

Abstract
Autophagy is a regulated process of intracellular catabolism required for normal cellular maintenance, as well as serving as an adaptive response under various stress conditions, including starvation. The molecular regulation of autophagy in mammalian cells remains incompletely understood. Here we demonstrate a role for protein acetylation in the execution and regulation of autophagy. In particular, we demonstrate that the p300 acetyltransferase can regulate the acetylation of various known components of the autophagy machinery. Knockdown of p300 reduces acetylation of Atg5, Atg7, Atg8, and Atg12, although overexpressed p300 increases the acetylation of these same proteins. Furthermore, p300 and Atg7 colocalize within cells, and the two proteins physically interact. The interaction between p300 and Atg7 is dependent on nutrient availability. Finally, we demonstrate that knockdown of p300 can stimulate autophagy, whereas overexpression of p300 inhibits starvation-induced autophagy. These results demonstrate a role for protein acetylation and particularly p300 in the regulation of autophagy under conditions of limited nutrient availability.
Macro-autophagy, herein referred to as autophagy, is an evolutionary conserved process first characterized in lower organisms (1). In yeast, over 20 separate genes (designated ATG1, ATG2, etc.) have been demonstrated to be essential to carry out the autophagy program. This process is thought to provide a mechanism for the efficient removal of both long lived proteins and damaged cellular organelles. This regulated degradation provides several essential functions for the cell. First, it allows for the removal of damaged and potentially harmful cellular contents. In addition, in breaking down various intracellular components, the autophagy process provides essential building blocks for the cell to use in the re-synthesis of necessary macromolecules. To accomplish this recycling effort, the coordinated actions of various Atg gene products are required. In particular, the Atg gene products together orchestrate the formation of a double membrane structure known as the autophagosome that engulfs the intended cellular cargo targeted for degradation. The autophagosome eventually fuses with the vacuole in yeast or the lysosome in mammals.
In both yeast and mammalian cells, autophagy can be stimulated by the withdrawal of nutrients. Under these conditions, autophagic degradation of nonessential components may be essential to meet ongoing energetic needs in the presence of limited extracellular nutrients. This point was underscored by the analysis of mice containing a targeted deletion of Atg5 (2). In the absence of Atg5, there is a lack of both basal and starvation-induced autophagy. Mice lacking Atg5 are born normally but succumb within the 1st day of life. This post-natal lethality is thought to be due in large part for the requirement of autophagy to supply the energetic needs of neonates. These needs are particularly critical during the small window of time where the animal no longer has a placental circulation and before the pup can begin to nurse and thus obtain external nutrients.
Relatively little is known regarding how signals such as nutrient availability are able to be transduced to ultimately regulate the level of cellular autophagy. One important pathway that impinges on the process is signaling thorough the target of rapamycin (TOR)2 network (3). Evidence suggests that TOR signaling inhibits autophagy, and indeed agents such as rapamycin that can inhibit TOR are known to result in increased autophagy. We recently have observed that in addition to this mode of regulation, the NAD-dependent deacetylase Sirt1 is also a regulator of autophagy in mammalian cells and tissues (4). In particular, we demonstrated that in the absence of Sirt1 levels of acetylation for various components of the autophagy machinery are increased and that starvation-induced autophagy is impaired. Interestingly, like the Atg5 knock-out animals, Sirt1-/- mice are also born normally but die within the few hours to days after birth. Consistent with a defect in autophagy, electron micrographs of hearts from Sirt1-/- mice demonstrated an accumulation of abnormal appearing organelles, including mitochondria, a phenotype previously observed in Atg-deficient animals (5). Here we have further characterized the role of acetylation in the regulation of autophagy, and in particular, we demonstrate a role for the p300 acetyltransferase in this process.
EXPERIMENTAL PROCEDURES
Cell Culture and DNA Constructs—HeLa cells (obtained from ATCC) were cultured at 37 °C in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). The GFP-LC3 plasmid and the various Myc-tagged Atg constructs have been described previously (4). The HA-tagged p300 expression vector was obtained from Addgene. siRNA directed against p300, CBP, or PCAF was obtained from Dharmacon.
GFP-LC3 Assays—Induction of autophagy was observed in HeLa cells transfected with GFP-LC3 using confocal microscopy (LSM 510 Meta, Zeiss). The number of GFP-LC3 dots per cell was assessed as described previously (4). A minimum of 30 random cells per condition were counted, and the data presented represent the results from three independent experiments (mean ± S.D.). For starvation conditions, HeLa cells were switched from full growth medium to culture medium consisting of Hanks' balanced salt solution (Invitrogen) for 2 h at 37 °C.
Immunoprecipitation and Immunoblotting—For immunoprecipitation analysis of transfected cells, protein lysates (2 mg) were prepared from HeLa cells and mixed with the indicated antibodies (4 °C overnight) followed by addition of 60 μl of protein G-Sepharose (Amersham Biosciences) for 2 h at 4 °C. The endogenous interaction between Atg proteins and p300 was analyzed in a similar fashion; however, we employed 4 mg of protein lysate. Immune complexes were washed five times with Lysis buffer (50 mm Tris, pH 7.4, 1% Triton X-100, 0.5% Nonidet P-40, 150 mm NaCl, protease and phosphatase inhibitor mixture (Sigma) and 10% glycerol). After boiling in 2× sample buffer, samples were subjected to SDS-PAGE analysis. Following transfer to nitrocellulose, membranes were immunoblotted with the indicated primary antibodies, including anti-HA (Roche Applied Science, catalog number 11867423001); Atg7 (ProSci, catalog number XW-7984); actin (Sigma, catalog number A2103); c-Myc (Santa Cruz Biotechnology, catalog number SC-40); Atg8/LC3 (Novus, catalog number NB100-2331, and Sigma, catalog number L8918); Atg12 (Cell Signaling Technology, catalog number 2010); p62 (Progen, catalog number GP62-C); p300 (Upstate, catalog number 05-257); CBP and PCAF (Santa Cruz Biotechnology, catalog numbers SC-369 and SC-13124); and acetyl-Lys (Cell Signaling Technology, catalog number 9441) followed by the appropriate horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology). Bands were visualized by enhanced chemiluminescence (Pierce). Where indicated, cells were incubated with the lysosomal protease inhibitors pepstatin A (Sigma) and E-64d (Sigma) for 30 min (10 μg/ml) prior to harvest (6).
Transfections and Immunofluorescence—HeLa cells were routinely transfected with Effectene (Qiagen) according to the manufacturer's recommendations. For the GFP-LC3 assays, cells were plated on chamber slides and then cotransfected where indicated with 0.6 μg of a p300 expression construct or the corresponding empty vector along with 0.4 μg of the pGFP-LC3 construct. Cells were visualized 24 h after transfection. For acetylation analysis of autophagy proteins, HeLa cells in 10-cm dishes were transfected with 3 μg of the indicated epitope-tagged Atg constructs and a similar amount of either the HA-tagged p300 vector or the corresponding empty vector as a control. Protein lysates were analyzed for acetylation levels 24 h after transfection.
To assess the requirement of acetyltransferase activity on autophagy induction, HeLa cells were transfected with either 300 nm siRNA directed against p300 or PCAF (Dharmacon) or a corresponding control nontargeting RNAi along with 0.8 μg of pGFP-LC3. In these sets of experiments, cells were transfected using Oligofectamine (Invitrogen) according to the manufacturer's protocol. Cells were analyzed 36 h after RNAi transfection. For detection of endogenous Atg7 acetylation, 10-cm dishes of HeLa cells were transfected with 300 nm of a control, p300, CBP, or PCAF siRNA. Protein acetylation was routinely analyzed 48 h after transfection.
For immunofluorescence analysis of the subcellular localization of both Atg7 and p300, HeLa cells were grown on Lab-Tek II dishes (Nunc) and transfected with Myc-Atg7 and HA-p300. Twenty four hours later, cells were washed with ice-cold PBS and then fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. Cells were subsequently permeabilized in 0.5% Triton X-100. To detect transfected Atg7 and p300, non-specific sites were first blocked by incubating cells with a solution of 5% bovine serum albumin and 0.05% Triton X-100 in PBS for 1 h. The cells were then incubated with the primary antibodies directed against either Atg7 (rabbit) or p300 (mouse), and both were used at a 1:100 dilution in 5% bovine serum albumin. After an overnight incubation at 4 °C, cells were washed with PBS and incubated with Alexa Fluor 594 goat anti-mouse IgG (1:250; Molecular Probes) and with Alexa Fluor 488 goat anti-rabbit IgG (1:250, Molecular Probes) for 1 h at room temperature. Images were recorded using a confocal laser scanning microscope.
RESULTS
p300 Acetyltransferase Regulates Acetylation of Autophagy Proteins—We recently demonstrated that Sirt1 can regulate the deacetylation of multiple essential proteins involved in autophagy and that Sirt1 activity was required for the induction of autophagy under starved conditions (4). These results suggested that besides cellular deacetylases, additional regulation of autophagy may be provided by cellular enzymes that catalyze the forward acetylation reaction. To test whether such regulation exists, we sought to identify the relevant acetyltransferases. To accomplish this goal we first examined whether the acetylation of endogenous Atg7 was altered by RNAi-mediated knockdown of three well characterized acetyltransferases as follows: p300, CBP, and PCAF. As demonstrated in Fig. 1, A and B, using an antibody-based detection strategy to identify internal acetyl-lysine residues, we noted that knockdown of p300, and to a lesser extent CBP, reduced endogenous Atg7 acetylation. Although somewhat less efficient, knockdown of PCAF in contrast appeared to have relatively little effect on the level of Atg7 acetylation (Fig. 1C).
FIGURE 1.

Knockdown of protein acetyltransferases reduces acetylation of endogenous Atg7. A, siRNA-mediated knockdown of p300 results in a reduction of endogenous Atg7 acetylation. Protein acetylation was determined by immunoprecipitation (IP) with an antibody recognizing internal acetyl-lysine residues followed by Western blotting (WB) for Atg7. Immunoprecipitation was performed with 2 mg of protein lysate from HeLa cells transfected with either a control (-) or p300-specific RNAi. Evaluation of the protein input (40 μg) revealed that the change observed in Atg7 acetylation was not accompanied by changes in the level of Atg7 protein expression. B, similar analysis for RNAi-mediated knockdown of CBP. C, RNAi-mediated knockdown of PCAF revealed no obvious change in Atg7 acetylation. Shown is one representative example of three similar experiments.
Given that Atg7 acetylation was most heavily influenced by p300 knockdown, we concentrated our future experiments on this enzyme. We next sought to assess whether the acetylation of other Atg gene products was also regulated by p300. Because antibodies are not readily available for all autophagy components, in these experiments we relied on transfected epitope-tagged constructs. Using Myc-tagged Atg5, -7, -8, and -12, we analyzed the effect of p300 knockdown on levels of protein acetylation. As noted in Fig. 2, A–D, and as previously observed with endogenous Atg7, knockdown of p300 reduced the level of acetylation observed for these various Atg constructs without affecting the total level of Atg expression. Using a similar approach, we also noted that transient increased expression of p300 augmented the level of Atg protein acetylation, again without affecting the total levels of Atg expression (Fig. 3).
FIGURE 2.
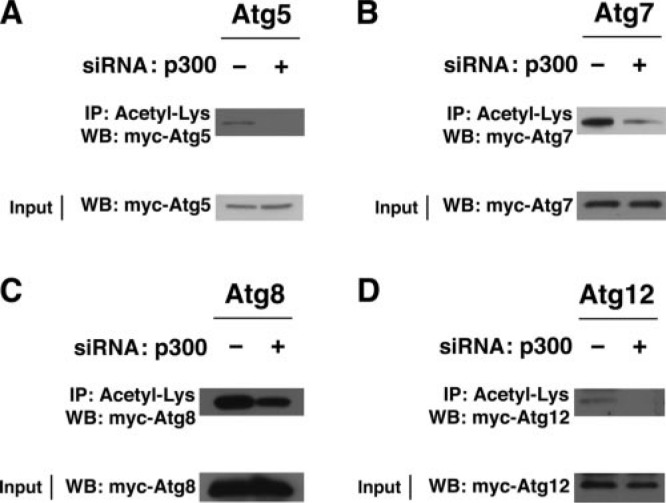
RNAi-mediated knockdown of p300 reduces acetylation of various Atg constructs. HeLa cells were assessed after control or p300-specific knockdown for the level of Atg acetylation. Various Myc-tagged Atg constructs were employed including the following: A, Atg5; B, Atg7; C, Atg8; and D, Atg12. Level of acetylation was determined by immunoprecipitation (IP) of transfected protein cell lysate (2 mg) with an acetyl-lysine antibody followed by Western blotting (WB) employing the Myc epitope. In each case, p300 knockdown reduced the level of Atg acetylation without altering the level of Atg expression.
FIGURE 3.
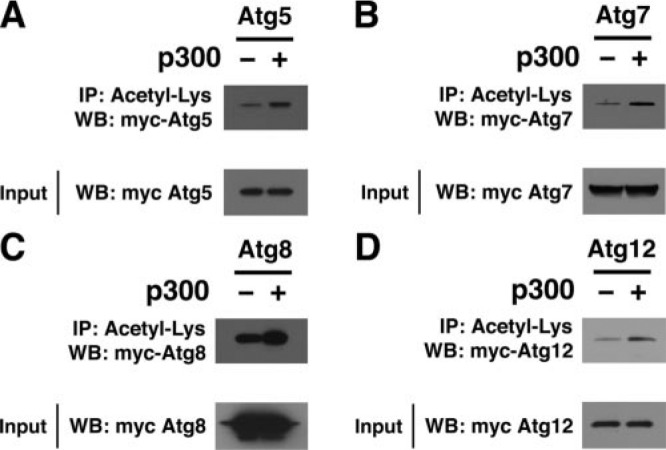
Increased expression of p300 augments acetylation of various essential autophagy components. HeLa cells were transiently transfected with the indicated construct encoding for a Myc-tagged Atg family member along with either a p300 expression vector or the empty vector alone (-). Levels of Atg5 (A), Atg7 (B), Atg8 (C), or Atg12 (D) acetylation were determined by immunoprecipitation (IP) of protein lysate (2 mg) using an antibody recognizing internal acetyl-lysine residues followed by Western blotting (WB) employing the Myc epitope. Expression levels of the various transfected myc-Atg constructs were assessed by Western blotting of the protein input (40 μg).
Atg7 Physically Associates with p300 in a Nutrient-dependent Fashion—We next asked whether p300 could physically associate with components of the autophagy machinery. We prepared protein HeLa cell lysates and assessed whether we could coimmunoprecipitate endogenous Atg7 and p300 in a complex. As demonstrated in Fig. 4A, immunoprecipitation of endogenous Atg7 readily coimmunoprecipitated endogenous p300. Similarly, the reciprocal immunoprecipitation of p300 could coimmunoprecipitate Atg7. This interaction appeared to be specific for Atg7 because under similar conditions we could not detect an interaction between Atg12 and p300 (supplemental Fig. 1). In addition, when we transiently transfected Atg7 and p300 into HeLa cells, we noted that a substantial portion of Atg7 and p300 appeared to localize within the same subcellular domains (Fig. 4B).
FIGURE 4.

Interaction of Atg7 and p300. A, physical interaction between endogenous p300 and Atg7 is demonstrated by coimmunoprecipitation. HeLa cell protein lysates (4 mg) were immunoprecipitated (IP) with an Atg7-specific antibody or a nonspecific IgG isotype-matched control antibody, and the level of coimmunoprecipitated p300 was determined. The reciprocal immunoprecipitation employing a p300-specific antibody or IgG control is also demonstrated. Levels of p300 and Atg7 in the input lysate (40 μg) are also shown. WB, Western blot. B, subcellular distribution in HeLa cells of transfected Atg7 (green), p300 (red), and their overlap (yellow) are shown.
We had previously demonstrated that levels of acetylation for various Atg proteins fell under nutrient-depleted conditions (4). We also have described that although Sirt1 associates with Atg7, this association is not apparently sensitive to nutrient availability (4). As such, it remained unclear what mediates the observed fall in Atg acetylation under starved conditions. Based on the preceding observations, we wondered whether, as opposed to our results involving Sirt1, the association between p300 and Atg7 was in fact sensitive to nutrient status. To test this, we observed the level of Atg7 acetylation under fed and starved conditions, as well as the corresponding strength of interaction between Atg7 and p300. As demonstrated in Fig. 5A, similar to what we had described previously, Atg7 acetylation decreased under starved conditions. Under these same conditions, we noted that the interaction between the p300 acetyltransferase and Atg7 was also reduced (Fig. 5B). This occurred even though the expression of both p300 and Atg7 remained unchanged during fed and starved conditions. Quantification of these effects suggested that the interaction between p300 and Atg7 was reduced ∼40% after 2 h of starvation (fed, 1.0 ± 0.2; starved, 0.6 ± 0.1; n = 3; p < 0.05).
FIGURE 5.
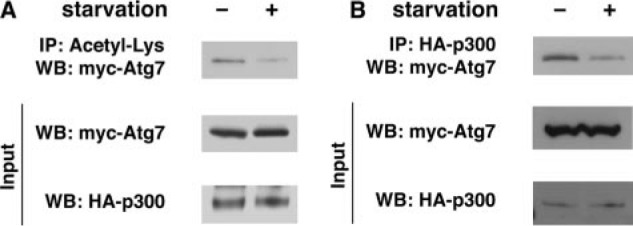
The interaction between p300 and Atg7 is regulated by external nutrients. A, levels of Atg7 acetylation fall under starved conditions. Atg7 acetylation was assessed under fed (-) conditions or following 2 h of starvation (+). Although total levels of Atg7 were unchanged, overall acetylation was reduced following nutrient withdrawal. B, interaction between p300 and Atg7 is reduced under starved conditions. Protein-protein interaction was determined by immunoprecipitation (IP) of epitope-tagged p300 and assessment of coimmunoprecipitated Atg7. Although levels of both p300 and Atg7 were unchanged under fed or starved conditions, the interaction between p300 and Atg7 was routinely reduced ∼40% (fed, 1.0 ± 0.2; starved, 0.6 ± 0.1; n = 3 separate experiments; p < 0.05). WB, Western blot.
p300 Acetyltransferase Regulates Autophagy—Given the role of p300 in modulating acetylation of autophagy components, we next asked whether manipulating p300 levels affected the induction and execution of autophagy. One useful marker for autophagy induction is the conversion of LC3-I to LC3-II. LC3-I, also known as Atg8, must be cleaved and post-translationally modified to a protein that migrates at an apparent lower molecular mass (LC3-II) prior to insertion into the autophagosome. Consistent with a role for p300 in regulating autophagy, we observed an increase in the level of LC3-II in cells where p300 was knocked down by siRNA (Fig. 6, A and B). Although we observed that the addition of lysosomal protease inhibitors increased LC3-II levels under basal fed conditions, this treatment did not appreciably alter steady state levels of LC3-II after p300 knockdown or in untransfected cells following starvation (supplemental Fig. 2). In addition to the LC3 conversion assay, the level of autophagy is often assessed by the steady state level of p62, a protein normally cleared through autophagy (7). Again, consistent with increased autophagy in p300 knockdown cells, levels of p62 expression were reduced when p300 was targeted for knockdown. To further assess the role of p300 in autophagy, we analyzed the incorporation of LC3 into autophagosomes by analyzing the subcellular distribution of a chimeric protein consisting of green fluorescent protein (GFP) fused to LC3 (GFP-LC3). This additional assay of autophagosome induction again suggested that autophagy was increased under both fed and starved conditions in cells where p300 had been knocked down (Fig. 6, C and D). In contrast, knockdown of another acetyltransferase PCAF that we observed does not affect acetylation of Atg7 (Fig. 1C) and had no significant effect on GFP-LC3 redistribution (Fig. 6E).
FIGURE 6.

Knockdown of p300 augments autophagy. HeLa cells were assessed after either control or p300-specific RNAi knockdown. A, induction of autophagy under fed conditions was monitored by the conversion of LC3-I to LC3-II, whereas steady state rates of autophagy were analyzed by Western blot (WB) analysis for p62 expression. Levels of p300 expression and actin (loading control) are also shown. B, quantification of LC3-II to LC3-I ratio in control versus the p300-specific knockdown cells. Shown are the results (mean ± S.D.) of three separate experiments (*, p < 0.05). C, representative confocal image of the localization of GFP-LC3 in cells transfected with a control siRNA, or D, in cells transfected with an siRNA directed against p300. E, quantification of “LC3 dots” using transient transfection of a construct encoding a GFP-LC3 chimeric protein along with the indicated RNAi. Knockdown of p300 appears to stimulate autophagosome formation under both fed and starved conditions. In contrast, knockdown of PCAF, which appeared to have little effect on Atg acetylation, also had little to no effect on autophagy.
To complement these observations, we also analyzed the effects of p300 overexpression on the induction of autophagy. As might be expected, using the previously described biochemical markers of autophagy induction and execution, we observed that under starved conditions, increased expression of p300 reduced the conversion of LC3-I to LC3-II (Fig. 7, A and B). Under these conditions, the addition of lysosomal protease inhibitors augmented the steady state level of LC3-II. Consistent with a role for p300 in regulating autophagic flux and not autophagy induction, levels of p62 also appeared to increase following p300 overexpression (Fig. 7A). Finally, analysis of GFP-LC3 also revealed that expression of p300 inhibited autophagosome formation under starved condition (Fig. 7C).
FIGURE 7.

Increased expression of p300 inhibits autophagy. HeLa cells were assessed following transfection with either an empty vector (-) or a vector encoding the p300 acetyltransferase. A, induction of autophagy was assessed by analyzing the conversion of LC3–1 to LC3-II under starved conditions. Steady state autophagy was monitored by assessing the level of p62 expression. Expression of the epitope-tagged p300 and actin (loading control) is also demonstrated. Protein levels were assessed either in the absence or presence of lysosomal protease inhibitors. WB, Western blot. B, autophagy induction as assessed by the ratio of LC3-II to LC3-I is demonstrated following p300 expression. Shown are the results (mean ± S.D.) of three separate experiments performed in the absence of lysosomal protease inhibitors (*, p < 0.05). C, expression of p300 suppresses starvation-induced autophagy as assessed by the GFP-LC3 dot assay.
DISCUSSION
There is a growing interest in the role of autophagy in normal health and in various disease states. Impairment of autophagy has been suggested to contribute to numerous conditions, including cancer and neurodegeneration (8). Surprisingly, relatively little is known regarding the molecular regulation of autophagy. Studies in yeast have implicated a role for both the TOR pathway and protein kinase A as important regulators of autophagy during starvation. These pathways appear to also be operational in mammalian cells because rapamycin, a pharmacological inhibitor of mammalian TOR, is also a well known activator of basal autophagy. In addition, a recent report has demonstrated that patients with Carney syndrome, an inherited disease caused by mutations in the regulatory subunit of protein kinase A, also manifest evidence of impaired autophagy (9).
We recently demonstrated that the NAD+-dependent deacetylase Sirt1 represented another aspect of potential autophagy regulation (4). In that study, we showed that in the absence of Sirt1, levels of Atg protein acetylation increased. We further noted a general similarity between Sirt1-/- mice and previously reported mice lacking Atg5 (2, 5). Here we have extended our observations by further exploring the role of protein acetylation. In particular, we have addressed what are the endogenous acetyltransferases that can regulate the autophagic process.
Previously we had demonstrated that the level of acetylation is reduced under starved conditions. Surprisingly, the association between the Sirt1 deacetylase and the various Atg proteins did not appear to be affected by nutrient availability (4). In contrast, here we demonstrate that the association of p300 with Atg7 is reduced under starved conditions. These results are therefore consistent with the notion that the fall in Atg acetylation seen with starvation is mostly dependent on a decreased interaction with acetyltransferases, whereas the interaction with deacetylases is less susceptible to nutrient fluxes. In both studies, increased acetylation of the various autophagy proteins, caused either by the absence of Sirt1 or the increased expression of p300, was associated with impairment in the induction of autophagy. These data suggest a model of nutrient control of autophagy wherein protein acetylation plays an important regulatory role. Furthermore, our results would suggest that the acetylation of the various Atg proteins involves the coordinated but opposing activity of the p300 acetyltransferase and the NAD+-dependent deacetylase, Sirt1.
Assessment of autophagy in mammalian cells represents a challenging task and an evolving science. Care must be used for instance in the interpretation of increased LC3-II levels or GFP-LC3 “dots” that can represent either a block in the final stage of autophagy or an indication of increased autophagic flux. One strategy to distinguish these two disparate possibilities is the use of lysosomal protease inhibitors that can increase LC3-II under conditions of decreased flux but would be predicted to have little effect under conditions where the final stages of autophagy are completely blocked (10). Similarly, levels of p62 are often used as an independent measure to assess autophagic flux. In our experimental conditions, lysosomal protease inhibitors did not appear to augment the level of LC3-II during starved conditions or following p300 knockdown (supplemental Fig. 2). The reason for this is not entirely clear but may reflect a near maximal rate of autophagic flux under these conditions. In contrast, we did observe a significant increase in the levels of LC3-II when lysosomal protease inhibitors were used in the setting of p300 overexpression (Fig. 7A). In addition, we noted that p300 overexpression increased levels of p62, whereas knockdown of p300 consistently reduced p62 levels. Thus, our results are most consistent for a role of p300 acetyltransferase in the regulation of autophagic flux.
Limited nutrients produce a number of physiological responses that provide the organism with an enhanced capacity to withstand starvation. Understanding these responses has been spurred by observations that caloric restriction regimens result in an increased life span for a wide range of organisms from yeast to mammals (11). Increased autophagy is clearly one of many important physiological responses to nutrient withdrawal. Consistent with these observations, evidence suggests that autophagy can regulate the life span of simple organisms (12–15). Similarly, in mammals, conditional knockouts of essential autophagy genes can sometimes reproduce pathologies that occur as a consequence of normal aging (16, 17). As such, there is a growing realization that there might be a convergence between nutrient availability, the induction of autophagy, and the regulation of aging and age-related pathologies. In this context, our results describing a Sirt1- and p300-dependent regulation of starvation-induced autophagy provide a further refinement of our understanding of the cellular and physiological response to limited nutrient availability.
Supplementary Material
Footnotes
This work was authored, in whole or in part, by National Institutes of Health staff. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2.
The abbreviations used are: TOR, target of rapamycin; PBS, phosphate-buffered saline; siRNA, small interfering RNA; HA, hemagglutinin; CBP, CREB-binding protein; RNAi, RNA interference; GFP, green fluorescent protein; PCAF, p300/CBP-associated factor.
References
- 1.Ohsumi, Y. (2001) Nat. Rev. Mol. Cell Biol. 2, 211-216 [DOI] [PubMed] [Google Scholar]
- 2.Kuma, A., Hatano, M., Matsui, M., Yamamoto, A., Nakaya, H., Yoshimori, T., Ohsumi, Y., Tokuhisa, T., and Mizushima, N. (2004) Nature 432, 1032-1036 [DOI] [PubMed] [Google Scholar]
- 3.Sarbassov, D. D., Ali, S. M., and Sabatini, D. M. (2005) Curr. Opin. Cell Biol. 17, 596-603 [DOI] [PubMed] [Google Scholar]
- 4.Lee, I. H., Cao, L., Mostoslavsky, R., Lombard, D. B., Liu, J., Bruns, N. E., Tsokos, M., Alt, F. W., and Finkel, T. (2008) Proc. Natl. Acad. Sci. U. S. A. 105, 3374-3379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Komatsu, M., Waguri, S., Ueno, T., Iwata, J., Murata, S., Tanida, I., Ezaki, J., Mizushima, N., Ohsumi, Y., Uchiyama, Y., Kominami, E., Tanaka, K., and Chiba, T. (2005) J. Cell Biol. 169, 425-434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanida, I., Minematsu-Ikeguchi, N., Ueno, T., and Kominami, E. (2005) Autophagy 1, 84-91 [DOI] [PubMed] [Google Scholar]
- 7.Bjorkoy, G., Lamark, T., Brech, A., Outzen, H., Perander, M., Overvatn, A., Stenmark, H., and Johansen, T. (2005) J. Cell Biol. 171, 603-614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shintani, T., and Klionsky, D. J. (2004) Science 306, 990-995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mavrakis, M., Lippincott-Schwartz, J., Stratakis, C. A., and Bossis, I. (2006) Hum. Mol. Genet. 15, 2962-2971 [DOI] [PubMed] [Google Scholar]
- 10.Klionsky, D. J., Abeliovich, H., Agostinis, P., Agrawal, D. K., Aliev, G., Askew, D. S., Baba, M., Baehrecke, E. H., Bahr, B. A., Ballabio, A., Bamber, B. A., Bassham, D. C., Bergamini, E., Bi, X., Biard-Piechaczyk, M., Blum, J. S., Bredesen, D. E., Brodsky, J. L., Brumell, J. H., Brunk, U. T., Bursch, W., Camougrand, N., Cebollero, E., Cecconi, F., Chen, Y., Chin, L. S., Choi, A., Chu, C. T., Chung, J., Clarke, P. G., Clark, R. S., Clarke, S. G., Clave, C., Cleveland, J. L., Codogno, P., Colombo, M. I., Coto-Montes, A., Cregg, J. M., Cuervo, A. M., Debnath, J., Demarchi, F., Dennis, P. B., Dennis, P. A., Deretic, V., Devenish, R. J., Di Sano, F., Dice, J. F., Difiglia, M., Dinesh-Kumar, S., Distelhorst, C. W., Djavaheri-Mergny, M., Dorsey, F. C., Droge, W., Dron, M., Dunn, W. A., Jr., Duszenko, M., Eissa, N. T., Elazar, Z., Esclatine, A., Eskelinen, E. L., Fesus, L., Finley, K. D., Fuentes, J. M., Fueyo, J., Fujisaki, K., Galliot, B., Gao, F. B., Gewirtz, D. A., Gibson, S. B., Gohla, A., Goldberg, A. L., Gonzalez, R., Gonzalez-Estevez, C., Gorski, S., Gottlieb, R. A., Haussinger, D., He, Y. W., Heidenreich, K., Hill, J. A., Hoyer-Hansen, M., Hu, X., Huang, W. P., Iwasaki, A., Jaattela, M., Jackson, W. T., Jiang, X., Jin, S., Johansen, T., Jung, J. U., Kadowaki, M., Kang, C., Kelekar, A., Kessel, D. H., Kiel, J. A., Kim, H. P., Kimchi, A., Kinsella, T. J., Kiselyov, K., Kitamoto, K., Knecht, E., Komatsu, M., Kominami, E., Kondo, S., Kovacs, A. L., Kroemer, G., Kuan, C. Y., Kumar, R., Kundu, M., Landry, J., Laporte, M., Le, W., Lei, H. Y., Lenardo, M. J., Levine, B., Lieberman, A., Lim, K. L., Lin, F. C., Liou, W., Liu, L. F., Lopez-Berestein, G., Lopez-Otin, C., Lu, B., Macleod, K. F., Malorni, W., Martinet, W., Matsuoka, K., Mautner, J., Meijer, A. J., Melendez, A., Michels, P., Miotto, G., Mistiaen, W. P., Mizushima, N., Mograbi, B., Monastyrska, I., Moore, M. N., Moreira, P. I., Moriyasu, Y., Motyl, T., Munz, C., Murphy, L. O., Naqvi, N. I., Neufeld, T. P., Nishino, I., Nixon, R. A., Noda, T., Nurnberg, B., Ogawa, M., Oleinick, N. L., Olsen, L. J., Ozpolat, B., Paglin, S., Palmer, G. E., Papassideri, I., Parkes, M., Perlmutter, D. H., Perry, G., Piacentini, M., Pinkas-Kramarski, R., Prescott, M., Proikas-Cezanne, T., Raben, N., Rami, A., Reggiori, F., Rohrer, B., Rubinsztein, D. C., Ryan, K. M., Sadoshima, J., Sakagami, H., Sakai, Y., Sandri, M., Sasakawa, C., Sass, M., Schneider, C., Seglen, P. O., Seleverstov, O., Settleman, J., Shacka, J. J., Shapiro, I. M., Sibirny, A., Silva-Zacarin, E. C., Simon, H. U., Simone, C., Simonsen, A., Smith, M. A., Spanel-Borowski, K., Srinivas, V., Steeves, M., Stenmark, H., Stromhaug, P. E., Subauste, C. S., Sugimoto, S., Sulzer, D., Suzuki, T., Swanson, M. S., Tabas, I., Takeshita, F., Talbot, N. J., Talloczy, Z., Tanaka, K., Tanaka, K., Tanida, I., Taylor, G. S., Taylor, J. P., Terman, A., Tettamanti, G., Thompson, C. B., Thumm, M., Tolkovsky, A. M., Tooze, S. A., Truant, R., Tumanovska, L. V., Uchiyama, Y., Ueno, T., Uzcategui, N. L., van der Klei, I., Vaquero, E. C., Vellai, T., Vogel, M. W., Wang, H. G., Webster, P., Wiley, J. W., Xi, Z., Xiao, G., Yahalom, J., Yang, J. M., Yap, G., Yin, X. M., Yoshimori, T., Yu, L., Yue, Z., Yuzaki, M., Zabirnyk, O., Zheng, X., Zhu, X., and Deter, R. L. (2007) Autophagy 4, 151-17518188003 [Google Scholar]
- 11.Bishop, N. A., and Guarente, L. (2008) Nat. Rev. Genet. 8, 835-844 [DOI] [PubMed] [Google Scholar]
- 12.Melendez, A., Talloczy, Z., Seaman, M., Eskelinen, E. L., Hall, D. H., and Levine, B. (2003) Science 301, 1387-1391 [DOI] [PubMed] [Google Scholar]
- 13.Bergamini, E., Cavallini, G., Donati, A., and Gori, Z. (2007) Ann. N. Y. Acad. Sci. 1114, 69-78 [DOI] [PubMed] [Google Scholar]
- 14.Toth, M. L., Sigmond, T., Borsos, E., Barna, J., Erdelyi, P., Takacs-Vellai, K., Orosz, L., Kovacs, A. L., Csikos, G., Sass, M., and Vellai, T. (2008) Autophagy 4, 330-338 [DOI] [PubMed] [Google Scholar]
- 15.Hansen, M., Chandra, A., Mitic, L. L., Onken, B., Driscoll, M., and Kenyon, C. (2008) PLoS Genet. 4, e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J. I., Tanida, I., Ueno, T., Koike, M., Uchiyama, Y., Kominami, E., and Tanaka, K. (2006) Nature 441, 819-820 [DOI] [PubMed] [Google Scholar]
- 17.Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., Yokoyama, M., Mishima, K., Saito, I., Okano, H., and Mizushima, N. (2006) Nature 441, 885-889 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.