

Abstract
Excess levels of protein in urine (proteinuria) is a hallmark of kidney disease that typically occurs in conjunction with diabetes, hypertension, gene mutations, toxins or infections but may also be of unknown cause (idiopathic)1. Systemic soluble urokinase plasminogen activator receptor (suPAR) is a circulating factor implicated in the onset and progression of chronic kidney disease (CKD)2, such as focal segmental glomerulosclerosis (FSGS)3,4. The cellular source(s) of elevated suPAR associated with future and progressing kidney disease is unclear, but is likely extra-renal, as the pathological uPAR is circulating and FSGS can recur even after a damaged kidney is replaced with a healthy donor organ. Here we report that bone marrow (BM) Gr-1lo immature myeloid cells are responsible for the elevated, pathological levels of suPAR, as evidenced by BM chimera and BM ablation and cell transfer studies. A marked increase of Gr-1lo myeloid cells was commonly found in the BM of proteinuric animals having high suPAR, and these cells efficiently transmit proteinuria when transferred to healthy mice. In accordance with the results seen in suPAR-associated proteinuric animal models, in which kidney damage is caused not by local podocyte-selective injury but more likely by systemic insults, a humanized xenograft model of FSGS resulted in an expansion of Gr-1lo cells in the BM, leading to high plasma suPAR and proteinuric kidney disease. Together, these results identify suPAR as a functional connection between the BM and the kidney, and they implicate BM immature myeloid cells as a key contributor to glomerular dysfunction.
FSGS is a common primary glomerular disease leading to kidney failure, necessitating dialysis or kidney transplantation5. It is characterized morphologically by segmental sclerosis in some glomeruli; clinically, it is characterized by proteinuria6,7. About 80% of FSGS cases are primary or idiopathic. FSGS recurs in newly transplanted kidneys in 30% of adults and even more frequently in children8. Because of the rapid onset of FSGS recurrence after transplantation, circulating factors have been considered as pathogenic causes9–12. We previously reported that suPAR is one such circulating factor in FSGS, and we demonstrated that suPAR binds to and activates β3 integrin on the podocyte membrane. This leads to podocyte foot process effacement and disrupted glomerular barrier function, resulting in proteinuria3,4. Furthermore, as relatively high levels of suPAR associate with lower kidney function, prospective cohort studies in humans were subsequently performed: through these, suPAR has recently emerged as a risk factor for the incidence and progression of CKD2.
Circulating suPAR can be generated by release from the membrane-bound form of urokinase plasminogen activator receptor (uPAR), a glycosylphosphatidylinositol (GPI)-anchored three-domain (DI, DII and DIII) signaling protein13,14. suPAR exists in multiple forms due to alternative splicing, protein glycosylation and enzymatic cleavage of the mature protein15. While mounting experimental and clinical evidence suggests that suPAR is involved in the pathogenesis of CKD, the cellular source(s) of elevated suPAR remains unknown. Thus, identifying the cellular source(s) of suPAR that are relevant to kidney disease is one essential step required for the exploration of potential therapeutics aimed at the treatment of suPAR-related renal dysfunction such as that seen in FSGS.
Experimental studies have shown that mice injected with lipopolysaccharides (LPS) as a model of glomerular injury display a transient proteinuria associated with podocyte foot process effacement3,4,16,17, as well as some renal lesions similar to FSGS in humans18. Based on our previous findings that uPAR deficiency protects against LPS-induced proteinuria and podocyte injury3,4, we first tested the contribution of hematopoietic cells on suPAR production and proteinuria development in the LPS model using a bone marrow transplantation (BMT) technique (Fig. 1a). We have successfully generated BM chimeric mice in which the recipient uPAR-deficient knockout (Plaur−/−, KO) mice were irradiated and reconstituted with BM cells of either uPAR wild-type (Plaur+/+, WT) or uPAR KO mice (Supplementary Fig. 1, WT→KO; 94.6% ± 3.7, KO→KO; 99.3% ± 0.7). As expected, KO→KO chimeric mice showed a strong defect in suPAR production (Fig. 1b,c) with a lack of proteinuria development (Fig. 1d) upon LPS stimulation. In contrast, the chimeric mice expressing uPAR selectively within hematopoietic cells (WT→KO) exhibited elevated suPAR levels in both blood and urine (Fig. 1b,c), as well as proteinuria (Fig. 1d), following LPS stimulation. These results suggest that hematopoietic cells are sufficient for the production of suPAR and the development of proteinuria in this model.
Figure 1.
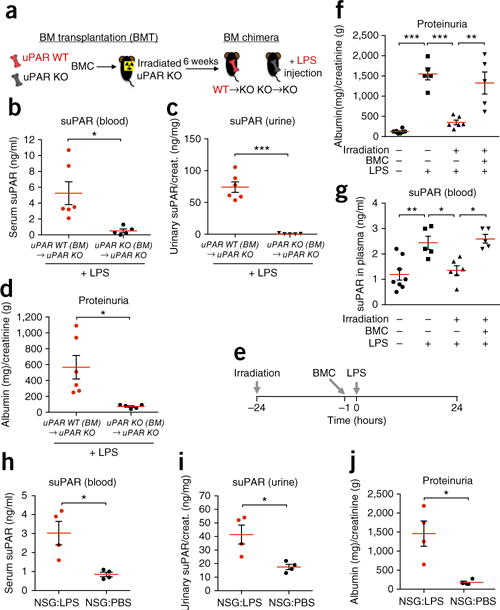
Hematopoietic cells are sufficient for suPAR-associated proteinuria. (a) Schematic diagram outlining the experimental design for BM chimera studies. (b–d) Examination of serum (b) and urinary (c) suPAR levels and proteinuria (d) in BM chimeric WT→KO (n = 6) and KO→KO (n = 5) mice that were injected with LPS. Urinary albumin-to-creatinine ratio (ACR) was calculated and used as a parameter to determine proteinuria. Data are shown as mean ± s.e.m.; unpaired two-tailed Student t-test, *P < 0.05, ***P < 0.001. (e) Schematic diagram outlining the experimental design for irradiation and BM reconstitution studies. (f,g) Proteinuria (f) and plasma suPAR levels (g) in BALB/c WT mice that were irradiated (+) or not (−), followed by injection of freshly isolated BMCs (+) or PBS (−), before LPS stimulation. The results are from two independent experiments (n = 8 for PBS, n = 5 for LPS and Irradiation + BMC + LPS, n = 6 for Irradiation + LPS). Data are shown as mean ± s.e.m. One-way ANOVA, followed up by Tukey’s multiple comparison test, *P < 0.05, **P < 0.01, ***P < 0.001. (h–j) Examination of serum (h) and urinary (i) suPAR levels and proteinuria (j) in NSG mice injected with either PBS or LPS (n = 4 per group from two independent experiments). Data are shown as mean ± s.e.m.; unpaired two-tailed Student t-test, *P < 0.05.
We further tested if the ablation of BM cells via irradiation could prevent suPAR-associated proteinuria following LPS treatment (Fig. 1e). Irradiation before LPS injection led to a marked reduction in the degree of proteinuria (Fig. 1f and Supplementary Fig. 2a,b) as well as in plasma suPAR levels (Fig. 1g) in BALB/c mice. However, when irradiated mice were reconstituted with unstimulated BM cells, they once again displayed proteinuria (Fig. 1f) with elevated plasma suPAR levels (Fig. 1g) upon LPS treatment.
Given that hematopoietic cells are categorized into lymphoid cells and myeloid (erythro-myeloid) cells, we next examined which cell lineages participate in suPAR-associated proteinuria. This was accomplished by taking advantage of NOD-scid IL-2rγnull (NSG) mice, in which the white blood cell population is comprised of predominantly myeloid-lineage cells due to the lack of mature lymphocytes: T cells, B cells and natural killer (NK) cells19. Despite impaired lymphoid populations, these NSG mice showed comparable levels of suPAR and proteinuria to BALB/c WT mice upon LPS administration (Fig. 1h–j). Based on these observations and previous findings16,17, we could rule out the contribution of mature lymphocytes on suPAR-associated proteinuria in this model and focus on cells of the myeloid lineage.
To define the nature of the myeloid cells in the LPS proteinuric mice, we studied the expressions of uPAR and of the mouse myeloid differentiation antigen Gr-1 in BM cells as well as in peripheral blood leukocytes. In LPS-treated animals, BM Gr-1+ myeloid cells—but not peripheral blood Gr-1+ cells—exhibited elevated levels of uPAR on their membrane when compared to phosphate-buffered saline (PBS)-treated mice (Fig. 2a). The surface expression of Gr-1 is representative of the maturation status of myeloid cells20. In the BM, the level of Gr-1 expression is low on myeloid progenitors or immature cells and increases as they mature to granulocytes. Of note, LPS stimulation led to a marked increase in the percentage of Gr-1lo cells in the BM of WT mice (Fig. 2b and Supplementary Fig. 3a,b). However, loss of granulocyte colony-stimulating factor (G-CSF) receptor (G-CSFR), a major regulator of myelopoiesis21, resulted in impaired expansion of Gr-1lo BM myeloid cells upon LPS treatment (Fig. 2b) and, accordingly, in lower levels of proteinuria (Fig. 2c). We therefore hypothesized that the reactive expansion of immature myeloid cell populations in BM may contribute to the pathogenic kidney process. To test this, we used two conditions—antibody-induced peripheral neutropenia and G-CSF treatment—as myeloid cell production can be stimulated either through feedback regulation in response to peripheral neutropenia or via direct supplementation with G-CSF. Injection of Ly6G-specific monoclonal antibody resulted in depletion of peripheral mature neutrophils (Supplementary Fig. 4a,b) and augmented the degree of proteinuria in LPS-challenged mice (Supplementary Fig. 4c). Similarly, G-CSF stimulated myelopoiesis (Supplementary Fig. 4d,e), accelerated proteinuria and elevated plasma suPAR levels as compared to LPS treatment alone (Supplementary Fig. 4f,g). Together, these data suggest that increased myelopoiesis amplifies suPAR-associated proteinuria in the LPS model.
Figure 2.
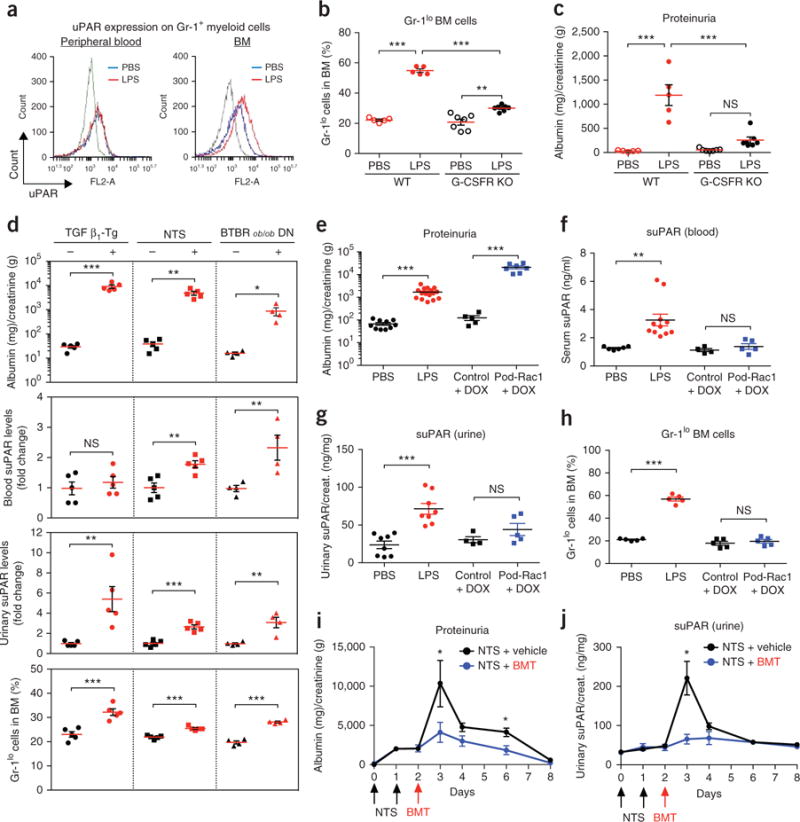
Expansion of Gr-1lo BM cells is involved in suPAR-associated proteinuria. (a) Representative overlay histograms of flow cytometric analysis showing the expression profiles of uPAR on the Gr-1+ myeloid cells isolated from the peripheral blood (left) and BM (right) of PBS-injected (n = 4) and LPS-injected (n = 5) mice. The results are representative of two independent experiments. Background fluorescence (gray line) was determined with an irrelevant isotype-matched antibody. Blue line, PBS; red line, LPS. (b,c) The percentages of Gr-1lo cells in BM (b) and the ACR (c) of C57BL/6 WT and G-CSFR KO mice that were injected with LPS or PBS. The results are from two independent experiments (n = 5 for WT PBS and WT LPS, n = 7 for KO PBS and KO LPS). Data are shown as mean ± s.e.m. One-way ANOVA, Tukey’s multiple comparison test, **P < 0.01, ***P < 0.001, NS, not significant. (d) Examination of proteinuria, suPAR levels, and the percentages of Gr-1lo cells in BM from three different animal models of proteinuria: (i) TGF β1-Tg mice (n = 5 per group), (ii) NTS-injected mice (n = 5 per group), and (iii) BTBR ob/ob DN mice (n = 4 per group). Their relevant controls were used as described in the Online Methods. Data are shown as mean ± s.e.m.; unpaired two-tailed Student t-test, *P < 0.05, **P < 0.01, ***P < 0.001, NS, not significant. (e–h) Examination of proteinuria (e), suPAR levels in the serum (f) and urine (g) and the percentages of Gr-1lo cells in BM (h) of LPS injected and Pod-Rac1 transgenic mice. Their relevant controls were used as described in the Online Methods. In e n = 10 for PBS, n = 16 for LPS, n = 5 for control+DOX, n = 7 for Pod-Rac1+DOX); in f n = 6 for PBS, n = 11 for LPS, n = 4 for control+DOX, n = 5 for Pod-Rac1+DOX; in g n = 8 for PBS, n = 8 for LPS, n = 4 for control+DOX, n = 5 for Pod-Rac1+DOX; in h n = 5 per group. Data are shown as mean ± s.e.m.; unpaired two-tailed Student t-test, **P < 0.01, ***P < 0.001, NS, not significant. (i,j) Proteinuria (i) and urinary suPAR levels (j) in the NTS-injected C57BL/6 mice receiving PBS (NTS + vehicle, n = 3) or BMT (NTS + BMT, n = 3). Data are shown as mean ± s.e.m.; unpaired two-tailed Student t-test, *P < 0.05.
Given the recent discovery of suPAR as a risk factor for CKD2, we reasoned that the expansion of Gr-1lo immature myeloid cells in the BM could be a common feature of suPAR-associated proteinuric kidney disease. We subsequently examined the levels of suPAR and Gr-1lo BM myeloid cells in additional animal models of proteinuria that included (i) TGF β1 Tg (transforming growth factor beta 1 transgenic), a mouse model that develops severe fibrosing kidney disease22,23, (ii) NTS (nephrotoxic serum nephritis), a rodent model of glomerulonephritis (GN)24, and (iii) BTBR ob/ob mice, a mouse model of diabetic nephropathy (DN)25. All tested animals exhibited proteinuria after their respective induction (Fig. 2d). Elevated suPAR levels were detected in the urine of TGF β1 Tg mice that typically have severe, nonselective proteinuria and in both blood and urine samples of NTS and DN models. These elevated suPAR levels were accompanied by an expansion in Gr-1lo BM myeloid cells (Fig. 2d) as seen in the LPS model.
Further testing with additional models of proteinuria shows a potential difference in the disease process between suPAR-mediated and suPAR-independent forms of disease. We tested blood and urine suPAR levels along with Gr-1lo cell populations in a genetic and a pharmacological model of podocyte injury. NEF-rtTA:Rac1 is a double transgenic mouse line where the expression of active Rac1 is induced in a podocyte-specific manner when animals are fed a diet that includes doxycycline (DOX) (hereafter referred as Pod-Rac1)26. Adriamycin (ADR) can induce nephropathy by causing toxin-mediated podocyte mitochondrial damage27. Unlike the suPAR-associated proteinuric models previously mentioned, the Pod-Rac1 (Fig. 2e) and ADR (Supplementary Fig. 5a) models, in which podocytes are the direct target of injury, did not exhibit elevated suPAR levels in the blood or urine (Fig. 2f,g and Supplementary Fig. 5b,c). Furthermore, there was no significant increase of the percentage of Gr-1lo myeloid cells in the BM (Fig. 2h and Supplementary Fig. 5d). Taken together, these results suggest that expansion of Gr-1lo BM myeloid cells could be a common upstream event that leads to systemic suPAR elevation, resulting in podocyte injury and proteinuria development in certain forms of kidney disease.
Next, we set out to test the therapeutic effect of BMT in suPAR-associated kidney dysfunction. NTS administration induced proteinuria and increased suPAR levels, reaching a peak at day 3 (Fig. 2i,j). However, the proteinuria following NTS injections was substantially ameliorated by irradiation and reconstitution with normal BM cells on day 2, and urinary suPAR levels were also reduced (Fig. 2i,j). BMT also ameliorated NTS-induced renal damage, including crescent formation and glomerular sclerosis (Supplementary Fig. 6a,b). These results support the notion that BM plays an important role in suPAR-associated glomerular dysfunction, acting as an upstream regulatory system.
Our group4 and others28 (GenBank accession NM_011113, without GPI anchor) have previously reported that injection either of full-length suPAR into uPAR KO mice or of a shorter form of suPAR (GenBank accession BC010309) into WT mice leads to rapid proteinuria. Subsequent studies29,30 have shown that full-length suPAR injected into WT mice does not elicit a rapid proteinuric response unless combined with some other podocyte-injurious molecule such as anti-CD40 autoantibody31. Given the recent insight that elevated suPAR predicts CKD several years before its onset, we tested the effect of chronic exposure of suPAR on podocyte injury by generating a novel transgenic mouse model (suPAR-Tg) that expresses mouse suPAR (full length: DIDIIDIII, without GPI anchor) under the control of the adipocyte protein 2 (aP2, also known as Fabp4) promoter. This model, in contrast to shorter timed suPAR exposure models29,30, allows for the analysis of long-term effects of high levels of suPAR (Supplementary Fig. 7a), as large amounts of suPAR can be generated from adipocytes of the transgenic mice fed a high-fat diet with a consequent release into the circulation (Supplementary Fig. 7a). At 16 weeks of age (and after being on a high-fat diet for the prior 8 weeks), suPAR-Tg mice exhibited a marked increase of suPAR expression in adipose tissues along with elevated plasma suPAR levels as compared to littermate controls (Supplementary Fig. 7b,c). Subsequently, these mice developed relatively mild but significant albuminuria (Supplementary Fig. 7d P = 0.016) with podocyte foot process effacement (Supplementary Fig. 7e), implying a disruption in kidney filter function.
Evidence continues to accumulate pointing to suPAR as being causative and not merely a biomarker for kidney disease4,28,31,32. Thus, we hypothesized that Gr-1lo BM myeloid cells harvested from proteinuric mice may be able to induce proteinuria when injected into healthy animals (Fig. 3a). To test this, BM cells isolated from PBS- or LPS-injected NSG mice were adoptively transferred into unchallenged NSG recipient mice. Indeed, transfer of LPS-challenged BM cells induced proteinuria in healthy recipient mice (Fig. 3a). Similarly, LPS-challenged WT C57BL/6 mouse BM cells were able to stimulate proteinuria (Fig. 3b and Supplementary Fig. 8a,b) and increase suPAR levels (Supplementary Fig. 8c,d) in recipient mice, whereas these effects were abrogated when Plaur−/− BM cells were transferred (Supplementary Fig. 8). However, the BM cells of Pod-Rac1 proteinuric mice did not cause proteinuria (Fig. 3b), suggesting that podocyte injury per se does not induce Gr-1lo BM myeloid cells to become kidney pathogenic; this interpretation is consistent with low systemic suPAR levels and low percentage of Gr-1lo myeloid cells in BM of Pod-Rac1 proteinuric mice.
Figure 3.
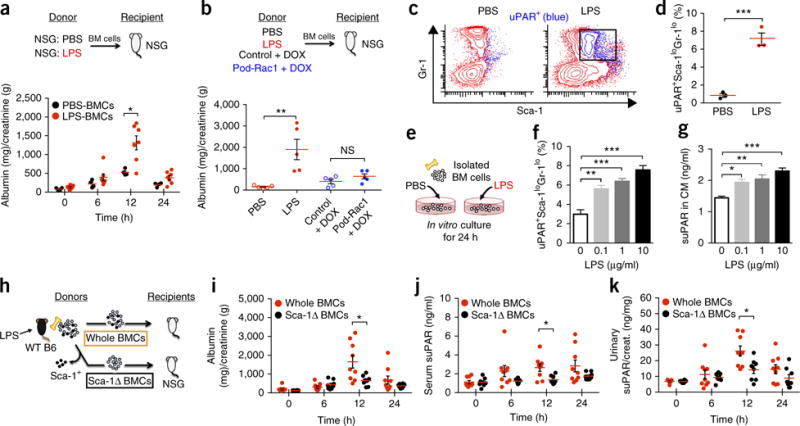
BM immature myeloid cells have an ability to transfer disease. (a) Examination of proteinuria in recipient NSG mice before (0) and 6, 12, and 24 h after adoptive transfer of BM cells obtained from PBS- or LPS-injected NSG mice. The results are from two independent experiments (n = 4 for PBS-BMCs; n = 7 for LPS-BMCs). Data are shown as mean ± s.e.m.; unpaired two-tailed Student t-test, *P < 0.05. (b) Examination of proteinuria in recipient NSG mice 12 h following adoptive transfer of BM cells obtained from LPS-challenged C57BL/6 WT and Pod-Rac1 mice. n = 5 per group. Data are shown as mean ± s.e.m. One-way ANOVA, Tukey’s multiple comparison test, **P < 0.01, NS, not significant. (c) Representative dot plots (chosen from a total of three generated) of triple-color stained (uPAR/Sca-1/Gr-1) total BM cells (red) isolated from PBS- or LPS-injected C57BL/6 mice (n = 3 per group). uPAR+ cells (blue) were gated and shown in these dot plots. (d) Quantitation for uPAR-expressing immature myeloid cells (uPAR+Sca-1loGr-1lo) cells shown in c (n = 3 per group). Data are shown as mean ± s.e.m.; unpaired two-tailed Student t-test, ***P < 0.001. (e–g) In vitro culture of total BM cells isolated from C57BL/6 WT mice with PBS or various concentrations of LPS (0.1, 1, and 10 μg/ml). (e) Experimental scheme. (f) In vitro induction of uPAR+Sca-1loGr-1lo cells determined by triple color-flow cytometric analysis. (g) suPAR secretion into culture medium (CM) measured by suPAR ELISA. Data are shown as mean ± s.e.m. One-way ANOVA, Tukey’s multiple comparison test, *P < 0.05, **P < 0.01, ***P < 0.001. (h) Schematic diagram outlining the experimental design of the BM cell transfer used in i–k. (i–k) Examination of proteinuria (i), serum (j) and urinary suPAR levels (k) in recipient NSG mice before (0) and 6, 12, and 24 h after adoptive transfer of either whole BM cells or Sca-1+ cell-depleted (Sca-1Δ) BM cells of LPS-challenged C57BL/6 WT mice. The results are from three independent experiments (n = 9 for whole BMCs; n = 8 for Sca-1ΔBMCs). Data are shown as mean ± s.e.m.; unpaired two-tailed Student t-test, *P < 0.05.
Since Gr-1lo myeloid cells in the BM are heterogeneous, we sought to determine which subsets of uPAR-expressing Gr-1lo cells are responsible for the development of proteinuria. Given that stem cell antigen-1 (Sca-1) is expressed on mouse hematopoietic stem/progenitor cells (HSPCs)33 and the expression is markedly enhanced by particularly primitive myeloid and granulopoietic progenitors during bacterial infection34, we examined Sca-1 expression in uPAR-expressing Gr-1lo BM cells. LPS stimulation increased a uPAR-expressing BM cell population (shown in blue in Fig. 3c) that is Sca-1loGr-1lo, suggesting that uPAR-expressing cells are myeloid-lineage-committed cells (defining immature myeloid cells) (Fig. 3c,d). To test whether the immature myeloid cells are capable of secreting suPAR, we studied suPAR secretion using in vitro BM cell culture under LPS stimulation (Fig. 3e). Consistent with the results from our in vivo studies, LPS stimulated the expansion of uPAR-expressing immature myeloid cells (Fig. 3f) as well as suPAR secretion (Fig. 3g) into the culture medium (CM) in a dose-dependent manner. To further evaluate if these cells can transfer disease when injected into healthy mice, Sca-1+ cells were depleted from LPS-challenged BM cells by magnetic separation before transfer into NSG mice (Fig. 3h and Supplementary Fig. 9). The removal of Sca-1+ BM cells lowered both proteinuria (Fig. 3i) and suPAR levels (Fig. 3j,k) when compared with that in non-depleted control BM cells. These results emphasize the primary role of BM immature myeloid cells in the regulation of suPAR production and proteinuria development.
In an effort to translate our findings to human disease, we used a humanized mouse approach35–37. A model commonly used is the immunodeficient mouse engrafted with human hematopoietic cells, such as peripheral blood mononuclear cells (PBMCs) or hematopoietic stem/progenitor cells (HSPCs). A previous report38 demonstrated that transfer of CD34+ HSPCs from individuals with glomerular diseases into healthy mice can induce albuminuria, but offered no mechanism. Thus, we hypothesized that human CD34+ cells isolated from individuals with recurrent FSGS might induce proteinuria via suPAR-driven podocyte injury. To test this hypothesis, we introduced whole PBMCs or PBMCs depleted of CD34+ cells (CD34Δ PBMCs) derived either from individuals with recurrent FSGS or from healthy individuals into the NSG mice (Fig. 4a). Engraftment rate of human cells was determined by the percentage of human CD45+ cells in the blood and BM of the recipient xenograft animals (Supplementary Fig. 10a). Mice receiving patient whole PBMCs, but not CD34Δ PBMCs, developed proteinuria (Fig. 4b) in association with high mouse suPAR levels in both blood and urine (Fig. 4c,d). These mice also had a higher percentage of Gr-1lo population in BM (Fig. 4e), similar to what we observed in the suPAR-associated proteinuric animal models. This disease phenotype was prevented by depleting CD34+ cells from the samples of individuals with FSGS (Fig. 4b–e). Proteinuria in mice receiving patient whole PBMCs was accompanied by mild glomerular sclerosis (Supplementary Fig. 10b) and podocyte foot process effacement (Fig. 4f). These mice also exhibited elevated blood urea nitrogen (BUN) levels, increased kidney weight and enlarged, loosely structured glomeruli (Supplementary Fig. 10c–e) reminiscent of human FSGS. It is unlikely that the observed impairment in kidney function was due to graft-versus-host disease (GVHD) since PBMCs from the healthy individuals, as well as those depleted of CD34+ cells derived from individuals with recurrent FSGS, did not cause kidney injury. In contrast, using a mouse model of GVHD, we observed both weight loss and a mortality rate of 80% within 3 weeks in the mice receiving allogeneic BM cells (Supplementary Fig. 11a,b). The xenograft mice did not exhibit body weight loss (Supplementary Fig. 12a). Moreover, only negligible glomerular deposits of human IgGs were observed (Supplementary Fig. 12b), as well as very mild lymphocyte infiltration in the skin and liver of the xenograft mice (Supplementary Fig. 12c). Since GVHD is mainly caused by donor T cells, we repeated the xenograft experiment with PBMCs depleted of T cells to avoid the risk of GVHD in the xenograft model. Consistently, we found that T-cell-depleted PBMCs from individuals with recurrent FSGS were sufficient to induce proteinuria and increase suPAR levels in mice (Fig. 4g–i). Taken together, these data suggest that engraftment of CD34+ cells derived from individuals with recurrent FSGS into NSG mice caused expansion of Gr-1lo BM myeloid cells, leading to suPAR-associated podocyte injury that results in proteinuria.
Figure 4.
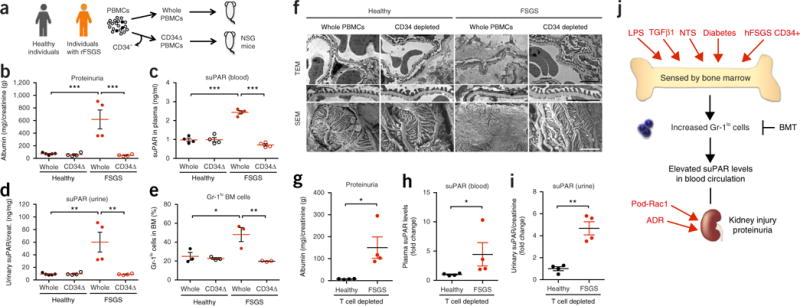
hFSGS CD34+ cells induce suPAR-associated proteinuria in mice. (a) Schematic diagram outlining the studies of humanized mice. rFSGS, recurrent FSGS. (b–e) Proteinuria (b), mouse suPAR levels in plasma (c) and in urine (d), percentages of Gr-1lo cells in BM (e) of the xenograft mice after 10–12 weeks post-engraftment. The results are from two independent experiments. In b–d, n = 4 per group, except n = 5 for healthy whole PBMCs; in e, n = 3 per group. Data are shown as mean ± s.e.m. One-way ANOVA, followed up by Tukey′s multiple comparison test, *P < 0.05, **P < 0.01, ***P < 0.001. (f) Transmission and scanning electron microscope (TEM, 10,000×, and SEM, 15,000×) analysis of kidney glomeruli of the xenograft mice. TEM images displaying podocyte foot processes were enlarged and highlighted. SEM images show a podocyte cell body, primary processes and interdigitating foot processes. Scale bars, 2 μm. (g–i) T-cell-depleted PBMCs of individuals with recurrent FSGS or healthy donors were injected into NSG mice. Proteinuria (g), mouse suPAR levels in plasma (h) and in urine (i) of the xenograft mice after 16 weeks post-engraftment (n = 4 per group). Data are shown as mean ± s.e.m. Student’s t-test, *P < 0.05, **P < 0.01. (j) Model depicting a role for Gr-1lo BM immature myeloid cells in suPAR-driven podocyte injury and proteinuria. Systemic immunological proteinuric models—LPS, hFSGS xenograft, TGF β1 transgenic (fibrosing nephrotic syndrome), NTS (serum nephritis) and BTBR ob/ob (diabetic nephropathy)—converge at the expansion of Gr-1lo cells in BM and high blood suPAR levels.
Here we demonstrate that BM myeloid lineage cells that are Gr-1lo and Sca-1lo are a common source for the overproduction of suPAR that fuels the development of proteinuric kidney disease. The systemic models (LPS, TGF β1-Tg, NTS and DN) and the xenograft mouse model of human FSGS converge at the expansion of uPAR-expressing immature myeloid cells in BM and high suPAR levels, unlike podocyte selective injury models (Pod-Rac1 and ADR) (Fig. 4j). Given that bacterial infection induces massive myeloid cell production in BM, it seems that the LPS model is a preferentially immune-related model as compared to other models tested. Indeed, the LPS model showed the highest percentage of Gr-1lo BM myeloid cells among the suPAR-associated proteinuric animal models. The pathogenesis of these other models may be undoubtedly more complex and heterogeneous, resulting in differing disease states and/or severity. In addition, transfer of these pathogenic myeloid cells was sufficient to cause proteinuria in healthy mice. Consistently, BMT substantially reduced suPAR levels and subsequently improved kidney function in proteinuric mice. In this study, we expand the role of BM from being merely a place for hematopoietic cell production to also being a key regulator of kidney function by expanding suPAR-generating cells. The expansion of suPAR-expressing immature myeloid cells could be a common upstream event that leads to suPAR-associated kidney injury seen in FSGS and possibly relevant to other proteinuric kidney diseases. Given the observation of increased Gr-1lo BM cells in animal models of primary and secondary forms of glomerular disease, this finding may represent a dogma change, and this discovery may also shift the focus away from the kidney to common upstream regulatory systems with broad relevance to general forms of CKD. In accordance with the notion of a very upstream mediator of kidney disease, suPAR levels predicted incidence of CKD in people many years ahead of the onset of symptoms or the diagnosis of disease2.
Although we have identified uPAR-expressing BM immature myeloid cells (Sca-1loGr-1lo) as cellular sources of suPAR responsible for damaging kidney podocytes in mice, the equivalent type of cell subset in humans remains to be determined and is not immediately identifiable given the lack of human homologs for Gr-1 or Sca-1. Thus, the identification of cellular sources of suPAR in patients followed by a relevant therapeutic regimen for proper modification could be the basis for a novel treatment of proteinuric kidney diseases that are characterized by high suPAR levels.
We engineered a xenograft mouse model of FSGS and further revealed that CD34+ HSPCs derived from individuals with recurrent FSGS are critical for development of a suPAR-associated glomerulopathy in mice. This was surprising given the rarity of CD34+ cells among PBMCs: a frequency of only 0.05–0.2%. However, the mechanism by which CD34+ cells from individuals with FSGS expand the murine myeloid cells remains elusive. According to our observations, it seems that CD34+ cells from people with suPAR-associated kidney disease are functionally different from those from healthy individuals, and these HSPCs carry and transfer information on disease pathogenesis. Similarly, Gr-1lo BM myeloid cells generated in the systemic inflammatory models such as the LPS model increased suPAR levels and caused proteinuria, whereas Gr-1lo BM myeloid cells produced by normal myelopoiesis did not. Supporting our hypothesis, it has been shown that HSPCs are significantly expanded and functionally altered in lupus model mice, with skewing toward myeloid cell production39. It has also been shown that undifferentiated HSPCs produce a wide range of cytokines upon stimulation and these cells are significantly more potent cytokine secretors than are mature immune cells40. It will be of interest to further investigate whether CD34+ cells from individuals with FSGS produce distinct cytokines that are capable of stimulating the mouse hematopoietic system to become pathogenic, leading to an increase of suPAR and induce proteinuria.
In conclusion, this study further demonstrates a direct pathogenic role of suPAR as a circulating mediator of kidney disease, such as FSGS. Given its specific cellular source, suPAR-associated renal diseases may originate as a primary hematopoietic stem cell disorder, providing novel functions for BM in extrarenal regulation of the glomerular filtration barrier. In addition, our findings serve as a prototype for the pathogenesis of other diseases in which BM progenitors may regulate organ function via soluble mediators presently categorized as ‘idiopathic’. Stem cell transplantation may prove to be a viable approach to treat diseases such as suPAR-associated kidney disease.
METHODS
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
ONLINE METHODS
Mice
BALB/c, C57BL/6, Plaur−/− (uPAR KO), B6.SJL (CD45.1), csf3r−/− (G-CSFR null), BTBR/ob heterozygotes (BTBRob+/−; BTBR.V(B6)-Lepob/WiscJ) and NOD-scid IL2rγnull (NSG) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). The Plaur−/− mice were originally on a mixed background of 75% C57BL/6 and 25% 129, but backcrossed to C57BL/6 mice for more than ten generations before use. EGFP_CA-Rac1 transgenic, and Nphs1-rtTA (NEF-rtTA) mice were kindly provided by A.S. Shaw (Washington University School of Medicine, Missouri)26. Albumin TGF β1 transgenic and littermate mice were kindly provided by M. Bitzer (University of Michigan)22. Animal experiments were carried out according to the National Institutes of Health Guide for Care and Use of Experimental Animals and approved by the Rush University Institutional Animal Care and Use Committee (IACUC). All groups of mice were age- and sex-matched. The numbers of animals used in each experiment are described in the figure legends. Five experimental animal models of proteinuria were used in this study. i) LPS injected mice: Proteinuria was induced with a single injection of LPS as described previously3,4. Briefly, 10- to 12-week-old female and male C57BL/6 mice were injected with LPS (Sigma, L3129) intraperitoneally (i.p.) at a dose of 10 mg per kg body weight. PBS injected mice were used as a control. 24 h after LPS treatment, urine and blood samples were collected and kidney, femurs and tibias were harvested from killed mice. ii) Pod-Rac1, inducible EGFP_CA-Rac1 transgenic mice: EGFP_CA-Rac1 transgenic mice were crossed to Nphs1-rtTA (NEF-rtTA) inducer mice to generate double transgenic (dTG, NEF × Rac1) mice. Male mice were used in this study. To induce transgene expression, regular chow was replaced with doxycycline (DOX)-supplemented chow (2,000 ppm; TestDiet). Single transgenic (EGFP_CA-Rac1) mice were used as a control. 12 d after DOX administered diet, urine and blood samples were collected and kidney, femurs and tibias were harvested from killed mice. iii) Adriamycin (ADR) injected mice: Proteinuria was induced with a single injection of ADR as described previously40. Briefly, male BALB/c mice aged 10–12 weeks were injected with ADR (Doxorubicin Hydrochloride, Sigma, D1515) via the tail vein at a dose of 11 mg per kg body weight. 6 d after ADR injection, urine and blood samples were collected and kidney, femurs and tibias were harvested from killed mice. iv) TGF β1 transgenic mice: transgenic mice expressing an active form of TGF β1 under the control of the murine albumin promoter developed proteinuria as described previously22. Since the transgene is on the Y chromosome, only male mice (aged <4 weeks) with a severe phenotype were used. Littermate female mice were used as a control. v) Nephrotoxic serum (NTS) nephritis model: Proteinuria was induced by injection of NTS as described previously24. Briefly, male C57BL/6 mice aged 8 weeks were injected i.p with sheep anti-rabbit-glomerular basement membrane antibody at a dose of 500 mg per kg body weight on consecutive days. PBS injected mice were used as a control. 7 d after injection, urine and blood samples were collected and kidney, femurs and tibias were harvested from killed mice. vi) BTBR ob/ob diabetic nephropathy (DN) model: the mouse strain BTBR with the ob/ob leptin-deficiency mutation developed severe type 2 diabetes with heavy proteinuria as described previously25. Wild-type (BTBR ob+/+), heterozygous (BTBR ob+/−) and homozygous (BTBR ob−/−) mice were obtained by mating heterozygous (BTBR ob+/−) mice. For the DN model, male and female homozygotes aged 16 weeks (BTBR ob−/−) were used. Wild-type (BTBR ob+/+) or heterozygote (BTBR ob+/−) littermates were used as controls.
Generation of BM chimeric mice
We used female uPAR KO mice aged 10–12 weeks as recipients. uPAR KO (CD45.2) mice were irradiated with a dose of 9.5 Gy using Gammacell 40 and reconstituted via retro-orbital injection with 1 × 107 BM cells of either uPAR WT (B6.SJL, CD45.1) or uPAR KO (CD45.2) mice. Mice were administered antibiotic-treated water and used for experiments at 6 weeks after BM transplantation. Engraftment of the donor cells was evaluated by flow cytometric analysis of peripheral blood leukocytes stained with fluorescence-conjugated antibodies against CD45.1 (eBioscience, 12-0453) and CD45.2 (eBioscience, 11-0454).
Detection of circulating and urinary suPAR levels
Mouse suPAR levels from plasma (and/or serum) and urine were evaluated by enzyme-linked immunosorbent assay (ELISA) kit (R&D systems, DY531) following the manufacturer’s protocol.
Albumin creatinine ratio (ACR) measurement
Mouse urine samples were collected and urinary albumin and creatinine were measured by mouse albumin ELISA (Bethyl Labs, E99-134), and creatinine assay (Cayman Chemical, 500701) kits according to manufacturers’ protocols. The ratio of urinary albumin to creatinine was then calculated.
Purification of mouse PBLs and BMCs
To collect blood, mice were anesthetized, then the blood was drawn from posterior vena cava into acid-citrate-dextrose (ACD, Sigma, C3821) solution-containing 1 ml syringe. After lysis of red blood cells (RBCs), blood leukocytes were washed and counted. For BMCs purification, femurs and tibias were taken from killed mice and flushed with a syringe filled with Hank’s balanced salt solution (HBSS, Life Technologies, 14170112) containing 0.1% bovine serum albumin and 20 mM HEPES (pH 7.4). Following RBC lysis, the BM cell suspensions were filtered through a 40 μm cell strainer (Falcon).
Adoptive transfer of BMCs
To explore the role of BMCs in production of suPAR in proteinuric mice, female BALB/c mice aged 10–12 weeks were lethally irradiated with 9.5 Gy using Gammacell 40. On the next day, these mice received syngeneic donor BMCs, which were labeled with Calcein-AM (Molecular Probes, C3100MP), via the retro-orbital route (5 × 106 cells per mouse). To induce proteinuria, LPS was injected into the recipient mice 1 h after transfer of BMCs. 24 h after LPS treatment, urine and blood samples were collected and kidney, femurs and tibias were harvested from the killed mice.
Flow cytometric analysis
Isolated peripheral blood leukocytes (PBLs) or BMCs were resuspended with PBS and single cell suspensions were first incubated with rat anti-mouse CD16/CD32 (eBioscience, 14-0161) at 5 μg/ml for 30 min on ice to block Fc receptors. The cells were then labeled for 30 min on ice with fluorescence-conjugated antibodies at 2 μg/ml: Gr-1-Alexa Fluor 647 (BioLegend, 108418), uPAR-phycoerythrin (PE) (R&D Systems, FAB531P), Sca-1- fluorescein isothiocyanate (FITC) (eBioscience, 11-5981), c-kit-allophycocyanin (APC) (eBioscience, 17-1171) and irrelevant IgG isotype control anti-bodies. Data acquisition and analysis were performed on an Accuri C6 flow cytometer (BD Biosciences).
In vivo depletion
Anti-Ly6G monoclonal antibody (1A8, BioXcell) or control rat IgG2a antibody (2A3, BioXcell) was injected i.p. into female BALB/c mice aged 10–12 weeks (500 μg per mouse) 48 h and 1 h before LPS injection. Neutrophil depletion was confirmed by flow cytometric analysis.
Treatment with granulocyte colony stimulating factor (G-CSF)
Recombinant mouse G-CSF (Shenandoah Biotechology, 200-14) was administrated by daily i.p. injection into BALB/c mice (female, aged 10–12 weeks) at a dose of 4 μg per 20 g body weight for two consecutive days. Blood cell counts were determined using a Hemavet 950FS (Drew Scientific).
Intervention study (NTS model)
Male C57BL/6 mice aged 8 weeks were injected i.p with NTS (250 mg per kg body weight) for two consecutive days (day 0 and day 1). The next day (day 2), proteinuric animals were divided into two groups having an equal ACR mean level; one group was irradiated (9.5 Gy) and received BM cells (1 × 107 in 100 μl PBS, retro-orbital injection) collected from naive male C57BL/6 mice as a treatment group (NTS + BMT). The other group received PBS (100 μl, retro-orbital injection) as a control (NTS + vehicle). Urine and blood samples were collected and proteinuria and serum suPAR levels were evaluated before and after BMT treatment.
Generation of suPAR transgenic mice
We generated a suPAR transgenic mouse (suPAR-Tg) model overexpressing the soluble form of mouse full-length suPAR (corresponding to NP_035243, DIDIIDIII without GPI anchor) in adipose tissue using the adipocyte fatty acid binding protein (AP2) promoter. The suPAR transgene (DIDIIDIII) is secreted into the circulation owing to the absence of a cell-tethering GPI anchor. Non-transgenic (non-Tg) littermate mice were used as a control. To enhance suPAR production, regular rodent diet was replaced by high fat food when the mice were aged at least 2 months, and the animals were kept on the high fat diet for 8 weeks. Both suPAR-Tg and non-Tg control animals showed comparable body weight. After 8 weeks of high fat diet feeding, urine and blood samples were collected and kidney and visceral fat tissues were harvested from the killed mice. Adipose-tissue-specific expression of the suPAR transgene was confirmed by performing ELISA using lysates of visceral fat tissues of the suPAR-Tg and non-Tg littermate controls.
BM cell transfer from proteinuric animal models into NSG mice
To evaluate if Gr-1lo BM myeloid cells from mice with proteinuria are able to induce proteinuria, the BM cells were isolated from different proteinuric animal models, then transferred into 10- to 12-week-old female NSG mice. 12 h following transfer of donor BM cells, ACR was measured from the urine samples of the recipient mice. To test the ability of BM cells to develop proteinuria directly, donor (WT or Plaur−/−) mice were injected with LPS, 24 h later, BM cells were isolated and transferred into NSG recipient mice (female, aged 10–12 weeks). ACR and suPAR levels were monitored in a time course as indicated in the relevant figure. To determine if Sca-1+ cells are required for suPAR-associated proteinuria, BM cells were isolated from LPS-challenged C57BL/6 WT mice (female, aged 10–12 weeks) and divided into 2 groups, whole BM cells and Sca-1+ cell depleted BM cells. These two groups of cells were then transferred into female NSG mice aged 10–12 weeks. ACR and suPAR levels were monitored over time.
In vitro LPS stimulation on isolated BM cells
The BM cells were isolated from C57BL/6 WT mice (female, aged 10–12 weeks) and treated with PBS or various concentrations of LPS (0.1, 1, and 10 μg/ml). Following 24-h incubation at 37 °C in a CO2 incubator (5%), the cells and culture supernatants were collected. The cells were stained for uPAR, Sca-1 and Gr-1. The in vitro induction of uPAR+Sca-1loGr-1lo cells in total BM cells was determined by triple color-flow cytometric analysis. The culture supernatants were used for suPAR measurement.
Cell depletion methods
To study adoptive transfer of Sca-1+ cell depleted BMCs, Sca-1+ cells were removed from whole BMCs using EasySep mouse cell isolation kits (Stemcell technologies, 18556). In brief, LPS-challenged WT BM cells were isolated and labeled with biotinylated anti-mouse Sca-1 antibody and then incubated with streptavidin-magnetic micro beads. By collecting unbound cells in the magnetic field, Sca-1+ cell-depleted BM cells were prepared. For the humanization study, human CD34+ or CD3+ cells were depleted from PBMCs obtained from individuals with recurrent FSGS and healthy donors, using EasySep human CD34 positive selection kit (Stemcell technologies, 18056) or EasySep human CD3 positive selection kit II (Stemcell technologies, 17851).
Human subjects
Peripheral blood was drawn from healthy volunteers and individuals with recurrent FSGS in accordance with guidelines on human research and approval of the Institutional Review Board of Rush University Medical Center. Informed consent was obtained from the blood donors. All individuals with recurrent FSGS participated in this study have undergone kidney transplantation and shown a recurrence in the allograft.
Engraftment of human PBMCs into NSG mice
PBMCs were isolated from peripheral blood of healthy volunteers and individuals with recurrent FSGS by standard ficoll separation. The purified PBMCs were transferred via i.p. injection into female NSG mice aged 6–8 weeks (5 × 106 cells per mouse) on day 0. The engrafted mice were monitored overtime for the development of FSGS-like phenotypes by monitoring proteinuria and high suPAR levels in the blood and urine, respectively. We used freshly isolated PBMCs for transfer as the freeze-thawed PBMCs showed poor engraftment in our experimental settings. For measurement of reconstitution rates of human cells, peripheral blood was drawn from the mice 10–12 weeks post engraftment and blood leukocytes were stained with fluorescence-labeled antibodies against human CD45 (Beckman Coulter, IM2473U) and mouse CD45 (BD Pharmingen, 553081). Following RBC lysis, the cells were analyzed by flow cytometry. To rule out the risk of GVHD, donor T cells were removed from PBMCs before engraftment. In brief, human CD3+ cells were depleted from PBMCs obtained from individuals with FSGS and healthy donors. The T-cell depleted PBMCs were injected via i.p into female NSG mice aged 8 weeks (3.5 × 106 cells per mouse) on day 0. ACR and suPAR levels were measured at 16 weeks post-engraftment.
Histology
Kidneys were dissected from the xenograft mice and NTS injected animals (NTS and NTS + BMT). Renal tissues were fixed overnight in formalin and embedded in paraffin. The sections were cut at 3- to 4-μm thickness and stained with hematoxylin and eosin (H&E) or periodic acid–Schiff (PAS). Glomerular crescent formation and sclerosis was evaluated in a blinded fashion on PAS-stained paraffin sections and scored using a semiquantitative scale (0–3), in which 0 represented no abnormalities, and 1, 2 and 3 represented mild, moderate and severe abnormalities, respectively. Ten glomeruli were assessed for each animal (n = 3 per group).
Electron microscopy
Following dissection, mouse kidneys were cut down to 23–33 mm pieces. For scanning electron microscopy (SEM), tissues were fixed in Trumps Fixative (Electron Microscopy Sciences, 11750), dehydrated with graded ethanol, dried using a 850 Critical Point Dryer (Electron Microscopy Sciences) and gold coated on a Cressington 108 Auto Sputter Coater (Ted Pella). For transmission electron microscopy (TEM), renal tissues were fixed and processed as described above then post fixed in 1% OsO4 for 1 h on ice. Tissues were washed, dehydrated and embedded in Epon812 (Electron Microscopy Sciences). Ultrathin kidney sections (70 nm) were obtained using an EM UC7 Ultramicrotome (Leica) and collected on Formvar coated Ni slot grids (Electron Microscopy Sciences, FF-2010-Ni) then stained in 5% uranyl acetate and 0.1% lead citrate. Digital micrographs were taken using a Sigma HDVP Electron Microscope (Zeiss).
Immunofluorescence
Frozen kidney tissues of the xenograft mice were cut at 4 μm thickness and fixed with ice-cold acetone for 5 min. After blocking with blocking solution (2.5% donkey normal serum and 2.5% FBS (FBS) in PBS) for 1 h, samples were stained with goat anti-mouse synaptopodin (Santa Cruz Biotechnology, sc-21537) diluted at 1:300 with blocking solution, followed by Alexa Fluor 546-conjugated donkey anti-goat IgG (Molecular Probes, A11057) diluted at 1:1,000 with blocking solution. After washing, the samples were further incubated with Alexa Fluor 488-conjugated goat anti-human IgG (Jackson ImmunoResearch, 109-545-006) diluted at 1:300 with blocking solution. After washing, stained samples were mounted with ProLong Gold antifade reagent with DAPI (Molecular Probes, P36935). Images were obtained and analyzed using a Carl Zeiss LSM 700 confocal microscope.
A mouse model of MHC mismatched GVHD
BALB/c recipients (female, aged 10–12 weeks) were lethally irradiated (9.5 Gy) and received 107 BM cells and 5 × 106 spleen cells from C57BL/6 donors (MHC mismatched allogeneic transplant) via retro-orbital injection. As a control (syngeneic transplant), irradiated BALB/c recipients received BM cells and spleen cells from BALB/c donors. Mice were monitored for body weight and mortality.
Statistical analysis
Data are shown as mean ± s.d. or ± s.e.m. Statistical analysis was assessed using GraphPad Prism 5.0 (⋆P < 0.05, ⋆⋆P < 0.01, ⋆⋆⋆P < 0.001). Significance of the difference between two groups was assessed using the Student’s unpaired two-tailed t test. For multiple group comparisons, we performed one-way ANOVA with Tukey’s multiple comparison test. No statistical method was used to predetermine sample size in animal studies. Sample sizes were estimated on the basis of sample availability and previous experimental studies3,4,22–27. No samples or animals were excluded from the analyses. Animals were allocated at random to treatment groups. Investigators were not blinded during animal experiments.
Supplementary Material
Acknowledgments
We thank A.S. Shaw (Washington University School of Medicine) for the Rac1 transgenic mice; S. Shankland (University of Washington) for providing the nephrotoxic serum (NTS); A. Finnegan (Rush University Medical Center) for reagents and advice; B. Samelko (née Tryniszewska) (Rush University Medical Center) for technical assistance; and S. Mangos (Rush University Medical Center) for help with manuscript revisions. This research was supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) R01DK101350 (J.R.), R01DK106051(J.R.), R01DK107984(V.G.) and R01DK084195(V.G.).
Footnotes
Accession codes
GenBank accession code for Mus musculus plasminogen activator, urokinase receptor (Plaur): NM_011113
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
E.H. designed and performed experiments, and wrote the paper; I.F., J.L., and Y.C. performed animal experiments; N.J.T. generated electron micrographs; A.Z. and J.L. contributed to xenotransplantation experiment; C.W., M.T., S.W., V.P., S.S.H., C.O., M.B., V.G., S.S., D.B.S., and D.T.S. contributed to experiments; J.R. designed and supervised the study, and wrote the paper.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
References
- 1.Fogo AB. Mechanisms of progression of chronic kidney disease. Pediatr Nephrol. 2007;22:2011–2022. doi: 10.1007/s00467-007-0524-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hayek SS, et al. Soluble urokinase receptor and chronic kidney disease. N Engl J Med. 2015;373:1916–1925. doi: 10.1056/NEJMoa1506362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wei C, et al. Modification of kidney barrier function by the urokinase receptor. Nat Med. 2008;14:55–63. doi: 10.1038/nm1696. [DOI] [PubMed] [Google Scholar]
- 4.Wei C, et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med. 2011;17:952–960. doi: 10.1038/nm.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cravedi P, Kopp JB, Remuzzi G. Recent progress in the pathophysiology and treatment of FSGS recurrence. Am J Transplant. 2013;13:266–274. doi: 10.1111/ajt.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winn MP, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 7.D’Agati VD, Kaskel FJ, Falk RJ. Focal segmental glomerulosclerosis. N Engl J Med. 2011;365:2398–2411. doi: 10.1056/NEJMra1106556. [DOI] [PubMed] [Google Scholar]
- 8.Kitiyakara C, Eggers P, Kopp JB. Twenty-one-year trend in ESRD due to focal segmental glomerulosclerosis in the United States. Am J Kidney Dis. 2004;44:815–825. [PubMed] [Google Scholar]
- 9.Fogo AB. Causes and pathogenesis of focal segmental glomerulosclerosis. Nat Rev Nephrol. 2015;11:76–87. doi: 10.1038/nrneph.2014.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallon L, Leventhal J, Skaro A, Kanwar Y, Alvarado A. Resolution of recurrent focal segmental glomerulosclerosis after retransplantation. N Engl J Med. 2012;366:1648–1649. doi: 10.1056/NEJMc1202500. [DOI] [PubMed] [Google Scholar]
- 11.McCarthy ET, Sharma M, Savin VJ. Circulating permeability factors in idiopathic nephrotic syndrome and focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2010;5:2115–2121. doi: 10.2215/CJN.03800609. [DOI] [PubMed] [Google Scholar]
- 12.Savin VJ, et al. Circulating factor associated with increased glomerular permeability to albumin in recurrent focal segmental glomerulosclerosis. N Engl J Med. 1996;334:878–883. doi: 10.1056/NEJM199604043341402. [DOI] [PubMed] [Google Scholar]
- 13.Thunø M, Macho B, Eugen-Olsen J. suPAR: the molecular crystal ball. Dis Markers. 2009;27:157–172. doi: 10.3233/DMA-2009-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol. 2002;3:932–943. doi: 10.1038/nrm977. [DOI] [PubMed] [Google Scholar]
- 15.Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11:23–36. doi: 10.1038/nrm2821. [DOI] [PubMed] [Google Scholar]
- 16.Faul C, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 2008;14:931–938. doi: 10.1038/nm.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reiser J, et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest. 2004;113:1390–1397. doi: 10.1172/JCI20402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bertelli R, et al. LPS nephropathy in mice is ameliorated by IL-2 independently of regulatory T cells activity. PLoS One. 2014;9:e111285. doi: 10.1371/journal.pone.0111285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shultz LD, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174:6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 20.Basu S, Hodgson G, Katz M, Dunn AR. Evaluation of role of G-CSF in the production, survival, and release of neutrophils from bone marrow into circulation. Blood. 2002;100:854–861. doi: 10.1182/blood.v100.3.854. [DOI] [PubMed] [Google Scholar]
- 21.Liu F, Wu HY, Wesselschmidt R, Kornaga T, Link DC. Impaired production and increased apoptosis of neutrophils in granulocyte colony-stimulating factor receptor-deficient mice. Immunity. 1996;5:491–501. doi: 10.1016/s1074-7613(00)80504-x. [DOI] [PubMed] [Google Scholar]
- 22.Kopp JB, et al. Transgenic mice with increased plasma levels of TGF-beta 1 develop progressive renal disease. Lab Invest. 1996;74:991–1003. [PubMed] [Google Scholar]
- 23.Schiffer M, et al. Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin Invest. 2001;108:807–816. doi: 10.1172/JCI12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kistler AD, et al. Transient receptor potential channel 6 (TRPC6) protects podocytes during complement-mediated glomerular disease. J Biol Chem. 2013;288:36598–36609. doi: 10.1074/jbc.M113.488122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hudkins KL, et al. BTBR Ob/Ob mutant mice model progressive diabetic nephropathy. J Am Soc Nephrol. 2010;21:1533–1542. doi: 10.1681/ASN.2009121290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu H, et al. Rac1 activation in podocytes induces rapid foot process effacement and proteinuria. Mol Cell Biol. 2013;33:4755–4764. doi: 10.1128/MCB.00730-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Papeta N, et al. Prkdc participates in mitochondrial genome maintenance and prevents Adriamycin-induced nephropathy in mice. J Clin Invest. 2010;120:4055–4064. doi: 10.1172/JCI43721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alfano M, et al. Full-length soluble urokinase plasminogen activator receptor down-modulates nephrin expression in podocytes. Sci Rep. 2015;5:13647. doi: 10.1038/srep13647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cathelin D, et al. Administration of recombinant soluble urokinase receptor per se is not sufficient to induce podocyte alterations and proteinuria in mice. J Am Soc Nephrol. 2014;25:1662–1668. doi: 10.1681/ASN.2013040425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spinale JM, et al. A reassessment of soluble urokinase-type plasminogen activator receptor in glomerular disease. Kidney Int. 2015;87:564–574. doi: 10.1038/ki.2014.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Delville M, et al. A circulating antibody panel for pretransplant prediction of FSGS recurrence after kidney transplantation. Sci Transl Med. 2014;6:256ra136. doi: 10.1126/scitranslmed.3008538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang J, et al. Urinary soluble urokinase receptor levels are elevated and pathogenic in patients with primary focal segmental glomerulosclerosis. BMC Med. 2014;12:81. doi: 10.1186/1741-7015-12-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holmes C, Stanford WL. Concise review: stem cell antigen-1: expression, function, and enigma. Stem Cells. 2007;25:1339–1347. doi: 10.1634/stemcells.2006-0644. [DOI] [PubMed] [Google Scholar]
- 34.Shi X, et al. Toll-like receptor 4/stem cell antigen 1 signaling promotes hematopoietic precursor cell commitment to granulocyte development during the granulopoietic response to Escherichia coli bacteremia. Infect Immun. 2013;81:2197–2205. doi: 10.1128/IAI.01280-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol. 2012;12:786–798. doi: 10.1038/nri3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brehm MA, Shultz LD, Greiner DL. Humanized mouse models to study human diseases. Curr Opin Endocrinol Diabetes Obes. 2010;17:120–125. doi: 10.1097/MED.0b013e328337282f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ito R, Takahashi T, Katano I, Ito M. Current advances in humanized mouse models. Cell Mol Immunol. 2012;9:208–214. doi: 10.1038/cmi.2012.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sellier-Leclerc AL, et al. A humanized mouse model of idiopathic nephrotic syndrome suggests a pathogenic role for immature cells. J Am Soc Nephrol. 2007;18:2732–2739. doi: 10.1681/ASN.2006121346. [DOI] [PubMed] [Google Scholar]
- 39.Niu H, et al. The function of hematopoietic stem cells is altered by both genetic and inflammatory factors in lupus mice. Blood. 2013;121:1986–1994. doi: 10.1182/blood-2012-05-433755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao JL, et al. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell. 2014;14:445–459. doi: 10.1016/j.stem.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.