

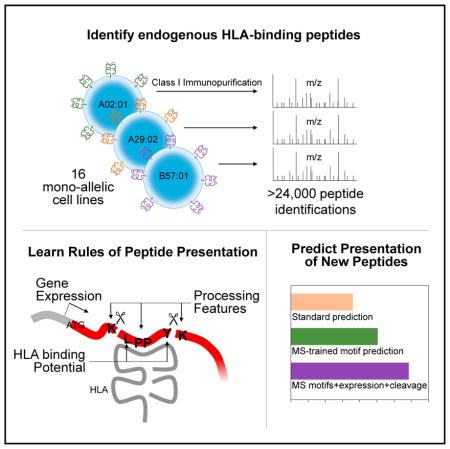
SUMMARY
Identification of human leukocyte antigen (HLA)-bound peptides by liquid chromatography-tandem mass spectrometry (LC-MS/MS) is poised to provide a deep understanding of rules underlying antigen presentation. However, a key obstacle is the ambiguity that arises from the co-expression of multiple HLA alleles. Here, we have implemented a scalable mono-allelic strategy for profiling the HLA peptidome. By using cell lines expressing a single HLA allele, optimizing immunopurifications, and developing an application-specific spectral search algorithm, we identified thousands of peptides bound to 16 different HLA class I alleles. These data enabled the discovery of subdominant binding motifs and an integrative analysis quantifying the contribution of factors critical to epitope presentation, such as protein cleavage and gene expression. We trained neural-network prediction algorithms with our large dataset (>24,000 peptides) and outperformed algorithms trained on data-sets of peptides with measured affinities. We thus demonstrate a strategy for systematically learning the rules of endogenous antigen presentation.
In Brief
HLA class I binding prediction has traditionally been based on biochemical binding experiments. Abelin and colleagues present an LC-MS/MS-based workflow and analytical framework that greatly accelerates gains in prediction performance. Key advances include the discovery of sequence motifs and improved quantification of the roles of gene expression and proteasomal processing.
INTRODUCTION
Human leukocyte antigen (HLA) class I glycoproteins (HLA-A, -B, and -C) are expressed on the surface of almost all nucleated cells in the human body and are required for presentation of short peptides for detection by T cell receptors. The HLA genes are the most polymorphic genes across the human population; more than 10,000 HLA class I allele variants have been identified to date (Robinson et al., 2015). Each HLA allele is estimated to bind and present ~1,000–10,000 unique peptides to T cells (Hunt et al., 1992; Rammensee et al., 1995; Vita et al., 2015), less than 0.1% of the estimated 10 million potential 9-mer peptides from human protein-coding genes. Given such diversity in HLA binding, accurate prediction of whether a peptide is likely to bind to a specific HLA allele is highly challenging.
Rules for peptide binding to HLA molecules have been studied extensively for a subset of HLA alleles (Vita et al., 2015) and have been encoded in modern advanced neural-network-based algorithms (Hoof et al., 2009; Lundegaard et al., 2008). However, the algorithms in common use today (Trolle et al., 2015) are trained almost exclusively on measurements of biochemical affinity of synthetic peptides. This imparts several disadvantages. First, the throughput of these methods is limited because only a very small percentage of peptides are expected to bind, and therefore researchers must synthesize and experimentally assess potentially 1,000s of negative examples to identify 10s of strong-binding positive examples. Biased sampling can improve these odds but carries the risk of skewing the results or missing subdominant motifs. Meanwhile, other unintentional forms of bias, such as pre-existing notions of the length distribution or limitations on peptide synthesis and solubility, are difficult to avoid. Most importantly, these approaches do not necessarily consider the endogenous processing and transport of peptides prior to HLA binding. Mass spectrometry (MS)-based approaches yield a large and relatively unbiased portrait of the population of processed and presented peptides and should theoretically address most of these problems. However, historically liquid chromatography-tandem mass spectrometry (LC-MS/MS) methods have required large cellular input, which limits throughput, and the multi-allelic nature of the data complicates productive motif learning.
In this study, we developed a biochemical and computational pipeline for LC-MS/MS analysis of endogenously processed HLA-associated peptides that requires less input material and provides single-allele resolution. Currently, the only single-allele approach available is based on the isolation of soluble HLA from cell lines grown in bioreactors, a setup that is not straightforward to implement and requires several orders of magnitude more input material (Hawkins et al., 2008; Trolle et al., 2016). Our approach, which isolates peptides from cells engineered to express a single HLA allele, provides a scalable means to improve the predictive power of algorithms for class I HLA-presented peptides and avoid in silico allelic deconvolution (Bassani-Stern-berg and Gfeller, 2016). Meanwhile, it leverages advances in instrumentation for rapid collection of high-resolution data and database search strategies that dynamically learn and leverage HLA peptide-binding motifs. The combination of direct antigen sequencing by LC-MS/MS and comprehensive bioinformatic analyses enabled the development of a predictor that outperformed current algorithms that are trained on peptide affinity data.
RESULTS
Workflow Uncovers Large Mono-allelic Peptide Repertoires with Minimal MS Bias
HLA-bound peptides can be directly identified via immunopurification and LC-MS/MS. We processed class I HLA-deficient cell lines (30 million–90 million B721.221-derived cells), each stably transduced to express one of 16 different class I HLA alleles (Figures 1A and 1B). High-quality tandem mass spectra (MS/MS) were subjected to iterative database searches, with stringent quality criteria (Experimental Procedures). The first round used no-enzyme specificity and no-variable peptide modifications, and the second round applied a database digestion specificity that leveraged the peptide-binding motif of individual HLA alleles determined from the first-round results and allowed peptide modifications (Experimental Procedures, Figure 1C). The second round of search typically increased identifications by an average of 14% (5%–40%) while maintaining a stringent 1% FDR cutoff (Figure S1A; Table S1).
Figure 1. An Efficient Sample-Processing and -Analysis Pipeline for HLA Peptide Sequencing.

(A) Overview of the standard multi-allele workflow. Cells (~500 million [M]) expressing multiple class I HLA alleles are lysed, and HLA-associated peptides are immunopurified with a pan-anti-HLA antibody. The complex mixture of HLA peptides is sequenced via LC-MS/MS, and the allele-binding assignments are inferred from previous knowledge.
(B) In our single-allele approach, B721.221 cells (~50 M), are transduced to express only one HLA allele. Immunopurified peptides are analyzed by LC-MS/MS and sequenced via an HLA-allele-specific database search.
(C) Schema of the HLA-specific database search strategy.
(D) HLA-class-I-associated peptide identifications from 16 single-HLA-expressing cell lines. Total numbers of unmodified (purple), modified (orange), and negative control (black) peptides identified per allele are shown. Allele frequencies among Caucasian, Asian, and Black populations are shown. An asterisk denotes alleles for which LC-MS/MS experiments have generated a greater number of peptides than what is reported in the Immune Epitope Database.
(E) To evaluate LC-MS/MS bias, we calculated the “MS observability index,” as measured by the ESP algorithm (Fusaro et al., 2009), for IEDB (blue) and MS (orange) peptide datasets. Distributions of the MS observability are displayed.
(F) Amino acid frequencies within peptides reported in our single-allele dataset are compared to amino acid frequencies in peptides reported in IEDB. See also Figure S1.
A total of 223 non-specifically bound peptides (negative controls) were identified by immunopurification from untransduced B721.221 cells or beads lacking antibody and represented ~3% of all peptide identifications (Table S1A). After filtering for these non-specific binders, we identified between 900 and 3550 unique peptides by LC-MS/MS for each HLA allele (median 1,505) (Figure 1D). Variation in surface presentation of HLA molecules on B721.221 cells, as compared to primary lymphocytes, appeared to explain most of the variation in observed peptide counts (Figure S1B). For common alleles (population frequency >1%), 74% percent of peptides were not reported in the Immune Epitope Database (IEDB); for rare alleles, nearly 100% were unreported (Figure S1C).
A high degree of peptide overlap was observed between biological replicates (~70%) and a previously reported B cell HLA-peptide dataset (Figure S1D) (Bassani-Sternberg et al., 2015). A median of 92% of presented peptides were unmodified, and 5% were modified, of which 3% were phosphorylated, and the remainder were consistent with oxidation, deamidation, and other handling artifacts (Figure S1E) (Berg et al., 2006). Comparisons among MS peptides and allele-matched synthetic peptides that were assigned as binders by IEDB (measured affinity < 500 nM; Figures 1E and 1F; Experimental Procedures) revealed only negligible peptide sequence biases related to our experimental procedures. The predicted MS observability of the HLA peptides and frequencies of individual amino acids between MS and IEDB peptides were highly similar, aside from underrepresentation of cysteine (Figures 1E and 1F). Free cysteine, which interferes with precursor fragmentation during LC-MS/MS, is underrepresented in other MS-based HLA-peptide datasets (Bassani-Sternberg et al., 2015; Trolle et al., 2016) (Figure S1F). We recovered cysteine-containing peptides when a third round of database search accounted for cysteinylation (Table S1C).
Mono-allelic MS-Derived Repertoires Reveal Motifs Underrepresented in IEDB
HLA-binding affinities of peptides sequenced with our LC-MS/ MS platform were predicted with NetMHCpan (Figure 2A) and assessed in terms of their length distributions (Figure 2B). For nearly every allele, most MS peptides scored <500 nM; however, most alleles also exhibited populations of peptides with poor predicted binding scores, suggesting that our data included motifs underrepresented in the NetMHCpan training set. Indeed, comparison of MS and IEDB peptides showed significant differences in amino acid frequencies at specific positions. For instance, enrichments of isoleucine, valine, and leucine (p < 1 × 10−5, chi-square test) were often observed at positions 5–7, suggesting the presence of secondary anchors (Figure 2C and Figures S2A–S2C). This was true for both sparsely studied alleles, such as HLA-A*02:07, and well-studied alleles, such as HLA-A*68:02 and HLA-B*57:01. We also noted specific alleles with length preferences not captured in IEDB, such as HLA-A*31:01 and HLA-B*51:01, which bind high proportions of 11-mers and 8-mers, respectively (Figure 2B).
Figure 2. HLA-Peptide Binding Motifs Enriched in LC-MS/MS Data Relative to IEDB.

(A) Distributions of NetMHCpan-2.8-predicted HLA-binding affinities of peptides identified by LC-MS/MS (“hits”; red) compared to 1 × 106 random 9-mer peptides from protein-coding genes (“decoys”; blue).
(B) Length distributions of HLA-associated peptides identified from single-HLA-expressing cell lines.
(C) Systematic evaluation of the frequencies of each amino acid (positions 1–9) within 9-mers sequenced by LC-MS/MS for the 14 of 16 HLA alleles for which sufficient IEDB data are available (orange, amino acids overrepresented in LC-MS/MS data; blue, amino acids underrepresented in LC-MS/MS data; scaling by p value).
(D) MS 9-mer peptides (orange) compared to IEDB 9-mer peptides (blue). Non-metric multidimensional scaling (NMDS) was used for visualization of pairwise peptide distances in two dimensions for each analyzed HLA allele. Peptide distance was defined on the basis of sequence similarity (Kim et al., 2009). The size of each circle corresponds to the NetMHCpan-predicted affinity score of the corresponding peptide. Synthesized peptides for 4/5 alleles are marked in and are numbered per the corresponding line in the table of measured and predicted binding affinities (for HLA-B35:01, see Figure S2J).
(E) MS peptides scoring in the bottom 10% by NetMHCpan 2.8 were selected for experimental validation. See also Figure S2.
To visualize the dominant and subdominant motifs within our data and among IEDB “binders” (measured affinity < 500 nM), we defined an entropy-weighted peptide distance and plotted the peptides in two-dimensional space such that “similar” peptides would be clustered closely and dissimilar peptides distantly (Experimental Procedures, Figure 2D and Figure S2D). In this manner, we discovered patterns not immediately evident by conventional sequence logo visualizations. Notably, MS-defined peptides clustered more closely to each other than to IEDB peptides themselves (Figure S2E), which suggests that MS recovers stronger binding motifs compared to a greater preponderance of weak binding peptides in the IEDB binder sets. Moreover, we found multiple peptide clusters that were highly enriched in MS relative to IEDB (Figures S2F and S2G), reflecting unique information in the MS datasets. MS technology-related biases did not appear to underlie these patterns: a similar analysis focused on only the subset of peptides from MS or IEDB with physico-chemical properties favorable for MS detection revealed similar distances and clustering patterns (Figures S2H and S2I) (Eyers et al., 2011; Fusaro et al., 2009; Muntel et al., 2015; Searle et al., 2015).
To experimentally validate these motifs, we selected sequences from clusters that were enriched within the MS datasets but that scored only within the bottom 10% when MS hits were evaluated by NetMHCpan 2.8 (Experimental Procedures). By competitive peptide-binding assays, 32 of 33 peptides were confirmed to be strong binders (median IC50 < 14 nM), even though only 14 of 33 were predicted as binders by NetMHCpan 2.8 (Figures 2D and 2E; Figure S2J).
Peptide Sequence Contexts Show a Distinct and Conserved Proteasomal Processing Signature
Because HLA-bound peptides have successfully undergone all processing steps—including proteasomal cleavage (Nielsen et al., 2005; Toes et al., 2001), transporter for antigen presentation (TAP) transport (Burgevin et al., 2008), and endoplasmic reticulum amino peptidase (ERAP)-mediated trimming (Evnouchidou et al., 2014; Saveanu et al., 2005; York et al., 2002)—prior to presentation, eluted peptide datasets are ideal for understanding how protein sequence context contributes to peptide processing and presentation. Nevertheless, the state of the art in cleavage prediction, NetChop (Keşmir et al., 2002; Nielsen et al., 2005), relies on a relatively small dataset of a few thousand HLA-bound peptides and focuses primarily on C-terminal cleavage. We sought to refine our understanding of processing rules by analyzing our large dataset of 24,000 allele-specific MS peptides and finding motifs in the upstream and downstream flanking sequences, as well as within the HLA-binding peptide.
We first focused on the sequence context around each HLA peptide within its source protein, which is not confounded by HLA binding (Figure 3A). Upstream of the peptide, at the first position (“U1”), arginine and lysine were highly enriched (relative to peptide decoys, consisting of random proteome 9-mers matched for their first two and last two amino acids), indicating a strong tryptic-like specificity at the N terminus (Figure 3A). Downstream of the peptide, arginine and lysine were also enriched in the first position (“D1”) (which suggests that peptides are trimmed at the C terminus after a tryptic-like cleavage that occurs after these basic residues), and acidic residues were depleted in this position. In addition, there was an enrichment of alanine at the U1 and D1 positions. A strong depletion of proline at both termini was observed to extend 3–5 residues upstream and downstream, which may relate to pro-line’s conformational rigidity. There was a very strong preference for peptides arising from the exact C terminus (labeled with a dash in Figure 3A) of their host protein, where only a single cleavage event is required. By comparing amino acid frequencies upstream, within, and downstream of each peptide, we also observed depletion of “cleavable” amino acids (K, R, and A) and enrichment of “non-cleavable” proline within peptides (Figure 3B). Thus, avoidance of internal cleavage appears to be a key feature of HLA ligands. Finally, we considered whether protein sequence features, such as alpha helices and beta strands, might influence processing potential (Figure S3A). LC-MS/MS peptides were twice as likely as gene-matched decoys to arise from signal peptide sequences; other features were significant but did not show an effect size greater than ±15%.
Figure 3. Analysis of Peptide Cleavage Signatures and HLA-Binding Registers.

(A) Heatmap of amino acids frequencies (percent change relative to background) in the protein sequence context (upstream: U10-U1; downstream D1-D10) of HLA peptides identified from single-HLA-expressing B721.221 cell lines. Colors of heatmap cells indicate directionality (red: enriched; blue: depleted) and p value (see key).
(B–H) Amino acid frequency ratios for cleavage-influencing amino acids upstream of, downstream of, and within peptides derived from LC-MS/MS-identified peptides compared to random proteome 9-mers (B). Heatmaps of amino acid frequencies calculated from external class HLA I datasets, including the breast cancer cell line HCC1937 (C), colorectal cell line HCT116 (D), fibroblasts (E), HeLa cells (Bassani-Sternberg et al., 2015) (F), and peripheral blood mononuclear cells (Caron et al., 2015) (G), as well as class II data from MUTZ3 (Mommen et al., 2016) (H).
(I) Percent change in amino acid frequency of top-scoring peptides (top 25%) compared to bottom-scoring peptides (bottom 25%) among 1,000,000 random proteome 9-mers evaluated by NetChop (Saxová et al., 2003). Color coding indicates directionality and magnitude of percent change (see key).
(J) Distribution of predicted affinities for the short isoforms (red) and long isoforms (yellow) of nested sets as well as for simulated long isoforms (random amino acids added at the beginning or end of the short isoforms). See also Figure S3.
To explore whether the processing signature was likely to be generalizable, we analyzed the gene expression of the protea-some and the immunoproteasome; both were expressed in B721.221 B cells at proportions comparable to those in blood and epithelial cancers included in the cancer genome atlas (TCGA) (Figure S3B). When we examined the HLA-bound peptide repertoires previously recovered from cells of other lineages, including breast and colon cancer cells (Bassani-Sternberg et al., 2015), fibroblasts (Bassani-Sternberg et al., 2015), HeLa cells (Trolle et al., 2016), and peripheral blood mononuclear cells (Caron et al., 2015) (Figures 3C–3G), all the key features observed for B721.221 cells were likewise consistently observed for these other cell types. Applying this same analytic approach to reported class II peptides isolated from dendritic cells (Mommen et al., 2016) (MUTZ3 cell line), we observed a starkly different signature exhibiting preference for hydrophobic residues in the D1 position and a lack of the previously observed associations for lysine, arginine, and alanine (Figure 3H). Finally, we note that the HLA class I signature that we derived only modestly resembled that obtained by comparison of peptides with high versus low NetChop scores. Our analyses thus identify a common HLA class I cleavage signature that dramatically differs from that predicted by a widely used tool.
Although HLA-class-I-bound peptides are typically thought to be cleanly “tucked” within the HLA class I binding groove, a recent crystal structure (McMurtrey et al., 2016) and the detection of nested peptide sets within our dataset (8% of total peptides; Table S3) motivated us to explore whether some peptides might be bound with overhang. We hypothesized that if long iso-forms of nested sets overhang, then the additional amino acids need not provide new anchors. On the other hand, if both short and long isoforms bind in tucked conformation, then extensions force the binding register to shift, and only certain amino acid additions can be tolerated. We observed that long isoforms indeed gain suitable new anchor sites (providing binding potential on par with the short isoforms); random amino acid extensions of short isoforms have uniformly worse binding potential (Figure 3J). This suggests that most peptides bind in the canonical tucked conformation.
Gene Expression and Binding Affinity Demonstrate a Simple, Multiplicative Relationship
In addition to the derivation of peptide binding and cleavage rules, as shown above, we also systematically evaluated the relative roles of gene expression, source-protein localization, and other source-protein characteristics to peptide presentation.
First, we evaluated the impact of gene expression. Peptide presentation is statistically associated with expression (RNA and protein) (Bassani-Sternberg et al., 2015; Juncker et al., 2009); however, there remains a lack of a standard approach for weighing expression against affinity during epitope prioritization. As a result, expression is routinely ignored in epitope selection or treated as a binary variable (Linnemann et al., 2015). To clarify this relationship, we binned each of our MS peptides by source transcript(s) RNA-Seq expression and NetMHCpan-predicted affinity. We also binned random 9-mers from the genome, enabling the calculation of relative enrichment ratios (Figure 4A). This revealed a multiplicative relationship between expression and affinity, in which a 10-fold increase in expression could approximately compensate for a 90% decrease in binding potential. To rule out the possibility that this finding might be an artifact of MS detection limits, we compared the peptides with the highest versus lowest MS signal intensity and compared them in terms of RNA-Seq expression and predicted affinity. Low-intensity binders had lower expression and weaker affinity, showing that MS detection is not simply reflecting underlying protein abundance but also reflects relative binding strength (Figure 4B). Although a simple kinetic model of peptide on- and off-rates may have predicted this, limitations in expression data quality and depth and the use of multi-allelic data (for which prediction of affinity is more difficult) have previously obscured this finding. The presence of multiple upstream open reading frames in the 5′ UTR of a transcript is associated with reduced presentation potential for its associated peptides (Figure 4C), suggesting that accurate measurements of translational efficiencies may enhance epitope selection further.
Figure 4. Evaluation of HLA-Peptide Characteristics that Impact HLA-Binding Predictions.

(A) Hits and decoys binned according to source transcript expression (per RNA-Seq; y axis) and predicted affinity (x axis) for each allele. Per bin, hit (top) and decoy (bottom) counts are reported. Color is according to the hit:decoy ratio (red = enriched for hits; blue = depleted of hits).
(B) MS peptides with high (red) and low (blue) MS1 ion intensities (top and bottom 10%, respectively), plotted by their NetMHCpan-predicted affinity and source transcript expression.
(C) Each LC-MS/MS-identified peptide was matched to ten random proteome 9-mer decoys with approximately equal expression but different source genes. The observed count of MS peptides divided by the expected count (based on decoy frequencies) is shown as a function of the number of upstream ATGs. P values were calculated by t test.
(D) The observed count of LC-MS/MS-identified HLA peptides mapping to each localization (Uniprot) relative to the expected count based on random 9-mer decoys (left) or expression-matched decoys (right).
(E) The ratio of observed to expected peptides at each distance lag from the source protein N terminus (blackline). The expected counts were determined under the assumption that each peptide was equally likely to have arisen from any position in its source protein. Frequent premature translation abortion would be expected to create an N-terminal bias (dashed red line).
(F) Observed versus expected HLA-peptide counts (determined from expression-matched decoys) as a function of source protein instability index (Guruprasad et al., 1990). P values were calculated by t test.
(G) Similar analysis to (F) showing enrichments as a function of the amount of intrinsically disordered sequence within each peptide’s source protein.
(H) Enrichments according to the count of ubiquitination sites, as previously observed (Krönke et al., 2015; Krönke et al., 2014; Udeshi et al., 2012), within the source protein.
(I) Approximately 200 protein-protein interaction experiments (Behrends et al., 2010; Christianson et al., 2011 Sowa et al., 2009), each yielding a set of 50–100 high-confidence interacting proteins for a given bait (usually a known protein-turnover-pathway gene) were scored according to their enrichment for LC-MS/MS-observed peptides, here depicted as a histogram. Each block corresponds to one experiment and is colored according to the directionality and significance (chi-square test) of the enrichment (see key). The bait protein used in outlier experiments (SQSTM1, PIK3C3, and OTUD4) is marked along with the corresponding p value. See also Figure S4.
Source-Protein Localization and Sequence Features Show Significant, but Modest, Predictive Value
To determine whether the HLA class I processing pathway has cellular localization biases, we investigated whether there was enrichment in protein localization in our MS peptides relative to random 9-mer decoys from protein-coding genes (Figure 4D, left) and relative to expression-matched 9-mer decoys (Figure 4D, right). Without controlling for expression, we found that the differences were dramatic: secreted proteins showed an unexpected enrichment. However, the expression-corrected analysis eliminated most of these differences (aside from a persistent enrichment of peptides from proteins associated with the late endosome). Lack of expression correction may help to explain why previous analyses of this question have reached inconsistent conclusions (Bassani-Sternberg et al., 2015; Rock et al., 2014).
Studies of peptide presentation kinetics have suggested that specialized pathways specifically target aborted translation products and misfolded proteins (Bourdetsky et al., 2014; Yew-dell, 2011). Consistent with recent analyses (Bourdetsky et al., 2014; Kim et al., 2013), we did not see an enrichment of peptides at the N-termini of their source proteins (Figure 4E), which would be expected if a meaningful fraction of peptides arose from aborted translation products. We also considered whether peptides from proteins with a high instability index (Guruprasad et al., 1990) or a high fraction of intrinsically disordered sequence (Experimental Procedures) were enriched in our MS data (Figures 4F and 4G) because we supposed that these would be more likely to trigger an unfolded protein response. The opposite trend was observed, suggesting either our measures of “foldability” were insufficient or that other unobserved variables potentially confound the signal.
Finally, we considered whether pathways of normal protein turnover were tied to presentation likelihood. The count of ubiquitination sites (previously observed in KG-1, Jurkat, or MM1S cells (Krönke et al., 2015; Krönke et al., 2014; Udeshi et al., 2012, 2013), was positively associated with HLA-peptide presentation, consistent with the known role for ubiquitin in delivering proteins to the proteasome (Figure 4H). Additionally, we queried a collection of 200 IP-MS/MS experiments, each profiling the physical interaction partners of a protein involved in deubiquitination, autophagy, or ER-associated degradation (Behrends et al., 2010; Christianson et al., 2011; Sowa et al., 2009) (Figure 4I). Most of these gene sets were positively enriched in our data. Several outliers include PIK3C3, ATG12, and OTUD4, whose interaction partners were most strongly enriched. Meanwhile, the interaction partners of the autophago-some cargo protein SQSTM1 were most depleted. Collectively, these analyses may help to point to turnover pathways with privileged access to the HLA presentation pathway.
Multivariate Models Quantify the Predictive Contributions of Distinct Processing Steps
Given the consistent patterns for cleavability and expression on presented peptide sequences, we sought to determine whether integrating multiple variables into a single predictor would improve epitope selection accuracy. To measure model performance, we assessed our ability to discern MS peptides among a 999-fold excess of decoy peptides; the top-scoring 0.1% of peptides were selected as positives. Because there are approximately 10 million 9-mers in the human proteome, and each allele presents approximately 10,000 of these, the 1:1000 ratio closely mimics the reality of the epitope selection problem. This positive predictive value (PPV) metric, which measures the percentage of predicted positives that were indeed observed in the MS, contrasts with the standard AUC value (area under receiver operator characteristic curve), which integrates performance over all possible target:decoy ratios and thus considers unrealistic scenarios in the final score (Experimental Procedures and Figure S4).
Averaging across 16 alleles, an affinity-only model (per NetMHCpan, model “A”) achieved a PPV of 28% (Figure 5A; see Table S4A for individual allele results). A stability-only model (NetMHCPanStab (Jørgensen et al., 2014), model “S”) performed nearly as well; however, joint prediction (model “AS”) showed minor synergism. On the other hand, adding RNA-Seq or iBAQ-based (Ishihama et al., 2005) protein expression (models “ASR” and “ASP”) improved PPV dramatically—to 39% and 47%, respectively. Adding cleavability prediction (per a de novo predictor trained on other MS data; Supplemental Experimental Procedures) provided a 7.9% boost (prediction with NetChop yields 3.1%). The incremental benefit of localization was less than 1%; other putative processing variables (stability index, disordered sequence content, count of ubiquitin sites, and sequence features such as alpha helices and beta strands) likewise showed incremental improvements less than 1%.
Figure 5. Evaluation of MS-Data-Based HLA-Peptide Binding Predictors.

(A) Positive predictive value of linear models used for discerning 9-mer MS peptides among a 999-fold excess of 9-mer decoys (averaging across 16 alleles). Models included one or more predictor variables (A = affinity, S = stability, R = RNA-Seq expression, P = protein expression (iBAQ), C = cleavability score, and L = source protein localization).
(B) Explanatory contributions of predictor variables derived from the cumulative improvement in predictive value as predictors are added.
(C) Cartoon representation of the neural-network model architecture. The 215 MSIntrinsic inputs included amino acid dummy variables (180 nodes), amino acid properties (27 nodes), and peptide properties (8 nodes). The 182 MSIntrinsicEC inputs included the amino acid dummy variables, expression (1 node), and cleavability (1 node).
(D) External evaluation. MS-binding data from two published datasets (Bassani-Sternberg et al., 2015; Trolle et al., 2016) were used for comparing the positive predictive value of MSIntrinsic and MSIntrinsicEC against NetMHCpan 2.8 and NetMHC 4.0 in identifying presented peptides among a 999-fold excess of random decoy 9-mers. Peptides were excluded from the evaluation if they were highly likely to bind an allele other than the one being evaluated. See also Figure S5.
By exhaustively testing all possible predictor combinations (Table S4A), we found the order of variable addition that added the most predictive value earliest; we tracked the incremental PPV improvement provided by each variable and assigned this as the variable’s “explanatory contribution” (Figure 5B). Affinity and expression dominate the analysis, although notably, iBAQ-based protein expression provided negligible contribution beyond RNA-Seq. For the 45% of MS peptides that were missed in the full model, it was not known how much this related to the suboptimal quality of the affinity and cleavage predictions, to unknown variables, or to stochasticity in the MS detection. The two genes with the most false negative calls per unit length were ubiquitin B and C, which suggests that improved understanding of protein turnover dynamics may be a key missing component.
De Novo Predictors Based on Mono-allelic MS Data Perform Better Than Affinity-Trained Predictors
To define whether these collective findings could be used to develop improved epitope-presentation predictors, we developed two single-layer artificial networks for each of the 16 alleles (Figure 5C). The first, MSIntrinsic, was trained exclusively on our MS data and used peptide-intrinsic features only; the second, MSIntrinsicEC, additionally accounted for RNA-Seq expression and the cleavability of the protein sequence context (Experimental Procedures).
We evaluated performance of these models on internal and external datasets by using PPV and AUC. We found that MSIntrinsic and MSIntrinsicEC outperformed both NetMHC 4.0 and NetMHCpan 2.8 in a 5-fold cross validation by an average PPV of 20 and 30 percentage points, respectively (Table S4B). Accordingly, despite the fact that all models achieved an average AUC > 0.98, MSIntrinsic and MSIntrinsicEC consistently reached higher true-positive rates at minimal false-positive rates; we note that performance plateaued at ~500 training peptides (Figures S5A–S5C). Most importantly, we predicted which peptides would be identified in the LC-MS/MS data of other groups (Bassani-Sternberg et al., 2015; Trolle et al., 2016), which comprised observations from six alleles and seven cell lines. In comparison to that of NetMHC 4.0 and NetMHCpan 2.8, the positive predictive value for MSIntrinsic was 1.4-fold better on average and worse than NetMHC in only one instance; MSIntrinsicEC was 1.9-fold better on average (Figure 5D). MS bias did not appear to account for these changes: NetMHC-based predictors that additionally accounted for MS observability (ESP) score and cysteine count did not show appreciable improvement (Table S4F). Thus, we expect that these approaches could roughly double the number of correct epitope identifications in a vaccine. In addition, we used our algorithms to predict immunogenic HIV epitopes (Llano et al., 2013). Although the dataset is of modest size, the rank position of the top scoring HIV epitope was higher than both NetMHC predictors or equal to one of them for 9/12 alleles overlapping with our dataset.
Because our neural-network approach is closely modeled after NetMHC and because we did not outperform NetMHC when we trained and evaluated on IEDB peptides (Figure S5D), our advances are likely to reflect the underlying high quality of our MS data and conceptual advances in data integration, rather than changes in neural-network design. Our results thus demonstrate that large MS datasets of endogenous HLA-binding peptides can greatly improve our understanding of antigen-processing rules and the power of algorithms to predict which peptides will be presented by specific HLA alleles.
DISCUSSION
The majority of LC-MS/MS studies of the HLA peptidome have used cells expressing multiple HLA molecules, which requires peptides to be assigned to one of up to six class I alleles through the use of pre-existing bioinformatics predictors, or “deconvolution” (Bassani-Sternberg and Gfeller, 2016). Thus, peptides that do not closely match known motifs cannot confidently be reported as binders to a given HLA allele. By contrast, we used a rapid approach to generate a high-quality LC-MS/MS dataset of >24,000 endogenous peptides whose assignment to specific HLA alleles was unambiguous. Because we knew the allele assignment a priori, we greatly enhanced our analyses depth. We discovered allele-specific binding motifs and proteasomal cleavage rules and discerned the effects of transcript abundance on presentation. By training models on these data, we more effectively predicted presentation of endogenous peptides than we did with models trained on IEDB binding measurement.
Given the availability of our large dataset, we sought to quantify in a systematic and unbiased fashion the role of different factors in predicting HLA-presented peptides. Prior proteomic studies have inconsistently discerned associations between protein abundance and HLA-peptide presentation (Bassani-Sternberg et al., 2015; Hickman et al., 2004; Milner et al., 2006). Although Bassani-Sternberg et al. provide convincing evidence of positive association between HLA-peptide presentation and source-protein expression, their analysis was limited by reliance on MS-based protein quantification that had a high amount of missing observations. Therefore, we utilized RNA sequencing, a genome-wide characterization with a very low limit of detection (< 1 PPM) and with far greater accessibility for expression analysis than comparable proteomic approaches. We conclude that gene expression is a highly predictive variable and is more productively captured with RNA-Seq than with MS-based proteomic quantitation. Moreover, we have presented an analytic framework that enables the joint use of expression and affinity as variables to select epitopes in a principled manner, which is highly implementable in routine epitope-selection efforts in clinical settings.
The contribution of cleavability in our analyses was large but not dominant, suggesting that proteasomes and other endogenous peptidases have a promiscuous specificity or are difficult to predict (Toes et al., 2001). Cellular localization played a weak role in presentation, indicating that HLA class I peptides are derived from endogenous proteins throughout the cell. The additional impact of stability prediction is modest, and affinity-based NetMHCpan outperforms stability-based NetMHCStab in a direct head-to-head. These results suggest that motifs learned from stability assays are not substantially different. Finally, we developed a neural network, MSIntrinsicEC, that integrates these different variables and improves prediction performance of endogenous peptides relative to neural networks trained on non-MS datasets (Andreatta and Nielsen, 2016; Hoof et al., 2009; Lundegaard et al., 2008). An open question is how additional data types (e.g., protein translation rates and modes of protein turnover), improved data quality, a larger collection of validated non-binding peptides, and more powerful machine-learning-based predictions would boost our ability to predict antigen presentation and immunological responses.
Our methodologies provide a path toward addressing challenges relating to HLA-peptide presentation. First, the application of our single HLA workflows to class II heterodimers should improve class II prediction because confident, allele-specific peptide-binding assignments can be made. Having a single-allele system is especially important for class II because the length distribution obscures the binding register, making deconvolution approaches more challenging. Second, a rapid pipeline should enable identification of HLA-associated peptides from patient-derived cell lines or primary tumor samples, providing a unique opportunity for more personalized therapies against cancer. Third, recent observations of CD8+ T cells targeting mutated antigens in tumors (Schumacher and Schreiber, 2015) have inspired cancer immunotherapy trials aimed at inducing personalized T cells responses targeting an individual’s tumor. More effective prediction of HLA-associated peptides from a collection of candidate antigens should contribute to the improvement of personalized cancer vaccines.
Overall, we expect these advances in unbiased identification and prediction of endogenous HLA-associated peptides to impact all areas of immunology, especially the identification of antigens driving autoimmunity and the design of more effective vaccines for infections and cancer.
EXPERIMENTAL PROCEDURES
Cell Culture and HLA-Peptide Immunopurification
We tested mono-allelic B cells generated by transduction of B721.221 cells with a retroviral vector coding a single class I HLA allele as described previously (Reche et al., 2006) (cells expressing HLA-A*02:01, -A*24:02, and -B*44:03 were purchased from the Fred Hutchinson Research Cell Bank, University of Washington; cells expressing HLA-A*03:01 were a gift from Dr. Marcus Altfeld and Dr. Wilfredo F. Garcia-Beltran, Ragon Institute; others were a gift from Dr. E.L. Reinherz, Dana Farber Cancer Institute). Cell lines were confirmed by standard molecular typing (Brigham and Women’s Hospital Tissue Typing Laboratory). HLA-peptide immunopurifcation is described in the Supplemental Experimental Procedures.
HLA-Peptide Sequencing by Tandem Mass Spectrometry
All nanoLC-ESI-MS/MS analyses employed the same LC separation conditions, instrument parameters, and data analytics described in the Supplemental Experimental Procedures. The original mass spectra may be downloaded from MassIVE (http://massive.ucsd.edu) under the identifier MassIVE: MSV000080527. The data are directly accessible via ftp://massive.ucsd.edu/MSV000080527.
Sequence Properties of MS-Identified Peptides Compared to IEDB
A curated set of previously identified class I HLA-bound peptides was downloaded from the Immune Epitope Database (IEDB) at http://www.iedb.org/ (accessed on 10/26/2015) (Vita et al., 2015). For each allele, IEDB peptides with a measured affinity <500 nM were compared to MS peptides in terms of their length and positional amino acid frequencies. In addition, a metric was defined for the pairwise “distance” between 9-mers (a Hamming distance calculated with an amino acid substitution matrix [Kim et al., 2009] and inversely weighted according to positional entropy) and used for clustering MS and IEDB peptides in a two-dimensional representation. A machine-learning approach (Supplemental Experimental Procedures) identified peptides with motifs favored in the MS but that were poor-scoring according to NetMHCpan 2.8; the MHC-binding affinities for these peptides were determined by competitive binding per gel filtration protocol (Sidney et al., 2001).
Peptide Processing Analyses
For each MS hit, the upstream ten amino acids and downstream ten amino acids were determined. Sequence context was likewise determined for decoy peptides (100 per hit; selected randomly from the proteome and matched according to their first two and last two amino acids). Relative amino acid frequencies were determined at each position upstream and downstream of hits and decoys. Additional previously published MS datasets were analyzed in the same manner. For comparison, peptides with high and low NetChop scores (top 25% and bottom 25% of 1 million randomly selected sites in the genome) were compared, and the motif most favored by NetChop was derived.
Relationship between Expression and Affinity
RNA was isolated from B721.221 cells expressing HLA-A*29:02, B*51:01, B*54:01, and B*57:01 (RNeasy mini kit, QIAGEN), processed to cDNA (Nextera XT kit; Smart-seq2 protocol), sequenced (HiSeq2500, Rapid Run mode; 50 bp paired-end), and aligned (bowtie2-2.2.1 (Langmead and Salzberg, 2012); UCSC hg19 annotation). We averaged transcript expression (RSEM-1.2.19 [Li and Dewey, 2011]; GEO: GSE93315) across the four cell lines and made adjustments by dropping non-coding transcripts and rescaling TPM values to sum to one million. We determined expression of each peptide source protein by summing all transcripts containing the peptide.
Impact of Processing Pathways
MS peptides were compared to decoys (ten decoys per MS peptide; each from a different gene; matched per transcript expression) in terms of various features potentially related to peptide processing: UNIPROT localization (www.uniprot.org), distance from protein N terminus, source protein stability index (Guruprasad et al., 1990), intrinsically disordered sequence content (http://d2p2.pro) (Oates et al., 2013), count of known ubiquitination sites (Eichmann et al., 2014; Krönke et al., 2015; Udeshi et al., 2012), and physical interaction with known protein turnover regulators (Behrends et al., 2010).
Development of New Epitope-Selection Algorithms
For each allele, we trained neural-network classifiers (one hidden layer with 50 units) (by using Theano (Theano Development Team, 2016); 5-fold cross-validation) to differentiate MS 9-mers from random decoy 9-mers by using different input feature schemes: dummy encoding, BLOSUM62, PMBEC (Kim et al., 2009), biochemical properties (Bremel and Homan, 2010), and peptide-level features (Osorio et al., 2014); we averaged the results of these models to obtain a single prediction (called MSIntrinsic). We made a second prediction (MSIntrinsicEC) by adding expression and MS-trained cleavability. We validated performance on external data by measuring PPV (fraction of true MS peptides among the top-scoring 0.1%, where decoys are present at 999:1). For multi-allelic datasets, the evaluation excluded any MS peptides that obviously belonged to an HLA-A or HLA-B allele other than the one in question (e.g., when predicting for A01:01 for a cell line with genotype A01:01/A02:01/B35:01/B44:02, we excluded MS-observed peptides with NetMHCPan 2.8 scores worse than 1,000 nM for A01:01 and better than 150 nM for A02:01, B35:01, or B44:02).
Supplementary Material
Highlights.
24,000 HLA class I peptides were identified through a scalable MS-based pipeline.
Mono-allelic data revealed binding motifs that were validated biochemically.
Comprehensive analyses provide an updated portrait of antigen processing rules.
Neural networks were trained for 16 alleles and outperform standard by 2-fold.
Acknowledgments
The work was supported by the Blavatnik Family Foundation (N.H. and C.W.), National Cancer Institute (NCI) R01 CA155010 (C.W.), NCI CPTAC U24 CA160034 (S.A.C, and K.C.), NHGRI T32 HG002295 (S.S.), and Dana-Farber/Harvard Cancer Center Kidney Cancer SPORE 2P50CA101942-11A1 (D.B.K.). C.W. is a Scholar of the Leukemia and Lymphoma Society. J.G.A. and M.S.R. are employees and shareholders of Neon Therapeutics; N.H. and C.J.W. are founders of Neon Therapeutics and members of its scientific advisory board.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, five figures, and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.immuni.2017.02.007.
AUTHOR CONTRIBUTIONS
Conceptualization, J.G.A., D.B.K., S.S., K.R.C., N.H., M.S.R., S.A.C., C.J.W.; methodology, J.G.A., D.B.K., S.S., K.R.C., G.L.Z., N.H., M.S.R., S.A.C., C.J.W.; investigation, J.G.A., D.B.K., S.S., C.R.H., W.Z., J.Si., J.St., W.L., G.L.Z., T.M.E., K.R.C, N.H., M.S.R., S.A.C., C.J.W.; writing—original draft, J.G.A., D.B.K., S.S., K.R.C., N.H., M.S.R., S.A.C., C.J.W.; writing—review & editing, J.G.A., D.B.K., S.S., G.L.Z., K.R.C., N.H., M.S.R., S.A.C., C.J.W.; funding acquisition, N.H., S.A.C., C.J.W.; resources, N.H., S.A.C., C.J.W.; supervision, K.R.C., N.H., M.S.R., S.A.C., C.J.W.
References
- Andreatta M, Nielsen M. Gapped sequence alignment using arti-ficial neural networks: application to the MHC class I system. Bioinformatics. 2016;32:511–517. doi: 10.1093/bioinformatics/btv639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani-Sternberg M, Gfeller D. Unsupervised HLA peptidome deconvolution improves ligand prediction accuracy and predicts cooperative effects in peptide-HLA interactions. J Immunol. 2016;197:2492–2499. doi: 10.4049/jimmunol.1600808. [DOI] [PubMed] [Google Scholar]
- Bassani-Sternberg M, Pletscher-Frankild S, Jensen LJ, Mann M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol Cell Proteomics. 2015;14:658–673. doi: 10.1074/mcp.M114.042812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg M, Parbel A, Pettersen H, Fenyö D, Björkesten L. Detection of artifacts and peptide modifications in liquid chromatography/ mass spectrometry data using two-dimensional signal intensity map data visualization. Rapid Commun Mass Spectrom. 2006;20:1558–1562. doi: 10.1002/rcm.2476. [DOI] [PubMed] [Google Scholar]
- Bourdetsky D, Schmelzer CEH, Admon A. The nature and extent of contributions by defective ribosome products to the HLA peptidome. Proc Natl Acad Sci USA. 2014;111:E1591–E1599. doi: 10.1073/pnas.1321902111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremel RD, Homan EJ. An integrated approach to epitope analysis I: Dimensional reduction, visualization and prediction of MHC binding using amino acid principal components and regression approaches. Immunome Res. 2010;6:7. doi: 10.1186/1745-7580-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgevin A, Saveanu L, Kim Y, Barilleau E, Kotturi M, Sette A, van Endert P, Peters B. A detailed analysis of the murine TAP transporter substrate specificity. PLoS ONE. 2008;3:e2402. doi: 10.1371/journal.pone.0002402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron E, Espona L, Kowalewski DJ, Schuster H, Ternette N, Alpízar A, Schittenhelm RB, Ramarathinam SH, Lindestam Arlehamn CS, Chiek Koh C, et al. An open-source computational and data resource to analyze digital maps of immunopeptidomes. eLife. 2015;4 doi: 10.7554/eLife.07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, Richter CM, Tyler RE, Greenblatt EJ, Harper JW, Kopito RR. Defining human ERAD networks through an integrative mapping strategy. Nat Cell Biol. 2011;14:93–105. doi: 10.1038/ncb2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichmann M, de Ru A, van Veelen PA, Peakman M, Kronenberg-Versteeg D. Identification and characterisation of peptide binding motifs of six autoimmune disease-associated human leukocyte antigen-class I molecules including HLA-B*39:06. Tissue Antigens. 2014;84:378–388. doi: 10.1111/tan.12413. [DOI] [PubMed] [Google Scholar]
- Evnouchidou I, Weimershaus M, Saveanu L, van Endert P. ERAP1-ERAP2 dimerization increases peptide-trimming efficiency. J Immunol. 2014;193:901–908. doi: 10.4049/jimmunol.1302855. [DOI] [PubMed] [Google Scholar]
- Eyers CE, Lawless C, Wedge DC, Lau KW, Gaskell SJ, Hubbard SJ. CONSeQuence: prediction of reference peptides for absolute quantitative proteomics using consensus machine learning approaches. Mol Cell Proteomics. 2011;10:003384. doi: 10.1074/mcp.M110.003384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusaro VA, Mani DR, Mesirov JP, Carr SA. Prediction of high-responding peptides for targeted protein assays by mass spectrometry. Nat Biotechnol. 2009;27:190–198. doi: 10.1038/nbt.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruprasad K, Reddy BVB, Pandit MW. Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. 1990;4:155–161. doi: 10.1093/protein/4.2.155. [DOI] [PubMed] [Google Scholar]
- Hawkins OE, Vangundy RS, Eckerd AM, Bardet W, Buchli R, Weidanz JA, Hildebrand WH. Identification of breast cancer peptide epi-topes presented by HLA-A*0201. J Proteome Res. 2008;7:1445–1457. doi: 10.1021/pr700761w. [DOI] [PubMed] [Google Scholar]
- Hickman HD, Luis AD, Buchli R, Few SR, Sathiamurthy M, VanGundy RS, Giberson CF, Hildebrand WH. Toward a definition of self: Proteomic evaluation of the class I peptide repertoire. J Immunol. 2004;172:2944–2952. doi: 10.4049/jimmunol.172.5.2944. [DOI] [PubMed] [Google Scholar]
- Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61:1–13. doi: 10.1007/s00251-008-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt DF, Henderson RA, Shabanowitz J, Sakaguchi K, Michel H, Sevilir N, Cox AL, Appella E, Engelhard VH. Characterization of peptides bound to the class I MHC molecule HLA-A2.1 by mass spectrometry. Science. 1992;255:1261–1263. doi: 10.1126/science.1546328. [DOI] [PubMed] [Google Scholar]
- Ishihama Y, Oda Y, Tabata T, Sato T, Nagasu T, Rappsilber J, Mann M. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics. 2005;4:1265–1272. doi: 10.1074/mcp.M500061-MCP200. [DOI] [PubMed] [Google Scholar]
- Jørgensen KW, Rasmussen M, Buus S, Nielsen M. NetMHCstab—Predicting stability of peptide-MHC-I complexes; impacts for cytotoxic T lymphocyte epitope discovery. Immunology. 2014;141:18–26. doi: 10.1111/imm.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juncker AS, Larsen MV, Weinhold N, Nielsen M, Brunak S, Lund O. Systematic characterisation of cellular localisation and expression profiles of proteins containing MHC ligands. PLoS ONE. 2009;4:e7448. doi: 10.1371/journal.pone.0007448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keşmir C, Nussbaum AK, Schild H, Detours V, Brunak S. Prediction of proteasome cleavage motifs by neural networks. Protein Eng. 2002;15:287–296. doi: 10.1093/protein/15.4.287. [DOI] [PubMed] [Google Scholar]
- Kim Y, Sidney J, Pinilla C, Sette A, Peters B. Derivation of an amino acid similarity matrix for peptide: MHC binding and its application as a Bayesian prior. BMC Bioinformatics. 2009;10:394. doi: 10.1186/1471-2105-10-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Yewdell JW, Sette A, Peters B. Positional bias of MHC class I restricted T-cell epitopes in viral antigens is likely due to a bias in conservation. PLoS Comput Biol. 2013;9:e1002884. doi: 10.1371/journal.pcbi.1002884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343:301–305. doi: 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krönke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, Chamberlain PP, Mani DR, Man HW, Gandhi AK, et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature. 2015;523:183–188. doi: 10.1038/nature14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnemann C, van Buuren MM, Bies L, Verdegaal EME, Schotte R, Calis JJA, Behjati S, Velds A, Hilkmann H, Atmioui DE, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. 2015;21:81–85. doi: 10.1038/nm.3773. [DOI] [PubMed] [Google Scholar]
- Llano A, Williams A, Overa A, Silva-Arrieta S, Brander C. Best-characterized HIV-1 CTL epitopes: The 2013 update. HIV Mol Immunol. 2013:3–25. [Google Scholar]
- Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 2008;36:W509–12. doi: 10.1093/nar/gkn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurtrey C, Trolle T, Sansom T, Remesh SG, Kaever T, Bardet W, Jackson K, McLeod R, Sette A, Nielsen M, et al. Toxoplasma gondii peptide ligands open the gate of the HLA class I binding groove. eLife. 2016;5:e12556. doi: 10.7554/eLife.12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner E, Barnea E, Beer I, Admon A. The turnover kinetics of major histocompatibility complex peptides of human cancer cells. Mol Cell Proteomics. 2006;5:357–365. doi: 10.1074/mcp.M500241-MCP200. [DOI] [PubMed] [Google Scholar]
- Mommen GPM, Marino F, Meiring HD, Poelen MCM, van Gaansvan den Brink JAM, Mohammed S, Heck AJR, van Els CACM. Sampling from the proteome to the human leukocyte antigen-DR (HLA-DR) ligandome proceeds via high specificity. Mol Cell Proteomics. 2016;15:1412–1423. doi: 10.1074/mcp.M115.055780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntel J, Boswell SA, Tang S, Ahmed S, Wapinski I, Foley G, Steen H, Springer M. Abundance-based classifier for the prediction of mass spectrometric peptide detectability upon enrichment (PPA) Mol Cell Proteomics. 2015;14:430–440. doi: 10.1074/mcp.M114.044321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, Lundegaard C, Lund O, Keşmir C. The role of the proteasome in generating cytotoxic T-cell epitopes: Insights obtained from improved predictions of proteasomal cleavage. Immunogenetics. 2005;57:33–41. doi: 10.1007/s00251-005-0781-7. [DOI] [PubMed] [Google Scholar]
- Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztányi Z, Uversky VN, Obradovic Z, Kurgan L, et al. D2P2: Database of disordered protein predictions. Nucleic Acids Res. 2013;41:D508–D516. doi: 10.1093/nar/gks1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio D, Rondón-Villarreal P, Torres R. Peptides: Calculate indices and theoretical physicochemical properties of peptides and protein sequences. 2014 http://CRAN.R-project.org/package=Peptides. R Package Version 1.1.0.
- Rammensee HG, Friede T, Stevanoviíc S. MHC ligands and peptide motifs: first listing. Immunogenetics. 1995;41:178–228. doi: 10.1007/BF00172063. [DOI] [PubMed] [Google Scholar]
- Reche PA, Keskin DB, Hussey RE, Ancuta P, Gabuzda D, Reinherz EL. Elicitation from virus-naive individuals of cytotoxic T lymphocytes directed against conserved HIV-1 epitopes. Med Immunol. 2006;5:1. doi: 10.1186/1476-9433-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh SGE. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res. 2015;43:D423–D431. doi: 10.1093/nar/gku1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock KL, Farfán-Arribas DJ, Colbert JD, Goldberg AL. Reexamining class-I presentation and the DRiP hypothesis. Trends Immunol. 2014;35:144–152. doi: 10.1016/j.it.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saveanu L, Carroll O, Lindo V, Del Val M, Lopez D, Lepelletier Y, Greer F, Schomburg L, Fruci D, Niedermann G, van Endert PM. Concerted peptide trimming by human ERAP1 and ERAP2 aminopeptidase complexes in the endoplasmic reticulum. Nat Immunol. 2005;6:689–697. doi: 10.1038/ni1208. [DOI] [PubMed] [Google Scholar]
- Saxová P, Buus S, Brunak S, Keşmir C. Predicting proteasomal cleavage sites: A comparison of available methods. Int Immunol. 2003;15:781–787. doi: 10.1093/intimm/dxg084. [DOI] [PubMed] [Google Scholar]
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- Searle BC, Egertson JD, Bollinger JG, Stergachis AB, MacCoss MJ. Using data independent acquisition (DIA) to model high-responding peptides for targeted proteomics experiments. Mol Cell Proteomics. 2015;14:2331–2340. doi: 10.1074/mcp.M115.051300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidney J, Southwood S, Oseroff C, del Guercio MF, Sette A, Grey HM. Measurement of MHC/peptide interactions by gel filtration. Curr Protoc Immunol Chapter. 2001;18:3. doi: 10.1002/0471142735.im1803s31. [DOI] [PubMed] [Google Scholar]
- Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theano Development Team. Theano: A Python framework for fast computation of mathematical expressions. 2016 ArXiv https://arxiv.org/abs/1605.02688.
- Toes RE, Nussbaum AK, Degermann S, Schirle M, Emmerich NP, Kraft M, Laplace C, Zwinderman A, Dick TP, Muller J, et al. Discrete cleavage motifs of constitutive and immunoproteasomes revealed by quantitative analysis of cleavage products. J Exp Med. 2001;194:1–12. doi: 10.1084/jem.194.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trolle T, Metushi IG, Greenbaum JA, Kim Y, Sidney J, Lund O, Sette A, Peters B, Nielsen M. Automated benchmarking of peptide-MHC class I binding predictions. Bioinformatics. 2015;31:2174–2181. doi: 10.1093/bioinformatics/btv123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trolle T, McMurtrey CP, Sidney J, Bardet W, Osborn SC, Kaever T, Sette A, Hildebrand WH, Nielsen M, Peters B. The length distribution of class I–restricted T cell epitopes is determined by both peptide supply and MHC allele-specific binding preference. J Immunol. 2016;196:1480–1487. doi: 10.4049/jimmunol.1501721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udeshi ND, Mani DR, Eisenhaure T, Mertins P, Jaffe JD, Clauser KR, Hacohen N, Carr SA. Methods for quantification of in vivo changes in protein ubiquitination following proteasome and deubiquitinase inhibition. Mol Cell Proteomics. 2012;11:148–159. doi: 10.1074/mcp.M111.016857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udeshi ND, Svinkina T, Mertins P, Kuhn E, Mani DR, Qiao JW, Carr SA. Refined preparation and use of anti-diglycine remnant (K-ε-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol Cell Proteomics. 2013;12:825–831. doi: 10.1074/mcp.O112.027094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, Cantrell JR, Wheeler DK, Gabbard JL, Hix D, Sette A, Peters B. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015;43:D405–D412. doi: 10.1093/nar/gku938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewdell JW. DRiPs solidify: Progress in understanding endogenous MHC class I antigen processing. Trends Immunol. 2011;32:548–558. doi: 10.1016/j.it.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York IA, Chang SC, Saric T, Keys JA, Favreau JM, Goldberg AL, Rock KL. The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8–9 residues. Nat Immunol. 2002;3:1177–1184. doi: 10.1038/ni860. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.