

Abstract
Vacuolar H+‐ATPase (V‐ATPase) is a large, multisubunit membrane protein complex responsible for the acidification of subcellular compartments and the extracellular space. V‐ATPase activity is regulated by reversible disassembly, resulting in cytosolic V 1‐ATPase and membrane‐integral V 0 proton channel sectors. Reversible disassembly is accompanied by transient interaction with cellular factors and assembly chaperones. Quantifying protein‐protein interactions involving membrane proteins, however, is challenging. Here we present a novel method to determine kinetic constants of membrane protein–protein interactions using biolayer interferometry (BLI). Yeast vacuoles are solubilized, vacuolar proteins are reconstituted into lipid nanodiscs with native vacuolar lipids and biotinylated membrane scaffold protein (MSP) followed by affinity purification of nanodisc‐reconstituted V‐ATPase (V 1 V 0ND). We show that V 1 V 0ND can be immobilized on streptavidin‐coated BLI sensors to quantitate binding of a pathogen derived inhibitor and to measure the kinetics of nucleotide dependent enzyme dissociation.
Keywords: vacuolar ATPase, lipid nanodiscs, biolayer interferometry, protein–protein interaction, inhibitor binding, membrane protein
Abbreviations
- BLI
biolayer interferometry
- ConA
concanamycin A
- MSP
membrane scaffold protein
- V‐ATPase
vacuolar ATPase
- V1V0ND
lipid nanodisc reconstituted vacuolar ATPase.
Introduction
The vacuolar H+‐ATPase (V‐ATPase; V 1 V 0‐ATPase) is an ATP‐dependent proton pump present in all eukaryotic cells. The enzyme is typically localized to the endomembrane system and functions in acidifying organelles for essential cellular functions including pH homeostasis, membrane trafficking, endocytosis, hormone secretion, and lysosomal degradation.1 In certain specialized cell types such as osteoclasts, renal tubular cells and cells of the male reproductive tract, the V‐ATPase is found on the plasma membrane where it acidifies the extracellular environment. Complete loss of V‐ATPase activity is embryonic lethal in mammals,2, 3 highlighting the essential nature of the enzyme. Not surprisingly therefore, aberrant V‐ATPase displaying hyper‐ or hypo‐activity has been implicated in a number of widespread human diseases such as osteoporosis,4 renal tubular acidosis,5 deafness,6 male infertility,7 viral infection,8 diabetes,9 and cancer,10 and it has been suggested that V‐ATPase may represent a valuable drug target for the development of therapies against these diseases.11, 12
V‐ATPase is a membrane integral, multi‐subunit protein complex composed of a cytosolic ATP hydrolyzing sector, V 1, and a membrane‐integral ion translocating sector, V 0 (Fig. 1). V‐ATPase is highly conserved from yeast to human and thanks to the relative ease of genetic manipulation, yeast has been a powerful model system to study the structure and mechanism of the eukaryotic enzyme. In yeast, V 1 is composed of subunits A3B3(C)DE3FG3H13 and V 0 contains subunits a,c8,c′,c″,d,e,f.14, 15 V‐ATPase belongs to the family of rotary motor enzymes that also includes F‐ and A‐ATPase/synthase found in bacteria, mitochondria, chloroplasts and Archaea. ATP hydrolysis in V 1 is coupled to rotation of a membrane integral ring of c subunits (“proteolipid” ring, composed of c8,c′,c″) in V 0, which results in an uphill transport of protons across the membrane.16 However, unlike F‐ and A‐type motors, V‐ATPase is regulated in vivo by a unique mechanism referred to as “reversible disassembly”, a process first described in yeast17 and insect.18 Upon nutrient deprivation in yeast, for example, V 1 disengages from V 0 with concomitant silencing of the enzyme's ATPase and proton translocation activities. Reversible enzyme dissociation is conserved in higher organisms19, 20, 21, 22, 23 and it has been proposed that the process provides a unique opportunity for therapeutic intervention by allowing for modulation of V‐ATPase activity (rather than complete inhibition) in a tissue specific manner.24 Reversible disassembly has been extensively studied in yeast25 and insect26 and it is known that the process requires an ATP hydrolysis competent enzyme25, 27 in addition to transient interaction with cellular effectors28 and chaperones.29 Understanding these interactions and their role in reversible disassembly would greatly benefit from a controlled in vitro system employing purified components. However, the limited stability of detergent solubilized V‐ATPase and the difficulties with analyzing protein–protein interactions in presence of detergent have limited such studies thus far.
Figure 1.
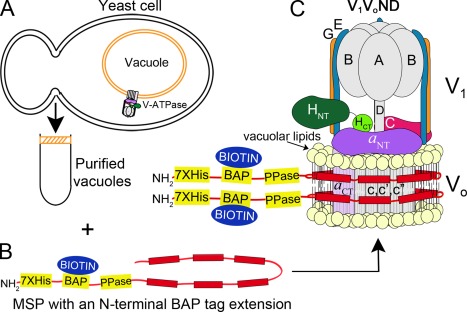
Purification and reconstitution strategy. (A) Yeast cells are lysed and vacuoles purified by flotation on a Ficoll gradient. (B) MSP with an N‐terminal extension containing a BAP tag for in vivo biotinylation and a Prescission protease cleavage site (PPase) is expressed in E. coli. Vacuoles are detergent solubilized and total vacuolar membrane proteins are nanodisc‐reconstituted using vacuolar lipids and biotinylated MSP. From this mixture of nanodisc‐reconstituted membrane proteins, V‐ATPase‐containing nanodiscs are purified by affinity chromatography using a FLAG‐tag at the G subunit N‐terminus. (C) V‐ATPase reconstituted in biotinylated and vacuolar lipid containing nanodiscs is schematically depicted as V 1 V 0ND. V‐ATPase is composed of a cytosolic V 1‐ATPase sector and a membrane integral V 0 proton channel sector.
Here we have developed a method for reconstituting V‐ATPase into lipid nanodiscs using native vacuolar lipids and membrane scaffold protein (MSP) engineered to contain an N‐terminal biotin acceptor protein (BAP) tag for biotinylation in E. coli. We show that lipid nanodisc‐reconstituted V 1 V 0 (V 1 V 0ND) is highly active and that the preparation can be immobilized on streptavidin‐coated biosensors for binding studies using biolayer interferometry (BLI). We further demonstrate that the system can be used to study the kinetics of the interaction between V‐ATPase and cellular factors known to bind to the enzyme and for monitoring the nucleotide‐dependent dissociation of the complex in real time.
Results
Preparation and characterization of lipid nanodisc‐reconstituted V‐ATPase
We initially attempted to first affinity purify V‐ATPase followed by nanodisc reconstitution using E. coli polar lipids as described for P‐glycoprotein.30 However, the tendency of the detergent solubilized enzyme to dissociate precluded this approach. We therefore developed a procedure that minimizes prolonged detergent exposure and allows for retention of native vacuolar lipids, some of which have been shown to be essential for enzyme function.31 To that end, total vacuolar proteins are detergent solubilized, reconstituted into nanodiscs with native vacuolar lipids (without the addition of exogenous lipids) followed by affinity purification of nanodisc‐reconstituted V 1 V 0 (V 1 V 0ND) (Fig. 1). The resulting V 1 V 0ND preparation is highly purified, showing bands for all the V‐ATPase subunits that can be resolved on SDS‐PAGE gels as seen in earlier preparations of detergent solubilized enzyme32 besides a band for MSP [Fig. 2(A)]. V 1 V 0ND displays robust MgATPase activity of ∼7.3 ± 0.3 U/mg (three independent purifications), 98% of which is inhibited by Concanamycin A [Fig. 2(B), red trace]. Concanamycin A (ConA) is a specific inhibitor of the V‐ATPase that binds to V 0 and inhibits rotation of the proteolipid ring, in turn inhibiting ATPase activity in the coupled V 1.33 The specific activity of purified vacuoles was ∼2.9 ± 0.9 U/mg (nine independent purifications), consistent with previously published results.34 Upon solubilization of vacuoles with dodecyl maltoside (DDM), the specific activity remained unchanged. However, while ∼85% of the ATPase activity of vacuoles was inhibited by ConA [Fig. 2(B), purple trace], inhibition of detergent‐solubilized enzyme was only ∼58% [Fig. 2(B), blue trace], suggesting that detergent causes partial uncoupling of the enzyme as has been described for the related F‐ATPase.35 Therefore, the virtually complete inhibition of V 1 V 0ND's ATPase activity by ConA indicates tight coupling of ATPase and proton pumping activities in lipid nanodisc‐reconstituted V 1 V 0. To verify the structural integrity of purified V 1 V 0ND, the preparation was subjected to glycerol gradient centrifugation followed by negative stain electron microscopy (EM). As can be seen from the silver stained SDS‐PAGE gel of the glycerol gradient fractions, the majority of the preparation migrated in a single peak on the gradient with only trace amounts of free subunits in fractions near the top of the gradient [Fig. 2(C)]. Negative stain EM analysis of the peak fraction of the glycerol gradient shown in Figure 2(C) showed monodisperse particles with the characteristic dumbbell shape of V‐ATPase complexes [Fig. 2(D)]. Taken together, reconstitution of V‐ATPase into lipid nanodiscs using native vacuolar lipids allows purification of a stable, highly active, and monodisperse enzyme preparation suitable for structural and biophysical studies.
Figure 2.
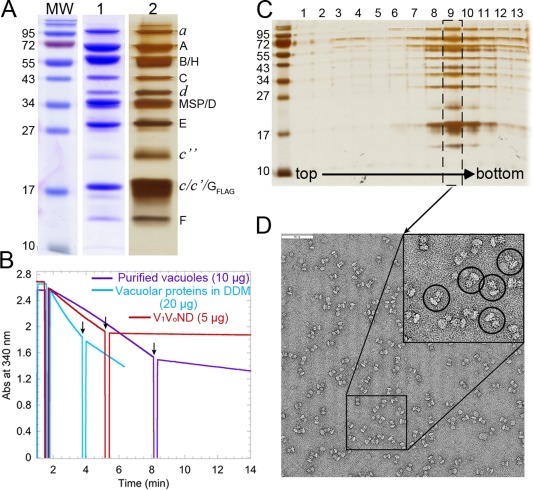
Characterization of V‐ATPase in lipid nanodiscs. (A) SDS‐PAGE of purified V 1 V 0ND showing the subunits of the V‐ATPase along with MSP. The three lanes show molecular weight markers (MW) and V 1 V 0ND, both Coomassie blue (1) and silver stained (2). (B) Representative ATPase activity measurements using an ATP regenerating assay with 10 μg purified vacuoles (purple trace), 20 μg DDM solubilized vacuoles (blue trace), and 5 μg purified V 1 V 0ND (red trace). The black arrows indicate points where 0.4 μM ConA was added in each case. The change in absorbance measured as a function of time was used to calculate the specific activity before and after addition of ConA. (C) Silver stained SDS‐PAGE of the fractions from the glycerol gradient. (D) Negative stain electron micrograph of purified V 1 V 0ND (fraction 9 of the glycerol gradient in panel (C)), showing monodisperse and assembled enzyme complexes. A 2× enlargement of the boxed region is shown as an inset and single V 1 V 0ND particles are highlighted by the black circles. Bar is 90 nm.
Biolayer interferometry of V 1 V 0ND for analysis of protein binding
The MSP used for reconstitution of the V‐ATPase into lipid nanodiscs contained an engineered N‐terminal BAP tag for specific attachment of a single biotin during protein expression in E. coli (Fig. 1). Initial experiments revealed that the level of biotinylation of MSP was close to stoichiometric (Supporting Information Fig. S1), that the biotin tag in V 1 V 0ND was accessible for binding to streptavidin‐coated BLI sensors, and that a concentration of ∼5 µg/mL V 1 V 0ND produced sufficient BLI signal for subsequent binding or dissociation experiments (data not shown). As a proof‐of‐principle, we then tested binding of the well characterized V‐ATPase inhibitor SidK to immobilized V 1 V 0ND for determining kinetic constants of the interaction (Fig. 3). SidK is a ∼60 kDa soluble polypeptide expressed by the pathogenic organism Legionella pneumophilia that specifically targets host cell V‐ATPases to inhibit phagosome acidification and subsequent digestion of the bacterium. It was previously shown that SidK inhibits yeast V‐ATPase by binding to the catalytic A subunit of the complex.36 BLI sensors with immobilized V 1 V 0ND were dipped in wells containing different concentrations of purified GST‐tagged SidK. The SidK–V 1 V 0ND interaction exhibited concentration dependence and the association and dissociation rates were used to estimate a K d of the interaction of ∼ 3.5 nM (Fig. 3). In another experiment, we tested binding of a monoclonal antibody directed against the catalytic A subunit of the enzyme and fitting of the binding data revealed a tight interaction in the sub‐nM range (Supporting Information Fig. S2). In summary, the binding experiments described above demonstrate that V‐ATPase reconstituted into nanodiscs using native lipids and biotinylated MSP can be used for BLI to study the kinetics of inhibitor and antibody binding.
Figure 3.
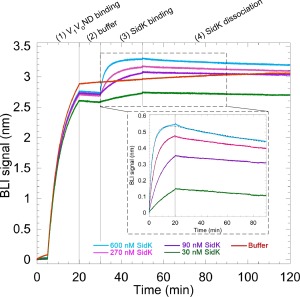
Biolayer interferometry with V 1 V 0ND. 5 μg/mL of biotinylated V 1 V 0ND was used to load streptavidin sensors (step 1) followed by dipping the sensors in buffer (step 2). The sensors were then dipped into wells containing either buffer, 30, 90, 270, or 600 nM of GST‐tagged SidK to measure association rates (step 3). All the sensors were then dipped into buffer containing wells to measure dissociation rates (step 4). GST‐SidK showed concentration dependent interaction with V 1 V 0ND as evident from the association and dissociation phases (enlarged box). The signal obtained in buffer (red) was subtracted from the GST‐SidK signal and the resultant curves were globally fit to a one site binding equation (black dotted traces). The on rate was found to be 1.25 × 104 ± 19 s−1 and the off rate was 4.4 × 10−5 ± 9.7 × 10−8 s−1, resulting in a K d of ∼3.5 nM. Representative experiment from two repeats is shown.
Dissociation of V 1 from V 0 monitored by biolayer interferometry
As mentioned earlier, under conditions of starvation, V 1 dissociates from V 0, causing silencing of the enzymes ATPase and proton translocation activities. We used immobilized V 1 V 0ND for BLI as an experimental setup to understand the kinetics of the dissociation of V 1 from V 0. From experiments with yeast vacuoles, it is known that V 1 can be released from membrane integral V 0 by treatment with chaotropic agents such as KNO3 or KI in presence of MgATP.37, 38 To determine the rate of chaotrope‐induced dissociation, BLI sensors with immobilized V 1 V 0ND were dipped into wells containing KNO3 with or without 1 mM MgATP (Fig. 4). Under these conditions, the BLI signal rapidly decreased but only when MgATP and the chaotrope were present together, consistent with the earlier experiments using yeast vacuoles.37, 38 Upon chaotrope treatment in presence of MgATP, the BLI signal decreased by only ∼15%, an unexpected finding considering that the size of the V 1 sector (∼600 kDa) is comparable to the size of V 0 in nanodiscs (∼400 kDa without lipids). However, incomplete stripping of V 1 from V 0 had also been observed after chaotrope treatment of yeast vacuoles.37, 38 It is also possible that the majority of the BLI signal was due to the nanodisc bound V 0 as the method of detection could be more sensitive to the nanodiscs bound close to the sensor surface. As a control, chaotrope treated sensors were dipped into wells containing Prescission protease. The close to complete loss of signal due to proteolytic cleavage of the protease site engineered between the BAP tag and the N‐terminus of MSP [Fig. 1(B)] confirmed that the majority of the initial BLI signal was due to specific binding of V 1 V 0ND via biotinylated MSP (Fig. 4).
Figure 4.
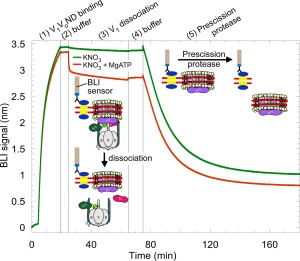
Biolayer interferometry to monitor the dissociation of V 1 from V 0 in real time. 5 μg/mL of biotinylated V 1 V 0ND was used to load the streptavidin sensors (step 1) followed by dipping the sensors in buffer (step 2). One sensor was dipped in 300 mM KNO3 (green trace) while the other sensor was dipped in 300 mM KNO3 with 1 mM MgATP (red trace) (step 3). In the presence of both KNO3 and MgATP, V 1 was released from V 0 as indicated by a fast drop in BLI signal (red trace, step 3). The sensors were then dipped in buffer (step 4). The remaining V 1 V 0ND and V 0ND molecules on the sensors were then released by dipping the sensors in 0.2 U/μL of Prescission protease (step 5). The experiment has been illustrated using schematics of V 1 V 0ND and its components. Representative experiment from two repeats is shown.
Of course, chaotrope induced dissociation is not physiological and to estimate the propensity of the enzyme to dissociate under more native conditions, we tested the effect of nucleotides and known V‐ATPase inhibitors on the dissociation of V 1 from V 0 (Fig. 5). To that purpose, BLI sensors with immobilized V 1 V 0ND were dipped in wells containing MgATP (Fig. 5, pink trace), MgATP with ConA (V 0 inhibitor) (Fig. 5, orange trace), MgATP with N‐ethylmaleimide (NEM; V 1 inhibitor), which covalently modifies an essential cysteine in the P‐loops of the catalytic A subunits, thereby preventing substrate binding to active sites39 (Fig. 5, red trace), MgAMPPNP, which is a non‐hydrolyzable substrate analog of MgATP (V 1 inhibitor) (Fig. 5, blue trace) and MgATP with KNO3 as a control (Fig. 5, purple trace). We found that V 1 was released from V 0 at a slow rate (k off ∼ 1 × 10−3 s−1) in the presence of MgATP, suggesting that rotary catalysis destabilizes the V 1/V 0 interface. Dissociation of V 1 from V 0 in the presence of MgATP has also been observed for the purified detergent solubilized insect V‐ATPase,27 suggesting that destabilization of the V 1/V 0 interface due to ATP hydrolysis is a conserved feature of the enzyme. Consistent with earlier findings that V‐ATPase must be ATP hydrolysis‐competent for in vivo dissociation to occur,25, 40 we observed that inhibition of V 1 by MgAMPPNP or NEM prevented dissociation (Fig. 5). Interestingly, release of V 1 from V 0 under conditions of ATP hydrolysis did not appear to be impaired in presence of ConA, an observation that seems inconsistent with the fact that V 1 V 0ND's ATPase activity is close to zero in presence of the inhibitor [Fig. 2(B)].
Figure 5.

Effect of nucleotides and inhibitors on the dissociation of V 1 from V 0. V 1 V 0ND immobilized on streptavidin sensors (step 1) was dipped into buffer (step 2), and then into wells containing either buffer, 1 mM MgATP, 1 mM MgAMPPNP, 2 mM NEM/1 mM MgATP, 0.2 μM Concanamycin A/1 mM MgATP, or 300 mM KNO3/1 mM MgATP (step 3) followed by buffer (step 4). Following dissociation of V 1 from V 0 in step 3, the remaining V 1 V 0ND or V 0ND was released from the sensors using 0.2 U/μL of Prescission protease (step 5). Inset: Enlarged view of the dissociation of V 1 from V 0 (step 3). In the presence of MgATP and MgATP/ConA (pink and orange traces, respectively), V 1 is released from V 0 at a slow rate. The two curves were fit to the equation for a single exponential decay (black dotted trace) and the off rates were found to be 1 × 10−3 ± 3.3 × 10−6 s−1 in MgATP and 9 × 10−4 ± 3.3 × 10−6 s−1 in ConA/MgATP. In the presence of KNO3/MgATP, V 1 was released from V 0 with an initial fast rate of 2.1 × 10−2 ± 1 × 10−6 s−1 followed by a slow off rate of 7.5 × 10−5 ± 1.6 × 10−7 s−1. In the presence of buffer (green), MgAMPPNP (blue) and NEM/MgATP (red), V 1 remains associated with V 0 over the duration of the experiment. Representative experiment from two repeats is shown.
Taken together, the results show that BLI of immobilized V 1 V 0ND can be used to monitor the dissociation of the complex in real time and while the observed rate of dissociation is significantly lower than the rate observed in live yeast,25 above experiments indicate that V 1 V 0ND is disassembly competent during catalytic turnover.
Discussion
Biophysical characterization of membrane proteins and membrane protein–protein interaction is challenging due to the presence of detergents that are required for keeping membrane proteins water soluble. Detergents are known to destabilize multisubunit membrane protein complexes35, 41 and the light scattering from detergent micelles can interfere with binding assays that depend on optical detection methods. The results summarized above indicate that there are two major advantages of using the “nanodisc‐reconstitution before purification” strategy. First, the lipids used for the process of reconstitution were derived directly from yeast vacuoles and therefore represent the native environment of the enzyme, an important aspect as it has been shown that in vivo V‐ATPase activity depends on low abundance, long chain fatty acid sphingolipids.31 Second, V‐ATPase is held together by multiple low‐affinity protein‐protein interactions for efficient enzyme regulation by reversible disassembly.42 These low‐affinity interactions are susceptible to breakage when exposed to detergent for long periods of time. Multiple attempts were made to first extract and purify the V‐ATPase from pelleted membranes followed by reconstitution into E. coli lipid‐containing nanodiscs as described for the enzyme's V 0 membrane sector.43 However, likely due to the prolonged exposure to detergent required for purifying the complex, the resultant preparation showed a significant fraction of disassembled V 1 and V 0 particles with little or no measurable ATPase activity (data not shown). Using the “nanodisc‐reconstitution before purification” strategy to obtain V 1 V 0ND, we found that the resulting preparation consisted of close to completely assembled V‐ATPase with high specific ATPase activity. The ATPase activity was 98% inhibited by Concanamycin A, suggesting tight coupling between V 1 V 0ND's V 1 and V 0 sectors.
For immobilization on streptavidin‐coated BLI sensors, we engineered a BAP tag at the N‐terminus of MSP for site directed biotinylation during expression in E. coli. The high affinity of the streptavidin‐biotin interaction allows immobilization of V 1 V 0ND at a relatively low concentration with little risk of nonspecific binding. We first showed that our method can be used successfully to study the kinetics of interaction of the V‐ATPase with binding partners such as an anti‐subunit A monoclonal antibody and the pathogen derived inhibitor, SidK. Both examples allowed an estimate of the K ds of the interactions ranging from the sub‐nM (anti‐A mAb) to the low nM (SidK) range. In addition, we showed that the dissociation of V 1 from V 0 under different conditions could be monitored in real time. The data indicate that ATP hydrolysis is required for disassembly to occur, consistent with in vivo and in vitro experiments conducted with yeast cells25 and detergent solubilized insect V‐ATPase,27 respectively. However, the observation that the V 0 specific inhibitor ConA did not prevent or slow down disassembly was surprising, but it is possible that residual turnover and/or the conformational changes induced by rapid binding and release of MgATP are sufficient to destabilize the complex. It should be noted that the rate of V 1 V 0 disassembly upon glucose withdrawal in live yeast occurs within minutes and is therefore significantly faster than the rate observed here. Thus, while the experiment shows that MgATP hydrolyzing V‐ATPase has the propensity to dissociate, additional events induced by, for example, nutrient withdrawal are required to accelerate the process. The experimental setup developed here will allow for a systematic analysis of the cellular factors that may be involved in promoting or impeding the disassembly process in vivo such as phosphorylation,26 variations in pH44 and the interaction with mTORC,22 phosphoinositides45 or glycolytic enzymes.28
Conclusion and Future Perspective
We have developed a procedure to reconstitute yeast V‐ATPase into native lipid containing nanodiscs for binding and dissociation assays using biolayer interferometry. Recent progress with the structure determination of the assembled V‐ATPase15 along with those of the disassembled and silenced V 1 46 and V 0 43, 47 suggest that following disassembly, there is a mismatch in the subunit conformations between V 1 and V 0 that may act to prevent spontaneous reassembly under conditions when enzyme activity is not desired.43, 46 To overcome the kinetic barriers for reassembly created by the conformational mismatch, subunit rearrangements at the V 1/V 0 interface facilitated by effector and chaperone proteins such as the RAVE complex48 are required. The here developed tool of immobilizing V 1 V 0ND for BLI can be used to study the function of these effector and chaperone proteins in the (re)assembly process. The method can also be extended to the discovery of small molecules that will modulate enzyme assembly, a strategy that may ultimately lead to the development of therapies designed to overcome aberrant V‐ATPase activity in human disease.
Materials and Methods
Materials
The plasmid encoding 6XHis‐tagged membrane scaffold protein, pMSP1E3D1, was a gift from Dr. Stephen Sligar (Addgene plasmid # 20066).49 Molecular biology reagents were purchased from Takara Bio Inc. Zymolyase 100T was from amsbio. An N‐terminal extension of MSP encoding the 7XHis tag, BAP tag and Prescission protease cleavage site was synthesized and subcloned into the pET28a vector (pMSP1E3D1) by Bio Basic Inc., resulting in plasmid pHBPMSP1E3D1. All other reagents were of analytical grade.
Strains
A yeast strain deleted for the VMA10 gene (subunit G), BY4741 vma10Δ:: KanMX was a kind gift from Dr. Patricia Kane, SUNY Upstate Medical University. The 3′ and 5′ UTR regions of VMA10 along with the KanMX selection marker was amplified from genomic DNA using the following primers: Vma10delKanfwd: 5′ gcc aca ccc ttc cct att aac tgg act cc tat tcc agc tca tc 3′ and Vma10delKanrev: 5′ ggc tga atg gat aaa gcg aga gtc gta aag acc gaa tgc aat gtc 3′. The PCR product was gel purified and transformed into a wild‐type yeast strain SF838‐5Aα50 for homologous recombination. Transformants were selected based on their resistance to G418 and their inability to grow on pH 7.5 and Ca2+. Deletion of the VMA10 gene was confirmed by DNA sequencing. For affinity purification of V 1 V 0ND, SF838‐5Aα vma10Δ::KanMX was transformed with a pRS315 plasmid encoding the G subunit with an N‐terminal FLAG‐tag.51
Purification of biotinylated MSP
A plasmid encoding the BirA gene for in vivo biotinylation of the BAP tag (pBirAcm52) was a kind gift from Dr. Thomas Duncan, SUNY Upstate Medical University. E. coli strain BL21 (DE3) was co‐transformed with pHBPMSP1E3D1 and pBirAcm. Cells were grown in rich broth supplemented with 0.1 mM d‐biotin, 34 µg/mL chloramphenicol and 30 µg/mL kanamycin at 37°C to an OD595 of ∼0.5 followed by induction with 0.5 mM IPTG at 37°C for 3–4 h. Biotinylated MSP was purified as described.43 The extent of biotinylation of MSP was estimated by performing pull down assays with streptactin beads as follows. MSP eluted from the Ni column was diluted 1:2 and 1:3 in streptactin buffer (15 mM Tris, pH 7.8, 150 mM NaCl, 0.5 mM EDTA) and incubated with 100 µL streptactin beads at 4°C for 1 h. The beads were washed for 1 h at 4°C in streptactin buffer, boiled in cracking buffer for 10 min and the beads, supernatant and wash were resolved using SDS‐PAGE. 100 µL of streptactin buffer served as a control.
Isolation of vacuoles
Vacuoles were isolated by flotation on Ficoll density gradients as described with the following modifications.53 Briefly, yeast strain SF838‐5Aα vma10Δ transformed with pRS315 encoding FLAG tagged G subunit51 was grown to an OD595 of ∼1.0 in YPD, pH 5 media and harvested by centrifugation at 5,000×g for 30 min. Cells from 12 l of culture were washed and resuspended in 100 mL of 1.2M sorbitol supplemented with ∼15 mg of zymolyase. Spheroplasts were recovered in 100 mL of 2× YPD and 100 mL of 2.4M sorbitol followed by resuspension in buffer A (10 mM MES–Tris pH 6.9, 0.1 mM MgCl2, and 12% Ficoll 400) supplemented with 1 mM PMSF and 2 µg/mL each of leupeptin, pepstatin, and chymostatin. The suspension was homogenized in a Dounce homogenizer using ten strokes and centrifuged for 40 min at 71,000×g. Vacuole wafers on top of the gradient were resuspended in buffer B containing 8% Ficoll 400, homogenized and centrifuged for 40 min at 71,000×g. Wafers were collected and resuspended in 1.5 mM Mes–Tris, pH 7.0, 4.8% glycerol, 1 mM BME. Vacuoles were analyzed for Concanamycin A sensitive ATPase activity using an ATP regenerating assay46 and protein concentration was determined with a modified BCA assay.54 Vacuoles were frozen in liquid nitrogen until further use.
Extraction of V‐ATPase and reconstitution into lipid nanodiscs
Typically three batches of vacuoles (12 liters each) were thawed, combined and supplemented with 1 mM PMSF and 2 µg/mL each of leupeptin, pepstatin, and chymostatin. 1.2 mg of n‐dodecyl β‐d‐maltopyranoside (DDM) per 1 mg of vacuolar membrane protein was added to the vacuoles and the mixture was rotated for 1 h at 4°C. Detergent solubilized vacuoles were centrifuged at 100,000×g for 15 min. The pellet was discarded and the supernatant was used for reconstitution into lipid nanodiscs. To the detergent solubilized vacuole sample containing vacuolar lipids, purified biotinylated MSP was added in a molar ratio of 1:50 (vacuolar protein: biotinylated MSP) and the mixture was rotated at 4°C for 1 h followed by the addition of 1.5 g of activated bio‐beads and rotation of the sample at 4°C for 2 h to remove detergent. V‐ATPase containing lipid nanodiscs were then separated by anti‐FLAG affinity chromatography (Fig. 1). The FLAG column eluate was concentrated using a Vivaspin concentrator (100,000 MWCO) and subjected to a 50–20% glycerol gradient density centrifugation as described.53 400 μL fractions were collected from top to bottom and analyzed by SDS‐PAGE. For negative stain electron microscopy, 5 μL of V 1 V 0ND (1:10 diluted from fraction 9 of the density gradient) was applied to glow‐discharged carbon‐coated copper grids and stained with 1% uranyl formate. Micrographs were collected on a JEOL JEM‐2100 at a magnification of 60,000× and a defocus of −1.5 μm.
Purification of GST‐SidK
The plasmid for overexpression of GST‐tagged SidK (pzl797) was a kind gift from Zhao‐Qing Luo, Purdue University.36 E. coli Rosetta2 cells (Novagen) were transformed with pzl797 and the cells were grown in rich broth supplemented with 34 μg/mL Ampicillin and 34 µg/mL Chloramphenicol. Cells were induced with 0.5 mM IPTG at an OD595 of ∼0.5 and grown for 6 h at 25°C. Cells were pelleted by centrifugation at 3000×g, resuspended in GST buffer (10 mM Tris pH 7.8, 150 mM NaCl, 0.5 mM EDTA, 1 mM DTT) and stored at −20°C until use. Cells were lysed by sonication in the presence of 20 µg/mL DNase, 1 mg/mL lysozyme and 1 mM PMSF. Lysed cells were centrifuged at 13,000×g for 40 min and the supernatant was subjected to affinity chromatography using a pre‐equilibrated GST column. The column was washed with ten column volumes of GST buffer and eluted in ten 2 mL fractions using 10 mM reduced glutathione. To improve purity, eluted GST‐SidK was subjected to size exclusion chromatography using a Superdex 200 1.6 cm × 50 cm column attached to an Äkta FPLC (GE Healthcare). The peak fractions were pooled, analyzed by SDS‐PAGE and used for biolayer interferometry.
Biolayer interferometry
An Octet‐RED system with streptavidin‐coated sensors (FortéBio, SA biosensors, catalog number 18‐5019) were used for biolayer interferometry, a technique similar to surface plasmon resonance.55 In all BLI experiments described, the buffer used was 20 mM Tris, pH 7.4 with 150 mM NaCl, 0.5 mM EDTA, 1 mM β mercaptoethanol and 0.5 mg/mL BSA. All steps were conducted at 22°C with each biosensor stirred in 0.2 mL of sample at 1000 rpm and a standard measurement rate of 5 s−1. In each case, the biosensors were pre‐wetted in BLI buffer for 10 min and then dipped in wells containing 5 μg/mL V 1 V 0ND. Control experiments were performed to show that the buffer components did not interact with the streptavidin sensors. Details of individual BLI experiments are given in their respective figure legends. FortéBio's data analysis software (version 6.4) was used for subtraction of reference sensors, Savitsky–Golay filtering and global or local fitting of the kinetic rates where applicable.
Conflict of Interest
The authors declare that they have no conflicts of interest with the contents of this article.
Supporting information
Supporting Information
Acknowledgments
We thank Dr. Patricia Kane for yeast strains and reagents, Dr. Thomas Duncan for help with biolayer interferometry and Dr. Rebecca Oot, Mr. Sergio Couoh‐Cardel, and Mr. Nicholas Stam for helpful discussions and for critically reading the manuscript.
Narrative: The enzymatic activity of the proton pumping vacuolar ATPase (V‐ATPase) is regulated by reversible disassembly in vivo, a process that can be exploited for therapeutic intervention. We have developed a novel method to reconstitute V‐ATPase into native lipid containing nanodiscs for biolayer interferometry. We demonstrate that the method can be used to determine binding constants and to study V‐ATPase disassembly in real time.
References
- 1. Forgac M (2007) Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol 8:917–929. [DOI] [PubMed] [Google Scholar]
- 2. Inoue H, Noumi T, Nagata M, Murakami H, Kanazawa H (1999) Targeted disruption of the gene encoding the proteolipid subunit of mouse vacuolar H(+)‐ATPase leads to early embryonic lethality. Biochim Biophys Acta 1413:130–138. [DOI] [PubMed] [Google Scholar]
- 3. Sun‐Wada G, Murata Y, Yamamoto A, Kanazawa H, Wada Y, Futai M (2000) Acidic endomembrane organelles are required for mouse postimplantation development. Dev Biol 228:315–325. [DOI] [PubMed] [Google Scholar]
- 4. Thudium CS,VKJ, Karsdal MA, Henriksen K (2012) Disruption of the V‐ATPase functionality as a way to uncouple bone formation and resorption—a novel target for treatment of osteoporosis. Curr Prot Pept Sci 13:141–151. [DOI] [PubMed] [Google Scholar]
- 5. Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, Hulton SA, Sanjad SA, Al‐Sabban EA, Lifton RP, Scherer SW, Karet FE (2000) Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116‐kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet 26:71–75. [DOI] [PubMed] [Google Scholar]
- 6. Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez‐Soriano J, Santos F, Cremers CW, Di Pietro A, Hoffbrand BI, Winiarski J, Bakkaloglu A, Ozen S, Dusunsel R, Goodyer P, Hulton SA, Wu DK, Skvorak AB, Morton CC, Cunningham MJ, Jha V, Lifton RP (1999) Mutations in the gene encoding B1 subunit of H+‐ATPase cause renal tubular acidosis with sensorineural deafness. Nature Genet 21:84–90. [DOI] [PubMed] [Google Scholar]
- 7. Breton S, Smith PJ, Lui B, Brown D (1996) Acidification of the male reproductive tract by a proton pumping (H+)‐ATPase. Nat Med 2:470–472. [DOI] [PubMed] [Google Scholar]
- 8. Lu X, Yu H, Liu SH, Brodsky FM, Peterlin BM (1998) Interactions between HIV1 Nef and vacuolar ATPase facilitate the internalization of CD4. Immunity 8:647–656. [DOI] [PubMed] [Google Scholar]
- 9. Sun‐Wada GH, Toyomura T, Murata Y, Yamamoto A, Futai M, Wada Y (2006) The a3 isoform of V‐ATPase regulates insulin secretion from pancreatic β‐cells. J Cell Sci 119:4531–4540. [DOI] [PubMed] [Google Scholar]
- 10. Sennoune SR, Bakunts K, Martínez GM, Chua‐Tuan JL, Kebir Y, Attaya MN, Martínez‐Zaguilán R (2004) Vacuolar H+‐ATPase in human breast cancer cells with distinct metastatic potential: distribution and functional activity. Am J Physiol Cell Physiol 286:1443–1452. [DOI] [PubMed] [Google Scholar]
- 11. Fais S, De Milito A, You H, Qin W (2007) Targeting vacuolar H+‐ATPases as a new strategy against cancer. Cancer Res 67:10627–10630. [DOI] [PubMed] [Google Scholar]
- 12. Kartner N, Manolson MF (2014) Novel techniques in the development of osteoporosis drug therapy: the osteoclast ruffled‐border vacuolar H+‐ATPase as an emerging target. Expert Opin Drug Discov 9:505–522. [DOI] [PubMed] [Google Scholar]
- 13. Kitagawa N, Mazon H, Heck AJ, Wilkens S (2008) Stoichiometry of the peripheral stalk subunits E and G of yeast V1‐ATPase determined by mass spectrometry. J Biol Chem 283:3329–3337. [DOI] [PubMed] [Google Scholar]
- 14. Powell B, Graham LA, Stevens TH (2000) Molecular characterization of the yeast vacuolar H+‐ATPase proton pore. J Biol Chem 275:23654–23660. [DOI] [PubMed] [Google Scholar]
- 15. Zhao J, Benlekbir S, Rubinstein JL (2015) Electron cryomicroscopy observation of rotational states in a eukaryotic V‐ATPase. Nature 521:241–245. [DOI] [PubMed] [Google Scholar]
- 16. Stewart AG, Sobti M, Harvey RP, Stock D (2013) Rotary ATPases: models, machine elements and technical specifications. Bioarchitecture 3:2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kane PM (1995) Disassembly and reassembly of the yeast vacuolar H(+)‐ATPase in vivo . J Biol Chem 270:17025–17032. [PubMed] [Google Scholar]
- 18. Sumner JP, Dow JA, Earley FG, Klein U, Jager D, Wieczorek H (1995) Regulation of plasma membrane V‐ATPase activity by dissociation of peripheral subunits. J Biol Chem 270:5649–5653. [DOI] [PubMed] [Google Scholar]
- 19. Trombetta ES, Ebersold M, Garrett W, Pypaert M, Mellman I (2003) Activation of lysosomal function during dendritic cell maturation. Science 299:1400–1403. [DOI] [PubMed] [Google Scholar]
- 20. Lafourcade C, Sobo K, Kieffer‐Jaquinod S, Garin J, van der Goot FG (2008) Regulation of the V‐ATPase along the endocytic pathway occurs through reversible subunit association and membrane localization. PLoS One 3:e2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sautin YY, Lu M, Gaugler A, Zhang L, Gluck SL (2005) Phosphatidylinositol 3‐kinase‐mediated effects of glucose on vacuolar H+‐ATPase assembly, translocation, and acidification of intracellular compartments in renal epithelial cells. Mol Cell Biol 25:575–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zoncu R, Bar‐Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM (2011) mTORC1 senses lysosomal amino acids through an inside‐out mechanism that requires the vacuolar H(+)‐ATPase. Science 334:678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stransky LA, Forgac M (2015) Amino acid availability modulates vacuolar H+‐ATPase assembly. J Biol Chem 290:27360–27369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kane PM (2012) Targeting reversible disassembly as a mechanism of controlling V‐ATPase activity. Curr Prot Pept Sci 13:117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Parra KJ, Kane PM (1998) Reversible association between the V1 and V0 domains of yeast vacuolar H+‐ATPase is an unconventional glucose‐induced effect. Mol Cell Biol 18:7064–7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Voss M, Vitavska O, Walz B, Wieczorek H, Baumann O (2007) Stimulus‐induced phosphorylation of vacuolar H(+)‐ATPase by protein kinase A. J Biol Chem 282:33735–33742. [DOI] [PubMed] [Google Scholar]
- 27. Huss M, Wieczorek H (2007) Influence of ATP and ADP on dissociation of the V‐ATPase into its V(1) and V(O) complexes. FEBS Lett 581:5566–5572. [DOI] [PubMed] [Google Scholar]
- 28. Lu M, Ammar D, Ives H, Albrecht F, Gluck SL (2007) Physical interaction between aldolase and vacuolar H+‐ATPase is essential for the assembly and activity of the proton pump. J Biol Chem 282:24495–24503. [DOI] [PubMed] [Google Scholar]
- 29. Smardon AM, Nasab ND, Tarsio M, Diakov TT, Kane PM (2015) Molecular interactions and cellular itinerary of the yeast RAVE (Regulator of the H+‐ATPase of Vacuolar and Endosomal Membranes) complex. J Biol Chem 290:27511–27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ritchie TK, Kwon H, Atkins WM (2011) Conformational analysis of human ATP‐binding cassette transporter ABCB1 in lipid nanodiscs and inhibition by the antibodies MRK16 and UIC2. J Biol Chem 286:39489–39496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Finnigan GC, Ryan M, Stevens TH (2011) A genome‐wide enhancer screen implicates sphingolipid composition in vacuolar ATPase function in Saccharomyces cerevisiae . Genetics 187:771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang Z, Zheng Y, Mazon H, Milgrom E, Kitagawa N, Kish‐Trier E, Heck AJ, Kane PM, Wilkens S (2008) Structure of the yeast vacuolar ATPase. J Biol Chem 283:35983–35995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huss M, Ingenhorst G, Konig S, Gassel M, Drose S, Zeeck A, Altendorf K, Wieczorek H (2002) Concanamycin A, the specific inhibitor of V‐ATPases, binds to the V(o) subunit c. J Biol Chem 277:40544–40548. [DOI] [PubMed] [Google Scholar]
- 34. Charsky CM, Schumann NJ, Kane PM (2000) Mutational analysis of subunit G (Vma10p) of the yeast vacuolar H+‐ATPase. J Biol Chem 275:37232–37239. [DOI] [PubMed] [Google Scholar]
- 35. Tsunoda SP, Aggeler R, Noji H, Kinosita K, Jr. , Yoshida M, Capaldi RA (2000) Observations of rotation within the F(o)F(1)‐ATP synthase: deciding between rotation of the F(o)c subunit ring and artifact. FEBS Lett 470:244–248. [DOI] [PubMed] [Google Scholar]
- 36. Xu L, Shen X, Bryan A, Banga S, Swanson MS, Luo ZQ (2010) Inhibition of host vacuolar H+‐ATPase activity by a Legionella pneumophila effector. PLoS Pathog 6:e1000822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Parra KJ, Kane PM (1996) Wild‐type and mutant vacuolar membranes support pH‐dependent reassembly of the yeast vacuolar H+‐ATPase in vitro . J Biol Chem 271:19592–19598. [DOI] [PubMed] [Google Scholar]
- 38. Kane PM, Yamashiro CT, Stevens TH (1989) Biochemical characterization of the yeast vacuolar H(+)‐ATPase. J Biol Chem 264:19236–19244. [PubMed] [Google Scholar]
- 39. Feng Y, Forgac M (1992) Cysteine 254 of the 73‐kDa A subunit is responsible for inhibition of the coated vesicle (H+)‐ATPase upon modification by sulfhydryl reagents. J Biol Chem 267:5817–5822. [PubMed] [Google Scholar]
- 40. MacLeod KJ, Vasilyeva E, Baleja JD, Forgac M (1998) Mutational analysis of the nucleotide binding sites of the yeast vacuolar proton‐translocating ATPase. J Biol Chem 273:150–156. [DOI] [PubMed] [Google Scholar]
- 41. Yang Z, Wang C, Zhou Q, An J, Hildebrandt E, Aleksandrov LA, Kappes JC, DeLucas LJ, Riordan JR, Urbatsch IL, Hunt JF, Brouillette CG (2014) Membrane protein stability can be compromised by detergent interactions with the extramembranous soluble domains. Protein Sci 23:769–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Oot RA, Wilkens S (2012) Subunit interactions at the V1–Vo interface in yeast vacuolar ATPase. J Biol Chem 287:13396–13406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stam NJ, Wilkens S (2016) Structure of nanodisc reconstituted vacuolar ATPase proton channel: definition of the interaction of rotor and stator and implications for enzyme regulation by reversible dissociation. J Biol Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dechant R, Binda M, Lee SS, Pelet S, Winderickx J, Peter M (2010) Cytosolic pH is a second messenger for glucose and regulates the PKA pathway through V‐ATPase. EMBO J 29:2515–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li SC, Diakov TT, Xu T, Tarsio M, Zhu W, Couoh‐Cardel S, Weisman LS, Kane PM (2014) The signaling lipid PI(3,5)P(2) stabilizes V(1)‐V(o) sector interactions and activates the V‐ATPase. Mol Biol Cell 25:1251–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Oot RA, Kane PM, Berry EA, Wilkens S (2016) Crystal structure of yeast V1‐ATPase in the autoinhibited state. EMBO J 35:1694–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mazhab‐Jafari MT, Rohou A, Schmidt C, Bueler SA, Benlekbir S, Robinson CV, Rubinstein JL (2016) Atomic model for the membrane‐embedded VO motor of a eukaryotic V‐ATPase. Nature 539:118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Smardon AM, Tarsio M, Kane PM (2002) The RAVE complex is essential for stable assembly of the yeast V‐ATPase. J Biol Chem 277:13831–13839. [DOI] [PubMed] [Google Scholar]
- 49. Denisov IG, Baas BJ, Grinkova YV, Sligar SG (2007) Cooperativity in cytochrome P450 3A4: linkages in substrate binding, spin state, uncoupling, and product formation. J Biol Chem 282:7066–7076. [DOI] [PubMed] [Google Scholar]
- 50. Stevens TH, Rothman JH, Payne GS, Schekman R (1986) Gene dosage‐dependent secretion of yeast vacuolar carboxypeptidase Y. J Cell Biol 102:1551–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang Z, Charsky C, Kane PM, Wilkens S (2003) Yeast V1‐ATPase: affinity purification and structural features by electron microscopy. J Biol Chem 278:47299–47306. [DOI] [PubMed] [Google Scholar]
- 52. Shah NB, Hutcheon ML, Haarer BK, Duncan TM (2013) F1‐ATPase of Escherichia coli: the ε‐ inhibited state forms after ATP hydrolysis, is distinct from the ADP‐inhibited state, and responds dynamically to catalytic site ligands. J Biol Chem 288:9383–9395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Uchida E, Ohsumi Y, Anraku Y (1985) Purification and properties of H+‐translocating, Mg2+‐adenosine triphosphatase from vacuolar membranes of Saccharomyces cerevisiae . J Biol Chem 260:1090–1095. [PubMed] [Google Scholar]
- 54. Couoh‐Cardel S, Milgrom E, Wilkens S (2015) Affinity purification and structural features of the yeast vacuolar ATPase Vo membrane sector. J Biol Chem 290:27959–27971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shah NB, Duncan TM (2014) Bio‐layer interferometry for measuring kinetics of protein–protein interactions and allosteric ligand effects. J Vis Exp 84:e51383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information