

Abstract
Depression is a common, devastating illness. Current pharmacotherapies help many patients, but there are high rates of partial- or non-response and the delayed onset of the effects of antidepressant leave many patients inadequately treated. However, new insights into the neurobiology of stress and human mood disorders have shed light on mechanisms underlying the vulnerability of individuals to depression and have pointed to novel antidepressants. Environmental events and other risk factors contribute to depression through converging molecular and cellular mechanisms that disrupt neuronal function and morphology, resulting in dysfunction of the circuitry essential for mood regulation and cognitive function. Although current antidepressants such as serotonin reuptake inhibitors produce subtle changes that take effect in weeks or months, new agents have recently shown improvement in mood ratings within hours of dosing in patients resistant to typical antidepressants. These new agents have also been shown to reverse the synaptic deficits caused by stress within a similar time scale.
Introduction
Depression is among the leading contributors to the global burden of disease1, affecting approximately 17 percent of the population in the United States. It is associated with enormous personal suffering and societal economic burden2. Further, depression can be a lethal illness resulting in elevated suicide risk3, as well as cardiac disease, cerebrovascular disorders, and other medical causes of mortality4. The magnitude of the clinical burden of depression reflects, in part, the limited effectiveness of present-day treatments. Currently available antidepressant medications, alone and in combination, are associated with high rates of partial- or non-response, delayed response onset of weeks to months, and limited duration of efficacy5. Certain approaches, notably electroconvulsive seizure therapy (ECT) have greater efficacy but also have significant side effects, notably retrograde amnesia. The development of truly novel medications that address these limitations has been hampered by a deficient understanding of the pathophysiology of depression. Gaining such understanding is particularly challenging given the clinical heterogeneity of MDD, the broad phenomenological criteria used to diagnose depression, and lack of reliable biomarkers of MDD and treatment response. Despite these complexities, progress is being made.
In this review, evidence will be presented in support of the hypothesis that depression is caused by disruption of functional and structural connections of the neural circuits that underlie the regulation of mood. We discuss the negative impact of stress-induced physiological changes and (or in combination with) other risk factors on synaptic connectivity of specific neural circuits and the molecular and cellular mechanisms underlying these effects. Lastly, we will consider positive modulators of synaptic connectivity (i.e., exercise, metabolic balance, anti-inflammatory agents) that oppose the effects of these pathophysiological mechanisms, as well as novel rapid acting antidepressants.
Risk factors
Susceptibility to depression, as well as other psychiatric illnesses is influenced by a variety of genetic, epigenetic, endocrine, and environmental risk factors (Fig. 1; Box 1,2). For example, susceptible individuals exposed to traumatic or stressful life events may develop depression; whether they develop disease may be influenced by differences in genetic make-up, prior stressful experiences, and other physiological parameters (e.g., gonadal hormone levels, metabolic imbalances). Conversely, some factors increase resilience and boost the ability to avoid the damaging effects of traumatic or chronic stress6. Resilience to depression can be the result of the absence of negative responses observed in susceptible individuals or from adaptive mechanisms that promote normal mood and emotion. Inter-individual susceptibility factors can also explain the heterogeneity of depression. Abnormal abundance or function of the hypothalamic-pituitary-adrenal (HPA) axis, neurotrophic factors, sex steroids, metabolic and/or inflammatory cytokines can lead to alterations in neurotransmitters, intracellular signaling, gene transcription, translation, and epigenetic changes that can contribute to short-term and long-lasting imbalances of neuronal function and behavior.
Figure 1. Heterogeneity of depression and influences on susceptibility to depression.
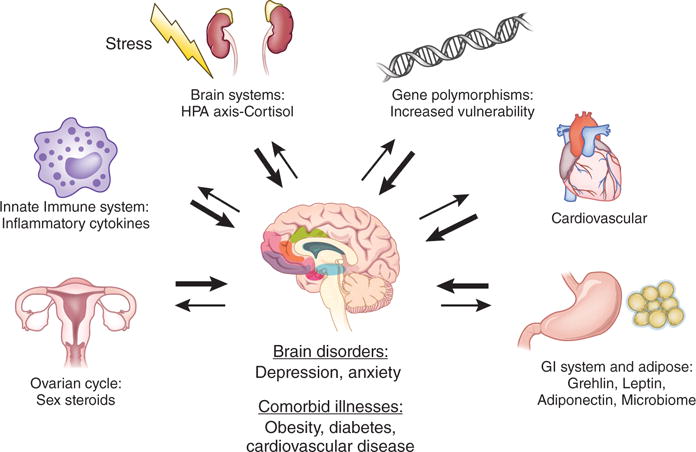
The heterogeneity of depression results from one or more pathological determinants. Notable effects include stress on brain neurotransmitter systems (NTs),activation of the HPA axis and cortisol, the innate immune system and inflammatory cytokines, fluctuations of ovarian steroids, the gastro intestinal (GI) system, adipose tissue and related peptides and microbiome, the cardiovascular system (e.g., VEGF or vascular endothelial growth factor), and gene polymorphisms that influence vulnerability and other organ systems as shown. These systems lead to increased incidence of depression as well as comorbid illnesses.
Box 1. Hereditary patterns of depression and Gene by Environment Interactions.
Genetic vulnerability accounts for approximately 35 to 40 percent of the variance in depression179. Early genetic studies focused on treatment related candidate genes (i.e., monoamine receptors and transporters), but were underpowered and/or have not been replicated180,181. More recent Genome Wide Association Studies (GWAS), which have been successful in identifying gene loci in schizophrenia, have not yet yielded replicable findings for MDD. This is thought to be due to the small number of major depressive disorder (MDD) cases in reported studies relative to the 75,000 to 100,000 cases estimated to be required to generate replicable significant findings182. The large sample sizes needed for these studies may be due to clinical heterogeneity, modest heritability (~40%), and the complexity of the genetic architecture for depression, i.e. a large number of interacting loci with small effect sizes182. Nonetheless, a recent GWAS pathway analysis of over 60,000 cases found significant associations of immune, neuronal signaling, synaptic density, and histone cascades in psychiatric disorders, including MDD, suggesting a clustering of risk variants in these pathways183. With the accumulating clinical samples, next generation sequencing may identify rare single nucleotide variants or rare copy number variants that contribute to the genetic risk for MDD181. Interactions of genetic vulnerability with environmental susceptibility factors most likely contribute to the complexity and heterogeneity of depression. These studies have focused primarily on early life stress or trauma, but gene by environment interaction studies are being expanded to include protective factors such as social support and intervention, as well as genes that increase resilience to depression 181,184.
Box 2. Epigenetics and depression.
Stress and environmental factors influence neuronal function and behavior through a variety of mechanisms, including acute signaling pathways that transiently regulate cell function. But alterations may also be persistent and contribute to the life-long stress sensitivity following early life stress. One of the best-studied mechanisms for persistent changes in gene expression is through epigenetic alterations that influence chromatin structure and gene transcriptional activity19,185. Chromatin structure is determined by histone modifications such as acetylation that relax the spacing between nucleosomes and thereby activate gene transcription, or by histone methylation that decreases activity. DNA can also be modified, notably via methylation that typically causes transcriptional repression. Studies in rodent models and in human depressed subjects, both brain tissue and blood cells, have reported epigenetic alterations, including histone and DNA modifications caused by stress and associated with depressive behaviors19,185. The potential significance of these epigenetic alterations is supported by studies demonstrating that histone deacetylase inhibitors produce antidepressant responses in rodent models19,185. In addition to alterations in brain, epigenetic modifications to germ cells may constitute a novel heritable form of neuroadaptation to stress186.
Depression: disruption of synapse number and function
Disruption of complex mood related circuitry has been implicated in depression (Box 3), but among the findings of altered brain structure and function in depression, the most consistent is reduced volume of the Prefrontal cortext (PFC) and hippocampus7,8. The extent of volume reduction is correlated with length of illness and time of treatment, and with the severity of depression. Recent postmortem studies also demonstrate reduced synapse number in PFC of depressed subjects9. Studies in rodent models have extended these human studies, and confirm that exposure to stress, like depression, causes atrophy and loss of neurons and glia in the PFC and hippocampus10,11,12.
Box 3. Neurocircuitry of depression.
Regions within the orbital and medial prefrontal cortices (oPFC, mPFC) appear to work as a coordinated unit to integrate sensory information, provide emotional salience, and to modulate visceral motor reactions and value-based decision processes187. The oPFC and ventral lateral PFC along with the dorsal anterior cingulate (dACC) are positioned at the interface of multimodal sensory networks mediating emotion and memory. These regions have connections with several sensory areas188 as well as inputs from the hypothalamus, amygdala, nucleus accumbens and hippocampus. Neurons in this region are capable of integrating multimodal stimuli with rewarding or aversive qualities189,190. The mPFC and the closely associated pre-genual and subgenual ACC are primarily considered modulators of emotion driven visceral reactions. The mPFC/pre-sub-genual ACC regions have multiple outputs to other cortical regions as well as hypothalamus, periaqueductal gray, locus coeruleus and autonomic nuclei within the brain stem allowing for modulation of vegetative and visceral functions191,192. In rodents, the infralimbic PFC (IL-PFC) is believed to carry out similar roles to the orbital/medial PFC networks by integrating information and modulating visceral reactions related to emotional processes through various connections with the amygdala, hypothalamus, and various brain stem nuclei193. Recent work suggests that the IL-PFC also modulates ventral tegmental area activation through effects on the amygdala and ventral subiculum, tying the region to subcortical reward processing networks194.
Disruption of the medial/orbital PFC networks and altered functional connectivity of the circuits in which they are contained has been tied to the changes in implicit emotional regulation and reward responsiveness, core components of depression195. Multiple studies provide evidence of reduced functional connectivity between the amygdala and mPFC and associated ACC regions. Other studies suggest elevated resting state activity in these regions, and enhanced amygdala response to emotional stimuli, especially negative valance stimuli (sad or fearful) in patients with mood disorders. These same brain regions are key components in a default mode node network, a functionally interconnected set of networks that are active at “rest” but relatively silenced by tasks requiring attention to external stimuli. Increased default mode network activity is associated with an introspective state, and a reduced ability to modulate the networks activity may impair an individuals ability to deploy attention from introspective processes to tasks requiring attention to external stimuli.
Anhedonia, especially deficits in non-consummatory reward behavior, is another core symptom of depression. Abnormal activity levels in the PFC/ACC, as well as the ventral and dorsal striatum have been reported in depressed patients with anhedonia196. Ventral striatal dysfunctions are hypothesized to reflect faulty coding of motivational significance and an impaired ability to accurately update predictions about expected reward based on experience, whereas impairments within dorsal striatial regions are believed to be more closely tied to defective action-reward contingency learning. Finally, abnormal function of medial/orbital PFC and ACC and their connections to the striatum have been associated with deficits in reward learning, effort valuation, and an impaired ability to generate adaptive responses to changes in environmental stimuli.
Synaptic plasticity represents one of the most fundamental, important functions of the brain: the ability to sense, assess and store complex information, and make appropriate, adaptive responses to subsequent related stimuli13–15. This critical brain function plays a key role in both short- and long-term memory, and the mechanisms underlying these changes have been linked to the pathophysiology and treatment of multiple neurobiological disorders, including depression.
Synaptogenesis is regulated by a complex interaction of signaling pathways, and disruption of many of the key pathways have been implicated in the susceptibility to depression, including loss of neurotrophic factor support, disruption of estradiol cycling, and elevation of inflammatory cytokines10 (Figure 3). Glia also play an important role in regulating the synthesis and reuptake of glutamate and thereby influence synaptic function and morphology16,17. Here we discuss the major systems implicated in the susceptibility and pathophysiology of depression and how they influence synaptic connections in depression circuitry.
Figure 3. The multiple heterogeneous signaling pathways that influence synapse formation and stability and that could contribute to loss of synapses in depression.
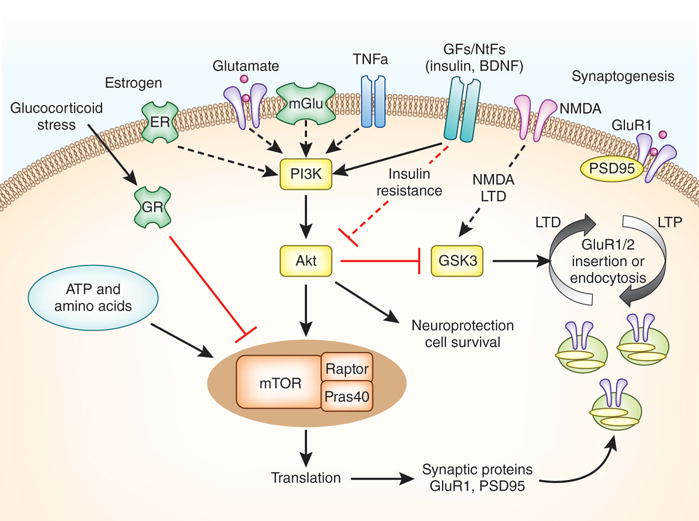
This includes neurotransmitters (i.e., glutamate), growth factors/neurotrophic factors (GFs/NTFs, cytokines (e.g., tumor necrosis factor α, TNFα), energy and metabolic factors (ATP, amino acids), sex steroids (e.g., estrogen), and the HPA axis (the glucocorticoid cortisol). These systems influence multiple intracellular signaling cascades that regulate all aspects of neuronal function. One of the key pathways of interest is the mTORC1 signaling cascade, which is a sensor of synaptic activity and multiple systems that can influence synaptic protein synthesis and plasticity as shown. Activation of mTORC1 signaling can occur via regulation of phosphatidylinositide 3 kinase (PI3K) and stimulation of protein kinase B (Akt). PI3K can be directly or indirectly (via multiple steps) stimulated by the different factors indicated, notably glutamate (via AMPA or mGlu receptors), estrogen (via estrogen receptors), BDNF, and other neurotrophins and growth factors. Stress and glucocorticoids via the glucocorticoid receptor (GR) can inhibit mTORC1 signaling via induction of factors that inhibit mTORC1 stability. Metabolic factors including ATP and amino acids that are required for protein synthesis, can also regulate mTORC1. Activation mTORC1 signaling leads to increased synthesis of proteins (e.g., GluA1 and PSD95) required for the maturation of existing synapses and formation of new ones. The insertion of GluA1 is also a point of regulation, notably by glycogen synthase kinase 3 (GSK3), and is involved in cellular models of learning and memory (i.e., LTP, long term potentiation; and LTD, long term depression).
Stress and depression: HPA axis and glucocorticoids
The most significant susceptibility factor for depression is acute traumatic or chronic stress. Exposure to stress could occur during early life, and cause long-lasting alterations that contribute to abnormal behavior (i.e., epigenetic alterations of DNA and/or histones; see Box 2) or could occur in adulthood.18,19,20 A hallmark feature of the stress response is activation of the HPA axis and increased circulating levels of glucocorticoids, designed to provide acute phase maximum physiological support for the fight or flight response. However, repeated stress and sustained elevation of glucocorticoids has deleterious effects on multiple organ systems, including the brain. Depression is often associated with elevated HPA axis activity and increased levels of glucocorticoids, as well as disruption of negative feedback mechanisms21–23.
Elevated levels of glucocorticoids act at multiple levels to influence neuronal function and behavior. Notably, chronic exposure of rodents to adrenal-glucocorticoids decreases synaptic number and function and causes atrophy of neurons in the prefrontal cortex and hippocampus, regions undergoing atrophy and disruption of connectivity in depressed subjects24,25. Acute stress also increases levels of extracellular glutamate in rodents, suggesting that excitotoxicity could contribute to neuronal atrophy26,27. Recent studies in rodents and humans also demonstrate a role for glucocorticoid regulation of molecular signaling pathways that influence gene transcription via epigenetic mechanisms, including regulation of the glucocorticoid receptor itself28. Variations of genes in the HPA axis, including CRF receptor 1 and FKBP5, have been suggested to interact with stressful life events and/or childhood abuse and/or trauma, although further studies are needed to confirm these effects29. In addition to disruption of neurotrophic factor signaling (see below), there is new evidence from rodent and human studies that stress and glucocorticoids directly influence the expression of factors that negatively regulate the translation of synaptic proteins (Figure 3)30.
The adaptive, evolutionary significance of disrupted synaptic plasticity in response to stress is not clear. One hypothesis is that extrasynaptic NMDA receptors act as neural sensors for metabolic or oxidative stress, causing regression of dendritic spines and branches in the service of protecting neuronal viability17,31. However, it is possible that these adaptive changes also serve functions not yet well understood at the neural network and whole animal level, perhaps related to a coordinated organismal stress response. Thus, it is likely that chronic stress, combined with genetic and environmental factors, results in short-term adaptive changes (mobilization of glucose, activation of immunity, etc.) that may have deleterious long-term consequences for synapses, the brain as a whole, and overall health and viability (i.e., multiple other stress-related medical disorders). Further, transgenerational epigenetic transmission may help to prepare newborns to cope in specific ways with adverse environments to which the parent had been exposed. As is the case with many stress-related disorders, such as posttraumatic stress disorder, the long-standing epigenetic adaptations to extremes become dysfunctional when the organism returns to a more congenial environment.
Reduced neurotrophic factor expression in stress and depression
Neurotrophic/growth factors, most notably brain derived neurotrophic factor (BDNF), but also vascular endothelial growth factor (VEGF), fibroblast growth factor 2 (FGF2), and insulin like growth factor 1 (IGF1), have been implicated in having a role in depression. Stress and depression decrease the expression and function of BDNF (Figure 2) in prefrontal cortex and hippocampus, structures implicated in depression, as well as decrease the levels in the blood of depressed patients21,32–35. Conversely, typical antidepressant treatments (e.g., SSRIs) increase BDNF expression and block the deficits in growth factor expression caused by stress and depression. Reduced neurotrophic/growth factor levels may be particularly relevant to the structural alterations caused by stress and depression, as these factors, particularly BDNF, are required for activity dependent formation and maintenance of synaptic connections13,36.
Figure 2. Chronic stress causes atrophy of neuronal processes and decreases synapse number.
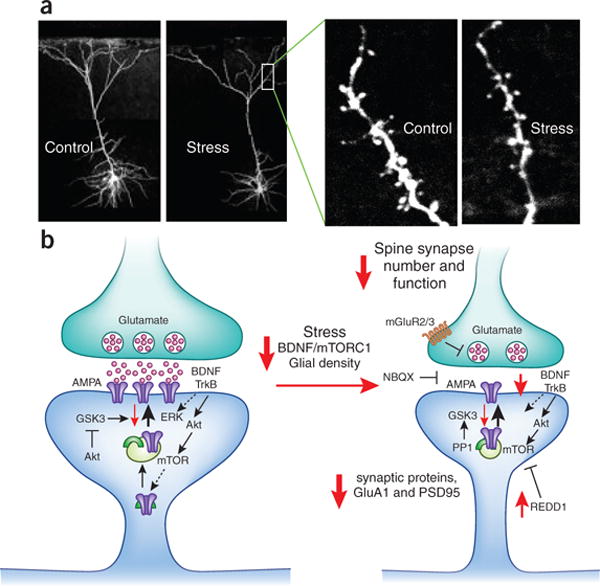
(a) The influence of repeated restraint stress (7 d) on pyramidal neurons (layer V) in the medial prefrontal cortex (mPFC) of rat. Pyramidal neurons in sections of mPFC are visualized after filling with neurobiotin and two-photon laser scanning microscopy. The left panels show the effects of repeated stress on the entire reconstructed pyramidal neurons, demonstrating a reduction in the number and length of apical dendrites. The higher power images on the right show a segment of dendrite decorated with spines (arrows), the point of synaptic contacts with neuronal inputs to the mPFC; repeated stress significantly decreases the number and function (determined electrophysiologically) of spine synapses. The lower panel shows the signaling pathways that lead to decreased numbers of synapses in response to stress, including decreased BDNF and mTORC1 signaling. Under normal conditions the upon stimulation the excitatory synapse releases glutamate and resulting in activation of postsynaptic glutamate AMPA receptors and depolarization; this causes activation of multiple intracellular pathways, including BDNF-TrkB signaling (and the downstream kinases Akt and ERK) and activation of the mTORC1 pathway. These pathways are essential for regulation of synaptic plasticity, a fundamental adaptive learning mechanism that includes maturation (increased spine head diameter) and number of synapses. This process requires mTORC1 mediated new protein synthesis of synaptic proteins, including glutamate GluA1 AMPA receptors and postsynaptic density protein PSD95. Repeated stress decreases BDNF and mTORC1 signaling in part via up-regulation of the negative regulator REDD1 (regulated in DNA damage and repair), which decreases the synthesis of synaptic proteins and thereby contributes to decreased number of spine synapses. Other pathways involved in the regulation of synaptic plasticity are GSK3 (glycogen synthase kinase 3) and PP1 (protein phosphatase 1).
Studies of a human BDNF polymorphism, Val66Met found in approximately 25 percent of the population have been insightful. The Val66Met allele, which blocks the processing and release of mature BDNF, is sufficient to cause atrophy of neurons in the hippocampal37 and mPFC of mice with this allele38. Heterozygous deletion of BDNF also decreases spine density and dendrite length of hippocampal and PFC neurons, decreases hippocampal volume, and occludes the effects of chronic stress39,40. These findings suggest that stress could cause atrophy via inhibition of BDNF, or that BDNF is required for neuronal remodeling. Mutant mouse studies also demonstrate that BDNF is required for the behavioral actions of antidepressants, and that deletion increases vulnerability to depression41,42. In humans, individuals with the Val66Met allele have reduced episodic memory and executive function, and reduced hippocampal volume. In addition, carriers of the Val66Met allele are at increased risk for depression when exposed to early life stress or trauma43–45.
The intracellular signaling pathways that mediate the actions of neurotrophic factors on synaptic connections, as well as neuronal survival and function include tyrosine kinase receptor activation of the kinases PI-3K-Akt and Raf-MEK-ERK46–48 (Figure 3). These pathways have been linked with multiple downstream targets that influence many aspects of neuronal function, including the protection and survival of neurons and the induction of synaptic plasticity. A key downstream convergence pathway for activity dependent synaptic plasticity and translation of synaptic proteins is the mechanistic target of rapamycin complex 1 (mTORC1)46,49. The mTORC1 pathway is regulated by neurotrophic factor signaling, but also by endocrine, metabolic, nutritional, and energy status and could therefore provide a nexus for multiple susceptibility factors (Figure 3)49.
In this way, mTORC1 serves as a neuronal sensor of activity dependent demand for new protein synthesis and synaptogenesis, balanced against the metabolic health of the neuron and organism. It is interesting to note that the expression and function of mTORC1 signaling proteins is reduced in postmortem PFC of depressed subjects, which could contribute to decreased synthesis of synaptic proteins in PFC of depressed subjects50,51. Conversely, rapid acting antidepressants (discussed below) stimulate mTORC1 signaling in the prefrontal cortex52,53. Postmortem studies demonstrated that the expression of REDD1, a negative regulator of mTORC1, is increased in PFC of depressed subjects30. Rodent studies also demonstrated that chronic stress decreases mTORC1 signaling proteins30,54,55, that over expression of REDD1 causes loss of synapses in the PFC, and that REDD1 deletion mutant mice are resilient to the synaptic and behavioral deficits caused by chronic stress30, further implicating mTORC1 signaling in stress, depression and antidepressant responses
Sex differences in susceptibility to depression
Depression is approximately twice as common in women compared to men2 and fluctuations of gonadal steroids (i.e., estrogen and progesterone) associated with the reproductive life cycle (puberty, menstrual cycle, childbirth, and menopause) contribute to depression vulnerability56,57. Mood and/or depressive symptoms in these disorders are associated with a precipitous drop in estradiol58. Sex steroids affect many aspects of neuronal function that may contribute to the risk for depression58–60.
Estrogen influences neurotransmitter activity, neurogenesis, and neurotrophic factor expression, as well as many aspects of glial function58,61,62. Notably, BDNF levels fluctuate with the estrous cycle and estrogen administration can increase the expression of BDNF in the PFC and hippocampus63–66. Dendrite complexity and spine synapse density also fluctuate with the ovarian cycle, and estrogen administration increases spine density in the hippocampus and PFC67–69. Estrogen and increased spine density are also associated with improvements in learning and memory in rodent models70–72. The BDNF Met polymorphism interacts with estrogen/estrous cycle resulting in disruption of memory and signaling73. Estrogen administration also has antidepressant actions in rodent behavioral models, and there has been clinical evidence of estrogen’s antidepressant effects in humans, although there have also been negative reports57,59. Estrogen also blocks neuronal atrophy caused by stress and glucocorticoids72, consistent with the hypothesis that disruption of estrogen signaling could result in synaptic deficits and depressive behaviors.
In addition to the well-characterized nuclear estrogen receptors (ER) that regulate transcription, estrogen acts rapidly on several pathways, including PI3K-Akt and MAPK-ERK, and mTORC1 signaling that are regulated by growth factors and that could contribute to the neuroprotective and synaptic effects of estrogen59,74,75 (Figure 3). These pathways have also been linked with estrogen enhancement of memory in rodent models and in human studies58,59,76,77. In addition to regulation of these signaling pathways, regulation of the serotonin system could contribute to the synaptic and antidepressant actions of estrogen59. Together these findings demonstrate molecular mechanisms through which fluctuations in gonadal steroid levels, particularly decreases in estrogen, may contribute to the increased incidence of depression in women.
Metabolic imbalance and diabetes: peptides and related signaling pathways
Metabolic disorders such as obesity and diabetes are associated with elevated rates of depression and share risk factors such as social and traumatic stress78–82,83(Figure 4). Elevations of glucocorticoids and inflammatory cytokines are associated with obesity, as well as depression84,85,86. Obesity and diabetes are also associated with disruption of PFC circuits and neurotransmitter systems (i.e., serotonin, dopamine, endocannabinoids, opioids) involved in motivation, reward, and anxiety that overlap with depression circuits80,87–89. Circulating peptides, including leptin and adiponectin (from adipose tissue) and ghrelin (from stomach), which influence feeding behavior and/or energy homeostasis90, are also regulated by stress and influence depression and anxiety behavior in rodent models91–93,94. Feeding, energy homeostasis, endocrine and neurocrine systems are also influenced by the gut microbiome, which has been implicated in healthy behavior while an imbalanced microbiome-brain interaction has been linked with psychiatric illnesses, including depression and anxiety95–97. Stress and immune/inflammatory systems can also interact with the gut microbiome and contribute to psychiatric as well as metabolic diseases. However, it should be pointed out that the data linking diabetes, obesity and depression are correlative and further studies are needed to identify direct functional evidence linking these disorders.
Figure 4. Mechansisms of action of the fast acting antidepressant ketamine in the medial prefrontal cortex.
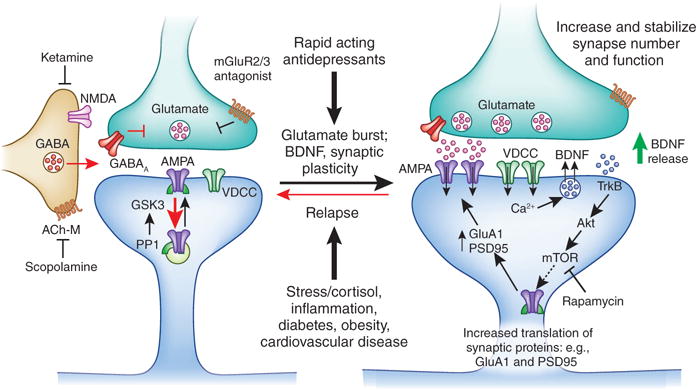
Ketamine causes a burst of glutamate that is thought to occur via disinhibition of GABA interneurons; the tonic firing of these GABA interneurons is driven by NMDA receptors, and the active, open channel state allows ketamine to enter and block channel activity. The resulting glutamate burst stimulates AMPA receptors causing depolarization and activation of voltage dependent Ca2+ channels, leading to release of BDNF and stimulation of TrkB-Akt that activates mTORC1 signaling leading to increased synthesis of proteins required for synapse maturation and formation (i.e., GluA1 and PSD95). Under conditions where BDNF release is blocked (BDNF Met knockin mice) or neutralized (BDNF neutralizing antibody) or when mTORC1 signaling is blocked (rapamycin infusion into the mPFC) the synaptic and behavioral actions of ketamine are blocked. Scopolamine also causes a glutamate burst via blockade of acetylcholine muscarinic M1 (ACh-M1) receptors on GABA interneurons. Antagonists of glutamate metabotropic 2/3 receptors (mGluR2/3) also produce rapid antidepressant actions via blockade of presynaptic autoreceptors that inhibit the release of glutamate. Relapse to a depressive state is associated with reduction of synapses on mPFC neurons, which could occur via stress and imbalance of endocrine (cortisol), estrogen, inflammatory cytokines, metabolic, and cardiovascular illnesses.
Dysfunction of energy metabolism and cellular respiration are also associated with depression, as well as obesity and diabetes. Abnormalities of mitochondrial energetics have been reported in mood disorders, including depression and bipolar disorder, as well as in obesity80,98–100. Further evidence of metabolic dysregulation is provided by studies demonstrating that impaired peripheral glucose regulation is associated with cognitive decline and depression, especially in obese subjects and patients with Type 2 diabetes101. High fat diet (HFD) leads to a metabolic syndrome, insulin resistance, and type 2 diabetes, conditions that are associated with cognitive deficits and neurodegenerative disorders102,103. HFD can also lead to anxiety and depressive behaviors in rodent models that are associated with disruption of neurotransmitter circuit connections103,104.
One of the consequences of insulin resistance is altered neuronal function, either as a result of disrupted energy metabolism or loss of insulin actions on neuronal function. HFD or experimental diabetes and insulin resistance cause neuronal atrophy in cortical and limbic structures and are associated with reduced synaptic plasticity104–106. BDNF also plays a central role in energy metabolism and cellular respiration107–109 and exerts significant control over feeding behavior and body mass110.
There are multiple interactions of signaling pathways for metabolic factors, energy metabolism, and stress/depression systems, notably the growth factor signaling pathways (Figure 3). HFD causes insulin resistance in cortical and limbic structures, including decreased insulin stimulation of Akt, S6K, GSK3β, and mTORC1 signaling, while AMP-activated protein kinase (AMPK) stimulation is increased104. These pathways also mediate the actions of growth factors and are disrupted by BDNF reductions that are caused by diet and stress80. Energy metabolism also regulates AMPK, another nodal regulator of mTORC1 signaling and synaptic protein synthesis. The possibility that insulin resistance and disruption of these signaling pathways could contribute to psychiatric illnesses is provided by studies of brain specific deletion of insulin receptors: these mice are characterized by disruption of mitochondrial function and imbalance of brain monoamine metabolism that are associated with increased anxiety and depression behaviors111.
These findings suggest that the high rates of comorbidity between depression, diabetes, and obesity in the developed world result in part from insulin resistance, abnormal energy metabolism and nutrient delivery to the brain, processes that are exacerbated by the damaging effects of adrenal glucocorticoids and inflammatory cytokines. In contrast, exercise produces dramatic beneficial effects, including increased BDNF, mTORC1 signaling, and muscle derived factors that increase neural plasticity and resistance to chronic stress exposure49,112,113.
Innate immune system, the inflammasome, and inflammatory cytokines
Psychological and social stressors can increase levels of inflammatory cytokines in humans, and cytokine infusions (e.g., interferon) can produce sickness behavior with characteristics of depression114–117. Serum levels of the pro-inflammatory cytokines interleukin-1β (IL-1β), IL-6, and tumor necrosis factor α (TNF-α) are increased in depressed patients, and levels are normalized by antidepressant treatment118,119. Recent studies demonstrate a role for inflammasome activation in the effects of stress115,120,121.
Inflammatory cytokines derived from microglia, the brains resident innate immune cells, influence synaptic plasticity and spine synapse formation under physiological conditions122–126. Notably, low levels of TNF-α and IL-1β support synaptic plasticity via regulation of PI3K-Akt signaling. However, stress, aging, and inflammation induce abnormal elevations of inflammatory cytokines that have the opposite effect via regulation of p38 and NFκB, canonical cytokine signaling pathways. Microglia are involved in activity dependent synaptic pruning during development127–129, and may be recruited during stress to participate in activity dependent synaptic loss. Together these studies demonstrate that normal brain function requires low levels of inflammatory cytokines but that elevated levels contribute to damage, atrophy and loss of spine synapses in response to stress and depression.
A role for inflammatory cytokines in the pathophysiology and treatment of depression in humans is supported by recent studies demonstrating that neutralization of TNF-α in patients with psoriasis also reduces depressive symptoms130. A second study supports and extends these findings, demonstrating that only patients with elevated C reactive protein, an inflammation biomarker, show an antidepressant response to the TNF-α antagonist infliximab131. Also interesting is evidence that patients with normal cytokine levels have worsened depressive symptoms with TNF-α neutralization, consistent with the notion that cytokines are needed for normal brain function.
Antidepressants and synaptic plasticity
Chronic administration of typical antidepressants that block the reuptake and breakdown of monoamines increase synaptic plasticity at several levels including increased birth of new neurons in the adult hippocampus, increased neurotrophic factor expression, and regulation of synapse formation10,132,133. Stress induced deficits of adult hippocampal neurogenesis have been implicated in the cognitive deficits associated with stress and depression, and are reversed by antidepressant treatmentss134. Different classes of antidepressants, including SSRIs, dual and triple reuptake inhibitors, dopaminergic agents, tricyclics, and ECT increase BDNF expression and the behavioral actions of these agents are blocked in BDNF deletion mutant mice135–137, 138. In particular, ECT causes a large induction of BDNF in the hippocampus and PFC that could be related to the greater therapeutic efficacy of this treatment compared with typical antidepressants (e.g., SSRIs); ECT is still considered the gold standard for difficult to treat depression. The ability of ECT to enhance synaptic plasticity, including increased connectivity and even structural alterations, has been directly linked to the greater therapeutic response to this treatment139,140.
In addition to studies of ECT, the consequences of increased BDNF expression on synaptic plasticity to typical antidepressants have been examined, particularly for the SSRI fluoxetine. Chronic administration of fluoxetine increases synaptic plasticity in cellular models and enhances plasticity in ocular dominance fields and extinction of fear conditioning in adult animals, effects that are dependent on BDNF141–143. There is also evidence, albeit limited that chronic administration of a typical antidepressant can increase spine density144 or block the effects of chronic stress145,146.
However, the therapeutic limitations of these pharmacological agents, notably the substantial time lag, low rates of efficacy, and side effects highlight major unmet needs (Table 1). These limitations may be due to targeting of monoamine neurotransmitters that predominately serve a modulator role and do not substantially influence synaptopgenesis. Regarding BDNF, typical antidepressants only increase the expression of BDNF, however activity dependent synapse formation and plasticity require BDNF release into the synapse in combination with activation of other cellular pathways that contribute to plasticity21,33,47,147. The induction of BDNF expression by typical antidepressants, which still requires weeks of drug administration, only produces subtle, alterations of BDNF release and function that seem to be inadequate for the level of synaptic plasticity required to alleviate depressive symptons. ECT produces a rapid, synaptic activity dependent induction of BDNF in rodents, although it still requires several weeks of treatment to produce a therapeutic response; this delay could be related to the intensity and broad depolarizing effects on the entire brain that could lead to activation of negative feedback pathways that oppose the neurotrophic actions of ECT (e.g., effects that could contribute to retrograde amnesia).
Table 1.
Therapeutic agents for the treatment of depression.
| Drug | Mechanism | Response time | Clinical Use |
|---|---|---|---|
| Early agents: Tricyclic Reuptake Inhibitor (RI), Monoamine oxidase inhibitor (MAOI), others | |||
| Imipramine | NE/5HT RI | weeks-months | FDA approved |
| Amitriptyline | NE/5HT RI | weeks-months | FDA approved |
| Desipramine | NE RI | weeks-months | FDA approved |
| Doxepin | NE RI/H1 antagonist | weeks-months | FDA approved |
| Amoxapine | NE/5HT RI/DA antagonist | weeks-months | FDA approved |
| Protriptyline | NE/5HT RI | weeks-months | FDA approved |
| Maprotiline | NE RI/H1 antagonist | weeks-months | FDA approved |
| Trimipramine | 5HT RI/ H1 antagonist | weeks-months | FDA approved |
| Tranylcypromine | MAOI | weeks-months | FDA approved |
| Phenelzine | MAOI | weeks-months | FDA approved |
| Isocarboxazid | MAOI | weeks-months | FDA approved |
| Selegiline | MAOI (MAO-B) | weeks-months | FDA approved |
| Bupropion | NE/DA RI | weeks-months | FDA approved |
| Trazadone | 5HT2A antagonist/5HT RI | weeks-months | FDA approved |
| Nefazodone | 5HT2A antagonist /5HT RI | weeks-months | FDA approved |
| Mirtazapine | α2AR/5-HT2A antagonist | weeks-months | FDA approved |
| Later generation reuptake inhibitors | |||
| Fluoxetine | SSRI | weeks-months | FDA approved |
| Sertraline | SSRI | weeks-months | FDA approved |
| Citalopram | SSRI | weeks-months | FDA approved |
| Paroxetine | SSRI | weeks-months | FDA approved |
| Vortioxetine | SSRI/variable 5HT effects | weeks-months | FDA approved |
| Vilazodone | SSRI/5HT1A partial agonist | weeks-months | FDA approved |
| Duloxetine | NE/5HT dual RI | weeks-months | FDA approved |
| Venlafaxine | NE/5HT dual RI | weeks-months | FDA approved |
| Levomilnacipran | NE/5HT dual RI | weeks-months | FDA approved |
| Atypical Antipsychotics (approved for use as add-on therapies for patients already taking an SSRI) | |||
| Quetiapine+SSRI | 5HT2/D2 antagonist | days-weeks | FDA approved |
| Olanzapine+fluoxetine | 5HT2/D2 antagonist | days-weeks | FDA approved |
| Aripiprazole+SSRI | 5HT2 antag/D2 partial agonist | days-weeks | FDA approved |
| Brexpiprazole+SSRI | 5HT2 antag/D2 partial agonist | days-weeks | FDA approved |
| Brain Stimulation Modalities | |||
| ECT | BDNF, circuit plasticity | weeks | FDA approved |
| TMS | Circuit plasticity | weeks-months | FDA approved |
| VNS | Circuit plasticity | months | FDA approved |
| Rapid Acting Agents | |||
| Ketamine | NMDA channel blocker | hrs-days | clinical trials |
| Lanicemine | NMDA channel blocker | hrs-days | clinical trials |
| CP 101,606 | NMDA-NR2B NAM | hrs-days | clinical trials |
| GLYX-13 | NMDA-Glycine NAM | hrs-days | clinical trials |
| AV-101 | NMDA-Glycine modulator | unknown | clinical trials |
| Scopolamine | ACh-muscarinic antagonist | hrs-days | experimental |
| Other novel treatments | |||
| Tianeptine | restores glutamate balance | weeks-months | EMA approved |
| ALKS-5461 | κ–opiate antagonist | weeks-months | clinical trials |
| L-Methylfolate | cofactor monoamine synthesis | weeks-months | dietary supplement |
| SAM-e | methyl group donor | weeks-months | dietary supplement |
| Acetyl-l-carnitine | fatty acid transfer | weeks-months | dietary supplement |
FDA approved, for depression; EMA, European Medicines Agency; NE, norepinephrine; 5-HT, serotonin; DA, dopamine; H1, histamine H1 receptor; SSRI, selective serotonin RI; D2, dopamine D2 receptor; ECT, electroconvulsie therapy; TMS, transcranial magnetic stimulation; VNS, vagal nerve stimulation; NAM, negative allosteric modulator; ACh, acetylcholine; SAM-e, S-adenosyl-methionine.
The discovery of agents that possess rapid-acting antidepressant properties is changing our understanding of antidepressant treatment. A single, intravenous or intranasal sub-anesthetic dose of ketamine, a non-competitive NMDA glutamate receptor channel blocker, produces a rapid onset of antidepressant response that can last several days in the majority of treatment resistant unipolar and bipolar depressed patients participating in a growing number of clinical trials148–150. Recent studies report that ketamine also reduces suicide ideation, a major advance over typical antidepressants with low efficacy and delayed onset of action151,152. In addition to ketamine, there is evidence that low doses of scopolamine, a nonselective cholinergic muscarinic receptor antagonist, also produces rapid antidepressant actions in depressed patients, providing early evidence for class of rapid acting agent different from ketamine153. This raises the possibility that the anticholinergic actions of tricyclic reuptake inhibitors could contribute to the therapeutic actions of these agents. The rapid antidepressant and anti-suicide actions of ketamine and scopolamine, by mechanisms completely different from typical monoamine reuptake inhibitors, represents a significant discovery for the treatment of mood disorders. Additional large-scale clinical studies are needed to further substantiate the clinical efficacy of ketamine and scopolamine in different populations of depressed patients154,155.
Rapid acting antidepressants: mechanism of action
Clinical reports of rapid antidepressant actions have lead to basic research studies of ketamine and scopolamine, but the molecular and cellular mechanisms underlying the rapid and efficacious actions of these agents are more complicated than simple NMDA and muscarinic receptor blockade. Rapid responses in treatment resistant depressed patients suggest a mechanism that results in fast changes in synaptic function and plasticity. In contrast to the effects of stress, ketamine and other NMDA receptor antagonists increase mTORC1 signaling via activation of Akt and ERK and increase synaptic number and function in the PFC52,156–158(Figure 4). This leads to increased synthesis of synaptic proteins that are required for synapse formation and maturation, effects that are blocked by pre-administration of the selective mTORC1 inhibitor rapamycin52,156,159. It is notable that the acute dissociative effects of ketamine subside within approximately one hour whereas the synaptic changes persist for a week or more, and these long-lasting structural changes correlate with its persistent antidepressant behavioral effects. Ketamine’s acute activation of mTORC1 and dendritic mRNA translation of synaptic proteins can be seen as a trigger for the subsequent persistent synaptogenic and behavioral actions of ketamine.
The mechanisms by which an NMDA receptor antagonist leads to induction of mTORC1 and synaptogenesis occur through indirect pathways. Ketamine-induction of mTORC1 signaling and antidepressant behavior is dependent on glutamate transmission and AMPA receptor activation52,160. There is evidence that NMDA receptor blockade increases glutamate transmission in rodents and humans via blockade of NMDA receptors on GABAergic interneurons27,161,162. There is also evidence that AMPA receptor activation stimulates mTORC1 signaling in cultured neurons via release of BDNF and activation of Akt and ERK signaling36,163. This possibility is supported by recent reports that ketamine-induction of antidepressant behavioral responses are blocked in BDNF null mice164,165.
The rapid induction of mTORC1 signaling and synaptogenesis could serve to reverse the loss of connections in depressed patients and thereby reinstate the function of PFC and appropriate inhibitory control of amygdala and emotion. This possibility is supported by studies in a mouse chronic stress model of depression, in which exposure to stress for several weeks causes atrophy of PFC neurons and anhedonia, a hallmark feature of depression55. These morphological and behavioral deficits are rapidly reversed by a single dose of ketamine. Together these studies demonstrate that NMDA receptor antagonists rapidly increase mTORC1 signaling and synaptogenesis and reverse the deficits caused by stress and depression. Brain imaging studies support this possibility demonstrating that ketamine increases connectivity between the PFC and other limbic structures in depressed patients166,167.
In contrast to the actions of NMDA antagonists, acute or chronic administration of typical antidepressants (e.g., SSRIs) does not increase mTORC1 signaling52. These findings suggest that alteration of mTORC1 signaling and synaptogenesis is important in the rapid and efficacious treatment of depression.
Novel targets for rapid acting antidepressants
If the rapid onset of antidepressant actions of ketamine can be confirmed by future clinical trials, it will represent a major advance for the treatment of mood disorders. However, its abuse potential and neurotoxicity associated with high, repeated dosing pose challenges to its broader use in clinical settings and highlight the need for safer drugs that produce similar effects.
Ketamine is a nonselective NMDA receptor channel blocker, and targeting a specific NMDA receptor subtype or a non-channel blocker or modulator of the NMDA receptor could result in antidepressant effects with fewer side effects. Preclinical and clinical studies demonstrate that selective NR2B receptor antagonists and non-selective, low trapping NMDA receptor antagonists, as well as allosteric modulators of NMDA channels such as GLYX-13 produce antidepressant responses in rodent models and in humans, supporting this possibility52,55,160,168,169. Other potential targets that regulate glutamate transmission include antagonists of presynaptic mGlu2/3 inhibitory autoreceptors that increase glutamate release, and postsynaptic AMPA receptor potentiating agents that directly increase receptor function170,171. There are also intracellular pathway molecules (e.g., GSK3 and MKP1) that negatively regulate mTORC1 and related signaling cascades that that could be targeted46,172. However,recent reports of negative phase II clinical trials for depression with some of these drugs targeting the NMDA and mGlu2/3 receptors raise concerns about the general efficacy of some of these targets, and remind us of the difficulty in conducting clinical trials in this population.
In addition to glutamatergic agents, there is emerging evidence that low doses of scopolamine, a nonselective cholinergic muscarinic receptor antagonist, produces rapid antidepressant actions in depressed patients153. Scopolamine also acutely stimulates mTORC1 signaling, leading to increased synaptogenesis in the PFC53 (Figure 4). Studies to identify the receptor subtype indicate that a selective muscarinic 1 antagonist produces antidepressant actions similar to scopolamine in rodent models173,174. Preclinical studies also report that other putative antidepressant agents increase or are dependent on mTORC1 signaling175–178. Together, these studies provide additional supporting evidence that the mTORC1 dendritic translational cascade and synapse formation may represent a common final pathway for a wide range of rapid acting, efficacious antidepressants.
Summary and Conclusions
Together, clinical and basic research studies demonstrate an emerging focus on the glutamate synapse as a major target for stress, depression and novel rapid-acting antidepressants. The atrophy of neurons and loss of glutamatergic synaptic connections caused by stress are key contributors to the symptoms of depression. In addition to the HPA axis, synaptic number and function is altered by other factors that have been implicated in depression and other mood disorders, notably neurotrophic factors, fluctuations of ovarian steroids, metabolic factors, and inflammatory cytokines (Figures 3–4). Interactions between these factors can also lead to increased susceptibility to depression. Genetic studies have been hampered by the heterogeneity of depression, by low heritability, and contribution of multiple, low impact gene loci; progress will require consideration of these heterogeneous factors as well as other environmental conditions. The discovery of a new class of antidepressant agents that produce rapid antidepressant effects has had a major impact on the field, not only providing a sorely needed rapid and efficacious treatment, but also a strategy to identify additional therapeutic response targets. Importantly, in preclinical models these new agents rapidly increase synaptic connections and reverse the loss of synapses caused by stress, thereby directly targeting the pathophysiology underlying depression (Figure 4). These findings provide a roadmap for ongoing and future drug discovery efforts to identify agents that restore glutamate synaptic connectivity as novel rapid and efficacious treatments with limited side effects. In addition, studies are under way to identify other therapeutic strategies that can naturally increase, strengthen and stabilize synapses in depression circuits, including adjunctive drug treatments and cognitive behavioral therapy. Together these findings provide enormous promise for a new generation of therapeutic strategies for pervasive and devastating mood disorders.
References
- 1.Whiteford HA, et al. Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet. 2013;382:1575–1586. doi: 10.1016/S0140-6736(13)61611-6. [DOI] [PubMed] [Google Scholar]
- 2.Kessler RC, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R) JAMA. 2003;289:3095–3105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- 3.Trivedi M, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psych. 2006;163:28–40. doi: 10.1176/appi.ajp.163.1.28. [DOI] [PubMed] [Google Scholar]
- 4.Walker ER, McGee RE, Druss BG. Mortality in Mental Disorders and Global Disease Burden Implications: A Systematic Review and Meta-analysis. JAMA Psychiatry. 2015 doi: 10.1001/jamapsychiatry.2014.2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaynes B, et al. What did STAR*D teach us? Results from a large-scale, practical, clinical trial for patients with depression. Psychiatr Serv. 2009;60:1439–1445. doi: 10.1176/ps.2009.60.11.1439. [DOI] [PubMed] [Google Scholar]
- 6.Russo SJ, Murrough JW, Han MH, Charney DS, Nestler EJ. Neurobiology of resilience. Nat Neurosci. 2012;15:1475–1484. doi: 10.1038/nn.3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacQueen GM, Yucel K, Taylor VH, Macdonald K, Joffe R. Posterior hippocampal volumes are associated with remission rates in patients with major depressive disorder. Biol Psychiatry. 2008;64:880–883. doi: 10.1016/j.biopsych.2008.06.027. [DOI] [PubMed] [Google Scholar]
- 8.Savitz J, Drevets WC. Bipolar and major depressive disorder: neuroimaging the developmental-degenerative divide. Neurosci Biobehav Rev. 2009;33:699–771. doi: 10.1016/j.neubiorev.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang HJ, et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat Med. 2012;18:1413–1417. doi: 10.1038/nm.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duman R, Aghajanian G. Synaptic Dysfunction in Depression: Novel Therapeutic Targets. Science. 2012;338:68–72. doi: 10.1126/science.1222939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McEwen BS, Eiland L, Hunter RG, Miller MM. Stress and anxiety: Structural plasticity and epigenetic regulation as a consequence of stress. Neuropharmacology. 2012;62:3–12. doi: 10.1016/j.neuropharm.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morrison JH, Baxter MG. The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci. 2012;13:240–250. doi: 10.1038/nrn3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- 14.Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshihara Y, De Roo M, Muller D. Dendritic spine formation and stablization. Curr Opin Neurobiol. 2009;19:146–153. doi: 10.1016/j.conb.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 16.Sanacora G, Zarate CA, Krystal JH, Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov. 2008;7:426–437. doi: 10.1038/nrd2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2012;13:22–37. doi: 10.1038/nrn3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun H, Kennedy PJ, Nestler EJ. Epigenetics of the depressed brain: role of histone acetylation and methylation. Neuropsychopharmacology. 2013;38:124–137. doi: 10.1038/npp.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menke A, Binder EB. Epigenetic alterations in depression and antidepressant treatment. Dialogues Clin Neurosci. 2014;16:395–404. doi: 10.31887/DCNS.2014.16.3/amenke. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weaver IC, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- 21.Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902. doi: 10.1038/nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manji HK, Drevets WC, Charney DS. The cellular neurobiology of depression. Nature Medicine. 2001;7:541–547. doi: 10.1038/87865. [DOI] [PubMed] [Google Scholar]
- 23.Nemeroff CB, Owens MJ. Treatment of mood disorders. Nat Neurosci. 2002;5(Suppl):1068–1070. doi: 10.1038/nn943. [DOI] [PubMed] [Google Scholar]
- 24.Liu RJ, Aghajanian GK. Stress blunts serotonin- and hypocretin-evoked EPSCs in prefrontal cortex: role of corticosterone-mediated apical dendritic atrophy. Proc Natl Acad Sci U S A. 2008;105:359–364. doi: 10.1073/pnas.0706679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Magarinos A, BS M. Stress-induced atrophy of apical dendrites of hippocampal CA3c neuronos: involvement of glucocorticoid secretion and excitatory amino acid receptors. Neurosci. 1995;69:89–98. doi: 10.1016/0306-4522(95)00259-l. [DOI] [PubMed] [Google Scholar]
- 26.Lowry M, Wittenberg L, Yamamoto B. Effect of acute stress on hippocampal glutamate levels and spectrin proteolysis in young and aged rats. J Neurochem. 1995;65:268–274. doi: 10.1046/j.1471-4159.1995.65010268.x. [DOI] [PubMed] [Google Scholar]
- 27.Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang T, Labonté B, Wen X, Turecki G, Meaney M. Epigenetic mechanisms for the early environmental regulation of hippocampal glucocorticoid receptor gene expression in rodents and humans. Neuropsychopharmacol. 2013;38:111–123. doi: 10.1038/npp.2012.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hosang GM, Shiles C, Tansey KE, McGuffin P, Uher R. Interaction between stress and the BDNF Val66Met polymorphism in depression: a systematic review and meta-analysis. Bmc Medicine. 2014;12:7. doi: 10.1186/1741-7015-12-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ota K, et al. REDD1 is essential for stress-induced synaptic loss and depressive behavior. Nat Med. 2014;20:531–535. doi: 10.1038/nm.3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bocchio-Chiavetto L, et al. Serum and plasma BDNF levels in major depression: a replication study and meta-analyses. World J Biol Psychiatry. 2010;11:763–773. doi: 10.3109/15622971003611319. [DOI] [PubMed] [Google Scholar]
- 33.Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 34.Dwivedi Y. Brain-derived neurotrophic factor: role in depression and suicide. Neuropsychiatr Dis Treat. 2009;5:433–449. doi: 10.2147/ndt.s5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turner CA, Akil H, Watson SJ, Evans SJ. The fibroblast growth factor system and mood disorders. Biol Psychiatry. 2006;59:1128–1135. doi: 10.1016/j.biopsych.2006.02.026. [DOI] [PubMed] [Google Scholar]
- 36.Jourdi H, et al. Positive AMPA receptor modulation rapidly stimulates BDNF release and increases dendritic mRNA translation. J Neurosci. 2009;29:8688–8697. doi: 10.1523/JNEUROSCI.6078-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen H, Pandey GN, Dwivedi Y. Hippocampal cell proliferation regulation by repeated stress and antidepressants. Neuroreport. 2006;17:863–867. doi: 10.1097/01.wnr.0000221827.03222.70. [DOI] [PubMed] [Google Scholar]
- 38.Liu R, et al. BDNF Met allele decreases the density and function of spine/synapses in the prefrontal cortex and blocks synaptogenic and antidepressant effects of ketamine. Biol Psych. 2011;71:996–1005. [Google Scholar]
- 39.Liu RJ, et al. GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology. 2013;38:2268–2277. doi: 10.1038/npp.2013.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Magariños A, et al. Effect of brain-derived neurotrophic factor haploinsufficiency on stress-induced remodeling of hippocampal neurons. Hippocampus. 2010;21:253–264. doi: 10.1002/hipo.20744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Autry AE, Adachi M, Cheng P, Monteggia LM. Gender-specific impact of brain-derived neurotrophic factor signaling on stress-induced depression-like behavior. Biol Psychiatry. 2009;66:84–90. doi: 10.1016/j.biopsych.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duman C, Schlesinger L, Kodama M, Russell D, Duman R. A role for MAPK signaling in behavioral models of depression and antidepressant treatment. Biol Psych. 2007b;61:661–670. doi: 10.1016/j.biopsych.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 43.Gatt JM, et al. Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Molecular Psychiatry. 2009;14:681–695. doi: 10.1038/mp.2008.143. [DOI] [PubMed] [Google Scholar]
- 44.Kaufman J, et al. Brain-derived neurotrophic factor-5-HTTLPR gene interactions and environmental modifiers of depression in children. Biol Psychiatry. 2006;59:673–680. doi: 10.1016/j.biopsych.2005.10.026. [DOI] [PubMed] [Google Scholar]
- 45.Kim JM, et al. Interactions between life stressors and susceptibility genes (5-HTTLPR and BDNF) on depression in Korean elders. Biological Psychiatry. 2007;62:423–428. doi: 10.1016/j.biopsych.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 46.Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu Y, Christian K, Lu B. BDNF: a key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol Learn Mem. 2007;89:312–323. doi: 10.1016/j.nlm.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Minichiello L. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci. 2009;10:850–860. doi: 10.1038/nrn2738. [DOI] [PubMed] [Google Scholar]
- 49.Watson K, Baar K. mTOR and the health benefits of exercise. Semin Cell Dev Biol. 2014;36:130–139. doi: 10.1016/j.semcdb.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 50.Feyissa AM, Chandran A, Stockmeier CA, Karolewicz B. Reduced levels of NR2A and NR2B subunits of NMDA receptor and PSD-95 in the prefrontal cortex in major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:70–75. doi: 10.1016/j.pnpbp.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jernigan CS, et al. The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Progress in Neuro-Psychopharmacology & Biological Psychiatry. 2011;35:1774–1779. doi: 10.1016/j.pnpbp.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li N, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Voleti B, et al. Scopolamine rapidly increases mTORC1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psych. 2013;74:742–749. doi: 10.1016/j.biopsych.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chandran A, et al. Reduced phosphorylation of the mTOR signaling pathway components in the amygdala of rats exposed to chronic stress. Prog Neuropsychopharmacol Biol Psychiatry. 2013;40:240–245. doi: 10.1016/j.pnpbp.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li N, et al. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. 2011;69:754–761. doi: 10.1016/j.biopsych.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bloch M, Daly RC, Rubinow DR. Endocrine factors in the etiology of postpartum depression. Compr Psychiatry. 2003;44:234–246. doi: 10.1016/S0010-440X(03)00034-8. [DOI] [PubMed] [Google Scholar]
- 57.Rubinow D, Girdler SS. Hormones heart disease health: individualized medicine versus throwing the baby out with the bathwater. Depress Anxiety. 2011;28:E1–E15. doi: 10.1002/da.20833. [DOI] [PubMed] [Google Scholar]
- 58.Borrow AP, Cameron NM. Estrogenic mediation of serotonergic and neurotrophic systems: implications for female mood disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2014;54:13–25. doi: 10.1016/j.pnpbp.2014.05.009. [DOI] [PubMed] [Google Scholar]
- 59.Hughes Z, et al. Estrogen receptor neurobiology and its potential for translation into broad spectrum therapeutics for CNS disorders. Curr Mol Pharmacol. 2009;2:215–236. doi: 10.2174/1874467210902030215. [DOI] [PubMed] [Google Scholar]
- 60.Kangaspeska S, et al. Transient cyclical methylation of promoter DNA. Nature. 2008;452:112–115. doi: 10.1038/nature06640. [DOI] [PubMed] [Google Scholar]
- 61.Licznerski P, Duman RS. Remodeling of Axo-Spinous Synapses in the Pathophysiology and Treatment of Depression. Neuroscience. 2013;251:33–50. doi: 10.1016/j.neuroscience.2012.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Karki P, Smith K, Johnson J, Jr, Lee E. Astrocyte-derived growth factors and estrogen neuroprotection: role of transforming growth factor-alpha in estrogen-induced upregulation of glutamate transporters in astrocytes. Mol Cell Endocrinol. 2014;389:58–64. doi: 10.1016/j.mce.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barouk S, et al. 17β-Estradiol increases as- trocytic vascular endothelial growth factor (VEGF) in adult female rat hippocampus. Endocrinol. 2011;152:1745–1751. doi: 10.1210/en.2010-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cavus I, Duman RS. Influence of estradiol, stress, and 5-HT2A agonist treatment on brain-derived neurotrophic factor expression in female rats. Biol Psychiatry. 2003;54:59–69. doi: 10.1016/s0006-3223(03)00236-1. [DOI] [PubMed] [Google Scholar]
- 65.Kiss A, et al. 17beta-estradiol replacement in young, adult and middle-aged female ovariectomized rats promotes improvement of spatial reference memory and an antidepressant effect and alters monoamines and BDNF levels in memory- and depression-related brain areas. Behav Brain Res. 2012;227:100–108. doi: 10.1016/j.bbr.2011.10.047. [DOI] [PubMed] [Google Scholar]
- 66.Scharfman HE, MacLusky NJ. The influence of gonadal hormones on neuronal excitability, seizures, and epilepsy in the female. Epilepsia. 2006;47:1423–1440. doi: 10.1111/j.1528-1167.2006.00672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hao J, et al. Estrogen alters spine number and morphology in prefrontal cortex of aged female rhesus monkeys. J Neurosci. 2006;26:2571–2578. doi: 10.1523/JNEUROSCI.3440-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shansky RM, Morrison JH. Stress-induced dendritic remodeling in the medial prefrontal cortex: effects of circuit, hormones and rest. Brain Res. 2009;1293:108–113. doi: 10.1016/j.brainres.2009.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Woolley CS, McEwen BS. Estradiol mediates fluctuation in hippocampal synapse density during the estrous cycle in the adult rat. J Neurosci. 1992;12:2549–2554. doi: 10.1523/JNEUROSCI.12-07-02549.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fan L, et al. Estradiol-induced object mem- ory consolidation in middle-aged female mice requires dorsal hippocampal extracel- lular signal-regulated kinase and phosphatidylinositol 3-kinase activation. J Neurosci. 2010;30:4390–4400. doi: 10.1523/JNEUROSCI.4333-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Packard M, Teather L. Intra-hippocampal estradiol infusion enhances memory in ovari- ectomized rats. Neuroreport. 1997;8:3009–3013. doi: 10.1097/00001756-199709290-00004. [DOI] [PubMed] [Google Scholar]
- 72.Wei J, et al. Estrogen protects against the detrimental effects of repeated stress on glutamatergic transmission and cognition. Mol Psych. 2014;19:588–598. doi: 10.1038/mp.2013.83. [DOI] [PubMed] [Google Scholar]
- 73.Spencer JL, Waters EM, Milner TA, Lee FS, McEwen BS. BDNF variant Val66Met interacts with estrous cycle in the control of hippocampal function. Proc Natl Acad Sci U S A. 2010;107:4395–4400. doi: 10.1073/pnas.0915105107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nilsson S, Gustafsson JA. Estrogen receptors: therapies targeted to receptor subtypes. Clin Pharmacol Ther. 2011;89:44–55. doi: 10.1038/clpt.2010.226. [DOI] [PubMed] [Google Scholar]
- 75.Spencer JL, et al. Uncovering the mechanisms of estrogen effects on hippocampal function. Front Neuroendocrinol. 2008;29:219–237. doi: 10.1016/j.yfrne.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fortress AM, Fan L, Orr PT, Zhao ZR, Frick KM. Estradiol-induced object recognition memory consolidation is dependent on activation of mTOR signaling in the dorsal hippocampus. Learning & Memory. 2013;20:147–155. doi: 10.1101/lm.026732.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fortress AM, Kim J, Poole RL, Gould TJ, Frick KM. 17beta-Estradiol regulates histone alterations associated with memory consolidation and increases Bdnf promoter acetylation in middle-aged female mice. Learn Mem. 2014;21:457–467. doi: 10.1101/lm.034033.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Leone T, Coast E, Narayanan S, de Graft Aikins A. Diabetes and depression comorbidity and socio-economic status in low and middle income countries (LMICs): a mapping of the evidence. Global Health. 2012;8:39. doi: 10.1186/1744-8603-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Luppino FS, et al. Overweight, obesity, and depression: a systematic review and meta-analysis of longitudinal studies. Arch Gen Psychiatry. 2010;67:220–229. doi: 10.1001/archgenpsychiatry.2010.2. [DOI] [PubMed] [Google Scholar]
- 80.Mansur R, Brietzke E, McIntyre R. Is there a “metabolic-mood syndrome”? A review of the relationship between obesity and mood disorders. Neurosc Biohehav Rev. 2015 doi: 10.1016/j.neubiorev.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 81.van Dooren FE, et al. Depression and risk of mortality in people with diabetes mellitus: a systematic review and meta-analysis. PLoS One. 2013;8:e57058. doi: 10.1371/journal.pone.0057058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vogelzangs N, et al. Association of depressive disorders, depression characteristics and antidepressant medication with inflammation. Transl Psychiatry. 2012;2:e79. doi: 10.1038/tp.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rethorst C, Bernstein I, Trivedi M. Inflammation, obesity, and metabolic syndrome in depression: analysis of the 2009–2010 National Health and Nutrition Examination Survey (NHANES) J Clin Psychiatry. 2014;75:1428–1432. doi: 10.4088/JCP.14m09009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Choi J, Joseph L, Pilote L. Obesity and C-reactive protein in various populations: a systematic review and meta-analysis. Obesity Reviews. 2013;14:232–244. doi: 10.1111/obr.12003. [DOI] [PubMed] [Google Scholar]
- 85.Pasquali R. The hypothalamic-pituitary-adrenal axis and sex hormones in chronic stress and obesity: pathophysiological and clinical aspects. Ann N Y Acad Sci. 2012;1264:20–35. doi: 10.1111/j.1749-6632.2012.06569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moulton C, Pickup J, Ismail K. The link between depression and diabetes: the search for shared mechanisms. Lancet Diabetes Endocrinol. 2015;3:461–471. doi: 10.1016/S2213-8587(15)00134-5. [DOI] [PubMed] [Google Scholar]
- 87.Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nat Rev Neurosci. 2013;14:609–625. doi: 10.1038/nrn3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Volkow ND, Wang GJ, Baler RD. Reward, dopamine and the control of food intake: implications for obesity. Trends Cogn Sci. 2011;15:37–46. doi: 10.1016/j.tics.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Volkow ND, Wang GJ, Fowler JS, Telang F. Overlapping neuronal circuits in addiction and obesity: evidence of systems pathology. Philos Trans R Soc Lond B Biol Sci. 2008;363:3191–3200. doi: 10.1098/rstb.2008.0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hendrickx H, McEwen BS, Ouderaa F. Metabolism, mood and cognition in aging: the importance of lifestyle and dietary intervention. Neurobiol Aging. 2005;26(Suppl 1):1–5. doi: 10.1016/j.neurobiolaging.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 91.Lu XY, Kim CS, Frazer A, Zhang W. Leptin: a potential novel antidepressant. Proc Natl Acad Sci U S A. 2006;103:1593–1598. doi: 10.1073/pnas.0508901103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lutter M, et al. The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nat Neurosci. 2008;11:752–753. doi: 10.1038/nn.2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Spencer SJ, Emmerzaal TL, Kozicz T, Andrews ZB. Ghrelin’s Role in the Hypothalamic-Pituitary-Adrenal Axis Stress Response: Implications for Mood Disorders. Biol Psychiatry. 2014 doi: 10.1016/j.biopsych.2014.10.021. [DOI] [PubMed] [Google Scholar]
- 94.Wędrychowicz A, Zając A, Pilecki M, Kościelniak B, Tomasik P. Peptides from adipose tissue in mental disorders. World J Psych. 2014;4:103–111. doi: 10.5498/wjp.v4.i4.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mayer E, Knight R, Mazmanian S, Cryan J, Tillisch K. Gut microbes and the brain: paradigm shift in neuroscience. J Neurosci. 2014;34:15490–15496. doi: 10.1523/JNEUROSCI.3299-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Petra A, et al. Gut-Microbiota-Brain Axis and Its Effect on Neuropsychiatric Disorders With Suspected Immune Dysregulation. Clin Ther. 2015;37:984–995. doi: 10.1016/j.clinthera.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhou L, Foster JA. Psychobiotics and the gut-brain axis: in the pursuit of happiness. Neuropsychiatr Dis Treat. 2015;11:715–723. doi: 10.2147/NDT.S61997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gardner A, Boles RG. Beyond the serotonin hypothesis: mitochondria, inflammation and neurodegeneration in major depression and affective spectrum disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:730–743. doi: 10.1016/j.pnpbp.2010.07.030. [DOI] [PubMed] [Google Scholar]
- 99.Manji H, Kato T, Di Prospero NA. Impaired mitochondrial function in 1587 psychiatric disorder. Nat Rev Neurosci. 2012;13:293–307. doi: 10.1038/nrn3229. [DOI] [PubMed] [Google Scholar]
- 100.Abdallah C, et al. Glutamate metabolism in major depressive disorder. Am J Psych. 2014;171:1320–1327. doi: 10.1176/appi.ajp.2014.14010067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tabák A, Akbaraly T, Batty G, Kivimäki M. Depression and type 2 diabetes: a casual association? Lancet Diabetes Endocrinol. 2013;2:236–245. doi: 10.1016/S2213-8587(13)70139-6. [DOI] [PubMed] [Google Scholar]
- 102.Kassi E, Pervanidou P, Kaltsas G, Chrousos G. Metabolic syndrome: definitions and controversies. BMC Med. 2011;9:48. doi: 10.1186/1741-7015-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Reaven GM. The metabolic syndrome: time to get off the merry-go-round? J Intern Med. 2011;269:127–136. doi: 10.1111/j.1365-2796.2010.02325.x. [DOI] [PubMed] [Google Scholar]
- 104.Arnold SE, et al. High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol Dis. 2014;67:79–87. doi: 10.1016/j.nbd.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Magarinos AM, McEwen BS. Experimental diabetes in rats causes hippocampal dendritic and synaptic reorganization and increased glucocorticoid reactivity to stress. Proc Natl Acad Sci U S A. 2000;97:11056–11061. doi: 10.1073/pnas.97.20.11056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stranahan AM, et al. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus. 2008;18:1085–1088. doi: 10.1002/hipo.20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Markham A, Cameron I, Bains R. Brain-derived neurotrophic factor- 1597 mediated effects on mitochondrial respiratory coupling and neuroprotection 1598 share the same molecular signalling pathways. Eur J Neurosci. 2012;35:366–374. doi: 10.1111/j.1460-9568.2011.07965.x. [DOI] [PubMed] [Google Scholar]
- 108.Marosi K, Mattson MP. BDNF mediates adaptive brain and body responses 1600 to energetic challenges. 2014;25:89–98. doi: 10.1016/j.tem.2013.10.006. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schwartz E, Mobbs CV. Hypothalamic BDNF and obesity: found in translation. Nature Medicine. 2012;18:496–497. doi: 10.1038/nm.2716. [DOI] [PubMed] [Google Scholar]
- 110.Unger TJ, Calderon GA, Bradley LC, Sena-Esteves M, Rios M. Selective deletion of Bdnf in the ventromedial and dorsomedial hypothalamus of adult mice results in hyperphagic behavior and obesity. J Neurosci. 2007;27:14265–14274. doi: 10.1523/JNEUROSCI.3308-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kleinridders A, et al. Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. PNAS. 2015;112:3463–3468. doi: 10.1073/pnas.1500877112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Agudelo LZ, et al. Skeletal muscle PGC-1alpha1 modulates kynurenine metabolism and mediates resilience to stress-induced depression. Cell. 2014;159:33–45. doi: 10.1016/j.cell.2014.07.051. [DOI] [PubMed] [Google Scholar]
- 113.Voss MW, Vivar C, Kramer AF, van Praag H. Bridging animal and human models of exercise-induced brain plasticity. Trends Cogn Sci. 2013;17:525–544. doi: 10.1016/j.tics.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dinan TG. Inflammatory markers in depression. Curr Opin Psychiatry. 2009;22:32–36. doi: 10.1097/YCO.0b013e328315a561. [DOI] [PubMed] [Google Scholar]
- 115.Iwata M, Ota KT, Duman RS. The inflammasome: pathways linking psychological stress, depression, and systemic illnesses. Brain Behav Immun. 2013;31:105–114. doi: 10.1016/j.bbi.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65:732–741. doi: 10.1016/j.biopsych.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27:24–31. doi: 10.1016/j.it.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dowlati Y, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67:446–457. doi: 10.1016/j.biopsych.2009.09.033. [DOI] [PubMed] [Google Scholar]
- 119.Howren M, Lamkin DM, Suls J. Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosomatic Medicine. 2009;71:171–186. doi: 10.1097/PSY.0b013e3181907c1b. [DOI] [PubMed] [Google Scholar]
- 120.Raison CL, Miller AH. Malaise, melancholia and madness: the evolutionary legacy of an inflammatory bias. Brain Behav Immun. 2013;31:1–8. doi: 10.1016/j.bbi.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Iwata M, et al. Psychological stress activates the inflammasome via release of ATP and stimulation of the P2X7 receptor. Biol Psych. 2016 doi: 10.1016/j.biopsych.2015.11.026. Epub ahead of press. [DOI] [PubMed] [Google Scholar]
- 122.Boulanger LM. Immune proteins in brain development and synaptic plasticity. Neuron. 2009;64:93–109. doi: 10.1016/j.neuron.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 123.Goshen I, et al. A dual role for interleukin-1 in hippocampal-dependent memory processes. Psychoneuroendocrinology. 2007;32:1106–1115. doi: 10.1016/j.psyneuen.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 124.Khairova RA, Machado-Vieira R, Du J, Manji HK. A potential role for pro-inflammatory cytokines in regulating synaptic plasticity in major depressive disorder. Int J Neuropsychopharmacol. 2009;12:561–578. doi: 10.1017/S1461145709009924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Murray CA, Lynch MA. Evidence that increased hippocampal expression of the cytokine interleukin-1b is a common trigger for age- and stress-induced impairments in long-term potentiation. J Neurosci. 1998;18:2974–2981. doi: 10.1523/JNEUROSCI.18-08-02974.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- 127.Paolicelli RC, et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science. 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 128.Schafer DP, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012;35:369–389. doi: 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- 130.Krishnan V, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 131.Raison CL, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry. 2013;70:31–41. doi: 10.1001/2013.jamapsychiatry.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Casren E, Hen R. Neuronal plasticity and antidepressant actions. Trends Neurosci. 2013;36:259–267. doi: 10.1016/j.tins.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Krishnan V, Nestler EJ. Linking molecules to mood: new insight into the biology of depression. Am J Psychiatry. 2010;167:1305–1320. doi: 10.1176/appi.ajp.2009.10030434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Miller BR, Hen R. The current state of the neurogenic theory of depression and anxiety. Curr Opin Neurobiol. 2015;30:51–58. doi: 10.1016/j.conb.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chen ZY, Jing D, Bath KG, Leraci A, Khan T, Siao C-J, Herrara DG, Toth M, Yang C, McEwen BS, Hampstead BL, Lee FS. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006b;314:140–143. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Duman RS, Voleti B. Signaling pathways underlying the pathophysiology and treatment of depression: novel mechanisms for rapid-acting agents. Trends Neurosci. 2012;35:47–56. doi: 10.1016/j.tins.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yu H, et al. Variant brain-derived neurotrophic factor Val66Met polymorphism alters vulnerability to stress and response to antidepressants. J Neurosci. 2012;32:4092–4101. doi: 10.1523/JNEUROSCI.5048-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jiang B, et al. SKF83959 produces antidepressant effects in a chronic social defeat stress model of depression through BDNF-TrkB pathway. Int J Neuropsychopharmacol. 2014;18 doi: 10.1093/ijnp/pyu1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Abbott CC, et al. Hippocampal structural and functional changes associated withelectroconvulsive therapy response. Transl Psych. 2014;4:e483. doi: 10.1038/tp.2014.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dukart J, et al. Electroconvulsive therapy-induced brain plasticity determines therapeutic outcome in mood disorders. Proc Natl Acad Sci U S A. 2014;111:1156–1161. doi: 10.1073/pnas.1321399111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bath KG, et al. BDNF Val66Met impairs fluoxetine-induced enhancement of adult hippocampus plasticity. Neuropsychopharmacology. 2012;37:1297–1304. doi: 10.1038/npp.2011.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Karpova NN, et al. Fear Erasure in Mice Requires Synergy Between Antidepressant Drugs and Extinction Training. Science. 2011;334:1731–1734. doi: 10.1126/science.1214592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Maya Vetencourt JF, et al. The antidepressant fluoxetine restores plasticity in the adult visual cortex. Science. 2008;320:385–388. doi: 10.1126/science.1150516. [DOI] [PubMed] [Google Scholar]
- 144.Ampuero E, et al. Chronic fluoxetine treatment induces structural plasticity and selective changes in glutamate receptor subunits in the rat cerebral cortex. Neuroscience. 2010;169:98–108. doi: 10.1016/j.neuroscience.2010.04.035. [DOI] [PubMed] [Google Scholar]
- 145.Bessa J, et al. Hippocampal neurogenesis induced by antidepressant drugs: an epiphenomenon in their mood-improving actions. Mol Psych. 2009;14:739. doi: 10.1038/mp.2008.119. [DOI] [PubMed] [Google Scholar]
- 146.Magarinos A, McEwen B. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: comparison of stressors. Neurosci. 1995a;69:83–88. doi: 10.1016/0306-4522(95)00256-i. [DOI] [PubMed] [Google Scholar]
- 147.Castren E. Is mood chemistry? Nat Rev Neurosci. 2005;6:241–246. doi: 10.1038/nrn1629. [DOI] [PubMed] [Google Scholar]
- 148.Berman RM, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 149.Diazgranados N, et al. A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch Gen Psychiatry. 2010;67:793–802. doi: 10.1001/archgenpsychiatry.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zarate CA, Jr, et al. Arch Gen Psychiatry. 2006;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 151.DiazGranados N, et al. Rapid Resolution of Suicidal Ideation After a Single Infusion of an N-Methyl-D-Aspartate Antagonist in Patients With Treatment-Resistant Major Depressive Disorder. Journal of Clinical Psychiatry. 2010;71:1605–1611. doi: 10.4088/JCP.09m05327blu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Price RB, Nock MK, Charney DS, Mathew SJ. Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biol Psychiatry. 2009;66:522–526. doi: 10.1016/j.biopsych.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Drevets WC, Furey ML. Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol Psychiatry. 2010;67:432–438. doi: 10.1016/j.biopsych.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.DeWilde K, Levitch C, Murrough J, Mathew S, Losifescu D. The promise of ketamine for treatment-resistant depression: current evidence and future directions. Ann Ny Acad Sci. 2015;1345:47–58. doi: 10.1111/nyas.12646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sanacora G, Schatzberg AF. Ketamine: promising path or false prophecy in the development of novel therapeutics for mood disorders? Neuropsychopharmacol. 2015;40:1307. doi: 10.1038/npp.2014.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Miller OH, et al. GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. Elife. 2014;3:e03581. doi: 10.7554/eLife.03581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Paul RK, et al. (R,S)-norketamine and (2S,6S)-hydroxynorketamine increase the mammalian target of rapamycin function. Anesthesiology. 2014;121:149–159. doi: 10.1097/ALN.0000000000000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhou W, et al. Ketamine-induced antidepressant effects are associated with AMPA receptors-mediated upregulation of mTOR and BDNF in rat hippocampus and prefrontal cortex. Eur Psychiatry. 2014;29:419–423. doi: 10.1016/j.eurpsy.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 159.Liu R-L, Ota KT, Dutheil S, Duman RS, Aghajanian G. Ketamine strengthens CRF-activated amygdalar synaptic inputs to basilar dendrites in mPFC layer V pyramidal cells in the prelimbic but not infralimbic subregion, a key suppressor of stress responses. Neuropsychopharmacol. 2015 doi: 10.1038/npp.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Maeng S, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63:349–352. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 161.Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Stone JM, et al. Ketamine effects on brain GABA and glutamate levels with 1H-MRS: relationship to ketamine- induced psychopathology. Molecular Psychiatry. 2012;17:664–665. doi: 10.1038/mp.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Takei N, et al. Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J Neurosci. 2004;24:9760–9769. doi: 10.1523/JNEUROSCI.1427-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Autry AE, et al. NMDA receptor blockade at rest triggers rapid behavioural anatidepressant responses. Nature. 2011;475:91–95. doi: 10.1038/nature10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Liu RJ, et al. Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol Psychiatry. 2012;71:996–1005. doi: 10.1016/j.biopsych.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gass N, et al. Sub-anesthetic ketamine modulates intrinsic BOLD connectivity within the hippocampal-prefrontal circuit in the rat. Neuropsychopharmacol. 2014;39:895–906. doi: 10.1038/npp.2013.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Murrough JW, et al. Regulation of neural responses to emotion perception by ketamine in individuals with treatment-resistant major depressive disorder. Transl Psychiatry. 2015;5:e509. doi: 10.1038/tp.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Preskorn S, et al. A placebo-controlled trial of the NR2B subunit specific NMDA antagonist CP-101,606 plus paroxetine for treatment resistant depression (TRD) APA. 2007 [Google Scholar]
- 169.Sanacora G, et al. Lanicemine: a low-trapping NMDA channel blocker produces sustained antidepressant efficacy with minimal psychotomimetic adverse effects. Mol Psychiatry. 2014;19:978–985. doi: 10.1038/mp.2013.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Arai AC, Kessler M. Pharmacology of ampakine modulators: from AMPA receptors to synapses and behavior. Curr Drug Targets. 2007;8:583–602. doi: 10.2174/138945007780618490. [DOI] [PubMed] [Google Scholar]
- 171.Pilc A, Chaki S, Nowak G, Witkin JM. Mood disorders: regulation by metabotropic glutamate receptors. Biochem Pharmacol. 2007;75:997–1006. doi: 10.1016/j.bcp.2007.09.021. [DOI] [PubMed] [Google Scholar]
- 172.Duric V, et al. Negative regulator of MAP kinase is increased in depression and is necessary and sufficient for depressive behavior. Nat Med. 2010;16:1328–1332. doi: 10.1038/nm.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Navarria A, et al. Rapid antidepressant actions of scopolamine: Role of medial prefrontal cortex and M1-subtype muscarinic acetylcholine receptors. Neurobiol Dis. 2015 doi: 10.1016/j.nbd.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Witkin JM, et al. M1 and m2 muscarinic receptor subtypes regulate antidepressant-like effects of the rapidly acting antidepressant scopolamine. J Pharmacol Exp Ther. 2014;351:448–456. doi: 10.1124/jpet.114.216804. [DOI] [PubMed] [Google Scholar]
- 175.Luoni A, M F, Papp M, Molteni R, Riva MA. Lurasidone exerts antidepressant properties in the chronic mild stress model through the regulation of synaptic and neuroplastic mechanisms in the rat prefrontal cortex. Int J Neuropsychopharmacol. 2014;18:1–12. doi: 10.1093/ijnp/pyu061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Osborn M, et al. Antidepressant-like effects of erythropoietin: a focus on behavioural and hippocampal processes. PLoS One. 2013;8:e72813. doi: 10.1371/journal.pone.0072813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Palucha-Poniewiera A, Szewczyk B, Pilc A. Activation of the mTOR signaling pathway in the antidepressant-like activity of the mGlu5 antagonist MTEP and the mGlu7 agonist AMN082 in the FST in rats. Neuropharmacology. 2014;82:59–68. doi: 10.1016/j.neuropharm.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 178.Zhong P, et al. Monoacylglycerol lipase inhibition blocks chronic stress-induced depressive-like behaviors via activation of mTOR signaling. Neuropsychopharmacology. 2014;39:1763–1776. doi: 10.1038/npp.2014.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sullivan PF, Neale MC, K KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psych. 2000;157:1552–1562. doi: 10.1176/appi.ajp.157.10.1552. [DOI] [PubMed] [Google Scholar]
- 180.Lopez-Leon S, et al. Meta-analyses of genetic studies on major depressive disorder. Mol Psychiatry. 2008;13:772–785. doi: 10.1038/sj.mp.4002088. [DOI] [PubMed] [Google Scholar]
- 181.Dunn E, et al. Genetic determinants of depression: recent findings and future direction. Harv Rev Psych. 2014;23:1–18. doi: 10.1097/HRP.0000000000000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Levinson D, et al. Genetic studies of major depressive disorder: Why are there no genome-wide association study findings and what can we do about it? Biol Psych. 2014;76:510–512. doi: 10.1016/j.biopsych.2014.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.PGC, P.A.S. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci. 2015 doi: 10.1038/nn.3922. epub, ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kaufman J, et al. Social supports and serotonin transporter gene moderate depression in maltreated children. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:17316–17321. doi: 10.1073/pnas.0404376101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Nestler EJ. Epigenetic mechanisms of depression. JAMA Psychiatry. 2014;71:454–456. doi: 10.1001/jamapsychiatry.2013.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Dias BG, Maddox SA, Klengel T, Ressler KJ. Epigenetic mechanisms underlying learning and the inheritance of learned behaviors. Trends Neurosci. 2015;38:96–107. doi: 10.1016/j.tins.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wallis JD. Cross-species studies of orbitofrontal cortex and value-based decision-making. Nature neuroscience. 2012;15:13–19. doi: 10.1038/nn.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Carmichael ST, Price JL. Sensory and premotor connections of the orbital and medial prefrontal cortex of macaque monkeys. J Comp Neurol. 1995;363:642–664. doi: 10.1002/cne.903630409. [DOI] [PubMed] [Google Scholar]
- 189.Haber SN, Kunishio K, Mizobuchi M, Lynd-Balta E. The orbital and medial prefrontal circuit through the primate basal ganglia. The Journal of Neuroscience : the official journal of the Society for Neuroscience. 1995;15:4851–4867. doi: 10.1523/JNEUROSCI.15-07-04851.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ongur D, Ferry AT, Price JL. Architectonic subdivision of the human orbital and medial prefrontal cortex. The Journal of ComparativeNneurology. 2003;460:425–449. doi: 10.1002/cne.10609. [DOI] [PubMed] [Google Scholar]
- 191.Price JL, Drevets WC. Neurocircuitry of mood disorders. Neuropsychopharmacology. 2010;35:192–216. doi: 10.1038/npp.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Phillips ML, Ladouceur CD, Drevets WC. A neural model of voluntary and automatic emotion regulation: implications for understanding the pathophysiology and neurodevelopment of bipolar disorder. Mol Psychiatry. 2008;13(829):833–857. doi: 10.1038/mp.2008.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Vertes RP. Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse. 2004;51:32–58. doi: 10.1002/syn.10279. [DOI] [PubMed] [Google Scholar]
- 194.Patton MH, Bizup BT, Grace AA. The infralimbic cortex bidirectionally modulates mesolimbic dopamine neuron activity via distinct neural pathways. The Journal of Neuroscience: the official journal of the Society for Neuroscience. 2013;33:16865–16873. doi: 10.1523/JNEUROSCI.2449-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Phillips ML, et al. Identifying predictors, moderators, and mediators of antidepressant response in major depressive disorder: neuroimaging approaches. Am J Psychiatry. 2015;172:124–138. doi: 10.1176/appi.ajp.2014.14010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Pizzagalli DA. Depression, stress, and anhedonia: toward a synthesis and integrated model. Annu Rev Clin Psychol. 2014;10:393–423. doi: 10.1146/annurev-clinpsy-050212-185606. [DOI] [PMC free article] [PubMed] [Google Scholar]