (see Table S1 in the supplemental material) were tested by HEV RT-qPCR. Since all 16 constructs generated cycle threshold (CT) values and dissociation curves with melting temperature (Tm) values ranging from 57.7°C to 68.2°C (Table S2), the primers and probe used for HEV RT-qPCR demonstrated specific detection of this diverse group of HEV sequence variants, including representative sequences of HEV genotypes 1 through 4.
Analytical sensitivity and linearity of RT-qPCR.
Replicate testing performed with dilutions of the WHO HEV international standard at levels of 500, 200, 50, 20, 10, and 0 IU/ml demonstrated an analytical sensitivity (95% detection rate) of 25.2 IU/ml (95% confidence interval [CI], 19.2 to 44.1 IU/ml) (see Table S3 in the supplemental material). Among these replicates, dissociation curve analysis produced a mean Tm of 68.4°C (range of 68.1°C to 68.9°C). Tenfold dilutions of SynTura HEV (2.00 log10 to 7.00 log10 IU/ml) were each tested 5 times in 3 separate assay runs. Linear regression produced a slope of 1.039 (R2 = 0.998), with an overall mean VL difference of 0.06 log10 IU/ml and individual differences ranging from −0.02 log10 to 0.27 log10 IU/ml (Fig. S1). The within-run SD at various levels ranged from 0.07 log10 to 0.20 log10 IU/ml, while the between-run SD at these various levels ranged from 0.03 log10 to 0.14 log10 IU/ml. These results demonstrated good assay sensitivity, linearity, and reproducibility over a VL range spanning 5 logs.
Calibrators and controls for RT-qPCR.
Assay calibrators (SynTura HEV) and controls were tested over 10 assay runs. The efficiencies of these qPCRs were determined as follows: efficiency = 10(−1/slope) − 1. Acceptable qPCR efficiencies were expected to range from 90% to 100%, with corresponding slopes ranging between −3.6 and −3.3, respectively. Data from the 10 assay runs demonstrated excellent efficiency (mean slope = −3.33; SD = 0.03) (see Table S4 in the supplemental material). The mean results for the high- and low-positive controls were 4.83 log10 IU/ml (SD = 0.04 log10 IU/ml) and 2.80 log10 IU/ml (SD = 0.05 log10 IU/ml), respectively. The performance of the HEV calibrators was evaluated by testing replicate dilutions (at 2.70 log10 and 4.00 log10 IU/ml) of the WHO HEV international standard by HEV RT-qPCR using these assay calibrators to perform VL assignments, followed by a comparison of expected and observed results. Observed mean VLs for the low (2.70 log10 IU/ml) and high (4.00 log10 IU/ml) dilutions of the WHO HEV international standard were 2.66 log10 IU/ml (range of 2.62 log10 to 2.72 log10 IU/ml) and 3.96 log10 IU/ml (range of 3.93 log10 to 3.98 log10 IU/ml), respectively. Consistent and reproducible results for these calibrators and controls prepared with SynTura HEV were observed over multiple assay runs, supporting an HEV RT-qPCR quantification range extending from 100 (2.00 log10) IU/ml to 5,000,000 (6.70 log10) IU/ml.
Qualitative detection of HEV RNA.
Results obtained from 125 clinical serum specimens tested by HEV RT-qPCR and a previously described nested RT-PCR assay (33) showed generally good qualitative agreement (Table 1). Complete agreement was observed among those specimens known to contain HEV RNA (n = 35; all genotype 4) and those specimens from patients with suspected viral hepatitis (n = 68). In contrast, mixed results were observed among a group of HEV IgM-positive specimens (n = 22) evaluated in this study: 10/22 (45.5%) specimens showed no evidence of HEV RNA by either assay, 8/22 (36.4%) specimens yielded detectable HEV RNA by both assays (6 containing genotype 1and 2 containing genotype 3 HEV RNA), and 4/22 (18.2%) specimens yielded evidence of HEV RNA by HEV RT-qPCR only. Among the 8 IgM-positive specimens with detectable HEV RNA by both assays, the mean VL was 801,000 IU/ml (range of 943 to 5,110,000 IU/ml), while the mean VL among the 4 IgM-positive specimens with detectable HEV RNA by HEV RT-qPCR only was 238 IU/ml (range of 129 to 313 IU/ml). The specific detection of HEV RNA was confirmed by dissociation curve analysis for each of the 4 specimens with HEV RNA detectable by HEV RT-qPCR only (Table 1), but no additional testing of these specimens was possible due to an insufficient specimen volume. The failure of nested RT-PCR to detect HEV RNA in these 4 specimens may be due, at least in part, to the relatively low HEV RNA levels (i.e., <500 IU/ml) observed among these specimens. A failure rate of 2.4% (3/125) for the MS2 RNA internal control was observed among these specimens tested by HEV RT-qPCR.
TABLE 1.
Qualitative detection of HEV RNA in clinical serum specimens
| HEV RT-qPCR result category | No. of specimens with nested RT-PCR result |
|
|---|---|---|
| Positive | Negative | |
| Not detected | 0 | 78a |
| Detected but at <100 IU/ml | 1b | 0 |
| Quantifiable (100 to 5,000,000 IU/ml) | 42 | 4c |
| Detected but at >5,000,000 IU/ml | 0 | 0 |
Three specimens required repeat testing (1:10 dilution) by HEV RT-qPCR due to MS2 RNA internal control failure (i.e., PCR inhibition) during initial testing.
Dilution in normal human serum (1:6 dilution) was performed prior to testing by HEV RT-qPCR due to an insufficient sample volume.
HEV RT-qPCR results of 129, 249, 260, and 313 IU/ml, with the specificity of amplification being confirmed by dissociation curve analysis (Tm values of 68.7°C, 68.7°C, 68.8°C, and 68.9°C, respectively).
Agreement of HEV RNA results.
All 11 members of the WHO HEV reference panel (code 8578/13) (comprised of HEV genotypes 1a, 1a, 1e, 2a, 3, 3b, 3c, 3e, 3f, 4c, and 4g) were detected by HEV RT-qPCR, with Tm values ranging from 68.2°C to 68.9°C and falling within the Tm range (68.1°C to 68.9°C) observed for the WHO HEV international standard (Fig. 1). HEV RT-qPCR also generated VL results that were comparable to the VL results obtained by WHO collaborative study participants with this diverse group of HEV strains, including those genotypes associated with chronic infections in immunocompromised subjects.
FIG 1.
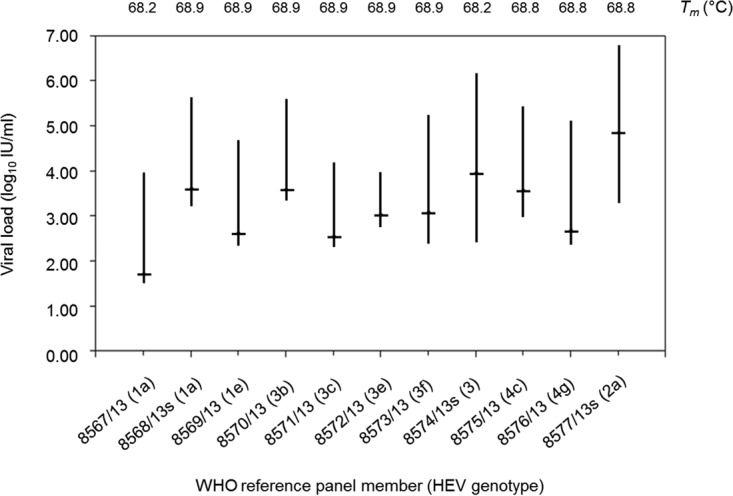
Results of members of the WHO international reference panel for HEV genotypes (code 8578/13) tested by HEV RT-qPCR. Viral load ranges for individual panel members, based on combined data obtained by laboratories participating in the initial evaluation of this HEV genotype panel (see Table 12 in reference 42), are indicated by vertical lines, while values obtained by HEV RT-qPCR are indicated by horizontal lines. Tm values were obtained by dissociation curve analysis performed in conjunction with HEV RT-qPCR.
Serial VL monitoring in SOT recipients.
Serial VL testing confirmed active HEV infection and demonstrated rapid clearance of detectable HEV RNA with normalization of the serum alanine aminotransferase (ALT) level in the orthotopic liver transplant (OLT) recipient after a 3-month course of ribavirin therapy (Fig. 2A). Despite a rapid decline in the HEV VL of >3 logs, accompanied by a temporary normalization of the ALT level, HEV RNA remained detectable in a kidney/pancreas transplant (KPT) recipient during and after a 6-month course of ribavirin therapy (Fig. 2B). Both of these HEV strains were identified as genotype 3 strains. ALT level and HEV VL appeared to be well correlated in these patients; however, results of HEV serological test results did not correlate well with the presence or absence of HEV RNA in these SOT recipients.
FIG 2.
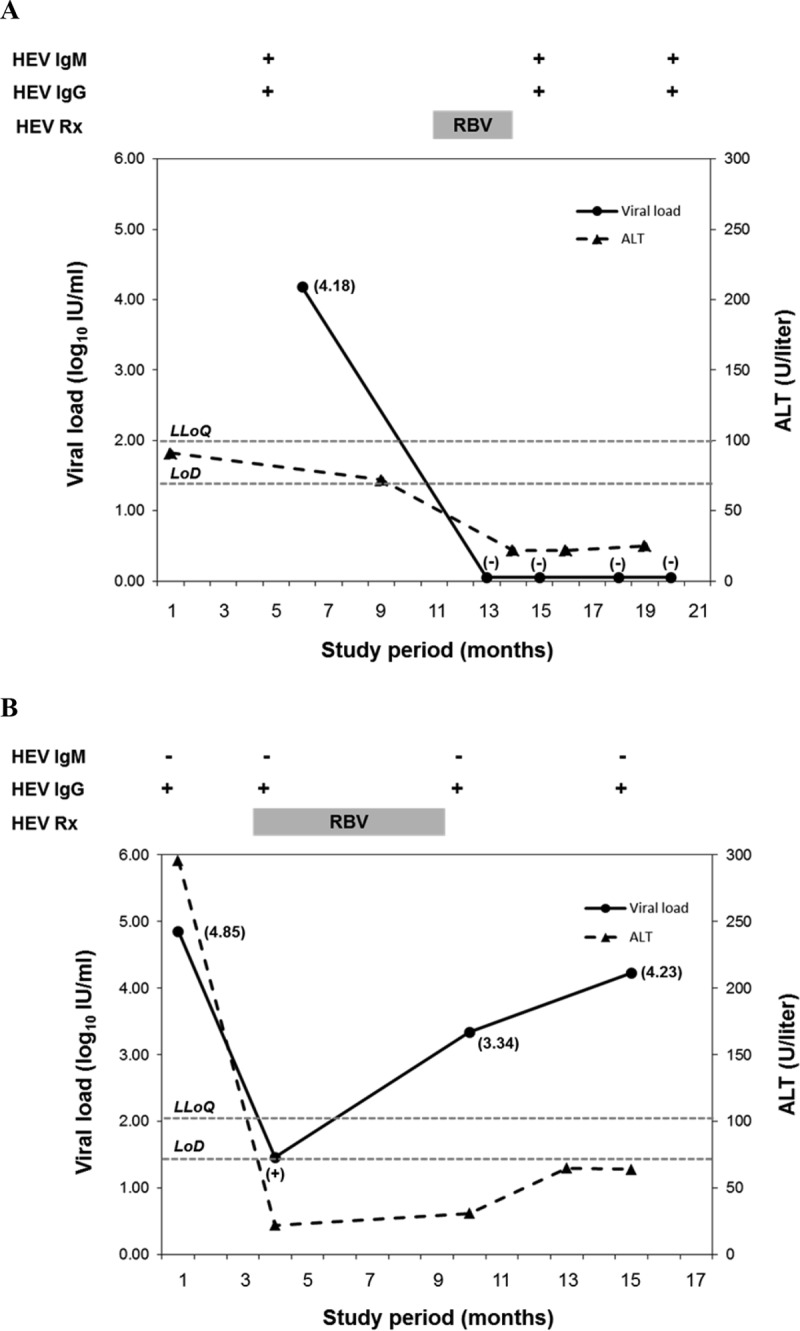
Viral loads generated from sera with HEV RT-qPCR demonstrating clearance of detectable HEV RNA from a patient following treatment with ribavirin (A) and persistently detectable HEV RNA in a patient following treatment with ribavirin (B). Detectable HEV RNA levels of <100 IU/ml are indicated by a plus sign, while undetectable HEV RNA levels are indicated by a minus sign. The lower limit of quantification (LLoQ) and limit of detection (LoD) for HEV RT-qPCR are represented by horizontal dotted lines. RBV, ribavirin; Rx, treatment.
DISCUSSION
The emergence of autochthonous HEV infection as a cause of hepatitis among SOT recipients has highlighted the need for standardized, sensitive, and specific assays for the detection and quantification of HEV RNA in clinical serum or plasma specimens. Real-time PCR has become the preferred tool for those diagnostic laboratories involved in molecular assay development because of its ability to detect as well as quantify the target of interest, in addition to its relative ease of use, rapid turnaround time, and reduced risk of contamination. Despite continued advances in real-time PCR technology, the design and optimization of molecular assays remain challenging due to a variety of issues ranging from target selection to probe design (32, 34–39).
The use of a broadly reactive, but relatively well-conserved, HEV primer/probe set targeting the overlapping region of ORF2 and ORF3 (25, 27, 30) was critical to the successful development of an RT-qPCR assay for the detection and quantification of HEV RNA. However, the potential for novel single nucleotide polymorphisms (SNPs) within the primer and probe binding regions still posed a potential risk for detection failure or underquantification of HEV RNA levels due to failed or poor hybridization of primers and/or probe sequences to the complementary target sequence (29, 40). Adaptation of the original probe sequence using a Pleiades probe design (28) enhanced the stability of probe binding through use of a minor groove binder (MGB) moiety, minimized potential background fluorescence, and allowed the probe to remain intact for dissociation curve analysis following PCR amplification. Because the dissociation curve analysis allowed probe annealing at a lower temperature (35°C), the use of this probe design also permitted fluorescence detection despite the presence of SNPs that were not accounted for in the original TaqMan probe design.
As key characteristics for any real-time PCR assay used in diagnostic testing, accuracy and reproducibility are critical to the successful development of quantitative molecular assays. Although the availability of the first WHO HEV international standard (code 6329/10) marked an important step toward making standardized VL testing for HEV possible (24), this standard contained a relatively low concentration (250,000 IU/ml) of HEV RNA and was available in only limited quantities. These limitations necessitated the development of a secondary HEV standard that could be produced at a higher concentration and in larger quantities for routine use for testing clinical serum or plasma specimens.
The use of SynTura technology (41) to develop assay calibrators had several distinct advantages over the use of plasmids or RNA transcripts containing the HEV target sequence. Unlike a DNA-containing plasmid or RNA transcript, SynTura HEV is a packaged, recombinant bovine viral diarrhea virus (BVDV) containing the HEV RNA target sequence. Therefore, SynTura HEV more closely mimicked the actual virus and offered a better accounting of the cumulative effects of the entire assay procedure on VL measurement, including the impact of the RNA extraction and reverse transcription steps. Since this secondary standard contains a relatively short segment of the HEV genome (121-nucleotide [nt] sequence of overlapping HEV ORF2 and ORF3), it is suitable for use only in molecular assays that share this relatively small target region. In the present study, SynTura HEV proved to be a reliable source of biosynthetic HEV RNA suitable for the development of a secondary standard, which was then used successfully in assay verification studies (e.g., linearity and reproducibility), and for the development of assay calibrators and controls supporting an assay quantification range spanning nearly 5 logs (100 to 5,000,000 IU/ml).
The overall performance characteristics of HEV RT-qPCR were favorable for its use for the routine detection and quantification of HEV RNA in human serum. Evaluation of qualitative results obtained from 125 clinical serum specimens (Table 1) suggests a relatively low inhibition rate (2.4%) among clinical serum specimens tested by HEV RT-qPCR along with a higher sensitivity than that of a previously described nested RT-PCR protocol (33). The observed sensitivity of our assay (limit of detection [LoD] of 25.2 IU/ml; 95% CI, 19.2 to 44.1 IU/ml) was consistent with data from other reports that recognized the relative insensitivity of some nested RT-PCR protocols for the detection of HEV RNA (24). Furthermore, VL results obtained by testing members of the WHO HEV genotype reference panel yielded satisfactory agreement with VL results generated by collaborative study participants among representative strains of all 4 major HEV genotypes (Fig. 1). Of particular importance, the narrow range of Tm values (68.2°C to 68.9°C) associated with the amplification products generated from individual panel members suggests that unanticipated SNPs with the potential to affect probe binding efficiency were not present in any of the nucleotide sequences of these 11 HEV strains. Although our assay was capable of amplifying and detecting all diluted DNA oligonucleotides representing worst-case mismatching with assay primer and probe sequences (based on reported HEV sequences available from GenBank), underquantification of HEV RNA levels in clinical specimens containing such HEV strains could occur, as suggested by the higher CT and lower Tm values observed with some of these oligonucleotides (see Table S2 in the supplemental material).
The accurate detection and quantification of HEV RNA have become important tools for the diagnosis of chronic HEV infection and monitoring of HEV VL in SOT recipients undergoing antiviral treatment. In the present study, serial monitoring of HEV VL in the 2 SOT recipients demonstrated a rapid decline in HEV VL following the initiation of ribavirin therapy. While a relatively good correlation was observed between ALT level and HEV VL, direct measurement of HEV RNA levels currently offers the preferred means of assessing viral kinetics in an infected patient.
In summary, our use of Pleiades probe chemistry, together with a broadly reactive HEV primer/probe set and synthetic assay calibrators and controls (SynTura HEV), enabled the development of an HEV RT-qPCR assay that is sensitive, accurate, and reproducible, in addition to being able to detect and quantify HEV strains of all 4 major genotypes. The use of secondary standards, like SynTura HEV, calibrated to the WHO HEV international standard can improve the overall performance and standardization of nucleic acid-based assays used for the detection and accurate quantification of HEV RNA in clinical practice.
MATERIALS AND METHODS
Clinical specimens.
A total of 125 serum specimens were selected for evaluation by HEV RT-qPCR, including 68 specimens from patients with elevated serum transaminase levels and suspected viral hepatitis, 22 specimens from patients with serological evidence of recent HEV infection (HEV IgM antibody enzyme-linked immunosorbent assay [ELISA] kit; Anogen-Yes Biotech Laboratories, Mississauga, Ontario, Canada), and 35 specimens known to contain HEV RNA. In addition to testing by HEV RT-qPCR, all 125 specimens were tested for HEV RNA by a nested RT-PCR assay as previously described (33). The 691-bp products amplified by the nested RT-PCR assay for all HEV RNA-positive specimens were also sequenced to determine the HEV genotype as previously described (5, 33). Finally, serial serum specimens obtained from 2 SOT recipients, a 60-year-old male OLT recipient (n = 5) and a 56-year-old male KPT recipient (n = 4), with serological evidence of HEV infection (recomWell HEV IgG, IgM; Mikrogen GmbH, Neuried, Germany) were evaluated by HEV RT-qPCR. Genotypes of the HEV strains detected in these 2 SOT recipients were determined by Sanger sequencing of segments of ORF1 and ORF2 performed at the Laboratory Branch of the Division of Viral Hepatitis, Centers for Disease Control and Prevention, Atlanta, GA. This research study protocol was reviewed and approved by the Mayo Clinic institutional review board.
HEV reference materials.
The first WHO international standard for HEV RNA for nucleic acid amplification technique (NAT)-based assays (code 6329/10) (29), consisting of HEV genotype 3a at a reconstituted concentration of 250,000 IU/ml, was diluted (500, 200, 50, 20, 10, and 0 IU/ml) in normal human serum for evaluation of analytical sensitivity. A total of 12 aliquots (600 μl each) were prepared at each level and tested by HEV RT-qPCR. The first WHO international reference panel for HEV genotypes for NAT-based assays (code 8578/13) (42), comprising 11 members and HEV genotypes 1a, 1a, 1e, 2a, 3, 3b, 3c, 3e, 3f, 4c, and 4g, was used to evaluate HEV genotype inclusivity and VL agreement with HEV RT-qPCR results.
HEV sequence variants (DNA oligonucleotides).
To evaluate the ability of HEV RT-qPCR to detect atypical or variant HEV sequences, DNA oligonucleotide constructs corresponding to 16 HEV sequences retrieved from the GenBank database and exhibiting high-level nucleotide mismatching within the HEV ORF2/3 primer and probe binding regions (see Table S1 in the supplemental material) were synthesized (Sigma-Aldrich Inc., St. Louis, MO), PAGE purified, quantified spectrophotometrically, and diluted to ∼1,000 copies/ml prior to testing by HEV RT-qPCR. Data from dissociation (melt) curve analyses were used to define a Tm acceptance range for use for the confirmation of HEV amplification products.
HEV secondary standard (SynTura HEV).
A custom recombinant HEV reference material (SynTura HEV) containing a 121-bp sequence corresponding to HEV genotype 3a (GenBank accession number AF060669; nt 5258 to 5378) was developed in partnership with Thermo Scientific Quality Controls (Fremont, CA). This material consisted of a BVDV construct modified by the introduction of 2 unique restriction sites, SnaBI and PacI, enabling the insertion of the target sequence (41). After in vitro transcripts of the recombinant viral genome were generated, RNA was introduced into Madin-Darby bovine kidney (MDBK) cells, and recombinant BVDV-HEV was grown over several passages in the laboratory. Viral progeny were sequenced and amplified by real-time PCR to ensure that the insert contained the intended HEV sequence of the correct size. The SynTura HEV reference material was value assigned and traceable to the WHO HEV international standard (43). A working assay calibrator, the AcroMetrix primary standard (APS), was value assigned to the WHO HEV international standard. Subsequently, a custom manufacturing stock, the AcroMetrix standardized manufacturing stock (ASMS), was value assigned by using the APS. All lots of SynTura HEV reference material were made by using the ASMS and tested against the original APS to ensure consistency and traceability to this higher-order standard.
SynTura HEV calibrators and controls.
SynTura HEV reference material previously calibrated to the WHO HEV international standard was diluted with normal human serum to prepare 5-member calibration panels (at HEV RNA levels of 500, 5,000, 50,000, 500,000, and 5,000,000 IU/ml) and assay controls at 3 levels (0, 750, and 75,000 IU/ml). Calibrators and controls prepared by the manufacturer and shipped frozen as single-use aliquots were stable for a minimum of 1 year when stored at −70°C (data not shown). An acceptable performance of the HEV calibrators was confirmed by testing replicate dilutions (2.70 and 4.00 log10 IU/ml) of the WHO HEV international standard by HEV RT-qPCR and comparing expected and observed results.
Nucleic acid extraction.
The MagNA Pure LC high-performance total nucleic acid isolation kit, the MagNA Pure LC instrument, and the Total NA HS 500 extraction protocol (Roche Diagnostics Corp., Indianapolis, IN) were used for sample preparation with input and elution volumes of 500 μl and 75 μl, respectively. A bacteriophage MS2 RNA internal control (MGB Alert MS2 RNA template; ELITechGroup Inc., Bothell, WA) was added directly to the MagNA Pure LC lysis/binding buffer (1:12,000 ratio) just prior to the start of automated sample processing to protect it from RNase digestion in the unprocessed serum specimens. Assuming 100% recovery of the template, each reaction mixture contained ∼250 copies of the MS2 RNA internal control.
RT-qPCR primers and probes.
The primer and probe sequences used for HEV RT-qPCR were based on an adaptation of a previously described and broadly reactive primer set that amplifies a 69-bp segment of the HEV ORF2/3 nucleotide sequence (30). The final assay design utilizing MGB Pleiades probe chemistry (28) was developed in collaboration with ELITechGroup Inc. (Bothell, WA) and was used in conjunction with an MS2 RNA internal control primer/probe set as outlined in Table 2.
TABLE 2.
HEV and internal control RT-qPCR primers and Pleiades probes
| Target and primer or probe | Nucleotide sequencea | Concn (1×) (μM) |
|---|---|---|
| HEV ORF 2/3 | ||
| HEV forward primer | 5′-AATAAATCATAAGGTGGTTTCTGGGGTGAC-3′ | 0.250 |
| HEV reverse primer | 5′-AATAAATCATAAGGGGTTGGTTGGATGAA-3′ | 1.000 |
| HEV probe | 5′-MGB-FAM-G*TGATTCTCAGCCCTTCG-NFQ-3′ | 0.400 |
| MS2 RNA internal control | ||
| MS2 forward primer | 5′-CCA*TCAAA*GTCGA*GGTGCCTAAAGTG-3′ | 0.075 |
| MS2 reverse primer | 5′-ACGAACGCCATGCGGCTACAGGAAGCTC-3′ | 0.075 |
| MS2 probe | 5′-MGB-AP525-G*CTGTTGGTGGTGTAGAGC-NFQ-3′ | 0.200 |
Underlined nucleotides represent 5′ flaps added to enhance RT-qPCR amplification (31). G*and A* represent super G and super A bases (ELITechGroup Inc.) incorporated to enhance stability (32). AP525, proprietary fluorescent dye (ELITechGroup Inc.); FAM, 6-carboxyfluorescein; MGB, minor grove binder; NFQ, nonfluorescent quencher; ORF, open reading frame.
RT-qPCR amplification.
Each 50-μl reaction mixture combined 25 μl of the sample eluate with primers and probes and a custom 5× LibertyTaq One-Step RT-qPCR master mix containing heat-labile uracil-N-glycosylase (UNG) and ROX passive reference dye (Thermo Fisher Scientific, Waltham, MA). Amplification and detection were performed on an Applied Biosystems 7900HT Fast real-time PCR system (Thermo Fisher Scientific) using ROX reference dye normalization, 100% ramp rates, and the following thermal cycling profile: 20 min at 30°C for one cycle, 5 min at 55°C for one cycle, 30 min at 49°C for one cycle, and 2 min at 95°C for one cycle, followed by 50 repetitions of a three-step cycle (15 s at 95°C, 30 s at 56°C, and 30 s at 72°C). This amplification profile was followed by dissociation curve analysis, which consisted of a 15-s hold time each at 95°C, 35°C, and 95°C with 100% ramp rates, except for a 5% ramp rate during the final ramp from 35°C to 95°C. Fluorescent signals for HEV (6-carboxyfluorescein [FAM] dye [absorption wavelength {λ} = 496 nm; emission λ = 517 nm]) and the MS2 RNA internal control (AP525 dye [absorption λ = 527 nm; emission λ = 549 nm]) were acquired during each annealing step of the amplification and throughout the final ramp of the dissociation step. The maximum change in normalized fluorescence (ΔRn) values obtained from the sera of 50 healthy blood donors without evidence of HEV infection were used to determine the mean background ΔRn and to establish a standardized CT value of 0.08 ΔRn (mean background ΔRn plus 10 SD) for the assessment of HEV target and MS2 RNA internal control amplification curves along with a CT acceptance range (30 to 42 cycles) for the MS2 RNA internal control (data not shown).
Statistical analysis.
Results from testing of the 12 replicate HEV RNA dilution panels (500, 200, 50, 20, and 0 IU/ml) prepared from the WHO HEV international standard were evaluated by Probit regression analysis using a 95% detection rate.
Supplementary Material
ACKNOWLEDGMENTS
No external funding was used in support of this work.
We are grateful to Mair Hughes (Thermo Fischer Scientific) for development of the SynTura HEV material used in this work and to Kymberly D. Watt (Mayo Clinic) and Brian Kim (Keck School of Medicine, University of Southern California) for the clinical history of the solid-organ transplant recipients. We acknowledge the assistance of Jan Drobeniuc and Alexandra Tejada (Laboratory Branch of the Division of Viral Hepatitis, Centers for Disease Control and Prevention, Atlanta, GA) for assistance in determining the HEV genotypes in the SOT recipients.
We dedicate this publication to Irina A. Afonina (ELITechGroup Inc.), who was instrumental in this work and passed away in 2015.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JCM.02334-16.
REFERENCES
- 1.Cao D, Meng XJ. 2012. Molecular biology and replication of hepatitis E virus. Emerg Microbes Infect 1:e17. doi: 10.1038/emi.2012.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Panda SK, Thakral D, Rehman S. 2007. Hepatitis E virus. Rev Med Virol 17:151–180. doi: 10.1002/rmv.522. [DOI] [PubMed] [Google Scholar]
- 3.Purcell RH, Emerson SU. 2008. Hepatitis E: an emerging awareness of an old disease. J Hepatol 48:494–503. doi: 10.1016/j.jhep.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 4.Hoofnagle JH, Nelson KE, Purcell RH. 2012. Hepatitis E. N Engl J Med 367:1237–1244. doi: 10.1056/NEJMra1204512. [DOI] [PubMed] [Google Scholar]
- 5.Dong C, Meng J, Dai X, Liang JH, Feagins AR, Meng XJ, Belfiore NM, Bradford C, Corn JL, Cray C, Glass GE, Gordon ML, Hesse RA, Montgomery DL, Nicholson WL, Pilny AA, Ramamoorthy S, Shaver DD, Drobeniuc J, Purdy MA, Fields HA, Kamili S, Teo CG. 2011. Restricted enzooticity of hepatitis E virus genotypes 1 to 4 in the United States. J Clin Microbiol 49:4164–4172. doi: 10.1128/JCM.05481-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meng XJ. 2010. Hepatitis E virus: animal reservoirs and zoonotic risk. Vet Microbiol 140:256–265. doi: 10.1016/j.vetmic.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nelson KE, Kmush B, Labrique AB. 2011. The epidemiology of hepatitis E virus infections in developed countries and among immunocompromised patients. Expert Rev Anti Infect Ther 9:1133–1148. doi: 10.1586/eri.11.138. [DOI] [PubMed] [Google Scholar]
- 8.Lee GH, Tan BH, Teo EC-Y, Lim SG, Dan YY, Wee A, Aw PP, Zhu Y, Hibberd ML, Tan CK, Purdy MA, Teo CG. 2016. Chronic infection with camelid hepatitis E virus in a liver transplant recipient who regularly consumes camel meat and milk. Gastroenterology 150:355.e3–357.e3. doi: 10.1053/j.gastro.2015.10.048. [DOI] [PubMed] [Google Scholar]
- 9.Kamar N, Selves J, Mansuy JM, Ouezzani L, Peron JM, Guitard J, Cointault O, Esposito L, Abravanel F, Danjoux M, Durand D, Vinel JP, Izopet J, Rostaing L. 2008. Hepatitis E virus and chronic hepatitis in organ-transplant recipients. N Engl J Med 358:811–817. doi: 10.1056/NEJMoa0706992. [DOI] [PubMed] [Google Scholar]
- 10.Kamar N, Garrouste C, Haagsma EB, Garrigue V, Pischke S, Chauvet C, Dumortier J, Cannesson A, Cassuto-Viguier E, Thervet E, Conti F, Lebray P, Dalton HR, Santella R, Kanaan N, Essig M, Mousson C, Radenne S, Roque-Afonso AM, Izopet J, Rostaing L. 2011. Factors associated with chronic hepatitis in patients with hepatitis E virus infection who have received solid organ transplants. Gastroenterology 140:1481–1489. doi: 10.1053/j.gastro.2011.02.050. [DOI] [PubMed] [Google Scholar]
- 11.Kamar N, Mansuy JM, Cointault O, Selves J, Abravanel F, Danjoux M, Otal P, Esposito L, Durand D, Izopet J, Rostaing L. 2008. Hepatitis E virus-related cirrhosis in kidney- and kidney-pancreas-transplant recipients. Am J Transplant 8:1744–1748. doi: 10.1111/j.1600-6143.2008.02286.x. [DOI] [PubMed] [Google Scholar]
- 12.Gerolami R, Moal V, Colson P. 2008. Chronic hepatitis E with cirrhosis in a kidney-transplant recipient. N Engl J Med 358:859–860. doi: 10.1056/NEJMc0708687. [DOI] [PubMed] [Google Scholar]
- 13.Yoo N, Bernstein J, Caldwell C, Dong C, Drobeniuc J, Kamili S, Landry ML. 2013. Hepatitis E virus infection in a liver transplant recipient: delayed diagnosis due to variable performance of serologic assays. Transpl Infect Dis 15:E166–E168. doi: 10.1111/tid.12096. [DOI] [PubMed] [Google Scholar]
- 14.Sue PK, Pisanic N, Heaney CD, Mixson-Hayden T, Kamili S, Nelson K, Schwarz KB, Forman M, Valsamakis A, Ticehurst J, Karnsakul W. 2015. Variability of hepatitis E serologic assays in a pediatric liver transplant recipient: challenges to diagnosing hepatitis E virus infection in the United States. Transpl Infect Dis 17:284–288. doi: 10.1111/tid.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drobeniuc J, Meng J, Reuter G, Greene-Montfort T, Khudyakova N, Dimitrova Z, Kamili S, Teo CG. 2010. Serologic assays specific to immunoglobulin M antibodies against hepatitis E virus: pangenotypic evaluation of performances. Clin Infect Dis 51:e24–e27. doi: 10.1086/654801. [DOI] [PubMed] [Google Scholar]
- 16.Abravanel F, Chapuy-Regaud S, Lhomme S, Miedouge M, Peron JM, Alric L, Rostaing L, Kamar N, Izopet J. 2013. Performance of anti-HEV assays for diagnosing acute hepatitis E in immunocompromised patients. J Clin Virol 58:624–628. doi: 10.1016/j.jcv.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 17.Kamar N, Rostaing L, Abravanel F, Garrouste C, Lhomme S, Esposito L, Basse G, Cointault O, Ribes D, Nogier MB, Alric L, Peron JM, Izopet J. 2010. Ribavirin therapy inhibits viral replication on patients with chronic hepatitis E virus infection. Gastroenterology 139:1612–1618. doi: 10.1053/j.gastro.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 18.Gerolami R, Borentain P, Raissouni F, Motte A, Solas C, Colson P. 2011. Treatment of severe acute hepatitis E by ribavirin. J Clin Virol 52:60–62. doi: 10.1016/j.jcv.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 19.Alric L, Bonnet D, Beynes-Rauzy O, Izopet J, Kamar N. 2011. Definitive clearance of a chronic hepatitis E virus infection with ribavirin treatment. Am J Gastroenterol 106:1562–1563. doi: 10.1038/ajg.2011.158. [DOI] [PubMed] [Google Scholar]
- 20.Kamar N, Izopet J, Tripon S, Bismuth M, Hillaire S, Dumortier J, Radenne S, Coilly A, Garrigue V, D'Alteroche L, Buchler M, Couzi L, Lebray P, Dharancy S, Minello A, Hourmant M, Roque-Afonso AM, Abravanel F, Pol S, Rostaing L, Mallet V. 2014. Ribavirin for chronic hepatitis E virus infection in transplant recipients. N Engl J Med 370:1111–1120. doi: 10.1056/NEJMoa1215246. [DOI] [PubMed] [Google Scholar]
- 21.Ambrosioni J, Mamin A, Hadengue A, Bernimoulin M, Samii K, Landelle C, Negro F, Kaiser L. 2014. Long-term hepatitis E viral load kinetics in an immunocompromised patient treated with ribavirin. Clin Microbiol Infect 20:O718–O720. doi: 10.1111/1469-0691.12576. [DOI] [PubMed] [Google Scholar]
- 22.Kamar N, Lhomme S, Abravanel F, Cointault O, Esposito L, Cardeau-Desangles I, Del Bello A, Dorr G, Lavayssiere L, Nogier MB, Guitard J, Ribes D, Goin AL, Broue P, Metsu D, Saune K, Rostaing L, Izopet J. 2015. An early viral response predicts the virological response to ribavirin in hepatitis E virus organ transplant patients. Transplantation 99:2124–2131. doi: 10.1097/TP.0000000000000850. [DOI] [PubMed] [Google Scholar]
- 23.Dao Thi VL, Debing Y, Wu X, Rice CM, Neyts J, Moradpour D, Gouttenoire J. 2016. Sofosbuvir inhibits hepatitis E virus replication in vitro and results in an additive effect when combined with ribavirin. Gastroenterology 150:82.e4–85.e4. doi: 10.1053/j.gastro.2015.09.011. [DOI] [PubMed] [Google Scholar]
- 24.Baylis SA, Hanschmann KM, Blumel J, Nubling CM, HEV Collaborative Study Group. 2011. Standardization of hepatitis E virus (HEV) nucleic acid amplification technique-based assays: an initial study to evaluate a panel of HEV strains and investigate laboratory performance. J Clin Microbiol 49:1234–1239. doi: 10.1128/JCM.02578-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abravanel F, Sandres-Saune K, Lhomme S, Dubois M, Mansuy JM, Izopet J. 2012. Genotype 3 diversity and quantification of hepatitis E virus RNA. J Clin Microbiol 50:897–902. doi: 10.1128/JCM.05942-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abravanel F, Chapuy-Regaud S, Lhomme S, Dubois M, Peron JM, Alric L, Rostaing L, Kamar N, Izopet J. 2013. Performance of two commercial assays for detecting hepatitis E virus RNA in acute or chronic infections. J Clin Microbiol 51:1913–1916. doi: 10.1128/JCM.00661-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mokhtari C, Marchadier E, Haim-Boukobza S, Jeblaoui A, Tesse S, Savary J, Roque-Afonso AM. 2013. Comparison of real-time RT-PCR assays for hepatitis E virus RNA detection. J Clin Virol 58:36–40. doi: 10.1016/j.jcv.2013.06.038. [DOI] [PubMed] [Google Scholar]
- 28.Lukhtanov EA, Lokhov SG, Gorn VV, Podyminogin MA, Mahoney W. 2007. Novel DNA probes with low background and high hybridization-triggered fluorescence. Nucleic Acids Res 35:e30. doi: 10.1093/nar/gkl1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baylis SA, Blumel J, Mizusawa S, Matsubayashi K, Sakata H, Okada Y, Nubling CM, Hanschmann KM. 2013. World Health Organization international standard to harmonize assays for detection of hepatitis E virus RNA. Emerg Infect Dis 19:729–735. doi: 10.3201/eid1905.121845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jothikumar N, Cromeans TL, Robertson BH, Meng XJ, Hill VR. 2006. A broadly reactive one-step real-time RT-PCR assay for rapid and sensitive detection of hepatitis E virus. J Virol Methods 131:65–71. doi: 10.1016/j.jviromet.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 31.Afonina I, Ankoudinova I, Mills A, Lokhov S, Huynh P, Mahoney W. 2007. Primers with 5′ flaps improve real-time PCR. Biotechniques 43:770, 772, 774. doi: 10.2144/000112631. [DOI] [PubMed] [Google Scholar]
- 32.Hymas W, Atkinson A, Stevenson J, Hillyard D. 2007. Use of modified oligonucleotides to compensate for sequence polymorphisms in the real-time detection of norovirus. J Virol Methods 142:10–14. doi: 10.1016/j.jviromet.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 33.Dai X, Dong C, Zhou Z, Liang J, Dong M, Yang Y, Fu J, Tian H, Wang S, Fan J, Meng J, Purdy MA. 2013. Hepatitis E virus genotype 4, Nanjing, China, 2001-2011. Emerg Infect Dis 19:1528–1530. doi: 10.3201/eid1909.130013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raymaekers M, Smets R, Maes B, Cartuyvels R. 2009. Checklist for optimization and validation of real-time PCR assays. J Clin Lab Anal 23:145–151. doi: 10.1002/jcla.20307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stevenson J, Hymas W, Hillyard D. 2005. Effect of sequence polymorphisms on performance of two real-time PCR assays for detection of herpes simplex virus. J Clin Microbiol 43:2391–2398. doi: 10.1128/JCM.43.5.2391-2398.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hymas W, Stevenson J, Taggart EW, Hillyard D. 2005. Use of lyophilized standards for the calibration of a newly developed real time PCR assay for human herpes type six (HHV6) variants A and B. J Virol Methods 128:143–150. doi: 10.1016/j.jviromet.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 37.Hymas WC, Aldous WK, Taggart EW, Stevenson JB, Hillyard DR. 2008. Description and validation of a novel real-time RT-PCR enterovirus assay. Clin Chem 54:406–413. doi: 10.1373/clinchem.2007.095414. [DOI] [PubMed] [Google Scholar]
- 38.Stevenson J, Hymas W, Hillyard D. 2008. The use of armored RNA as a multi-purpose internal control for RT-PCR. J Virol Methods 150:73–76. doi: 10.1016/j.jviromet.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hymas WC, Hillyard DR. 2009. Evaluation of Nanogen MGB Alert Detection Reagents in a multiplex real-time PCR for influenza virus types A and B and respiratory syncytial virus. J Virol Methods 156:124–128. doi: 10.1016/j.jviromet.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 40.Garson JA, Ferns RB, Grant PR, Ijaz S, Nastouli E, Szypulska R, Tedder RS. 2012. Minor groove binder modification of widely used TaqMan probe for hepatitis E virus reduces risk of false negative real-time PCR results. J Virol Methods 186:157–160. doi: 10.1016/j.jviromet.2012.07.027. [DOI] [PubMed] [Google Scholar]
- 41.Baroth M, Peters Y, Schonbrunner ER, Behrens SE. 2010. Stable recombinants of bovine viral diarrhea virus containing a hepatitis C virus insert. J Gen Virol 91:1213–1217. doi: 10.1099/vir.0.016998-0. [DOI] [PubMed] [Google Scholar]
- 42.Expert Committee on Biological Standardization. 2015. Collaborative study to establish the 1st World Health Organization international reference panel for hepatitis E virus RNA genotypes for nucleic acid amplification technique (NAT)-based assays; document WHO/BS/2015.2264. World Health Organization, Geneva, Switzerland: http://www.who.int/biologicals/expert_committee/BS2264_Establishment_HEV_genotypes_1st_IRP_for_NAT.pdf Accessed 1 November 2016. [Google Scholar]
- 43.International Organization for Standardization Technical Committee CEN/TC. 2003. Measurement of quantities in biological samples—metrological traceability of values assigned to calibrators and control materials; document EN ISO17511:2003. International Organization for Standardization, Geneva, Switzerland: https://www.iso.org/obp/ui/#iso:std:iso:17511:ed-1:v1:en Accessed 1 November 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.