

Abstract
Objective:
To assess the potential effect of variants in genes encoding molecules that are implicated in leukocyte trafficking into the CNS on the clinical phenotype of multiple sclerosis (MS).
Methods:
A total of 389 Greek MS cases and 336 controls were recruited in 3 MS centers from Cyprus and Greece. We genotyped 147 tagging single nucleotide polymorphisms (SNPs) in 9 genes encoding for P-selectin (SELP), integrins (ITGA4, ITGB1, and ITGB7), adhesion molecules (ICAM1, VCAM1, and MADCAM1), fibronectin 1 (FN1), and osteopontin (SPP1) involved in lymphocyte adhesion and trafficking into the CNS. Clinical end points of the study were age at MS onset and MS severity as measured by the Multiple Sclerosis Severity Score. Permutation testing was applied to all analyses.
Results:
SNPs rs6721763 of the ITGA4 and rs6532040 of the SPP1 were found to significantly influence disease severity (permutation p values: 3.00e-06 and 0.009884, respectively). SNP rs1250249 of the FN1 had a dose-dependent effect on age at disease onset (permutation p value: 0.0002).
Conclusions:
This study provides evidence implicating variants encoding adhesion molecules, responsible for lymphocyte adhesion and trafficking within the CNS, as modifiers of MS disease severity. These genetic biomarkers, which can be available at the time of diagnosis, may be used to assess the biological aggressiveness of the disease and thus guide decisions on treatment.
Multiple sclerosis (MS) is considered to result from an interplay between environmental exposures and genetic background.1–3 The majority of genetic studies have focused on susceptibility variants, and this effort has yielded more than 110 genetic risk factors of MS.4 In addition, there is evidence that genetic factors may influence disease phenotype such as age at onset, disease severity, and the clinical course.5–7 However, 3 genome-wide association studies (GWASs) investigating MS severity have failed to reach a genome-wide significance threshold.8–10 A recent meta-analysis of 10 cohorts in 7,125 patients with MS also failed to detect any significant signal.11
Disability in MS, either resulting from relapses or accumulating during the progressive phase of the disease, is partly a result of immune attack from adaptive and innate immunity. A number of molecules are important in leukocyte adhesion and diapedesis through the blood-brain barrier, as well as their migration through the extracellular matrix to the target tissue. The use of natalizumab, a monoclonal antibody against α4 integrin, was found to markedly reduce relapses, new lesion formation, and disability progression12,13 by blocking lymphocyte adhesion and migration through the vascular endothelial cells and by blocking interaction with extracellular matrix proteins. The impressive effectiveness of natalizumab argues strongly in favor of the significance of this pathogenic pathway.
This study was designed to test the effect of genetic variants of molecules, that are implicated in leukocyte trafficking through blood vessel walls into the CNS, on disease severity as measured by the Multiple Sclerosis Severity Score (MSSS).
METHODS
Study population.
Greek patients with MS and controls were recruited from 3 MS centers: The Cyprus Institute of Neurology and Genetics in Cyprus, the University Hospital of Larissa, Greece, and the AHEPA Hospital of Aristotle University in Thessaloniki, Greece.14,15 In total, 389 MS cases and 336 controls were recruited for the study. Patients were included in the study if they had clinically definite MS according to the 2005 revised McDonald criteria, were aged ≥18 years, and had disease duration of ≥5 years. Cases with primary progressive MS were excluded.
Clinical and demographic data were collected using the same criteria in each center. Neurologists specialized in MS collected data such as sex, current age, and age at disease onset, as age at the first neurologic symptom suggestive of MS. Use of disease-modifying therapies (DMTs) and treatment duration were also recorded. Patients were receiving one of the following DMTs: interferon-β, glatiramer acetate, or natalizumab. Disability status was assessed in the absence of relapse for at least 6 months, using the Kurtzke Expanded Disability Status Scale (EDSS). Disease duration was defined as the time (in years) from the first symptom to the last EDSS assessment. The rate of disease progression was expressed by the progression index, which is the ratio of EDSS to MS duration. Finally, disease severity in each individual was assessed by means of MSSS which is a useful algorithm for disability distribution in MS groups evaluated cross-sectionally.16 The control group consisted of healthy volunteers from the same ethic regions and was matched with cases for age and sex.
Standard protocol approvals, registrations, and patient consents.
The study protocol was approved by the local ethics committees, and written informed consent was obtained from all patients.
Isolation of DNA, SNP selection, and genotyping.
DNA was isolated from peripheral blood samples using the Puregene DNA purification kit (Qiagen, Valencia, CA). Based on the study rationale, we chose to genotype genes that encode for the following adhesion and extracellular matrix molecules: (1) P-selectin (OMIM #173610) encoded by the SELP gene located on chromosome 1q23-q25, (2) integrin α-4 (OMIM #1929750, ITGA4, 2q31-q32), (3) integrin-β–1 (OMIM #135630; ITGB1, 10p11.2), (4) integrin-β–7 (OMIM #147559, ITGB7, 12q13.13), (5) intercellular adhesion molecule 1 (OMIM #147840, ICAM1, 19p13.3-p13.2), (6) vascular cell adhesion molecule 1 (OMIM #192225, VCAM1, 1p32-p31), (7) mucosal vascular addressin cell adhesion molecule 1 (OMIM #102670, MADCAM1, 19p13.3), (8) fibronectin 1 (OMIM #135600, FN1, 2q34), and (9) osteopontin (OMIM #166490, SPP1, 4q21-q25).
To cover most of the polymorphic variability along with the above-mentioned genes, we used the linkage disequilibrium tagger algorithm of the HapMap project for selection of tag-SNPs. Selection of tagging single nucleotide polymorphisms (SNPs) was based on the HapMap Release 27 database (Phase II + III, Feb09, on NCBI B36 assembly, dbSNP b126) for CEU population, using the criteria of pairwise r2 values ≥0.8 and minor allele frequency >0.05. This approach retrieved 32 tagging SNPs in SELP gene, 36 in ITGA4 gene, 16 in ITGB1, 3 in ITGB7, 5 in ICAM1, 15 in VCAM1, 3 in MADCAM1, 38 in FN1, and 8 in SPP1 which sum to 156 SNPs. Chromosome position of each SNP, the minor allele frequency for CEU population and TSI population (Tuscan in Italy), which is considered closer to the Greek population, and the minor allele frequency (MAF) of our control group are presented in table e-1 at Neurology.org/nn.
TaqMan 5′-nuclease assays were used to genotype SNPs by acquiring specific assays for each SNP and then performing allelic discrimination assays on the ABI 7900HT Real-Time System (Applied Biosystems, Foster City, CA). Genotype success rates were above 96%. However, 9 SNPs as shown in table e-1 failed to be genotyped and therefore were not included in the analysis.
Statistical analysis.
Quality control was performed by evaluating the genotype distributions of all SNPs in the control samples for the Hardy-Weinberg equilibrium using an exact test17 with a threshold of p ≤ 0.05. MAFs were estimated and used for quality control (threshold of MAF <0.05%). Linear regression analysis for the age at disease onset and MSSS were performed for association testing. The cutoff threshold for significance was a priori set to a permutation-derived p value of at most 0.01. Analyses for age at onset were adjusted for sex and site. Analyses with the MSSS, as a dependent variable, were additionally adjusted for age at onset and treatment status. For treatment status, the duration of treatment with DMT in months was used as a covariate to ensure that any detected effect was not driven by treatment duration. Permutation testing was applied to adjust for multiple testing for known or unknown factors that may affect the balance of the distribution of the data. A total of 106 permutations were performed by using a high-performance computing platform.
Label-swapping permutations were used with 2 sets of empirical significance p values to estimate individual SNPs significance. The minimum possible p value depends on the number of tests performed; in our case, it was 10−6. Because the permutation scheme followed preserves the correlational structure between SNPs, there is no assumption of independence unlike Bonferroni, which assumes all tests are independent. As this is a candidate gene study with relatively dense coverage, there is a likelihood of dependence between neighboring SNPs due to linkage disequilibrium; therefore, permutation offers a better alternative for adjusting for multiple testing.
RESULTS
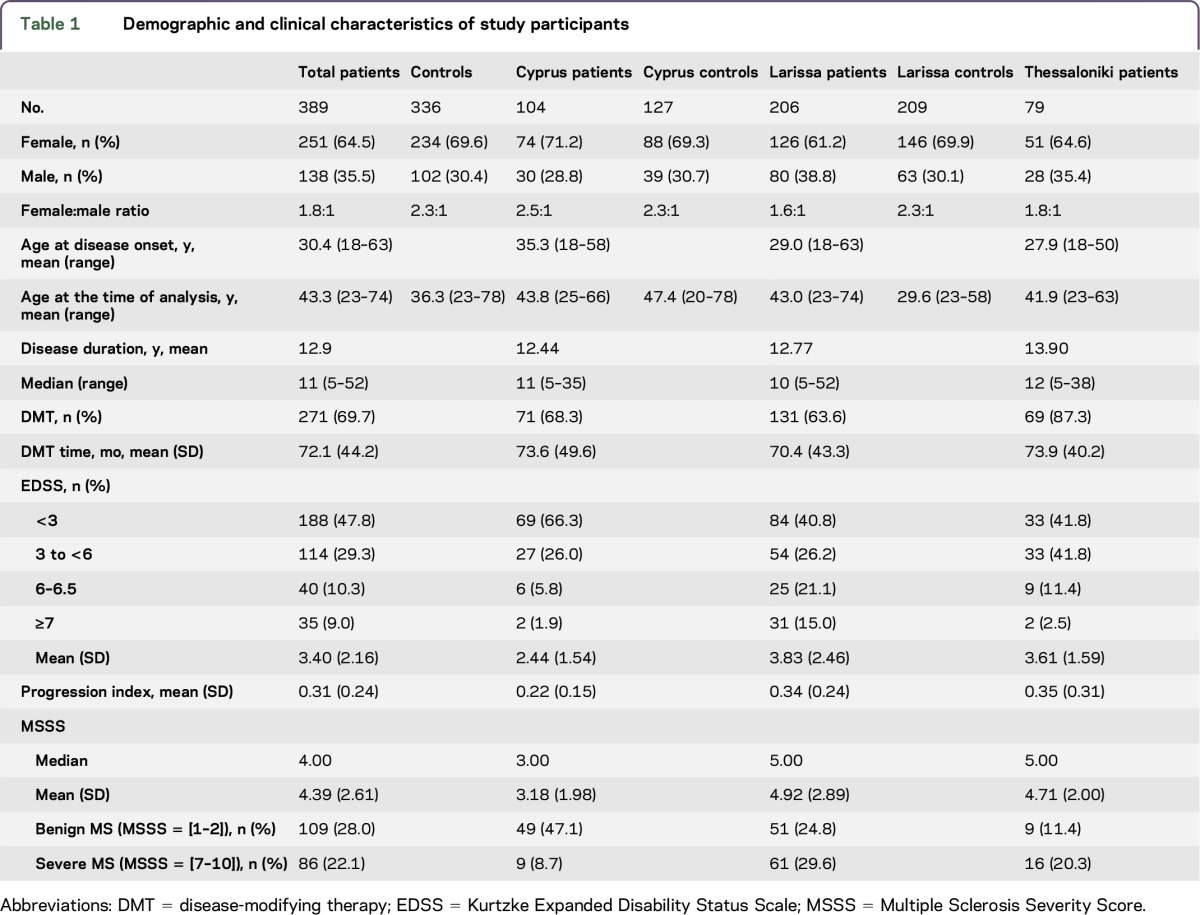
Clinical and demographic data of controls and patients as a group and in each MS center are presented in table 1. More than two-thirds of the patients were women. The mean disease duration was 12.9 years, and most patients (70%) were under DMT. The mean EDSS was 3.4, whereas half of the patients had EDSS less than 3. Severe accumulation of disability (MSSS: 7–10) was observed in approximately 20% of the patients. Patients recruited from the Larissa and Thessaloniki sites presented a more severe phenotype regarding EDSS, progression index, and MSSS scores compared with the patients recruited from Cypriot (p < 0.05).
Table 1.
Demographic and clinical characteristics of study participants
The distribution of SNP alleles in patients, their respective scores in MSSS, and the results of linear regression analysis are presented in table e-2. Significant associations with p < 0.05 were found with 4 ITGA4 polymorphisms (rs10930969, rs2124440, rs3770136, and rs6721763), in rs3780873 ITGB1, in 2 FN1 polymorphisms (rs1250258 and rs13432972), and in rs9138 of SPP1. Permutation analysis was performed to adjust for multiple testing (table 2). Significant associations were found with rs6721763 of ITGA4 (permutation p value: 3.00e-06) and rs6532040 of SPP1 (permutation p value: 0.009884). A dose-dependent effect of minor allele on MSSS was noted with worse scores in minor allele homozygotes for both SNPs. In particular, for rs6721763 of ITGA4, the mean MSSS increased from 3.191 in AA homozygotes to 3.94 in Ab heterozygotes and finally 5.012 in bb homozygotes. The same trend was observed for rs6532040 of SPP1 with an increase from a mean MSSS of 3.538 to 4.034 and 4.176 in AA, Ab, and bb genotypes, respectively.
Table 2.
Permutation analysis results in MSSS

Permutation analyses of the effects of SNP alleles on age at onset are shown in table e-3. The permutation p value significance threshold of 0.01 was exceeded by rs1250249 of FN1 (p = 0.0002). A dose-dependent effect of this polymorphism on age at onset was observed: age at onset dropped from 32.3 years in AA homozygotes to 28.1 years in Ab heterozygotes and to 26.3 years in bb homozygotes.
DISCUSSION
Despite the considerable progress in identifying genetic susceptibility variants,4 there is limited evidence in identifying genetic factors that may contribute to MS disease severity. Genetic association studies have focused on candidate genes such as APOE, genes related to immune function, autoimmunity, neurobiology, myelin structure, and the human leukocyte antigen (HLA), which resulted in conflicting data.18,19 In addition, 3 GWAS investigating MS severity failed to surpass a genome-wide significance threshold.8–10 However, analysis of SNPs in genes involved in glycosylation and calcium and glutamate signaling that may represent disease modifiers reached levels close to genome-wide significance threshold. The effect of HLA on disease onset was relatively consistent among studies.20,21 Carriers of the DRB1*15:01 allele had an earlier onset of MS, with each copy accounting for a decrease in age at onset by about 10.6 months.22
This study detected a statistically significant association between rs6721763 of ITGA4 gene and disease severity as measured with the MSSS in a dose-dependent manner. ITGA4 encodes for the α4 subunit of α4β1 integrin, which is involved in leukocyte adhesion and migration through the blood-brain barrier.23 The monoclonal antibody, natalizumab, which is approved for the treatment of relapsing-remitting MS, exerts its favorable effects on relapses and disability progression by targeting α4 integrin.24 Polymorphisms in ITGA4 have been linked so far only to MS susceptibility.25–27 In particular rs1143676, a nonsynonymous polymorphism (R878Q) located in exon 24 of ITGA4 has been reported to be associated with disease susceptibility in Slovaks25 but not in other populations.26,27 This polymorphism is located 9.3 kb distant from our polymorphism. The second ITGA4 polymorphism that was linked so far to disease susceptibility in a Basque group is the intronic SNP rs1449263,26 which however is 66.5 kb away from our polymorphism. Finally, rs155100 (36 kb far from rs6721763) has been reported to influence the risk of autism.28 SNP rs6721763 is an intronic polymorphism (intron 17–18) located only just 929 bp from the splicing site of exon 18 of ITGA4.
The second genetic modifier of disease course identified in our study is rs6532040 of SPP1. The presence of the minor allele worsens the disease disability course. Osteopontin is a secreted matrix protein with pleiotropic, but mainly, proinflammatory functions involved in T-cell activation, proliferation, and migration. A part of its actions is mediated by binding to integrins. In addition, intracellular osteopontin activates intracellular signaling pathways.29 Osteopontin transcripts are upregulated in MS brain lesions and in normal-appearing white matter. In the mouse model of experimental autoimmune encephalomyelitis, osteopontin was implicated in disease severity and activity.30 Genetic polymorphisms altered osteopontin levels.31 A genetic association study showed that a G/A polymorphism located in the 3′ untranslated region of exon 7 (position 9583) had a significant effect on the age at onset.32 SNP rs6532040, which emerged as significant in our study, is located in intron 5–6, which is only 1.5 kb distant from the start of exon 7. In another study, rs9138 of the osteopontin gene, located at exon 7 and 2.14 kb distant from our polymorphism, showed a modest trend for association. In particular, patients with the wild-type genotype were less likely to have a mild course (p = 0.08) and had increased risk of having a secondary-progressive course of the disease (p = 0.05).33 In addition, a haplotype study of 4 SNPs (rs4754, rs1126616, rs1126772, and rs9138; 0.49 kb, 1.65 kb, 1.98 kb, and 2.14 kb away from our polymorphism, respectively) showed that homozygotes combining the wild-type alleles of all 4 SNPs displayed a milder disease course, slower accumulation of disability, and delayed conversion from relapsing-remitting to secondary-progressive disease course.31 The same research group replicated these findings in an independent group and also detected similar effects on a polymorphism located in the promoter region of the osteopontin gene (rs7687316, 5.5 kb distant from our polymorphism).34 However, a study of 8 SNPs located in exons 6 and 7 failed to detect any significant association with MS severity.35
Finally, our study found that rs1250249 of FN1 exerts a dose-dependent effect on disease onset. The presence of the minor allele predisposes to an earlier age at onset. Fibronectin 1 is an extracellular matrix glycoprotein that interacts with a number of integrins mediating a variety of functions including cytoskeleton organization, cell adhesion, and migration.36 Polymorphisms of FN1 have been associated so far among others with schizophrenia37 and the development of intracranial aneurysms.38
The difficulty in identifying genetic modifiers of MS course and severity may reflect either the complexity or diversity of inflammatory and neurodegenerative mechanisms that contribute to the clinical expression or some inherent flaws in measuring relevant clinical endpoints. In addition, the advent of immunomodulatory treatments has proved to alter the course of the disease.39,40 Early initiation and timely escalation of disease-modifying drugs are known to prevent the accumulation of disability. Therefore, the inclusion in a genetic association study of patients with MS without any treatment along with those who may or may not receive the best medical treatment is an important confounding factor that may obscure the detection of significant genetic effects.
Our study included a clinically well-characterized and ethnically homogenous group. Disease severity in each individual was assessed by means of MSSS, a robust and validated measure, that shows stability over time. In addition, patients were only included if they had at least 5 years duration of the disease, as it is known that the first 5 years of the disease represent a significant outcome milestone of MS. Also, a number of meaningful clinical predictors that may have an effect on long-term severity were collected and included as covariates in the analysis. Moreover, genetic variability across the tested genes was covered by a large number of tagging SNPs. Finally, permutation analysis, a powerful unbiased test, was implemented to adjust for multiple testing of known and unknown factors.
In brief, our study provides evidence implicating variants in genes that encode adhesion molecules, responsible for lymphocyte trafficking and activation, in the severity of MS. Replication of these associations in other independent populations will render our findings more robust. Such biomarkers, if validated, may become valuable prognostic tools at an individual level at the time of diagnosis. Furthermore, therapies targeting adhesion molecules may be worth exploring in the future.
Supplementary Material
GLOSSARY
- DMT
disease-modifying therapy
- GWAS
genome-wide association study
- MAF
mean allele frequency
- PI
progression index
- SNP
single nucleotide polymorphism
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
Efthimios Dardiotis, Georgios M. Hadjigeorgiou, and Theodoros Kyriakides: original idea, study design, clinical management and cohort recruitment, interpretation of results, and drafting of the manuscript. Elena Panayiotou, Kyproula Christodoulou, and Andreas Hadjisavvas: experimental work. Antonios Provatas, Athanasios Lourbopoulos, Marios Pantzaris, and Nikolaos Grigoriadis: clinical management and cohort recruitment. Athos Antoniades: statistical analysis.
STUDY FUNDING
The study was supported by the Project YGEIA/BIOS/0609 (BIE/01), which is co-financed by the European Regional Development Fund and the Republic of Cyprus through the Research Promotion Foundation.
DISCLOSURE
E. Dardiotis, E. Panayiotou, and A. Provatas report no disclosures. K. Christodoulou received research support from Research Promotion Foundation (Cyprus), Telethon Grants (Cyprus), and Norwegian Financial Mechanism, EU. A. Hadjisavvas reports no disclosures. A. Antoniades is the CEO and Head of Research and Development of Stemble Ventures Ltd. A. Lourbopoulos reports no disclosures. M. Pantzaris received travel funding and/or speaker honoraria from Sanofi-Genzyme Greece, Genesis Pharma Greece. N. Grigoriadis served on the scientific advisory board for Biogen, Nayer, Novartis, Celgene, Genesis Pharma, Merck Serono, Sanofi-Aventis, Paradig MS, Navigating MS, and Helani and has consulted for Biogen, Bayer, Novartis, Celgene, Genesis Pharma, Merck Serono, and Sanofi-Aventis. G.M. Hadjigeorgiou served on the scientific advisory board for Genesis Pharma, Genzyme, Merck Serono, Bayer Hellas, and TEVA and received speaker honoraria from Genesis Pharma, Genzyme, Merck Serono, Bayer Hellas, and TEVA. T. Kyriakides reports no disclosures. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Compston A, Sadovnick AD. Epidemiology and genetics of multiple sclerosis. Curr Opin Neurol Neurosurg 1992;5:175–181. [PubMed] [Google Scholar]
- 2.Sokratous M, Dardiotis E, Tsouris Z, et al. . Deciphering the role of DNA methylation in multiple sclerosis: emerging issues. Auto Immun Highlights 2016;7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Efthymiou G, Dardiotis E, Liaskos C, et al. . Anti-hsp60 antibody responses based on Helicobacter pylori in patients with multiple sclerosis: (ir)relevance to disease pathogenesis. J Neuroimmunol 2016;298:19–23. [DOI] [PubMed] [Google Scholar]
- 4.Sawcer S, Franklin RJ, Ban M. Multiple sclerosis genetics. Lancet Neurol 2014;13:700–709. [DOI] [PubMed] [Google Scholar]
- 5.DeLuca GC, Ramagopalan SV, Herrera BM, et al. . An extremes of outcome strategy provides evidence that multiple sclerosis severity is determined by alleles at the HLA-DRB1 locus. Proc Natl Acad Sci USA 2007;104:20896–20901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gray OM, Abdeen H, McDonnell GV, Patterson CC, Graham CA, Hawkins SA. An investigation of susceptibility loci in benign, aggressive and primary progressive multiple sclerosis in Northern Irish population. Mult Scler 2009;15:299–303. [DOI] [PubMed] [Google Scholar]
- 7.Kroner A, Mehling M, Hemmer B, et al. . A PD-1 polymorphism is associated with disease progression in multiple sclerosis. Ann Neurol 2005;58:50–57. [DOI] [PubMed] [Google Scholar]
- 8.Baranzini SE, Wang J, Gibson RA, et al. . Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Hum Mol Genet 2009;18:767–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brynedal B, Wojcik J, Esposito F, et al. . MGAT5 alters the severity of multiple sclerosis. J Neuroimmunol 2010;220:120–124. [DOI] [PubMed] [Google Scholar]
- 10.International Multiple Sclerosis Genetics Consortium. Genome-wide association study of severity in multiple sclerosis. Genes Immun 2011;12:615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.George MF, Briggs FB, Shao X, et al. . Multiple sclerosis risk loci and disease severity in 7,125 individuals from 10 studies. Neurol Genet 2016;2:e87 doi: 10.1212/NXG.0000000000000087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Polman CH, O'Connor PW, Havrdova E, et al. . A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2006;354:899–910. [DOI] [PubMed] [Google Scholar]
- 13.Tsivgoulis G, Katsanos AH, Mavridis D, et al. . The efficacy of natalizumab versus fingolimod for patients with relapsing-remitting multiple sclerosis: a systematic review, indirect evidence from randomized placebo-controlled trials and meta-analysis of observational head-to-head trials. PLoS One 2016;11:e0163296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aggelakis K, Zacharaki F, Dardiotis E, et al. . Interleukin-1B and interleukin-1 receptor antagonist gene polymorphisms in Greek multiple sclerosis (MS) patients with bout-onset MS. Neurol Sci 2010;31:253–257. [DOI] [PubMed] [Google Scholar]
- 15.Hadjigeorgiou G, Dardiotis E, Tsivgoulis G, et al. . Correction: observational study assessing demographic, economic and clinical factors associated with access and utilization of health care services of patients with multiple sclerosis under treatment with interferon beta-1b (EXTAVIA). PLoS One 2015;10:e0140308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roxburgh RH, Seaman SR, Masterman T, et al. . Multiple Sclerosis Severity Score: using disability and disease duration to rate disease severity. Neurology 2005;64:1144–1151. [DOI] [PubMed] [Google Scholar]
- 17.Wigginton JE, Cutler DJ, Abecasis GR. A note on exact tests of Hardy-Weinberg equilibrium. Am J Hum Genet 2005;76:887–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramagopalan SV, Deluca GC, Degenhardt A, Ebers GC. The genetics of clinical outcome in multiple sclerosis. J Neuroimmunol 2008;201–202:183–199. [DOI] [PubMed] [Google Scholar]
- 19.Ramagopalan SV, Deluca GC, Morrison KM, et al. . Analysis of 45 candidate genes for disease modifying activity in multiple sclerosis. J Neurol 2008;255:1215–1219. [DOI] [PubMed] [Google Scholar]
- 20.Hensiek AE, Sawcer SJ, Feakes R, et al. . HLA-DR 15 is associated with female sex and younger age at diagnosis in multiple sclerosis. J Neurol Neurosurg Psychiatry 2002;72:184–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masterman T, Ligers A, Olsson T, Andersson M, Olerup O, Hillert J. HLA-DR15 is associated with lower age at onset in multiple sclerosis. Ann Neurol 2000;48:211–219. [PubMed] [Google Scholar]
- 22.International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium 2, Sawcer S, et al. . Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011;476:214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 2007;7:678–689. [DOI] [PubMed] [Google Scholar]
- 24.Rudick RA, Sandrock A. Natalizumab: alpha 4-integrin antagonist selective adhesion molecule inhibitors for MS. Expert Rev Neurother 2004;4:571–580. [DOI] [PubMed] [Google Scholar]
- 25.Durmanova V, Shawkatova I, Javor J, et al. . VLA4 gene polymorphism and susceptibility to multiple sclerosis in Slovaks. Folia Biol (Praha) 2015;61:8–13. [PubMed] [Google Scholar]
- 26.O'Doherty C, Roos IM, Antiguedad A, Aransay AM, Hillert J, Vandenbroeck K. ITGA4 polymorphisms and susceptibility to multiple sclerosis. J Neuroimmunol 2007;189:151–157. [DOI] [PubMed] [Google Scholar]
- 27.Andreoli V, Cittadella R, Valentino P, et al. . The role of VLA4 polymorphisms in multiple sclerosis: an association study. J Neuroimmunol 2007;189:125–128. [DOI] [PubMed] [Google Scholar]
- 28.Correia C, Coutinho AM, Almeida J, et al. . Association of the alpha4 integrin subunit gene (ITGA4) with autism. Am J Med Genet B Neuropsychiatr Genet 2009;150B:1147–1151. [DOI] [PubMed] [Google Scholar]
- 29.Rittling SR, Singh R. Osteopontin in immune-mediated diseases. J Dent Res 2015;94:1638–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chabas D, Baranzini SE, Mitchell D, et al. . The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science 2001;294:1731–1735. [DOI] [PubMed] [Google Scholar]
- 31.Chiocchetti A, Comi C, Indelicato M, et al. . Osteopontin gene haplotypes correlate with multiple sclerosis development and progression. J Neuroimmunol 2005;163:172–178. [DOI] [PubMed] [Google Scholar]
- 32.Niino M, Kikuchi S, Fukazawa T, Yabe I, Tashiro K. Genetic polymorphisms of osteopontin in association with multiple sclerosis in Japanese patients. J Neuroimmunol 2003;136:125–129. [DOI] [PubMed] [Google Scholar]
- 33.Caillier S, Barcellos LF, Baranzini SE, et al. . Osteopontin polymorphisms and disease course in multiple sclerosis. Genes Immun 2003;4:312–315. [DOI] [PubMed] [Google Scholar]
- 34.Comi C, Cappellano G, Chiocchetti A, et al. . The impact of osteopontin gene variations on multiple sclerosis development and progression. Clin Dev Immunol 2012;2012:212893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hensiek AE, Roxburgh R, Meranian M, et al. . Osteopontin gene and clinical severity of multiple sclerosis. J Neurol 2003;250:943–947. [DOI] [PubMed] [Google Scholar]
- 36.Goossens K, Van Soom A, Van Zeveren A, Favoreel H, Peelman LJ. Quantification of fibronectin 1 (FN1) splice variants, including two novel ones, and analysis of integrins as candidate FN1 receptors in bovine preimplantation embryos. BMC Dev Biol 2009;9:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakata K, Ujike H, Sakai A, et al. . Association study between the fibronectin gene and schizophrenia. Am J Med Genet B Neuropsychiatr Genet 2003;116B:41–44. [DOI] [PubMed] [Google Scholar]
- 38.Ruigrok YM, Rinkel GJ, van't Slot R, Wolfs M, Tang S, Wijmenga C. Evidence in favor of the contribution of genes involved in the maintenance of the extracellular matrix of the arterial wall to the development of intracranial aneurysms. Hum Mol Genet 2006;15:3361–3368. [DOI] [PubMed] [Google Scholar]
- 39.Tsivgoulis G, Katsanos AH, Grigoriadis N, et al. . The effect of disease modifying therapies on disease progression in patients with relapsing-remitting multiple sclerosis: a systematic review and meta-analysis. PLoS One 2015;10:e0144538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsivgoulis G, Katsanos AH, Grigoriadis N, et al. . The effect of disease-modifying therapies on brain atrophy in patients with clinically isolated syndrome: a systematic review and meta-analysis. Ther Adv Neurol Disord 2015;8:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.