

Abstract
Objective:
Dravet syndrome is a rare neurodevelopmental disorder characterized by seizures and other neurologic problems. SCN1A mutations account for ∼80% of cases. Animal studies have implicated mutation-related dysregulated cortical inhibitory networks in its pathophysiology. We investigated such networks in people with the condition.
Methods:
Transcranial magnetic stimulation using single and paired pulse paradigms was applied to people with Dravet syndrome and to 2 control groups to study motor cortex excitability.
Results:
Short interval intracortical inhibition (SICI), which measures GABAergic inhibitory network behavior, was undetectable in Dravet syndrome, but detectable in all controls. Other paradigms, including those testing excitatory networks, showed no difference between Dravet and control groups.
Conclusions:
There were marked differences in inhibitory networks, detected using SICI paradigms, while other inhibitory and excitatory paradigms yielded normal results. These human data showing reduced GABAergic inhibition in vivo in people with Dravet syndrome support established animal models.
Dravet syndrome (DS; OMIM #607208) is an epileptic encephalopathy, characterized by seizures that are often resistant to treatment, and onset is typically with complex febrile seizures, usually in the first year of life. Substantial developmental delay is a common feature; further problems, such as gait disorder, feeding or appetite difficulties, behavioral problems, and sleep disturbance, also occur. Development is normal prior to seizure onset.1,2 Structural brain imaging is usually normal, but various structural abnormalities have been described, including focal brain atrophy, cortical dysplasia, and hippocampal sclerosis.3 EEG may be normal at seizure onset, but later typically shows multifocal epileptiform abnormalities and a disturbed background rhythm.4
Mutations in SCN1A, encoding the α subunit of a voltage-gated sodium channel (Nav1.1), have been found in >80% of affected individuals; the majority of these mutations arise de novo,5 and the SCN1A variants include both missense and truncating mutations in addition to whole gene deletion, suggesting a loss of function mechanism.6
The mechanisms by which SCN1A mutations cause the features of DS have been investigated using knockout mouse models (Scn1a+/− and Scn1a−/−). Homozygous Scn1a−/− mice had seizures and ataxia, leading to death by day 15, while heterozygous Scn1a+/− mice showed spontaneous seizures, but some survived past 15 weeks.7 Whole-cell sodium current recordings from mouse neurons suggest that DS-causing mutations do not affect sodium currents in excitatory pyramidal cells, a finding that may be explained by upregulation of other sodium channels, such as Nav1.3. Recordings from GABAergic inhibitory interneurons using the same mouse models show reduced sodium currents.7,8 This leads to an increased threshold for action potential generation in affected inhibitory cells.6 It has been suggested that DS-causing SCN1A mutations reduce the activity of cortical inhibitory networks, with the resultant imbalance of neuronal activity leading to seizures. Apart from seizures, Scn1a+/− mice exhibit behavioral problems such as hyperactivity, cognitive difficulties, and impaired social interaction. These may be attributable to reduced GABAergic inhibition in frontal cortical networks.9 A recent in vivo study of inhibitory interneurons in the same mouse model of DS, however, demonstrated normal spontaneous firing patterns, suggesting that the picture may be more complex.10
While GABAergic inhibition has been well-studied in mouse models of DS, little has been done to understand how inhibitory networks are affected in humans with DS. An EEG-fMRI study of 10 people with DS could not identify a common network underlying epileptiform discharges.11
In the current study, transcranial magnetic stimulation (TMS) was used to probe cortical excitability in people with DS. TMS can be used to stimulate motor cortex noninvasively, triggering a motor evoked potential (MEP) in a target muscle. The resting motor threshold (rMT) measures the ease of activation of motor cortex via excitatory networks, while paired pulse paradigms with variable interstimulus intervals test inhibitory and excitatory intracortical circuitry.
We hypothesized that GABAergic inhibition would be reduced in people with DS compared to control groups. GABAergic inhibition can be measured using inhibitory paired pulse paradigms such as short interval intracortical inhibition (SICI) and long interval intracortical inhibition (LICI).12 We also hypothesized that rMT and intracortical facilitation (ICF), which are less influenced by GABAergic inhibition, would be normal in DS.
METHODS
Standard protocol approvals, registrations, and patient consents.
The project was approved by the local ethical review board (National Research Ethics Service Committee London: Camden and Islington); TMS parameters were within current safety guidelines.13 A safety questionnaire was completed for each participant. Participants gave written informed consent. In those lacking capacity, a relative was asked to give assent.
People with DS were identified using an existing clinical registry. Inclusion criteria for people with DS were as follows: clinical criteria for diagnosing DS (normal developmental skills before onset; generalized, or unilateral, or alternating unilateral hemiclonic, febrile, and afebrile seizures beginning in the first year of life, with subsequent myoclonic and generalized tonic-clonic seizures and, sometimes, partial seizures; absence of epileptiform EEG discharges in the initial EEG studies, if available, with later generalized spike-wave and polyspike-wave discharges, focal abnormalities, and possible early photosensitivity; evidence of delayed development from the second year of life onward1,14) and the presence of an SCN1A mutation confirmed in a clinically accredited laboratory.
Six people with DS were recruited. Five had full TMS testing and 1 only tolerated part of the protocol. Control data were gathered from 10 people with non-DS epilepsy taking antiepileptic medication and 10 healthy participants.
The technical details of the hardware and software used in this study have been published, as have the techniques used to identify the motor hot spot for the adductor pollicis brevis (APB) muscle.15
The following paradigms were tested: rMT, SICI, ICF, and LICI. Exact definitions and experimental details of the paradigms used have been published.15 The following interstimulus intervals were used: for SICI, 2 and 5 ms; for ICF, 10 and 15 ms; for LICI, 100, 150, 200, and 250 ms.
rMT is a measure of motor cortex excitability, with high rMT implying low excitability.12 SICI is mediated by GABAAergic intracortical circuits16 while ICF is mediated by glutamatergic intracortical circuits, possibly alongside a reduction in GABAergic inhibition.12 LICI is mediated by GABABergic intracortical circuits.12
Statistical analysis.
rMT for cases and the 2 control groups was compared using univariate analysis of variance (ANOVA). SICI, ICF, and LICI results for cases and controls were compared using multivariate ANOVA for each paradigm. p Values of 0.05 or lower were interpreted as statistically significant. Post hoc analysis was performed using Tukey honest significant difference (HSD) test. All analysis was performed using IBM (Armonk, NY) SPSS Statistics, version 22.0.
RESULTS
Participants.
Six adults with typical DS were recruited (D1–D6); 5 completed all TMS paradigms, while the 6th subject was unwilling to continue after initial measurement of rMT. All participants with DS had a known mutation in SCN1A (table 1). Control participants included 10 consecutive people with treated non-DS epilepsy (E1–E10) and 10 healthy controls (H1–H10). Our healthy control data have been reported.15
Table 1.
Age, sex, mutation (patients with Dravet syndrome only), epilepsy diagnosis (epilepsy controls only), transcranial magnetic stimulation paradigms tested, medication at time of testing, and seizure frequency for all participants
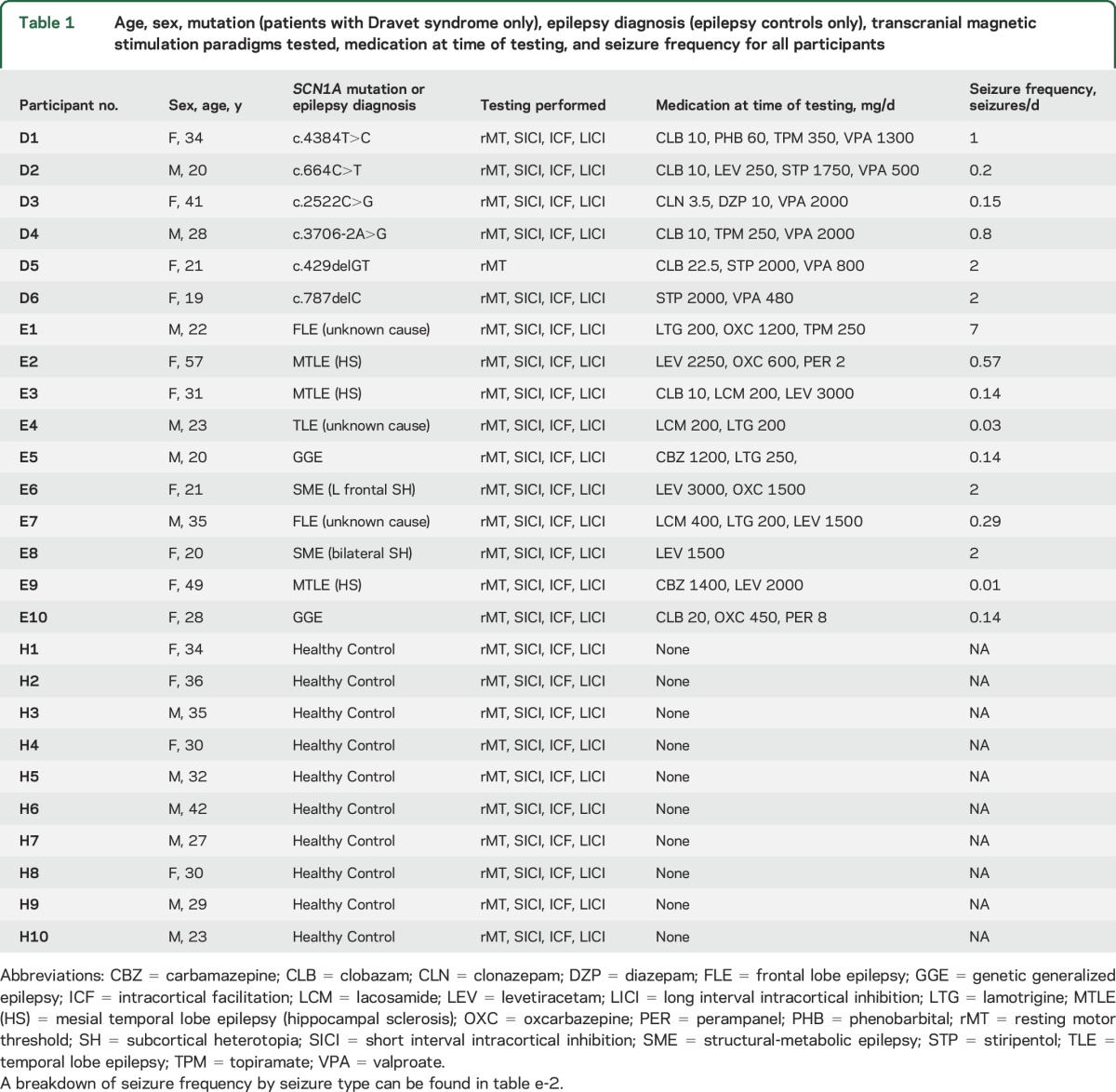
Table 1 shows demographic information, genetic diagnosis (for those with DS), epilepsy diagnosis (for epilepsy controls), medication at time of testing, and seizure frequency. There were no significant differences in age or sex among the 3 groups (age: univariate ANOVA p = 0.61; sex: Fisher exact test p = 0.68).
Medication and seizure frequency.
Those with DS were taking a mean of 3.2 medications each, while the epilepsy controls were taking a mean of 2.5 medications each (table 1); there was no significant difference (unpaired, 2-tailed t test, p = 0.14). Drugs used in the 2 groups do not match exactly; 3/6 with DS were taking stiripentol, which is rarely used outside of DS, while some epilepsy controls were taking drugs such as carbamazepine and lamotrigine, which are typically avoided in DS. Mean reported seizure frequency in the DS group was 1.3 seizures per day, while epilepsy controls reported a mean of 1.0 seizure per day; there was no significant difference (unpaired, 2-tailed t test, p = 0.76). Healthy controls were taking no medication, and reported no seizures.
Resting motor threshold.
rMT was measured in 6 people with DS. Average rMT was 74.7% of maximum machine output, SD 10.8%, range 55%–85% (figure 1). This average and range is similar to that seen in epilepsy controls (75.5%, SD 12.9%, range 59%–91%; unpaired, 2-tailed t test p = 0.90), implying similar baseline cortical excitability in the groups. Meaningful comparison between epilepsy groups and healthy controls is difficult, since antiepileptic medication is known to affect rMT. A univariate ANOVA comparing rMT in all 3 groups showed significant differences (p = 0.03); post hoc analysis confirmed a significant difference only between epilepsy controls and healthy controls (Tukey HSD test; p = 0.04).
Figure 1. Resting motor threshold (rMT) in all groups.
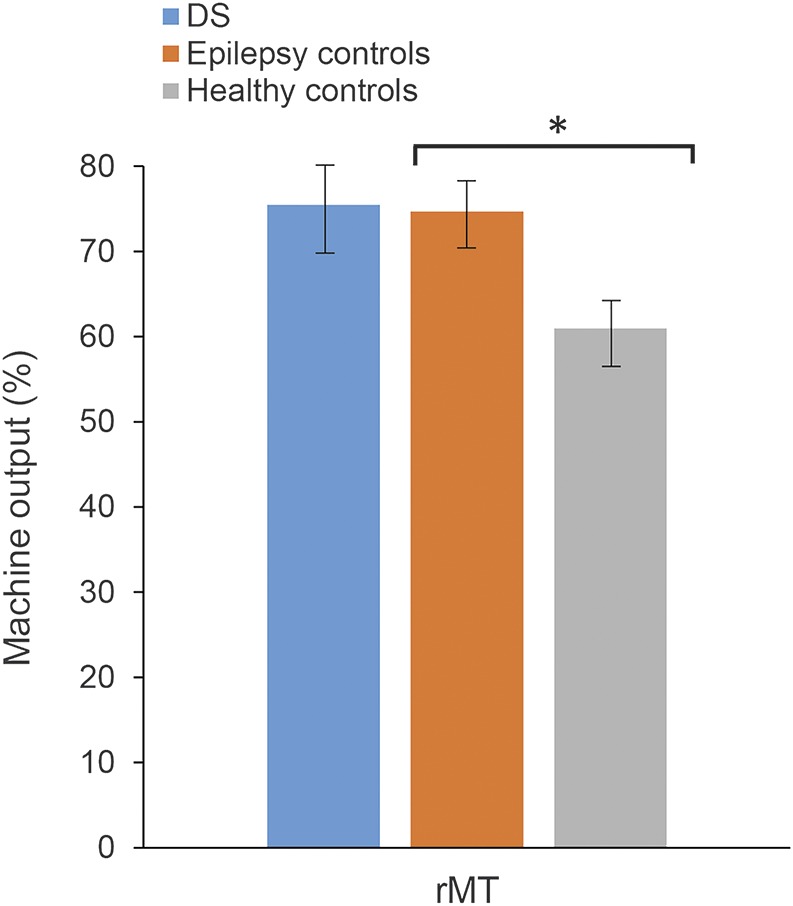
Mean results for rMT for people with Dravet syndrome (DS), epilepsy controls, and healthy controls. There was a significant difference (indicated by *) between healthy controls and epilepsy controls (univariate analysis of variance and post hoc Tukey honest significant difference test; p = 0.04), but no significant difference between DS and either control group. Error bars show standard error.
Short interval intracortical inhibition.
SICI paradigms using ISI of 2 and 5 ms did not cause inhibition in our DS group. Instead, there was significant facilitation, with mean MEP responses of 2.18 (SD 0.47) at 2 ms and 1.74 (SD 1.03) at 5 ms normalized to unconditioned responses. By contrast, all 20 controls, including those with and without epilepsy, showed inhibition at an ISI of 2 ms (mean normalized response 0.44, SD 0.15), and most also showed inhibition at an ISI of 5 ms (mean normalized response 0.86, SD 0.32). Data for SICI across DS and control groups are shown in figure 2. A multivariate ANOVA comparing SICI at both ISIs in DS to all controls showed a significant difference (p < 0.0001). Post hoc analysis with adjustment for multiple comparisons (Tukey HSD test) confirmed a significant difference between DS and each control group at both ISIs (2 ms: DS vs epilepsy controls, p < 0.0001; DS vs healthy controls, p < 0.0001; 5 ms: DS vs epilepsy controls, p = 0.02; DS vs healthy controls, p = 0.01). There was no significant difference between non-DS epilepsy controls and healthy controls at either ISI (2 ms, p = 0.38; 5 ms, p = 0.92).
Figure 2. Short interval intracortical inhibition (SICI), intracortical facilitation (ICF), and long interval intracortical inhibition (LICI) in all groups.

Mean results for SICI, ICF, and LICI for people with Dravet syndrome (DS), epilepsy controls, and healthy controls. There was a significant difference (indicated by *) between DS and both control groups in SICI only. Error bars show standard error. MEP = motor evoked potential.
There was no significant difference in ICF or LICI between any group (figure 2) (multivariate ANOVA, ICF p = 0.58, LICI p = 0.65).
DISCUSSION
Our key finding is the lack of GABA-mediated SICI in people with DS, consistent with our main hypothesis. There was no inhibition at 2 ms ISI in any people with DS, while inhibition was detected in all 20 controls; the difference was significant. There was also significantly less inhibition in the DS group at 5 ms ISI.
Instead of the expected normal inhibition of the MEP at ISIs of 2 and 5 ms, the DS group showed facilitation. In healthy participants, a subthreshold conditioning stimulus causes some activity in both excitatory intracortical and inhibitory intracortical neurons. In controls, as in the extensive TMS literature on paired pulse stimulation, the inhibitory effects were dominant at shorter ISIs (SICI) while the excitatory effects were dominant at longer ISIs (intracortical facilitation). In our DS group, the excitatory effects appeared to be dominant at shorter ISIs. This may be because the subthreshold conditioning stimulus failed to activate a sufficient number of inhibitory interneurons to trigger detectable inhibition.
LICI, which is believed to be mediated by the same GABAergic interneurons, was robustly detectable in all groups, with no difference detected between DS and controls.
Aside from a different ISI, an obvious difference between SICI and LICI paradigms is that the conditioning pulse in SICI is given at subthreshold intensity, while that in LICI is given at suprathreshold intensity.
There is a striking parallel between our findings in people with DS and the data from mouse models of the condition. Scn1a+/− mouse inhibitory interneurons showed a reduction in sodium currents, leading to an increased threshold for action potential generation in these cells6–8; subthreshold conditioning stimuli in our SICI paradigm failed to generate any detectable inhibition on subsequent MEPs in people with DS. Suprathreshold stimuli were still able to generate action potentials in Scn1a+/− mouse inhibitory interneurons; the suprathreshold conditioning stimuli in LICI triggered an inhibitory effect on the MEP similar to that seen in controls.
The presence of normal LICI suggests that inhibitory cortical networks are functionally intact in people with DS, as indeed they are in the Scn1a+/− mouse.10 The lack of SICI, however, suggests that these inhibitory networks may show reduced sensitivity.
It is worth noting that a TMS impulse activates axons directly, while physiologic activation of the same neurons would occur synaptically. The spreading depolarization ahead of a synaptically triggered action potential is much larger than needed to discharge the membrane and therefore might be relatively insensitive to small changes in membrane threshold. Thus, the relative insensitivity of inhibitory neurons to subthreshold magnetic stimuli in DS may not imply an equally significant reduction in the physiologic firing of these neurons. A pattern of reduced sodium channel function but relatively preserved spontaneous firing patterns has been shown in animal models.7,10
The fact that our patients with DS were all on antiepileptic drug (AED) treatment implies that the therapies used did not fully neutralize the pathologic effects of the SCN1A mutation. This is consistent with their clinical presentation; none of our DS group members was seizure-free.
There are other potential explanations for the lack of SICI and the presence of LICI in DS. SICI is believed to be mediated via GABAA receptors on corticospinal neurons, while LICI is mediated via GABAB receptors.16 It is possible that expression or activity of these receptors might be affected unequally in DS, such that GABAB-mediated inhibition functions normally and GABAA-mediated inhibition does not. Another possibility is that the longer ISIs in LICI paradigms are important; for example, the SCN1A mutation might delay release of GABA from the inhibitory cortical neurons, thus preventing inhibition at the shorter ISIs of SICI. A further explanation would be that the mechanisms controlling SICI are normal in people with DS, but the inhibition is not detectable as the SCN1A mutation leads to extra facilitation not found in controls. Facilitation can be found at short ISIs in healthy people, a process termed short ICF, but this is typically triggered by a suprathreshold conditioning stimulus.17 ICF uses a subthreshold stimulus, and was similar in DS and controls in our study. None of these alternative explanations is supported by the animal data.
rMT was lower in healthy controls compared to either the DS group or epilepsy controls, although the comparison between DS and healthy controls did not reach statistical significance. The difference is most likely due to effects of the AEDs used by our DS and epilepsy populations. There was no difference in rMT between the DS group and epilepsy controls, who were on a similar number of medications. Likewise, ICF was similar in DS and controls. These findings are in keeping with our hypothesis that DS would not affect rMT or ICF.
AEDs are known to modify TMS results, with different drugs affecting different measures. Our inclusion of a control group with epilepsy is helpful in this regard, as it demonstrates that SICI is robustly detectable in people with epilepsy of various types despite long-term AED treatment, which aids a meaningful comparison between cases and controls. It was not possible to match exactly the AED exposure of our patients with DS and epilepsy controls. Sodium channel-blocking AEDs, including commonly used AEDs like lamotrigine and carbamazepine, may exacerbate DS; they are typically avoided in this population. Conversely, stiripentol is in common use in DS, but is rarely used to treat other types of epilepsy. Given the impossibility of matching drugs exactly, we instead compared the number of AEDs. Moreover, although sodium channel blocking AEDs are widely recognized to increase rMT, they do not affect SICI.18 We consider that disparities between drug types cannot explain the unusual pattern of SICI in our DS group.
People with DS commonly have substantial learning difficulties and may have other behavioral problems; performing TMS can be challenging in this population. However, this seems an unlikely explanation for the robust differences seen in SICI between our DS and control groups, since other TMS paired pulse measurements (ICF and LICI) were no different vs controls. We have previously reported TMS results in people with alternating hemiplegia of childhood, a neurodevelopmental disorder associated with learning and behavioral difficulties of comparable severity to DS; this group had normal SICI,15 supporting the specificity of our results for DS.
Our DS group is smaller than our control groups. DS is a rare condition, and many people with DS are not able to undergo TMS testing, due to learning and behavioral difficulties. People with a more severe phenotype were not approached for this study, since it was believed unlikely that they would tolerate testing; this is a potential bias (table e-1 at Neurology.org). All our participants are adults, in whom the electrophysiologic profile may differ from that in children.19 Only small numbers of people with DS could be recruited. The differences in SICI between the DS and control groups are statistically significant, but need cautious interpretation and replication.
We used TMS to study motor cortex excitatory and inhibitory networks in people with DS. There were clear abnormalities in inhibitory networks, detected using SICI paradigms, while other inhibitory and excitatory paradigms yielded normal results. These human data show reduced GABAergic inhibition in vivo in DS, supporting established animal models. The specific absence of SICI, in the presence of normal LICI, suggests that inhibitory networks in people with DS are functionally intact, but have a reduced sensitivity to low-intensity stimuli. TMS measures may provide rapid-readout biomarkers for cortical excitability and its modulation by treatment in DS.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the participants and their families.
GLOSSARY
- AED
antiepileptic drug
- ANOVA
analysis of variance
- APB
adductor pollicis brevis
- DS
Dravet syndrome
- HSD
honest significant difference
- ICF
intracortical facilitation
- LICI
long interval intracortical inhibition
- MEP
motor evoked potential
- rMT
resting motor threshold
- SICI
short interval intracortical inhibition
- TMS
transcranial magnetic stimulation
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
W.M.S. designed and carried out the experiments, wrote the first draft of the manuscript, and performed the statistical analysis. J.W.S. provided supervision for the experiments and contributed to the manuscript. J.C.R. provided technical expertise on the experimental techniques and contributed to the manuscript. S.M.S. provided supervision for the experiments and contributed to the manuscript.
STUDY FUNDING
This work was undertaken at University College London Hospitals/University College London, which received a proportion of funding from the Department of Health's National Institute for Health Research Biomedical Research Centres funding scheme. This study was funded by the Epilepsy Society. The Article Processing Charge was paid by Research Councils UK (BBSRC, grant no. BB/N016793/1).
DISCLOSURE
W. Stern reports no disclosures relevant to this manuscript. J. Sander has received research grants and honoraria from UCB, Eisai, Teva, Lundbeck, and GSK, which are involved in the manufacturing of AEDs. J. Rothwell reports no disclosures. S. Sisodiya has received institutional research grants or honoraria from UCB, Eisai, GSK, Vitaflo, and Nutricia. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Dravet C. The core Dravet syndrome phenotype. Epilepsia 2011;52:3–9. [DOI] [PubMed] [Google Scholar]
- 2.Hurst DL. Epidemiology of severe myoclonic epilepsy of infancy. Epilepsia 1990;31:397–400. [DOI] [PubMed] [Google Scholar]
- 3.Guerrini R, Striano P, Catarino C, Sisodiya SM. Neuroimaging and neuropathology of Dravet syndrome. Epilepsia 2011;52:30–34. [DOI] [PubMed] [Google Scholar]
- 4.Bureau M, Bernardina BD. Electroencephalographic characteristics of Dravet syndrome. Epilepsia 2011;52:13–23. [DOI] [PubMed] [Google Scholar]
- 5.Marini C, Scheffer IE, Nabbout R, et al. The genetics of Dravet syndrome. Epilepsia 2011;52:24–29. [DOI] [PubMed] [Google Scholar]
- 6.Rubinstein M, Han S, Tai C, et al. Dissecting the phenotypes of Dravet syndrome by gene deletion. Brain J Neurol 2015;138:2219–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu FH, Mantegazza M, Westenbroek RE, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci 2006;9:1142–1149. [DOI] [PubMed] [Google Scholar]
- 8.Ogiwara I, Miyamoto H, Morita N, et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci 2007;27:5903–5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han S, Tai C, Westenbroek RE, et al. Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature 2012;489:385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stasi AMD, Farisello P, Marcon I, et al. Unaltered network activity and interneuronal firing during spontaneous cortical dynamics in vivo in a mouse model of severe myoclonic epilepsy of infancy. Cereb Cortex 2016;26:1778–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moehring J, von Spiczak S, Moeller F, et al. Variability of EEG-fMRI findings in patients with SCN1A-positive Dravet syndrome. Epilepsia 2013;54:918–926. [DOI] [PubMed] [Google Scholar]
- 12.Terao Y, Ugawa Y. Basic mechanisms of TMS. J Clin Neurophysiol 2002;19:322–343. [DOI] [PubMed] [Google Scholar]
- 13.Rossi S, Hallett M, Rossini PM, Pascual-Leone A. Safety, ethical considerations, and application guidelines for the use of transcranial magnetic stimulation in clinical practice and research. Clin Neurophysiol 2009;120:2008–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guerrini R, Oguni H. Borderline Dravet syndrome: a useful diagnostic category? Epilepsia 2011;52(suppl 2):10–12. [DOI] [PubMed] [Google Scholar]
- 15.Stern WM, Desikan M, Hoad D, et al. Spontaneously fluctuating motor cortex excitability in alternating hemiplegia of childhood: a transcranial magnetic stimulation study. PLoS One 2016;11:e0151667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rothwell JC, Day BL, Thompson PD, Kujirai T. Short latency intracortical inhibition: one of the most popular tools in human motor neurophysiology. J Physiol 2009;587:11–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rothwell JC. Paired-pulse investigations of short-latency intracortical facilitation using TMS in humans. Electroencephalogr Clin Neurophysiol Suppl 1999;51:113–119. [PubMed] [Google Scholar]
- 18.Ziemann U. TMS and drugs. Clin Neurophysiol 2004;115:1717–1729. [DOI] [PubMed] [Google Scholar]
- 19.Kim SH, Nordli DR, Berg AT, Koh S, Laux L. Ictal ontogeny in Dravet syndrome. Clin Neurophysiol 2015;126:446–455. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.