

Abstract
The potential for infection by coronaviruses (CoVs) has become a serious concern with the recent emergence of Middle East respiratory syndrome and severe acute respiratory syndrome (SARS) in the human population. CoVs encode two large polyproteins, which are then processed into 15–16 nonstructural proteins (nsps) that make significant contributions to viral replication and transcription by assembling the RNA replicase complex. Among them, nsp9 plays an essential role in viral replication by forming a homodimer that binds single‐stranded RNA. Thus, disrupting nsp9 dimerization is a potential anti‐CoV therapy. However, different nsp9 dimer forms have been reported for alpha‐ and beta‐CoVs, and no structural information is available for gamma‐CoVs. Here we determined the crystal structure of nsp9 from the avian infectious bronchitis virus (IBV), a representative gamma‐CoV that affects the economy of the poultry industry because it can infect domestic fowl. IBV nsp9 forms a homodimer via interactions across a hydrophobic interface, which consists of two parallel alpha helices near the carboxy terminus of the protein. The IBV nsp9 dimer resembles that of SARS‐CoV nsp9, indicating that this type of dimerization is conserved among all CoVs. This makes disruption of the dimeric interface an excellent strategy for developing anti‐CoV therapies. To facilitate this effort, we characterized the roles of six conserved residues on this interface using site‐directed mutagenesis and a multitude of biochemical and biophysical methods. We found that three residues are critical for nsp9 dimerization and its abitlity to bind RNA.
Keywords: coronaviruses, nonstructural proteins, infectious bronchitis virus, Nsp9, dimerization
Short abstract
PDB Code(s): 5C94
Importance and Impact Statement
In this study, the dimeric nsp9 crystal structure of avian infectious bronchitis virus (IBV) was solved at 2.5 Å resolution. A hydrophobic region and two parallel α‐helices were found to be essential for nsp9 dimerization. The dimeric state was independently evaluated using multiple biochemical methods, including mutagenesis, circular dichroism, size exclusion chromatography with in‐line multi‐angle light scattering analysis (SEC‐MALs), electrospray ionization mass spectrometry (ESI‐MS), and analytical ultracentrifugation. This is the first time an nsp9 structure for a gamma‐coronavirus has been determined.
Introduction
Coronaviruses (CoVs) are enveloped, positive‐strand RNA viruses that cause a broad spectrum of diseases in humans and animals.1, 2 In the 1930s, the avian infectious bronchitis virus (IBV) was the first CoV identified,3 and since that time CoVs have been shown to infect diverse hosts, including humans.1, 2, 4, 5 Recent outbreaks of severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) have focused attention on CoVs, since pathogenic CoVs can be fatal to humans.6, 7, 8, 9 Although progress has been made in limiting CoV infection over the past decade, scientists and physicians still lack a complete understanding of CoV pathogenicity, particularly from the molecular and structural points of view. On the basis of genome organization and phylogenetic analysis, CoVs can be divided into four distinct genera, termed alpha‐, beta‐, gamma‐ and delta‐CoVs.10 In the past, human CoV infections have all resulted from alpha‐ and beta‐CoVs (i.e., SARS and MERS are beta‐CoVs), with no cases involving gamma‐ or delta‐CoVs repreted so far. However, the risk of cross‐species infection from gamma and delta genera should not be ignored, and research concerning these two genera is critical to enable quick and efficient responses to novel animal‐to‐human CoV infections in the future.
Avian IBV is a gamma‐CoV that causes a highly contagious disease in domestic fowl. As such, avian IBV is one of the primary sources of economic loss in the poultry industry.11 New viral serotypes continue to emerge due to viral mutation and recombination, making this virus very difficult to control.12 The genome of IBV consists of single‐stranded, positive‐sense RNA that is 27.6 kb in length.13 Upon infection, viral replicase proteins are translated from the genomic RNA, initially generating two precursor polyproteins, pp1a and pp1ab. These polyproteins are then processed into 15 individual nonstructural proteins (nsp2–16) by two virus‐encoded proteinases, 3C‐like and papain‐like proteinases.14, 15 These nsps assemble into a replicase complex responsible for viral transcription and replication.16 To understand the molecular mechanisms of IBV replication, great effort has been made to characterize nsp structures. To date, only a few nsp structures from IBV have been determined, including nsp2a, the ADRP (adenosine diphosphate‐ribose‐1″‐phosphatase) domain of nsp3, and nsp5 main protease (PDB:3LD1,3EWO,3EJF,3EKE,4X2Z).17, 18, 19
Nsp9 is essential for viral infection, as its deletion halts virus propagation.20 Nsp9 is a member of the oligosaccharide/oligonucleotide binding (OB)‐fold superfamily and a variety of methods have been used to show that nsp9 can bind both RNA and DNA.21, 22 Interestingly, nsp9 forms a homodimer, and its dimeric form is believed to be critical for RNA binding and for viral infection. The dimeric structure of nsp9 for SARS‐CoV was first reported by Egloff et al.21 They observed that the nsp9 dimer is stabilized by hydrophobic interactions between two parallel α‐helices (we call this Form‐A). The SARS‐CoV nsp9 contains a protein‐protein association motif “GXXXG” (residues G100–G104) on the parallel α‐helices of the nsp9 dimer.23, 24 Mutation of a single residue (G104E) within this motif terminates viral assembly in vivo. This mutation may alter the α‐helical interaction and disrupt nsp9 dimerization.25 This indicates that the dimeric form of nsp9 is essential for viral infection and also suggests that the disruption of nsp9 dimerization may be an effective strategy for combating CoV‐associated diseases. Although Form A seems to be the proper dimeric form of nsp9, other researchers have observed two different nsp9 dimers. Sutton et al. also solved the SARS‐CoV nsp9 structure, confirming that it forms a dimer in two different space groups.22 However, they also described a different dimer for SARS‐CoV nsp9, which is formed by an interaction between β‐strands (β5)—one from each subunit (we call this Form‐B). Later, Ponnusamy et al. determined the nsp9 structure for HCoV‐229E, revealing a third homodimeric form (Form‐C), which is formed via interactions between two anti‐parallel α‐helices, with a disulfide‐linkage between the two Cys69s within the interacting helices.26 This structure formed a trimer of dimers in the crystal, which is also different from the two structures previously determined for SARS‐CoV nsp9. These observations raise an important question; whether there is a common and conserved dimeric form for all CoV nsp9s, or whether there are different dimeric forms for diferent CoV nsp9s. Moreover, no structural information is available for nsp9s of other CoV genera (particularly gamma‐CoVs). It is important to determine which type of dimer these nsp9s form, and whether there is structural and functional conservation among these nsp9s from different CoV genera.
In this study, we selected IBV nsp9 as a representative of gamma‐CoVs and investigated its structure and oligomerization state. We resolved the crystal structure of IBV nsp9 at 2.5 Å, which revealed a Form‐A dimeric conformation (similar to SARS‐CoV nsp9). The homodimeric interface was formed by two parallel α‐helices, which was further surrounded by a hydrophobic shell. The residues involved in this hydrophobic interaction are conserved across species, suggesting that nsp9 homodimerization in Form A is essential for all CoVs. We further characterized six conserved residues within the dimeric interface of IBV nsp9 by site‐directed mutagenesis and biochemical approaches, and found that residues F73, I95, and G98 were critical for both dimerization and RNA interactions.
Results
Sequence alignment and phylogenetic analysis of CoV nsp9s
To study the evolutionary relationships between CoV nsp9 proteins, we analyzed 34 nsp9 sequences from different species (derived from the NCBI database) using MUSCLE (MEGA6).27 Pairwise sequence alignment revealed that sequence similarity ranged from 24.8% to 98.2% for all nsp9 sequences. This large range in conservation is notable, as nsp9 is crucial for viral replication and therefore a higher level of conservation was expected. Two features account for this variation. First, there are two main gaps in the nsp9 alignment (i.e., stretches of little sequence identity) located at loops connecting β‐strands β1–β2 and β4–β5, based upon solved nsp9 structures. Second, the low similarity of pairwise sequence alignment was mainly derived from Beluga Whale CoV (similarity ranged from 24.8% to 56.8%). However, the C‐terminal halves of nsp9 proteins (residues 67–111), which include β‐strands β6–β7 and helix α1, displayed much higher sequence similarity than the rest of the proteins [Fig. 1(C)]. There are 16 conserved residues located in this region, of which ∼70% are hydrophobic [Fig. 1(D)], suggesting that this region is vital for the physiological function of nsp9.
Figure 1.
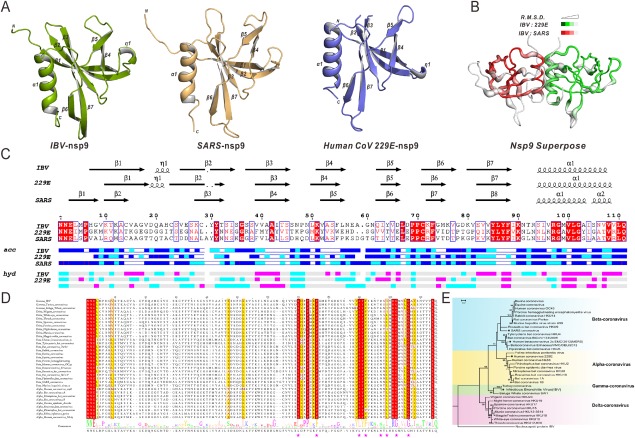
Nsp9 crystal structures(monomer), structural superimposition, multiple sequence alignment, and phylogenetic tree. (A) Ribbon representation of IBV nsp9(green), HCoV‐229E nsp9(orange), and SARS‐CoV nsp9(purple). (B) Superposition of IBV, HCoV‐229E, and SARS‐CoV nsp9. IBV and SARS‐CoV(red) are superimposed(RMSD = 0.875Å), IBV and HCoV‐229E(green) are superimposed(RMSD = 1.409Å). (C) Secondary structure elements and multiple sequence alignments of IBV, HCoV‐229E, and SARS‐CoV. (D) Multiple sequence alignment of the nsp9 protein with representatives from all four CoV genera. The seven residues(F73, L86, F88, I95, G98, and G102) are marked with red asterisks. (E) Phylogenetic analysis of the CoV nsp9 family is presented.
The phylogenetic tree was generated according to sequence alignment of nsp9 [Fig. 1(E)]. Overall, the tree segregated each genus of CoVs, as previously described.28 The Poisson Correction distance indicated a very limited difference between each genus (Supporting Information Table S1). The topology of the CoV nsp9 tree generally agreed with the currently accepted view of the organismal phylogeny of CoVs, based upon the evolutionary relationships of RdRp (RNA‐dependent RNA polymerase), S and N proteins.29, 30
Crystal structure of IBV nsp9 reveals a form‐a homodimer
IBV nsp9 crystals belong to space group I432, with unit cell dimensions a = b = c = 123.4 Å, α = β = γ = 90°. There is one molecule in the asymmetric unit. The final R‐factor for the structural model is 18.7%, and the Rfree factor is 23.6%. In total, 95.4% of the amino acids fell in favored regions of the Ramachandran plot, whereas none fell in disallowed regions. Data collection and refinement statistics are detailed in Table 1. The two‐fold axis of the IBV nsp9 homodimer is coincident with a crystallographic two‐fold axis of symmetry in the protein crystal. Each protomer has seven β‐strands (β1–β7), which form a partial barrel‐like domain, followed by one α‐helix (α1) at the C‐terminus. Similar structures are seen for nsp9 monomers from HCoV‐229E (PDB code: 2J97)26 and SARS‐CoV (PDB code: 1UW7)22 [Fig. 1(A,C)]. The backbone root‐mean‐square deviation (RMSD) of monomeric nsp9 between IBV and HCoV‐229E (green) is 1.409 Å (for 75 Cα atoms) and 0.875 Å (for 75 Cα atoms) between IBV and SARS‐CoV (red), respectively [Fig. 1(B)].
Table 1.
IBV‐nsp9 Data‐collection and Refinement Statistics
| Values in parentheses are for the highest resolution shell | |
|---|---|
| Data collection | |
| Wavelength (Å) | 1.5418 |
| Space group | I432 |
| Unit‐cell parameters(Å, °) | a = b = c = 123.4, α = β = γ = 90.0 |
| Resolution (Å) | 50.00–2.44(2.50–2.44) |
| Rmerge a (%) | 8.9(45.6) |
| Rpim b (%) | 2.6(13.5) |
| Average I/σ(I) | 16.4(2.4) |
| No. of observed reflections | 73760 |
| No. of unique reflections | 5952 |
| Completeness (%) | 98.8(91.2) |
| Multiplicity | 12.4(2.3) |
| Matthews coefficient (Å3Da−1) | 3.26 |
| Solvent content (%) | 62.34 |
| Molecules per asymmetric unit | 1 |
| Refinement | |
| Resolution (Å) | 22.53–2.44 |
| Rwork/Rfree | 0.187/0.236 |
| Ramachandran favored (%) | 95.37 |
| Ramachandran outliers (%) | 0 |
| No. of atoms | |
| Protein | 862 |
| Water | 165 |
| Wilson B value | 30.94 |
| R.m.s. Deviations | |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.422 |
R merge= , where is an individual intensity measurement and is the average intensity for all i reflections.
R pim is approximated estimated by multiplying the Rmerge value by the factor , where N is the overall redundancy of the data set.
For IBV nsp9, two protomers form a dimer [Fig. 2(B)], with the two α‐helices positioned at a 44° angle relative to one another. The main chains of the α‐helices are very close to each other, as the distance between vectors in the center of each helix is only 5.8 Å (calculated by PyMOL31 script “helix_angle.py”, written by Dr. Robert L. Campbell, Queen's University) . These two α‐helices are surrounded by a hydrophobic interface composed of residues from β‐strands β2–β3 and β6–β7, and an N‐terminal loop from each nsp9 protomer. This dimeric form resembles the SARS‐CoV nsp9 dimer. However, the total buried surface area in the IBV nsp9 dimer is 668 Å2 (calculated by PyMOL), which is smaller than seen for the SARS‐CoV nsp9 dimer (903 Å2) (PDB code: 1QZ8)21 (Supporting Information Table S2), but is similar to that of HCoV‐229E (693 Å2) and larger than the minimum value (368 Å2) of homodimeric interface reported by Jones S.32
Figure 2.
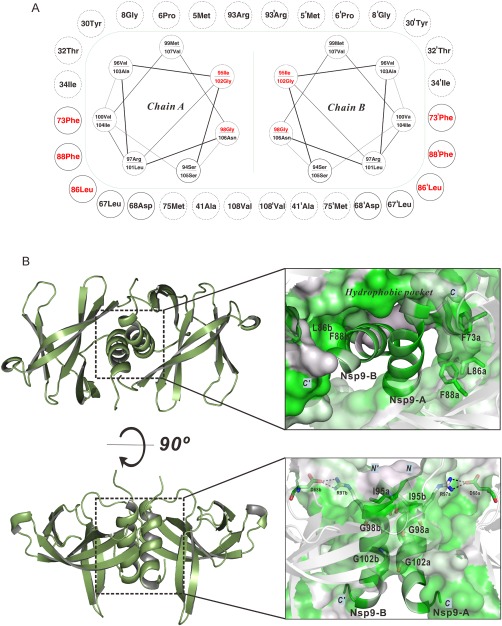
Mutational analysis of the IBV nsp9 protein. (A) Sketch map for the IBV nsp9 parallel α‐helices and the hydrophobic pocket. The green dash represents the hydrophobic contact interface. Circles connected by spirals represent parallel α‐helices. Circles outside the green dash represent hydrophobic residues and critical polar residues surrounding the parallel α‐helices. Solid circles indicate conserved residues. Dashed circles indicate non‐conserved residues. Mutant residues are colored red in the parallel α‐helices and surrounding pocket. (B) Parallel α‐helices and the hydrophobic region in the crystal structure of the nsp9 dimer. Surface and cartoon representations are colored according to the level of residue hydrophobicity from white (polar) to green (hydrophobic). The side chains of conserved hydrophobic residues (F73, L86, and F88) are shown. Hydrophobic interactions in parallel α‐helices highlighting residues I95, G98, and G102 are shown.
Site‐directed mutagenesis of the nsp9 dimerization interface does not affect nsp9 secondary structure
Since the dimerization interface for IBV nsp9 consists mostly of the highly conserved C‐terminal α‐helices and β6–β7, we next evaluated the role of both the hydrophobic interface and the α‐helices in stabilizing nsp9 dimerization. On the basis of the sequence alignment and structural analysis, we generated a set of nsp9 mutants, targeting six conserved amino acids (F73, L86, F88, I95, G98, and G102) [Fig. 2(A,B)]. Residues I95, G98, and G102, which are located in the C‐terminal α‐helix (α1) [Fig. 2(B)], were replaced with negatively charged residues (aspartates) to disrupt the interaction between the two α‐helices. Residues F73, L86, and F88, which are part of the hydrophobic shell that surrounds the two α‐helices [Fig. 2(B)], were replaced by glycines to disrupt the hydrophobic environment.
Circular dichroism (CD) spectroscopy was used to detect potential conformation changes induced by each of these mutations. The fractional content of secondary structures in wild‐type nsp9 was calculated to be 12.6% helix, 55.0% sheets/turns, and 32.4% non‐repetitive secondary structure, which is in good agreement with our crystal structure (Supporting Information Table S3). The CD spectra and data analysis associated with the nsp9 mutants are described in Supporting Information Figure S2. No changes were detected for the F73G, I95D, G98D, and G102D mutants compared with wild‐type nsp9, but changes in secondary structure were detected for the L86G and F88G mutants using the far‐UV CD spectra. However, assessed by near‐UV CD spectrum, the overall tertiary structures of all mutants were similar, indicating that the L86G and F88G mutations did not significantly disrupt protein structure.
Mutations within the nsp9 dimeric interface disrupt dimer formation
After confirming that these six mutations within the dimer interface had little effect on secondary protein structure, we next asked whether these mutations affected nsp9 dimeric organization. At first, size exclusion chromatography (SEC) was applied to wild‐type and mutant IBV nsp9s to evaluate their oligomerization states. Molecular weights were calculated from a standard calibration curve. SEC analysis revealed that wild‐type nsp9 had a molecular weight of 20 kDa, close to the theoretical molecular weight of a nsp9 dimer (25 kDa) [Fig. 3(A)]. However, the nsp9 mutants (F73G, L86G, F88G, I95D, G98D, and G102D) exhibited a larger retention volume, which corresponds to a smaller molecular weight (Fig. 3, Supporting Information Fig. S3). These results indicate that mutations to the dimeric interface of nsp9 disrupt dimer formation. We also studied it by a second approach, glutaraldehyde crosslinking of nsp9 proteins (both wile‐type and mutants). However, the results from this study were not very clear, although a decreased dimeric portion for mutant G98D was observed with wild‐type nsp9 (Supporting Information Fig. S4 and Table S7)
Figure 3.
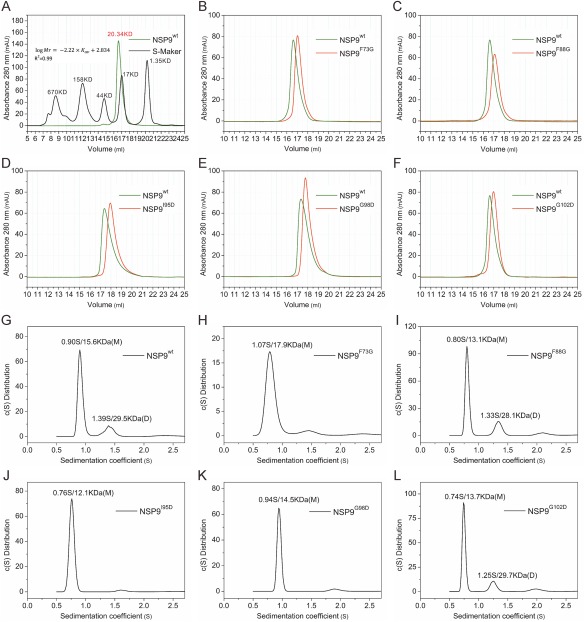
Biochemical characterization of IBV nsp9 dimerization. (A) The SEC assay is used to evaluate the molecular weight of wild‐type nsp9. The standard equation “ ”, Adj.R2 is used (Adjusted R2 is 0.99). The black line is from commercial molecular weight standards, and the wild‐type nsp9 chromatogram is colored green. (B‐F) Comparison of SEC results for wild‐type nsp9 and different mutants. (G‐L) Sedimentation velocity ultracentrifugation results for wild‐type nsp9 and nsp9 mutants. Sedimentation coefficients (S) and molecular weight (MW) are indicated. “M” and “D” stand for the monomer and dimer.
The failure of the second method to give a consistent result may be because it is influenced by a number of factors, such as pH, temperature, concenteations of reactants,etc., and generally hard to control. Since the above two commonly used methods did not give us a clear and consistent conclusion, we continued pursuing more sensitive and quantitative approaches. Electrospray ionization mass spectrometry (ESI‐MS) was used as a third way. It verified the molechular weight of all recombinant proteins.However, mixtures of dimers and monomers were detected in all samples (Supporting Information Table S4 and Fig. S5), indicating that the dimer and monomer forms may be in an equilibrium. This also implied that ESI‐MS is such a sensitive technique that even a tiny fraction of any oligomeric state can be detected.We further adopted analytical ultracentrifugation,which is a method not widely available but mor accurate, to precisely characterize the monomer‐dimer equilibrium for wild‐type and mutant nsp9 (F73G, L86G, F88G, I95D, G98D, and G102D) (Supporting Information Fig. S6 and Table S5). Sedimentation coefficients for the F73G, G98D, and I95D mutants revealed that these proteins are less able to form a dimer than the wild‐type protein, and therefore tend to remain in the monomeric state. The other mutants (L86G, F88G, and G102D) had oligomerization states similar to wild‐type nsp9. These data imply that F73 (from the hydrophobic surface), as well as I95 and G98 (from α1) are more important for IBV nsp9 dimerization than the other tested positions (L86, F88, and G102).
Nsp9 mutations that disrupt dimer formation also affect nucleic acid binding
The electrophoretic mobility shift assay (EMSA) was used to detect protein‐nucleic acid complexes in solution. Using a typical EMSA protocol, we did not detect a nucleic acid mobility shift when wild‐type nsp9 was mixed with FAM‐labeled RNA (Supporting Information Fig. S7). This is commonly seen when a macromolecular complex is in dynamic equilibrium. We therefore used a modified zone‐interference gel electrophoresis method to assess interactions between nsp9 and nucleic acids.26, 33 This analysis revealed that both wild‐type nsp9 and the F73G mutant were able to bind nucleic acids [Fig. 4(A)]. These mobility shifts were more dramatic with RNA than with single‐stranded DNA under the same conditions [Fig. 4(B)]. However, the I95D and G98D mutants did not seem to bind nucleic acids.
Figure 4.
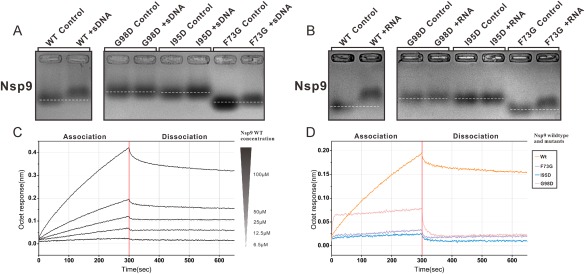
Oligonucleotide binding analysis of wild‐type and mutant IBV nsp9. (A,B) EMSA illustrating wild‐type and mutants (F73G, I95D, and G98D) IBV nsp9 association with ssDNA/RNA(20‐mer). The white dotted lines represent the position of the bands of IBV nsp9 wild‐type and mutants (F73G, I95D, and G98D) without reaction with ssDNA/RNA. (C) Biolayer interferometry analysis of wild‐type nsp9 with RNA. Biotinylated 20‐mer RNA is immobilized on SA biosensors. Binding curves show association and dissociation for different protein concentrations. (D) Sensorgrams obtained using biosensors loaded with nsp9 mutants. The variation of the response is recorded with mutants at a concentration of 50 uM.
Because mobility shift assays can be influenced by alterations in residue charge and conformation changes, which may obscure the formation of a protein nuclear‐acid complex, we further tested the ability of wild‐type and mutant nsp9 to bind RNA using two additional approaches that are more sensitive and based on completely different principles. The first approach was biolayer interferometry (BLI). In this assay, biotinylated RNA was immobilized on streptavidin biosensor tips34, 35 and nsp9 was added at various concentrations (6.25–100 μM) to establish association and dissociation rates [Fig. 4(C,D)]. The BLI data provided an overall affinity value (Kd) of 28 μM for the interaction between wild‐type nsp9 and RNA. These biosensor binding data also showed that the F73G, G98D, and I95D mutants could not bind RNA [Fig. 4(D)]. These data suggest that the dimeric form of nsp9 is required for nsp9‐RNA interactions. The second method we used was surface plasmon resonance (SPR),21 which is also a chip‐based immobilized affinity detection technique similar to BLI, but using a different detector. Using SPR, we measured wild‐type nsp9‐RNA binding affinity at 3.6 μM, very similar to the data from BLI measurements [Supporting Information Fig. S7(C)]. In addition, the three nsp9 mutants (F73D, I95D, and G98D) exhibited dramatically reduced RNA association rates [Supporting Information Fig. S7(D)], which is consistent with the BLI observations. In summary, these data indicate that nsp9 mutants (F73D, I95D, and G98D) that cannot dimerize also lose the ability to bind RNA and single‐stranded DNA.
Discussion
Nsp9 proteins are essential for CoV replication, reproduction, and virulence.25 For several CoVs, the formation of nsp9 homodimers is critical for viral replication, as disruption of nsp9 dimer formation hinders viral assembly. This suggests that disrupting the dimerization interface of nsp9 is a potential anti‐viral strategy.36 However, the structural mechanisms underlying nsp9 dimerization have not been clarified. At least three dimeric forms of nsp9 have been reproted, and it is unclear whether there is a conserved dimeric form for all CoV nsp9s, or whether individual nsp9s adopt unique dimer forms. In addition, all reported nsp9 structures belong to the alpha and beta genera, whereas there is no structural information for nsp9s of gamma‐CoVs. Thirdly, a detailed characterization of specific residues within the nps9 dimeric interface has not yet been performed. This analysis is critical for developing potential anti‐viral strategies that are based on disrupting nsp9 dimerization.
In this study, we characterized nsp9 from the IBV CoV using structural, biophysical, and biochemical approaches. IBV is a representative gamma‐CoV that affects the economy of the poultry industry by infecting domestic fowl. We determined IBV nsp9 structure and found that its monomer resembled those reported for nsp9s from SARS‐CoV (PDB code: 1UW7)22 and HCoV‐229E (PDB code: 2J97).26 In addition, IBV nsp9 formed a Form‐A dimer similar to SARS‐CoV nsp9. The region forming the dimer interface of nsp9 is highly conserved based on sequence alignment and structural analysis. This strongly supports the hypothesis that there is a conserved dimeric form among nsp9s of all CoVs. As such, designing ways to disrupt nsp9 dimerization (a potential anti‐viral strategy) may be quite straightforward, as only the conserved dimeric interface must be considered. This strategy may also be quite powerful, for it may work against all of these CoVs. To facilitate the developent of this potential anti‐viral strategy, we further characterized six conserved residues (F73, L86, F88, I95, G98, and G102) associated with the dimeric interface and the conserved hydrophobic region. We used site‐directed mutagenesis and diverse biophysical and biochemical approaches. We found that three conserved residues (F73, I95, and G98) are critical for stabilizing the dimeric conformation of nsp9 and for nsp9 to bind RNA.
Diversified nsp9 dimer models exist in CoV
Nsp9 can adopt several different dimeric conformations.21, 22, 26 For IBV nsp9 dimers, only Form‐A was observed in the crystal lattice. This dimeric model has been seen in three different crystal structures of nsp9, including SARS‐CoV nsp9, and 229E‐CoV nsp9 C69A mutant (PDB code: 2J98, 1UW7, 1QZ8) (Supporting Information Fig. S1).21, 22, 26 Form‐B, which is seen with interactions between β sheets of the SARS‐CoV nsp9 dimer, was not found in the IBV nsp9 dimeric structure because the IBV nsp9 β5s are too far from each other. For Form‐B, the lack of sequence conservation in the critical protein region, and the small interaction surface, make it unlikely that Form‐B is a conserved dimeric conformation of CoV. Form‐C, which is seen in 229E‐CoV nsp9 wild‐type dimers, is characteristic of a novel disulfide‐linked homodimer formed by two antiparallel α‐helixes. Noticeably, Cys69 is conserved in nsp9s of alpha‐, beta‐ and gamma‐CoVs (including IBV and SARS‐CoV). However, this disulfide bond is not observed in IBV and SARS nsp9 dimeric structures. Based on our results, we propose that the dimeric Form‐A conformation is the universal state for the nsp9 dimer.
Helix‐helix association stabilizes dimeric nsp9
Crystallographic studies have previously found that the helix‐helix association (α1‐α′1) is key to stabilizing the nsp9 homodimer. To investigate the possibility that IBV nsp9 dimerization could be prevented by a single mutation in the α‐helix, three conserved residues (I95, G98, and G102) involved in the helix‐helix association were mutated. All three nsp9 mutants (I95D, G98D, and G102D) exhibited a monomer‐dimer equilibrium in solution, as shown by analytical ultracentrifugation and ESI‐MS. This implies that each point mutation lessened the degree of nsp9 dimerization, but did not completely eliminate it. This is particularly true for the I95 and G98 nsp9 mutants. Therefore, nsp9 homodimerization may be disrupted by targeting conserved residues within the helix‐helix interface. These data provide further evidence that the Form‐A conformation is the common state of nsp9 dimers. Zachary et al. also showed that mutations to the α‐helix in SARS‐CoV nsp9 were lethal to the virus.25 Although the same mutations would presumably affect the interaction between the C‐terminal α‐helices in both Forms A and C, because Form C involves a disulfide bridge which is uncommon to nsp9s in four CoV genera, we consider that Form‐A state of nsp9 is biologically relevant.
A hydrophobic shell stabilizes the inter‐helical interaction at the dimerization interface of nsp9
As discussed earlier, there is a hydrophobic shell that surrounds the two C‐terminal α‐helices of IBV nsp9. We explored the role of this hydrophobic region in the formation of the nsp9 dimer by mutating three conserved residues to glycine (F73G, L86G, and F88G). Each mutant displayed a delayed elution peak by gel‐filtration chromatography, when compared with wild‐type nsp9. Among the mutated proteins, F73G nsp9 was very rarely in a dimeric form, as measured by analytical ultracentrifuge. From these data, we infer that the hydrophobic shell helps stabilize the nsp9 dimer. These data reveal that the hydrophobic surface, in addition to the two parallel α‐helices, functions to stabilize nsp9 Form‐A dimerization.
Dimerization is required for nsp9 to interact with nucleic acids
Wild‐type nsp9 preferentially adopts a dimeric form in solution,25 whereas the nsp9 mutants (F73G, I95D, and G98D) retained their monomeric form. Thus, we further analyzed the interaction between nucleic acids and the different forms of nsp9. EMSAs revealed that wild‐type and F73G nsp9s could associate with both single‐stranded DNA and RNA. The G98D and I95D mutants could not bind nucleic acids, indicating that the residues near the GXXXG motif (within the C‐terminal α‐helix) are critical for RNA binding. These data conflict with Zachary et al., who performed Non‐Radioactive EMSAs after cross‐linking nucleic acid‐protein mixtures at 254nm by UV‐light and showed that each of the SARS‐CoV nsp9 wild‐type and mutants (G100E, G104E, and G104V) SARS‐CoV was able to bind RNA.25 However, disrupting the nsp9 dimer did result in a 5‐ to 12‐fold decrease in affinity by FA measurements.25 These conflicting data could be explained by the relatively low‐affinity binding constants, and suggests that nsp9 may not have a specific binding sequence, although interactions with a stem‐loop in the 3′ region of the genome were reported recently.25, 37
To obtain kinetic information and to quantitatively compare the ability of wild‐type and mutant versions of nsp9 to bind RNA, we performed BLI and SPR experiments. Although wild‐type nsp9 showed a strong RNA‐binding signal, the single‐residue mutants (F73G, I95D, and G98D) exhibited dramatically decreased signals. This indicates that the dimeric nsp9 state is required for RNA binding, a conclusion supported by both BLI and SPR measurements.
Conclusions
In this study, we solved the IBV nsp9 dimeric structure at 2.5 Å resolution. The dimeric state was independently evaluated and validated by an assortment of biochemical methods, including mutagenesis, circular dichroism, SEC‐MALs, ESI‐MS, and ultracentrifugation. Our results indicate that a hydrophobic region, along with two parallel α‐helices, play fundamental roles in stabilizing nsp9 dimers in solution. The conserved residues F73, I95, and G98, which are found in the C‐terminal region of nsp9, are pivotal for dimerization. We also showed that nsp9 dimerization is critical for nsp9 to bind RNA. In conclusion, we have revealed structural and biochemical forces that control nsp9 dimerization, providing a valuable resource for assessing the role of the dimeric state in viral genome transcription and replication in vivo.
Materials and Methods
Alignment and phylogenetic analysis
Amino acid sequence data for CoV nsp9s retrieved from the NCBI database were analyzed using MEGA 6.06 software.27 In total, 34 nsp9 amino acid sequences from diverse CoV species of the Coronaviridae subfamily were aligned using the MUSCLE (MEGA6) UPGMA clustering method. Nucleocapsid protein from IBV was used as the out‐group. A phylogenetic tree was created using the maximum likelihood method and bootstrap full heuristic analysis, with 1000 bootstrap replications. Bootstrap values for condensed tree cut off < 50%. The phylogenetic distances between groups were calculated with passion correction method using MEGA6.
Generation of wild‐type and correlated mutant constructs
The cDNA encoding IBV strain M41 ORF1a polyprotein was provided by Professor Ming Liao (South China Agricultural University, People's Republic of China). IBV nsp9 was cloned into a pGEX‐6P‐1 plasmid (GE Healthcare) using BamHI and XhoI restriction sites. The F73G, L86G, F88G, I95D, G98D, and G102D single‐site mutations were generated using primers encoding the mutated amino acids. Mutagenesis constructs were confirmed by commercial DNA sequencing (Sangon Biotech).
Protein purification, crystallization, and structure determination
Wild‐type IBV nsp9 was expressed, purified, and crystallized as described.38 Crystals of IBV‐CoV nsp9 diffracted to 2.5 Å resolution and a complete dataset was collected in‐house using a Rigaku Cu Kα rotating‐anode X‐ray generator (MM007) operating at 40 kV and 20 mA (λ = 1.5418 Å) with a Rigaku R‐AXIS IV++ image plate detector at 100 K maintaining by an Oxford Cryosystem. Using the monomeric structure of SARS‐CoV nsp9 (PDB code: 1QZ8)21 as an initial search model, one copy of nsp9 was located by the molecular replacement method with Phaser 39 and was refined to 2.5 Å resolution. Structure superimposition and calculation of r.m.s. deviations were carried out using PyMOL.31 Coordinate and structure factor file of IBV‐CoV nsp9 were submitted to the Protein Data Bank (PDB code: 5C94).
Size‐exclusion chromatography
Size‐exclusion chromatography (SEC) analysis for investigating the oligomeric nature of IBV nsp9 and nsp9 mutants was performed using a Superdex 200 10/300 GL column (GE Healthcare). The column was pre‐equilibrated with 1× PBS buffer (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4, pH 7.3) and protein samples analyzed at a flow rate of 0.5 mL/min at room temperature. The protein samples were prepared at a concentration of ∼1 mg/mL.
Dynamic light scattering
Measurements were taken using a Zetasizer μV (Malvern Instruments, Worcestershire, UK) with a 1 mg/mL solution of IBV‐CoV nsp9 in 1× PBS buffer (500 μL, 25°C). The results were analyzed using software provided by the manufacturer. Experimental errors were estimated as standard deviations calculated from ten independent measurements per sample.
Chemical cross‐linking assay
Wild‐type and mutant nsp9 proteins were purified using a Superdex 75 10/300 GL column (GE Healthcare). Glutaraldehyde (25%, Sigma) was diluted to a series of concentrations (0.2%, 0.4%, 0.6%, 0.8%, and 1%) in distilled water. The protein in 1× PBS buffer was incubated with glutaraldehyde on ice for 30 min. The reaction was then stopped by adding 2× loading buffer and heated at 100°C for 10 min before SDS‐PAGE analysis.The crosslinking SDS‐PAGE analyzed by Gelpro software for calculating the dimer and monomer proporations.
CD spectroscopy
Before CD measurement, all proteins were purified using a Superdex 75 10/300 GL column in 1× PBS running buffer. The CD spectra were recorded on a Jasco J‐715 Spectropolarimeter (JASCO, Maryland, US) using an average of three scans over the 190–240 nm wavelength range in a 10‐mm path length quartz cuvette at 25°C. Data for raw ellipticity, θ obs (in millidegrees) were converted to mean residue ellipticity, [θ] (in millidegrees) using the formula,
where θ is ellipticity in millidegrees, l is the path length of the cuvette in cm, MRW is the mean residue weight, and c is the concentration in mg/mL.40 Data were analyzed with CONTINLL in CDPro,41 using the reference sets 10(SMP56) including membrane proteins, 4(SP43) including 43 soluble proteins, and 7(SDP48) including SP43 with five denatured proteins. Good fits were obtained by averaging results for the fractional content of secondary structures of nsp9 wild‐type calculated from X‐ray structure data by DSSP 2.0.42
Sedimentation velocity analysis
Sedimentation velocity experiments were carried out using Rayleigh interference optical systems in a Beckman‐Coulter XL‐I analytical ultracentrifuge (Brea, CA). Samples were prepared in 1× PBS buffer and loaded into the cells. A Beckman An‐50 Ti rotor was equilibrated under vacuum at 20°C for about 1 h and then accelerated to 55,000 rpm. Absorbance scans at 280 nm were acquired at 4.5‐min intervals until the main boundary had reached the bottom of the cell. The SINV data were analyzed using SEDFIT or SEDPHAT.43 The solvent density and viscosity were calculated using SEDNTERP.44
ESI mass spectrometry
Aliquots of each sample (20 μL) were analyzed on a Waters Synapt G1 high‐definition mass spectrometer (Milford, MA). Mass spectra were acquired within a mass range of 400–4000 m/z, with capillary voltage 3.0 kV in positive‐ion mode, sample cone voltage 45 V, extraction cone voltage 4.0 V, desolvation temperature 300°C, source temperature 100°C, and desolvation gas flow 400 L/h. External calibration with a solution of sodium iodide achieved mass accuracy within 10 ppm.
Electrophoretic mobility shift assay (EMSA)
EMSAs were performed to detect nucleic acid affinity of wild‐type and mutant nsp9s by mixing proteins with RNA. In the standard assay, a concentration gradient of nsp9 wild‐type samples (0.01, 0.1, 0.2, 0.5, 1.0, and 2.0 nmol) was mixed with 0.01 nmol 6‐carboxyfluorescein‐labeled RNA (5′FAM‐CGACUCAUGGACCUUGGC*A*G, Takara) on ice for 30 min before loading onto native‐PAGE for electrophoresis. RNA residues with phosphorothioate linkages (the last two residues on the 3′ terminus, labeled *) were resistant to nuclease. To detect the weak affinity between nsp9 and RNA, a modified zone interference gel electrophoresis was used. Oligonucleotide RNA (5′‐CGACUCAUGGACCUUGGC*A*G‐3′, Takara) and single‐strand DNA (5′‐CGACTCATGGACCTTGGCAG‐3′, TsingKe) were dissolved at a concentration of 3 nmol before loading onto a 1% agarose gel. The gel was run for 60 min with running buffer (20 mM MOPS, 2 mM NaOAc, 1 mM EDTA, pH 7.0) at 100 mA, 4°C. With the poles of the electrodes interchanged, 1.5 nmol nsp9 protein was loaded and run for another 60 min. The nucleic acids in the gels were stained with gel view (BioTeke) for 5 min. The proteins in the gels were stained with Coomassie brilliant blue R‐250 and visualized under UV with Quantum ST5 (Vilber).
Surface plasmon resonance (SPR)
SPR measurements were performed using a Biacore 3000 (GE Healthcare). The short RNA (5′biotin‐CGACUCAUGGACCUUGGCAG‐3′) was diluted to a final concentration of 10 nM in running buffer (20 mM HEPES, 100 mM NaCl, 0.005% Tween‐80) and immobilized on the SA sensor chip (GE Healthcare). The sensor chip contained four flow cells. Two different surface capacities were prepared on each flow cell with 300 RU of RNA and 410 RU of RNA respectively, while the other two flow cells served as reference. For detecting the interactions between RNAs and nsp9, running buffer was used as the basal buffer with a constant flow rate of 30 μL/min. The association time with the RNA sensor was set to 60 s, and the dissociation time to 180 s. Nsp9 proteins were injected from low to high concentrations to observe interactions with the oligonucleotide. To examine the role of individual residues in RNA binding, a series of nsp9 mutants were injected at the same concentration of 25 μΜ in the sensor chip. Between trials, the sensor chips were regenerated with 0.2% SDS to remove any trace of nsp9 proteins.
Biolayer interferometry (BLI) binding analysis
An Octet RED 96 (ForteBio) instrument was used to perform BLI measurements. Labeled RNA (5′biotin‐CGACUCAUGGACCUUGGC*A*G‐3′, Takara) was diluted to 30 nM in running buffer (20 mM HEPES, 100 mM NaCl, 0.005% Tween‐80). SA sensors were immersed in running buffer for 10 min. Labeled RNA was loaded onto the SA sensors for 120 s to 0.6 nm with vibration at 1000 rpm. The nsp9‐RNA associations were performed using a gradient of wild‐type nsp9 concentrations (6.25 μM, 12.5 μM, 25 μM, 50 μM, and 100 μM) for 300 s and the dissociations were performed for 360 s. Nsp9 mutants were diluted to 50 μM and loaded onto the sensors under the same conditions. Results were analyzed with Octet Data Analysis Software 7.0.
Supporting information
Supporting Information.
Acknowledgments
We gratefully acknowledge the core facility staff members of West China Hospital. We thank D. O'Keefe for scientific editing. We are grateful to Dr. Georges Mer at Mayo Clinic for careful reading of the manuscript. We also thank Qingfei Huang in the Department of Chemistry, Sichuan University, for CD spectrum analysis, Shuang Guo in the Department of Life Science, Nankai University, for sedimentation velocity data analysis, and Lixin Li at Tianjin International Joint Academy of Biomedicine for ESI‐MS analysis. We are grateful to Yuanyuan Che, zhenwei Yang at Institute of Biophysics for technical help with ITC and BLI experiments.
Grant sponsor: National Science Foundation of China; Grant numbers: 31370735, 31300150, and 31670737; Grant sponsor: Key Technology Research and Development Program of Sichuan Province; Grant number: 14zc1822; Grant sponsor: Sichuan Province Science Foundation for Youths; Grant number: 2015JQ0029; Grant sponsor: Specialized Research Fund for the Doctoral Program of Higher Education of China; Grant number: 20130032120090; Grant sponsor: Tianjin Municipal Natural Science Foundation; Grant number: 13JCYBJC42500; Grant sponsor: National Key Basic Research Program of China (973 program); Grant number: 2015CB859800.
Protein Data Bank Accession Codes: The Atomic coordinate and structure factors of IBV nsp9 were submitted to RCSB Protein Data Bank, with accession code 5C94.
Contributor Information
Haitao Yang, Email: yanght@tju.edu.cn.
Dan Su, Email: sudan@scu.edu.cn.
References
- 1. Spaan WJM, Cavanagh D (2004) Virus Taxonomy VIIIth Report of the ICTV. London: Elsevier/Academic Press; 7: 945–962. [Google Scholar]
- 2. Knipe DM, Fields BN. Fields virology In: Knipe DM, Ed. (2001) Lippincott Williams & Wilkins. [Google Scholar]
- 3. Hudson CB, Beaudette FR (1932) Infection of the Cloaca with the virus of infectious nronchitis. Science 76:34. [DOI] [PubMed] [Google Scholar]
- 4. Siddell SG, Ziebuhr J, Snijder EJ (2005) Topley and Wilson's microbiology and microbia infections. Virology 10:823–856. [Google Scholar]
- 5. Pereira HG. Reoviridae. Andrewes' Viruses of Vertebrates In: Porterfield JS, Ed. (1989) London: BaiIlibre Tindall; pp 42–57. [Google Scholar]
- 6. Peiris JS, Lai ST, Poon LL, Guan Y, Yam LY, Lim W, Nicholls J, Yee WK, Yan WW, Cheung MT, Cheng VC, Chan KH, Tsang DN, Yung RW, Ng TK, Yuen KY group Ss (2003) Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361:1319–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA, Berger A, Burguiere AM, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra JC, Muller S, Rickerts V, Sturmer M, Vieth S, Klenk HD, Osterhaus AD, Schmitz H, Doerr HW (2003) Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 348:1967–1976. [DOI] [PubMed] [Google Scholar]
- 8. Bermingham A, Chand MA, Brown CS, Aarons E, Tong C, Langrish C, Hoschler K, Brown K, Galiano M, Myers R, Pebody RG, Green HK, Boddington NL, Gopal R, Price N, Newsholme W, Drosten C, Fouchier RA, Zambon M (2012) Severe respiratory illness caused by a novel coronavirus, in a patient transferred to the United Kingdom from the Middle East, September 2012. Euro Surveill 17:20290. [PubMed] [Google Scholar]
- 9. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA (2012) Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367:1814–1820. [DOI] [PubMed] [Google Scholar]
- 10. Sawicki SG, Sawicki DL, Siddell SG (2007) A contemporary view of coronavirus transcription. J Virol 81:20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DC (2003) Severe acute respiratory syndrome vaccine development: experiences of vaccination against avian infectious bronchitis coronavirus. Avian Pathol 32:567–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen Y, Jiang L, Zhao W, Liu L, Zhao Y, Shao Y, Li H, Han Z, Liu S (2017) Identification and molecular characterization of a novel serotype infectious bronchitis virus (GI‐28) in China. Veterinary Microbiol 198:108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boursnell ME, Brown TD, Foulds IJ, Green PF, Tomley FM, Binns MM (1987) Completion of the sequence of the genome of the coronavirus avian infectious bronchitis virus. J Gen Virol 68:57–77. [DOI] [PubMed] [Google Scholar]
- 14. Liu DX, Tibbles KW, Cavanagh D, Brown TD, Brierley I (1995) Identification, expression, and processing of an 87‐kDa polypeptide encoded by ORF 1a of the coronavirus infectious bronchitis virus. Virology 208:48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lim KP, Ng LF, Liu DX (2000) Identification of a novel cleavage activity of the first papain‐like proteinase domain encoded by open reading frame 1a of the coronavirus Avian infectious bronchitis virus and characterization of the cleavage products. J Virol 74:1674–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Snijder EJ, Bredenbeek PJ, Dobbe JC, Thiel V, Ziebuhr J, Poon LL, Guan Y, Rozanov M, Spaan WJ, Gorbalenya AE (2003) Unique and conserved features of genome and proteome of SARS‐coronavirus, an early split‐off from the coronavirus group 2 lineage. J Mol Biol 331:991–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu Y, Cong L, Chen C, Wei L, Zhao Q, Xu X, Ma Y, Bartlam M, Rao Z (2009) Crystal structures of two coronavirus ADP‐ribose‐1″‐monophosphatases and their complexes with ADP‐Ribose: a systematic structural analysis of the viral ADRP domain. J Virol 83:1083–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Piotrowski Y, Hansen G, Boomaars‐van der Zanden AL, Snijder EJ, Gorbalenya AE, Hilgenfeld R (2009) Crystal structures of the X‐domains of a Group‐1 and a Group‐3 coronavirus reveal that ADP‐ribose‐binding may not be a conserved property. Protein Sci 18:6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kong L, Shaw N, Yan L, Lou Z, Rao Z (2015) Structural view and substrate specificity of papain‐like protease from avian infectious bronchitis virus. J Biol Chem 290:7160–7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Frieman M, Yount B, Agnihothram S, Page C, Donaldson E, Roberts A, Vogel L, Woodruff B, Scorpio D, Subbarao K, Baric RS (2012) Molecular determinants of severe acute respiratory syndrome coronavirus pathogenesis and virulence in young and aged mouse models of human disease. J Virol 86:884–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Egloff MP, Ferron F, Campanacci V, Longhi S, Rancurel C, Dutartre H, Snijder EJ, Gorbalenya AE, Cambillau C, Canard B (2004) The severe acute respiratory syndrome‐coronavirus replicative protein nsp9 is a single‐stranded RNA‐binding subunit unique in the RNA virus world. Proc Natl Acad Sci U S A 101:3792–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sutton G, Fry E, Carter L, Sainsbury S, Walter T, Nettleship J, Berrow N, Owens R, Gilbert R, Davidson A, Siddell S, Poon LL, Diprose J, Alderton D, Walsh M, Grimes JM, Stuart DI (2004) The nsp9 replicase protein of SARS‐coronavirus, structure and functional insights. Structure 12:341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gimpelev M, Forrest LR, Murray D, Honig B (2004) Helical packing patterns in membrane and soluble proteins. Biophys J 87:4075–4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kleiger G, Grothe R, Mallick P, Eisenberg D (2002) GXXXG and AXXXA: common alpha‐helical interaction motifs in proteins, particularly in extremophiles. Biochemistry 41:5990–5997. [DOI] [PubMed] [Google Scholar]
- 25. Miknis ZJ, Donaldson EF, Umland TC, Rimmer RA, Baric RS, Schultz LW (2009) Severe acute respiratory syndrome coronavirus nsp9 dimerization is essential for efficient viral growth. J Virol 83:3007–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ponnusamy R, Moll R, Weimar T, Mesters JR, Hilgenfeld R (2008) Variable oligomerization modes in coronavirus non‐structural protein 9. J Mol Biol 383:1081–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. YPC, Woo YH, Lau SKP, Yuen KY (2010) Coronavirus genomics and bioinformatics analysis. Viruses 2:1804–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Woo PC, Lau SK, Yip CC, Huang Y, Tsoi HW, Chan KH, Yuen KY (2006) Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1. J Virol 80:7136–7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Patrick CY, Woo YH, Susanna KP Lau, Kwok‐Yung Yuen (2010) Coronavirus Genomics and Bioinformatics Analysis. Viruses 2:1804–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. DeLano WL (2002) The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific. [Google Scholar]
- 32. Jones S, Thornton JM (1995) Principles of protein‐protein interactions. Proc Natl Acad Sci U S A 93:13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Abrahams JP, Kraal B, Bosch L (1988) Zone‐interference gel electrophoresis: a new method for studying weak protein‐nucleic acid complexes under native equilibrium conditions. Nucleic Acids Res 16:10099–10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guenaga J, de Val N, Tran K, Feng Y, Satchwell K, Ward AB, Wyatt RT (2015) Well‐ordered trimeric HIV‐1 subtype B and C soluble spike mimetics generated by negative selection display native‐like properties. PLoS Pathog 11:e1004570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ciesielski GL, Hytonen VP, Kaguni LS (2016) Biolayer interferometry: A novel method to elucidate protein‐protein and protein‐DNA interactions in the mitochondrial DNA replisome. Methods Mol Biol 1351:223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen B, Fang S, Tam JP, Liu DX (2009) Formation of stable homodimer via the C‐terminal alpha‐helical domain of coronavirus nonstructural protein 9 is critical for its function in viral replication. Virology 383:328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zust R, Miller TB, Goebel SJ, Thiel V, Masters PS (2008) Genetic interactions between an essential 3′ cis‐acting RNA pseudoknot, replicase gene products, and the extreme 3′ end of the mouse coronavirus genome. J Virol 82:1214–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ma Y, Chen C, Wei L, Yang Q, Liao M, Li X (2010) Crystallization and preliminary X‐ray diffraction studies of infectious bronchitis virus nonstructural protein 9. Acta Cryst F66:706–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McCoy AJ, Grosse‐Kunstleve RW, Storoni LC, Read RJ (2005) Likelihood‐enhanced fast translation functions. Acta Cryst D61:458–464. [DOI] [PubMed] [Google Scholar]
- 40. Yang JT, Wu CS, Martinez HM (1986) Calculation of protein conformation from circular dichroism. Methods Enzymol 130:208–269. [DOI] [PubMed] [Google Scholar]
- 41. Sreerama N, Woody RW (2000) Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem 287:252–260. [DOI] [PubMed] [Google Scholar]
- 42. Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 43. Schuck P (2000) Size‐distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 78:1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Laue T, Shah B, Ridgeway T, Pelletier S. Analytical Ultracentrifugation in Biochemistry and Polymer Science In: Harding SE, Rowe AJ, Horton JC, Ed. (1992) Cambridge: Royal Society of Chemistry. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information.