

Abstract
The importance of the heterocyclic core elements with peripheral phenolic and alkyl substituents as a dominant structural motif of ligands for estrogen receptor (ER) has been well recognized. Here, we expand the structural diversity of core elements by preparing selenium-containing heterocycles and exploring the activities of these selenophenes on the two ERs, ERα and ERβ. Careful structure-activity relationship analysis of their ER binding affinities showed that most selenophenes are ERβ-selective, with the position of the phenol substituents on the selenophene-core and the nature of these substituents having in a marked effect on their binding affinities. The compound bis(2-fluoro-4-hydroxyphenyl)selenophene (2f) has the highest relative binding affinity (RBA) of 24.3 for ERβ. In transcription assays, most of selenophenes exhibit partial to full agonist activity for both ER subtypes, with compounds bis(2-methyl-4-hydroxyphenyl)selenophene (2b), bis(4-fluoro-3-hydroxyphenyl) 3-bromoselenophene (6f), 2,3,5-tris(hydroxyphenyl)-thiophenes (8b and 8d) profiling as superagonists for ERα, but several compounds display a range of ERα or ERβ antagonistic activities. A few selenophenes exhibited antiproliferative activity, with compound 8c showing antiproliferative effects comparable to that of 4OHT in breast cancer MCF-7 cells while being nontoxic to normal VERO cells. These new ligands could act as models for the development of novel agents leading to improved therapeutics that target the estrogen receptor.
Keywords: selenophenes, selective estrogen receptor modulators, diversity of structure, relative binding affinity (RBA), breast cancer
Add a New Element!
This is the first report of selenophene-core compounds as ER ligands. In transcription assays, several selenophenes profiling as superagonists for ERα; moreover, a few selenophenes exhibited antiproliferative activity comparable to that of 4OHT in breast cancer MCF-7 cells while being nontoxic to normal VERO cells. These new ligands could act as models for the development of novel agents leading to improved therapeutics that target the estrogen receptor.
Introduction
Selenium is known to play important roles in the development and maintenance of various physiological functions in humans, and recent research on this trace element support its protective effect against various types of cancer.[1–2] Recently, the biological effects of organoselenium compounds have attracted much attention,[3–5] as has their application as chiral catalysts.[6–8] In particular, many diarylselenides possess anticancer, antiviral, antimicrobial, as well as antioxidant properties.[9–11] In addition, a number of biologically active selenaheterocycles, such as cytoprotective agent ebselen, have recently been discovered.[12] Moreover, we and other found that a few selenophene compounds inhibited the motility and development of stages of the comparable nematode Haemonchus contortus[13]. More recently, fluselenamyl, a novel planar benzoselenazole derivative, was used as a PET imaging agent for amyloid in Alzheimer’s disease (Figure 1). [14]
Figure 1.
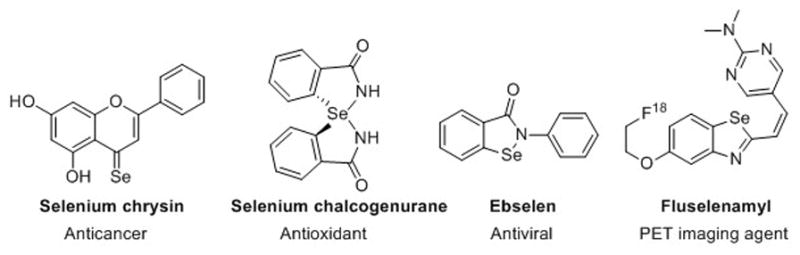
Representative examples of selenium-containing bioactive agents.
Estrogens play important roles in the development and maintenance of both reproductive and non-reproductive tissues in both women and men.[15] Estrogens are required and can provide health benefits in some tissues, such as the reproductive system, bone, vasculature, and brain, but they can also drive breast and endometrial cancers.[16–18] The broad spectrum of estrogen effects in diverse tissues provides an intriguing opportunity for the creation of tissue-selective estrogen receptor ligands. The multiple actions of estrogens are known to be mediated by two subtypes of the estrogen receptor (ER), ERα and ERβ.[19] Although structurally related, these two receptor subtypes have distinct tissue distributions; for example, ERα is mainly found in the female reproductive system, and ERβ is mainly expressed in prostate, colon, cardiovascular and central nervous systems.[20] Even when both ER subtypes are present in the same cells, estrogen action through these receptors leads to differential gene regulation, resulting in distinct physiological outcomes having non-overlapping and even opposing functions.[21–24] Efforts to obtain tissue-selective estrogens have focused on compounds, termed SERMs (for selective estrogen receptor modulators), that showed different levels of partial agonist/antagonist activity (i.e., intrinsic activity) in different target tissues. The selectivity of SERMs is believed to arise from their differential engagement of coregulator proteins in a target cell- and gene-specific manner,[25–27] and compounds such as tamoxifen, raloxifene, lasofoxifene, and bazedoxifene were found to be largely agonistic in bone and antagonistic in breast, but to have varying activity in the uterus. However, the above-mentioned ER-targeted therapies are limited by on-target side-effects, such as breast cancer risk with hormone replacement therapy,[28] or thromboembolic events, endometrial proliferation and hot flushes with the ERα ligand, such as tamoxifen.[29]
In the development of both SERMs and subtype-selective ligands, extensive investigation has been made of non-steroidal compounds having heterocyclic cores. Typically, the peripheral substituents, such as phenols, simple alkyl groups, and basic and polar phenyl substituents, have generally remained the same. By contrast, a wide variety of heterocyclic cores has been explored, often in the context of SERM development in which both five- and to a lesser extent six-membered ring heterocycles have been used.[30–34] Some examples of the 5-membered ring heterocycle series are presented below (Figure 2).
Figure 2.

Structure and binding affinity data of the most promising five-membered ring heterocycles and title selenophene compounds.
As part of our ongoing interest in ER ligands with different core structures and biological activity,[35–38] we recently reported on a series of novel ER ligands based on a thiophene core, and we found that most of the 2,5- and 2,4-bis(hydroxyphenyl)-thiophenes were ERβ selective, whereas the bulkier 2,3,5-tris(hydroxyphenyl)-thiophenes were ERα selective.[34] Analogous furan-core compounds, prepared for comparison, generally had lower affinity and/or less selectivity than their congeneric thiophenes. Intriguingly, some of the 2,5- and 2,4-diarylthiophenes showed distinct superagonist activity in reporter gene assays, giving maximal activities 2–3 times that of estradiol; superagonist activity was ascribed to the somewhat larger overall length of these thiophene core molecules compared to heterocycles having only second row elements.[34]
ER ligands having heterocyclic cores have been built from second row elements (O and N) and the third row element (S); thus far, however, there are no reports of using the fourth row element (Se). The well recognized importance of combining heterocyclic core elements with phenolic and alkyl peripheral substituents in the design of estrogen receptor (ER) ligands led us to prepare ligands having selenophene cores. As relatively unexplored structural elements of drugs, these selenium-containing heterocycles expand the structure diversity of heterocycle core elements, and we studied their binding affinity and cellular potencies and efficacies for ERα and ERβ, finding that some of them act as superagonists. We also investigated their anti-proliferative activities in cancer and normal cell lines. Our results, together with molecular modeling, offer a framework for understanding the structural basis for superagonist activity, and reveal what might be a general strategy for obtaining superagonists for other members of the nuclear receptor superfamily.
Results and Discussion
Chemical Synthesis
Four series of novel selenophene-core compounds, shown in Schemes 1–2, were prepared from selenophene according to the synthetic procedures established in our laboratory for producing thiophene-core ER ligands. In the synthesis of compounds 2a–j (Scheme 1A), key intermediates 1a–j were obtained by the Suzuki cross-coupling of aryl boronic acid with 2,5-dibromoselenophene 9 in the presence of sodium carbonate and 1,1′-bis(diphenylphosphino)ferrocene (dppf)-coordinated palladium chloride as a catalyst at reflux.[39] The key intermediate selenophene precursor, 9, was prepared by bromination of the selenophene using N-bromosuccinimide (NBS). Initially, Suzuki cross-coupling was investigated using Pd(OAc)2/PPh3 as the catalyst; however, the yield was below 5%. Further studies with other palladium catalysts, including Pd2(dba)2, Pd(CN)2Cl2, Pd(PPh3)Cl2, gave no improvement, an exception being Pd(dppf)Cl2, which furnished modest to good yields. Finally, treatment of the compounds 1a–j with boron tribromide gave the final 2,5-disubstituted diphenolic selenophenes 2a–j in good yields.[40]
Scheme 1.

Synthesis of 2,5-substituted selenophene compounds. Reagents and conditions: (a) NBS, DMF; (b) [Pd] catalyst (Pd(dppf)Cl2 for Series I; Pd(OAc)2/PPh3 for Series II), Na2CO3, toluene/water (1:1), reflux, 24 h; (c) BBr3, CH2Cl2, −20 °C to rt, 4 h; (d) Br2, AcOH, rt, 18h; (e) Zn, AcOH/water (1:1), 37 °C, 12 h. To simplify comparisons between compounds in closely related series, we designate locant positions of the substituents on the phenyl groups with respect to the selenophene core; for clarity, locant positions on the selenophene core itself are given by numbers in italics.
Scheme 2.

Synthesis of trisubstituted selenophene compounds. Reagents and conditions: (a) Br2, DMF; (b) [Pd] catalyst (Pd(dppf)Cl2 for Series III; Pd(PPh3)4) for Series IV, Na2CO3, toluene/water (1:1), reflux, 24 h; (c) BBr3, CH2Cl2, −20 °C to rt, 4 h. To simplify comparisons between compounds in closely related series, we designate locant positions of the substituents on the phenyl groups with respect to the selenophene core; for clarity, locant positions on the selenophene core itself are given by numbers in italics.
The key intermediates, 3a–d, for the synthesis of 3,4-disubstituted selenophene derivatives 4a–d (Scheme 1B) were prepared by treating 3,4-dibromoselenophene 11 with aryl boronic acid by using Pd(OAc)2/PPh3 instead of Pd(dppf)Cl2 as the catalyst. The 3,4-dibromoselenophene intermediate (11) cannot be obtained by bromination of selenophene, but it was synthesized by debromination of 2,3,4,5-tetrabromoselenophene (10) using Zn and AcOH in water.[41] 2,3,4,5-Tetrabromoselenophene (10) was synthesized by bromination of selenophene using CHCl3 and AcOH as solvent, with dropwise addition of Br2.[42] The amount of bromine used and the rate of addition proved to be important for the outcome of this reaction: With large excess of bromine (>4 equiv.), 2,3,4,5-tetrabromoselenophene (10) was the major product, whereas with less than 2 equivalents of bromine, 2,5-dibromoselenophene (9) could also be obtained in good yield. Subsequent cleavage of the methoxyl groups of 3a–d by BBr3 yielded the desired products 4a–h in 51–68% yields.
With the aim of further investigating the effect of a third substituent on the selenophene core, we prepared a series of trisubstituted selenophene compounds 6a–h (Scheme 2A). First, 1 equiv of 2,3,5-tribromoselenophene was treated with 4 equiv of aryl boronic acid using Pd(dppf)Cl2 to afford 5a–h, followed by ether cleavage to give compounds 6a–h directly.
During our prior work on furan- and thiophene-core ER ligands, we found that triaryl, especially triphenolic compounds, showed higher binding affinity and subtype selectivity than the corresponding bisphenolic compounds.[34] Thus, we also attempted to introduce a third phenol ring into the selenophene core to obtain triaryl-substituted selenophene analogues 8a–8d (Scheme 2B). Reaction of 1 equiv of 2,3,5-tribromoselenophene with 5 equiv of aryl boronic acid using standard Suzuki coupling conditions afforded the key esters 7a–d, which were subsequently reacted with BBr3 at −20 °C to give the hydroxyl derivatives 8a–8d. The starting material 2,3,5-tribromoselenophene (12) was synthesized by bromination of selenophene using Br2 in DMF.
The structures of compounds 1j and 7a were unambiguously confirmed by X-ray crystallography.
Relative Binding Affinities
The binding affinities of selenophene compounds for both ERα and ERβ were determined by a competitive fluorometric receptor-binding assay and are summarized in Table 1. These affinities are presented as relative binding affinity (RBA) values, where estradiol (E2) has an affinity of 100%.
Table 1.
Relative Binding Affinity (RBA) of series I (2a–j), series II (4a–e), series III (6a–g) and series IV (8a–d) for ERα and ERβα.
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Entry | compound | Core | R | ERb | ERb | β/α |
| 1 | 2a |
|
H | 0.61 ± 0.03 | 2.87 ± 0.20 | 4.70 |
| 2 | 2b | 2-Me | 5.60 ± 0.43 | 11.1 ± 0.73 | 1.98 | |
| 3 | 2c | 3-Me | <0.01 | 0.94 ± 0.05 | >94 | |
| 4 | 2d | 3,5-diMe | <0.01 | 1.21 ± 0.02 | >121 | |
| 5 | 2e | 2,6-diMe | 4.09 ± 0.05 | 4.12 ± 0.15 | 1.00 | |
| 6 | 2f | 2-F | 5.90 ± 0.90 | 24.3 ± 0.52 | 4.11 | |
| 7 | 2g | 2-Cl | 6.11 ± 0.05 | 12.7 ± 3.66 | 2.07 | |
| 8 | 2h | 3-Cl | 0.29 ± 0.02 | 1.52 ± 0.19 | 5.10 | |
| 9 | 2i | 2-F | 2.02 ± 0.19 | 1.83 ± 0.30 | 0.90 | |
| 10 | 2j | 4-F | 0.92 ± 0.07 | 3.10 ± 0.73 | 3.37 | |
|
| ||||||
| 11 | 4a |
|
H | 0.32 ± 0.04 | 2.01 ± 0.11 | 6.28 |
| 12 | 4b | 2-Me | 0.27 ± 0.06 | 6.70 ± 0.49 | 24.81 | |
| 13 | 4c | 3-Me | 0.71 ± 0.09 | 0.62 ± 0.12 | 0.87 | |
| 14 | 4d | 3-F | 0.58 ± 0.14 | 1.56 ± 0.23 | 2.67 | |
| 15 | 4e | 2-F | 0.89 ±0.32 | 1.95 ± 0.58 | 2.19 | |
|
| ||||||
| 16 | 6a |
|
H | 5.04 ± 0.40 | 11.7 ± 0.01 | 2.32 |
| 17 | 6b | 2-Me | 14.6 ± 2.84 | 10.8 ± 0.77 | 0.74 | |
| 18 | 6c | 3-Me | 1.39 ± 0.71 | 6.12 ± 2.40 | 4.40 | |
| 19 | 6d | 3,5-diMe | 2.17 ± 0.71 | 0.93 ± 0.07 | 0.43 | |
| 20 | 6e | 3-Cl | 2.98 ± 0.24 | 0.74 ± 0.20 | 0.25 | |
| 21 | 6f | 4-F | 1.31 ± 0.05 | 0.37 ± 0.03 | 0.28 | |
| 22 | 6g | 2-F | 3.35 ± 0.83 | 4.36 ± 0.49 | 1.30 | |
|
| ||||||
| 23 | 8a |
|
H | 6.38 ± 0.23 | 7.39 ± 1.30 | 1.15 |
| 24 | 8b | 3-Me | 1.25 ± 0.26 | 12.4 ± 1.47 | 9.92 | |
| 25 | 8c | 3,5-diMe | 4.91 ± 0.34 | 2.73 ± 0.11 | 0.55 | |
| 26 | 8d | 3-Cl | 1.62 ± 0.31 | 1.71 ± 0.24 | 1.06 | |
| 27 | 4OHT | 13.8 ± 0.2 | 12.6 ± 1.7 | 0.91 | ||
To simplify comparisons between compounds in closely related series, we designate locant positions of the substituents on the phenyl groups with respect to the selenophene core; locant positions on the selenophene core itself are given by numbers in italics.
Relative Binding Affinity (RBA) values are determined by competitive fluorometric binding assays and are expressed as IC50estradiol/IC50compound × 100 ± the range (RBA, estradiol = 100%). In these assays, the Kd value of estradiol is 3.1 nM for ERα and 3.4 nM for ERβ, respectively. For details, see Experimental Section.
As an overall observation, it is notable that a large number of the selenophene-core derivatives show moderate levels of ERβ selectivity, which is agreement with our previous studies with furan- and thiophene-core compounds.[34] In addition, compared to furan- and thiophene-core compounds, these selenophene-core derivatives generally demonstrate better binding affinity for ERβ. Second, the addition of substituents in the phenol rings, as well as the position of the hydroxyl group, have significant effects on the binding affinity and selectivity of the ligands. Compared to 4OHT (RBA values were 13.8 for ERα[43] and 12.6 for ERβ; β/α was 0.91), compounds 2f, 2g, 8b had high binding affinity for ERβ and higher ERβ selectivity. Compound 6b also retained high binding affinity for ERα. Among the 2,5-bisphenol selenophene (series I), those substituted at C2 (meta to the phenol; 2b, 2e, 2f and 2g) demonstrate a better binding affinity than the corresponding non-substituted compound 2a, with RBA values ranging from 4 to 24. In fact, bis(2-fluoro-4-hydroxyphenyl)selenophene (2f) has the highest binding affinity for ERβ among all of the analogues, binding 8-fold better to ERβ than does 2a; this increased affinity, however, comes at a cost in ERβ subtype selectivity (Table 1, entries 2, 5–7 vs 1). Interestingly, when the Me or Cl substituents are present in the C3 position, next to the phenolic hydroxyl group, as in compounds 2c, 2d and 2h, binding affinities for both ER subtypes were dramatically decreased (Table 1, entries 2 vs 3, 4; 7 vs 8). Some interesting things were also observed with hydroxyl analogues 2i and 2j. The marked effect of the position of the hydroxyl groups on ER binding affinities is evident from entries 6 vs 9 and 10 (Table 1): when this group was moved from the C4 to the C3-position of the phenyl ring, binding affinities were also diminished.
We have previously observed that 3,4-disubstituted diphenolic furan and thiophenes showed lower binding affinities than corresponding 2,5-disubstituted compounds.[33] Similar trends were also observed for selenophene-core compounds (series II), where affinities for ERα were always less than 0.8%. Binding to ERβ was mostly higher than to ERα, with again the C2-substituted analog (4b) being the best.
To further assess the role of an additional substituent on the selenophene core, we prepared the trisubstituted selenophene analogues of 6a–g (series III, bromine substituted) and 8a–d (series IV, arene substituted). The trisubstituted compounds showed an obvious increase in binding affinity for ERα, with RBA values ranging from 1 to 14, but by contrast, ERβ affinity underwent no significant change. These trends are also similar to those we reported earlier for furan- and thiophene-core ligands.[34] Bromine substitution at the 3-position of the selenophene core (6a) resulted in an 8-fold increase for ERα over 2a, and, as in series I and II, substituents at the C2-position of phenol ring (6b, 6g) increased ERα affinity, whereas those at C3 (compounds 6c and 6e) or 3,5-disubstituted compound 6d did the opposite, but less so than in the series I compounds. In fact, the C2 methyl analog, 6b, had RBA values of 14.6 and 10.8 for ERα and ERβ; it was the highest affinity ligand for ERα, binding more than 23 times better than that of the parent compound 2a. The low binding affinity of compound 6f, which had hydroxyl groups at C3-position instead of C4-position, had lower ER binding affinities, as was the case in series I.
Compared to the compounds of series III, introduction of a third phenol moiety on the selenophene core (series IV, compounds 8a–d) did not cause a significant increase in binding affinity for both ERα and ERβ. Nevertheless, compounds 8a–d, with three phenolic groups on selenophene core, showed higher binding affinity for both ERs than did the corresponding bisphenolic group derivatives 2 (Figure 3). Overall then, the positions of the substituent and hydroxyl group on the phenols are of great importance for binding, with optimal affinity coming with appropriate substitution at the C2-position on the phenols and a hydroxyl group at the C4-position. In earlier work, we suggested that the beneficial effect of small substituents at the C-2 position of phenolic substituents on ligand binding was likely due to a twisting of the phenol with respect to the ligand core, a motion that increased the surface area of the ligand without greatly increasing its steric size, thereby improving productive contact between the ligand and residues in the ligand binding pocket.[44]
Figure 3.
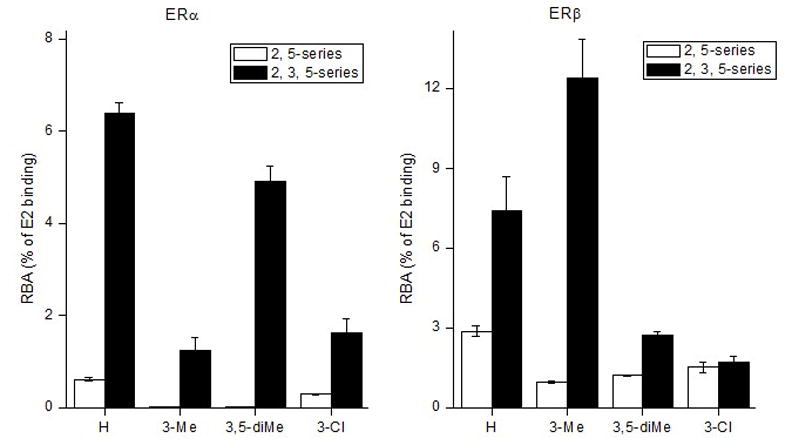
Graphical display of the RBA values for 2,5-disubstituted diphenolic selenophenes (2a, 2c–d, 2h) and corresponding 2,3,5-triphenolic derivatives (8a–d).
Comparisons of congeneric selenophene, thiophene and furan-core ER ligands in ERα and ERβ binding affinities have been made in Table 2, which revealed the structure–affinity relationships of the disubstituted selenophene, thiophenes and furans. As a global comparison, the importance of the selenophene core on enhancing affinity is evident. In nearly all cases, selenophenes show better binding affinity compared to their corresponding thiophene and furan analogues, except compound 2f, which still gives the high binding affinity for ERα (5.9) and for ERβ (24.3). In addition, the importance of C2 substitution for enhancing affinity is evident (entries 2 vs 3, and 7 vs 8). This has been noted in our prior work on ER ligands,[34, 36–38] and in a recent report by Gust and colleagues on pyrroles.[33] Besides, the binding affinity of analogues containing halogens (F and Cl) is better than the unsubstituted and Me analogues (entries 2 vs 5, 3 vs 6). Other trends showed when changing a substituent from C3- into C2-position, the binding affinity will be better.
Table 2.
Comparisons of congeneric selenophene, thiophene and furan-core ER ligands in ERα and ERβ binding affinitiesa.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Selenophenes
|
Thiophenes
|
Furans
|
||||||
| Entry | R | compd | RBA
|
RBA
|
RBA
|
|||
| ERa | ERb | ERa | ERb | ERa | ERb | |||
|
|
|
|
||||||
| 1 | H | 2a | 0.61 ± 0.034 | 2.87 ±0.20 | 0.04 ± 0.004 | 0.79 ± 0.11 | 0.08 ± 0.007 | 0.34 ± 0.10 |
| 2 | 2-Me | 2b | 5.60 ± 0.43 | 11.1 ± 0.73 | 1.43 ± 0.19 | 1.74 ± 0.26 | 0.78 ± 0.12 | 0.24 ± 0.02 |
| 3 | 3-Me | 2c | <0.01 | 0.94 ± 0.05 | <0.01 | <0.01 | -- -- | -- -- |
| 4 | 2-F | 2f | 5.90 ± 0.90 | 24.3 ± 0.52 | 2.03 ± 0.19 | 33.1 ± 5.8 | 6.29 ± 0.73 | 32.2 ± 1.0 |
| 5 | 2-CI | 2g | 6.11 ± 0.049 | 12.7 ± 3.66 | 6.7 ± 1.3 | 10.0 ± 2.4 | 2.73 ± 0.41 | 6.9 ± 1.7 |
| 6 | 3-CI | 2h | 0.29 ± 0.02 | 1.52 ± 0.19 | 0.009 ± 0.001 | 0.036 | -- -- | -- -- |
|
|
|
|||||||
| 7 | 5-F | 2i | 2.02 ± 0.19 | 1.83 ± 0.30 | <0.01 | 0.066 ± 0.006 | -- -- | -- -- |
| 8 | 4-F | 2j | 0.92 ± 0.071 | 3.10 ± 0.73 | <0.01 | 0.010 ± 0.002 | -- -- | -- -- |
|
|
|
|
||||||
| 9 | H | 4a | 0.32 ± 0.04 | 2.01 ± 0.11 | 0.57 ± 0.12 | 1.69 ± 0.45 | 0.22 ± 0.06 | 0.69 ± 0.07 |
| 10 | 2-Me | 4b | 0.27 ± 0.06 | 6.70 ± 0.49 | 2.16 ± 0.54 | 4.9 ± 1.3 | 0.79 ± 0.21 | 2.28 ± 0.34 |
| 11 | 3-Me | 4c | 0.71 ± 0.09 | 0.62 ± 0.12 | 0.830 | 0.297 | -- -- | -- -- |
|
|
|
|
||||||
In each table, the RBA values are given for binding to ERα and ERβ for the various biaryl selenophene, thiophenes and furans we have studied. The disposition of the aryl groups with respect to the heteroatom (Se, S or O) is indicated by the numbers in parentheses.
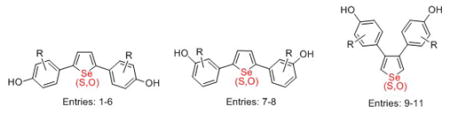
The effects of the heterocycle geometry changes with the nature of the heteroatom on the binding affinity of the ligands were also investigated. Comparisons of bond lengths, atomic distances and bond angles among the 2,5-disubstituted selenophene, thiophenes and furans can be viewed more easily in Figure 4. While it is a speculation, the higher binding affinity might be the consequence of the increased HO–OH distance between the two phenols in the selenophenes (13.5 Å) compared to other ligands, which is 12.9 Å in the thiophenes and 12.0 Å in the furans. The O–Se distance in the selenophenes is also considerably longer than in the alternate-position in the thiophene and furan bisphenols, due to the greater length of the C–Se bonds (1.9 Å) vs the C-S (1.47 Å) and C–O bonds (1.2 Å) in the heterocyclic core (This bond length difference also affects to a lesser degree, the display angle of the two phenols on the heterocyclic core). Thus, the alternate-position bisphenol selenophenes might be too long to form adequate hydrogen bonds with His-524. When the hydroxyl group was moved from the C4 to the C3-position of the phenyl ring, the atomic distance of HO–OH is 11.7 Å, 11.2 Å and 10.5 Å in selenophene, thiophene and furan, respectively (data not shown), and the binding affinities of the corresponding ligands were diminished (Figure 4), presumably because the atomic HO–OH distances of HO–OH are too short.
Figure 4.
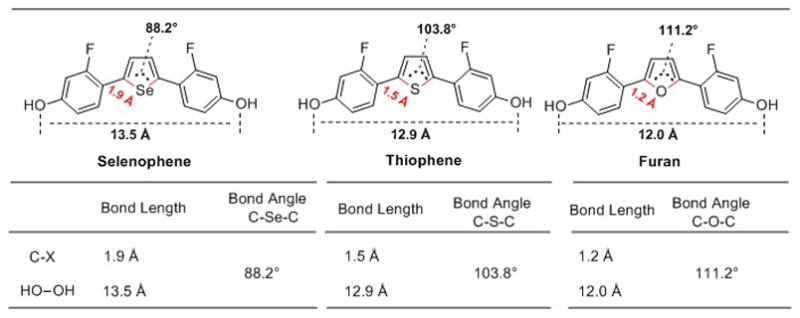
Comparisons of congeneric selenophene, thiophene and furan-core ER ligands in bond length, atomic distances and bond angle. The bond length between the carbon and heteroatom in selenophene, thiophene and furan, and atomic distances between the two oxygen atoms in hydroxyl groups. The X is representative of heteroatom (Se, S or O).
Transcription Activation Assays
Various selenophene-core compounds, generally selected for their binding affinities or for structural comparisons, were tested for their ability to stimulate the transcriptional activities of ERα and ERβ in ER-responsive luciferase reporter gene assays. These assays were conducted in HEK293T cells transfected with an expression vector for either ERα or ERβ, together with a widely used 3 × ERE-luciferase reporter. With each compound, dose-response assays were run in both an agonist mode (compound only) and an antagonist mode (in the presence of 10 nM E2). The results from both assays, summarized in Table 3, are expressed as EC50 (agonist mode) or IC50 (antagonist mode) values, together with efficacies values (Eff (% E2)). The latter represents the ultimate activity value at the highest concentration, compared to that for E2; high Eff values indicate agonist activity, low values indicate antagonist character, and intermediate Eff values indicate partial agonist/antagonist activity. (Note, it is difficult to establish EC50 values when the efficacy is low in agonist mode, and IC50 values when efficacy is high in antagonist mode. Hence, some values are missing) Dose-response curves for a few compounds are shown as examples in Figure 5.
Table 3.
Effects of selenophene-core compounds on the transcriptional activities of Estrogen Receptora and antiproliferative activity in MCF-7 cells.
| series | entry | cmpd | Agonist Modeb
|
Antagonist Modec, d
|
MCF-7Inhibition(%)e | VEROIC50 (μM)f | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ERα
|
ERβ
|
ERα
|
ERβ
|
|||||||||
| EC50 (μM) | Eff (%E2) | EC50 (μM) | Eff (%E2) | IC50 (μM) | Eff (%E2) | IC50 (μM) | Eff (%E2) | |||||
| I | 1 | 2a | - | 16 ± 2 | - | 11 ± 4 | 0.52 | 44 ± 10 | 0.028 | 35 ± 4 | 0 | 16.6 ± 3.89 |
| 2 | 2b | 1.36 | 157 ± 12 | 0.079 | 82 ± 7 | - | 96 ± 13 | 0.3 | 50 ± 11 | 3.4 | >50g | |
| 3 | 2c | - | 33 ± 6 | - | 24 ± 3 | - | 103 ± 28 | - | 98 ± 14 | 9.3 | >50 | |
| 4 | 2d | - | 12 ± 4 | - | 38 ± 6 | - | 148 ± 2 | - | 86 ± 13 | 0 | >50 | |
| 5 | 2e | - | −7 ± 3 | 0.434 | 60 ± 4 | 0.425 | −22 ± 8 | 5.391 | 66 ± 3 | 19.3 | >50 | |
| 6 | 2f | 0.23 | 68 ± 2 | 0.17 | 71 ± 1 | - | 59 ± 10 | - | 91 ± 6 | 10.5 | >50 | |
| 7 | 2g | 1.28 | 44 ± 13 | - | 14 ± 5 | - | 73 ± 8 | - | 74 ± 5 | 19.6 | >50 | |
| 8 | 2h | 0.04 | 49 ± 3 | - | 22 ± 8 | - | 100 ± 14 | −0.25 | 51 ± 16 | 5.3 | >50 | |
| 9 | 2i | - | 25 ± 8 | 0.13 | 55 ± 5 | - | 75 ± 7 | - | 89 ± 6 | 17.9 | >50 | |
| 10 | 2j | - | 25 ± 8 | 0.15 | 42 ± 10 | 0.72 | 68 ± 2 | - | 79 ± 3 | 4.4 | >50 | |
|
| ||||||||||||
| II | 11 | 4a | - | 7 ± 3 | - | 16 ± 7 | - | 57 ± 2 | 0.475 | 49 ± 9 | 0 | >50 |
| 12 | 4b | - | 19 ± 4 | 0.034 | 62 ± 8 | - | 80 ± 10 | - | 94 ± 10 | 0 | >50 | |
| 13 | 4c | 0.32 | 32 ± 8 | 1.147 | 54 ± 10 | - | 46 ± 9 | 0.341 | 50 ± 5 | 0 | >50 | |
| 14 | 4d | - | 21 ± 6 | - | 21 ± 3 | - | 33 ± 6 | 0.001 | 10 ± 7 | 0 | >50 | |
| 15 | 4e | - | 35 ± 5 | - | 17 ± 2 | - | 67 ± 3 | - | 58 ± 6 | 0 | >50 | |
|
| ||||||||||||
| III | 16 | 6a | 0.015 | 69 ± 11 | 0.013 | 40 ± 5 | - | 161 ± 22 | 2.53 | 75 ± 4 | 0 | >50 |
| 17 | 6b | 0.79 | 21 ± 5 | 0.009 | 69 ± 9 | - | 93 ± 11 | 2.96 | 25 ± 8 | 2.5 | >50 | |
| 18 | 6c | - | −3 ± 1 | 0.441 | 46 ± 1 | 1.33 | 8 ± 1 | 0. 51 | 21 ± 5 | 26.1 | >50 | |
| 19 | 6d | - | 16 ± 4 | - | 15 ± 6 | 1.58 | 26 ± 2 | - | 18 ± 13 | 26.5 | >50 | |
| 20 | 6e | - | −23 ± 3 | - | 27 ± 2 | 0.561 | 19 ± 2 | - | 125 ± 20 | 11.3 | >50 | |
| 21 | 6f | 0.12 | 226 ± 28 | 0.3 | 31 ± 7 | - | 127 ± 42 | - | 69 ± 16 | 0 | >50 | |
| 22 | 6g | - | 73 ± 9 | - | 50 ± 3 | - | 87 ± 6 | - | 59 ± 8 | 8.1 | >50 | |
|
| ||||||||||||
| IV | 23 | 8a | - | −4 ± 3 | - | 12 ± 3 | 10.4 | −7 ± 2 | 0.111 | 23 ± 3 | 29.6 | >50 |
| 24 | 8b | 0.32 | 112 ± 29 | 0.033 | 77 ± 8 | - | 104 ± 29 | - | 82 ± 2 | 16.6 | >50 | |
| 25 | 8c | 0.27 | 74 ± 4 | 0.017 | 69 ± 5 | 0.87 | 27 ± 1 | - | 79 ± 7 | 75.3 | >50 | |
| 26 | 8d | 1.8 | 148 ± 2 | 0.061 | 73 ± 14 | - | 74 ± 19 | - | 91 ± 8 | 18.6 | >50 | |
| 27 | 4OHT | 0.008 | 35 ± 3 | - | −1 ± 1 | 0.003 | 35 ± 3 | 0.001 | −20 ± 2 | 81.7 | 15.1 ± 5.2 | |
Luciferase activity was measured in HEK293T cells transfected with 3 × ERE-driven luciferase reporter and expression vectors encoding ERα or ERβ and treated in triplicate.
In the agonist mode, increasing concentrations of compounds alone (up to 10−5 M) are given. EC50 values and standard deviations (mean ± SD from three replicates) represent the concentrations producing 50% of the maximal response to the compound on ERα or ERβ. Eff (% E2) is the maximal response to the compound as a percentage of the response to 10−8 M 17β-estradiol (E2).
In the antagonist mode, increasing concentrations of compounds (up to 10−5 M) are given together with a maximally stimulating dose of E2 (10−8 M). IC50 values and standard deviations (mean ± SD from three replicates) represent the concentrations that reduce the E2 response by 50%. Eff (% E2) is the response to the compound at the highest concentration as a percentage of the activity of 10−8 M 17β-estradiol (E2).
ERs have considerable basal activity in HEK293T cells; compounds with inverse agonist activity are given negative efficacy values. Omitted EC50 or IC50 values were too high to be determined accurately; omitted Eff (%E2) values were too low to be accurately determined.
Relative inhibition rate of antiproliferative potencies determined by MTT assays. The final concentration of the compounds is 10 μM.
IC50 values are an average of at least three independent experiments ± standard deviation (mean ± SD).
IC50 not determinable up to highest concentrations tested.
Figure 5.
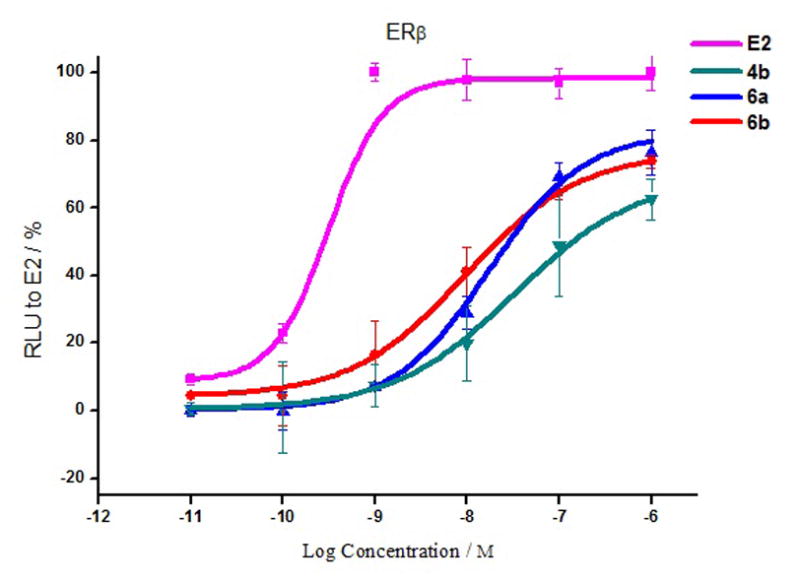
Illustrative dose-response curves for the ERβ agonist effects of E2, and three selenophene-core compounds 4b, 6a and 6b. Efficacy values (given in Table 3) are the mean ± SD from three experiments. For details, see the Experimental Section.
The interesting activities were seen in series I compounds, the 2,5-disubstituted diphenolic selenophenes. These ligands displayed a wide range of activities at both ERα and ERβ. Compound 2a was a partial antagonist of ERα and ERβ, with nanomolar range IC50 values. Addition of a methyl group giving 2b, however, shifted activity substantially from an antagonist to agonist (2b); in fact, 2b profiled as a superagonist on ERα and an agonist ERβ, showing efficacy on ERα 1.5 times greater than that of estradiol (Table 3, entry 2). It is of note that we also encountered superagonist activity in our earlier work on thiophene-core ligands.[34] When the methyl group was shifted from the C2- to C3-position (2c) or a second methyl group was added (2d), the compounds are inactive on ER, as 2c and 2d have low binding affinities to ERs.
Moreover, replacing the methyl group (2a) with a halogen group also had obvious effects on the transcriptional activity of the ER subtypes. The fluorine-substituted compound 2f profiled as an ERα agonist, being about 6-fold more potent than 2b, but with reduced efficacy. Also the chloro analogue 2g showed a similar effect on ERα compared to 2b. However, when the chloro was changed from the C2- to C3-position, compound 2h was still an ERα partial agonist with higher potency compared to 2b despite its lower affinity (Table 1, entries 8 vs 2). Surprisingly, when the hydroxyl group was moved from the C4- to the C3-position of the phenyl ring, compounds 2i, 2j acted as a partial agonist at ERβ but showed less activity on ERα, with the position of the substituent having only a minor effect on transcriptional activity (Table 3, entries 9 and 10).
Comparisons of the position of phenolic groups in the selenophene core indicated that the 3,4-disubstituted compounds (series II) had decreased ERα binding affinity (Table 1, 4a–d) and also showed decreased efficacy as ERα agonists (Table 3, entries 11 vs 1; 12 vs 2). Interestingly, 4b had improved ERβ binding affinity, as well as potency as an ERβ agonist, whereas 4a, and 4c–d profiled as an ERβ antagonist with IC50 values in the nanomolar (nM) range.
Addition of a third substituent onto the selenophene core also had major effects on transcriptional activity. Introduction of the bromine atom to 2a, converted it from antagonist into an ERα full agonist/ERβ partial agonist (compound 6a, Table 3, entries 1 vs 16). In fact, 6a displayed low nanomolar potency for activating both ERs. However, when selenophenes had substituents on the phenolic group, the trend of transcriptional activity is still similar to compounds 2 (Table 3, entries 17 vs 2). For example, 2-methyl derivative 6b was also a substantial ERβ antagonist activity, and analogues 6f also profiled as a very effective ERα agonist, and analogues 6c–e also profiled as ERα antagonists. Of note is that compound 6a showed about a 10-fold improved binding affinity for ERα and increased efficacy, compared to 2a and 4a. By contrast, the C3-hydroxyl analogue 6f showed improved ERα binding affinity (Tables 1, entries 21 vs 10), as well as efficacy; actually, 6f profiled as superagonist on ERα, showing efficacy in ERα 2.2 times greater than that of estradiol (Table 3, entry 21).
The most interesting activities were seen with the triaryl-substituted selenophenes. When the bromo substituent was replaced by a phenol moiety, the transcriptional activity was almost increased. Thus, 8a profiled as an antagonist on the ERs, showing efficacy in ERα about 2 times greater than that of 2,5-diphenolic analogue 2a (Table 3, entries 23 vs 1); 8b–d profiled as agonists on both ERs, with increased efficacy compared to corresponding the 2,5-diphenolic derivatives, with only 8c also showing some antagonistic activity in ERα. 8b and 8d having substituents (Me or Cl) at C3-position of phenolic group, were more efficacious than estradiol, being ERα superagonists.
Antiproliferative Activity on Breast Cancer and Normal Cells
To evaluate the antiproliferative activity of these compounds, most of the selenophene-core compounds were screened against MCF-7 breast cancer cells, in which 4OH-tamoxifen acts as an reference compound and also in normal (non-cancerous and ER-negative) kidney epithelial cells (VERO[45]); the results are summarized in Table 3.
In general, all the compounds in Table 3 that showed no obvious antagonistic activities on ERα or ERβ on transcriptional activities, exhibited weak or no inhibition of MCF-7 cells. Compounds 2b and 2j, which displayed certain antagonist activity on ERα, were essentially inactive, presumably due to their poor binding affinities for ERα, with RBA values were less than 1%. Trisubstituted selenophenes 6a–b, which possess high binding affinities, however, show weak inhibition for MCF-7 cells. This suggests that binding affinity and antiproliferative potency are independent. Interestingly, modifications of position of substituent (compound 6c) or addition of a second methyl group (compound 6d) led to compounds having antiproliferative activity. Furthermore, introduction of the third phenolic group (compounds 8a–d) resulted in compounds having even more promising antiproliferative activity. However, the most potent compound in suppressing the proliferation of MCF-7 cell was 8c with an inhibitory ratio of 75.3%, which was close to that of 4OH-tamoxifen, probably due to its excellent antagonistic potency on ERα.
Healthy VERO cells, commonly used for toxicity studies on normal cells, were used for toxicity studies[46–48]; epithelial cells from normal (non-cancerous) breast are very difficult to culture. Notably, most of selenophene-core compounds, except 2a, were nontoxic to healthy VERO cells, while 4OHT showed considerable toxicity, consistent with the antiproliferative effect of compound 8c being through ERα.
Models for Selenophenes Binding to the Estrogen Receptor α Ligand Binding Pocket and a Possible Model for Superagonism
E2 fits into the ERα ligand-binding pocket in an orientation in which the A-ring phenolic hydroxyl has hydrogen bonding interactions with helix 3 residue Glu353 and helix 6 residue Arg394, and more variably, through hydrogen bonding of the D-ring hydroxyl group with helix 11 residue His524 (Figure 6A). This ligand-binding orientation allows helix 12 to dock across helices 3 and 11, where it forms one side of a hydrophobic groove for the transcriptional coactivator binding site on the surface that constitutes the functional core of AF2. In contrast, SERMs such as 4-hydroxytamoxifen, directly or indirectly obstruct the agonist position of helix 12, relocating it out of this position and thereby blocking the recruitment of transcriptional coactivators.
Figure 6.
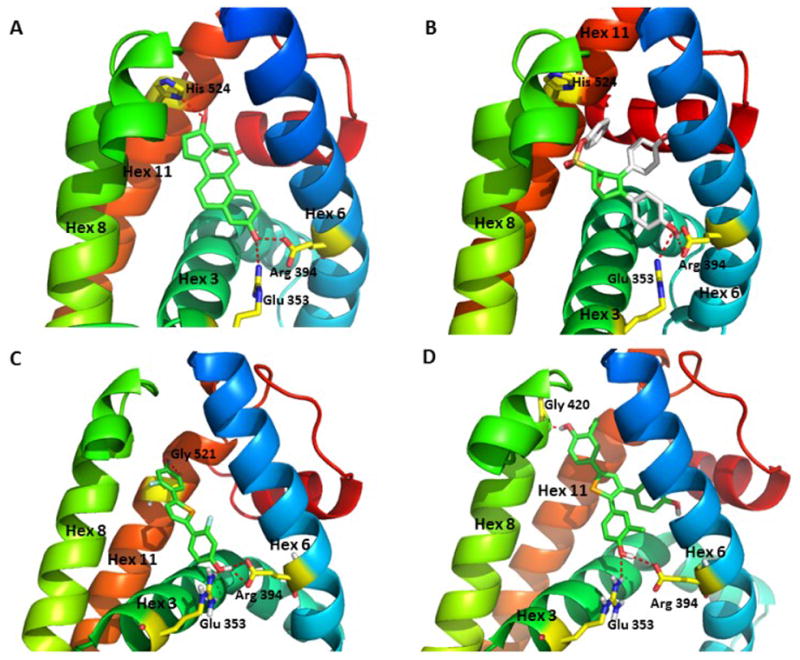
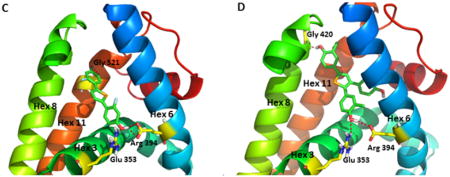
Model of selenophene ligands bound to ERα and comparisons with estradiol and OBHS. (A) Crystal structure of the ERα LBD in complex with E2 (PDB: 1ERE). The A-ring phenolic hydroxyl group of E2 forms hydrogen bonding interactions with helix 3 residue Glu353 and helix 6 residue Arg394, while the D-ring 17β alcohol interacts with the helix 11 residue His524. (B) Crystal structure of the ERα LBD in complex with oxabicyclic heptane sulfonate (OBHS) bound ERα. OBHS H-bonds to the conserved Glu353 and Arg394 residues. The phenyl sulfonate extends outward between helices 8 and 11.39 (C) Computer-developed model of 2f bound to ERα with conserved H-bonding to Glu353 and Arg394. The second phenolic group H-bonds to Gly521 on helix 11, which stabilizes helix 12 in the agonist conformation. (D) Computer-developed model of 8b bound to ERα with the conserved H-bonding to Glu353 and Arg394, and the second phenolic group with H-bonds to Gly420 on helix 8, avoiding the clash with helix 11.
We recently reported on the crystal structure of oxabicyclic compound OBHS, where one of phenols, which mimics the role of A-ring phenol of E2, engages in strong hydrogen bonding with the helix 3 residue Glu353 and the helix 6 residue Arg394. Beyond this, however, the large non-polar phenyl sulfonate group in OBHS extends between helices 8 and 11, into a region in which it makes strong steric clashes with helix 11 and indirectly modulates the conformation of the critical helix 12 (Figure 6B), giving it partial antagonist activity, but limited agonist activity.39
With reference to these modes of ligand binding, our modeling shows that 2,5-disubstituted diphenolic selenophene 2f can similarly form hydrogen bonds between one of the phenolic hydroxyl groups with helix 3 residue Glu353 and helix 6 residue Arg394; the second phenol also makes interaction with helix 11, but it does so by forming hydrogen bond contacts with the helix 11 residue Gly521 (Figure 6C). In contrast to OBHS, however, compound 2f lacks ERα antagonist activity and is a full ERα agonist. Based on our model, we suggest that the hydrogen bonding interaction with Gly521 constrains helix 11 in a manner that does not further destabilize helix 12; rather, it mimics the role of the D-ring phenol E2, which stabilizes helix 12 in the agonist conformation. In support of this conjecture, we recently published on a series of thiophene ligands, in which one of the phenol groups also extended between helices 8 and 11. These thiophenes showed pronounced agonist activity, which on further study also appeared to stem from interactions with helix 11 that stabilized helix 12 in the agonist conformation.38
Interestingly, when a third phenol moiety is introduced into the selenophene core, as in the triphenolic selenophene 8b (Figure 6D), our modeling reveals a possible novel mechanism of agonism via the second phenol, which makes an interaction with residue Gly420 of helix 8, thereby avoiding the clash with helix 11, but this does not explain its superagonist activity. We presume that the superagonist activity of this compound was due to an “improved” positioning of helix 8 that further stabilizes helix 12 in the agonist conformation to an even greater degree than does the binding of E2. Therefore, contact with helix 8 may represent a novel epitope to generate an ERα superagonist.
Conclusions
To further explore anti-breast cancer drugs that might have superior efficacy and fewer side effects than tamoxifen or other therapeutic agents, we present the synthesis of a novel series of selenophene-core compounds. This is the first report of selenophene-core compounds as ER ligands, and we have investigated their ER binding affinities, estrogen response element-driven transcriptional activity, and cell antiproliferative activity. Ligands in this selenophene-core compound class can be easily prepared by Suzuki coupling reactions between various bromoselenophenes and various aryl boronic acids. Careful analysis of their ER binding affinity showed that the selenophene core ligands are largely ERβ-selective, which is similar to that reported earlier for ligands having thiophene and furan cores. As before, the position of the phenolic group has a marked effect on their binding affinity for ERβ, but in terms of absolute affinity, most of the selenophenes have higher RBA values for ERβ than those of the earlier classes. In transcription assays, most of selenophenes exhibit partial or full ERβ agonist activity, whereas on ERα, these ligands display a wide range of activities, covering antagonists and agonists, with some selenophenes even profiling as superagonists. Molecular modeling suggests that interaction of one of the phenolic hydroxyl groups with a residue in helix 8 might be responsible for the superagonism of members of this ligand class, and this represents a novel epitope for the design ERα superagonists. Compared with the approved anti-breast cancer drug tamoxifen, 8c compounds exhibited substantial anti-proliferative potency in breast cancer MCF-7 cell lines. Compound 2f has the highest binding affinity for ERβ, but its anti-proliferative activity in breast cancer MCF-7 cells is modest. Aside from compound 2a, no compounds were toxic to non-transformed VERO cells, while tamoxifen showed, at least to some extent, inherent toxicity on these cells.
Our generation of this new selenophene series of ER ligands provides important insights into the diversity of structures that function as ER ligands and might lead to improved therapeutics that target the estrogen receptor. While ER remains an important pharmaceutical target for the treatment of breast cancer, the quest for the “ideal” drugs for this disease will certainly remain a challenge due to the fact that breast cancer is a high complex disease.
Experimental Section
General
Starting materials were purchased from Aldrich, Acros, Aladdin-reagent, and Alfa-Asar and were used without purification. Toluene was freshly distilled from sodium, and dichloromethane was distilled from anhydrous CaH2. Glassware was oven-dried, assembled while hot, and cooled under an inert atmosphere. Unless otherwise noted, all reactions were conducted in an inert atmosphere. All reactions were performed under an argon atmosphere unless otherwise specified. Reaction progress was monitored by analytical thin-layer chromatography (TLC). Visualization was achieved by UV light (254 nm). The characterization data for compounds 1, 3, 5, 7, and 9–12 were given in Supporting Information.
1H NMR and 13C NMR spectra were measured on a Bruker Biospin AV400 (400 MHz, 1H NMR; 100 MHz, 13C NMR) instrument. Chemical shifts are reported in ppm (parts per million) and are referenced to either tetramethylsilane or the solvent. Melting points were determined on an X-4 Beijing Tech melting point apparatus, and the data were uncorrected. The purity of all compounds for biological testing was determined by HPLC method (see Supporting Information), confirming >95% purity.
General procedure for Suzuki Coupling
Under Ar atmosphere, a mixture of bromoselenophene (1 equiv), arylboronic acid (3 equiv for disubstituted, 4 equiv for trisubstituted selenophenes), Pd catalyst (0.1 equiv), sodium carbonate (2 equiv) in an oxygen-free toluene/water (1:1) solution was stirred at 120 °C for 24 h, after which, the reaction mixture was cooled to room temperature. The aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous Na2SO4 and then filtered and concentrated in vacuum. The product was purified by column chromatography (CC).
General procedure for Ether Cleavage
Under Ar atmosphere, to a solution of methoxyphenyl selenophene derivative (1 equiv) in dry dichloromethane, boron tribromide (3 equiv per methoxy function) was added dropwise at −20 °C. The mixture was allowed to stir for 4 h, and quenched with MeOH. The reaction mixture was poured into water, and extracted with ethyl acetate. The extracts were dried (Na2SO4) and evaporated. The residue was purified by silica gel column chromatography (CC).
2,5-Bis(4-hydroxyphenyl)selenophene (2a)
Compound 2a was prepared by 2,5-bis(4-methoxyphenyl)selenophene (1a) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a blue solid, 72% yield, mp 137–139°C. 1H NMR (400 MHz, Acetone-d6) δ 7.46 (d, J = 8.8 Hz, 4H), 7.36 (s, 2H), 6.89 (m, 4H). 13C NMR (100 MHz, Acetone-d6) δ 158.22, 149.14, 129.01, 127.89, 125.60, 116.70. HRMS (MALDI/DHB) calcd for C16H13O2Se (M + H+) 316.0053, found 316.0057.
2,5-Bis(4-hydroxy-2-methylphenyl)selenophene (2b)
Compound 2b was prepared by 2,5-bis(4-methoxy-2-methylphenyl)selenophene (1b) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a yellow solid, 86% yield, mp 176–178 °C. 1H NMR (400 MHz, Acetone-d6) δ 8.48 (s, 2H), 7.25 (d, J = 8.4 Hz, 2H), 7.13 (s, 2H), 6.79 (d, J = 8.8 Hz, 2H), 6.73 (d, J = 8.4 Hz, 2H), 2.40 (d, J = 8.4 Hz, 6H). 13C NMR (100 MHz, Acetone-d6) δ 157.95, 149.73, 137.59, 132.39, 129.06, 128.40, 118.33, 113.92, 21.61. HRMS (MALDI/DHB) calcd for C18H17O2Se (M + H+) 344.0444, found 344.0448.
2,5-Bis(4-hydroxy-3-methylphenyl)selenophene (2c)
Compound 2c was prepared by 2,5-bis(4-methoxy-3-methylphenyl)selenophene (1c) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a blue solid, 88% yield, mp 194–196 °C. 1H NMR (400 MHz, Acetone-d6) δ 8.46 (s, 2H), 7.37 (d, J = 8.0 Hz, 2H), 7.33 (s, 2H), 7.27 (d, J = 8.8 Hz, 2H), 6.85 (d, J = 8.4 Hz, 2H), 2.24 (s, 6H). 13C NMR (100 MHz, Acetone-d6) δ 156.19, 149.19, 129.12, 129.05, 125.77, 125.43, 125.20, 116.00, 16.27. HRMS (MALDI/DHB) calcd for C18H17O2Se (M + H+) 344.0444, found 344.0448.
2,5-Bis(4-hydroxy-3,5-dimethylphenyl)selenophene (2d)
Compound 2d was prepared by 2,5-bis(4-methoxy-3,5-dimethylphenyl)selenophene (1d) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a blue solid, 86% yield, mp 268–270 °C. 1H NMR (400 MHz, Acetone-d6) δ 7.50 (s, 2H), 7.34 (s, 2H), 7.22 (s, 4H), 2.27 (s, 12H). 13C NMR (100 MHz, Acetone-d6) δ 154.14, 149.24, 129.00, 126.74, 125.39, 125.36, 16.67. HRMS (MALDI/DHB) calcd for C20H21O2Se (M + H+) 372.0757, found 372.0761.
2,5-Bis(4-hydroxy-2,6-dimethylphenyl)selenophene (2e)
Compound 2e was prepared by 2,5-bis(4-methoxy-2,6-dimethylphenyl)selenophene (1e) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a yellow solid, 85% yield, mp 213–215 °C. 1H NMR (400 MHz, Acetone-d6) δ 8.31 (s, 2H), 6.90 (s, 2H), 6.63 (s, 4H), 2.18 (s, 12H). 13C NMR (100 MHz, Acetone-d6) δ 157.87, 149.82, 139.52, 129.88, 127.91, 115.07, 21.16. HRMS (MALDI/DHB) calcd for C20H21O2Se (M + H+) 372.0754, found 372.0761.
2,5-Bis(2-fluoro-4-hydroxyphenyl)selenophene (2f)
Compound 2f was prepared by 2,5-bis(2-fluoro-4-methoxyphenyl)selenophene (1f) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a blue solid, 78% yield, mp 206–208 °C. 1H NMR (400 MHz, Acetone-d6) δ 8.02 (s, 2H), 7.19 (d, J = 8.4 Hz, 2H), 6.91 (d, J = 8.8 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H). 13C NMR (100 MHz, Acetone-d6) δ 159.26, 129.84, 129.78, 127.37, 127.73, 113.24, 104.21, 103.96. HRMS (MALDI/DHB) calcd for C16H11O2F2Se (M + H+) 351.9942 found 351.9946.
2,5-Bis(2-chloro-4-hydroxyphenyl)selenophene (2g)
Compound 2g was prepared by 2,5-bis(2- chloro-4-methoxyphenyl)selenophene (1g) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a blue solid, 74% yield, mp 160–162 °C. 1H NMR (400 MHz, Acetone-d6) δ 9.21 (s, 2H), 7.52 (d, J = 8.8 Hz, 2H), 7.44 (s, 2H), 7.03 (d, J = 8.8 Hz, 2H), 6.90 (d, J = 8.4 Hz, 2H). 13C NMR (100 MHz, Acetone-d6) δ 155.03, 153.51, 153.01, 149.92, 131.55, 129.93, 129.42, 128.54, 127.62, 126.83, 126.53, 126.13, 118.09, 117.64, 117.25. HRMS (MALDI/DHB) calcd for C16H11F2O2Se (M + H+) 384.9351 found 384.9355.
2,5-Bis(3-chloro-4-hydroxyphenyl)selenophene (2h)
Compound 2h was prepared by 2,5-bis(3-chloro-4-methoxyphenyl)selenophene (1h) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a blue solid, 73% yield, mp 168–170 °C. 1H NMR (400 MHz, CD3OD) δ 7.46 (d, J = 8.4 Hz, 1H), 7.40 (d, J = 8.8 Hz, 1H), 7.30 (d, J = 8.4 Hz, 1H), 7.26 (d, J = 8.0 Hz, 1H), 7.23 (t, J = 8.0 Hz, 1H), 7.21 (s, 2H), 6.87 (d, J = 8.4 Hz, 1H), 6.79 (t, J = 8.4 Hz, 2H). 13C NMR (100 MHz, CD3OD) δ 158.63, 147.03, 132.73, 132.66, 129.62, 127.03, 117.84, 115.89. HRMS (MALDI/DHB) calcd for C16H11Cl2O2Se (M + H+) 384.1154 found 384.1156.
2,5-Bis(2-fluoro-5-hydroxyphenyl)selenophene (2i)
Compound 2i was prepared by 2,5-bis(2-fluoro-5-methoxyphenyl)selenophene (1i) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a yellow solid, 70% yield, mp 206–208 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.65 (s, 2H), 7.15 (s, 2H), 7.14 (m, 4H), 6.75 (d, J = 8.8 Hz, 2H). 13C NMR (100 MHz, Acetone-d6) δ 154.82, 129.17, 124.79, 124.65, 117.85, 116.64, 116.55, 114.41. HRMS (MALDI/DHB) calcd for C16H11F2O2Se (M + H+) 351.9947 found 351.9946.
2,5-Bis(4-fluoro-5-hydroxyphenyl)selenophene (2j)
Compound 2j was prepared by 2,5-bis(4-fluoro-5-methoxyphenyl)selenophene (1j) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a yellow solid, 72% yield, mp 175–177 °C. 1H NMR (400 MHz, Acetone-d6) δ 8.94 (s, 2H), 7.50 (m, 2H), 7.26 (d, J = 8.4 Hz, 2H), 7.17 (m, 4H). 13C NMR (100 MHz, Acetone-d6) δ 152.14, 149.24, 146.13, 134.02, 127.46, 118.48, 117.48, 115.93. HRMS (MALDI/DHB) calcd for C16H11F2O2Se (M + H+) 351.9947 found 351.9946.
3,4-Bis(4-hydroxyphenyl)selenophene (4a)
Compound 4a was prepared by 3,4-bis(4-methoxyphenyl)selenophene (3a) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a blue solid, 61% yield, mp 144–147°C. 1H NMR (400 MHz, Acetone-d6) δ 8.24 (s, 2H), 7.76 (s, 2H), 6.85 (d, J = 8.4 Hz, 4H), 6.59 (d, J = 8.4 Hz, 4H). 13C NMR (100 MHz, Acetone-d6) δ 157.31, 144.98, 131.00, 128.54, 115.75. HRMS (MALDI/DHB) calcd for C16H13O2Se (M + H+) 317.2253, found 317.2259.
3,4-Bis(4-hydroxy-2-methylphenyl)selenophene (4b)
Compound 4b was prepared by 3,4-bis(4-methoxy-2-methyl phenyl)selenophene (3b) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a yellow solid, 51% yield, mp 151–153 °C. 1H NMR (400 MHz, Acetone-d6) δ 8.20 (s, 2H), 7.84 (s, 2H), 6.83 (d, J = 8.0 Hz, 2H), 6.58 (d, J = 7.6 Hz, 2H), 6.52 (d, J = 7.2 Hz, 2H), 6.50 (d, J = 7.2 Hz, 6H), 2.01 (s, 6H). 13C NMR (100 MHz, Acetone-d6) δ 157.14, 145.56, 138.21, 132.37, 128.53, 117.31, 112.95, 20.63. HRMS (MALDI/DHB) calcd for C18H17O2Se (M + H+) 345.0394, found 345.0398.
3,4-Bis(4-hydroxy-3-methylphenyl)selenophene (4c)
Compound 4c was prepared by 3,4-bis(4-methoxy-3-methylphenyl)selenophene (3c) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether : ethyl acetate = 3:1) gave the title compound as a blue solid, 68% yield, mp 148–151 °C. 1H NMR (400 MHz, Acetone-d6) δ 8.21 (s, 2H), 7.88 (s, 2H), 6.98 (s, 2H), 6.75 (d, J = 8.4 Hz, 2H), 6.69 (d, J = 8.4 Hz, 2H), 2.14 (s, 6H). 13C NMR (100 MHz, Acetone-d6) δ 155.20, 145.11, 132.33, 130.84, 128.33, 128.21, 124.55, 114.97, 16.35. HRMS (MALDI/DHB) calcd for C18H17O2Se (M + H+) 345.0394, found 345.0395.
3,4-Bis(3-fluoro-4-hydroxyphenyl)selenophene (4d)
Compound 4d was prepared by 3,4-bis(3-fluoro-4-methoxy phenyl)selenophene (3d) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a blue solid, 58% yield, mp 167–169 °C. 1H NMR (400 MHz, CDCl3) δ 8.76 (s, 2H), 8.01 (s, 2H), 6.95 (d, J = 8.4 Hz, 2H), 6.91 (d, J = 7.6 Hz, 1H), 6.67 (d, J = 7.2 Hz, 1H), 6.82 (d, J = 8.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 151.79, 144.66, 143.56, 131.39, 129.92, 126.19, 118.35, 117.38. HRMS (MALDI/DHB) calcd for C16H11F2O2Se (M + H+) 352.9892, found 352.9895.
3,4-Bis(2-fluoro-4-hydroxyphenyl)selenophene (4e)
Compound 4e was prepared from 3,4-bis(2-fluoro-4-methoxy phenyl)selenophene (3e) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a blue solid, 54% yield, mp 168–171 °C. 1H NMR (400 MHz, Acetone-d6) δ 8.08 (s, 2H), 7.01 (s, 2H), 6.57 (d, J = 8.4 Hz, 1H), 6.55 (d, J = 8.4 Hz, 1H), 6.52 (d, J = 8.4 Hz, 2H), 13C NMR (100 MHz, CDCl3) δ 163.73, 161.32, 155.95, 140.16, 132.01, 131.91, 105.39, 102.64. HRMS (MALDI/DHB) calcd for C16H11F2O2Se (M + H+) 352.9892, found 352.9895.
3-Bromo-2,5-bis(4-hydroxyphenyl)selenophene (6a)
Compound 6a was prepared by 3-bromo-2,5-bis(4-methoxyphenyl)selenophene (5a) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a blue solid, 68% yield, mp 193–195 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.91 (s, 1H), 9.89 (s, 1H), 7.56 (s, 1H), 7.52 (d, J = 8.0 Hz, 2H), 7.49 (d, J = 8.4 Hz, 2H), 7.48 (d, J = 8.0 Hz, 1H), 6.92 (d, J = 8.4 Hz, 2H), 6.87 (d, J = 8.0 Hz, 2H). 13C NMR (100 MHz, CD3OD) δ 159.10, 158.82, 149.74, 141.81, 131.61, 131.42, 128.85, 128.10, 127.36, 116.88, 116.43, 116.33, 108.08. HRMS (MALDI/DHB) calcd for C16H12BrO2Se (M + H+) 394.9237 found 394.9240.
3-Bromo-2,5-bis(4-hydroxy-2-methylphenyl)selenophene (6b)
Compound 6b was prepared by 3-bromo-2,5-bis(4-methoxy-2-methylphenyl)selenophene (6b) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a yellow solid, 79% yield, mp 52–54 °C. 1H NMR (400 MHz, Acetone-d6) δ 8.59 (s, 2H), 7.26 (d, J = 8.4 Hz, 1H), 7.15 (d, J = 7.6 Hz, 2H), 6.81 (t, J = 8.8 Hz, 2H), 6.75 (d, J = 8.4 Hz, 2H), 2.41 (s, 3H), 2.23 (s, 3H). 13C NMR (100 MHz, Acetone-d6) δ 154.89, 154.57, 149.50, 141.47, 129.88, 128.46, 127.44, 126.79, 126.68, 125.59, 125.07, 107.38, 16.67, 16.62. HRMS (MALDI/DHB) calcd for C18H16BrO2Se (M + H+) 422.9548 found 422.9553.
3-Bromo-2,5-bis(4-hydroxy-3-methylphenyl)selenophene (6c)
Compound 6c was prepared by 3-bromo-2,5-bis(4-methoxy-3-methylphenyl)selenophene (5c) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a blue solid, 82% yield, mp 155–157 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.68 (s, 2H), 7.21 (d, J = 8.4 Hz, 2H), 7.10 (s, 1H), 6.72 (s, 2H), 6.68 (d, J = 8.0 Hz, 1H), 6.66 (d, J = 8.4 Hz, 1H), 2.36 (s, 3H), 2.17 (s, 3H). 13C NMR (100 MHz, Acetone-d6) δ 156.93, 156.64, 149.47, 141.43, 132.30, 129.07, 128.63, 128.49, 128.41, 127.51, 126.05, 125.52, 125.24, 116.09, 115.62, 107.50, 16.27, 16.23. HRMS (MALDI/DHB) calcd for C18H16BrO2Se (M + H+) 422.9548 found 422.9553.
3-Bromo-2,5-bis(4-hydroxy-3,5-dimethylphenyl)selenophene (6d)
Compound 6d was prepared by 3-bromo-2,5-bis(4-methoxy-3,5-dimethoxyphenyl)selenophene (5d) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a blue solid, 86% yield, mp 199–201 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.64 (d, J = 8.4 Hz, 2H), 7.39 (s, 1H), 7.25 (s, 4H), 2.29 (s, 6H), 2.28 (s, 6H). 13C NMR (100 MHz, Acetone-d6) δ 154.88, 154.56, 149.50, 141.48, 129.89, 128.46, 127.45, 126.69, 125.59, 125.07, 107.38, 16.70, 16.65. HRMS (MALDI/DHB) calcd for C20H20BrO2Se (M + H+) 450.9864 found 450.9866.
3-Bromo-2,5-bis(3-chloro-4-hydroxyphenyl)selenophene (6e)
Compound 6e was prepared by 3-bromo-2,5-bis(3-chloro-4-methoxy-phenyl)selenophene (5e) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a gray solid, 72% yield, mp 98–100 °C. 1H NMR (400 MHz, Acetone-d6) δ 9.33 (s, 2H), 7.64 (d, J = 8.8 Hz, 2H), 7.52 (s, 1H), 7.44–7.39 (m, 2H), 7.08 (d, J = 8.8 Hz, 2H). 13C NMR (100 MHz, Acetone-d6) δ 156.19, 149.19, 129.12, 129.05, 125.77, 125.43, 125.20, 116.00. HRMS (MALDI/DHB) calcd for C16H10Cl2O2Se (M + H+) 462.8326 found 450. 8327.
3-Bromo-2,5-bis(4-fluoro-5-hydroxyphenyl)selenophene (6f)
Compound 6f was prepared by 3-bromo-2,5-bis(4-fluoro-5-methoxyphenyl)selenophene (5f) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a yellow solid, 70% yield, mp 80–82 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.26 (s, 1H), 10.22 (s, 1H), 7.63 (s, 1H), 7.26 (d, J = 8.8 Hz, 1H), 7.24 (d, J = 8.4 Hz, 1H), 7.16 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 8.4 Hz, 1H), 7.03 (d, J = 8.4 Hz, 1H), 7.01 (d, J = 8.4 Hz, 2H). 13C NMR (100 MHz, Acetone-d6) δ 148.0, 145.7, 143.3, 141.0, 130.2, 121.5, 119.4, 118.4, 117.5, 117.3, 117.1, 116.9, 116.8, 116.6, 116.0. HRMS (MALDI/DHB) calcd for C16H10BrF2O2Se (M + H+) 430.1023 found 430.1025.
3-Bromo-2,5-bis(2-fluoro-4-hydroxyphenyl)selenophene (6g)
Compound 6g was prepared from 3-bromo-2,5-bis(4-fluoro-5-methoxyphenyl)selenophene (5g) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a yellow solid, 75% yield, mp 80–82 °C. 1H NMR (400 MHz, CD3OD) δ 7.57 (m, 2H), 7.35 (d, J = 8.4 Hz, 1H), 7.06 (d, J = 9.9 Hz, 2H), 6.93 (t, J = 8.3 Hz, 2H), 6.76 (d, J = 10.0 Hz, 1H), 6.65 (m, 2H). 13C NMR (101 MHz, Acetone-d6) δ 161.10, 159.87, 159.18, 158.64, 132.56, 132.52, 128.94, 128.84, 112.48, 111.64, 103.26, 103.01. HRMS (MALDI/DHB) Calcd for C16H9BrF2O2Se ( M + H+) 430.9053 found 430.9052
2,3,5-Tris(4-hydroxyphenyl)selenophene (8a)
Compound 8a was prepared by 2,3,5-tris(4-methoxyphenyl)selenophene (7a) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a blue solid, 88% yield, mp 114–116 °C. 1H NMR (400 MHz, CD3OD) δ 7.42 (m, 2H), 7.36 (s, 1H), 7.10 (m, 4H), 6.80 (m, 2H), 6.71 (m, 4H). 13C NMR (100 MHz, CD3OD) δ 158.43, 157.85, 157.38, 148.47, 142.57, 141.19, 131.43, 131.35, 130.84, 129.47, 129.33, 128.81, 128.12, 116.72, 116.22, 116.12. HRMS (MALDI/DHB) calcd for C22H17O3Se (M + H+) 407.0388 found 407.0397.
2,3,5-Tris(4-hydroxy-3-methylphenyl)selenophene (8b)
Compound 8b was prepared by 2,3,5-tris(4-methoxy-3-methylphenyl)selenophene (7b) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a black solid, 84% yield, mp 38–40 °C. 1H NMR (400 MHz, Acetone-d6) δ 8.48 (s, 1H), 8.34 (s, 1H), 8.26 (s, 1H), 8.12 (s, 1H), 7.30 (d, J = 8.4 Hz, 2H), 7.14 (d, J = 8.0 Hz, 1H), 6.99 (d, J = 8.0 Hz, 2H), 6.85 (d, J = 8.4 Hz, 2H), 6.71 (d, J = 8.4 Hz, 1H), 6.57 (d, J = 8.4 Hz, 2H), 2.25 (s, 3H), 2.16 (s, 3H), 2.14 (s, 3H). 13C NMR (100 MHz, Acetone-d6) δ 156.31, 155.70, 155.25, 147.91, 141.98, 140.70, 132.37, 132.25, 130.27, 129.10, 128.89, 128.84, 128.77, 128.42, 128.33, 125.77, 125.24, 125.15, 124.88, 116.01, 115.47, 115.34, 16.30, 16.25, 16.22. HRMS (MALDI/DHB) calcd for C25H23O3Se (M + H+)450.0857 found 450.0866.
2,3,5-Tris(4-hydroxy-3,5-dimethylphenyl)selenophene (8c)
Compound 8c was prepared by 2,3,5-tris(4-methoxy-3,5-dimethylphenyl)selenophene (7c) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a blue solid, 83% yield, mp 76–78 °C. 1H NMR (400 MHz, Acetone-d6) δ 7.53 (s, 1H), 7.45 (s, 1H), 7.43 (s, 1H), 7.31 (s, 1H), 7.26 (s, 2H), 6.93 (d, J = 8.4 Hz, 4H), 2.28 (s, 6H), 2.17 (s, 6H), 2.14 (s, 6H). 13C NMR (100 MHz, Acetone-d6) δ 167.50, 155.89, 153.51, 153.45, 131.03, 128.15, 126.23, 121.83, 121.73, 121.63, 112.56, 112.49, 112.40, 112.33, 35.49, 31.44. HRMS (MALDI/DHB) calcd for C28H29O3Se (M + H+) 491.1328 found 491.1336.
2,3,5-Tris(3-chloro-4-methoxyphenyl)selenophene (8d)
Compound 8d was prepared by 2,3,5-tris(3-chloro-4-methoxyphenyl)selenophene (7d) and boron tribromide according to general procedure for ether cleavage. Purification by CC (petroleum ether:ethyl acetate = 3:1) gave the title compound as a yellow solid, 78% yield, mp 48–50 °C. 1H NMR (400 MHz, Acetone-d6) δ 10.48 (s, 2H), 10.29 (s, 1H), 7.70 (d, J = 8.8 Hz, 1H), 7.67 (s, 1H), 7.45 (d, J = 8.4 Hz, 1H), 7.36 (d, J = 8.4 Hz, 1H), 7.27 (d, J = 8.8 Hz, 1H), 7.10–7.06 (m, 2H), 7.05 (d, J = 8.4 Hz, 1H), 6.99 (d, J = 8.4 Hz, 1H), 6.88 (d, J = 8.8 Hz, 1H). 13C NMR (100 MHz, Acetone-d6) δ 153.76, 153.46, 153.02, 147.33, 141.84, 140.22, 131.13, 131.09, 130.91, 129.93, 129.79, 129.58, 127.79, 126.75, 121.82, 121.25, 121.06, 118.11, 117.75, 117.52. HRMS (MALDI/DHB) calcd for C22H14Cl3O3Se (M + H+), 510.9223 found 510.9228.
Gene Clone and Protein Purification
Human ERα or ERβ ligand binding domain (LBD) genes were amplified by PCR from plasmid pVP-16-ERα and pVP-16-ERβ. The PCR product was cloned into plasmid, and PGEx-KG E. coli BL21(DE3) used for the overexpression of ER-LBD. The cells were induced by IPTG (10 μM) for 2 h, then cells were harvested, frozen, and thawed in phosphate-buffered saline (PBS), containing 1 mM EDTA and 1 mM DTT. After being ultrasonicated in an icy bath, the supernatant was applied to a column of GSH-resin. The collection was dialyzed in ice buffer for 4 h. After being checked by a combination of sodium dodecyl sulfate–polyacrylamide gel electrophoresis and Western blotting, the protein was prepared as a 10-mM stock in potassium phosphate and stored at −80 °C.[49]
Estrogen Receptor Binding Affinity
Relative binding affinities were determined by a competitive fluorometric binding assay as previously described. Briefly, 40 nM fluorescence tracer (coumestrol, Sigma-Aldrich, MO) and 0.8 μM purified human ERα or ERβ ligand binding domain (LBD) were diluted in 100 mM potassium phosphate buffer (pH 7.4), containing 100 μg/mL bovine gamma globulin (Sigma-Aldrich, MO). Incubations were for 2h at room temperature (25 °C). Fluorescence polarization values were then measured. The binding affinities are expressed as relative binding affinity (RBA) values with the RBA of 17-β estradiol set to 100%. The values given are the average ± range of two independent determinations. IC50 values were calculated according to equations described previously.[50]
Gene Transcriptional Activity
The human embryonic kidney cell lines, HEK 293T, was maintained in Dulbecco’s Minimum Essential Medium (DMEM) (Gibco by Invitrogen Corp., CA) with 10% fetal bovine serum (FBS) (Hylcone by Thermo Scientific, UT). Cells were plated in phenol red-free DMEM with 10% FBS. HEK 293T cells were transfected with 25 μL mixture per well, containing 300 ng of 3 × ERE-luciferase reporter, 100 ng of ERα or ERβ expression vector, 125 mM calcium chloride (GuoYao, China) and 12.5 μL 2 × HBS. The next day, the cells were treated with increasing doses of ER ligands diluted in phenol red-free DMEM with 10% FBS. After 24h, luciferase activity was measured using Dual-Luciferase Reporter Assay System (Promega, MI) according to the manufacturer’s protocol.
Cell Culture and Cell Viability Assay
The human breast cancer cell lines MCF-7 was obtained from ATCC. Cells were maintained in DMEM with 10% FBS. For all experiments, cells were grown in 96-well microtiter plates (Nest Biotech Co., China) with appropriate ligand triplicate for 72h. MTT colormetric tests (Biosharp, China) were employed to determine cell viability per manufacturer instructions. IC50 values were calculated according to the following equation using Origin software: Y = 100% inhibition + (0% inhibition - 100% inhibition)/(1 + 10[(LogIC50-X)×Hillslope]), where Y = fluorescence value, X = Log [inhibitor]. [50]
Molecular Modeling
Crystal structure of ER LBD in complex with E2 was downloaded from the protein data bank (PDB ID: 1ERE). Compounds 2f, and 8b were docked into the three-dimension structure of ERα LBD with AutoDock software (version 4.2).[51–52] Crystallographic coordinates of the 2f, and 8b were created by Biochemoffice. The crystal structure of ERα LBD was obtained from the OBHS-bound ER crystal structure and all water molecules were removed.[35] Preparations of all ligands and the protein were performed with AutoDockTools (ADT). A docking cube with the edge of 60 Å, 60 Å, 58 Å in X, Y, Z dimension respectively (a grid spacing of 0.375 Å), which encompassed the whole active site, was used throughout docking. On the basis of the Lamarckian genetic algorithm (LGA), 80 runs were performed for each ligand with 500 individuals in the population. The figures were prepared using PyMOL.
Supplementary Material
Acknowledgments
We are grateful to the NSFC (81373255, 31371331, 81573279), Key Scientific Research Project of Ministry of Education of China (313040), Hubei Province’s Outstanding Medical Academic Leader Program, and Hubei Province Engineering and Technology Research Center for Fluorinated Pharmaceuticals and to the National Institutes of Health (PHS 5R01 DK015556 to J.A.K.) for support of this research.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Early DS, Hill K, Burk R, Palmer I. Am J Gastroenterol. 2002;97:745–748. doi: 10.1111/j.1572-0241.2002.05558.x. [DOI] [PubMed] [Google Scholar]
- 2.Kornitzer M, Valente F, Bacquer DD, Neve J, Backer GD. Eur J Clin Nutr. 2004;58:98–104. doi: 10.1038/sj.ejcn.1601754. [DOI] [PubMed] [Google Scholar]
- 3.Terazawa R, Garud DR, Hamada N, Fujita Y, Itoh T, Nozawa Y, Nakane K, Deguchi T, Koketsu M, Ito M. Bioorg Med Chem. 2010;18:7001–7008. doi: 10.1016/j.bmc.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 4.Kumar BS, Kunwar A, Singh BG, Ahmad A, Priyadarsini KI. Biol Trace Elem Res. 2011;140:127–138. doi: 10.1007/s12011-010-8692-3. [DOI] [PubMed] [Google Scholar]
- 5.Filho CB, Fabbro LD, Boeira SP, Furian AF, Savegnago L, Soares LC, Braga AL, Jesse CR. Cell Biochem Funct. 2013;31:152–158. doi: 10.1002/cbf.2869. [DOI] [PubMed] [Google Scholar]
- 6.Mukherjee AJ, Zade SS, Singh HB, SRB Chem Rev. 2010;110:4357–4416. doi: 10.1021/cr900352j. [DOI] [PubMed] [Google Scholar]
- 7.Andrade LH, Silva AV, Milani P, Koszelewski D, Kroutil W. Org Biomol Chem. 2010;8:2043–2051. doi: 10.1039/b920946h. [DOI] [PubMed] [Google Scholar]
- 8.Godoi M, Paixão MW, Braga AL. Dalton Trans. 2011;40:11347–11155. doi: 10.1039/c1dt11022e. [DOI] [PubMed] [Google Scholar]
- 9.Back TG, Moussa Z. J Am Chem Soc. 2003;125:13455–13460. doi: 10.1021/ja0357588. [DOI] [PubMed] [Google Scholar]
- 10.Sarma BK, Mann D, Minoura M, Mugesh G. J Am Chem Soc. 2010;132:5364–5374. doi: 10.1021/ja908080u. [DOI] [PubMed] [Google Scholar]
- 11.Martins IL, Charneira C, Gandin V, Ferreira da Silva JL, Justino GC, Telo JP, Vieira AJ, Marzano C, Antunes AM. J Med Chem. 2015;58:4250–4265. doi: 10.1021/acs.jmedchem.5b00230. [DOI] [PubMed] [Google Scholar]
- 12.Mukherjee S, Weiner WS, Schroeder CE, Simpson DS, Hanson AM, Sweeney NL, Marvin RK, Ndjomou J, Kolli R, Isailovic D, Schoenen FJ, Frick DN. ACS Chem Biol. 2014;9:2393–2403. doi: 10.1021/cb500512z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Preston S, Luo J, Zhang Y, Jabbar A, Crawford S, Baell J, Hofmann A, Hu M, Zhou HB, Gasser RB. Parasites & Vectors. 2016;9:1–11. doi: 10.1186/s13071-016-1612-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sundaram GSM, Dhavale DD, Prior JL, Ping Y, Cirrito J, Rath NP, Laforest R, Cairns NJ, Lee JM, Kotzbauer PT. Scientific Reports. 2016;6:35636. doi: 10.1038/srep35636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 16.Katzenellenbogen BS, Choi I, Delage-Mourroux R, Ediger TR, Martini PG, Montano M, Sun J, Weis K, Katzenellenbogen JA. J Steroid Biochem Mol Biol. 2000;74:279–285. doi: 10.1016/s0960-0760(00)00104-7. [DOI] [PubMed] [Google Scholar]
- 17.Katzenellenbogen BS, Katzenellenbogen JA. Science. 2002;295:2380–2381. doi: 10.1126/science.1070442. [DOI] [PubMed] [Google Scholar]
- 18.Mendelsohn ME, Karas RH. Science. 2005;308:1583–1587. doi: 10.1126/science.1112062. [DOI] [PubMed] [Google Scholar]
- 19.Katzenellenbogen JA. J Med Chem. 2011;54:5271–5282. doi: 10.1021/jm200801h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swedenborg E, Power KA, Cai W, Pongratz I, Rueqq J. Cell Mol Life Sci. 2009;66:3873–3894. doi: 10.1007/s00018-009-0118-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gong P, Madakerdogan Z, Li J, Cheng J, Greenlief CM, Helferich W, Katzenellenbogen JA, Katzenellenbogen BS. Nuclear Receptor Signaling. 2014;12:e001–e001. doi: 10.1621/nrs.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang EC, Frasor J, Komm B, Katzenellenbogen BS. Endocrinology. 2006;147:4831–4842. doi: 10.1210/en.2006-0563. [DOI] [PubMed] [Google Scholar]
- 23.Chang EC, Charn TH, Park SH, Helferich WG, Komm B, Katzenellenbogen JA, Katzenellenbogen BS. Mol Endocrinol. 2008;22:1032–1043. doi: 10.1210/me.2007-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Madak-Erdogan Z, Charn TH, Jiang Y, Liu ET, Katzenellenbogen JA, Katzenellenbogen BS. Mol Syst Biol. 2013;9:676. doi: 10.1038/msb.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jordan VC. J Med Chem. 2003;46:883–908. doi: 10.1021/jm020449y. [DOI] [PubMed] [Google Scholar]
- 26.McDonnell DP, Wardell SE. Curr Opin Pharmacol. 2010;10:620–628. doi: 10.1016/j.coph.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jordan VC. J Med Chem. 2003;46:1081–1111. doi: 10.1021/jm020450x. [DOI] [PubMed] [Google Scholar]
- 28.Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J. J A M A. 2002;288:321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 29.Benson JR. J Natl Cancer Inst. 2001;93:1493–1494. doi: 10.1093/jnci/93.19.1493. [DOI] [PubMed] [Google Scholar]
- 30.Stauffer SR, Coletta CJ, Tedesco R, Nishiguchi G, Carlson K, Sun J, Katzenellenbogen BS, Katzenellenbogen JA. J Med Chem. 2000;43:4934–4947. doi: 10.1021/jm000170m. [DOI] [PubMed] [Google Scholar]
- 31.Mortensen DS, Rodriguez AL, Carlson KE, Sun J, Katzenellenbogen BS, Katzenellenbogen JA. J Med Chem. 2001;44:3838–3848. doi: 10.1021/jm010211u. [DOI] [PubMed] [Google Scholar]
- 32.Zhou HB, Carlson KE, Stossi F, Katzenellenbogen BS, Katzenellenbogen JA. Bioorg Med Chem Lett. 2009;19:108–110. doi: 10.1016/j.bmcl.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schäfer A, Wellner A, Strauss M, Schäfer A, Wolber G, Gust G. J Med Chem. 2012;55:9607–9618. doi: 10.1021/jm300860j. [DOI] [PubMed] [Google Scholar]
- 34.Min J, Wang P, Srinivasan S, Nwachukwu JC, Guo P, Huang MJ, Carlson KE, Katzenellenbogen JA, Nettles KW, Zhou HB. J Med Chem. 2013;56:3346–3366. doi: 10.1021/jm400157e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng YF, Zhu MH, Srinivasan S, Nwachukwu JC, Cavett V, Min J, Carlson KE, Wang PC, Dong C, Katzenellenbogen JA, Katzenellenbogen BS, Nettles KW, Zhou HB. Chem Med Chem. 2012;7:1094–1100. doi: 10.1002/cmdc.201200048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu MH, Zhang C, Nwachukwu JC, Srinivasan S, Cavett V, Zheng YF, Carlson KE, Dong C, Katzenellenbogen JA, Nettles KW, Zhou HB. Org Biomol Chem. 2012;10:8692–8700. doi: 10.1039/c2ob26531a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liao ZQ, Dong C, Carlson KE, Srinivasan S, Nwachukwu JC, Chesnut RW, Sharma A, Nettles KW, Katzenellenbogen JA, Zhou HB. J Med Chem. 2014;57:3532–3545. doi: 10.1021/jm500268j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang PC, Min J, Nwachukwu JC, Cavett V, Carlson KE, Guo P, Zhu MH, Zheng YF, Dong C, Katzenellenbogen JA, Nettles KW, Zhou HB. J Med Chem. 2012;55:2324–2341. doi: 10.1021/jm201556r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amb CM, Rasmussen SC. Eur J Org Chem. 2008;2008:801–804. [Google Scholar]
- 40.van Leeuwen PW, Kamer PC, Reek JN, Dierkes P. Chem Rev. 2000;100:2741–2770. doi: 10.1021/cr9902704. [DOI] [PubMed] [Google Scholar]
- 41.Dell’Erba C, Spinelli D, Garbarino G, Leandri G. J Heterocyclic Chem. 1968;5:45–48. [Google Scholar]
- 42.Tùng ĐT, Villinger A, Langer P. Adv Synth Catal. 2008;350:2109–2117. [Google Scholar]
- 43.Pigeon P, Top S, Vessières A, Huché M, Hillard EA, Salomon E, Jaouen G. J Med Chem. 2005;48:2814–2821. doi: 10.1021/jm049268h. [DOI] [PubMed] [Google Scholar]
- 44.Zhou HB, Comninos JS, Stossi F, Katzenellenbogen BS, Katzenellenbogen JA. J Med Chem. 2005;48:7261–7274. doi: 10.1021/jm0506773. [DOI] [PubMed] [Google Scholar]
- 45.Kocsis Z, Marcsek ZL, Jakab MG, Szende B, Tompa A. Int J Hyg Envir Heal. 2004;208:211–218. doi: 10.1016/j.ijheh.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 46.Gryder BE, Rood MK, Johnson KA, Patil V, Raftery ED, Yao LPD, Rice M, Azizi B, Doyle DF, Oyelere AK. J Med Chem. 2013;56:5782–5796. doi: 10.1021/jm400467w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang HZ, Zhang H, Kemnitzer W, Tseng B, Cinatl J, Michaelis M, Doerr HW, Cai SX. J Med Chem. 2006;49:1198–1201. doi: 10.1021/jm0507678. [DOI] [PubMed] [Google Scholar]
- 48.Sanz A, Gomez-Contreras F, Navarro P, Sanchez-Moreno M, Boutaleb-Charki S, Campuzano J, Pardo M, Osuna A, Cano C, Yunta M. J Med Chem. 2008;51:1962–1966. doi: 10.1021/jm701179m. [DOI] [PubMed] [Google Scholar]
- 49.Wang C, Li C, Zhou H, Huang J. J Biomol Screen. 2014;19:253–258. doi: 10.1177/1087057113502673. [DOI] [PubMed] [Google Scholar]
- 50.Tang C, Li CH, Zhang SL, Hu ZY, Wu J, Dong C, Huang J, Zhou HB. J Med Chem. 2015;58:4550–4572. doi: 10.1021/acs.jmedchem.5b00099. [DOI] [PubMed] [Google Scholar]
- 51.Huey R, Morris GM, Olson AJ, Goodsell DS. J Comput Chem. 2007;28:1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 52.Audie J. Biophys Chem. 2009;139:84–91. doi: 10.1016/j.bpc.2008.10.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.