

Abstract
Human metallothionein 1a, a protein with two cysteine‐rich metal‐binding domains (α with 11 Cys and β with 9), was analyzed in its metal‐free form by selective, covalent Cys modification coupled with ESI‐MS. The modification profiles of the isolated β‐ and α‐fragments reacted with p‐benzoquinone (Bq), N‐ethylmalemide (NEM) and iodoacetamide (IAM) were compared with the full length protein using ESI‐mass spectral data to follow the reaction pathway. Under denaturing conditions at low pH, the reaction profile with each modifier followed pathways that resulted in stochastic, Normal distributions of species whose maxima was equal to the mol. eq. of modifier added. Our interpretation of modification at this pH is that reaction with the cysteines is unimpeded when the full protein or those of its isolated domains are denatured. At neutral pH, where the protein is expected to be folded in a more compact structure, there is a difference in the larger Bq and NEM modification, whose reaction profiles indicate a cooperative pattern. The reaction profile with IAM under native conditions follows a similar stochastic distribution as at low pH, suggesting that this modifier is small enough to access the cysteines unimpeded by the compact structure. The data emphasize the utility of residue modification coupled with electrospray ionization mass spectrometry for the study of protein structure.
Keywords: metallothionein, cysteine modification, ESI‐MS, protein structure, iodoacetamide, N‐ethylmalemide, p‐benzoquinone, thiol reactions, post‐translational modification
Introduction
Since their discovery in 1957,1 metallothioneins (MTs) have been of interest due to their many unique structural and metal binding properties. These special properties include binding of up to 7 Zn2+ and Cd2+, forming two metal‐thiolate clustered domains (the α and β‐domains) that are joined by a short linker sequence.2, 3 While the biological function(s) of this family of proteins are still debated, it is generally agreed that their functions are connected with zinc and copper homeostasis,4, 5, 6, 7 redox signalling,8, 9, 10 and toxic metal sequestration.11, 12, 13, 14, 15 Despite these many proposed functions, and their capacity to bind so many metals,16, 17 MTs are surprisingly small, flexible proteins that lack optically active structural features. This lack of formal secondary and tertiary structure in the absence of bound metal ions precludes traditional structural analysis via spectroscopic methods.18, 19 In addition, the dynamic nature of the coordinated metals in the metal‐binding sites makes characterization even more difficult for metalation states other than the fully saturated protein.2, 20
The structures of metalated metallothioneins have been probed with a wide range of methods, including fluorescent resonance energy transfer (FRET),21 ion‐mobility mass‐spectrometry (IM‐MS)22 and, following cysteine modification, electrospray ionization mass spectrometry (ESI‐MS) analysis.23, 24, 25 Traditional ESI‐MS studies have resulted in a large library of mass spectrometric data being reported that clearly distinguish between metalation states present in solution. Through the semi‐quantitative properties of ESI‐MS, the distribution of those species can be analyzed.26 In addition to the extent of metal‐saturation, ESI‐mass spectral data can also determine the relative concentrations of the many differentially modified protein species simultaneously.24 The abundance of easily modifiable cysteine residues (20 in mammalian MTs) offers an opportunity to exploit this reactivity to obtain information on the solution structure of MTs based on the relative reactivity of individual cysteinyl thiols.25, 27, 28 This experiment is similar to the use of H/D exchange in identifying regions of proteins that are more or less exposed to the solvent.29, 30 Analysis of charge state distributions31 and ion drift in IM‐MS gives additional information about the overall folded state of the protein.32
ESI‐mass spectral data have been used to determine structural properties of apo‐MT25 and MT partially metalated with As3+ 24 and Cd2+.28, 33 ESI‐MS data can also be used to determine quantitative biochemical parameters as recently demonstrated in the determination of arsenic metalation kinetics,34 cadmium and zinc equilibrium, and kinetic constants28, 35, 36 and copper binding affinity.37 The key to these semi‐quantitative data is the assumption that apo‐MTs and those with different numbers of metals coordinated exhibit very similar ionization efficiencies; this assumption has been supported by the literature, especially for metalation of metallothioneins.38 While it is true that the different MT‐isoforms will have slightly varying ionization efficiencies,39 when analyzing modifications of the same isoform, the behavior of all species in solution has been demonstrated to be predictable and the metal‐binding parameters determined align closely with those determined by other methods.36, 40, 41
Previous studies using cysteine modification agents to probe the solution structure and metal binding properties of MTs have focused on one modifier at a time, leading us to question whether there was a bias based on the size and relative solvation properties of the modifying molecule.
In this article, we use three cysteine modification agents of varying size and hydrophobicity, p‐benzoquinone (Bq), N‐ethylmalemide (NEM), and iodoacetamide (IAM), to probe MT solution structure as a function of the accessibility of its cysteine residues. We carried out the reactions of the full‐length human MT1a and its isolated α‐ and β‐domains under native and denaturing conditions. By analyzing the characteristic modification profiles of folded and unfolded MTs (obtained at neutral and low pH) as well as changes in charge state distribution, we describe the modification properties of the folded, globular apo‐metallothionein structure under native conditions and compare it under denaturing conditions. The ESI‐mass spectral data provide modification profiles and charge state distributions that we use to determine the relative accessibility of the cysteinyl thiols to the modifiers, which leads to conclusions about the overall compactness of the proteins.
Results
The isolated α‐ and β‐domain fragments of MT were reacted with each of the three cysteine modifying agents to probe the compactness of their structures at neutral (native conditions) and low pH (denaturing conditions). The fragments were studied separately from the full‐length protein to test whether cysteinyl thiols would be buried to a different extent within the smaller volumes of the individual fragments, especially the small β‐domain.
Modification of the isolated β‐domain fragment of metallothionein
Figure 1 shows the reaction profile of the β‐domain fragment (N‐terminus fragment) with p‐benzoquinone (Bq). The data show the distinct cysteine access differences between the extended conformation state of the fragment at low pH and a more compact, native conformation state at neutral pH. At low pH the modifications follow a systematic and stochastic trend as the number of molar equivalents of Bq is increased. At pH 6.8, the pattern is very different, with both the unmodified (0 Bq) apo‐fragment and the fully modified (9 Bq) fragment coexisting. While some adducts are present in the low pH spectra, they did not add to the shielding of cysteine residues and a Normal distribution of modifications is seen.
Figure 1.
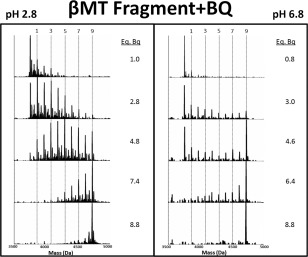
Representative deconvoluted ESI‐MS spectra from the modification of the β‐domain fragment of MT1a with p‐benzoqinone (Bq). The reaction at pH 2.8 (left panel) and 6.7 (right panel) gave markedly different reaction products labelled with vertical lines for the 1, 3, 5, and 9 Bq bound masses. Note that the apo‐fragment is not indicated, its mass lies to the left of the “1” line. The fully modified fragment is indicated by the “9” line. The number of molar equivalents of Bq added in the stepwise titration is listed to the right of each panel up to 8.8.
In the second test, the more hydrophobic and slightly larger modifier, NEM, was used. The reaction profile of the β‐domain fragment with NEM is shown in Figure 2 under native and denaturing conditions. At low pH, the data show an incremental increase in modification species as the molar equivalents of NEM is increased. At pH 7.4, NEM modified fragments from apo‐(0 NEM) to fully modified (9 NEM) are present until the end; a stark departure from the pattern than observed at low pH and remarkably similar to the Bq modification pattern at neutral pH. At all points during the reaction either apo‐ or NEM9‐β‐MT are the most abundant species.
Figure 2.
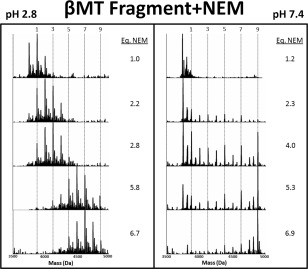
Representative deconvoluted ESI‐MS spectra from the stepwise modification of the βMT fragment by NEM. The extent of the modification in terms of number of covalent NEM modifications on the MT is shown by the vertical lines for the 1, 3, 5, and 9 NEM bound masses. Note that the apo‐fragment is not indicated, its mass lies to the left of the “1” line. The fully modified fragment is indicated by the “9” line. The number of molar equivalents of NEM added in the stepwise titration is listed to the right of each panel up to 8.8. The NEM was added stepwise with the mol eq as shown to the right of each panel at pH 2.8 (left panel) and pH 7.4 (right panel).
The reaction profiles for the NEM and Bq modification reactions of the β‐domain fragment of MT1a (Figs. 1 and 2) are strikingly similar at low pH. Both exhibit a stochastic, Normal distribution of intermediate species under these denaturing conditions. Under more native conditions for both modifiers the starting apo‐β‐MT and end product, Mod9β‐MT are the two dominant species in solution with minor abundance of intermediate species. It appears that the modification of the β‐domain fragment leads to the protein unfolding, promoting subsequent modification reactions due to the disruption of native structure and exposure of previously buried Cys residues to the solvent. The series of spectra show that throughout the stepwise addition, the apo and the fully modified species are the most abundant. The spectra around the 4.0 NEM mol. eq. point demonstrate the differences between conditions most clearly. To further understand the properties of this metal‐free, folded state of MT under these conditions, the charge state distributions were analyzed.
The charge states of both isolated domain fragments were similar following complete modification with both Bq and NEM. The charge state distributions for the NEM modifications of the β‐domain fragment are shown in Figure 3(C, D). The charge state average shifts higher following the Cys modification reaction, indicating an increase in surface area. However, this weighted average shifts only slightly for the β‐domain. Also the average charge state is essentially unchanged when going from neutral to low pH indicating there is no dramatic volume change between compact and extended conformations.
Figure 3.
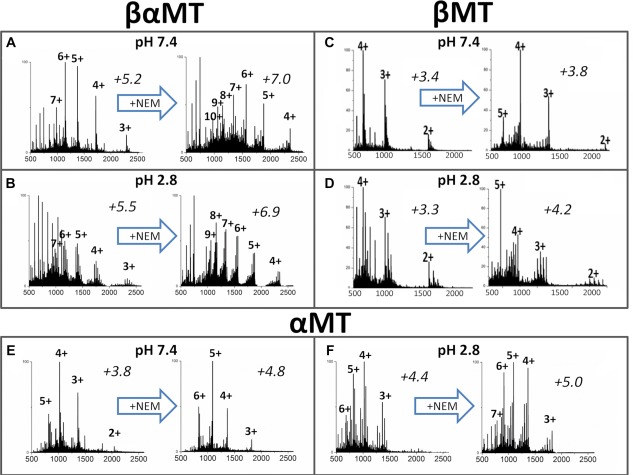
Representative ESI‐MS charge state manifolds measured during cysteine modification of βαMT1a and its isolated fragments. Spectra at approx. 1.6 mol. eq. of NEM added are shown on the left side of each panel and those near the end of the titration on the right side. The weighted average charged state is shown above each spectrum in italics.
Modification of the isolated α‐domain fragment of metallothionein
The α‐domain fragment reacted in a very similar manner to the β‐domain when modified with both Bq and NEM. The data for the reaction of the α‐domain fragment of MT1a with Bq has been published previously.25 The stepwise NEM modification reaction of the α‐domain fragment under native and denaturing conditions is shown in Figure 4.
Figure 4.
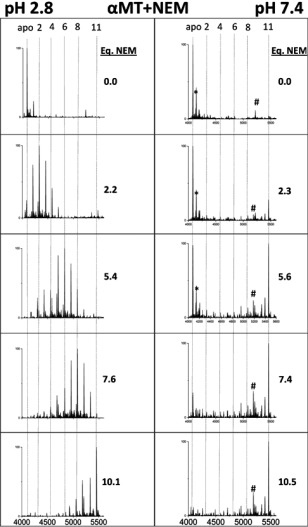
Representative deconvoluted ESI‐MS spectra of the modification of α‐MT fragment with NEM. The extent of the covalent NEM modification (0 −11) is shown by vertical lines (apo (0), 2, 4, 6, 8, and 11). NEM was added stepwise, with the molar equivalents added shown to the right of each panel, at pH 7.4 (left panel) and 2.8 (right panel). *‐+60 Da adduct, #‐unknown contaminant at 5183 Da not corresponding to MT or any modified species.
The NEM reaction at pH 2.8 follows the trend for the β‐domain fragment and also the previously reported modification reaction of the α‐domain fragment by Bq.25 Under denaturing conditions, where the peptide is expected to be unfolded in an extended conformation, modification follows a stochastic, Normal distribution of modification intermediates. At neutral pH it can be seen that the α‐domain fragment modification by NEM results in very low abundance of the partially‐modified intermediates (NEM1‐10α‐MT), even when compared with the β‐domain reaction. The series of spectra in Figure 4 (right) indicate that the modification of the apo‐fragment is initially sterically unfavorable compared with the low pH reactions. However, once modification has begun, those peptides unfold, exposing buried cysteinyl thiols and allowing subsequent modifications to proceed rapidly, culminating in the growing presence of the fully modified NEM11α‐fragment. The throttling effect of the initial steric hindrance results in the very low abundances of the partially modified fragment. The implication is that for the α‐domain fragment, the compact conformation under native conditions is resistant to modification. When comparing fragments, the lower abundance of intermediates for the α‐domain suggests that it adopts a more compact conformation than the β‐domain under native conditions.
The charge state analyses in Figure 3(E‐F) show a slight increase in average charge state upon reduction of pH, indicating a slight expansion between compact and extended conformations. This is a departure from the smaller β‐fragment where the average does not significantly change between conditions. The charge state average increases dramatically at neutral pH upon NEM modification from 3.8 to 4.8 and this is more similar to the large change seen in βα‐MT modification (net change of +1.8) than the β‐MT modification (net change of +0.4). The large charge state shift seen in α‐MT following the modification reaction is indicative of a more extended conformer with a larger surface area.
Modification of the full length protein (βα‐MT)
The full length protein, βα‐MT, consists of the two isolated domain fragments discussed in the previous sections, joined by a short linker sequence. The protein is much larger with 20 cys in the sequence. Much like the isolated domain fragments, the full protein exhibits a stochastic, Normal distribution of modification intermediates (NEM1‐19βα‐MT) under denaturing conditions as can be seen in Figure 5. Both NEM and Bq modifiers exhibit nearly identical patterns and this is consistent across all three peptides studied. Despite the differences in size and hydrophobicity, both NEM and Bq reactions cause similar changes to the structure of the proteins under native conditions.
Figure 5.
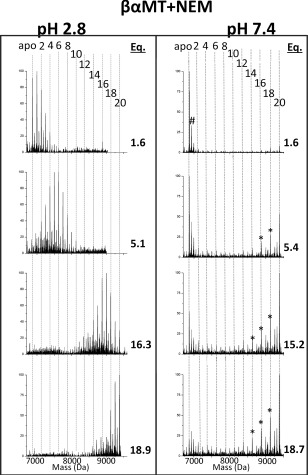
Deconvoluted mass spectra of NEM modifications of βα‐MT under native (pH 7.4, right) and denaturing (pH 2.8, left) conditions. The masses and numbers of the covalent modifications are shown by the vertical lines 0 (apo)−20. The mol. eq. of NEM reacted are shown to the right of each spectrum. *‐indicates the presence of partially oxidized MT species #‐+60 Da adduct.
The results of the modification reactions at pH 7.4 are different when compared with the isolated domains fragments. In particular, the charge state distribution significantly changes during the modification reaction at pH 7.4, which can be seen in Figure 3(A, B). Two new charge states (+9 and +8) emerge for the fully modified protein (NEM20βα‐MT) compared with only a slight shift in charge state abundance for the modified isolated α‐ and β‐domain fragments. This can be explained by a larger number of covalent modifications, resulting in a much larger peptide that also has a very different electrostatic surface and by the more drastic surface area difference between the compact and extended conformations.
For βα‐MT the native condition average charge state is 5.2 and increases marginally to 5.5 when the pH is lowered [left spectra, Fig. 3(A, B)]. There is a more dramatic shift in charge states upon modification in all cases. The largest differences in average charge state occurs under native conditions in the βα‐MT modification reaction [Fig. 3(A)]. While the appearance of new charge states indicates unfolding, the effect in the small MT‐protein and the even smaller fragments is more subtle than in classic studies, such as that for myoglobin unfolding.42 Thus it is difficult to make conclusions about the folded state of MT from charge states alone. However, when combined with the ESI‐MS data for the modification reaction patterns the conformational picture of MT becomes clearer.
Despite having different properties, it is clear that in the case of the full length protein, cysteine modification with Bq and NEM follows a similar pathway, resulting in the same types of reaction profiles. In order to contrast the reactions of these hydrophobic molecules, we also used iodoacetamide (IAM) to covalently modify the cysteine residues of the full length protein. IAM being smaller and more hydrophillic, is expected to be able to penetrate the compact conformation more easily and result in a less dramatic conformation change upon reaction. Thus, in Figure 6, we show the results of the IAM modification of the full length protein which has the largest volume for residue burial and would be the most likely to shield cysteinyl thiols from a small, hydrophillic modifier.
Figure 6.
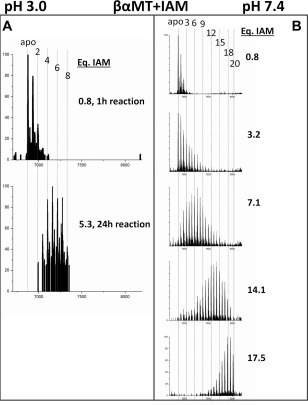
Deconvoluted mass spectra of the βα‐MT modification with IAM under denaturing conditions at pH 3.0 (A) and native conditions at pH 7.4 (B). At low pH, the kinetics of the reaction is very slow so only the spectrum at 1 h (top) and the incompletely modified spectrum at 24 h (bottom) are shown in (A).
Figure 6 shows the results of the IAM modification experiment. Under both denaturing (left panel) and native (right panel) conditions, IAM results in a Normal distribution of modified MT species, although the distribution is wider under the native conditions. At low pH, the modification reaction is much slower than for the other two modifiers tested, due to the difference in reaction mechanism, so excess IAM was reacted with the apo‐MT solution overnight. However, even with a large excess of IAM, after 24 h only 5.8 molar equivalents of IAM had reacted but the general pattern of modification is apparent and matches that of the other modifiers. It is also similar to the neutral pH reaction in terms of modification profile and distribution of modified species, although a smaller range of modified species are present at any given time. The IAM modification did not result in the cooperative modification patterns indicative of structure disruption in the full length protein.
Discussion
The challenges of investigating the structure of apo‐metallothioneins are numerous. The fluxional nature of a disorganized peptide and the lack of good chromophores make traditional methods of structure determination difficult or impossible. Proton NMR experiments showed a disordered structure for apo‐human, horse and bovine MT2a, however, the backbone chemical shifts were similar to the zinc and cadmium analogues.43 FRET studies have shown that the overall volumes of the full length protein, as well as its isolated domains, are largely unchanged between the metal‐free and metal‐saturated states.21, 44, 45 Ion‐mobility mass‐spectrometry has confirmed the presence of a range of conformations in the apo‐protein and inherent fluxionality before metal binding occurs.22 In previous reports, we have begun to probe the structure via global reactivity toward cysteine modifying agents.24, 25, 27, 31 Other groups have also probed the reactivity of the fully metalated protein towards NEM to investigate properties of demetalation and stability of cadmium‐MT clusters.33
In this study we sought to probe the metal‐free solution structures of βα‐MT and its isolated domain fragments using three different commonly used cysteine modifiers and analyze the reaction profiles using ESI‐MS. Our report includes the reaction profiles for modification of the isolated domain fragments and contrasts those differential modification data with the reactivity of the full length protein. The isolated domains being smaller, having less overall volume and a smaller surface area than the full protein were predicted to exhibit a smaller change in average charge state and this is observed in Figure 3. Although the average charge states only changed slightly in the isolated domain fragments (α increased 1.0 and 0.6 and β 0.4 and 0.9 at neutral and low pH, respectively), reaction profiles for NEM and Bq followed a cooperative pathway under native conditions at neutral pH in contrast with a non‐cooperative mechanism under denaturing conditions (Figs. 1, 2, 4).
We have previously described the cooperative pattern arising from unequal solvent access of the cysteines in apo‐MTs.25 The modification proceeds rapidly once the compact conformation initially proposed many years ago from early molecular dynamics calculations19 is unfolded by the initial modifications. This indicates that both isolated fragments are capable of adopting a more compact conformation to shield cysteine residues from the solvent despite their small size (3.7 and 4.08 kDa). This is surprising as the metal‐free β‐domain structure predicted by molecular dynamics (MD) simulation is a more open conformation.46 We have previously reported through MD/MM simulation that when demetalation is taken into account, the conformation adopted to accommodate the metalated state remains somewhat intact.47 Despite great advancements in our structure predicting abilities in silico, diverging simulations occur and experimental evidence must be relied upon, even when that evidence is difficult to obtain as in the case of disordered proteins.48, 49
In Figures 1, and 4, it is clear that during the reaction of both apo‐α and apo‐β, the unmodified apo‐fragments coexist with the fully modified species during the step‐wise addition of the modifier. This is counter‐intuitive for a protein with 9 or 11 cysteinyl thiols that remain unreacted while other individual MT molecules, with only a few free cysteines, react to become fully modified. This can be explained by the compact conformers adopted at neutral pH (6.8–7.4) making the cysteines sterically inaccessible. Conceptually, it can be understood that modifiers of any size or hydrophobicity should be at least somewhat hindered in their reaction and that is what we observe. The data we have measured are essentially “snapshots” of the reaction, the progress reports that would be observed if excess modifier were added and ultra‐high speed spectra recorded. Instead we use a step‐wise addition where we run out of modifier and the distribution of modified species is recorded at each point. The modification status is governed by the relative kinetics of the reaction to modify each cysteine. The reaction kinetics are governed by two major factors: the intrinsic rate of the chemical modification of the cysteine and the inhibition effect of the steric hindrance of the surrounding protein structure. Since the reactions are irreversible, the reverse equilibrium reaction has no effect on the distribution of the products.
Under denaturing conditions at low pH, all three modifiers (NEM, Bq, and IAM) followed pathways that resulted in a stochastic, Normal distribution of species that summed to the mol. eq. of modifier added. Under these conditions the protein adopts a more extended conformation and it is reasonable to conclude that all cysteines are essentially equally exposed to the solvent and incoming modifier molecules. This results in a purely statistical reaction pathway where individual MT molecules with more unreacted cysteinyl thiols are more likely to be modified than those with more modifications already present. Therefore, the intrinsic properties of the modifier are unimportant to the overall reaction profile. Under denaturing conditions, more consideration should be given to the reaction mechanism of the cyteine modifier which, in the case of IAM, may extend experimental equilibration times dramatically at lower pH.
A very different picture emerges during modification under native conditions at neutral pH. The larger, more hydrophobic modifiers (NEM and Bq) give rise to a cooperative‐like pattern. Under native conditions, the more compact conformations shield most of the cysteinyl thiols, allowing only a fraction of the apo‐protein to react, becoming increasingly unfolded as modifications proceed. Russell and coworkers showed a small fraction of apo‐MT2a exists as a more disordered and open conformer, at least in the gas phase for the +5 charge state.22 These conformers are likely more accessible for modification and are the ones that fully react at first, leaving the more compact conformers of apo‐MT with unmodified thiols. To visualize the equilibrium between apo‐MT conformers, molecular dynamics simulations were used to generate a range of possible structures (Fig. 8). Because the reaction with the modifiers is controlled by access to the cysteinyl thiols, the thiols in the more compact conformers remain less reactive than those species whose structure has been disrupted by modification. This results in a large fraction of unreacted apo‐MT, small amounts of partially modified protein and the accumulation of the fully modified protein as seen in Figure 7(B) for NEM and Bq modifications.
Figure 8.
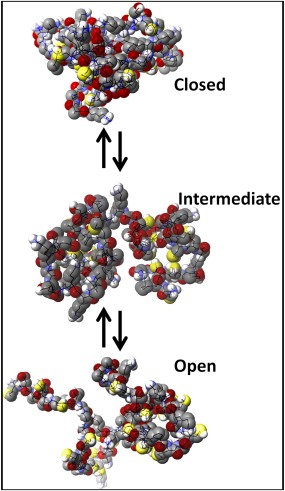
Molecular dynamics simulated structures of apo‐βα‐MT under native conditions. The “closed” structure is one where the Cys residues are more buried within the interior of a bundled protein, “intermediate” where there is more access to the Cys residues and “open” where there is the most unhindered access.
Figure 7.
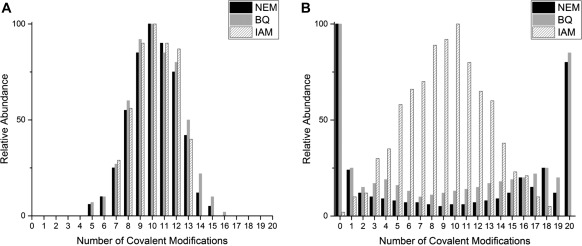
Comparison of the reaction profiles shown by the relative abundances of each modified βαMT‐species at approximately the halfway point, for the three cysteine modifiers used. (A) Relative abundances under denaturing conditions at pH 2.8–3.0 and (B) relative abundances under native conditions at pH 6.8–7.4. The relative abundances shown in this figure were extracted from raw deconvoluted ESI‐MS data at approximately 10 mol. eq. of each modifier.
It is interesting that this cooperative‐like pattern is not observed in the mass spectral data from the reaction of the smaller and more hydrophilic IAM modifier [Fig. 7(B)]. Instead, a wider stochastic distribution is observed and the width of the distribution of the IAM reaction profile is compared in Figure 9(C). The wider distribution is likely due to the unequal solvent accessibility of the cysteinyl thiols in the compact configuration. IAM is less disruptive to the compact conformers of MT than the larger and more hydrophobic Bq and NEM modifiers. This has implications for the choice of modifier when examining properties of cysteine‐rich proteins like MTs. IAM may be more suitable if native structure needs to remain largely intact, whereas Bq and NEM are better in producing a more dramatic change in reaction profile depending on the protein conformation. In addition, NEM is able to be used over a wide range of pH, 2.8–7.4 tested here, where IAM is unreliable at low pH (<4) and Bq unreliable at basic pH (>7) ranges.
Figure 9.
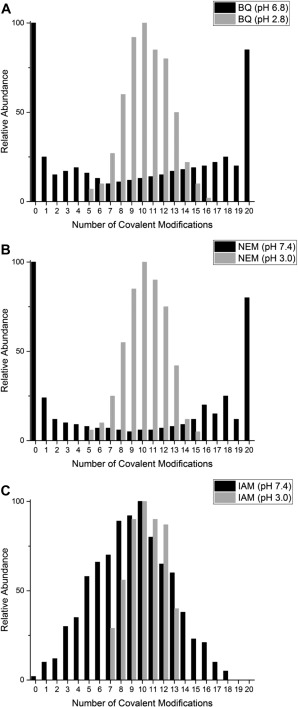
Extracted ESI‐MS reaction profiles for all three cysteine modifiers used. In this Figure, the relative abundances of each of the βαMT‐species are compared after 10 mol. eq. of each modifier has been reacted under native (neutral pH) and denaturing (low pH) conditions. (A) Comparison of the BQ, (B) NEM and (C) IAM reaction at low (gray bars) and neutral pH (black bars).
Figure 9 compares the reaction profiles at the halfway point (10 mol. eq.) under native and denaturing conditions. The Bq and NEM reaction profiles show how the reactivity of apo‐MT has been dramatically changed by the switch between compact and extended conformers at neutral and low pH respectively. However, these profiles are contrasted by the reaction profile of the IAM species, Figure 9(C), where the profiles follow stochastic distributions under both denaturing and native conditions, although the low pH distribution is much narrower. This may be a function of slow kinetics of the reaction under these conditions or may speak toward the equal accessibility of the cysteines and even less interference by the surrounding peptide due to the small size of the IAM molecule.
Our systematic approach in probing the structures of βα‐MT and its isolated domains have provided further evidence for the adoption of a globular or compact structure by the metal‐free protein. These data summarized in Table 1, indicate that even the smaller, isolated fragments behave in the same fashion. To completely understand the metalation mechanisms of MTs, a starting point must be established which up until recently was a poorly defined apo‐structure.
Table 1.
Summary of the Reaction Profiles of the Three Cysteine Modifiers the Full Length Protein, βαMT, and the two Isolated Fragments Under Native and Denaturing Conditions
| Protein construct | Modifier | Native reaction profile | Denatured reaction profile |
|---|---|---|---|
| βαMT1a | NEM | Cooperative | Non‐cooperative, stochastic |
| Bq31 | Cooperative | Non‐cooperative, stochastic | |
| IAM | Non‐cooperative, stochastic, wide distribution | Non‐cooperative, stochastic, narrow distribution | |
| αMT1a | NEM | Cooperative | Non‐cooperative, stochastic |
| Bq25 | Cooperative | Non‐cooperative, stochastic | |
| IAM | Non‐cooperative, stochastic | Non‐cooperative, stochastic | |
| βMT1a | NEM | Cooperative | Non‐cooperative, stochastic |
| Bq | Cooperative | Non‐cooperative, stochastic | |
| IAM | Non‐cooperative, stochastic | Non‐cooperative, stochastic |
The biological significance of apo‐metallothioneins remains a source of controversy within the community. Some have indicated that apo‐MT comprises a significant portion of the cellular MT pool,50, 51, 52 although there remains some controversy regarding the role and significance of cellular apo‐MTs. Regardless, upon ribosomal translation the nascent MT will be in its apo‐form and it remains a mystery how specificity and quantity of MT metalation occurs. The redox properties of MT have also been a source of controversy. While it is clear gluathione is the major source of reducing thiols in the cell,53, 54 it is unclear to what extent MTs also play a role.55, 56, 57 The “hidden” nature of most of the thiols in MT demonstrated in this article may cast doubt on how integral a role MT1a plays in cellular redox chemistry.
Conclusions
In this article, we describe the cysteine modification reactions of metallothionein and its isolated domain fragments with well‐known modification reagents (Table 1). All three peptides (α‐ and β‐domain fragments and full length βα‐MT) gave similar reaction profiles under native and denaturing conditions, indicating all three adopt compact conformations, burying selective cysteine residues. The larger modifiers, Bq and NEM, showed the most drastic difference between conditions, going from a stochastic, non‐cooperative pattern when denatured to a more cooperative one under native conditions. The smaller IAM only showed a broader distribution of modified cysteine residues under native conditions but the overall pattern was similar to the denatured conditions. The larger modifiers were better able to probe the ill‐defined and fluxional conformations of apo‐MT as they caused larger disruptions to the native, compact conformer. This highlights a new way to probe intrinsically disordered, or “less ordered”, protein structure by monitoring residue modification by ESI‐MS.
Materials and Methods
Protein preparation
Recombinant human metallothionein 1a (βα: MGKAAAACSC ATGGSCTCTG SCKCKECKCN SCKKCC SCCPMSCAKC AQGCVCKGAS EKCSCCK KA, α: GSMGKAAAACCSC CPMSCAKCAQGCVC KGASEKCSCCKKAAAA, β: GSMGKAAAA CSCATGGSCTCTGSC KCKECKCNSCKKAAAA) was expressed with an S‐tag in BL21 E. coli cells which has been described in detail elsewhere.58 In brief, cells containing the plasmid constructs for the full protein (βα‐MT1a) and for the isolated domains (β‐MT1a and α‐MT1a) were plated on to growth media containing kanamycin from a stock culture stored at −80°C and grown for 16 h at 37°C. The cells were then transferred into 1L broth cultures enriched with 50 μL of 1 M cadmium and incubated in a shaker for 4 h until OD 600 absorbance was between 0.6 and 0.8. Isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) was then added to induce expression of MT and 30 min later 150 µL of 1 M cadmium sulfate was added. The cells were collected 3.5 h after induction, centrifuged, and stored at −80°C.
The recombinant cells were lysed using a cell disruptor (Constant Systems, UK) at 20K psi. The cell lysate was centrifuged for 1 h to remove cellular debris. The supernatant was filtered and loaded on to an SP ion exchange column (GE Healthcare) with a total volume of 10 mL. The columns were washed with pH 7.4 10 mM Tris (tris‐hydroxymethylaminomethane) buffer for approximately 2 h to remove loosely bound proteins and other organic materials. MT was eluted using an increasing gradient of 1 M NaCl+ 10 mM Tris buffer at pH 7.4. The eluted MT was concentrated down to <20 mL and the S‐tag cleaved using a Thrombin Clean‐Cleave kit as per the manufacturers' instructions (Sigma‐Aldrich). The S‐tag was separated using an ion‐exchange column since the S‐tag does not bind as strongly as MT and thus elutes at lower salt concentrations. The eluted, cut Cd‐MT was concentrated to approximately 120 µM and stored at −20°C. In this article, we refer to the recombinant human liver MT1a isoform as “MT,” but all other isoforms are referred to with their complete isoform and subisoform descriptors.
To prepare MT for the modification experiments, aliquots were first demetalated and desalted using centrifugal filter tubes with a 3 kDa membrane (Millipore) and a 10 mM pH 2.8 ammonium formate buffer. The low pH solutions contained 1 mM dithiothreitol (DTT) to prevent oxidation of the free thiols in MT. The pH was raised by buffer exchange with argon saturated, pH 7.0 10 mM ammonium formate solutions that did not contain reductant. The final concentration of the protein solutions were determined by remetalation of a small aliquot with cadmium using the metal‐to‐ligand charge transfer band at 250 nm (ε250 = 89,000 Lmol−1 cm−1). The solutions were also monitored for oxidation using UV‐visible absorption spectroscopy to monitor absorption corresponding to 280 nm from oxidized disulfide. Once demetalated and desalted, the MT concentration was determined, all concentrations were between 40 and 90 µM to ensure good signal to noise ratios In the ESI‐MS experiment.
In addition to demetalating MT in the presence of DTT, the solutions were vacuum degassed and bubbled with Argon to displace any dissolved oxygen. This was also carried out for the 10 mM 1,4‐benzoquinone (Bq), N‐ethylmalemide (NEM) and iodoacetamide (IAM) solutions and the 0.5% NH4OH and 0.5% formic acid solutions as well to ensure no oxygen was introduced into the system during the modification reaction or pH adjustment. Great care was taken to reduce the possibility of oxidation of the protein especially at neutral pH. All modification agents were obtained from Sigma‐Aldrich (USA).
ESI‐MS spectra collection
Mass spectra were measured with a micrOTOF II electrospray‐ionization time‐of‐flight mass spectrometer (Bruker Daltonics) in the positive ion mode. NaI was used as the mass calibrant. The scan conditions for the spectrometer were: end plate offset, −500 V; capillary, +4200 V; nebulizer, 2.0 bar; dry gas flow, 8.0 L min−1; dry temperature, 30°C; capillary exit, 180 V; skimmer 1, 22.0 V; hexapole 1, 22.5 V; hexapole RF, 600 Vpp; skimmer 2, 22 V; lens 1 transfer, 88 μs; lens 1 pre‐pulse storage, 23 μs. The mass range was 500.0–3000.0 m/z. Spectra were assembled and deconvoluted using the Bruker Compass data analysis software package. ESI‐mass spectrometry was used to monitor all stages of the modification reaction. Approximately 1 molar equivalent of the modifying agent (Bq, NEM or IAM) was added stepwise and a spectrum recorded after each addition. The ESI‐mass spectra were recorded and averaged over 2 min.
Molecular models
MM3/MD calculations were carried out using Scigress Software (Fujitsu, Poland) and parametrized using the modified force field described by Chan et al.58 with the dielectric constant of 78 for water to obtain energy minimized structures of the Cu‐bound protein. The original apo‐MT1a structure was obtained from Rigby et al.19 A cycle of MM3 minimizations followed by MD calculations gave energy‐minimized apo‐MT structures reported here. The structures were first energy minimized using the MM3 calculation followed by an MD simulation at 500 K for 10 ps and then another MD simulation at the same temperature for 1000 ps. Structures with closed, intermediate and open configurations were selected from energy minima as representations of the multiple conformations apo‐MT adopts in solution.
Acknowledgments
We would like to thank Drs. Tyler Pinter, Micheal Tiedemann and Duncan Sutherland for helpful discussions on MT/Bq modification, as well as NSERC and U.W.O for funding sources listed on the first page.
References
- 1. Margoshes M, Vallee BL (1957) A cadmium protein from equine kidney cortex. J Am Chem Soc 79:4813–4814. [Google Scholar]
- 2. Boulanger Y, Armitage I, Miklossy K, Winge D (1982) 113Cd NMR study of a metallothionein fragment. Evidence for a two‐domain structure. J Biol Chem 257:13717–13719. [PubMed] [Google Scholar]
- 3. Braun W, Vasak M, Robbins A, Stout C, Wagner G, Kägi J, Wüthrich K (1992) Comparison of the NMR solution structure and the X‐ray crystal structure of rat metallothionein‐2. Proc Natl Acad Sci USA 89:10124–10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maret W (2000) The function of zinc metallothionein: a link between cellular zinc and redox state. The Journal of Nutrition 130:1455S–1458S. [DOI] [PubMed] [Google Scholar]
- 5. Fukada T, Yamasaki S, Nishida K, Murakami M, Hirano T (2011) Zinc homeostasis and signaling in health and diseases. J Biol Inorg Chem 16:1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maret W (2011) Metals on the move: zinc ions in cellular regulation and in the coordination dynamics of zinc proteins. BioMetals 24:411–418. [DOI] [PubMed] [Google Scholar]
- 7. Miyayama T, Ishizuka Y, Iijima T, Hiraoka D, Ogra Y (2011) Roles of copper chaperone for superoxide dismutase 1 and metallothionein in copper homeostasis. Metallomics 3:693–701. [DOI] [PubMed] [Google Scholar]
- 8. Maret W (2003) Cellular zinc and redox states converge in the metallothionein/thionein pair. The Journal of Nutrition 133:1460S–1462S. [DOI] [PubMed] [Google Scholar]
- 9. Kang YJ (2006) Metallothionein redox cycle and function. Exp Biol Med 231:1459–1467. [DOI] [PubMed] [Google Scholar]
- 10. Gonzalez‐Iglesias H, Alvarez L, García M, Petrash C, Sanz‐Medel A, Coca‐Prados M (2014) Metallothioneins (MTs) in the human eye: a perspective article on the zinc–MT redox cycle. Metallomics 6:201–208. [DOI] [PubMed] [Google Scholar]
- 11. Nordberg G, Goyer R, Nordberg M (1975) Comparative toxicity of cadmium‐metallothionein and cadmium chloride on mouse kidney. Arch Pathol 99:192–197. [PubMed] [Google Scholar]
- 12. Dorian C, Gattone VH, Klaasen CD (1992) Renal cadmium deposition and injury as a result of accumulation of cadmium‐metallothionein (CdMT) by the proximal convoluted tubules—a light microscopic autoradiography study with 109 CdMT. Toxicol Appl Pharmacol 114:173–181. [DOI] [PubMed] [Google Scholar]
- 13. Satarug S, Baker J, Reilly PE, Moore M, Williams D (2001) Changes in zinc and copper homeostasis in human livers and kidneys associated with exposure to environmental cadmium. Hum Exp Toxicol 20:205–213. [DOI] [PubMed] [Google Scholar]
- 14. Person RJ, Ngalame NNO, Makia NL, Bell MW, Waalkes MP, Tokar EJ (2015) Chronic inorganic arsenic exposure in vitro induces a cancer cell phenotype in human peripheral lung epithelial cells. Toxicol Appl Pharmacol 286:36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Qu W, Waalkes MP (2015) Metallothionein blocks oxidative DNA damage induced by acute inorganic arsenic exposure. Toxicol Appl Pharmacol 282:267–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sutherland DE, Stillman MJ (2011) The “magic numbers” of metallothionein. Metallomics 3:444–463. [DOI] [PubMed] [Google Scholar]
- 17. Sutherland DE, Stillman MJ (2014) Challenging conventional wisdom: single domain metallothioneins. Metallomics 6:702–728. [DOI] [PubMed] [Google Scholar]
- 18. Olafson RW, McCUBBIN WD, Kay C (1988) Primary‐and secondary‐structural analysis of a unique prokaryotic metallothionein from a Synechococcus sp. cyanobacterium. Biochem J 251:691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rigby KE, Stillman MJ (2004) Structural studies of metal‐free metallothionein. Biochem Biophys Res Commun 325:1271–1278. [DOI] [PubMed] [Google Scholar]
- 20. Duncan KER, Stillman MJ (2006) Metal‐dependent protein folding: metallation of metallothionein. J Inorg Biochem 100:2101–2107. [DOI] [PubMed] [Google Scholar]
- 21. Hong S‐H, Hao Q, Maret W (2005) Domain‐specific fluorescence resonance energy transfer (FRET) sensors of metallothionein/thionein. Protein Eng Des Sel 18:255–263. [DOI] [PubMed] [Google Scholar]
- 22. Chen S‐H, Chen L, Russell DH (2014) Metal‐induced conformational changes of human metallothionein‐2A: A combined theoretical and experimental study of metal‐free and partially metalated intermediates. J Am Chem Soc 136:9499–9508. [DOI] [PubMed] [Google Scholar]
- 23. Yu X, Wu Z, Fenselau C (1995) Covalent sequestration of melphalan by metallothionein and selective alkylation of cysteines. Biochemistry 34:3377–3385. [DOI] [PubMed] [Google Scholar]
- 24. Irvine GW, Stillman MJ (2013) Topographical analysis of As‐induced folding of α‐MT1a. Biochem Biophys Res Commun 441:208–213. [DOI] [PubMed] [Google Scholar]
- 25. Irvine GW, Duncan KE, Gullons M, Stillman MJ (2015) Metalation kinetics of the human α‐metallothionein 1a fragment is dependent on the fluxional structure of the apo‐protein. Chem–Eur J 21:1269–1279. [DOI] [PubMed] [Google Scholar]
- 26. Palumaa P, Tammiste I, Kruusel K, Kangur L, Jörnvall H, Sillard R (2005) Metal binding of metallothionein‐3 versus metallothionein‐2: lower affinity and higher plasticity. Biochimica et Biophysica Acta 1747:205–211. [DOI] [PubMed] [Google Scholar]
- 27. Irvine GW, Summers KL, Stillman MJ (2013) Cysteine accessibility during As 3+ metalation of the α‐and β‐domains of recombinant human MT1a. Biochem Biophys Res Commun 433:477–483. [DOI] [PubMed] [Google Scholar]
- 28. Chen S‐H, Russell DH (2015) Reaction of human Cd7 metallothionein and N‐ethylmaleimide: Kinetic and structural insights from electrospray ionization mass spectrometry. Biochemistry 54:6021–6028. [DOI] [PubMed] [Google Scholar]
- 29. Konermann L, Pan J, Liu Y‐H (2011) Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem Soc Rev 40:1224–1234. [DOI] [PubMed] [Google Scholar]
- 30. Englander SW, Mayne L, Kan Z‐Y, Hu W (2016) Protein folding‐how and why: By hydrogen exchange, fragment separation, and mass spectrometry. Ann Rev Biophys 45:135–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Summers KL, Mahrok AK, Dryden MD, Stillman MJ (2012) Structural properties of metal‐free apometallothioneins. Biochem Biophys Res Commun 425:485–492. [DOI] [PubMed] [Google Scholar]
- 32. Bartman CE, Metwally H, Konermann L (2016) Effects of multidentate metal interactions on the dtructure of collisionally activated proteins: Insights from ion mobility spectrometry and molecular dynamics simulations. Anal Chem 88:6905–6913. [DOI] [PubMed] [Google Scholar]
- 33. Chen S‐H, Russell WK, Russell DH (2013) Combining chemical labeling, bottom‐up and top‐down ion‐mobility mass spectrometry to identify metal‐binding sites of partially metalated metallothionein. Anal Chem 85:3229–3237. [DOI] [PubMed] [Google Scholar]
- 34. Ngu TT, Easton A, Stillman MJ (2008) Kinetic analysis of arsenic − metalation of human metallothionein: Significance of the two‐domain structure. J Am Chem Soc 130:17016–17028. [DOI] [PubMed] [Google Scholar]
- 35. Pinter TB, Irvine GW, Stillman MJ (2015) Domain selection in metallothionein 1A: Affinity‐controlled mechanisms of zinc binding and cadmium exchange. Biochemistry 54:5006–5016. [DOI] [PubMed] [Google Scholar]
- 36. Pinter TB, Stillman M (2015) Putting the pieces into place: Properties of intact zinc metallothionein 1A determined from interaction of its isolated domains with carbonic anhydrase. Biochem J BJ20150676. [DOI] [PubMed] [Google Scholar]
- 37. Banci L, Bertini I, Ciofi‐Baffoni S, Kozyreva T, Zovo K, Palumaa P (2010) Affinity gradients drive copper to cellular destinations. Nature 465:645–648. [DOI] [PubMed] [Google Scholar]
- 38. Zaia J, Fabris D, Wei D, Karpel RL, Fenselau C (1998) Monitoring metal ion flux in reactions of metallothionein and drug‐modified metallothionein by electrospray mass spectrometry. Protein Sci 7:2398–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pérez‐Rafael S, Atrian S, Capdevila M, Palacios Ò (2011) Differential ESI‐MS behaviour of highly similar metallothioneins. Talanta 83:1057–1061. [DOI] [PubMed] [Google Scholar]
- 40. Yu X, Wojciechowski M, Fenselau C (1993) Assessment of metals in reconstituted metallothioneins by electrospray mass spectrometry. Anal Chem 65:1355–1359. [DOI] [PubMed] [Google Scholar]
- 41. Hathout Y, Fabris D, Fenselau C (2001) Stoichiometry in zinc ion transfer from metallothionein to zinc finger peptides. Int J Mass Spectrom 204:1–6. [Google Scholar]
- 42. Grandori R (2003) Origin of the conformation dependence of protein charge‐state distributions in electrospray ionization mass spectrometry. J Mass Spectrom 38:11–15. [DOI] [PubMed] [Google Scholar]
- 43. Vasak M, Galdes A, Hill HAO, Kaegi JH, Bremner I, Young BW (1980) Investigation of the structure of metallothioneins by proton nuclear magnetic resonance spectroscopy. Biochemistry 19:416–425. [DOI] [PubMed] [Google Scholar]
- 44. Hong S‐H, Maret W (2003) A fluorescence resonance energy transfer sensor for the β‐domain of metallothionein. Proc Natl Acad Sci USA 100:2255–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maret W (2009) Fluorescent probes for the structure and function of metallothionein. J Chromatogr B 877:3378–3383. [DOI] [PubMed] [Google Scholar]
- 46. Ni FY, Cai B, Ding ZC, Zheng F, Zhang MJ, Wu HM, Sun HZ, Huang ZX (2007) Structural prediction of the β‐domain of metallothionein‐3 by molecular dynamics simulation. Proteins Struct Funct Bioinf 68:255–266. [DOI] [PubMed] [Google Scholar]
- 47. Duncan R, Kelly E, Kirby CW, Stillman MJ (2008) Metal exchange in metallothioneins–a novel structurally significant Cd5 species in the alpha domain of human metallothionein 1a. febs J 275:2227–2239. [DOI] [PubMed] [Google Scholar]
- 48. Berweger CD, Thiel W, van Gunsteren WF (2000) Molecular‐dynamics simulation of the β domain of metallothionein with a semi‐empirical treatment of the metal core. Proteins Struct Funct Bioinf 41:299–315. [DOI] [PubMed] [Google Scholar]
- 49. Banci L (2003) Molecular dynamics simulations of metalloproteins. Curr Opin Chem Biol 7:143–149. [DOI] [PubMed] [Google Scholar]
- 50. Yang Y, Maret W, Vallee BL (2001) Differential fluorescence labeling of cysteinyl clusters uncovers high tissue levels of thionein. Proc Natl Acad Sci USA 98:5556–5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Haase H, Maret W (2004) A differential assay for the reduced and oxidized states of metallothionein and thionein. Anal Biochem 333:19–26. [DOI] [PubMed] [Google Scholar]
- 52. Petering DH, Zhu J, Krezoski S, Meeusen J, Kiekenbush C, Krull S, Specher T, Dughish M (2006) Apo‐metallothionein emerging as a major player in the cellular activities of metallothionein. Exp Biol Med 231:1528–1534. [DOI] [PubMed] [Google Scholar]
- 53. Maryon EB, Molloy SA, Kaplan JH (2013) Cellular glutathione plays a key role in copper uptake mediated by human copper transporter 1. Am J Physiol Cell Physiol 304:C768–C779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hernández LE, Sobrino‐Plata J, Montero‐Palmero MB, Carrasco‐Gil S, Flores‐Cáceres ML, Ortega‐Villasante C, Escobar C (2015) Contribution of glutathione to the control of cellular redox homeostasis under toxic metal and metalloid stress. J Exp Bot 66:2901–2911. [DOI] [PubMed] [Google Scholar]
- 55. Jiang L‐J, Maret W, Vallee BL (1998) The glutathione redox couple modulates zinc transfer from metallothionein to zinc‐depleted sorbitol dehydrogenase. Proc Natl Acad Sci USA 95:3483–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Maret W, Krężel A (2007) Cellular zinc and redox buffering capacity of metallothionein/thionein in health and disease. Mol Med 13:371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lynes MA, Hidalgo J, Manso Y, Devisscher L, Laukens D, Lawrence DA (2014) Metallothionein and stress combine to affect multiple organ systems. Cell Stress Chaperones 19:605–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chan J, Huang Z, Watt I, Kille P, Stillman MJ (2007) Characterization of the conformational changes in recombinant human metallothioneins using ESI‐MS and molecular modeling. Can J Chem 85:898–912. [Google Scholar]