

Abstract
Objective:
To investigate whether cortical superficial siderosis (cSS) is associated with increased risk of future first-ever symptomatic lobar intracerebral hemorrhage (ICH) in patients with cerebral amyloid angiopathy (CAA) presenting with neurologic symptoms and without ICH.
Methods:
Consecutive patients meeting modified Boston criteria for probable CAA in the absence of ICH from a single-center cohort were analyzed. cSS and other small vessel disease MRI markers were assessed according to recent consensus recommendations. Patients were followed prospectively for future incident symptomatic lobar ICH. Prespecified Cox proportional hazard models were used to investigate cSS and first-ever lobar ICH risk adjusting for potential confounders.
Results:
The cohort included 236 patients with probable CAA without lobar ICH at baseline. cSS prevalence was 34%. During a median follow-up of 3.26 years (interquartile range 1.42–5.50 years), 27 of 236 patients (11.4%) experienced a first-ever symptomatic lobar ICH. cSS was a predictor of time until first ICH (p = 0.0007, log-rank test). The risk of symptomatic ICH at 5 years of follow-up was 19% (95% confidence interval [CI] 11%–32%) for patients with cSS at baseline vs 6% (95% CI 3%–12%) for patients without cSS. In multivariable Cox regression models, cSS presence was the only independent predictor of increased symptomatic ICH risk during follow-up (HR 4.04; 95% CI 1.73–9.44, p = 0.001), after adjusting for age, lobar cerebral microbleeds burden, and white matter hyperintensities.
Conclusions:
cSS is consistently associated with an increased risk of future lobar ICH in CAA with potentially important clinical implications for patient care decisions such as antithrombotic use.
Cortical superficial siderosis (cSS) on T2*-weighted gradient recalled echo (T2*-GRE) or susceptibility-weighted imaging (SWI) is a strong hemorrhagic signature of cerebral amyloid angiopathy (CAA)1—a common small vessel disease characterized by cerebrovascular amyloid deposition affecting superficial cortical microvascular networks, leading to spontaneous lobar intracerebral hemorrhage (ICH). cSS results from bleeding episodes within or adjacent to cortical sulci, presumably from amyloid-laden superficial cortical and leptomeningeal arterioles. cSS is a common manifestation of CAA, being found in 40%–60% of patients, and has distinct clinical and prognostic implications in this setting.1
cSS increases the sensitivity of the Boston criteria for clinical–radiologic CAA diagnosis and is associated with characteristic clinical symptoms, including transient focal neurologic episodes (amyloid spells).2 In CAA cohorts with ICH, cSS was demonstrated to be the most important prognostic risk factor for future recurrent ICH.3,4 In these studies, cSS remained the main driver of ICH recurrence in CAA after adjusting for the presence of multiple lobar cerebral microbleeds (CMBs) (a putative hemorrhagic marker of CAA5,6 previously shown to influence ICH risk)7 and white matter hyperintensities (WMHs) (another small vessel damage biomarker).
Lobar ICH is the most common symptomatic CAA presentation and lobar ICH survivors are at high risk of a recurrent event (up to 10%–15% per year). CAA is now often diagnosed on MRI in the setting of isolated lobar CMBs or cSS in patients with non-ICH neurologic presentations in stroke or memory clinics.5,8 However, little is known about the effects of cSS on the risk of future first-ever lobar ICH in patients with CAA presenting with neurologic symptoms and without lobar ICH. Small case series indicate that in these patients with CAA, cSS might still be a warning sign for subsequent lobar ICH,9,10 but data from large cohort studies with sufficient power are lacking.
We tested the hypothesis that in patients with CAA presenting with neurologic symptoms without lobar ICH, cSS is associated with an increased risk of future incident (first-ever) symptomatic lobar ICH in a large cohort study.
METHODS
Study population and patient selection.
We analyzed prospectively collected data from consecutive patients meeting modified Boston criteria for probable CAA in the absence of ICH (symptomatic or asymptomatic) seen by the Massachusetts General Hospital Stroke Service (including stroke unit and outpatient clinics) or Memory Disorders Unit (March 2000–November 2015). Detailed inclusion criteria included (1) diagnosis of probable CAA by modified Boston criteria; (2) clinical presentation other than hemorrhagic (or ischemic) stroke; and (3) available MRI sequences, including T2*-weighted/SWI, T2-weighted, fluid-attenuated inversion recovery (FLAIR) sequences, and diffusion-weighted imaging. Patients without ICH but with strictly deep CMBs, mixed (deep and lobar) and cerebellar CMBs, or single strictly lobar CMBs were also excluded.
Full medical history, including demographic and clinical information, was obtained at presentation using standardized data collection forms. Baseline neurologic examination and symptoms were recorded prospectively. Diagnosis of mild cognitive impairment and dementia at presentation was determined based on the clinical assessment of daily living function status in line with the recommendations from the National Institute on Aging and Alzheimer's Association workgroup.11,12
Two fellows independently reviewed and classified baseline clinical presentations based on all available information as follows: transient focal neurologic episodes (TFNEs) (episodes of positive or negative neurologic symptoms lasting for minutes with subsequent complete resolution, without a cause other than CAA after adequate evaluation, including angiography studies and carotid imaging, as previously suggested),13,14 cognitive complaints, or nonfocal neurologic symptoms.
Standard protocol approvals, registrations, and patient consents.
This study was performed in accordance with the guidelines and with approval of the institutional review boards at our institution.
Neuroimaging data acquisition and analysis.
Images were obtained using a 1.5T MRI scanner and included whole brain T2-weighted, T2*-GRE (echo time [TE] 750/50 ms, 5 mm slice thickness, 1 mm interslice gap), and FLAIR (repetition time [TR]/TE 10,000/140 ms, inversion time 2,200 ms, 1 number of excitations, 5 mm slice thickness, 1 mm interslice gap). For a subset of patients (n = 77), the T2*-weighted MRI sequences included SWI, TR/TE 27/20 ms, 1.5 mm slice thickness. In the rare scenario of a patient having both T2*-GRE and SWI available, we reviewed the SWI. All MRI were reviewed blinded to clinical and follow-up data by 2 trained observers, according to Standards for Reporting Vascular Changes on Neuroimaging (STRIVE).15
CMB presence and number were evaluated on axial T2*-GRE or SWI images using current consensus criteria.16,17 cSS was defined and jointly assessed by 2 trained raters in line with recent consensus recommendations (interrater κ = 0.87).1 cSS was defined as curvilinear hypointensities following the cortical surface distinct from vessels, and was assessed on axial T2*-weighted sequences according to a validated scale: absent, focal (restricted to ≤3 sulci), or disseminated (affecting 4 or more sulci).18,19 cSS ratings were performed independently of the rating of other imaging markers.
Enlarged perivascular spaces (EPVS) were assessed on axial T2-weighted MRI separately in the basal ganglia and centrum semiovale (CSO), using a validated 4-point visual rating scale (0 = no EPVS, 1 = <10 EPVS, 2 = 11–20 EPVS, 3 = 21–40 EPVS and 4 = >40 EPVS).20 We prespecified a dichotomized classification of EPVS degree as high (score >2) or low (score ≤2) in line with previous studies.
WMH volumes were calculated on axial FLAIR sequences with a previously described semi-automated planimetric method using MRICron software.21 WMH were also classified using the 0–3 Fazekas scale.22 The antero-posterior ratio of WMH lesion distribution was computed using a validated approach,23 in which a lower score reflects more posteriorly distributed WMH lesions.23 Lacunes were defined according to STRIVE criteria.15
Follow-up data.
Prospective follow-up (including follow-up phone calls at 3 months after enrollment and every 6 months thereafter) was supplemented by a comprehensive systematic chart review of all available information (including discharge summaries, follow-up outpatient and general practitioner letters, and death certificates), using standardized data collection forms. We collected information on clinically symptomatic ICH, defined as a symptomatic stroke syndrome associated with neuroimaging evidence of a corresponding ICH. The anatomical location of ICH was defined as lobar if the hematoma was in the cerebral cortex or at the junction of the cortex and white matter (including subcortical white matter). Outcome events were assessed using all clinical and radiologic information available, blinded to the presence of cSS at baseline MRI or other imaging findings. All patients were followed from their date of baseline presentation until the occurrence of ICH, death, or the end of follow-up.
Statistics.
Baseline demographic, clinical, and neuroimaging characteristics of patients with probable CAA with vs without incident ICH during follow-up were compared in univariate analyses, using 2-sample t test, Wilcoxon rank sum, Pearson χ2, and Fisher exact tests, as appropriate.
We determined the presence of cSS as a univariable predictor of incident lobar ICH risk using the Kaplan-Meier plot with significance testing by the log-rank test. Survival time was calculated from date of baseline MRI scan until the date of lobar ICH at follow-up or the last known date without the outcome event of interest. For individuals experiencing multiple lobar ICHs during follow-up, data were censored at the time of first ICH. Data were also censored at the time of death from causes other than documented symptomatic ICH. Prespecified Cox proportional hazards regression analyses were performed to calculate the multivariable hazard ratio (HR) of cSS presence (and burden, i.e., focal and disseminated) in relation to first-ever CAA-related lobar ICH. In these models, we included biologically plausible potential predictors identified in previous studies for recurrent CAA-related lobar ICH, including age, lobar CMBs, and WMH burden.7,24 Other covariates demonstrating a univariable association with the outcome in Cox regression analysis (p < 0.1) were also considered for inclusion. The main full model included cSS presence, age at MRI (dichotomized by the median age of the cohort at 80 years), presence of >5 lobar CMBs, and severe (Fazekas 5–6) WMH. In a sensitivity analysis multivariable Cox regression model, we included cSS presence and continuous covariates (age, log-transformed lobar CMB number, and log-transformed WMH volume to assure normal distributions) to assess any potential dose-effect relationship between these covariates and the outcome. In further sensitivity analyses, we also adjusted for blood-sensitive MRI sequence parameters (SWI vs T2*-GRE). The proportional hazard assumption in unadjusted and adjusted models was tested using graphical checks and Schoenfeld residuals-based tests.
All tests of significance were 2-tailed and significance level was set at 0.05 for all analyses. Stata software (version 11.2, StataCorp., College Station, TX) was used for all analyses. The manuscript was prepared with reference to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guidelines.25
RESULTS
A total of 260 potentially eligible patients with probable CAA according to the Boston criteria were screened for this analysis. A flowchart of patient selection is provided in figure 1. Reliable follow-up data were available for 237 patients. Patients without follow-up information were not different from patients with CAA included in the longitudinal analysis in baseline clinical characteristics or imaging markers of the disease (all p > 0.05, data not shown). One patient was excluded post hoc because of a deep ICH during follow-up, leaving 236 patients for our analysis. Of these, 51 (22%) presented with TFNEs, 163 (68%) cognitive complaints, and 23 (10%) other neurologic symptoms prompting the baseline MRI investigation. Among these patients, 75 (32%) and 161 (68%) were referred to the Stroke Service and Memory Clinics, respectively. Eighty-one patients (34%) had cSS at baseline. cSS presence was associated with clinical presentation with TFNEs (odds ratio [OR] 2.74, 95% confidence interval [CI] 1.55–4.85; p = 0.001).
Figure 1. Flowchart of patient selection.

CAA = cerebral amyloid angiopathy; CMB = cerebral microbleed; ICH = intracerebral hemorrhage; MGH = Massachusetts General Hospital.
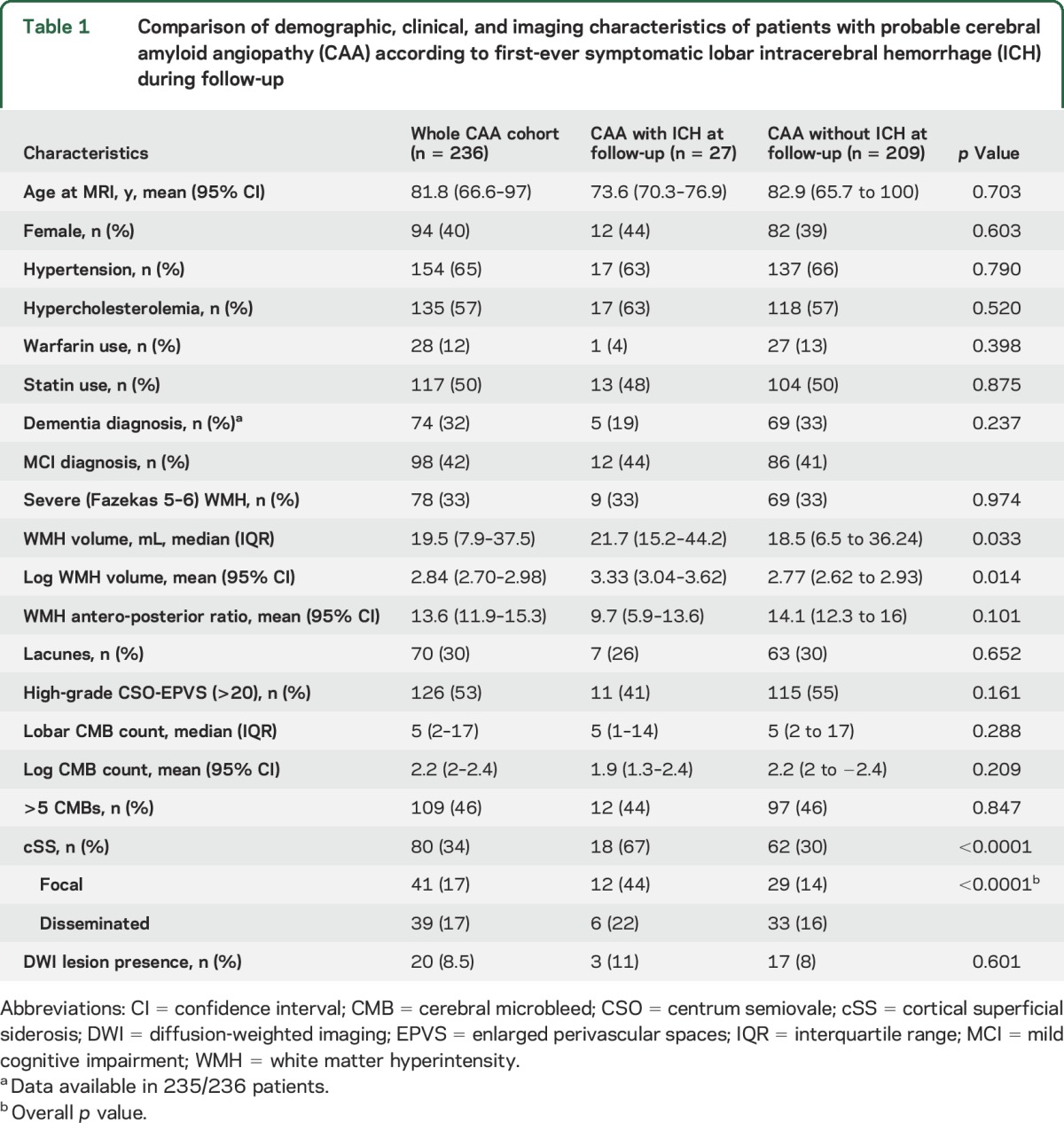
During a median follow-up time of 3.26 years (interquartile range 1.42–5.50 years; 1,878.8 person-years of follow-up), 27 of 236 patients (11.4%, 95% CI 7.7–16.2) experienced a first symptomatic CAA-related lobar ICH (incidence rate 1.44; 95% CI 0.99–2.10 per 100 patient-years). Three of these 27 patients experienced multiple (>1) sequential symptomatic lobar ICH during follow-up. The characteristics of our cohort per incident lobar ICH are summarized in table 1. No differences were found in demographic characteristics or vascular risk factors between the 2 groups. Patients who had a first-ever symptomatic ICH during follow-up had slightly higher WMH volumes and significantly higher prevalence of cSS (67% vs 30%; p < 0.0001) compared to patients with CAA free of ICH during follow-up (table 1). All other imaging markers of CAA and small vessel disease were comparable between the 2 patient groups.
Table 1.
Comparison of demographic, clinical, and imaging characteristics of patients with probable cerebral amyloid angiopathy (CAA) according to first-ever symptomatic lobar intracerebral hemorrhage (ICH) during follow-up
In Kaplan-Meier analysis, the presence of cSS at baseline MRI was a predictor of faster time until CAA-related lobar ICH (p = 0.0007, by the log-rank test) (figure 2A). The risk of symptomatic lobar ICH at 5 years of follow-up was 19% (95% CI 11%–32%) for patients with cSS at baseline vs 6% (95% CI 3%–12%) for patients without cSS. In univariable Cox regression analysis, cSS presence was a predictor of incident symptomatic lobar ICH (HR 3.68, 95% CI 1.64–8.25; p = 0.002); the HRs were similar for focal and disseminated cSS (3.84, 95% CI 1.60–9.23 and 3.41, 85% CI 1.20–9.65, respectively). None of the demographic, clinical, or imaging variables (including lobar CMB burden, severe Fazekas WMH, CSO/EPVS, or moderate to severe atrophy) showed an association with future lobar ICH in univariable analysis (all HR < 1 and p > 0.5). Only WMH volume on a continuous scale was associated with increased hazard of ICH during follow-up (HR 1.65, 95% CI 1.08–2.52; p = 0.021).
Figure 2. Time to incident first-ever symptomatic lobar intracerebral hemorrhage (ICH) during follow-up.
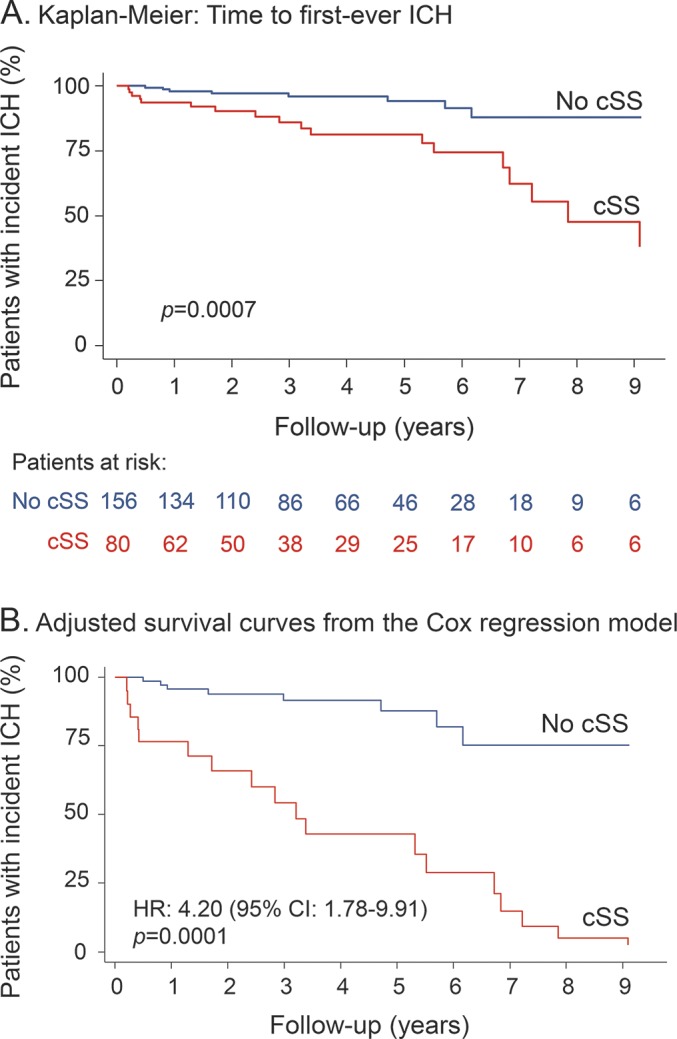
(A) Kaplan-Meier estimates of progression to symptomatic lobar ICH in the presence of cortical superficial siderosis (cSS) in patients with probable cerebral amyloid angiopathy. Testing of significance is by the log-rank test. (B) Adjusted survival curves from the Cox regression model on the effect of cSS presence on future lobar ICH risk (adjusting for baseline clinical and imaging characteristics). CI = confidence interval; HR = hazard ratio.
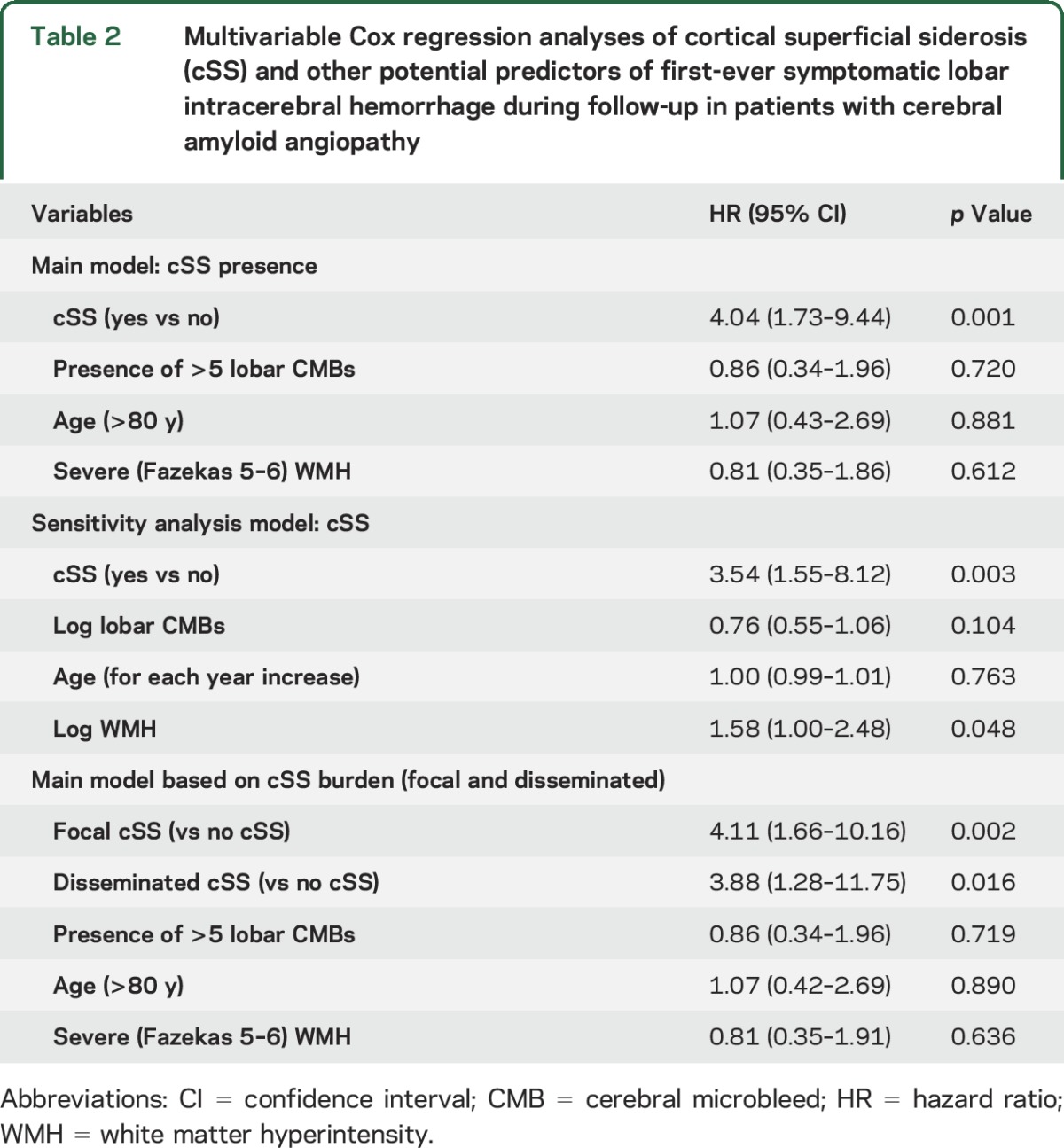
In prespecified multivariable Cox regression models adjusting for biologically significant risk factors (including age, CMB burden, and WMH), presence of cSS was the only independent predictor of increased symptomatic CAA-related lobar ICH risk during follow-up (table 2 and figure 2B for the adjusted Kaplan-Meier curves). These results remained consistent in sensitivity multivariable models using covariates as continuous variables (table 2) and if the single case of a deep ICH is included in the cohort (data not shown). The effect sizes were similar for focal and disseminated cSS included in comparable models (HR 4.10, 95% CI 1.66–10.16 and HR 3.88, 95% CI 1.28–11.75, respectively) (table 2). Further adjusting these models for blood-sensitive MRI sequence parameters (SWI vs T2*-GRE) did not change the effect sizes.
Table 2.
Multivariable Cox regression analyses of cortical superficial siderosis (cSS) and other potential predictors of first-ever symptomatic lobar intracerebral hemorrhage during follow-up in patients with cerebral amyloid angiopathy
DISCUSSION
In this large consecutive cohort of patients with probable CAA presenting with neurologic symptoms but without ICH, we found that cSS on T2*-GRE/SWI MRI is associated with an increased risk of future first-ever symptomatic lobar ICH (figure 3). The prognostic value of cSS in this setting was strong and independent of age and other neuroimaging markers of CAA severity, including lobar CMB burden and WMH. Hence, cSS may help stratify future bleeding risk even in symptomatic patients with CAA coming to medical attention without ICH, with implications for prognosis and treatment decisions.
Figure 3. Cortical superficial siderosis (cSS) and intracerebral hemorrhage (ICH).
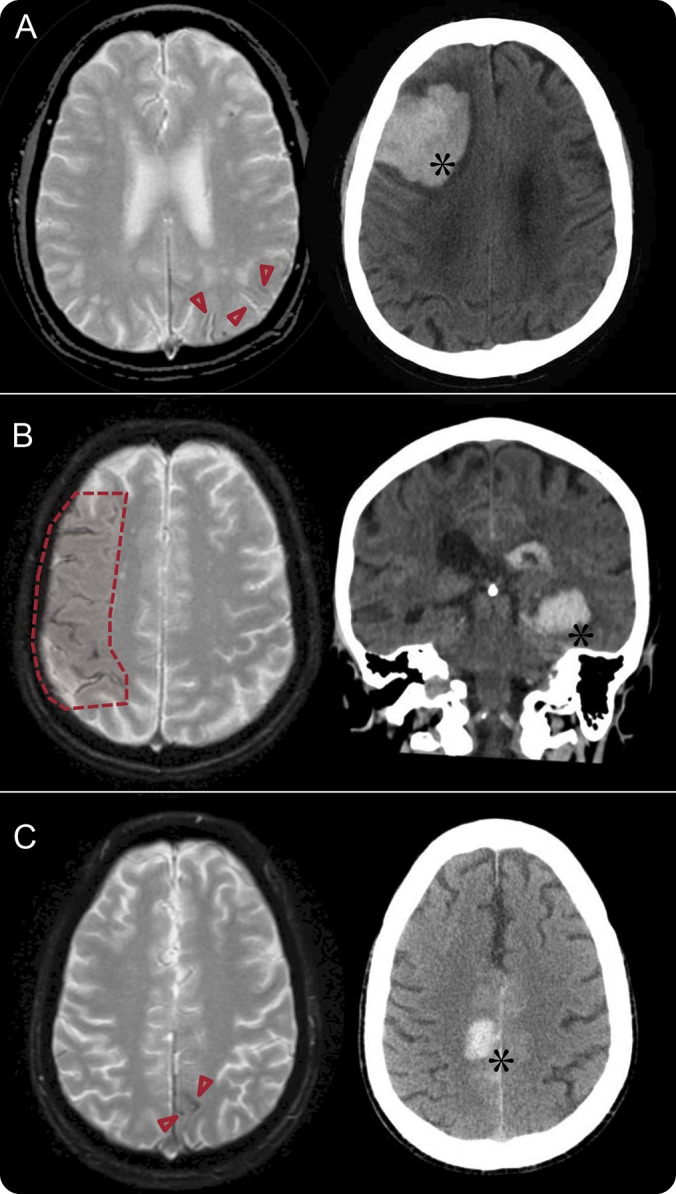
Representative examples of patients with probable cerebral amyloid angiopathy with preexisting cSS and subsequent first-ever ICH during follow-up. Arrowheads show focal cSS in A and C and the shaded area depicts disseminated cSS in B. Of note, overall in our cohort areas of cSS did not seem to predict the exact site of future lobar ICH.
cSS is a common hemorrhagic marker of CAA in the appropriate clinical setting (after other causes are excluded).1,26,27 Characteristically cSS affects the cerebral convexities and its imaging manifestation reflects blood breakdown products, including hemosiderin, that line the outermost surface of the cortex or lie in the subarachnoid space. One study reported cSS in up to 61% of patients (n = 38; mean age 71 years) with histopathologically confirmed CAA and no cSS in patients with ICH without histopathologic evidence of CAA (n = 22; mean age 55 years).18 A subsequent imaging study found cSS in 40% of patients with probable CAA with lobar ICH and fewer than 5% of patients with a strictly deep pattern of ICH, typically denoting non-CAA “hypertensive” hemorrhages.19 In another longitudinal cohort of probable CAA cases (most with ICH) (n = 84), the prevalence of cSS was also found to be 48%.28 While cSS prevalence is relatively low in memory clinic cohorts in general (being found in around 5% of cases),29,30 it is still common in those patients with strictly lobar CMBs suggesting underlying CAA (15%–20%).31 The prevalence of cSS in our cohort of patients without ICH with probable CAA is in line with these previous reports.
Much of the enduring interest in cSS is due to the possibility that this marker might reflect a more aggressive disease phenotype related to particularly high future ICH risk. A previous retrospective study including 51 patients with CAA-ICH with cSS observed new lobar ICHs in 18 patients (35%) during a median follow-up time period of 35.3 months.3 A multicenter cohort of probable or possible CAA (n = 118), mainly patients with lobar ICH at baseline, found that over a median follow-up time of 2 years, cSS was a strong independent predictor of recurrent ICH (HR 2.53, 95% CI 1.05–6.15).4 Our study extends these findings to patients without prior ICH seen in 2 common clinical settings: stroke services (where patients with CAA are often seen for TFNEs or other nonfocal neurologic symptoms) and memory clinics (where they are evaluated for cognitive symptoms or subjective cognitive complaints).11 It has been previously shown that patients with CAA first seen for acute neurologic symptoms often have recurrent symptoms and are at high risk of developing subsequent ICH.10,13,14 Our findings show that this risk is largely driven by cSS at baseline and reinforce the notion of cSS as a central hemorrhagic signature of CAA, providing insights into future ICH risk across the spectrum of CAA presentations.1,32 Of note, we did not find an association between lobar CMB burden and incident ICH risk, in line with a previous report8 and a multicenter study looking at risk of recurrent ICH in CAA-ICH.4 While multiple strictly lobar CMBs are useful as diagnostic markers of CAA in the context of the Boston criteria, cSS presence might be the strongest risk factor for future ICH, over and above CMB burden, across the spectrum of CAA subtypes.
The pathophysiologic mechanisms underlying cSS and its association with future ICH risk in CAA are not yet clear.1 Several lines of evidence support the prevailing hypothesis that cSS reflects repeated episodes of hemorrhage into the subarachnoid space from fragile superficial cortical or leptomeningeal CAA-laden vessels.1 cSS may thus be a marker of increased and widespread leptomeningeal small-vessel fragility and high CAA disease activity, heralding a high risk for lobar ICH.18,33,34 However, systematic neuropathologic studies specifically focusing on CAA-related cSS are currently lacking. A number of recent studies raised the interesting possibility that the APOE ε2 allele is more common in patients with CAA with vs without cSS and might influence pathophysiologic pathways and small vessel disease networks causing cSS.28,35,36 APOE ε2 is hypothesized to promote CAA vasculopathic changes (vessel cracking, vessel-within-vessel appearance, and fibrinoid necrosis), which can lead to vessel rupture,37 including cSS in multiple spatially separated foci.28,35
Key strengths of our study include the large sample size, the systematic evaluation of MRI using validated scales for the full spectrum of small vessel disease imaging markers, and the use of cSS definitions and rating methods in line with recent consensus recommendations.1 The current group of patients with probable CAA without ICH presents an opportunity to characterize and investigate unambiguously the role of primary in situ cSS as opposed to the possible secondary cSS that can occur as the result of lobar ICHs rupturing into the overlying subarachnoid space or ventricular system.1 The large sample size of consecutive patients with CAA without ICH combined with the long follow-up in our cohort allowed us to build robust multivariable survival models. A limitation is the potential selection bias due to the requirement for MRI performed as part of routine clinical care and the use of T2*-GRE/SWI sequences, which might have different sensitivity for cSS detection.27 Also, the lack of research quality T1-weighted isometric sequences precluded any volumetric analyses of cortical atrophy. Another potential bias might come from referral of advanced CAA cases that might be at highest risk of future ICH to our tertiary center, though we have made every effort to include consecutive patients across non-ICH presentations. Further larger cohorts will be needed to explore cSS and ICH risk stratified by presentation and patient referral patterns. An additional limitation is that it is impossible to score cSS without some unblinding to imaging findings, though raters were blinded to MRI markers on the non-T2*-weighted images and to all clinical and ICH recurrence data.
Taken together, our observations further support the notion that cSS is a central component of hemorrhagic CAA brain injury and can identify new mechanisms and distinct CAA phenotypes.2,38,39 The increased risk of future CAA-related lobar ICH associated with cSS may have important clinical implications for patient care, particularly in evaluating the risk vs benefit of antithrombotic agents, and for use of any future disease-modifying treatments.40
GLOSSARY
- CAA
cerebral amyloid angiopathy
- CI
confidence interval
- CMB
cerebral microbleed
- CSO
centrum semiovale
- cSS
cortical superficial siderosis
- EPVS
enlarged perivascular spaces
- FLAIR
fluid-attenuated inversion recovery
- HR
hazard ratio
- ICH
intracerebral hemorrhage
- OR
odds ratio
- STRIVE
Standards for Reporting Vascular Changes on Neuroimaging
- SWI
susceptibility-weighted imaging
- T2*-GRE
T2*-weighted gradient recalled echo
- TE
echo time
- TFNE
transient focal neurologic episode
- TR
repetition time
- WMH
white matter hyperintensity
AUTHOR CONTRIBUTIONS
A. Charidimou: study concept and design, data collection, imaging analysis, statistical analysis, writeup. G. Boulouis: study concept and design, data collection, imaging analysis, critical revisions. L. Xiong: data collection, critical revisions. M. Jessel: data collection and management. D. Roongpiboonsopit: critical revisions. A. Ayres: data collection and management. K.M. Schwab: data collection and management. J. Rosand: critical revisions. E.M. Gurol: critical revisions. S.M. Greenberg: funding, project concept and design, critical revisions. A. Viswanathan: funding, project concept and design, writeup, critical revisions.
STUDY FUNDING
This work was supported by NIH grant R01-AG026484 (S.M. Greenberg). Gregoire Boulouis was supported by a J. William Fulbright Scholarship and a Monahan Foundation Biomedical Research Grant. Andreas Charidimou receives postdoctoral support from the Bodossaki Foundation. This study is not industry-sponsored.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Charidimou A, Linn J, Vernooij MW, et al. Cortical superficial siderosis: detection and clinical significance in cerebral amyloid angiopathy and related conditions. Brain 2015;138:2126–2139. [DOI] [PubMed] [Google Scholar]
- 2.Greenberg SM, Vonsattel JP, Stakes JW, Gruber M, Finklestein SP. The clinical spectrum of cerebral amyloid angiopathy: presentations without lobar hemorrhage. Neurology 1993;43:2073–2079. [DOI] [PubMed] [Google Scholar]
- 3.Linn J, Wollenweber FA, Lummel N, et al. Superficial siderosis is a warning sign for future intracranial hemorrhage. J Neurol 2013;260:176–181. [DOI] [PubMed] [Google Scholar]
- 4.Charidimou A, Peeters AP, Jager R, et al. Cortical superficial siderosis and intracerebral hemorrhage risk in cerebral amyloid angiopathy. Neurology 2013;81:1666–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martinez-Ramirez S, Romero JR, Shoamanesh A, et al. Diagnostic value of lobar microbleeds in individuals without intracerebral hemorrhage. Alzheimers Dement 2015;11:1480–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Knudsen KA, Rosand J, Karluk D, Greenberg SM. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology 2001;56:537–539. [DOI] [PubMed] [Google Scholar]
- 7.Greenberg SM, Eng JA, Ning M, Smith EE, Rosand J. Hemorrhage burden predicts recurrent intracerebral hemorrhage after lobar hemorrhage. Stroke 2004;35:1415–1420. [DOI] [PubMed] [Google Scholar]
- 8.van Etten ES, Auriel E, Haley KE, et al. Incidence of symptomatic hemorrhage in patients with lobar microbleeds. Stroke 2014;45:2280–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calviere L, Cuvinciuc V, Raposo N, et al. Acute convexity subarachnoid hemorrhage related to cerebral amyloid angiopathy: clinicoradiological features and outcome. J Stroke Cerebrovasc Dis 2016;25:1009–1016. [DOI] [PubMed] [Google Scholar]
- 10.Ni J, Auriel E, Jindal J, et al. The characteristics of superficial siderosis and convexity subarachnoid hemorrhage and clinical relevance in suspected cerebral amyloid angiopathy. Cerebrovasc Dis 2015;39:278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charidimou A, Peeters A, Fox Z, et al. Spectrum of transient focal neurological episodes in cerebral amyloid angiopathy: multicentre magnetic resonance imaging cohort study and meta-analysis. Stroke 2012;43:2324–2330. [DOI] [PubMed] [Google Scholar]
- 14.Charidimou A, Law R, Werring DJ. Amyloid “spells” trouble. Lancet 2012;380:1620. [DOI] [PubMed] [Google Scholar]
- 15.Wardlaw JM, Smith EE, Biessels GJ, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 2013;12:822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greenberg SM, Vernooij MW, Cordonnier C, et al. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol 2009;8:165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gregoire SM, Chaudhary UJ, Brown MM, et al. The Microbleed Anatomical Rating Scale (MARS): reliability of a tool to map brain microbleeds. Neurology 2009;73:1759–1766. [DOI] [PubMed] [Google Scholar]
- 18.Linn J, Halpin A, Demaerel P, et al. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology 2010;74:1346–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Charidimou A, Jager RH, Fox Z, et al. Prevalence and mechanisms of cortical superficial siderosis in cerebral amyloid angiopathy. Neurology 2013;81:626–632. [DOI] [PubMed] [Google Scholar]
- 20.Doubal FN, MacLullich AM, Ferguson KJ, Dennis MS, Wardlaw JM. Enlarged perivascular spaces on MRI are a feature of cerebral small vessel disease. Stroke 2010;41:450–454. [DOI] [PubMed] [Google Scholar]
- 21.Rorden C, Karnath HO, Bonilha L. Improving lesion-symptom mapping. J Cogn Neurosci 2007;19:1081–1088. [DOI] [PubMed] [Google Scholar]
- 22.Fazekas F, Chawluk JB, Alavi A, Hurtig HI, Zimmerman RA. MR signal abnormalities at 1.5 T in Alzheimer's dementia and normal aging. AJR Am J Roentgenol 1987;149:351–356. [DOI] [PubMed] [Google Scholar]
- 23.Thanprasertsuk S, Martinez-Ramirez S, Pontes-Neto OM, et al. Posterior white matter disease distribution as a predictor of amyloid angiopathy. Neurology 2014;83:794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biffi A, Halpin A, Towfighi A, et al. Aspirin and recurrent intracerebral hemorrhage in cerebral amyloid angiopathy. Neurology 2010;75:693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.von Elm E, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Lancet 2007;370:1453–1457. [DOI] [PubMed] [Google Scholar]
- 26.Malhotra A, Schindler J, Mac Grory B, et al. Cerebral microhemorrhages and meningeal siderosis in infective endocarditis. Cerebrovasc Dis 2016;43:59–67. [DOI] [PubMed] [Google Scholar]
- 27.Zhao H, Wang J, Lu Z, et al. Superficial siderosis of the central nervous system induced by a single-episode of traumatic subarachnoid hemorrhage: a study using MRI-enhanced gradient echo T2 star-weighted angiography. PLoS One 2015;10:e0116632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shoamanesh A, Martinez-Ramirez S, Oliveira-Filho J, et al. Interrelationship of superficial siderosis and microbleeds in cerebral amyloid angiopathy. Neurology 2014;83:1838–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wollenweber FA, Buerger K, Mueller C, et al. Prevalence of cortical superficial siderosis in patients with cognitive impairment. J Neurol 2014;261:277–282. [DOI] [PubMed] [Google Scholar]
- 30.Zonneveld HI, Goos JD, Wattjes MP, et al. Prevalence of cortical superficial siderosis in a memory clinic population. Neurology 2014;82:698–704. [DOI] [PubMed] [Google Scholar]
- 31.Charidimou A, Ni J, Martinez-Ramirez S, et al. Cortical superficial siderosis in memory clinic patients: further evidence for underlying cerebral amyloid angiopathy. Cerebrovasc Dis 2016;41:156–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Linn J, Herms J, Dichgans M, et al. Subarachnoid hemosiderosis and superficial cortical hemosiderosis in cerebral amyloid angiopathy. AJNR Am J Neuroradiol 2008;29:184–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takeda S, Hinokuma K, Yamazaki K, et al. The hemorrhage caused by sporadic-type cerebral amyloid angiopathy occurs primarily in the cerebral sulci. Neuropathology 2012;32:38–43. [DOI] [PubMed] [Google Scholar]
- 34.Takeda S, Yamazaki K, Miyakawa T, et al. Subcortical hematoma caused by cerebral amyloid angiopathy: does the first evidence of hemorrhage occur in the subarachnoid space? Neuropathology 2003;23:254–261. [DOI] [PubMed] [Google Scholar]
- 35.Charidimou A, Martinez-Ramirez S, Shoamanesh A, et al. Cerebral amyloid angiopathy with and without hemorrhage: evidence for different disease phenotypes. Neurology 2015;84:1206–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Na HK, Park JH, Kim JH, et al. Cortical superficial siderosis: a marker of vascular amyloid in patients with cognitive impairment. Neurology 2015;84:849–855. [DOI] [PubMed] [Google Scholar]
- 37.Greenberg SM, Vonsattel JP, Segal AZ, et al. Association of apolipoprotein E epsilon2 and vasculopathy in cerebral amyloid angiopathy. Neurology 1998;50:961–965. [DOI] [PubMed] [Google Scholar]
- 38.Maia LF, Mackenzie IR, Feldman HH. Clinical phenotypes of cerebral amyloid angiopathy. J Neurol Sci 2007;257:23–30. [DOI] [PubMed] [Google Scholar]
- 39.Charidimou A. Convexity subarachnoid hemorrhage in cerebral amyloid angiopathy: the saga continues. J Cereb Blood Flow Metab 2015;35:707–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Charidimou A, Jager HR. Developing biomarkers for cerebral amyloid angiopathy trials: do potential disease phenotypes hold promise? Lancet Neurol 2014;13:538–540. [DOI] [PubMed] [Google Scholar]