

Abstract
Synthetic binding proteins are constructed using nonantibody molecular scaffolds. Over the last two decades, in‐depth structural and functional analyses of synthetic binding proteins have improved combinatorial library designs and selection strategies, which have resulted in potent platforms that consistently generate binding proteins to diverse targets with affinity and specificity that rival those of antibodies. Favorable attributes of synthetic binding proteins, such as small size, freedom from disulfide bond formation and ease of making fusion proteins, have enabled their unique applications in protein science, cell biology and beyond. Here, we review recent studies that illustrate how synthetic binding proteins are powerful probes that can directly link structure and function, often leading to new mechanistic insights. We propose that synthetic proteins will become powerful standard tools in diverse areas of protein science, biotechnology and medicine.
Keywords: protein engineering, protein–protein interaction, directed evolution, structure‐function relationship, biologic therapeutics
Introduction
Synthetic binding proteins are human‐made proteins that have been tailored to bind to a target molecule of interest. The capability of the immune system to generate antibodies that can bind virtually any antigens and the knowledge of the molecular mechanisms underlying this capability have inspired the genesis and subsequent development of the field of the design and engineering of synthetic binding proteins. Analogous to how natural antibodies for diverse antigens are made by altering portions of the immunoglobulin molecule, synthetic binding proteins are most commonly generated by altering portions of a functionally inert protein, referred to as a protein scaffold [Fig. 1(A)]. Synthetic binding protein systems are developed with the ultimate aim of generating binding proteins to diverse target molecules, rather than binding proteins to one specific target. These proteins are synthetic in that they have not been found in nature, although they are polypeptides consisting of natural amino acids and made using natural machinery for protein synthesis.
Figure 1.
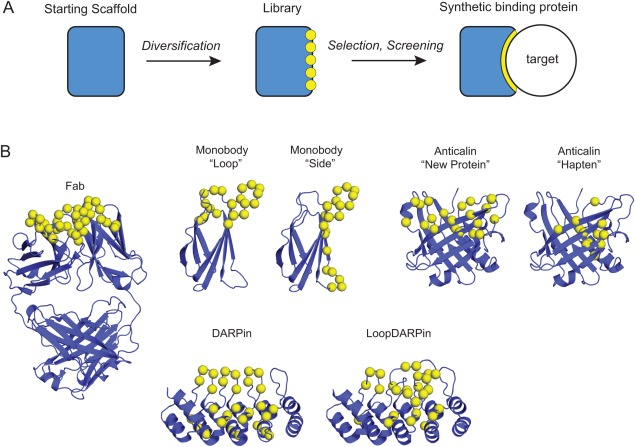
Generation of synthetic binding proteins using molecular scaffolds. (A) A schematic representation of the processes for generating synthetic binding proteins. The rectangle at the left indicates an inert scaffold. The yellow circles denote sequence diversity at chosen positions. The right is a binding protein‐target complex with the optimized interface shown in yellow. (B) The three‐dimensional structures of representative scaffold architectures, including the natural antibody for which only the Fab portion is shown. Only those scaffolds for which structure‐guided design of libraries have led to improved performance are shown. References are given in the main text.
As described in the following section, the challenge of generating a highly functional molecular recognition interface using a protein scaffold is essentially solved. Attention is now turning to whether these synthetic binding proteins can expand the scope of basic research and drug discovery beyond those enabled with conventional antibodies. One of the main motivations behind the continued development of synthetic binding proteins remains their therapeutic applications. Indeed, synthetic binding proteins are making steady progress in this area.1, 2, 3, 4, 5, 6, 7, 8 However, in this review in Protein Science, we will focus on applications of synthetic binding proteins to mechanistic studies of proteins in biochemical and cellular contexts. We will also emphasize examples with Monobodies, because there have been recent comprehensive review articles on other well‐established synthetic binding protein systems, i.e., Anticalin,9 Affibody,10 and DARPin,4 and also on Nanobody,11, 12 a natural single‐domain antibody system that shares many characteristics of synthetic binding proteins.
Generation of Synthetic Binding Proteins
Synthetic binding proteins are usually generated by introducing multiple mutations, typically 10‐20, in a protein scaffold [Fig. 1(A)]. Directed evolution approaches, in particular those utilizing molecular display technologies, enables one to efficiently generate a vast ensemble (“library”) of mutants and identify clones that bind to the target molecule of interest with high affinity. The starting scaffold systems are usually chosen with the hope of generating synthetic binding proteins with desirable functional and biophysical properties, including the ability to generate high‐performance molecular recognition interfaces for diverse targets, small size, high stability, ease of production and ease of use as a building block in fusion proteins. A number of successful platforms have been developed, and the reader is referred to many extensive reviews on this topic including additional scaffold systems and molecular display technologies.8, 13, 14, 15, 16, 17, 18, 19
Choosing an appropriate starting scaffold is an important step, but it is equally important to carefully choose how portions of the scaffold are diversified in a combinatorial library. Many practitioners in the field originally thought that, given the capacity of molecular display methods to test billions of sequences, it should be straightforward to produce high‐performance binding proteins by introducing amino acid diversity using a random mixture of all possible codons, such as NNN and NNK where N is a mixture of A, T, G and C and K is a mixture of T and G, at casually chosen positions. However, they quickly found that this was not the case. Binding proteins generated from such libraries often had low affinity and low specificity. A major breakthrough came from the work of Sidhu and colleagues on synthetic antibody libraries.20, 21, 22, 23, 24 They established that the utilization of a highly biased distribution of amino acids (with particular enrichment of Tyr) in synthetic libraries is highly effective in generating potent and specific antibodies. Parallel studies demonstrated that the equivalent approach is effective even in a much smaller synthetic scaffold, Monobody (see below). The reader is referred to reviews dedicated to this topic.25, 26
Among synthetic binding protein platforms, the most established systems include Affibodies, Anticalins, Monobodies and DARPins [Fig. 1(B)]. Affibodies are based on the Z domain of protein A from Staphylococcus aureus. They contain three α‐helices, no disulfides, and are among the smallest synthetic binders (∼6 kDa) that have been well characterized.27, 28, 29, 30 Anticalins, based on lipocalins, have a β‐barrel architecture with an attached α‐helix. While some lipocalins do contain disulfides, they are chosen due to their natural ability to bind to small molecules using their barrel and loops, and this mode of binding has been exploited for Anticalin libraries.31, 32, 33, 34 Monobodies are based on the fibronectin type III (FN3) domain that has an immunoglobulin fold, but no disulfide bonds.35 Following successes of Monobodies and their equivalence in the industry, Adnectins, several “Monobody mimics” have been successfully developed,5, 36 demonstrating the robustness of the FN3 scaffold for generating synthetic binding proteins. Designed ankyrin‐repeat proteins (DARPins) exploit repetitive structural units to form an extended binding surface.37 DARPins also lack disulfide bonds yet exhibit high thermodynamic stability.38, 39 Although these platforms are based on proteins with distinct folds, they have all produced high‐performance synthetic binding proteins against diverse targets. These numerous successes clearly show that the synthetic binding protein field has collectively established sufficient knowledge and technologies for developing an effective scaffold system.
Ubiquitin is a particularly noteworthy addition. Ubiquitin is a 76‐residue protein involved in many intracellular regulatory processes. Many enzymes involved in ubiquitin‐dependent pathways bind to ubiquitin with weak affinity. Combinatorial phage‐display libraries of ubiquitin have been developed from which ubiquitin variants were identified that have high affinity (K D in the 1–100 nM range) and are specific to a particular ubiquitin‐interacting protein.40 These results demonstrate that a promiscuous, low‐affinity binding protein can be evolved into a highly potent and selective one, in analogy to antibody maturation; and that more generally, the affinity and specificity of natural protein‐protein interactions are tuned for biological functions and can readily be re‐tuned for other purposes. Ubiquitin‐based binding proteins were also generated for the extradomain B of fibronectin, a protein that is not known to interact with ubiquitin.41 Similarly the use of the aforementioned ubiquilin libraries has been extended for generating binding proteins outside ubiquitin‐interacting proteins.42 However, it remains to be seen whether ubiquitin‐based single‐domain binding proteins for general purposes can achieve high potency comparable to the most advanced platforms, as their efficacy still seems quite low: concatenation of two ubiquitin units was required (and further dimerization leading to a total of four units in the latter case) in order to efficiently capture the endogenous target.
The developer of a scaffold system usually designs a combinatorial library with a particular mode of interaction in mind. For example, the original libraries for the Monobody system introduced amino acid diversity in loops located at one end of the molecule, a design that closely mimics the locations of diversified positions in natural immunoglobulins [Fig. 1(B)].35 Structural analyses of Monobody‐target complexes revealed that in addition to the intended mode of target interaction mediated by the diversified loops, a distinct mode was observed in which (unmutated) residues on the β‐sheet surface (“side” of the scaffold when we place the diversified loops at the “top”) contributed to target recognition. Inspired by this observation, a new library was constructed in which residues on a β‐sheet were diversified [Fig. 1(B)].43 Monobodies from the new “side” library presented a concave surface for recognition, as opposed to convex surfaces typically found for Monobodies from the original “loop” libraries, therefore expanding the diversity of binding site topography. As intended by the designs, these two distinct libraries show preferences toward differently shaped surfaces. The loop library tends to prefer binding into a concave epitope, whereas the side library prefers a flatter surface. For example, in an unbiased library selection experiment against the Abl SH2 domain, that is, a selection that did not involve a step that steer binders to a specific epitope, a dominant Monobody clone from the loop library bound to the concave, peptide‐binding groove, whereas a dominant clone from the side library bound to a flat surface on the opposite side of the SH2 domain.44, 45 A similar library design has been reported for another FN3‐based system, Centyrin, although no information was available for the epitopes of resulting molecules.5 These results illustrate the possibility of expanding the efficacy of a scaffold system by the use of distinct surfaces for presenting amino acid diversity and thereby expanding the types of epitopes that can be effectively recognized.
Conceptually similar to the Monobody libraries but a development in the opposite direction, a new library for the DARPin system was developed that also expanded binding site topography. The original design of DARPin libraries diversified positions primarily on α‐helices, presenting a concave surface. The new “LoopDARPin” library introduced extensive diversity in loops that line one edge of the scaffold [Fig. 1(B)].46 This design created protruding loops thereby complementing the original library design. Remarkably, high‐affinity DARPins were identified from the new library after a single round of selection, suggesting the potency of the library.
Structural analysis of Anticalins has also led to a second‐generation library in which positions for presenting amino acid diversity have been fine tuned for targeting large antigens such as proteins [Fig. 1(B)],47 supporting the effectiveness of structure‐guided improvement of combinatorial libraries.
The examples described above clearly illustrate that the field of protein engineering and design collectively has the knowledge and technical expertise that are sufficient for generating synthetic binding proteins using a protein scaffold. As already stated several years ago,14 the breadth and effectiveness of available scaffold systems suggest that establishing another molecular scaffold system will be an exercise of diminishing returns, unless it offers a clear advantage over existing ones.
Expanding Structural Biology
The use of antibody fragments as crystallization chaperones has made important contributions to the successes of challenging structural biology projects. Crystallization chaperones can increase the likelihood of producing macromolecular crystals suitable for diffraction studies through several potential mechanisms including (a) reducing the fraction of disordered regions (b) reducing the conformational heterogeneity and (c) providing surfaces that are conducive to forming crystal contacts.48, 49 Although antibody fragments such as Fab and Fv are still the most common crystallization chaperones, the ability to produce large quantities of stable, high‐affinity binding proteins in E. coli has made synthetic binding proteins attractive alternatives. Unlike Fab that exhibits substantial hinge bending motions between the variable and constant domains,50 synthetic binding proteins and also Nanobodies (single‐domain antibody fragments derived from the camelid heavy chain‐only antibodies) are single‐domain proteins and thus do not have such internal flexibility. This attribute seems to contribute to the ability of these single‐domain chaperones to help produce higher‐resolution structures. In the recent structure of the extracellular region of an adhesion GPCR, GPR56/ADGRG1, a Monobody simultaneously interacts with two domains of GPR56 via two separate regions on its opposite ends, presenting yet another way to reduce the inter‐domain motions.51 A combination of a Monobody chaperone and linking of heterodimer into a single‐chain construct was used to determine the structure of an otherwise ill‐behaving Prdm14‐Mtgr1 complex.52 Furthermore, their small sizes may be important for crystallizing integral membrane proteins using the lipid cubic phase method, because of the limited size of cavities that can accommodate water‐exposed portions of the protein system, i.e., the water‐exposed portion of the target protein plus the chaperone.53, 54, 55 Additional examples are discussed in a recent review and references therein.12, 56
Synthetic Binding Proteins, Particularly Monobodies, Target a Functional Site
Although these synthetic binding protein systems have been developed originally for the purpose of generating simple affinity reagents, ensuing research has revealed that many of them, particularly Monobodies, have a strong tendency to bind to a functional surface on the target molecule. This attribute makes them modulators of biological functions. Combined with high specificity, high affinity, simple design and ability to function regardless of redox potential of the environment, Monobodies offer unique capabilities beyond “just” affinity reagents. In the following section, we will review examples that illustrate this capability that have contributed to advancing mechanistic understanding.
In a typical project of synthetic binding protein generation, many clones are available at the end of the selection campaign, and the “best” clones among the candidates are chosen based on their affinity, specificity and amino acid sequences. However, these clones are chosen without the knowledge of where within the target molecule they bind (epitope). Although it is technically straightforward to direct binding proteins to a specific surface, such an approach is taken only in a project that starts with a detailed mechanistic understanding of the target molecule and clear descriptions of the desired properties of binding proteins. Despite this unbiased selection in terms of epitopes, synthetic binding proteins, particularly Monobodies, are found to bind to a functional site within the target molecule (Fig. 2).
Figure 2.
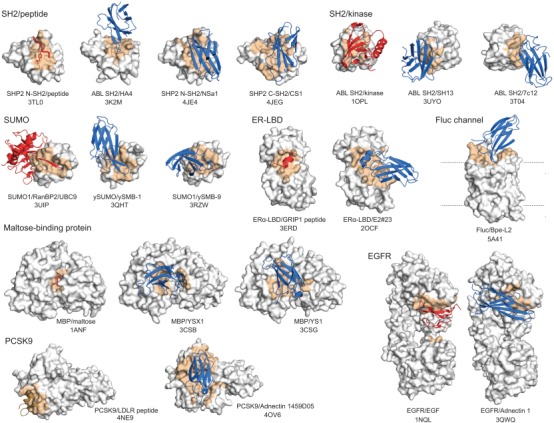
Examples of Monobodies and Adnectins binding to a functional site within the target protein. The target proteins are shown in gray with the epitope in orange. Natural ligands are in red, and Monobodies and Adnectins in blue. The identities of the target molecules and PDB entry codes are indicated. For the Fluc channel structure, the natural ligand, F– ion, is not shown because of its small size.
The strong tendency of binding to a functional site was first observed for the VHH/Nanobodies, and it was rationalized based on the geometric matching between the generally concave surfaces of protein functional sites and the compact prolate shape of the target‐recognition surface presented by the VHH scaffold.57 This mechanism of action seems to explain a number of cases for Monobodies that are structurally similar to VHH/Nanobody and often bind to a concave cleft (Fig. 3). However, as discussed below, recent examples show that Monobodies may also preferentially bind to a functional surface that is not strongly concave.
Figure 3.
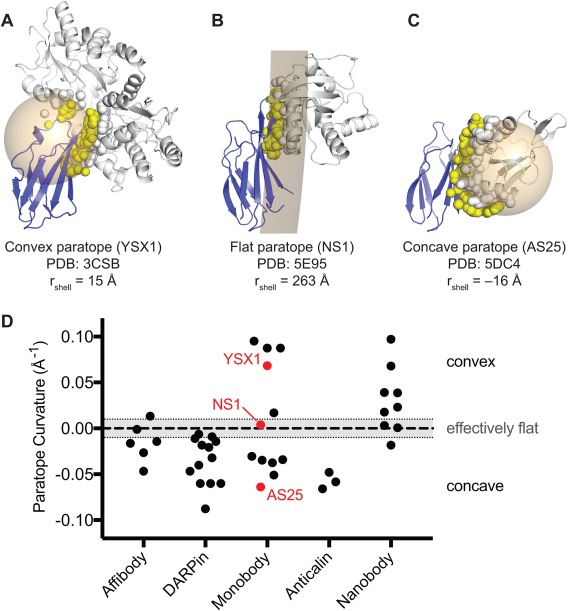
Concavity analysis of binding protein‐target interfaces. (A–C) Three representative structures of Monobody‐target complexes with different levels of concavity. For each crystal structure, a spherical shell (tan) was fit to all the atoms that compose the target‐contacting residues on the Monobody (i.e., the paratope; yellow spheres). A spherical shell with a large radius approximates a flat interaction. Spherical shells with smaller radii, centered within the Monobody or target represent convex or concave paratopes, respectively. To distinguish between the two orientations, the radii of shells corresponding to concave paratopes were assigned negative values. Monobody and target structures are shown as blue and gray cartoons, respectively. Atoms composing the Monobody‐contacting residues on the target (i.e., the epitope) are shown as gray spheres. (D) Concavity analysis on 34 synthetic binding protein‐target complex structures from the PDB. Nanobody complexes are also included for comparison. Curvature is defined as the inverse of the radius of the spherical shell as described above. An arbitrary threshold of |rshell| ≥ 100Å (|curvature| ≤ 0.01 Å−1) was defined as an effectively flat interface.
A Monobody, YSX1, derived from a loop library bound to a concave surface around the sugar‐binding cleft of maltose‐binding protein [Figs. 2 and 3(A)].58 Similarly, Monobody HA4 bound to the peptide‐binding cleft of the Abl SH2 domain.44 Although this epitope is convex, the observed binding mode can be rationalized by the fact that the Monobody mimics the natural peptide ligand. In contrast, Monobody AS25 derived from the side library bound to a convex surface on the opposite side of the SH2 domain that is used for intramolecular interaction with the kinase domain of Abl [Fig. 3(C)].45 This surface does not have a cleft for peptide binding, and the Monobody does not mimic the binding mode of the kinase domain. In another example, Monobody NS1, also generated in an unbiased manner from the side library, was bound to a nearly flat surface of H‐RAS that is involved in dimerization (see below for additional information about this interface) [Fig. 3(B)].59 These cases clearly show that the preference toward a functional site is not only due to the geometric matching between a functional cleft and a small globular binding protein. However, we note that the geometric matching is an important factor in the ability of these Monobodies presenting a concave binding surface to bind to a convex or flat surface of their target.
Then, what is the molecular basis of the strong preference of these small binding proteins toward a functional surface? Although the paucity of binding proteins directed to a clearly nonfunctional surface makes it impossible to elucidate the basis, we speculate that the key is the surface characteristic inherent to natural proteins. It is well established that functional surfaces of natural proteins are enriched with amino acids that are conducive to forming interactions such as Tyr, Trp, and Arg, whereas nonfunctional surfaces contain higher fractions of amino acids that tend to break interactions such as Glu and Lys.25, 60 Because synthetic binding proteins are generated in a short period under strong selection pressure for high affinity, it is not difficult to imagine that this approach should enrich clones that bind to surfaces that are more conducive to forming high‐affinity interactions, as opposed to other surfaces that have not evolved to interact with other molecules. Although Nanobodies are not fully synthetic, they are also generated in a short period under strong selection pressure of animal immunization and phage‐display selection. Thus, the Nanobody generation processes should also enrich those clones that bind to target surfaces conducive to forming interactions. Therefore, although shape complementarity is an important factor in epitope selection, the dominant determinant appears to be the surface chemical properties of natural proteins. The strong preference of Nanobodies toward concave surfaces may be due to the fact that the natural immune repertoires of Nanobodies produce mostly convex antigen‐binding site [Fig. 3(D)]. This notion in turn suggests the exciting possibility of controlling virtually all types of protein functions by utilizing synthetic binding proteins capable of presenting target‐binding site with diverse topography.
Advancing Mechanistic Understanding Underlying Molecular Recognition
Because synthetic binding proteins by definition bind to a particular target molecule, the structure of the complex of a target with a synthetic binding protein provides a direct approach to analyze and understand how molecular recognition is achieved. As described above, synthetic binding proteins often bind a functional site in the target protein, which provides opportunities to observe modes of molecular recognition for a natural functional site beyond those observed with natural ligands. Comparisons of interactions with natural ligands to those with synthetic binding proteins expand a mechanistic understanding of the properties of a functional site and reveals alternative strategies that the researcher can exploit to engineer interactions. Such knowledge deepens our understanding of the general principles governing molecular recognition and in return is useful for further improving the design of synthetic binding proteins. A more thorough review addressing the engineering aspects of molecular recognition is found elsewhere.26 Here, we review examples that have shed light on aspects of molecular recognition in specific biological systems.
Unsurprisingly, synthetic binders often closely mimic a natural ligand, particularly when the targeted site is involved in protein‐protein interaction. Naturally, scaffolds that use a flexible loop for presenting amino acid diversity, such as Monobody, are more capable of forming diverse protein backbone conformations than rigid scaffolds. For example, Monobodies that bind yeast SUMO closely mimic the interaction mode of natural peptides called SUMO‐interacting motifs (SIMs) (Fig. 2).61 Both Monobody ySMB‐1 and SIMs present a β‐strand that docks on and extends the anti‐parallel β‐sheet of SUMO. ySMB‐1 also mimics the chemistry of SIMs in that it presents a β‐strand that is predominantly hydrophobic and flanked with acidic residues. Monobody E2#23 targeting the estrogen receptor ligand‐binding domain presents a short helix that mimics the natural LXXLL motif (Fig. 2; PDB ID 2OCF).62 Monobody HA4 mimics the conformation of phosphotyrosine (pY)‐containing peptides.44 Even though HA4 does not contain pY, HA4 inserts a tyrosine into the pY‐binding pocket of the SH2 and, along with an inorganic phosphate molecule, mimics the pY moiety. Perhaps the most intriguing example is the mimicry of sugar hydroxyl positions by Tyr hydroxyls of a Monobody bound to maltose‐binding protein.63
A DARPin binding to caspase‐3 presents an interesting example of molecular recognition.64 Caspase‐3 contains four substrate‐binding pockets (S4‐S1) that accommodate the four‐residue recognition motif DEVD, respectively, running N‐ to C‐terminus [Fig. 4(A)]. XIAP is a natural inhibitor of the caspase‐3 binding site, however, XIAP presents a peptide fragment that runs in the opposite orientation of DEVD. In the XIAP/caspase‐3 complex, S4 is occupied by D148 of XIAP, similar in DEVD, but S3 is empty. XIAP presents a valine (V146) oriented towards the S2 pocket, but the pocket is shielded by Y204 of caspase‐3 to form van der Waal contacts with V146. Consequently, XIAP is angled away from S1 and solvent occupies this pocket. The DARPin inhibitor binding to the substrate‐binding pocket64 follows a similar molecular recognition pattern as XIAP: S4 occupied by aspartate, S3 empty, S2 shielded by Y204, S1 occupied by solvent. However, the DARPin achieves this by presenting two loops into the binding site that is opposite in orientation as XIAP. Thus, this DARPin is able to mimic a natural mode of side chain recognition using a completely different backbone motif.
Figure 4.
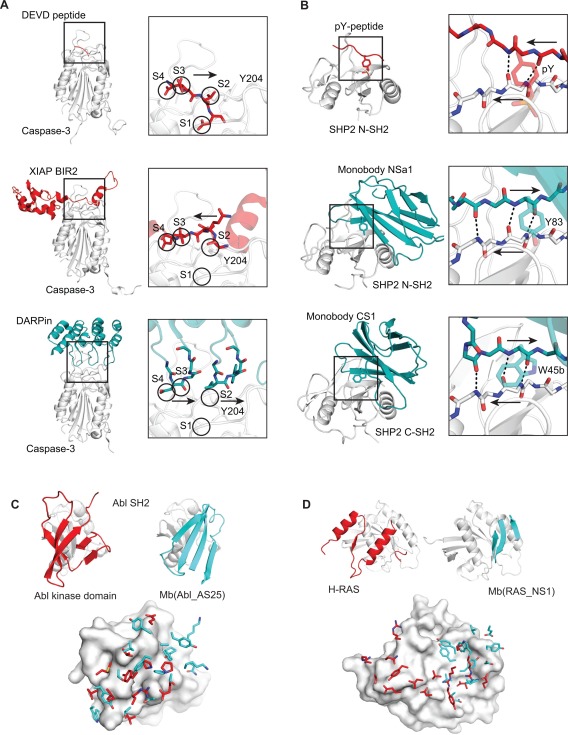
Synthetic binding proteins identify novel modes of protein‐protein interactions. Comparisons of the binding mode of a natural ligand (red) and that of a synthetic binding protein (blue) for Caspase‐3 (A), SHP2 SH2 domain (B), Abl SH2 (C) and H‐RAS (D) are shown. For C and D, the side chains of the ligand and synthetic binding protein located within 4.5 Å of the target protein are shown in the lower panels.
The above examples illustrate how synthetic binding proteins can closely mimic natural ligands, but broader insights can be gained when the modes of interaction do not follow those of natural ligands. SH2 domains recognize pY‐peptides using a conserved binding site that contains a highly‐charged phosphate‐binding pocket and a hydrophobic groove to accommodate residues C‐terminal of pY.65 The peptide, pY‐X‐X‐X, binds the SH2 domain in a canonical orientation that lies perpendicular to the central β‐sheet of the SH2 domain [Fig. 4(B)]. The aforementioned Monobody, HA4, closely mimics this mode. Interestingly, Monobodies generated towards the N‐ and C‐SH2 domains of the SHP2 phosphatase presented peptide fragments that run in the opposite direction.66 Thus, the Monobodies seem to have overcome restrictions that nature has imposed on natural ligands for binding to this particular site. These Monobodies do not contain a pY or a pY mimic, thus they cannot take advantage of the significant binding energy that derives from phosphate binding. Instead, the two Monobodies partially insert a Tyr or Trp residue into the pY‐binding pocket and these residues adopt energetically unfavorable side‐chain conformers, suggesting that these interactions are suboptimal at best. To compensate these interactions, these Monobodies present an extended segment antiparallel to a β‐strand of SH2 and extend the central β‐sheet. These β‐strands run in the opposite orientation to that of the canonical pY‐X‐X‐X peptide but enable the formation of more backbone H‐bonds. Additional interactions between this Monobody and SH2 domain further strengthen this binding mode.
In contrast to the above examples of Monobodies binding to a peptide‐binding site, Monobodies binding to a protein‐protein interaction (PPI) surface revealed distinctly different solutions to molecular recognition from those observed in natural PPIs. Monobody AS25 binds to the surface of the Abl SH2 domain that is involved in interaction with the kinase domain in the full‐length kinase. Although both AS25 and the kinase domain use β‐sheet surfaces for binding to the SH2 domain, the topology and directions of β‐strands are different between the two [Fig. 4(C)]. Furthermore, there is no discernable homology between the amino acid side chains involved in the recognition of the SH2 domain [Fig. 4(C)].
The difference between the natural PPI and a synthetic PPI is even more dramatic in the case of H‐RAS. Monobody NS1 binds to a homodimerization interface of H‐RAS [Fig. 4(D)]. H‐RAS uses primarily two α‐helices for dimerization. In contrast, the Monobody uses a β‐sheet and loops. Although the epitope for the Monobody is essentially a subset of the homodimerization interface, there is no conservation in the side chains used in recognizing the overlapping surfaces between the two structures. In both cases, conformational changes of the targets (SH2 or RAS) were minimal.
The modes of interactions among synthetic binding proteins for the identical target can be vastly different. Anticalins have been developed that bind specifically to the extra‐domain B (ED‐B) of the oncofetal isoform of the extracellular matrix protein fibronectin.47 They all have high affinity, with K D in the low nM range. The crystal structures of these three Anticalins show that, although they recognize largely overlapping epitopes of the ED‐B, they use distinctly different orientations, involving highly individual interfaces and side‐chain arrangements (A. Skerra et al. personal communication). These studies further support the view that there are many ways to recognize a protein surface and that natural PPIs represent a small subset of such possibilities.
Even though the interaction interfaces of synthetic binding proteins and their targets are synthetic and “unnatural,” they by definition fulfill the fundamental principles of molecular recognition by natural polypeptides. Thus, structural analyses of these interfaces expand the collection of productive “binding poses,” deepen our understanding of why natural ligands recognize their targets in a certain manner, and suggest unprecedented ways to create interfaces. Such knowledge greatly benefits our effort toward rational and computational design of molecular recognition surfaces. A clear message from these examples is that there are many ways to recognize the same surface of a protein and that one does not need to, or perhaps should not, try to mimic the binding poses of natural PPIs when one wishes to generate synthetic PPIs that are substantially superior to natural ones in terms of affinity and specificity.
Controlling Conformational Equilibrium Underlying Allostery
Allostery is a common mechanism underlying protein regulation.67, 68 Proteins can exist as an ensemble of different conformational states with different levels of function. Effector molecules preferentially bind to a subset of these states and hence bias the equilibrium, thereby allosterically activating or inhibiting the protein function. Because the binding sites for allosteric effectors are less likely to be conserved than the active site among the members of a protein family, e.g., protein kinases, one may be able to achieve high selectivity toward controlling the protein of interest by targeting an allosteric site. However, allosteric effectors do not exist for many proteins. Because proteins are fundamentally flexible, it is conceivable that one can develop synthetic allosteric effectors in the form of binding proteins, as the feasibility to generate conformation‐specific binding proteins is well established.56, 62, 69 Here, we review examples where synthetic binding proteins have been used to allosterically control protein functions.
A series of Monobodies have been developed that allosterically activate or inhibit ABL kinase and its oncogenic variant, BCR‐ABL. ABL is tightly regulated by its SH2 domain. In the auto‐inhibited state, the SH2 domain binds to the C‐terminal lobe (C‐lobe) of the kinase domain. In the activated state, the SH2 domain sits on the N‐lobe using a different surface. The HA4 Monobody,44 developed primarily as an inhibitor of the SH2‐phosphopeptide interaction, also sterically inhibits the interaction of SH2 with the C‐lobe but not its interaction with the N‐lobe, which relieves auto‐inhibition and thus allosterically activates ABL. Therefore, HA4 stabilizes and traps the active conformation of the wild‐type kinase. Mutational studies had established the importance of the interface between the SH2 domain and the N‐lobe70 but it was not clear whether this interface could be targeted in trans for allosteric regulation. A Monobody, 7c12, binds to this epitope and disrupts SH2/kinase interactions. A construct of 7c12 and HA4 fused in tandem, which reduced BCR‐ABL activity and, when expressed as a genetically encoded reagent, induced apoptosis in a chronic myelogenous leukemia (CML) cell line and in primary cells from CML patients. Recently more potent Monobodies directed to the SH2‐kinase interface have been developed that inhibit BCR‐ABL without the need to be fused with another Monobody [Fig. 5(A)].45
Figure 5.
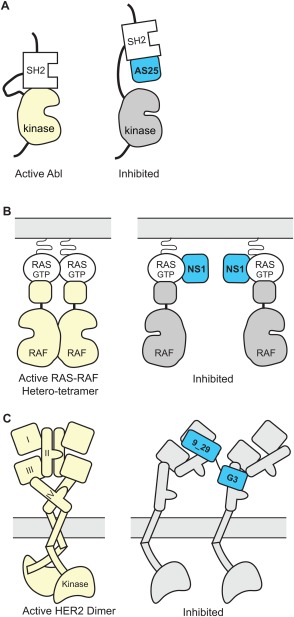
Schematics showing allosteric regulation using synthetic binding proteins. (A) Monobody binding to the SH2 domain of ABL kinase disrupts the intramolecular, domain‐domain interaction, which leads to kinase inhibition.45, 70 (B) Monobody binding to a dimerization interface of RAS disrupts the RAS‐RAF hetero‐trimer and inhibits RAS‐mediated activation of RAF.59 (C) Tandem DARPin disrupts the dimer formation of HER2, thereby inhibiting the kinase activity of HER2. Figure based on Jost et al.71.
The RAS‐binding Monobody, NS1, that was introduced in the preceding section is a potent inhibitor of RAS‐mediated signaling as tested in cell based assays, although it binds to a previously uncharacterized surface that is away from the so‐called switch regions involved in interactions with known effectors.59 As expected from the epitope location, the NS1 Monobody did not inhibit the interaction of RAS with Raf kinase. Instead, it inhibits RAS dimerization and nanoclustering on the membrane surface, which in turn inhibits Raf dimerization, i.e., homo‐dimerization of the RAS‐Raf heterodimers, resulting in the inhibition of RAS‐mediated signaling [Fig. 5(B)]. It is remarkable that this possibility of allosterically regulating RAS was discovered using the NS1 Monobody, although RAS has been intensely studied for several decades.
Allosteric inhibitors of the HER2 receptor have been developed using DARPins. A key event in eliciting HER2 signaling is the formation of proper homodimers or hetero‐dimers with other EGF receptor family members, and disrupting receptor dimerization and signal activation is a major goal of targeted therapies for HER2‐dependent malignancies. A tandem DARPin (termed 9_5_G) has been developed from DARPins 9_29 and G3 that bind to extracellular subdomains I and IV of HER2, respectively.71 This tandem DARPin showed higher cytotoxic effects than either DARPin alone or the monoclonal antibody trastuzumab on a HER2‐dependent cell line. Crystal structures and modeling of the single HER2 subdomain/DARPin complexes and full‐length HER2 extracellular domains ruled out the possibility that the tandem DARPin can bind intramolecularly to the same HER2 monomer. Instead, binding of the tandem DARPin probably forces the receptor into a nonnatural conformation that prohibits the intracellular kinase domains from coming together, thus blocking signaling [Fig. 5(C)]. It is interesting that the bispecific, tandem binding protein achieves conformational control. Because proteins involved in cellular regulation often contain multiple domains, the use of bispecific binding protein targeting different domains may prove to be broadly useful. Synthetic binding proteins are ideal for this approach because of their compact nature and the ease of making bispecific fusion molecules.
It is notable that, in these examples, allosteric regulation is achieved via the modulation of PPIs among modular domains and among protein complexes, rather than the modulation of the conformational equilibrium within a single globular protein. Because the complex behaviors of regulatory proteins are often produced by weak, multivalent interactions among multiple modular interaction domains and their ligands,72, 73 we speculate that synthetic binding proteins directed toward individual components will help the discovery of novel modes of allosteric control of cellular regulation.
Fine‐Tuning Specificity
In addition to the well‐established applications of synthetic binding proteins as affinity capture reagents, inhibitors and activators, they have been used to modulate the specificity of PPI and enzyme substrates, thereby further expanding their utility.
Monobodies have been used as a building block for making “affinity clamps,” a fusion protein of a natural modular recognition domain and a synthetic binding domain.74, 75, 76 In this approach, the Monobody unit is engineered to bind to the complex of a target peptide and the modular recognition domain, rather than the peptide or the recognition domain alone. By “clamping” on the peptide, the fusion protein achieves much higher binding affinity and/or specificity than the starting modular recognition domain.
Monobodies have also been used as a proxy modulator of the substrate specificity of transglycosylation reaction catalyzed by a β‐galactosidase.77 By iteratively generating Monobodies that bind to different sites within β‐galactosidase from Bacillus circulans, Tanaka et al. have identified a Monobody that does not inhibit the catalytic activity but limits the access of longer‐chain oligosaccharides. Consequently, with this proxy Monobody, the enzyme produces only shorter‐chain oligosaccharides instead of an uncontrolled, heterogeneous mixture of oligosaccharides.
Expanding Cell and Chemical Biology
Antibodies have been the dominant reagents for detecting and capturing proteins in cell biology research. However, their structural complexity has limited their utility in intracellular applications. Although it is routine to introduce proteins and their variants as genetically encoded reagents into cells using vector transfection and viral transduction, the same approach usually fails for antibodies and antibody fragments because of the dependence of antibody folding and assembly on disulfide formation, which is inefficient under the reducing environment of the cytoplasm and nucleus. Because an important motivation behind the development of synthetic binding protein platforms is to overcome precisely these unfavorable attributes of conventional antibodies, synthetic binding proteins are naturally suited as genetically encoded reagents for intracellular reagents used in live cells. Coupled with the capacity to generate specific and potent synthetic binding proteins to diverse targets, innovative approaches have been developed that expands the scope of cell biology investigation. Here, we will limit our discussion to those examples that target endogenous proteins. An extensive discussion on the use of binding proteins to tagged molecules, such as GFP fusions, is found in a recent review.12
Validating the specificity of binding proteins is critically important in in‐cell applications, because the high concentrations of numerous molecules inside cells presents a challenging environment for specifically recognizing the target molecule of interest. To properly interpret results of in‐cell monitoring and perturbation (see below), one needs to verify that the ability of a synthetic binding protein to specifically recognize its target in the context of the cell used for the actual application. Intracellular expression of Monobodies fused to epitope tags followed by affinity capture and mass spectroscopy‐based proteomic analysis has been used to validate the specificity of these molecules. The beauty of this approach is that one can use the same experimental system for both specificity validation and biological characterization.44, 52, 66 Whereas most of the Monobodies tested selectively capture their respective targets (e.g., only the targeted SH2 domain among ∼100 SH2 containing proteins), one Monobody developed for the SH2 domain of SHP2 exhibited substantial binding to a metabolic enzyme, an unanticipated off‐target.66 This example illustrates the importance of unbiased assessment of binding protein specificity, in addition to testing cross‐reactivity against a panel of expected off‐targets such as the closest homologs of the intended target.
Synthetic binding proteins can serve as a building block of genetically encoded tools for in‐cell imaging of endogenous proteins. Gross et al. developed a FingR (synonym for Monobody) fused to GFP that localized to endogenous PSD‐95 in neuronal postsynapses.78 Prior studies had revealed overexpression of PSD‐95 fused to fluorescent protein tags resulted in altered morphology of dendritic spines and synaptic behavior, indicating the system is sensitive to perturbation. The authors implemented transcriptional control to prevent significant perturbation of endogenous PSD‐95 by the FingR, as judged by cell morphology and behavior, and minimize background fluorescence. Synthetic binding proteins are therefore powerful tools for monitoring the localization of endogenous proteins in live cells, but one must exercise caution in minimizing perturbation because these reagents are likely to bind to a functional site as discussed earlier.
Genetic modulation techniques of intracellular proteins include gene knock down (RNA interference), gene knock out and knock in of mutations. Knock down and knock out deplete the entire protein molecule and thus they cannot address questions at a finer resolution at, for example the level of individual domains. Mutations often require multiple preparation steps each involving culturing cells for several generations during which cells can make compensatory changes to genetic modifications. Acute production of synthetic binding proteins in cells overcomes these limitations, although we acknowledge that the technology needs further improvements. Recent studies, in addition to those discussed under the section entitled Allostery,44, 45, 70 illustrate the effectiveness of this approach.
Two DARPins have been generated that bind to essentially the same epitope of the ERK2 kinase but recognize distinct conformational states (unphosphorylated versus phosphorylated).79 One DARPin inhibited ERK phosphorylation in COS7 cells whereas the other acted as an intracellular sensor of ERK2 activation after being stimulated or inhibited in response to a signal. In addition, DARPins have been generated that discriminate closely related isoforms of c‐Jun N‐terminal kinases, JNK1 and JNK2, and inhibited JNK activation in mammalian cells.80
The RAS‐binding Monobody described above exhibited its inhibitory function in cell‐based assays but not in biochemical assays using purified components.59 This example demonstrates that synthetic binding proteins with high potency and high specificity are powerful tools for discovering a novel mechanism of controlling cellular proteins.
SHP2 is a protein tyrosine‐phosphatase and a signaling adaptor that plays an important role in normal development and in oncogenesis. SHP2 contains two SH2 domains, termed N‐SH2 and C‐SH2, both of which are capable of binding to pY‐containing peptide motifs. Monobodies have been generated that inhibit each SH2 domain of SHP2.66 Inhibition of either SH2 domain by the Monobodies stopped phosphorylation events on SHP2, however only inhibition of the N‐SH2 but not the C‐SH2 significantly reduced SHP2‐GAB2 interactions. Furthermore, inhibition of either SH2 domain almost completely blocked downstream ERK activation in a lung cancer cell model.
A Monobody binding to the pre‐SET/SET domain of a transcriptional regulator, human Prdm14 (different from the crystallization chaperone described earlier) inhibits its interaction with a co‐repressor Mtgr1.52 When expressed in the nucleus of embryonic stem cell under an inducible promoter, it inhibited the progression to primordial germ cells.
These studies have convincingly demonstrated that synthetic binding proteins are powerful tools for dissecting the function of intracellular proteins at the level of individual domains without introducing perturbations to the endogenous proteins. Such knowledge is invaluable for identifying and validating potential drug targets. Thus, we propose that synthetic binding proteins will find broad utility in chemical biology and drug development outside the biologic therapeutics.
The strong tendency of Monobodies to bind to a functional site within the target protein should help discover previously unidentified functional sites. For example, the previously described GPR56‐targeted Monobody binds via two loops that each inserts a Trp residue to a cleft in the PLL and GAIN domains, respectively, and allosterically inhibits the receptor.51 These sites may be binding sites for endogenous ligands, although such molecules have not yet been found.
Conclusions
Synthetic binding proteins represent powerful tools for controlling protein functions and interactions in and outside the cell. Their ability to selectively stabilize a particular conformational state and accelerate structure determination of the captured conformation of a protein makes these reagents particularly powerful tools for bridging functional investigation and structural investigation, a challenging gap for conventional research approaches. Therefore, we envision that we will witness increasing cases of integrated protein science using synthetic binding proteins.
Conflict of Interest
S.K. is an inventor of patents and patent applications on certain technologies discussed in this article.
Acknowledgments
We thank Dr. Akiko Koide for helpful comments on the manuscript. This work was supported in part by the National Institutes of Health grants R01DA036887, R01CA194864 and U01MH109102 (S.K.) and by F30GM116455 (G.S.S.). We acknowledge that due to the rapid growth of this field in recent years and the limited space we are unable to cite all relevant literature.
References
- 1. Bloom L, Calabro V (2009) FN3: a new protein scaffold reaches the clinic. Drug Discov Today 14:949–955. [DOI] [PubMed] [Google Scholar]
- 2. Stumpp MT, Binz HK, Amstutz P (2008) DARPins: a new generation of protein therapeutics. Drug Discov Today 13:695–701. [DOI] [PubMed] [Google Scholar]
- 3. Wurch T, Pierre A, Depil S (2012) Novel protein scaffolds as emerging therapeutic proteins: from discovery to clinical proof‐of‐concept. Trends Biotechnol 30:575–582. [DOI] [PubMed] [Google Scholar]
- 4. Pluckthun A (2015) Designed ankyrin repeat proteins (DARPins): binding proteins for research, diagnostics, and therapy. Annu Rev Pharmacol Toxicol 55:489–511. [DOI] [PubMed] [Google Scholar]
- 5. Diem MD, Hyun L, Yi F, Hippensteel R, Kuhar E, Lowenstein C, Swift EJ, O'Neil KT, Jacobs SA (2014) Selection of high‐affinity Centyrin FN3 domains from a simple library diversified at a combination of strand and loop positions. Protein Eng Des Sel 27:419–429. [DOI] [PubMed] [Google Scholar]
- 6. Sachdev E, Gong J, Rimel B, Mita M (2015) Adnectin‐targeted inhibitors: rationale and results. Curr Oncol Rep 17:35. [DOI] [PubMed] [Google Scholar]
- 7. Gille H, Hulsmeyer M, Trentmann S, Matschiner G, Christian HJ, Meyer T, Amirkhosravi A, Audoly LP, Hohlbaum AM, Skerra A (2016) Functional characterization of a VEGF‐A‐targeting Anticalin, prototype of a novel therapeutic human protein class. Angiogenesis 19:79–94. [DOI] [PubMed] [Google Scholar]
- 8. Vazquez‐Lombardi R, Phan TG, Zimmermann C, Lowe D, Jermutus L, Christ D (2015) Challenges and opportunities for non‐antibody scaffold drugs. Drug Discov Today 20:1271–1283. [DOI] [PubMed] [Google Scholar]
- 9. Richter A, Eggenstein E, Skerra A (2014) Anticalins: exploiting a non‐Ig scaffold with hypervariable loops for the engineering of binding proteins. FEBS Lett 588:213–218. [DOI] [PubMed] [Google Scholar]
- 10. Feldwisch J, Tolmachev V (2012) Engineering of affibody molecules for therapy and diagnostics. Methods Mol Biol 899:103–126. [DOI] [PubMed] [Google Scholar]
- 11. Muyldermans S (2013) Nanobodies: natural single‐domain antibodies. Annu Rev Biochem 82:775–797. [DOI] [PubMed] [Google Scholar]
- 12. Helma J, Cardoso MC, Muyldermans S, Leonhardt H (2015) Nanobodies and recombinant binders in cell biology. J Cell Biol 209:633–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Skerra A (2007) Alternative non‐antibody scaffolds for molecular recognition. Curr Opin Biotechnol 18:295–304. [DOI] [PubMed] [Google Scholar]
- 14. Koide S. Design and engineering of synthetic binding proteins using nonantibody scaffolds In: Park SJ, Cochran JR, Eds. (2010) Protein Engineering and Design. Boca Raton, FL: CRC Press; pp. 109–130. [Google Scholar]
- 15. Sidhu SS, Koide S (2007) Phage display for engineering and analyzing protein interaction interfaces. Curr Opin Struct Biol 17:481–487. [DOI] [PubMed] [Google Scholar]
- 16. Gai SA, Wittrup KD (2007) Yeast surface display for protein engineering and characterization. Curr Opin Struct Biol 17:467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lipovsek D, Pluckthun A (2004) In‐vitro protein evolution by ribosome display and mRNA display. J Immunol Methods 290:51–67. [DOI] [PubMed] [Google Scholar]
- 18. Jost C, Pluckthun A (2014) Engineered proteins with desired specificity: DARPins, other alternative scaffolds and bispecific IgGs. Curr Opin Struct Biol 27:102–112. [DOI] [PubMed] [Google Scholar]
- 19. Park SJ, Cochran JR (2010) Protein engineering and design. Boca Raton, FL: CRC Press. [Google Scholar]
- 20. Birtalan S, Zhang Y, Fellouse FA, Shao L, Schaefer G, Sidhu SS (2008) The intrinsic contributions of tyrosine, serine, glycine and arginine to the affinity and specificity of antibodies. J Mol Biol 377:1518–1528. [DOI] [PubMed] [Google Scholar]
- 21. Fellouse FA, Barthelemy PA, Kelley RF, Sidhu SS (2006) Tyrosine plays a dominant functional role in the paratope of a synthetic antibody derived from a four amino acid code. J Mol Biol 357:100–114. [DOI] [PubMed] [Google Scholar]
- 22. Fellouse FA, Esaki K, Birtalan S, Raptis D, Cancasci VJ, Koide A, Jhurani P, Vasser M, Wiesmann C, Kossiakoff AA, Koide S, Sidhu SS (2007) High‐throughput generation of synthetic antibodies from highly functional minimalist phage‐displayed libraries. J Mol Biol 373:924–940. [DOI] [PubMed] [Google Scholar]
- 23. Fellouse FA, Li B, Compaan DM, Peden AA, Hymowitz SG, Sidhu SS (2005) Molecular recognition by a binary code. J Mol Biol 348:1153–1162. [DOI] [PubMed] [Google Scholar]
- 24. Lee CV, Liang WC, Dennis MS, Eigenbrot C, Sidhu SS, Fuh G (2004) High‐affinity human antibodies from phage‐displayed synthetic Fab libraries with a single framework scaffold. J Mol Biol 340:1073–1093. [DOI] [PubMed] [Google Scholar]
- 25. Koide S, Sidhu SS (2009) The importance of being tyrosine: lessons in molecular recognition from minimalist synthetic binding proteins. ACS Chem Biol 4:325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gilbreth RN, Koide S (2012) Structural insights for engineering binding proteins based on non‐antibody scaffolds. Curr Opin Struct Biol 22:413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nord K, Nilsson J, Nilsson B, Uhlen M, Nygren PA (1995) A combinatorial library of an alpha‐helical bacterial receptor domain. Prot Eng 8:601–608. [DOI] [PubMed] [Google Scholar]
- 28. Wikman M, Steffen AC, Gunneriusson E, Tolmachev V, Adams GP, Carlsson J, Stahl S (2004) Selection and characterization of HER2/neu‐binding affibody ligands. Protein Eng Des Sel 17:455–462. [DOI] [PubMed] [Google Scholar]
- 29. Sandstrom K, Xu Z, Forsberg G, Nygren PA (2003) Inhibition of the CD28‐CD80 co‐stimulation signal by a CD28‐binding affibody ligand developed by combinatorial protein engineering. Protein Eng 16:691–697. [DOI] [PubMed] [Google Scholar]
- 30. Nord K, Gunneriusson E, Ringdahl J, Stahl S, Uhlen M, Nygren PA (1997) Binding proteins selected from combinatorial libraries of an alpha‐helical bacterial receptor domain. Nat Biotechnol 15:772–777. [DOI] [PubMed] [Google Scholar]
- 31. Skerra A (2000) Lipocalins as a scaffold. Biochim Biophys Acta 1482:337–350. [DOI] [PubMed] [Google Scholar]
- 32. Beste G, Schmidt FS, Stibora T, Skerra A (1999) Small antibody‐like proteins with prescribed ligand specificities derived from the lipocalin fold. Proc Natl Acad Sci USA 96:1898–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mercader JV, Skerra A (2002) Generation of anticalins with specificity for a nonsymmetric phthalic acid ester. Anal Biochem 308:269–277. [DOI] [PubMed] [Google Scholar]
- 34. Schlehuber S, Beste G, Skerra A (2000) A novel type of receptor protein, based on the lipocalin scaffold, with specificity for digoxigenin. J Mol Biol 297:1105–1120. [DOI] [PubMed] [Google Scholar]
- 35. Koide A, Bailey CW, Huang X, Koide S (1998) The fibronectin type III domain as a scaffold for novel binding proteins. J Mol Biol 284:1141–1151. [DOI] [PubMed] [Google Scholar]
- 36. Gilbreth RN, Chacko BM, Grinberg L, Swers JS, Baca M (2014) Stabilization of the third fibronectin type III domain of human tenascin‐C through minimal mutation and rational design. Protein Eng Des Sel 27:411–418. [DOI] [PubMed] [Google Scholar]
- 37. Forrer P, Stumpp MT, Binz HK, Pluckthun A (2003) A novel strategy to design binding molecules harnessing the modular nature of repeat proteins. FEBS Lett 539:2–6. [DOI] [PubMed] [Google Scholar]
- 38. Binz HK, Stumpp MT, Forrer P, Amstutz P, Pluckthun A (2003) Designing repeat proteins: well‐expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J Mol Biol 332:489–503. [DOI] [PubMed] [Google Scholar]
- 39. Kohl A, Binz HK, Forrer P, Stumpp MT, Pluckthun A, Grutter MG (2003) Designed to be stable: crystal structure of a consensus ankyrin repeat protein. Proc Natl Acad Sci USA 100:1700–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ernst A, Avvakumov G, Tong J, Fan Y, Zhao Y, Alberts P, Persaud A, Walker JR, Neculai AM, Neculai D, Vorobyov A, Garg P, Beatty L, Chan PK, Juang YC, Landry MC, Yeh C, Zeqiraj E, Karamboulas K, Allali‐Hassani A, Vedadi M, Tyers M, Moffat J, Sicheri F, Pelletier L, Durocher D, Raught B, Rotin D, Yang J, Moran MF, Dhe‐Paganon S, Sidhu SS (2013) A strategy for modulation of enzymes in the ubiquitin system. Science 339:590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lorey S, Fiedler E, Kunert A, Nerkamp J, Lange C, Fiedler M, Bosse‐Doenecke E, Meysing M, Gloser M, Rundfeldt C, Rauchhaus U, Hanssgen I, Gottler T, Steuernagel A, Fiedler U, Haupts U (2014) Novel ubiquitin‐derived high affinity binding proteins with tumor targeting properties. J Biol Chem 289:8493–8507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Leung I, Jarvik N, Sidhu SS (2017) A highly diverse and functional naive ubiquitin variant library for generation of intracellular affinity reagents. J Mol Biol 429:115–127. [DOI] [PubMed] [Google Scholar]
- 43. Koide A, Wojcik J, Gilbreth RN, Hoey RJ, Koide S (2012) Teaching an old scaffold new tricks: monobodies constructed using alternative surfaces of the FN3 scaffold. J Mol Biol 415:393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wojcik J, Hantschel O, Grebien F, Kaupe I, Bennett KL, Barkinge J, Jones RB, Koide A, Superti‐Furga G, Koide S (2010) A potent and highly specific FN3 monobody inhibitor of the Abl SH2 domain. Nat Struct Mol Biol 17:519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wojcik J, Lamontanara AJ, Grabe G, Koide A, Akin L, Gerig B, Hantschel O, Koide S (2016) Allosteric inhibition of Bcr‐Abl kinase by high affinity monobody inhibitors directed to the Src homology 2 (SH2)‐kinase interface. J Biol Chem 291:8836–8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schilling J, Schoppe J, Pluckthun A (2014) From DARPins to LoopDARPins: novel LoopDARPin design allows the selection of low picomolar binders in a single round of ribosome display. J Mol Biol 426:691–721. [DOI] [PubMed] [Google Scholar]
- 47. Gebauer M, Schiefner A, Matschiner G, Skerra A (2013) Combinatorial design of an Anticalin directed against the extra‐domain b for the specific targeting of oncofetal fibronectin. J Mol Biol 425:780–802. [DOI] [PubMed] [Google Scholar]
- 48. Hunte C, Michel H (2002) Crystallisation of membrane proteins mediated by antibody fragments. Curr Opin Struct Biol 12:503–508. [DOI] [PubMed] [Google Scholar]
- 49. Koide S (2009) Engineering of recombinant crystallization chaperones. Curr Opin Struct Biol 19:449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wilson IA, Stanfield RL (1994) Antibody‐antigen interactions: new structures and new conformational changes. Curr Opin Struct Biol 4:857–867. [DOI] [PubMed] [Google Scholar]
- 51. Salzman GS, Ackerman SD, Ding C, Koide A, Leon K, Luo R, Stoveken HM, Fernandez CG, Tall GG, Piao X, Monk KR, Koide S, Arac D (2016) Structural basis for regulation of GPR56/ADGRG1 by its alternatively spliced extracellular domains. Neuron 91:1292–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nady N, Gupta A, Ma Z, Swigut T, Koide A, Koide S, Wysocka J (2015) ETO family protein Mtgr1 mediates Prdm14 functions in stem cell maintenance and primordial germ cell formation. Elife 4:e10150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stockbridge RB, Kolmakova‐Partensky L, Shane T, Koide A, Koide S, Miller C, Newstead S (2015) Crystal structures of a double‐barrelled fluoride ion channel. Nature 525:548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK (2011) Structure of a nanobody‐stabilized active state of the beta(2) adrenoceptor. Nature 469:175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK (2011) Crystal structure of the beta2 adrenergic receptor‐Gs protein complex. Nature 477:549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Steyaert J, Kobilka BK (2011) Nanobody stabilization of G protein‐coupled receptor conformational states. Curr Opin Struct Biol 21:567–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. De Genst E, Silence K, Decanniere K, Conrath K, Loris R, Kinne J, Muyldermans S, Wyns L (2006) Molecular basis for the preferential cleft recognition by dromedary heavy‐chain antibodies. Proc Natl Acad Sci USA 103:4586–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gilbreth RN, Esaki K, Koide A, Sidhu SS, Koide S (2008) A dominant conformational role for amino acid diversity in minimalist protein‐protein interfaces. J Mol Biol 381:407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Spencer‐Smith R, Koide A, Zhou Y, Eguchi RR, Sha F, Gajwani P, Santana D, Gupta A, Jacobs M, Herrero‐Garcia E, Cobbert J, Lavoie H, Smith M, Rajakulendran T, Dowdell E, Okur MN, Dementieva I, Sicheri F, Therrien M, Hancock JF, Ikura M, Koide S, O'Bryan JP (2017) Inhibition of RAS function through targeting an allosteric regulatory site. Nat Chem Biol 13:62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bogan AA, Thorn KS (1998) Anatomy of hot spots in protein interfaces. J Mol Biol 280:1–9. [DOI] [PubMed] [Google Scholar]
- 61. Gilbreth RN, Truong K, Madu I, Koide A, Wojcik JB, Li NS, Piccirilli JA, Chen Y, Koide S (2011) Isoform‐specific monobody inhibitors of small ubiquitin‐related modifiers engineered using structure‐guided library design. Proc Natl Acad Sci USA 108:7751–7756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Koide A, Abbatiello S, Rothgery L, Koide S (2002) Probing protein conformational changes by using designer binding proteins: application to the estrogen receptor. Proc Natl Acad Sci USA 99:1253–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Koide A, Gilbreth RN, Esaki K, Tereshko V, Koide S (2007) High‐affinity single‐domain binding proteins with a binary‐code interface. Proc Natl Acad Sci USA 104:6632–6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schroeder T, Barandun J, Flutsch A, Briand C, Mittl PR, Grutter MG (2013) Specific inhibition of caspase‐3 by a competitive DARPin: molecular mimicry between native and designed inhibitors. Structure 21:277–289. [DOI] [PubMed] [Google Scholar]
- 65. Waksman G, Shoelson SE, Pant N, Cowburn D, Kuriyan J (1993) Binding of a high affinity phosphotyrosyl peptide to the Src SH2 domain: crystal structures of the complexed and peptide‐free forms. Cell 72:779–790. [DOI] [PubMed] [Google Scholar]
- 66. Sha F, Gencer EB, Georgeon S, Koide A, Yasui N, Koide S, Hantschel O (2013) Dissection of the BCR‐ABL signaling network using highly specific monobody inhibitors to the SHP2 SH2 domains. Proc Natl Acad Sci USA 110:14924–14929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Monod J, Wyman J, Changeux JP (1965) On the nature of allosteric transitions: A plausible model. J Mol Biol 12:88–118. [DOI] [PubMed] [Google Scholar]
- 68. Kuriyan J, Eisenberg D (2007) The origin of protein interactions and allostery in colocalization. Nature 450:983–990. [DOI] [PubMed] [Google Scholar]
- 69. Gao J, Sidhu SS, Wells JA (2009) Two‐state selection of conformation‐specific antibodies. Proc Natl Acad Sci USA 106:3071–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Grebien F, Hantschel O, Wojcik J, Kaupe I, Kovacic B, Wyrzucki AM, Gish GD, Cerny‐Reiterer S, Koide A, Beug H, Pawson T, Valent P, Koide S, Superti‐Furga G (2011) Targeting the SH2‐kinase interface in Bcr‐Abl inhibits leukemogenesis. Cell 147:306–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jost C, Schilling J, Tamaskovic R, Schwill M, Honegger A, Pluckthun A (2013) Structural basis for eliciting a cytotoxic effect in HER2‐overexpressing cancer cells via binding to the extracellular domain of HER2. Structure 21:1979–1991. [DOI] [PubMed] [Google Scholar]
- 72. Findlay GM, Smith MJ, Lanner F, Hsiung MS, Gish GD, Petsalaki E, Cockburn K, Kaneko T, Huang H, Bagshaw RD, Ketela T, Tucholska M, Taylor L, Bowtell DD, Moffat J, Ikura M, Li SS, Sidhu SS, Rossant J, Pawson T (2013) Interaction domains of Sos1/Grb2 are finely tuned for cooperative control of embryonic stem cell fate. Cell 152:1008–1020. [DOI] [PubMed] [Google Scholar]
- 73. Pawson T, Nash P (2003) Assembly of cell regulatory systems through protein interaction domains. Science 300:445–452. [DOI] [PubMed] [Google Scholar]
- 74. Huang J, Koide A, Makabe K, Koide S (2008) Design of protein function leaps by directed domain interface evolution. Proc Natl Acad Sci USA 105:6578–6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Huang J, Makabe K, Biancalana M, Koide A, Koide S (2009) Structural basis for exquisite specificity of affinity clamps, synthetic binding proteins generated through directed domain‐interface evolution. J Mol Biol 392:1221–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yasui N, Findlay GM, Gish GD, Hsiung MS, Huang J, Tucholska M, Taylor L, Smith L, Boldridge WC, Koide A, Pawson T, Koide S (2014) Directed network wiring identifies a key protein interaction in embryonic stem cell differentiation. Mol Cell 54:1034–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tanaka S, Takahashi T, Koide A, Ishihara S, Koikeda S, Koide S (2015) Monobody‐mediated alteration of enzyme specificity. Nat Chem Biol 11:762–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gross GG, Junge JA, Mora RJ, Kwon HB, Olson CA, Takahashi TT, Liman ER, Ellis‐Davies GC, McGee AW, Sabatini BL, Roberts RW, Arnold DB (2013) Recombinant probes for visualizing endogenous synaptic proteins in living neurons. Neuron 78:971–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kummer L, Parizek P, Rube P, Millgramm B, Prinz A, Mittl PR, Kaufholz M, Zimmermann B, Herberg FW, Pluckthun A (2012) Structural and functional analysis of phosphorylation‐specific binders of the kinase ERK from designed ankyrin repeat protein libraries. Proc Natl Acad Sci USA 109:E2248–E2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Parizek P, Kummer L, Rube P, Prinz A, Herberg FW, Pluckthun A (2012) Designed ankyrin repeat proteins (DARPins) as novel isoform‐specific intracellular inhibitors of c‐Jun N‐terminal kinases. ACS Chem Biol 7:1356–1366. [DOI] [PubMed] [Google Scholar]