

Abstract
In human cells, thousands of predominantly neuronal genes are regulated by the repressor element 1 (RE1)‐silencing transcription factor/neuron‐restrictive silencer factor (REST/NRSF). REST/NRSF represses transcription of these genes in stem cells and non‐neuronal cells by tethering corepressor complexes. Aberrant REST/NRSF expression and intracellular localization are associated with cancer and neurodegeneration in humans. To date, detailed molecular analyses of REST/NRSF and its C‐terminal repressor complex have been hampered largely by the lack of sufficient amounts of purified REST/NRSF and its complexes. Therefore, the aim of this study was to express and purify human REST/NRSF and its C‐terminal interactors in a baculovirus multiprotein expression system as individual proteins and coexpressed complexes. All proteins were enriched in the nucleus, and REST/NRSF was isolated as a slower migrating form, characteristic of nuclear REST/NRSF in mammalian cells. Both REST/NRSF alone and its C‐terminal repressor complex were functionally active in histone deacetylation and histone demethylation and bound to RE1/neuron‐restrictive silencer element (NRSE) sites. Additionally, the mechanisms of inhibition of the small‐molecule drugs 4SC‐202 and SP2509 were analyzed. These drugs interfered with the viability of medulloblastoma cells, where REST/NRSF has been implicated in cancer pathogenesis. Thus, a resource for molecular REST/NRSF studies and drug development has been established.
Keywords: REST/NRSF, CoREST, LSD1, HDAC1, DNA transcription, transcriptional repression, histone deacetylation, histone demethylation, drug mechanism, medulloblastoma
Abbreviations
- aa
amino acid
- AOD
amino oxidase domain
- bp
base pair
- CE
cytoplasmic extract
- CRC
C‐terminal REST/NRSF‐CoREST‐LSD1‐HDAC1 transcriptional repressor complex
- DTT
dithiothreitol
- E
elution
- EDTA
ethylenediaminetetraacetic acid
- ELM2
Egl‐27‐MTA1 homology 2
- EMSA
electrophoretic mobility shift assay
- FBS
fetal bovine serum
- FT
flow‐through
- HDAC
histone deacetylase
- HEPES
4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid
- IPTG
isopropyl β‐D‐1‐thiogalactopyranoside
- kDa
kilo Dalton
- LB
lysogeny broth
- LSD1
lysine‐specific demethylase 1
- NE
nuclear extract
- NRSE
neuron‐restrictive silencer element
- NRSF
neuron‐restrictive silencer factor
- PAGE
polyacrylamide gel electrophoresis
- PMSF
phenylmethanesulfonyl fluoride
- P/S
penicillin/streptomycin
- RE1
repressor element 1
- REST
repressor element 1 silencing transcription factor
- RFU
relative fluorescence unit
- RLU
relative light unit
- SANT
switching‐defective protein 3 (Swi3)‐adaptor 2 (Ada2)‐nuclear receptor corepressor (N‐CoR)‐transcription factor IIIB
- SDS
sodium dodecyl sulfate
- SEM
standard error of the mean
- SWIRM
SWI3‐RSC8‐MOIRA
- TBE
tris/borate/EDTA
- W
wash
- WS
washes
- X‐Gal
5‐bromo‐4‐chloro‐3‐indolyl‐β‐D‐galactopyranoside
- YFP
yellow fluorescent protein
- ZF
zinc finger
Introduction
The expression of a developmental stage‐ and cell‐type‐specific gene profile is precisely controlled by regulatory factors such as transcription factors.1 A particularly powerful transcription factor is the repressor element 1 (RE1) silencing transcription factor/neuron‐restrictive silencer factor (REST/NRSF),2, 3 which can bind to thousands of sites in the human genome.4 These RE1/neuron‐restrictive silencer element (RE1/NRSE) sites comprise two RE1/NRSE half‐sites (i.e., a left and a right half‐site RE1/NRSE), which in the canonical RE1/NRSE type amounts to about 21 base pairs (bp).5, 6 A second type of REST/NRSF binding site comprises the noncanonical RE1/NRSE sites, which have a variable linker between the two half‐site RE1s/NRSEs,7 while a third type features half‐site RE1/NRSE with only one‐half of the canonical RE1/NRSE.7 Of note, variations of the RE1/NRSE sequence are associated with modulation of REST/NRSF affinity to the respective DNA site, allowing some RE1/NRSE sites to be occupied by REST/NRSF, while others may remain unbound in the same cell.8 RE1/NRSE sites are enriched in neuronal genes such as brain‐derived neurotrophic factor.9 In addition, mitosis‐related genes are regulated by REST/NRSF.10 During development, REST/NRSF represses RE1/NRSE‐harboring genes in stem cells, and these genes become derepressed upon neuronal differentiation.11
REST/NRSF is a Krüppel‐type zinc finger (ZF) protein and harbors a total of nine C2H2 ZF domains, eight of which are located in the central DNA‐binding domain and the remaining one is at the C‐terminus.2 Its N‐ and C‐termini comprise repressor domains to which the SIN3A/B12 and CoREST13 corepressor complexes (CRC), respectively, can be recruited. The CoREST corepressor thereby tethers two functional activities to RE1/NRSE sites, that is, the histone deacetylase 1/2 (HDAC1, HDAC2)14 and the lysine‐specific demethylase 1A (LSD1),15, 16 which facilitate a repressive chromatin environment. CoREST and LSD1 tightly interact primarily via CoREST's linker between its SANT domains and a helical insert of LSD1 between the two parts of the split amine oxidase domain (AOD), the core structure of which has been determined by X‐ray crystallography.17 CoREST binds to HDAC1/2 via its N‐terminal half including the ELM2 domain and the N‐terminal SANT domain.18 In contrast to REST/NRSF, the CoREST–LSD1 complex binds nonspecifically to DNA.19 Thus, REST/NRSF directs the repressor complex towards genomic target sites, while the CoREST complex brings enzymatic activities to the RE1/NRSE sites to establish a repressive chromatin state.
Aberrant REST/NRSF activity has been suggested to contribute to the pathogenesis of brain cancer, including medulloblastoma,20 neuroblastoma,21 and glioblastoma.22 Medulloblastoma has been reported to overexpress REST/NRSF.20 Induced expression of mouse Rest/Nrsf inhibits neural differentiation of myc‐transduced neural stem/progenitor cells and gives rise to tumors upon injection into the rodent cerebellum.23 In medulloblastoma, two main REST/NRSF forms are found: an approximately 125 kDa form found in the cytoplasm, and an apparently higher molecular weight form of about 220 kDa in the nucleus, with a variable contribution of the smaller REST/NRSF form. This differential localization of REST/NRSF forms also has been reported for the rat striatal neural progenitor cell line ST14A, the murine striatal knock‐in cell line STHdh7/7, and mouse adult brain, amongst others.24 Moreover, REST/NRSF has been linked to neurodegenerative disorders including Huntington disease.24 In Huntington disease, a mutation in the huntingtin (HTT) gene interferes with the formation of the REST/NRSF–HTT complex, which results in translocation of REST/NRSF into the nucleus of neurons, allowing REST/NRSF to repress neuronal gene expression.24
The development of drugs that target REST/NRSF could benefit from the availability of REST/NRSF and its interactors in quantities allowing molecular studies. Therefore, the aim of this study was to establish a coexpression system for functionally active REST/NRSF and its C‐terminal repressor complex as a resource for molecular characterization. In addition, whether or not the CRC can be used for molecular pharmacological studies was tested using the two small‐molecule inhibitors SP2509 and 4SC‐202.
Results
Establishing an expression system for recombinant expression of the C‐terminal REST/NRSF CRC
The aim of this study was to establish a coexpression system for the CRC that allows a stepwise increase in complexity by the addition of genes. Due to the large sizes of the involved proteins, these proteins are challenging targets for protein expression. It was hypothesized that coexpression of their physiological interaction partners may improve the protein yield; therefore, the MultiBac baculovirus/insect cell system25 was selected. To this end, the REST/NRSF coding region from a plasmid with human REST/NRSF and the insect cell‐codon‐optimized coding sequences of human CoREST, LSD1, and HDAC1 were subcloned into suitable acceptor and donor vectors [Fig. 1(A)]. The vectors facilitate expression under the strong polyhedrin and p10 promoters in insect cells. These plasmids were then recombined stepwise by using Cre recombinase and inserted into a bacmid using the Tn7 transposition.25
Figure 1.
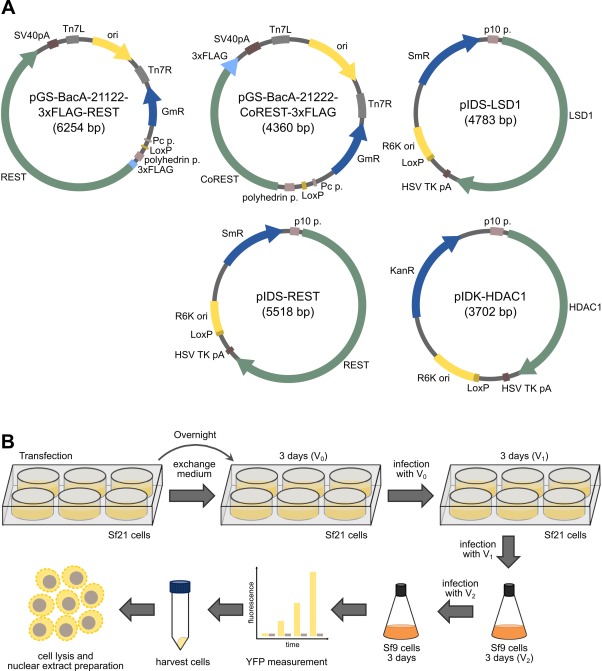
The expression and purification strategy. A: Representative donor and acceptor vectors constructed in this study. Relevant elements are indicated (ori, origin; R6K ori, R6K γ origin; Pc p., Pc promoter; polyhedrin p., polyhedrin promoter; p10 p., p10 promoter; pA, polyA signal; GmR, gentamycin resistance gene; SmR, spectinomycin resistance gene; KanR, kanamycin resistance gene). B: The protein production strategy used. Sf21 cells are transfected overnight with bacmids for baculovirus production in six‐well plates, followed by replacement of the medium. After 3 days, the supernatant (V0) is collected. Sf21 cells seeded in six‐well plates are infected with V0, and V1 is collected after 3 days. The V1 baculovirus stock is used to infect a culture of Sf9 cells in shaking flasks. After three more days, the V2 stock is collected and used to infect a preparative Sf9 culture. Protein production is monitored using YFP measurements, and cells are typically harvested after 3 days and lysed. For protein purification, the CE and the NE were prepared.
For virus production, Sf21 insect cells were transfected in a six‐well‐plate format using the isolated bacmid, and the supernatant (V0) was used to infect Sf21 cells seeded in six‐well plates [Fig. 1(B)]. The supernatant (V1) was then used to infect an Sf9 culture in a shaking flask for the production of V2. V2 was used to infect a preparative Sf9 culture for protein production. As an indicator of recombinant protein expression, the fluorescence of YFP, a reporter encoded on the bacmid,26 was monitored over time. Cells were typically harvested after 3 days, the time point that the YFP fluorescence peaked. Because it was hypothesized that REST/NRSF and its interactors might be enriched in the nucleus, the cells were lysed, and the cytoplasmic extract (CE) and nuclear extract (NE) were prepared for protein isolation.
Full‐length REST/NRSF can be isolated from NE as a high‐molecular‐weight form
First, the expression and purification of full‐length human REST/NRSF as an N‐terminally 3 × FLAG‐tagged protein were tested. Both wild‐type REST/NRSF and a Cys402Ser REST/NRSF mutant were expressed. The latter replaces a third cysteine residue within ZF domain 8 by a serine [Fig. 2(A)]. In the mouse Rest/Nrsf, this third cysteine residue, which is not involved in zinc ion coordination, has been suggested to interfere with folding.27 Both the wild‐type and Cys402Ser mutant REST/NRSF (denoted as RESTwt and RESTm) were well expressed in the baculovirus system ([Fig. 2(B,C)] for Coomassie‐stained sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and [Fig. 2(D,E)] for western blotting). Full‐length REST/NRSF was enriched in the nucleus [Fig. 2(D,E)], indicating that the protein is imported into the nucleus. Of note, wild‐type and Cys402Ser mutant REST/NRSF were primarily observed as high‐molecular‐weight proteins with an apparent molecular weight of about 220 kDa [Fig. 2(B–E)]. This observation is in line with the behavior of nuclear REST/NRSF in mammalian cells observed previously.24
Figure 2.
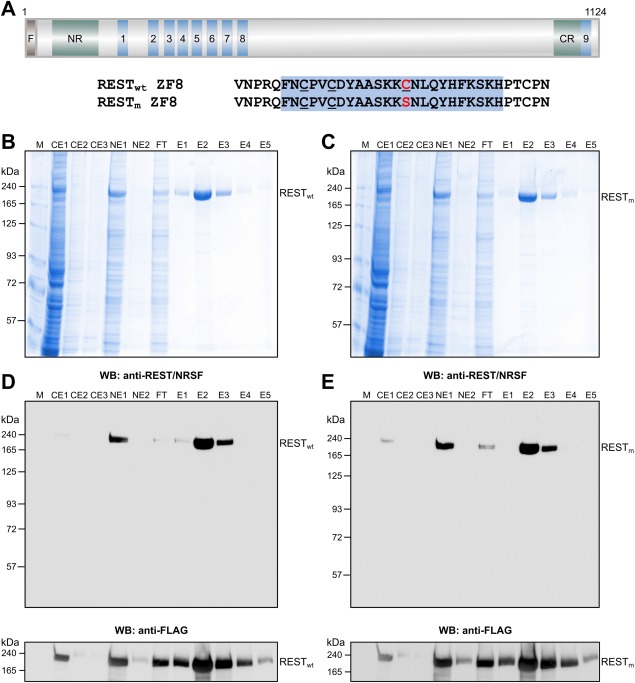
Expression and purification of full‐length human REST/NRSF. A: The domain structure of REST/NRSF showing the N‐terminal repressor domain (NR), the nine ZF domains (denoted 1–9), and the C‐terminal repressor domain (CR). REST/NRSF is expressed as an N‐terminally 3 × FLAG‐tagged (F) protein, providing a protein of 1124 amino acids. Both the wild‐type sequence (denoted RESTwt) with three cysteines within ZF8 (underlined) and a mutated REST/NRSF (denoted RESTm), in which the cysteine 402 (red) that is not involved in zinc binding has been mutated to encode a serine (red), are shown. B: Purification of RESTwt from insect cell NE using anti‐FLAG affinity selection. Shown is a Coomassie‐stained 7.5% SDS‐PAGE gel of CE1–3, NE1–2, flow‐through (FT), as well as elution fractions E1–5. RESTwt is indicated on the right. M, molecular weight marker in kDa. C: Same as (B), but for the purification of RESTm. D: Western blotting of RESTwt using anti‐REST/NRSF antibody (top) and anti‐FLAG antibody (bottom). E: Same as (D), but for RESTm.
Both full‐length wild‐type and Cys402Ser mutant REST/NRSF could be purified efficiently from insect cell extracts using a single‐step purification procedure [Fig. 2(B–E)]. To this end, REST/NRSF was enriched by anti‐FLAG affinity selection, and the proteins were eluted under mild conditions by the addition of 3 × FLAG peptide. The expression and purification efficiency were comparable for both REST/NRSF constructs and strongly enriched for the target proteins [Fig. 2(B,C)]. Thus, human full‐length REST/NRSF can be recombinantly expressed in a baculovirus system and efficiently purified as the nuclear form.
The CoRESTcore–LSD1core complex can be coexpressed in the nuclear fraction
As it was desired to assemble the CRC in a stepwise manner, the CoRESTcore–LSD1core complex was selected as the starting point [Fig. 3(A)]. This complex has been expressed previously as individual proteins and reconstituted by mixing purified CoRESTcore and LSD1core.17 This construct also included the core domain of human HDAC1. HDAC1core was not expected to be copurified as the HDAC interaction domain of CoREST18 was not included in the CoRESTcore construct and served here as a control. Therefore, this construct represents a model for the setup of the coexpression system.
Figure 3.
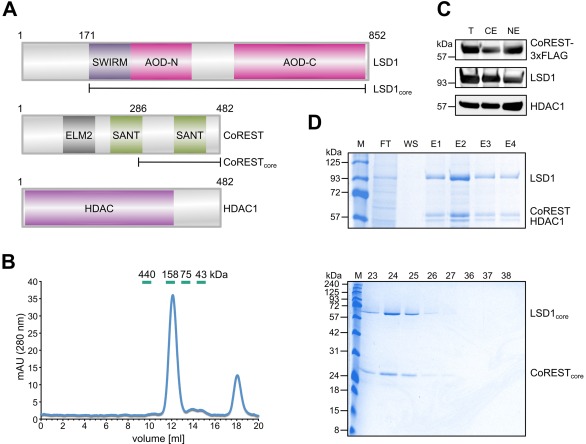
Coexpression of the CoREST–LSD1–HDAC1 complex. A: Domain structures of CoREST, LSD1, and HDAC1 with ELM2, SANT, SWIRM, and AOD split into N‐terminal and C‐terminal parts (AOD‐N and AOD‐C). The CoRESTcore and LSD1core fragments are indicated as well. A 3 × FLAG was fused to the C‐terminus of CoREST. ELM2, Egl‐27‐MTA1 homology 2; SANT, Switching‐defective protein 3 (Swi3)‐adaptor 2 (Ada2)‐nuclear receptor corepressor (N‐CoR) transcription factor (TF)IIIB; SWIRM, SWI3‐RSC8‐MOIRA; AOD, amino oxidase domain. B: Gel filtration of anti‐FLAG immunoaffinity‐selected CoRESTcore–LSD1core complexes. Core domains of CoREST, LSD1, and HDAC1 were coexpressed. HDAC1core does not bind to the CoRESTcore–LSD1core complex and serves as a control. Shown on the left is the 280 nm absorption profile, including an indication of the 500‐µL fractions. Migration of calibration proteins are shown in green. Coomassie‐stained SDS‐PAGE gel of gel filtration peak fractions (right) showing the comigration of CoRESTcore and LSD1core. Proteins are indicated on the right. M, molecular weight marker (kDa), 23–27 and 36–38, gel filtration fractions. C: Coexpression of CoREST, LSD1, and HDAC1 full‐length proteins as analyzed by immunoblotting against FLAG (for CoREST), LSD1, and HDAC1. Shown is the total cell extract (T), CE, and NE. D: Purification of the CoREST–LSD1–HDAC1 full‐length complex. Shown is a Coomassie‐stained SDS‐PAGE gel of the FT, wash (WS), and E1–4. The positions of LSD1, CoREST, and HDAC1 are indicated.
The CoRESTcore–LSD1core complex was well expressed and was purified using anti‐FLAG immunoaffinity chromatography; it was confirmed by gel filtration that CoRESTcore and LSD1core form a stable complex lacking HDAC1core [Fig. 3(B)]. The CoRESTcore–LSD1core complex peaked in the gel filtration at an apparent molecular weight of ∼145 kDa upon calibration with globular proteins, which is higher than expected for the nominal molecular weight of 100 kDa. The running behavior of the complex is, however, in line with the elongated shape17 of the CoRESTcore–LSD1core complex.28 Thus, CoRESTcore and LSD1core can be isolated as a complex.
The full‐length CoREST–LSD1–HDAC1 complex can be recombinantly coexpressed and copurified
As the next step, the CoREST–LSD1–HDAC1 complex was addressed. As HDAC1 binds to an N‐terminal domain of CoREST18 that is not included in CoRESTcore, a bacmid for coexpression of full‐length CoREST, LSD1, and HDAC1 was constructed [Fig. 3(A)]. All three proteins were expressed in our system as shown by immunoblotting of the total cell extracts [Fig. 3(C), lane T]. Fractionation into CE and NE indicated that the proteins of the complex located to both the cytoplasm and the nucleus, although CoREST and HDAC1 were more enriched in the nuclear fraction [Fig. 3(C)]. Therefore, for purification, both the CE and the NE were mixed, and the complex was selected by anti‐FLAG immunochromatography [Fig. 3(D)]. All three proteins coeluted from the column. Thus, HDAC1 can be copurified with the CoREST–LSD1 complex using full‐length genes.
Fully assembled CRC can be recombinantly coexpressed in the nucleus
Next, the recombinant expression of the fully assembled CRC composed of full‐length proteins was addressed using the baculovirus expression system [Fig. 4(A)]. All proteins were present in the CE and NE, although the proteins were enriched in the nucleus (data not shown). Therefore, the NE was used as the source for protein purification by anti‐FLAG affinity selection. The CRC could be strongly enriched by immunoaffinity purification [Fig. 4(A)]; all four proteins coeluted, indicating that the proteins form a complex. The apparently higher molecular weight form of REST/NRSF was copurified.
Figure 4.
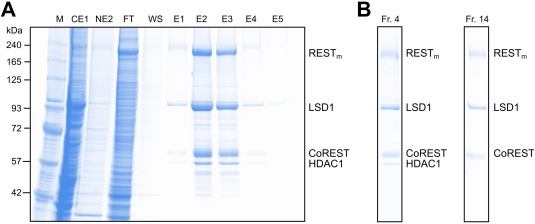
Coexpression and purification of the RESTm–CoREST–LSD1–HDAC1 full‐length complex, CRC. A: Coomassie‐stained SDS‐PAGE gel of a CRC purification. Positions of RESTm, LSD1, CoREST, and HDAC1 are indicated. B: Fractions of CRC isolates upon glycerol gradient ultracentrifugation (fraction 1, bottom; fraction 39, top). Shown is a Coomassie‐stained SDS‐PAGE gel of fractions 4 and 14. Positions of RESTm, LSD1, CoREST, and HDAC1 are labeled on the right.
The HDAC1 band, however, appeared to be underrepresented in the eluate. Therefore, glycerol gradient ultracentrifugation was used to separate the eluate. Two peaks were observed in the glycerol gradient (Fr. 1–4 and 14–15). In the higher Svedberg fraction, Fr. 4, all four proteins of the CRC comigrated; while in the lower Svedberg fraction, Fr. 14, only REST/NRSF–CoREST–LSD1 comigrated [Fig. 4(B)]. Thus, the fully assembled CRC as well as the REST/NRSF–CoREST–LSD1 form stable protein complexes that can be extracted from nuclei.
Isolated REST/NRSF and the CRC bind to RE1/NRSE
In contrast to CoREST–LSD1, which binds sequence‐nonspecifically to DNA,19 REST/NRSF binds specifically to RE1/NRSE sites in the genome.29 Therefore, electrophoretic mobility shift assay (EMSA) was performed to assess whether or not the recombinantly expressed REST/NRSF and CRC interact with RE1/NRSE sites. To this end, a 37‐bp probe, RE1‐37, which harbors a canonical RE1/NRSE site flanked by 11 and 5 bp, respectively, was designed [Fig. 5(A)].
Figure 5.
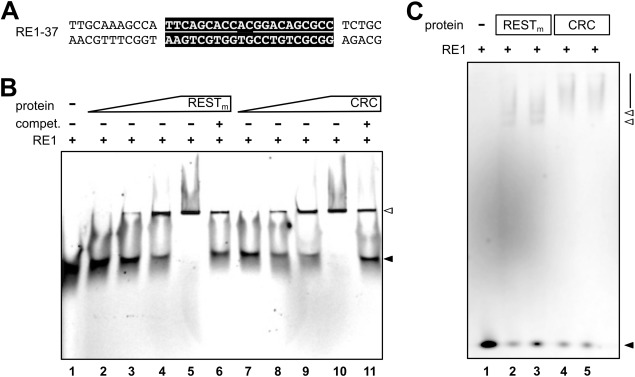
Recombinantly expressed RESTm full‐length protein and CRC bind to RE1/NRSE. A: Design of the RE1‐37 dsDNA. The canonical RE1/NRSE site is highlighted in black, and the left and right half site RE1/NRSE are indicated by a line. The motif is flanked by 11 and 5 bp, respectively. B: EMSA using native PAGE in 0.5× TBE buffer. Increasing amounts of purified full‐length RESTm and CRC were incubated with 5 nM Cy5‐labeled RE1‐37 dsDNA. Unlabeled competitor RE1‐37 dsDNA (1.5 µM) was added in some of the samples as indicated. Under these conditions, no difference in the running behavior of RESTm–RE1‐37 and CRC–RE1‐37 complexes is observed. Positions of the unbound RE1‐37 duplex (black arrowhead) and the RE1‐protein complexes (white arrowhead) are indicated. Lanes are numbered at the bottom. C: EMSA using native PAGE in Tris–glycine buffer. A total of 1 nM Cy5‐labeled RE1‐37 dsDNA was incubated with purified full‐length RESTm and the CRC for 30 min (lanes 2 and 4) and 60 min (lanes 3 and 5) and loaded together with Cy5‐labeled RE1‐37 dsDNA alone (lane 1) on a 3% native PAGE gel. Positions of the unbound RE1‐37 duplex (black arrowhead), the RE1‐RESTm complexes (white arrowheads), and RE1‐CRC (line) are indicated. Lanes are numbered at the bottom.
For isolated REST/NRSF, a protein concentration‐dependent band‐shift was observed [Fig. 5(B), lanes 2–5] (probe without protein in lane 1). The band‐shift could be competed by an excess of unlabeled RE1‐37 dsDNA [Fig. 5(B), lane 6]. A similar result was observed for the CRC [Fig. 5(B), lanes 7–11]. In particular, for the CRC, a band‐shift that was dependent on the protein–complex concentration was found [Fig. 5(B), lanes 7–10]. The presence of excess unlabeled RE1‐37 dsDNA reduced the band‐shift [Fig. 5(B), lane 11]. Note that under the experimental conditions used, a difference in the migration behaviors of the REST/NRSF–RE1‐37 form and of the CRC–RE1‐37 complex was not seen. In contrast, using 3% native PAGE in a Tris–glycine buffer system, a difference in the migration of the REST/NRSF–RE1‐37 and CRC–RE1‐37 complex was observed [Fig. 5(C)]. REST/NRSF–RE1‐37 produced two faster migrating band‐shifts [Fig. 5(C), lanes 2–3] as compared with the CRC–RE1‐37 complex, which resulted in a slower migrating shift [Fig. 5(C), lanes 4–5] (RE1‐37 alone in lane 1). Thus, both the REST/NRSF form and the CRC recognize RE1/NRSE dsDNA.
Targeting the HDAC activity of the CRC
The CRC harbors two enzymatic activities, HDAC1 and LSD1, which are druggable. To address whether or not the recombinantly expressed CRC is also active in terms of its deacetylase and demethylase activities and thus can be used for drug testing, a set of experiments was performed in the absence and presence of the recently developed combined HDAC/LSD1 inhibitor 4SC‐202 and the LSD1 inhibitor SP2509 [Fig. 6(A)].
Figure 6.
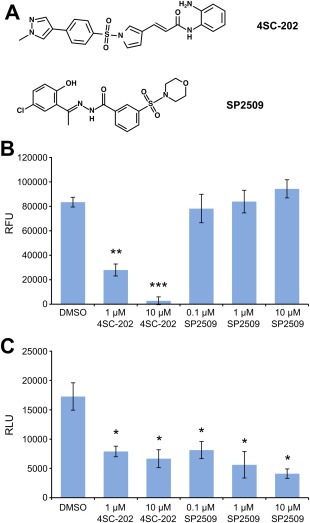
The small molecules 4SC‐202 and SP2509 inhibit the function of the CRC. A: Chemical structures of the drugs used. B: Histone deacetylase assay using the purified CRC exposed to 4SC‐202 and SP2509 at the concentrations indicated. As a control, DMSO exposure was used. Shown are the mean relative fluorescence units (RFU) ± SEM for three individual purifications (n = 3). C: LSD1 assay of purified CRC incubated with 4SC‐202 and SP2509. The concentrations of the inhibitors are indicated. Shown are the mean relative light units (RLU) ± SEM for three individual purifications (n = 3). * P < 0.05, ** P < 0.01, and *** P < 0.001.
Using an HDAC assay, it was confirmed that the CRC deacetylates the BOC‐Ac‐Lys‐AMC substrate [Fig. 6(B)] (DMSO), demonstrating that the deacetylase activity of the complex is functional. In contrast, the addition of 1 µM and 10 µM 4SC‐202 significantly reduced the deacetylase activity of the CRC, while SP2509 up to a concentration of 10 µM did not have a significant effect on the deacetylase activity [Fig. 6(B)]. Thus, the CRC is functionally active in terms of deacetylation, and 4SC‐202 can inhibit the activity of the recombinantly expressed and purified CRC.
Targeting the histone demethylase activity of the CRC
Using a dimethylated K4 histone 3 peptide, the activity of LSD1 could be tested. In this assay, the activity of LSD1 was measured as a function of H2O2 production, visualized by luminol.30 The purified CRC was functional in terms of demethylating lysine residues [Fig. 6(C)]. In contrast, incubation with 4SC‐202 or SP2509 resulted in a significant decrease of the demethylation activity [Fig. 6(C)]. Thus, fully assembled CRC has lysine demethylation activity, and its activity can be attenuated by using 4SC‐202 or SP2509.
SP2509 does not destabilize the CoREST–LSD1 interaction
Our activity assays suggest that SP2509 directly inhibits the enzymatic activity of LDS1 rather than interfering with the CoREST–LSD1 interaction suggested previously.31 To exclude that SP2509 interferes with the assembly of the CoREST–LSD1 complex, while not being able to dissociate a CoREST–LSD1 complex once it is formed, the insect cells were treated with the inhibitor during protein production, and anti‐FLAG affinity selection was used for purification. Independent of the absence or presence of SP2509 [Fig. 7(A)], the CRC could be isolated; based on the Coomassie‐stained SDS‐PAGE gels of the eluates, major differences in the LSD1/CoREST ratios between the purifications were not observed [Fig. 7(A)].
Figure 7.
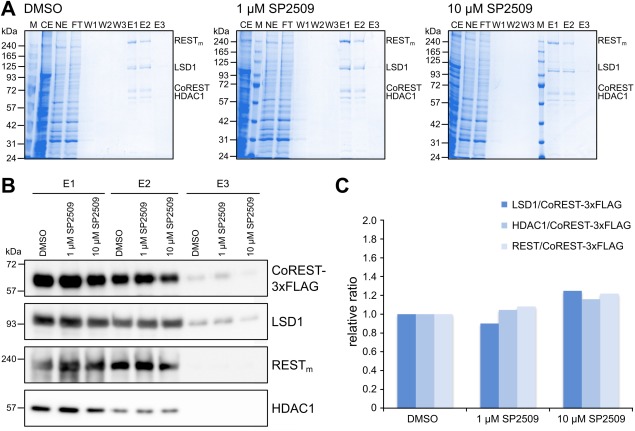
SP2509 does not interfere with CRC assembly. A: Anti‐FLAG affinity purification of the CRC from cells exposed to DMSO, 1 µM SP2509, and 10 µM SP2509. Protein identities are given on the right. Shown are Coomassie‐stained SDS‐PAGE gels of the CE, NE, FT, wash fractions W1–3, as well as E1–3. B: Western blotting of elution fractions E1–3 isolated from cells exposed to DMSO or SP2509, as indicated. Anti‐FLAG (for CoREST), anti‐LSD1, anti‐REST, and anti‐HDAC1 antibodies were used for detection. C: Relative quantification of western blot bands expressed as a ratio to FLAG (CoREST) and normalized to the DMSO sample. Shown is the average for E1 and E2, where the western blot bands could be identified clearly.
To corroborate these results, western blotting of the four proteins in the complex were performed, and the bands were quantified [Fig. 7(B,C)]. All proteins were readily detected by western blotting [Fig. 7(B)], and quantification of the band intensity ratios revealed that the coimmunopurification of LSD1 with CoREST was similar in all samples [Fig. 7(C)]. In addition, for HDAC1 and REST/NRSF, similar ratios were found [Fig. 7(C)]. Thus, SP2509 has no major destabilizing effect on the CRC, but rather inhibits the enzymatic activity of LSD1.
4SC‐202 and SP2509 interfere with medulloblastoma cell growth
Finally, whether or not the small‐molecule drugs also have an effect on cells was addressed. To this end, medulloblastoma cells were selected as a model because REST/NRSF has been implicated in medulloblastoma pathogenesis.20, 23 Three cell lines, namely Daoy, D283 Med, and ONS‐76, were exposed to 4SC‐202 and SP2509. While the DMSO‐exposed medulloblastoma cells showed the typical morphology, strong induction of cell death using 1 or 10 µM 4SC‐202 was observed [Fig. 8(A)]. For 0.1 µM SP2509, no morphological changes were found; however, at 1 or 10 µM SP2509, cell death was induced [Fig. 8(A)].
Figure 8.
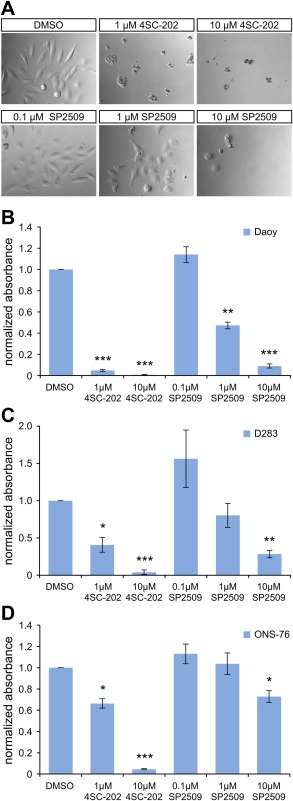
The small molecules 4SC‐202 and SP2509 interfere with viability of medulloblastoma cells. A: Morphology of the Daoy medulloblastoma cell line upon 3 days of exposure to DMSO, 4SC‐202, and SP2509, respectively, at the indicated concentrations. B–D: Shown are XTT assays for the cell lines Daoy (B), D283 Med (C), and ONS‐76 (D) treated with DMSO or the drugs at the indicated concentrations. Data were determined from three biological replicates with three replicates for each. * P < 0.05, ** P < 0.01, and *** P < 0.001.
To quantify the effect of the drugs, XTT viability assays were used. In the three medulloblastoma cell lines, 4SC‐202 significantly decreased the viability in a concentration‐dependent manner [Fig. 8(B–D)]. Daoy cells reacted most strongly to 4SC‐202 [Fig. 8(B)]. For SP2509, Daoy cells had significantly reduced viability at both 1 µM and 10 µM concentrations [Fig. 8(B)], while 10 µM SP2509 was required to significantly decrease the viability of the D283 and ONS‐76 cells [Fig. 8(C,D)]. Thus, the double‐specific drug 4SC‐202 inhibits medulloblastoma cell growth, and SP2509 likewise interferes with the viability of medulloblastoma cells.
Discussion
In this study, it was demonstrated that the human multiprotein CRC can be coexpressed efficiently as full‐length proteins in the baculovirus expression system and purified in amounts allowing molecular analyses. The possibility to recombine acceptor and donor vectors using the Cre/LoxP system25 enabled a stepwise reconstitution of the CRC, while taking advantage of coexpression of the protein's physiological interaction partners. Proteins were enriched in the nucleus, the physiological organelle of transcription factors. Using a set of functional assays including EMSAs, histone deacetylation assays, and histone demethylation assays, supplemented with small‐molecule inhibitor testing, it was demonstrated that the isolates are active. Importantly, our results show that the recombinantly expressed REST/NRSF binds both RE1/NRSE and the CoREST–LSD1–HDAC1 CRC.
The interaction of the CoRESTcore–LSD1core complex with DNA is DNA‐sequence independent and becomes destabilized at salt concentrations >50 mM KCl.19 Accordingly, the CoREST CRC benefits from an interaction with further DNA‐binding proteins that tether the CoREST complex to specific sites in the genome. In this respect, REST/NRSF binds sequence‐specifically to RE1/NRSE sites7, 29 and allows a band‐shift at 260 mM salt conditions (110 mM NaCl and 150 mM KCl) upon incubation of isolated REST/NRSF and the CRC shown herein. Thus, it is the sequence‐specificity of REST/NRSF that directs the CRC to RE1/NRSE sites.
The presence of two enzymatic activities in the CRC makes the REST/NRSF complex a valuable target for drug testing; thus, the CRC represents a resource for pharmacological studies. In contrast to other studies that use HDAC1 or LSD1 as a single component,32, 33 our system allows drug testing to be performed in the context of a macromolecular assembly, that is, the CRC, while taking advantage of a fully defined in vitro system. Of note, the presence of interactors can, in principle, modulate the activity of inhibitors towards their drug target. Such modulations are supported by chemoproteomics binding studies, which have demonstrated that HDAC‐containing complexes such as the HDAC1/2–CoREST complex and the HDAC1/2–SIN3 complex differ in their binding strength towards individual HDAC inhibitors34 despite that both complexes harbor HDAC1 subunits. The CRC established in our study can thus contribute to assess the effect of drugs in the relevant assembly.
Moreover, our system also offers advantages in terms of studying the mechanism of inhibition. It allows us to distinguish between inhibition of the enzymatic activity and interference of complex assembly by using enzyme assays and coimmunopurification. Thus, our system also allows capturing drugs that modulate protein–protein interactions, which cannot be effectively studied using individual proteins. Inhibitors that interfere with the assembly of protein complexes may, in principle, prevent the formation of protein–protein interactions during the assembly of a macromolecular complex or may disrupt protein–protein interactions within the assembled complex.35, 36 In our system, drugs can be added to the cells during protein production, or they can be added to the purified complex after purification; thus, both types of protein–protein interaction inhibitors can be studied in addition to inhibitors blocking catalytic activity.
Accordingly, two recently developed inhibitors, 4SC‐202 and SP2509, were tested. It has been suggested that the mechanism of inhibition of SP2509 is to interfere with the CoREST–LSD1 interaction.31 However, our immunopurification studies did not find support for this mechanism. The CRC was efficiently copurified, independent of whether or not the cells had been treated with SP2509. Furthermore, western blotting did not show any difference in the LSD1/CoREST ratio in the tested concentration range. In contrast, SP2509 inhibited the enzymatic activity of LSD1 rather than acting as a protein–protein interaction inhibitor and blocked the growth of medulloblastoma cells.
The effect of 4SC‐202 was also studied. The drug 4SC‐202 has been suggested to inhibit both HDACs (i.e. HDAC1, HDAC2, and HDAC3) and LSD1.37 Accordingly, it was demonstrated that the drug efficiently inhibited both HDAC1 and LSD1 in the CRC isolates. The drug 4SC‐202 has been shown to inhibit the growth of colorectal cancer cells,38 hepatocellular carcinoma cells,39 and urothelial carcinoma cells.40 Here, it was demonstrated that 4SC‐202 also blocks the growth of medulloblastoma cell lines, a cancer type with involvement of REST/NRSF.20, 23
In conclusion, our system provides a toolbox for the expression of the central transcriptional repressor complex, CRC, allowing the complex to be assembled in a stepwise manner. Proteins can be isolated efficiently using immuno‐affinity selection and are functionally active. Thus, our system can be used as a resource for both biochemical studies and drug development.
Materials and Methods
Oligonucleotides and plasmids
The oligonucleotide (Sigma, Haverhill, U.K.) sequences used in this study are given in Table S1 of the Supporting Information. The human full‐length CoREST, LSD1, and HDAC1 (including an additional leucine‐encoding codon originating from a subcloning step) coding sequences were codon‐optimized for insect cell expression and produced by gene synthesis (GeneCust, Dudelange, Luxembourg). Full‐length cDNA of human wild‐type and mutated REST/NRSF was obtained from the plasmids pGS0261‐RESTwt and pGS0261‐RESTm, respectively. The vectors pGS‐BacA‐21122 and pGS‐BacA‐21222, derivatives of pACEBac1, were used for N‐terminal and C‐terminal 3 × FLAG‐tagging. The MultiBac vectors pACEBac1, pIDS, and pIDK were purchased from Geneva Biotech (Geneva, Switzerland). The plasmids generated in this study are listed in Table S2 of the Supporting Information.
Small‐molecule drugs
SP2509 and 4SC‐202 were obtained from Selleckchem (Munich, Germany). Stock solutions were prepared by dissolving the drugs in dimethyl sulfoxide (DMSO; Sigma, St. Louis, MO), and working dilutions were prepared by serial dilutions.
Construction of plasmid vectors
Core and full‐length CoREST were amplified using Phusion HF MasterMix (Thermo Fisher Scientific, Waltham, MA) and the primer pairs CoREST‐C‐for/CoREST‐C‐rev and CoREST‐fl‐for/CoREST‐C‐rev, respectively. The polymerase chain reaction fragment and pGS‐BacA‐21222 vector were treated with EcoRI and XbaI (Thermo Fisher Scientific) and ligated using T4 DNA ligase (Thermo Fisher Scientific). Subsequently, the vector was transformed into DH5α E. coli cells (Thermo Fisher Scientific) and selected on lysogeny broth (LB)‐gentamycin plates. For construction of the remaining vectors, the following primers, plasmids, and restriction enzymes for subcloning were used: primers REST‐fl‐accept‐for/REST‐fl‐accept‐rev, pGS‐BacA‐21122 vector, as well as NheI and SalI for REST wt and REST m; primers REST‐fl‐donor‐for/REST‐fl‐donor‐rev, pIDS vector, as well as XhoI and NheI for REST wt and REST m; primers LSD1‐fl‐for/LSD1‐fl‐rev, pIDS vector, as well as XhoI and NheI for LSD1 fl; primers LSD1‐C‐for/LSD1‐C‐rev, pIDS vector, as well as XhoI and NheI for LSD1 core; primers HDAC1‐fl‐for/HDAC1‐fl‐rev, pIDK vector, as well as XhoI and KpnI for HDAC1fl; and primers HDAC1‐core‐for/HDAC1‐core‐rev, pIDK vector, as well as XhoI and KpnI for HDAC1 core. Colonies were screened by restriction enzyme digestion, followed by verification using DNA sequencing (Macrogen Europe, Amsterdam, The Netherlands).
Cre‐mediated recombination
For recombination, 1 µg of the respective plasmids were mixed with Cre buffer and 2 U of Cre recombinase (NEB, Ipswich, MA) in a final volume of 20 μL. Reactions were incubated at 37°C for 1 h, and thereafter inactivated at 70°C for 10 min. Reactions were transformed into chemically competent DH5α E. coli. A total of 400 μL of LB medium was added to the bacteria, and the bacteria were incubated at 37°C in a shaking incubator overnight for recovery. The E. coli were then plated on LB agar plates containing a relevant combination of antibiotics and incubated at 37°C overnight. Recombination was verified by restriction enzyme digestion of isolated plasmids.
Construction of bacmids
First, 10 ng of vector and 100 μL of electro‐competent DH10EMBacY cells (Geneva Biotech) were incubated on ice. Subsequent to electroporation (200Ω, 25 μF, 1.8 kV), the cells were incubated at 37°C overnight in a shaking incubator and plated on LB plates containing 50 μg/mL kanamycin, 7 μg/mL gentamycin, 10 μg/mL tetracyclin, 40 μg/mL 5‐bromo‐4‐chloro‐3‐indolyl‐β‐D‐galactopyranoside (X‐gal), and 40 μg/mL isopropyl β‐D‐1‐thiogalactopyranoside. Plasmid mini‐preparation kits (Omega bio‐tek, Norcross, GA) were used to extract recombinant bacmids. Bacmids were precipitated from cleared lysates using isopropanol instead of using kit columns.
Virus and protein production in insect cells
For transfection, 7 × 105 Sf21 cells (Thermo Fisher Scientific) were seeded in 3 mL of Sf‐900 III SFM medium (Thermo Fisher Scientific) per well of a six‐well plate, and the cells were allowed to attach at 27°C for 1 h. A total of 2 μg of bacmid was mixed with 75 μL of Sf‐900 III SFM medium and then added to 16 μL of PromoFectin‐Insect transfection reagent (PromoKine, Heidelberg, Germany) mixed with 75 μL of Sf‐900 III SFM medium. Both mixtures were mixed gently and incubated at room temperature for 15 min. Then, 800 µL of Sf‐900 III SFM medium was added to the transfection mixture. The Sf‐900 III SFM medium was exchanged by 2.5 mL of fresh medium prior to adding the transfection mixture to the cells. The cells were incubated at 27°C overnight. Thereafter, the medium was changed to Sf‐900 III SFM medium supplemented with 1.5% fetal bovine serum (FBS; GE Healthcare Hyclone, Logan, UT) and 1× penicillin/streptomycin (P/S; Sigma or Thermo Fisher Scientific). After 3 days, the supernatant (V0) was collected. To amplify the virus, 7 × 105 Sf21 cells in 3 mL of Sf‐900 III SFM medium supplemented with 1.5% FBS and 1× P/S were infected with 600 μL of V0 in six‐well plates and incubated for 3 days. V1 was collected, and 2.5 × 108 Sf9 cells (Thermo Fisher Scientific) in 50 mL of Sf‐900 III SFM medium supplemented with 1.5% FBS and 1× P/S were infected with 5 mL of V1. The cells were incubated at 27°C for 3 days in a shaking incubator at 125 rpm. V2 was collected and a total of 0.5 mL of supernatant (V2) was used to infect 2.5 × 108 Sf9 cells in 50 mL Sf‐900 III SFM medium supplemented with 1× P/S for protein production over ∼3 days. The protein expression level was monitored by measuring yellow fluorescent protein (YFP) fluorescence.
Alternatively, 1 × 106 Sf9 cells in 2.5 mL of Sf‐900 III SFM were used for transfection, and V0 was collected after 4 days. A volume of 2 mL of V0 was added to 1.35 × 107 Sf9 cells in 15 mL of Sf‐900 III SFM medium supplemented with 1.5% FBS and 1× P/S. The cells were cultured in a shaking incubator at 27°C for 4 days, after which the supernatant (V1) was collected by centrifugation. For protein production, 2.5 mL of V1 virus was added to 1 × 108 Sf9 cells in 25 mL of Sf‐900 III SFM medium supplemented with 1.5% FBS and 1× P/S. The cells were cultured at 27°C, and protein expression was monitored by YFP measurements over ∼3 days. For monitoring recombinant protein expression, a 200‐μL aliquot from the infected cell culture was centrifuged, and the cell pellet was resuspended in phosphate‐buffered saline supplemented with 1% Triton‐X100. The YFP signal was measured using a Qubit 3.0 fluorometer (Thermo Fisher Scientific).
NE preparation
Baculovirus‐infected insect cells were harvested by centrifugation. Cell pellets were resuspended in 10 mL of ice‐cold cell lysis buffer (10 mM HEPES pH 7.5, 10 mM KCl, 0.1 mM EDTA, 0.5% IGEPAL CA‐630, 0.5 mM phenylmethanesulfonyl fluoride [PMSF], 1× cOmplete ULTRA tablet [Roche, Mannheim, Germany]; supplemented with 1 mM DTT in some experiments) and allowed to swell on ice for 15–20 min with intermittent mixing. Centrifugation at 3,828 × g and 4°C for 10 min was used to separate the supernatant (CE1) and nuclei. The pelleted nuclei were washed twice with 10 mL of ice‐cold cell lysis buffer, and the supernatant was collected (CE2 and CE3) by centrifugation at 3,828 × g and 4°C for 10 min. The pelleted nuclei were resuspended in 10 mL of ice‐cold NE buffer (20 mM HEPES pH 7.5, 400 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1× complete ULTRA tablet; supplemented with 1 mM DTT in some experiments) and incubated on ice for 30 min. The supernatant (NE1) was obtained by centrifugation at 3,828 × g and 4°C for 15 min. The pellet was washed with NE buffer, and the supernatant (NE2) was collected by centrifugation in some experiments. In some purifications, a volume of 250 µL of n‐heptane was added to NE1, and the tube was gently inverted. Upon recentrifugation at 3,828 × g and 4°C for 10 min, the aqueous NE1 was collected.
Protein purification
A volume of 150 µL of anti‐FLAG M2 affinity gel (A2220; Sigma) per 50 mL of cell culture volume (2.5 × 108 cells) was resuspended in 10 mL of NE buffer, the mixture was centrifuged at 700 × g and 4°C for 10 min, and the supernatant was removed carefully. The resin was washed with 10 mL of NE buffer and centrifuged again at 700 × g and 4°C for 10 min; then the supernatant was discarded. This step was repeated once. Subsequently, NE or, in some experiments, an NE/CE mixture was added to the equilibrated anti‐FLAG gel and incubated on a head‐over‐tail rotator overnight at 4°C. NE or NE/CE‐resin was centrifuged at 700 × g and 4°C for 10 min, and the supernatant was collected as the flow‐through fraction (FT). The resin was washed with 10 mL of NE buffer (supplemented with 50 µM ZnCl2 in some experiments) three times, and each supernatant was collected as wash fractions 1–3 (WS1–3), respectively. Then, the washed resin was transferred into a gravity flow column and incubated with 400 µL of elution buffer (NE buffer supplemented with 125 µg/mL 3 × FLAG peptide, F4799, Sigma; and 50 µM ZnCl2 in some experiments) on ice for 30 min. Elution was done by gravity and collected as fraction E1. The resin was again incubated with 400 µL of elution buffer on ice for 30 min and eluted by gravity, yielding fraction E2. For fraction E3, 400 µL of NE buffer was added to the resin and immediately eluted by gravity. The protein content peaked in E2 and E3. In some experiments, additional elution fractions using NE buffer were collected. Alternatively, the anti‐FLAG resin was incubated in elution buffer for 50 min on ice, yielding elution fraction E1, followed by collection of elution fractions E2 and E3 as described above. Using this elution procedure, the protein content peaked in E1 and E2.
Gel filtration
Anti‐FLAG‐purified CoRESTcore–LSD1core complexes were loaded on a Superdex 200 10/300 GL column (GE Healthcare, Little Chalfont, U.K.) mounted in an Äkta Purifier system (GE Healthcare). The column was equilibrated in gel filtration buffer (20 mM HEPES, pH 7.9, 0.15 M NaCl), and 500‐µL fractions were collected. For calibration, the Gel Filtration HMW Calibration Kit (GE Healthcare) was used.
Glycerol gradient ultracentrifugation
Anti‐FLAG eluates were loaded on a 5–20% glycerol gradient in gradient buffer [20 mM HEPES (pH = 7.5), 400 mM NaCl] and ultracentrifuged at 45,300 rpm and 4°C for 17 h in a TH‐660 rotor (Thermo Fisher Scientific). Gradients were fractionated into 39 fractions from the bottom in 5 drops as described previously.41
SDS–PAGE and western blotting
Samples were mixed with SDS loading buffer, heated to 98°C for 10 min, loaded onto SDS–PAGE gels, and electrophoresed. Gels were stained with colloidal Coomassie Blue stain. For western blotting, a protocol described previously42 was used. The antibodies used in this study are given in Table S3 of the Supporting Information. Coomassie‐stained gels and western blot membranes were imaged using an ImageQuant 4010 instrument (GE Healthcare). ImageStudio Lite (LI‐COR, Lincoln, NE) was used for quantification of western blot bands.
EMSA
Equal amounts of RE1‐37‐s/RE1‐37‐as and cyanine 5 (Cy5)‐labeled Cy5‐RE1‐37‐s/RE1‐37‐as oligonucleotides were annealed in a thermocycler. Increasing amounts of purified RESTm and the full‐length C‐terminal repressor complex were diluted in EMSA buffer (20 mM HEPES‐KOH pH 7.5, 0.1% IGEPAL CA‐630, 1 mM DTT, 5 mM MgCl2, 150 mM KCl, 5% glycerol, 0.1 mM PMSF), and mixed with 5 or 1 nM Cy5‐labeled, annealed RE1‐37 oligonucleotide. The final salt concentration was about 260 mM (110 mM NaCl and 150 mM KCl). In some samples, 1.5 µM unlabeled RE1‐37 was added prior to addition of the Cy5‐labeled RE1‐37. The samples were incubated for 30 min at room temperature before loading on a prerun 5% PAGE gel in 0.5× Tris/borate/EDTA (TBE) buffer. The gels were run at 100 V and 4°C, and the running buffer was continuously mixed using a peristaltic pump during the run. Alternatively, the samples were loaded after 30 and 60 min of incubation on a 3% native PAGE gel in Tris–glycine buffer and electrophoresed at 200 V for 35 min. The gels were imaged using an ImageQuant 4010 instrument in Cy5‐fluorescence mode.
HDAC assay
HDAC activity was assayed by a fluorogenic method as described previously.43 For each 40‐µL reaction, 5 μL of eluate was mixed with 100 μM BOC‐Ac‐Lys‐AMC HDAC substrate (Sigma) in HDAC buffer (25 mM Tris‐HCl, pH 8.0, 2.7 mM KCl, 1 mM MgCl2). DMSO and inhibitors were added at the indicated concentrations as well. The samples were incubated at 37°C for 1 h, followed by digestion using 5 mg/mL trypsin (Sigma) in the presence of 1 μM trichostatin A (Sigma). Samples were measured in a black 96‐well plate (Greiner bio‐one, Kremsmünster, Austria) using an EnSpire multimode reader (PerkinElmer, Waltham, MA) at an excitation of 355 nm and an emission of 460 nm. The assay was performed using three individual purifications (n = 3) with three technical replicates for each.
Histone demethylase assay
To measure the activity of LSD1, a luminol‐based assay was used.30 A total of 5 µL of eluate was diluted in LSD1 reaction buffer (50 mM Tris, pH 8.5, 50 mM KCl, 5 mM MgCl2, 5 nmol luminol, 20 μg/ml horseradish peroxidase, 1% Triton X‐100), and 10 µM H3K4me2 (1–21 aa) peptide was added as the substrate (all from Sigma). DMSO and inhibitors were added as indicated. The total volume per reaction was 100 µL. The samples were measured in a white 96‐well plate (Corning, Corning, NY) using an EnSpire multimode plate reader at an emission wavelength of 428 nm. Three individual purifications (n = 3) were used for the assay, with three technical replicates for each.
Coimmunopurification upon SP2509 treatment
Sf9 cells in 25 mL of Sf‐900 III SFM medium were infected with baculovirus. SP2509 was added at final concentrations of 1 and 10 µM, respectively. As a mock control, DMSO was used. DMSO and SP2509 were added every 24 h over a period of 4 days, followed by anti‐FLAG immunoselection as described above.
XTT cell proliferation assay
Daoy and D283 Med cells were purchased from the American Type Culture Collection (LGC Standards GmbH, Wesel, Germany) and cultured as described.44 ONS‐76 cells were obtained from the Japanese Collection of Research Bioresources Cell Bank (National Institute of Biomedical Innovation, Health and Nutrition, Osaka, Japan) and cultured in RPMI medium 1640 (Thermo Fisher Scientific) supplemented with 10% FBS (Hyclone), 50 U/mL penicillin, and 50 µg/mL streptomycin. All medulloblastoma cell lines were kept in an incubator at 37°C in a 5% CO2/5% O2/90% N2 atmosphere with maximum humidity. XTT assays44 were performed in triplicates (n = 3) with three replicates for each using 1 × 103 Daoy cells/well, 4 × 103 D283 Med cells/well, and 1 × 103 ONS‐76 cells/well in 100 µL of medium at initial seeding in 96‐well plates.
Statistical Analysis
The two‐sample unequal variance, two‐tailed distribution Student's t‐test was used for statistical analyses. Data are presented as the mean ± standard error of the mean (SEM). Statistical significance levels were set as follows: * P < 0.05, ** P < 0.01, and *** P < 0.001.
Conflict of Interest
No competing financial interests related to this work exist.
Supporting information
Supporting Information
Acknowledgments
The authors wish to thank Golshah Ayoubi and Susanne N. Stubbe for technical assistance. They are grateful for access to laboratory facilities at the Danish Neuroscience Center House.
References
- 1. Stein GS, Zaidi SK, Stein JL, Lian JB, van Wijnen AJ, Montecino M, Young DW, Javed A, Pratap J, Choi JY, Ali SA, Pande S, Hassan MQ (2009) Transcription‐factor‐mediated epigenetic control of cell fate and lineage commitment. Biochem Cell Biol 87:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chong JA, Tapia‐Ramirez J, Kim S, Toledo‐Aral JJ, Zheng Y, Boutros MC, Altshuller YM, Frohman MA, Kraner SD, Mandel G (1995) REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell 80:949–957. [DOI] [PubMed] [Google Scholar]
- 3. Schoenherr CJ, Anderson DJ (1995) The neuron‐restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron‐specific genes. Science 267:1360–1363. [DOI] [PubMed] [Google Scholar]
- 4. Rockowitz S, Zheng D (2015) Significant expansion of the REST/NRSF cistrome in human versus mouse embryonic stem cells: potential implications for neural development. Nucleic Acids Res 43:5730–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kraner SD, Chong JA, Tsay HJ, Mandel G (1992) Silencing the type II sodium channel gene: a model for neural‐specific gene regulation. Neuron 9:37–44. [DOI] [PubMed] [Google Scholar]
- 6. Mori N, Schoenherr C, Vandenbergh DJ, Anderson DJ (1992) A common silencer element in the SCG10 and type II Na+ channel genes binds a factor present in nonneuronal cells but not in neuronal cells. Neuron 9:45–54. [DOI] [PubMed] [Google Scholar]
- 7. Johnson DS, Mortazavi A, Myers RM, Wold B (2007) Genome‐wide mapping of in vivo protein‐DNA interactions. Science 316:1497–1502. [DOI] [PubMed] [Google Scholar]
- 8. Bruce AW, Lopez‐Contreras AJ, Flicek P, Down TA, Dhami P, Dillon SC, Koch CM, Langford CF, Dunham I, Andrews RM, Vetrie D (2009) Functional diversity for REST (NRSF) is defined by in vivo binding affinity hierarchies at the DNA sequence level. Genome Res 19:994–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bruce AW, Donaldson IJ, Wood IC, Yerbury SA, Sadowski MI, Chapman M, Gottgens B, Buckley NJ (2004) Genome‐wide analysis of repressor element 1 silencing transcription factor/neuron‐restrictive silencing factor (REST/NRSF) target genes. Proc Natl Acad Sci U S A 101:10458–10463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guardavaccaro D, Frescas D, Dorrello NV, Peschiaroli A, Multani AS, Cardozo T, Lasorella A, Iavarone A, Chang S, Hernando E, Pagano M (2008) Control of chromosome stability by the beta‐TrCP‐REST‐Mad2 axis. Nature 452:365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G (2005) REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 121:645–657. [DOI] [PubMed] [Google Scholar]
- 12. Huang Y, Myers SJ, Dingledine R (1999) Transcriptional repression by REST: recruitment of Sin3A and histone deacetylase to neuronal genes. Nat Neurosci 2:867–872. [DOI] [PubMed] [Google Scholar]
- 13. Andres ME, Burger C, Peral‐Rubio MJ, Battaglioli E, Anderson ME, Grimes J, Dallman J, Ballas N, Mandel G (1999) CoREST: a functional corepressor required for regulation of neural‐specific gene expression. Proc Natl Acad Sci U S A 96:9873–9878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hakimi MA, Bochar DA, Chenoweth J, Lane WS, Mandel G, Shiekhattar R (2002) A core‐BRAF35 complex containing histone deacetylase mediates repression of neuronal‐specific genes. Proc Natl Acad Sci U S A 99:7420–7425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee MG, Wynder C, Cooch N, Shiekhattar R (2005) An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 437:432–435. [DOI] [PubMed] [Google Scholar]
- 16. Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y (2005) Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell 19:857–864. [DOI] [PubMed] [Google Scholar]
- 17. Yang M, Gocke CB, Luo X, Borek D, Tomchick DR, Machius M, Otwinowski Z, Yu H (2006) Structural basis for CoREST‐dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol Cell 23:377–387. [DOI] [PubMed] [Google Scholar]
- 18. You A, Tong JK, Grozinger CM, Schreiber SL (2001) CoREST is an integral component of the CoREST‐ human histone deacetylase complex. Proc Natl Acad Sci U S A 98:1454–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pilotto S, Speranzini V, Tortorici M, Durand D, Fish A, Valente S, Forneris F, Mai A, Sixma TK, Vachette P, Mattevi A (2015) Interplay among nucleosomal DNA, histone tails, and corepressor CoREST underlies LSD1‐mediated H3 demethylation. Proc Natl Acad Sci U S A 112:2752–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lawinger P, Venugopal R, Guo ZS, Immaneni A, Sengupta D, Lu W, Rastelli L, Marin Dias Carneiro A, Levin V, Fuller GN, Echelard Y, Majumder S (2000) The neuronal repressor REST/NRSF is an essential regulator in medulloblastoma cells. Nat Med 6:826–831. [DOI] [PubMed] [Google Scholar]
- 21. Palm K, Metsis M, Timmusk T (1999) Neuron‐specific splicing of zinc finger transcription factor REST/NRSF/XBR is frequent in neuroblastomas and conserved in human, mouse and rat. Brain Res Mol Brain Res 72:30–39. [DOI] [PubMed] [Google Scholar]
- 22. Conti L, Crisafulli L, Caldera V, Tortoreto M, Brilli E, Conforti P, Zunino F, Magrassi L, Schiffer D, Cattaneo E (2012) REST controls self‐renewal and tumorigenic competence of human glioblastoma cells. PLoS One 7:e38486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Su X, Gopalakrishnan V, Stearns D, Aldape K, Lang FF, Fuller G, Snyder E, Eberhart CG, Majumder S (2006) Abnormal expression of REST/NRSF and Myc in neural stem/progenitor cells causes cerebellar tumors by blocking neuronal differentiation. Mol Cell Biol 26:1666–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E (2003) Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE‐controlled neuronal genes. Nat Genet 35:76–83. [DOI] [PubMed] [Google Scholar]
- 25. Berger I, Fitzgerald DJ, Richmond TJ (2004) Baculovirus expression system for heterologous multiprotein complexes. Nat Biotechnol 22:1583–1587. [DOI] [PubMed] [Google Scholar]
- 26. Trowitzsch S, Bieniossek C, Nie Y, Garzoni F, Berger I (2010) New baculovirus expression tools for recombinant protein complex production. J Struct Biol 172:45–54. [DOI] [PubMed] [Google Scholar]
- 27. Zhang Y, Hu W, Shen J, Tong X, Yang Z, Shen Z, Lan W, Wu H, Cao C (2011) Cysteine 397 plays important roles in the folding of the neuron‐restricted silencer factor/RE1‐silencing transcription factor. Biochem Biophys Res Commun 414:309–314. [DOI] [PubMed] [Google Scholar]
- 28. Erickson HP (2009) Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol Proced Online 11:32–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schoenherr CJ, Paquette AJ, Anderson DJ (1996) Identification of potential target genes for the neuron‐restrictive silencer factor. Proc Natl Acad Sci U S A 93:9881–9886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang Y, Greene E, Murray Stewart T, Goodwin AC, Baylin SB, Woster PM, Casero RA Jr. (2007) Inhibition of lysine‐specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc Natl Acad Sci U S A 104:8023–8028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fiskus W, Sharma S, Shah B, Portier BP, Devaraj SG, Liu K, Iyer SP, Bearss D, Bhalla KN (2014) Highly effective combination of LSD1 (KDM1A) antagonist and pan‐histone deacetylase inhibitor against human AML cells. Leukemia 28:2155–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hu E, Dul E, Sung CM, Chen Z, Kirkpatrick R, Zhang GF, Johanson K, Liu R, Lago A, Hofmann G, Macarron R, de los Frailes M, Perez P, Krawiec J, Winkler J, Jaye M (2003) Identification of novel isoform‐selective inhibitors within class I histone deacetylases. J Pharmacol Exp Ther 307:720–728. [DOI] [PubMed] [Google Scholar]
- 33. Sakane C, Okitsu T, Wada A, Sagami H, Shidoji Y (2014) Inhibition of lysine‐specific demethylase 1 by the acyclic diterpenoid geranylgeranoic acid and its derivatives. Biochem Biophys Res Commun 444:24–29. [DOI] [PubMed] [Google Scholar]
- 34. Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, Schlegl J, Abraham Y, Becher I, Bergamini G, Boesche M, Delling M, Dumpelfeld B, Eberhard D, Huthmacher C, Mathieson T, Poeckel D, Reader V, Strunk K, Sweetman G, Kruse U, Neubauer G, Ramsden NG, Drewes G (2011) Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol 29:255–265. [DOI] [PubMed] [Google Scholar]
- 35. Park SH, Raines RT (2000) Genetic selection for dissociative inhibitors of designated protein‐protein interactions. Nat Biotechnol 18:847–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Patgiri A, Yadav KK, Arora PS, Bar‐Sagi D (2011) An orthosteric inhibitor of the Ras‐Sos interaction. Nat Chem Biol 7:585–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maes T, Carceller E, Salas J, Ortega A, Buesa C (2015) Advances in the development of histone lysine demethylase inhibitors. Curr Opin Pharmacol 23:52–60. [DOI] [PubMed] [Google Scholar]
- 38. Zhijun H, Shusheng W, Han M, Jianping L, Li‐Sen Q, Dechun L (2016) Pre‐clinical characterization of 4SC‐202, a novel class I HDAC inhibitor, against colorectal cancer cells. Tumour Biol 37:10257–10267. [DOI] [PubMed] [Google Scholar]
- 39. Fu M, Wan F, Li Z, Zhang F (2016) 4SC‐202 activates ASK1‐dependent mitochondrial apoptosis pathway to inhibit hepatocellular carcinoma cells. Biochem Biophys Res Commun 471:267–273. [DOI] [PubMed] [Google Scholar]
- 40. Pinkerneil M, Hoffmann MJ, Kohlhof H, Schulz WA, Niegisch G (2016) Evaluation of the Therapeutic Potential of the Novel Isotype Specific HDAC Inhibitor 4SC‐202 in Urothelial Carcinoma Cell Lines. Target Oncol 11:783–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sander B, Golas MM. Molecular Electron Microscopy in Neuroscience: An Approach to Study Macromolecular Assemblies In: Van Bockstaele E, Ed. (2016) Transmission Electron Microscopy Methods for Understanding the Brain. New York: Springer, 115, pp. 205–216. [Google Scholar]
- 42. Lin TY, Voronovsky A, Raabe M, Urlaub H, Sander B, Golas MM (2015) Dual tagging as an approach to isolate endogenous chromatin remodeling complexes from Saccharomyces cerevisiae. Biochim Biophys Acta 1854:198–208. [DOI] [PubMed] [Google Scholar]
- 43. Nott A, Watson PM, Robinson JD, Crepaldi L, Riccio A (2008) S‐Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature 455:411–415. [DOI] [PubMed] [Google Scholar]
- 44. Yuan J, Llamas N, Sander B, Golas MM (in revision) Synergistic anti‐cancer effects of epigenetic drugs on medulloblastoma cells. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information