

Abstract
Objective:
To examine whether a polygenic risk score (PRS) derived from APOE4, CLU, and ABCA7 is associated with CSF biomarkers of Alzheimer disease (AD) pathology and whether higher cardiorespiratory fitness (CRF) modifies the association between the PRS and CSF biomarkers.
Methods:
Ninety-five individuals from the Wisconsin Registry for Alzheimer's Prevention were included in these cross-sectional analyses. They were genotyped for APOE4, CLU, and ABCA7, from which a PRS was calculated for each participant. The participants underwent lumbar puncture for CSF collection. β-Amyloid 42 (Aβ42), Aβ40, total tau (t-tau), and phosphorylated tau (p-tau) were quantified by immunoassays, and Aβ42/Aβ40 and tau/Aβ42 ratios were computed. CRF was estimated from a validated equation incorporating sex, age, body mass index, resting heart rate, and self-reported physical activity. Covariate-adjusted regression analyses were used to test for associations between the PRS and CSF biomarkers. In addition, by including a PRS×CRF term in the models, we examined whether these associations were modified by CRF.
Results:
A higher PRS was associated with lower Aβ42/Aβ40 (p < 0.001), higher t-tau/Aβ42 (p = 0.012), and higher p-tau/Aβ42 (p = 0.040). Furthermore, we observed PRS × CRF interactions for Aβ42/Aβ40 (p = 0.003), t-tau/Aβ42 (p = 0.003), and p-tau/Aβ42 (p = 0.001). Specifically, the association between the PRS and these CSF biomarkers was diminished in those with higher CRF.
Conclusions:
In a late-middle-aged cohort, CRF attenuates the adverse influence of genetic vulnerability on CSF biomarkers. These findings support the notion that increased cardiorespiratory fitness may be beneficial to those at increased genetic risk for AD.
The APOE4 gene is a well-established genetic risk factor for cholesterol dysmetabolism1 and cerebral β-amyloidosis in late-onset Alzheimer disease (AD).2 Recently, genome-wide association studies have suggested additional candidate genes associated with AD,3 including clusterin (CLU) and ATP-binding cassette transporter A7 (ABCA7), which, like APOE4, are also involved in cholesterol metabolism.4 The implication of APOE4, CLU, and ABCA7 in AD and cholesterol metabolism suggests that aberrant cholesterol processing might contribute to AD susceptibility. Previous studies support this notion by demonstrating an effect of these risk variants on AD biomarkers.5–7
Although the study of genetic variants in isolation has provided information on the distinct effects of genetic risk factors, interrogating such variants in aggregate is critical for understanding the involvement of specific pathways in AD etiology.1,7–9 Indeed, pathways linked to late-onset AD, including cholesterol metabolism, serve as promising targets for pharmacologic and nonpharmacologic interventions.4,8
Emerging evidence indicates that modifiable lifestyle factors such as physical activity may protect against the effect of genetic susceptibility on AD pathophysiology.10 In addition, it is unknown whether increased cardiorespiratory fitness (CRF) can modify a pathway-specific genetic effect on biomarkers of AD.
Therefore, we investigated whether a cholesterol metabolism polygenic risk score (PRS) is associated with CSF biomarkers of AD in an asymptomatic late-middle-aged cohort. We further sought to examine whether this association varies as a function of CRF.
METHODS
Standard protocol approvals, registrations, and patient consents.
The University of Wisconsin Institutional Review Board approved all study procedures, and each participant provided signed informed consent before participation.
Participants.
Participants in this study were recruited from the Wisconsin Registry for Alzheimer’s Prevention (WRAP) study.11 The WRAP study consists of >1,500 individuals who either were recruited from the community or were adult children of patients with AD seen in various University of Wisconsin dementia clinics. Participants are compensated for their participation in the WRAP study and in affiliated biomarker studies such as the one from which we obtained data presented in this report. Table e-1 at Neurology.org details background characteristics of the full WRAP cohort vis-à-vis our study sample.
For our analyses, participants were included on the basis of having undergone genotyping for APOE4, CLU, and ABCA7; being of self-reported European ancestry, which was confirmed with principal components genetic analyses,12 to bolster the genetic homogeneity of the study sample, having self-reported physical activity (SR-PA) measurements, and having undergone lumbar puncture for collection of CSF. Additional inclusion criteria included intact performance on a comprehensive battery of neuropsychological tests (table e-2). Exclusion criteria included significant neurologic disease, psychiatric disorders, and untreated hypertension. These criteria resulted in a sample of 95 participants.
CRF measure.
Although graded exercise testing (GXT) is the gold standard for assessing CRF, barriers to its implementation among older adults remain.13 Therefore, recent work has focused on developing non-GXT estimates of CRF using variables that are both clinically available and known to have an effect on CRF. One widely used formula for deriving such non-GXT estimates of CRF is as follows: CRF = 18.07 + sex (2.77) − age (0.10) – BMI (0.17) − resting heart rate (0.03) + SR-PA, where BMI is body mass index and with sex coded as female = 0 and male = 1.13 As described previously,14 SR-PA was derived from the moderate intensity physical activity question from the Women's Health Initiative physical activities questionnaire.15,16 Briefly, this question inquires about the frequency and duration of engagement in moderate, “not exhausting,” physical activity per week. Following established protocol,15,16 frequency and duration were multiplied to create a minutes per week measure of moderate-intensity physical activity (range 0–240 minutes). We further scaled this SR-PA measure by 30 so that each additional unit (range 0–8) captured the incremental effect of 30 minutes of activity as opposed to the effect of 1 minute of activity.17 Lastly, this SR-PA variable had a skewed distribution that we corrected via Blom18 transformation before inclusion in the CRF estimate.
Genotyping.
DNA collection, genotyping, and quality control.
DNA was extracted from whole-blood samples with the PUREGENE DNA Isolation Kit (Gentra Systems, Inc, Minneapolis, MN). DNA concentrations were quantified with ultraviolet spectrophotometry (DU 530 Spectrophotometer; Beckman Coulter, Fullerton, CA). Single nucleotide polymorphisms (SNPs) for CLU (rs9331896), ABCA7 (rs4147929), and APOE4 (rs429358 and rs7412) were genotyped by LGC Genomics (Beverly, MA), implementing competitive allele-specific PCR-based KASP genotyping assays. Duplicate quality control samples were placed randomly throughout all 96-well plates. The genotype concordance rate was 99.89%. All discordant genotypes were set to missing. Further quality control was conducted with PLINK version 1.07.19 Variants examined in this analysis did not deviate from Hardy-Weinberg equilibrium with a Bonferroni-adjusted p value threshold of 0.05. Genetic variants were coded additively according to the number of risk alleles.
Polygenic risk score.
A PRS was calculated from the following equation: 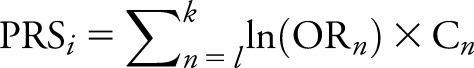 , where i represents the individual, whose score is calculated by summing over all SNPs; n is in the pathway ranging from l to k; OR is the odds ratio of the SNP n; and C is the individual's count of risk alleles for SNP n.20 If the minor allele was protective, as was the case for CLU, the inverse of the OR was used instead. SNP ORs for CLU and ABCA7 were taken from the International Genomics of Alzheimer's Project meta-analysis.3 Risk due to APOE4 was calculated additively with the use of an OR of 3.68, as reported in AlzGene.21 Furthermore, as a result of the inherent bimodal clustering of the PRS in our sample, we opted to dichotomize this variable into low and high PRS groups, PRS < 1.0 and PRS ≥ 1.0, respectively.
, where i represents the individual, whose score is calculated by summing over all SNPs; n is in the pathway ranging from l to k; OR is the odds ratio of the SNP n; and C is the individual's count of risk alleles for SNP n.20 If the minor allele was protective, as was the case for CLU, the inverse of the OR was used instead. SNP ORs for CLU and ABCA7 were taken from the International Genomics of Alzheimer's Project meta-analysis.3 Risk due to APOE4 was calculated additively with the use of an OR of 3.68, as reported in AlzGene.21 Furthermore, as a result of the inherent bimodal clustering of the PRS in our sample, we opted to dichotomize this variable into low and high PRS groups, PRS < 1.0 and PRS ≥ 1.0, respectively.
CSF assessment.
Lumbar puncture for collection of CSF was performed the morning after a 12-hour fast, with a Sprotte 24- or 25-gauge spinal needle at L3-4 or L4-5 using gentle extraction into polypropylene syringes. Each sample consisted of 22 mL CSF, which was then combined, mixed, and centrifuged at 2,000g for 10 minutes. Supernatants were frozen in 0.5-mL aliquots in polypropylene tubes and stored at −80°C.
CSF levels of β-amyloid 42 (Aβ42) and Aβ40 were quantified by electrochemiluminescence with an Aβ triplex assay (MSD Human Aβ peptide Ultra-Sensitive Kit, Meso Scale Discovery, Gaithersburg, MD). CSF Aβ42 was also quantified, along with total-tau (t-tau) and phosphorylated-tau (p-tau), with a sandwich ELISA (INNOTEST Aβ1-42, hTAU-Ag, and Phospho-Tau[181P], respectively; Fujirebio Europe, Ghent, Belgium). All measurements were performed in one round of analyses with one batch of reagents by board-certified laboratory technicians who were blinded to clinical data and used protocols accredited by the Swedish Board for Accreditation and Conformity Assessment, as previously described.22 We computed an Aβ42/Aβ40 ratio using the triplex assay values and t-tau/Aβ42 and p-tau/Aβ42 ratios using the INNOTEST assay values. The Aβ42/Aβ40 ratio, rather than Aβ42 by itself, was adopted as our measure of Aβ burden because it normalizes Aβ42 concentration to a measure of overall amyloidogenic processing by amyloid precursor protein, making it possible to detect low Aβ42 in high-Aβ producers and vice versa.23 The mean ± SD time interval between CSF sampling and the WRAP visit at which components of the CRF estimate were obtained was 6.5 ± 5.1 months.
Statistical analyses.
To investigate whether the cholesterol metabolism PRS is associated with CSF biomarkers of AD, we fitted a series of linear regression models, one for each CSF biomarker, that were adjusted for age and sex. Because CLU and ABCA7 are known to convey modest genetic effects compared with APOE4,8 we additionally sought to better understand the incremental utility of the PRS vs APOE4 alone by conducting sensitivity analyses.
Because we were interested in whether CRF attenuates the relationship between the cholesterol metabolism PRS and CSF biomarkers, we additionally refitted the original models while incorporating an interaction between the PRS and CRF. When significant, this interaction term would indicate that the deleterious effect of a higher PRS on the biomarker is modified by CRF.
To elucidate the relative contribution of each CRF component to the moderating effect of CRF, we examined models that tested interactions between each component of the CRF estimate (i.e., age, sex, resting heart rate, BMI, and SR-PA) and the PRS. For all analyses conducted, evaluations of assumptions for ordinary least squares were carried out via graphic analyses.24 All analyses were conducted with IBM SPSS, version 21.0. Only findings with values of p ≤ 0.05 (2-tailed) were considered to be significant.
RESULTS
Participant characteristics.
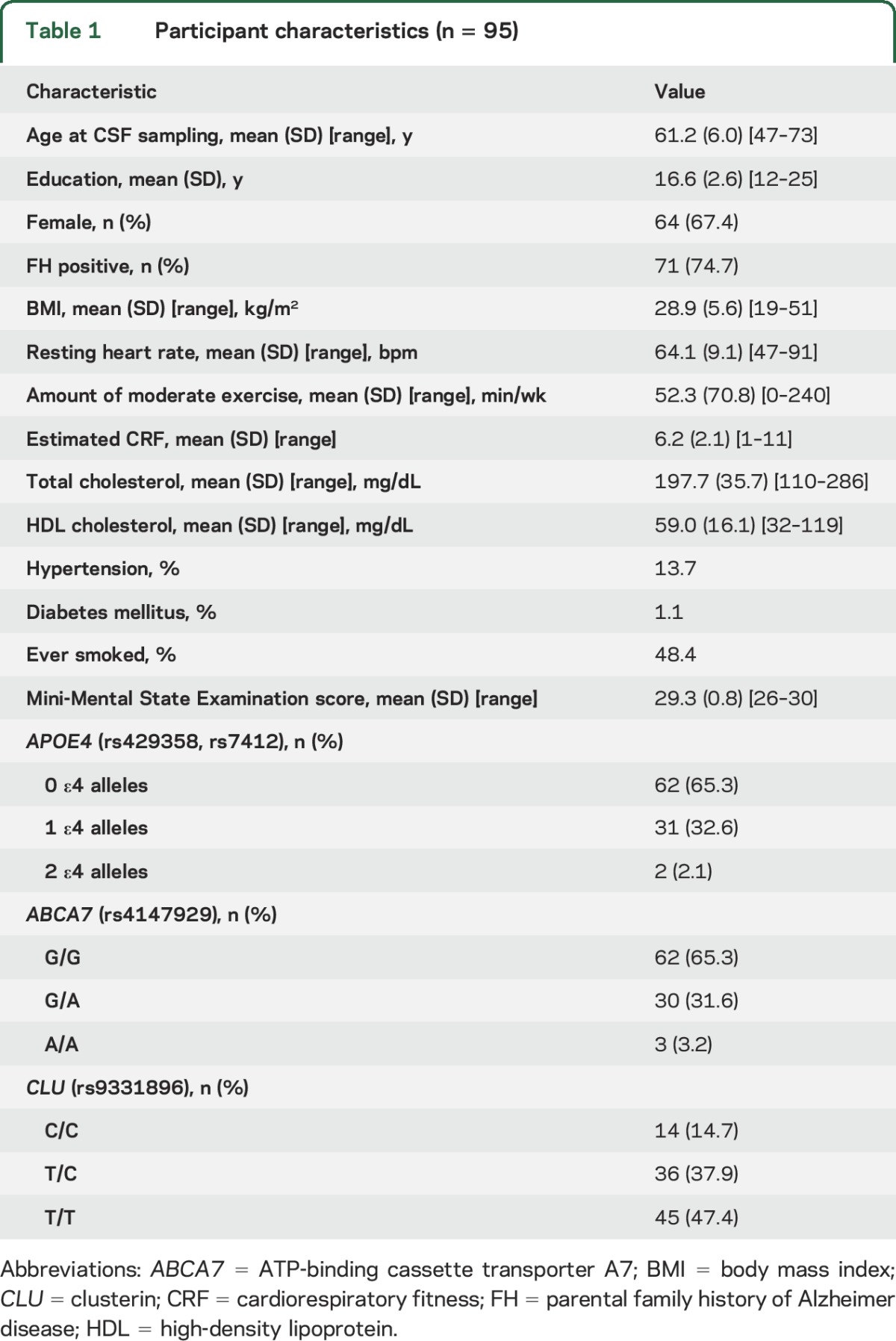
As shown in table 1, participants were predominately female (67.4%) with a mean ± SD age of 61.2 ± 6.0 years. Overall, participants were slightly overweight (BMI 28.9 ± 5.6 kg/m2) and reported engaging in 52.3 ± 70.8 minutes of moderate-intensity physical activity per week.
Table 1.
Participant characteristics (n = 95)
Associations between the PRS and CSF biomarkers.
The PRS was associated (B [95% confidence interval], p) with CSF measures of Aβ42/Aβ40 (−0.015 [−0.022 to −0.008], <0.001), t-tau/Aβ42 (0.126 [0.029–0.224], 0.012), and p-tau/Aβ42 (0.013 [0.001–0.026], 0.040) but not with t-tau (4.166 [−44.578 to 52.911], 0.866) or p-tau (−2.509 [−8.696 to 3.677], 0.422). Adjusted mean ± SE biomarker values for the high PRS group vis-à-vis the low PRS group are shown in figure 1, A–E.
Figure 1. Association between the PRS and CSF biomarkers of AD.

Panels A–E display estimated mean ± SE for CSF biomarkers after adjustment for age and sex. Aβ = β-amyloid; ABCA7 = ATP-binding cassette transporter A7; AD = Alzheimer disease; CLU = clusterin; p-tau = phosphorylated tau; PRS = polygenic risk score derived from APOE4, CLU, and ABCA7 risk variants; t-tau = total tau.
Sensitivity analyses.
For our sensitivity analyses, we first recalculated the PRS after excluding APOE4 to assess whether APOE4 could be driving the observed associations. When we tested the relationship between this recalculated PRS and the CSF biomarkers, we found no associations (p > 0.085). However, APOE4 alone was associated (B [95% confidence interval], p) with Aβ42/Aβ40 (−0.014 [−0.023 to −0.000], 0.004), t-tau/Aβ42 (0.124 [0.027–0.224], 0.013), and p-tau/Aβ42 (0.013 [0.000–0.026], 0.042). While these findings might suggest that APOE4 drove the initial findings, we additionally performed nested likelihood tests investigating the fit of the model with APOE4 vs the model without APOE4. These tests revealed that the model with APOE4 fits significantly better compared to the model without APOE4 for Aβ42/Aβ40 (χ2 [df = 1] = 15.45, p < 0.001) and t-tau/Aβ42 (χ2 [df = 1] = 5.70, p < 0.017). However, the model with APOE4 did not fit better than the model without APOE4 for p-tau/Aβ42 (χ2 [df = 1] = 3.73, p = 0.054).
CRF, PRS, and CSF biomarkers.
There were PRS × CRF interactions (B [95% confidence interval], p) for Aβ42/Aβ40 (0.005 [0.002–0.008], 0.003), t-tau/Aβ42 (−0.069 [−0.113 to −0.025], 0.003), and p-tau/Aβ42 (−0.010 [−0.015 to −0.004], 0.001), as reported in table 2. Covariate-adjusted means for low vs high PRS groups at low vs high CRF levels were plotted for each biomarker examined. These graphs, shown in figure 2, A–E, revealed that the adverse change in CSF biomarkers due to a higher PRS was evident in the low CRF group but muted in the high CRF group.
Table 2.
Cardiorespiratory fitness and PRS-related alterations in CSF biomarkers

Figure 2. CRF modifies the association between the PRS and CSF biomarkers of AD.

Panels A–E display estimated mean ±SE for CSF biomarkers after adjustment for age and sex. Although the analyses were performed using the full range of the CRF measures, for plotting purposes, the CRF variable was dichotomized at the median value to yield low and high groups. Aβ = β-amyloid; ABCA7 = ATP-binding cassette transporter A7; AD = Alzheimer disease; CLU = clusterin; CRF = cardiorespiratory fitness; p-tau = phosphorylated tau; PRS = polygenic risk score derived from APOE4, CLU, and ABCA7 risk variants; t-tau = total tau.
To investigate the extent to which these findings may be driven by other potential confounders, we reran these models after additionally adjusting for years of education, high-density lipoprotein cholesterol, history of smoking, diabetes mellitus, and hypertension. There remained PRS × CRF interactions (B [95% confidence interval], p) for Aβ42/Aβ40 (0.005 [0.002–0.009], 0.003), t-tau/Aβ42 (−0.071 [−0.118 to −0.023], 0.004), and p-tau/Aβ42 (−0.010 [−0.016 to −0.004], 0.002).
Prior studies have used alternative CRF estimate equations that included factors such as smoking history and ethnicity.25 To understand whether our results were dependent on the specific CRF estimate measure used, we recalculated our CRF estimate using an alternative formula,25 i.e., CRF = 77.41 − age (0.37) − BMI (0.91) − resting heart rate (0.07) + SR-PA quintile + ethnicity (8.032) − smoking status (1.976), with smoking status coded as never = 0 and ex-smoker or current smoker = 1 and ethnicity coded as nonwhite = 0 and white = 1. After we refitted our model with this recalculated CRF estimate, our results remained essentially unchanged. There were PRS × CRF interactions (B [95% confidence interval], p) for Aβ42/Aβ40 (0.001 [0.000–0.001], 0.037), t-tau/Aβ42 (−0.017 [−0.032 to −0.001], 0.034), and p-tau/Aβ42 (−0.003 [−0.005 to −0.001], 0.011) but not for t-tau (−3.212 [−11.061 to 4.637], 0.418) or p-tau (−0.686 [−1.675 to 0.303], 0.172).
Component analyses.
Our interrogation of the CRF components (table 3) revealed BMI × PRS interactions for t-tau/Aβ42 and p-tau/Aβ42; SR-PA × PRS interactions for Aβ42/Aβ40, t-tau/Aβ42, and p-tau/Aβ42; and a sex × PRS interaction for t-tau/Aβ42. In summary, having lower BMI, having higher SR-PA, and being male attenuated the deleterious effect of the PRS on CSF biomarkers.
Table 3.
Sex, BMI, and SR-PA attenuate the influence of the PRS on CSF biomarkers

Supplementary analyses.
In addition to our objectives of primary interest, we examined the following secondary objectives. First, we investigated whether there was a main effect of CRF on CSF biomarkers, without differentiating by genetic vulnerability. These analyses revealed no associations (p > 0.490). We also tested for, and failed to find, an association between the PRS and CRF (p = 0.576).
Lastly, because interactions between APOE4 and lifestyle measures have been previously studied,10 whereas the same is not true for ABCA7 and CLU, we sought to understand how CRF influences the association of ABCA7 and CLU independently on CSF biomarkers. To do this, we additionally fitted models to test an ABCA7 × CRF interaction and a CLU × CRF interaction on CSF biomarkers. We found that while CLU did not interact with CRF to predict CSF biomarkers (p > 0.192), ABCA7 and CRF had a modest interaction (B [95% confidence interval], p) for t-tau/Aβ42 (−0.035 [−0.072 to 0.001], 0.060) and p-tau/Aβ42 (−0.004 [−0.009 to 0.000], 0.064).
DISCUSSION
This study showed that, in a late-middle-aged cohort, a cholesterol metabolism PRS derived from APOE4, CLU, and ABCA7 is associated with hallmark pathophysiologic features of AD. Specifically, individuals with a higher PRS had reduced Aβ42/Aβ40 and elevated t-tau/Aβ42 and p-tau/Aβ42 compared to those with a lower PRS. Overall, these findings indicate that cholesterol metabolism genes may contribute additively to interindividual variation in these biomarkers. We observed that CRF modified this genetic-related alteration in these CSF biomarkers. That is, the adverse influence of the PRS on the CSF biomarkers was muted among those with higher CRF. We further found that BMI, SR-PA, and sex contributed to the moderating effect of CRF.
There is a well-established relationship between APOE4 allele carriership and Aβ accumulation.10,26–28 For example, a prior study28 found that cognitively normal APOE4 carriers had lower CSF Aβ42 than noncarriers. Likewise, in WRAP, we have shown that late-middle-aged APOE4 carriers had higher Aβ accumulation, measured by Pittsburgh compound B-PET imaging compared to noncarriers.16,29 Additionally, it is well established that APOE4 carriers have higher serum cholesterol levels than APOE3 or APOE2 carriers.30 Taken together, one proposed mechanism for the pathogenic contributions of APOE4 in AD is that it may influence Aβ accumulation through its role in central and peripheral cholesterol transport.1
Relatedly, CLU is involved in lipid transport and is associated with AD susceptibility in recent genome-wide association studies.3 However, its role in Aβ accumulation is less understood. A previous study found an association between CLU and CSF Aβ42 levels in individuals with AD, even after accounting for APOE4 status.6 In contrast, one study7 reported no association between a CLU risk variant and Aβ burden. The disparity in these findings might be due to the distinct populations examined; as our study suggests, perhaps it is a result of dissimilar CRF in these populations. Furthermore, ABCA7 encodes a protein that functions as a cholesterol efflux regulator, and there is recent evidence for an association between ABCA7 and AD pathophysiology. For example, in a population of cognitively normal elderly, the risk variant of ABCA7 was associated with a >2-fold increase in the odds of being Aβ positive.7
We extend these aforementioned findings by showing associations between a cholesterol metabolism PRS (comprising CLU, APOE4, and ABCA7 SNPs) and CSF biomarkers of AD. These data, in combination with the prior studies,6,7,31,32 strengthen the knowledge base suggesting that disordered cholesterol metabolism may play a crucial role in AD, beginning as early as late middle age. Our sensitivity analyses revealed that assessing the genetic role of cholesterol metabolism processes without APOE4 yielded no associations with CSF biomarkers. While these results may suggest that APOE4 plays the dominant role in the cholesterol metabolism pathway of AD, we further observed that the PRS derived from APOE4 and CLU increased model fitness for CSF measures relative to APOE4 alone. The findings underscore the potential value of examining the polygenic, rather than the individual, effect of genetic risk variants on CSF biomarkers.
There is emerging interest in the effect of a physically active lifestyle on genetic-related risk for AD. An earlier study10 reported that highly active APOE4 carriers had less Aβ deposition compared to their inactive peers. Additional groups have found that physical activity attenuates the detrimental effect of other genetic polymorphisms on cognitive function33,34 and brain volume.35 In the present study, we found that higher CRF counteracted the adverse influence of cholesterol metabolism polymorphisms on CSF biomarkers of AD. In addition, SR-PA, BMI, and sex were found to moderate the effect of the PRS on CSF biomarkers when we examined the components of the CRF estimate. These observed statistical interactions may reflect an underlying biological interaction between cholesterol processing and lifestyle factors (i.e., CRF, physical activity, and BMI) in the accumulation of AD pathophysiology. Collectively, the extant literature suggests that physical activity and higher CRF may provide resilience for individuals at increased genetic risk for AD. Our observation that lower BMI benefited individuals at greater genetic vulnerability for AD bolsters a recent report36 on the protective role of healthy BMI in midlife vis-a-vis AD in later life. In addition, our finding that men have diminished PRS-related alterations in CSF biomarkers is in agreement with epidemiologic studies showing that men are at decreased risk for AD.37
The current study was not without limitations, the most significant being the cross-sectional design, which raised the possibility of reverse causality. A prospective design will be critical in determining whether fitness exerts a causal influence on AD-related biomarkers in people at genetic risk of AD. Another potential limitation is our use of a nonexercise estimate of CRF in this study. However, we have previously shown a strong correspondence (r = 0.76, p < 0.001) between this nonexercise estimate and peak oxygen consumption, the gold standard for assessing CRF.14 Furthermore, while we adjusted for many common cardiovascular risk factors in our analyses, we may not have controlled for all potential confounds (e.g., dietary habits or family history of cardiovascular disease). In addition, while our findings highlight the potential benefits of investigating genetic polymorphisms in aggregate, from a clinical perspective, it may be infeasible to genotype patients for multiple genetic variants. However, our findings would be strengthened by replication in an independent sample. In addition, future studies examining whether higher CRF modifies the association between the PRS and other AD-related outcomes such as cognition or neuroimaging measures would complement our findings. Lastly, generalizability of our findings might be somewhat limited because of the restricted sample size and homogeneous demographic makeup (e.g., European ancestry).
Overall, we found that a cholesterol metabolism PRS was adversely associated with CSF biomarkers of AD and that this deleterious effect of the PRS is attenuated in individuals with higher CRF. These findings suggest that leading a physically fit lifestyle may be beneficial to those at increased genetic risk for AD.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the researchers and staff of the Clinical Neurochemistry Laboratory, Institute of Neuroscience and Physiology, The Sahlgrenska Academy at University of Gothenburg, Sweden, where the CSF assays took place; Caitlin A. Cleary, BSc, Sandra Harding, MS, Jennifer Bond, BA, Janet Rowley, BA, Amy Hawley, BS, Nancy Davenport-Sis, MS, and the WRAP study coordinators for helping with study data collection; Dr. Maritza Dowling for statistical consultation and assistance; and the participants in the WRAP for their continued dedication.
GLOSSARY
- Aβ
β-amyloid
- ABCA7
ATP-binding cassette transporter A7
- AD
Alzheimer disease
- BMI
body mass index
- CLU
clusterin
- CRF
cardiorespiratory fitness
- GXT
graded exercise testing
- OR
odds ratio
- p-tau
phosphorylated tau
- PRS
polygenic risk score
- SNP
single nucleotide polymorphism
- SR-PA
self-reported physical activity
- t-tau
total tau
- WRAP
Wisconsin Registry for Alzheimer's Prevention
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Study concept or design: Schultz, Okonkwo. Acquisition, analysis, or interpretation of data: Schultz, Boots, Darst, Zetterberg, Blennow, Edwards, Koscik, Carlsson, Gallagher, Bendlin, Asthana, Sager, Hogan, Hermann, Cook, Johnson, Engelman, Okonkwo. Drafting of manuscript: Schultz, Okonkwo. Critical revision of manuscript: Schultz, Boots, Darst, Zetterberg, Blennow, Edwards, Koscik, Carlsson, Gallagher, Bendlin, Asthana, Sager, Hogan, Hermann, Cook, Johnson, Engelman, Okonkwo. Statistical analysis: Schultz, Okonkwo. Obtaining funding: Carlsson, Asthana, Johnson, Okonkwo. Administrative, technical, or material support: Schultz, Boots, Darst, Zetterberg, Blennow, Edwards, Koscik, Gallagher, Bendlin, Sager, Hogan, Hermann, Cook, Engelman. Supervision: Okonkwo.
STUDY FUNDING
This work was supported by National Institute on Aging grants K23 AG045957 (O.C.O.), R01 AG027161 (M.A.S.), R01 AG031790 (C.M.C.), R01 AG021155 (S.C.J.), R01 AG037639 (B.B.B.), and P50 AG033514 (S.A.); by Veterans Administration Merit Review grant I01CX000165 (S.C.J.); and by a Clinical and Translational Science Award (UL1RR025011) to the University of Wisconsin, Madison. Portions of this research were supported by the Wisconsin Alumni Research Foundation, Helen Bader Foundation, Northwestern Mutual Foundation, Extendicare Foundation, and Veterans Administration, including facilities and resources at the Geriatric Research Education and Clinical Center of the William S. Middleton Memorial Veterans Hospital, Madison, WI. The funders' role was limited to providing funding for the study.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Di Paolo G, Kim TW. Linking lipids to Alzheimer's disease: cholesterol and beyond. Nat Rev Neurosci 2011;12:284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 1993;261:921–923. [DOI] [PubMed] [Google Scholar]
- 3.Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013;45:1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morgan K. The three new pathways leading to Alzheimer's disease. Neuropathol Appl Neurobiol 2011;37:353–357. [DOI] [PubMed] [Google Scholar]
- 5.Prince JA, Zetterberg H, Andreasen N, Marcusson J, Blennow K. APOE epsilon4 allele is associated with reduced cerebrospinal fluid levels of Abeta42. Neurology 2004;62:2116–2118. [DOI] [PubMed] [Google Scholar]
- 6.Elias-Sonnenschein LS, Helisalmi S, Natunen T, et al. Genetic loci associated with Alzheimer's disease and cerebrospinal fluid biomarkers in a Finnish case-control cohort. PLoS One 2013;8:e59676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hughes TM, Lopez OL, Evans RW, et al. Markers of cholesterol transport are associated with amyloid deposition in the brain. Neurobiol Aging 2014;35:802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karch CM, Goate AM. Alzheimer's disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry 2015;77:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leduc V, Jasmin-Belanger S, Poirier J. APOE and cholesterol homeostasis in Alzheimer's disease. Trends Mol Med 2010;16:469–477. [DOI] [PubMed] [Google Scholar]
- 10.Head D, Bugg JM, Goate AM, et al. Exercise engagement as a moderator of the effects of APOE genotype on amyloid deposition. Arch Neurol 2012;69:636–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sager MA, Hermann B, La Rue A. Middle-aged children of persons with Alzheimer's disease: APOE genotypes and cognitive function in the Wisconsin Registry for Alzheimer's Prevention. J Geriatr Psychiatry Neurol 2005;18:245–249. [DOI] [PubMed] [Google Scholar]
- 12.Engelman CD, Koscik RL, Jonaitis EM, et al. Interaction between two cholesterol metabolism genes influences memory: findings from the Wisconsin Registry for Alzheimer's Prevention. J Alzheimers Dis 2013;36:749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jurca R, Jackson AS, LaMonte MJ, et al. Assessing cardiorespiratory fitness without performing exercise testing. Am J Prev Med 2005;29:185–193. [DOI] [PubMed] [Google Scholar]
- 14.Boots EA, Schultz SA, Oh JM, et al. Cardiorespiratory fitness is associated with brain structure, cognition, and mood in a middle-aged cohort at risk for Alzheimer's disease. Brain Imaging Behav 2015;9:639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McTiernan A, Kooperberg C, White E, et al. Recreational physical activity and the risk of breast cancer in postmenopausal women: the Women's Health Initiative Cohort Study. JAMA 2003;290:1331–1336. [DOI] [PubMed] [Google Scholar]
- 16.Okonkwo OC, Schultz SA, Oh JM, et al. Physical activity attenuates age-related biomarker alterations in preclinical AD. Neurology 2014;83:1753–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kerr J, Marshall SJ, Patterson RE, et al. Objectively measured physical activity is related to cognitive function in older adults. J Am Geriatr Soc 2013;61:1927–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blom G. Statistical Estimates and Transformed Beta Variables. New York: Wiley; 1958. [Google Scholar]
- 19.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wineinger NE, Harper A, Libiger O, et al. Genomic risk models improve prediction of longitudinal lipid levels in children and young adults. Front Genet 2013;4:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hebert LE, Scherr PA, Beckett LA, et al. Age-specific incidence of Alzheimer's disease in a community population. JAMA 1995;273:1354–1359. [PubMed] [Google Scholar]
- 22.Palmqvist S, Zetterberg H, Blennow K, et al. Accuracy of brain amyloid detection in clinical practice using cerebrospinal fluid beta-amyloid 42: a cross-validation study against amyloid positron emission tomography. JAMA Neurol 2014;71:1282–1289. [DOI] [PubMed] [Google Scholar]
- 23.Lewczuk P, Lelental N, Spitzer P, Maler JM, Kornhuber J. Amyloid-beta 42/40 cerebrospinal fluid concentration ratio in the diagnostics of Alzheimer's disease: validation of two novel assays. J Alzheimers Dis 2015;43:183–191. [DOI] [PubMed] [Google Scholar]
- 24.Tabachnick BG, Fidell LS. Using Multivariate Statistics. 5th ed. Boston: Pearson/Allyn & Bacon; 2007. [Google Scholar]
- 25.O'Donovan G, Bakrania K, Ghouri N, et al. Nonexercise equations to estimate fitness in white European and South Asian men. Med Sci Sports Exerc 2016;48:854–859. [DOI] [PubMed] [Google Scholar]
- 26.Liang KY, Mintun MA, Fagan AM, et al. Exercise and Alzheimer's disease biomarkers in cognitively normal older adults. Ann Neurol 2010;68:311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mielke MM, Wiste HJ, Weigand SD, et al. Indicators of amyloid burden in a population-based study of cognitively normal elderly. Neurology 2012;79:1570–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vemuri P, Wiste HJ, Weigand SD, et al. Effect of apolipoprotein E on biomarkers of amyloid load and neuronal pathology in Alzheimer disease. Ann Neurol 2010;67:308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Racine AM, Adluru N, Alexander AL, et al. Associations between white matter microstructure and amyloid burden in preclinical Alzheimer's disease: a multimodal imaging investigation. Neuroimage Clin 2014;4:604–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bennet AM, Di Angelantonio E, Ye Z, et al. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA 2007;298:1300–1311. [DOI] [PubMed] [Google Scholar]
- 31.Jones L, Holmans PA, Hamshere ML, et al. Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer's disease. PLoS One 2010;5:e13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thambisetty M, Beason-Held LL, An Y, et al. Alzheimer risk variant CLU and brain function during aging. Biol Psychiatry 2013;73:399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Erickson KI, Banducci SE, Weinstein AM, et al. The brain-derived neurotrophic factor Val66Met polymorphism moderates an effect of physical activity on working memory performance. Psychol Sci 2013;24:1770–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferencz B, Laukka EJ, Welmer AK, et al. The benefits of staying active in old age: physical activity counteracts the negative influence of PICALM, BIN1, and CLU risk alleles on episodic memory functioning. Psychol Aging 2014;29:440–449. [DOI] [PubMed] [Google Scholar]
- 35.Brown BM, Bourgeat P, Peiffer JJ, et al. Influence of BDNF Val66Met on the relationship between physical activity and brain volume. Neurology 2014;83:1345–1352. [DOI] [PubMed] [Google Scholar]
- 36.Chuang YF, An Y, Bilgel M, et al. Midlife adiposity predicts earlier onset of Alzheimer's dementia, neuropathology and presymptomatic cerebral amyloid accumulation. Mol Psychiatry 2016;21:910–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hebert LE, Scherr PA, McCann JJ, Beckett LA, Evans DA. Is the risk of developing Alzheimer's disease greater for women than for men? Am J Epidemiol 2001;153:132–136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.