

Abstract
Polycythaemia vera (PV) is a myeloproliferative neoplasm classically characterized by an erythrocytosis and is associated with a high risk of thromboembolic events, constitutional symptoms burden and risk of transformation to myelofibrosis and acute myeloid leukaemia. Therapy is directed at the haematocrit (HCT) to reduce the risk of thrombotic events and usually comprises low-dose aspirin and phlebotomy to maintain HCT at >45%. Frequently in addition, cytoreductive therapy is indicated in high-risk patients for normalizing haematological parameters to mitigate the occurrence of thromboembolic events. Unfortunately, there is no clear evidence that current therapies reduce the risk of transformation to myelofibrosis and for some a risk of a therapy related complication is unknown for example leukaemia due to hydroxycarbamide (HC). First-line therapy for treating PV remains HC or interferon, the latter most often in younger patients, especially those of childbearing age. However, therapy related intolerance or resistance is a common feature and results in limited treatment options for such patients. The discovery of the JAK2 V617F mutation and consequently targeted therapy with Janus kinase inhibitors, in particular ruxolitinib, has extended the spectrum of agents that can be used as second or third line in PV. The findings of the phase II trial RESPONSE and the preliminary data from RESPONSE 2 trial have identified a role for ruxolitinib in PV patients who are resistant or intolerant to HC. In this article, using clinical cases we demonstrate our experience with ruxolitinib highlighting the clinical benefits and limitations we encountered in clinical practice.
Keywords: polycythaemia vera, ruxolitinib, treatment resistance
Introduction
Polycythaemia vera (PV) also known as Vasquez–Osler disease was first described in the 19th Century [Osler, 1908]. It is a myeloproliferative neoplasm (MPN) characterized by expansion of abnormal haematopoietic progenitor cells with an increased red cell mass, frequently in association with leucocytosis or thrombocytosis [Spivak, 2002; Tefferi et al. 2008]. PV is uncommon and has an estimated of prevalence of <60 cases per 100,000 persons [Mehta et al. 2014]. It is generally diagnosed in the 6th or 7th decade of life; however, almost 20% of diagnoses are made in patients aged under 40 years [Tibes and Mesa, 2013; Vannucchi, 2014]. PV is associated with a risk of thromboembolic events [Marchioli et al. 2005] in addition to a predisposition to transforming into myelofibrosis (MF) and acute myeloid leukaemia (AML) [Finazzi et al. 2005].
Over 95% of patients with PV have a mutation in exon 14 of JAK2, JAK2 V617F, and the majority of the remaining patients have one of a number of mutations in exon 12 of JAK2. The pathogenesis of PV is attributed to upregulation of the Janus kinase-signal transducers and activators of transcription (JAK-STAT) pathway resulting in erythrocytosis, leucocytosis and thrombocytosis which are the pathognomic features of PV. As therapy is focused at normalizing these haematological parameters, greater understanding of the role of the JAK-STAT pathway in PV has facilitated targeted therapy with JAK inhibitors such as ruxolitinib, the subject of this review.
The clinical presentation of PV can vary from the asymptomatic individual, or a patient with symptoms (classically itching or pruritus) to one who is diagnosed following presentation with a thromboembolic event. In 1213 PV patients followed for 20 years, 64% of arterial and venous thrombosis event occurred either at presentation or before diagnosis and there was a 3.4% per year incidence of thrombosis during follow up [Gruppo Italiano Studio, 1995]. We base our diagnosis of PV on the British Committee for Standards in Haematology (BCSH) guidelines, which are broadly similar to the revised World Health Organization (WHO) diagnostic criteria, as outlined in Table 1 [McMullin et al. 2005, 2007; Arber et al. 2016]. It important to obtain a thorough history and examination to exclude secondary causes of erythrocytosis in the diagnostic workup, as well as seeking cardiovascular risk factors.
Table 1.
Diagnosis of polycythaemia vera – The British Committee for Standards in Hematology (BCSH) and the revised World Health Organization (WHO) diagnostic criteria [McMullin et al. 2007; Arber et al. 2016].
| BCSH criteria | Revised WHO criteria | |
|---|---|---|
| Major criteria | Raised haematocrit (>52% in men or 48% in women) OR Raised red cell mass (>25% above predicted) |
1. Haemoglobin >10.5 g/dl in men; Haemoglobin >16.0 g/dl in women OR 2. Haematocrit >49% in men; Haematocrit >48% in women OR Increased red cell mass 3. BM biopsy showing hypercellularity for age with panmyelosis including prominent erythroid, granulocytic, and megakaryocytic proliferation with pleomorphic, mature megakaryocytes 4. JAK2 V617F or JAK2 exon 12 mutation |
| Minor criteria |
A1: Raised haematocrit (>60% in men or 56% in women) OR Raised red cell mass (>25% above predicted) A2: JAK2 mutation absent A3: No cause of secondary erythrocytosis A4: Palpable splenomegaly A5: Clonality marker (exclude BCR-ABL) B1: Thrombocytosis (platelet count > 400 × 109/l) B2: Neutrophil leucocytosis (neutrophil count > 10 × 109/l in nonsmokers; >12.5 × 109/l in smokers) B3: Splenomegaly on imaging B4: Endogenous erythroid colonies or low serum erythropoietin |
1. Suboptimal serum erythropoietin level |
| Diagnosis of PV requires: |
Both major criteria to be present OR: A1 + A2 + A3 + either another A or two B criteria |
All three major criteria OR The first two major criteria and the minor criterion |
BM, bone marrow; PV, polycythaemia vera.
Symptoms of PV are variable (Figure 1). The symptom burden can be considerable; common symptoms include fatigue, pruritus, facial flushing and headache in addition to nonspecific microvascular occlusion related symptoms such erythromelalgia [McMullin et al. 2005; Mesa et al. 2007; Vannucchi, 2014]. Patients can have a substantial disease burden that may interfere with their quality of life [Stein et al. 2014]. It is important to recognize the impact of symptoms and not only focus on correcting haematological parameters when treating patients.
Figure 1.

Frequency of symptoms reported by patients with polycythaemia vera [Geyer et al. 2016; Vannucchi, 2014].
Risk stratification and management
There is a need to review risk stratification for patients with PV as current models have been based upon retrospective cohorts of patients with variable treatment. In a recent large retrospective study, the median survival was over 25 years in the low-risk group and as low as 10 years in the high-risk cohort [Tefferi et al. 2013]. The intermediate-risk category is generally unclear, for it might refer for example to the younger patient who has cardiovascular risk factors in the absence of a thromboembolic event. In our practice, we usually manage this group of patients as high-risk [McMullin et al. 2005]. We follow guidance of the BCSH for risk stratification of our patients which incorporates a patient’s age and history of thromboembolic events; we also consider the presence of constitutional symptoms. As demonstrated in Figure 2, patients who are high risk require prompt initiation of cytoreductive therapy.
Figure 2.

Risk stratification in polycythaemia vera and recommended management approach in accordance with risk category [McMullin et al. 2005; Vannucchi, 2014; Tefferi et al. 2015].
Although the literature is conflicting, there is a trend that a higher JAK2 V617F allele burden is associated with splenomegaly, constitutional symptoms and transformation to MF [Passamonti et al. 2010]. Leucocytosis and thrombocytosis are also important to consider as both may have an impact on short and long-term outcomes; however, these do not feature in the standard risk stratification frameworks [Vannucchi, 2014]. In our clinical practice if a patient has a leucocytosis of 15–20 × 109/l, or the leucocyte count is rapidly increasing we would consider this a high-risk feature and discuss initiating cytoreductive therapy. There is no clear evidence that patients with JAK2 exon 12 mutations need to be managed differently, nor as yet a role for detecting additional epigenetic abnormalities.
Therapy goals in PV aim to reduce the risk of acquiring a thromboembolic event as well enhancing quality of life of patients who have constitutional symptoms. On diagnosis we invest time in discussing the long and short term implications of the diagnosis with the patient as well the importance of risk stratification. We advise all patients to adopt a healthier lifestyle and work with community doctors to optimize blood pressure and cholesterol control. We encourage smoking cessation and weight loss in those with raised body mass index.
Our target haematological parameters are reducing the haematocrit (HCT) to <45% (although this may differ at altitude and in pregnancy) and normalization of white cell and platelet count. Patients categorized as low risk are managed with low-dose aspirin 75 mg daily and phlebotomy, whilst cytoreductive therapy is additionally offered to high-risk patients. The Italian Cytoreductive Therapy in PV collaborative group demonstrated lower mortality and vascular events in patients with HCT <45% when compared with those whose HCT was 45–50% (mortality and vascular events were reposted as 1.3% versus 3.3% and 2.7% versus 9.8% respectively) [Marchioli et al. 2011, 2013]. Future goals of therapy aim to delay progression to post PV, MF or AML; the extent to which this is impacted by current therapy is unclear but is a major unmet need. In the long-term follow up of the French Polycythaemia Study Group study, Kiladjian and colleagues reported rates of progression to MF and AML were 32% and 24% at 20 years respectively in hydroxycarbamide (HC)-treated patients [Kiladjian et al. 2011]; there remains insufficient data on progression with interferon (IFN) use.
Phlebotomy
Hyperviscosity remains critical in the pathogenesis of the thromboembolic events, therefore reduction of HCT is an integral component of management [Kumar et al. 2009]. Our target HCT is 45% or less, as this was established to be associated with reduced thromboembolic complications [Marchioli et al. 2013] whether this should be lower in specific circumstances (e.g. pregnancy, splanchnic vein thrombosis or in women) is unclear. Phlebotomy unfortunately does not address the leucocytosis, thrombocytosis, or improve the constitutional symptom burden in many patients and in some subject it may not achieve adequate HCT control [Marchioli et al. 2011]. Phlebotomy is usually well tolerated specially in the younger cohort; however, it is important to recognize over 10% of patients may harbour anxiety related to needles and have a fear of phlebotomy [Deacon et al. 2006]. Furthermore, phlebotomy can result in iron deficiency; which may cause additional symptoms such as restless legs or exacerbate existing fatigue and pruritus. Therefore, phlebotomy may not be suitable for some subjects resulting in discontinuation as described in the CYTO-PV study [Marchioli et al. 2011; Prchal and Gordeuk, 2013]. Moreover, in a recent analysis from a Spanish registry, patients who required three or more phlebotomies per year in addition to cytoreductive therapy were at higher risk of thrombosis, highlighting the importance of maintaining good HCT control [Alvarez-Larrán et al. 2016].
Antiplatelet agents
Regardless of risk category the majority of PV patients receive low-dose aspirin 75 mg daily. Our practice is based on internationally agreed consensus following the reporting of the European Collaboration on Low-dose Aspirin in PV (ECLAP), which demonstrated lower rates of myocardial infarction, stroke, and pulmonary embolism in patients treated with low-dose aspirin with no significant increase in haemorrhagic events [Landolfi et al. 2004]. Cumulative rates of nonfatal thrombosis and cardiovascular mortality were 3.8 versus 1.5 events per 100 patient-years. Aspirin must be used with caution due to the risk of acquired Von Willebrand disease in PV patients with extreme thrombocytosis (platelet count > 1000 × 109/l) [Vannucchi, 2014].
Cytoreductive therapy
Our approach to cytoreductive therapy is individualized and we manage patients in accordance with their preference, tolerance and clinical needs. Hydroxycarbamide HC a ribonucleotide reductase inhibitor has an established history in haematological disorders and is the most commonly used agent in PV [Vannucchi, 2014]. A number of studies, including the phase II PV Study Group reported superior outcomes in reducing thrombotic events with HC when compared with phlebotomy [Fruchtman et al. 1997]. We use HC with caution in patients aged <60 years due to teratogenicity, proven risk of skin cancer and potential risks of AML [Finazzi and Barbui, 2008]. However, it is important to consider that numerous large trials have failed to demonstrate a significant risk of leukaemic transformation with HC [Tefferi et al. 2013]. HC is associated with side effects such as mucocutaneous ulceration, hair thinning, and fatigue resulting in intolerance [Sever et al. 2014].
Recombinant IFN has antiapoptotic, antiproliferative, and immunomodulatory properties and is widely employed as an antiviral and antineoplastic agent [Stein and Tiu, 2013]. IFN has been utilized in treatment of PV for over 20 years and has proved to induce haematological remission, achieve a morphological and molecular response manifested by reduction in the JAK2 V617F allele burden, with a complete molecular response sustained even after discontinuation of treatment in some patients [Silver, 1988; Kiladjian et al. 2008; Quintas-Cardama et al. 2009; Silver et al. 2013]. Interestingly patients who have the additional TET2 mutation, the clones commonly persist during IFN therapy despite eradication of the JAK2 V617F clone [Quintas-Cardama et al. 2013]. The use of IFN has been limited mainly by its side effect profile, mode of administration and lack of availability in many countries. We primarily offer it to patients under the age of 60 years and those of childbearing age [McMullin et al. 2005]. The side effect profile includes flu like symptoms, mood disturbances, fatigue, hair thinning, deranged liver function tests and thyroid dysfunction. The development of PEGylated IFN with better tolerability and less frequent administration has made it more of an attractive option for patients [Them et al. 2015]. Several studies are in progress which aim to evaluate IFN when compared with HC in treating PV. For example, the phase III PROUD-PV study [ClinicalTrials.gov identifier: NCT01949895] evaluated the use of ropeginterferon alpha (a novel monopegylated interferon alpha 2b) versus HC and reports both treatments to be equally well tolerated with dropout rates of 15% in each arm. In the preliminary pooled analysis, 45% of patients achieved a haematological response with a reduction in phlebotomy from 86% to 6% within 3 months [Gisslinger et al. 2016].
Busulfan (BU) has been used successfully in PV since 1958 [Louis, 1958]. In our practice, we reserve it for older (generally over 75 years) patients who are intolerant of HC and not suitable for IFN. The Spanish group showed BU was an effective option for PV patients intolerant or resistant to first-line therapies, with over 80% of patients achieving complete haematological response [Alvarez-Larran et al. 2014]. From our experience BU, is well tolerated and appreciated by patients who favour drug-free days. It can be associated with higher risk of leukaemia transformation, especially when used long term, therefore its use is avoided in younger patients [McMullin et al. 2005; Vannucchi, 2014]. We do not usually prescribe agents such as P32 or pipobroman due to the increased risk of leukaemic transformation [Tefferi et al. 2013].
Resistance and intolerance
In our experience, first-line or second-line options for the high-risk patients can be effective in the majority of PV patients. However, for a significant number adequate control is not gained because of resistance, intolerance or both. For example, HC is not tolerated or is ineffective in almost 25% of PV patients [Alvarez-Larran et al. 2012]. Alvarez-Larrán and colleagues were also able to report that HC resistant patients had higher rates of transformation to post polycythaemia vera myelofibrosis (PPV-MF) and AML with higher mortality [Alvarez-Larran et al. 2012]. The European Leukaemia Network (ELN) has developed a comprehensive criterion for resistance and intolerance to HC; we assess our patients using a modified version more amenable to everyday practice (Table 2). The availability of targeted therapy with the JAK1/2 inhibitor ruxolitinib has provided another treatment option for this group as will be discussed below.
Table 2.
European Leukaemia Network (ELN) criteria for resistance or intolerance to hydroxycarbamide (HC) adopted for our clinical practice [Barosi et al. 2010; Alvarez-Larran et al. 2012; McMullin et al. 2016].
| Definition | |
|---|---|
| Resistance intolerance | • Ongoing phlebotomy to Maintain HCT <45% after 3 months of at least 2 g/day of HC or uncontrolled myeloproliferation (i.e. platelet count 400 × 109/l AND white blood cell count 10 × 109/l) after 3 months of at least 2 g/day of HC or maximum tolerated dose • Failure to reduce massive splenomegaly by 50% as measured by palpation OR failure to completely relieve symptoms related to splenomegaly after 3 months of at least 2 g/day of HC or maximum tolerated dose • Failure to control disease-related symptoms (including but not limited to those relating to splenomegaly) • Thrombosis or haemorrhage related to disease despite therapy • Absolute neutrophil count 1.0 × 109/l OR platelet count 100 × 109/l OR haemoglobin 10 g/dl at the lowest dose of HC required to achieve a complete or partial clinic haematological response • Presence of leg ulcers or other unacceptable HC related nonhaematological toxicities, such as mucocutaneous manifestations, GI symptoms, pneumonitis, or fever at any dose of HC |
Italic text – modified criteria adopted for clinical practice.
GI, gastrointestinal; HC, hydroxycarbamide; HCT, haematocrit.
JAK inhibitors
The discovery of JAK2 V617F mutation in 2005 has changed the clinical arena of myeloproliferative neoplasms and has led to the development of the first JAK 1/2 inhibitor ruxolitinib, with promising clinical responses initially demonstrated in the MF patients in the COMFORT I and COMFORT II studies [Cervantes et al. 2013; Mesa et al. 2014]. It was then rational to trial the effectiveness of JAK inhibitors in PV due to the integral role the JAK-STAT pathway has in the pathogenesis of the disease. Use of ruxolitinib in PV was investigated by Verstovsek and colleagues in a phase II study [ClinicalTrials.gov identifier: NCT00726232] that enrolled 34 patients with HC resistance or intolerance for a median of 35 months [Verstovsek et al. 2014]. The study showed durable control of HCT, with cessation of phlebotomy in 97% of patients by week 24. Furthermore, in those with a palpable splenomegaly, there was at least a 50% reduction in spleen size in over 70% of patients within 24 weeks. An improvement in constitutional symptoms were observed within 4 weeks of initiating therapy with ruxolitinib. Interestingly, Verstovsek and colleagues also described a reduction in inflammatory cytokines and granulocyte activation which are thought to be the key drivers of the constitutional symptom burden [Hasselbalch and Bjorn, 2015].
Ruxolitinib has now been approved in the United States Food and Drug Administration and in Europe, by the European Medicines Agency for the treatment of patients with PV who have developed HC resistance or intolerance. This was based on the encouraging results of the phase III open label RESPONSE study [ClinicalTrials.gov identifier: NCT01243944], a multicentre study which compared ruxolitinib versus best available therapy (BAT) in patients with PV who were intolerant or resistant to HC and had splenomegaly with ongoing requirements for phlebotomy. The RESPONSE study’s primary endpoints evaluated were the combination of freedom from phlebotomy and spleen volume reduction >35%. The 32-week and 80-week outcome analysis were consistent, demonstrating superiority of ruxolitinib to BAT in controlling HCT, reducing spleen size and improving the constitutional symptoms [Vannucchi et al. 2015; Verstovsek et al. 2016].
At 32 weeks, 40% of patients in the ruxolitinib versus 1% in the BAT arm achieved at least 35% reduction in splenomegaly (p ⩽ 0.001). Furthermore, over 60% of patients on ruxolitinib versus 19% on BAT had gained control of HCT in the absence of phlebotomy. The constitutional symptom burden was significantly improved and complete haematological response (CHR) was achieved in almost 25% of patients in the ruxolitinib versus 9% in BAT. Here, CHR was defined as HCT < 45%, platelet < 400 × 109/l and white blood count < 10 × 109/l. At 32 weeks, 87% of patients in the BAT arm crossed over to ruxolitinib. Almost 70% of patients maintained the CHR at 80 weeks. At the 32-week evaluation, a five-fold higher number of thromboembolic events in the BAT arm compared with ruxolitinib was observed, and at 80-week the thromboembolic event rate per 100 patient-years was 1.8 in the ruxolitinib arm versus 8.2 in BAT [Vannucchi et al. 2015; Verstovsek et al. 2016]. This was not a predefined outcome at the start of the study.
Most of the haematological and nonhaematological adverse effects reported in the RESPONSE study were grade 1 or 2, and in the 80-week analysis there were no major changes in the adverse events profile [Verstovsek et al. 2016]. The most common nonhaematological adverse events were headache, diarrhoea dyspnoea, abdominal pain and fatigue. The rate of Herpes zoster was 5.3 per 100 patient-years in the ruxolitinib arm, versus none in the BAT cohort, although this was mostly grade 1 or 2 also and resolved with no long-term sequelae [Verstovsek et al. 2016]. Haematological toxicity commonly included grade 1 or 2 thrombocytopaenia. There was a two-fold increase in nonmelanoma skin cancer: 4.4 per 100 patient-years exposure with ruxolitinib versus 2.7 in BAT arm [Verstovsek et al. 2016]. At 80-weeks, there was a trend towards higher rates of transformation to PPV-MF and AML in the ruxolitinib arm when compared with BAT [Vannucchi et al. 2015; Verstovsek et al. 2016].
The RESPONSE study provided an invaluable evaluation for the role of ruxolitinib in a difficult cohort of PV patients, however, it had several shortfalls, firstly that only 20% of patients achieved the composite primary endpoint. Over 50% of patients on the BAT arm remained on HC despite resistance or intolerance, which raises the question of efficacy and compliance in the BAT arm. The study endpoints did not include risk of thromboembolic events and the findings of lower thromboembolic events in the ruxolitinib arm was not a predetermined endpoint. Similar to the COMFORT studies, a 35% reduction in spleen was a primary endpoint; however, a significant proportion of PV patients do not have an enlarged spleen, this factor was addressed by the RESPONSE 2 study. There needs to be long-term follow up of the RESPONSE trial patients to monitor the trends for transformation and thrombosis rates in the ruxolitinib cohort.
The RESPONSE 2 trial [ClinicalTrials.gov identifier: NCT02038036], a phase III open label multicentre trial evaluated the use of ruxolitinib in PV patients with intolerance or resistant to HC in the absence of splenomegaly [Passamonti et al. 2014]. The preliminary results were presented at the European Association Annual meeting in Copenhagen 2016, and suggests ongoing encouraging results in this cohort of difficult patients. HCT control was achieved in 60% of ruxolitinib arm versus 19% in BAT, with over 50% of ruxolitinib-treated patients reporting improvement in their symptoms versus 5% in the BAT arm and almost 25% of patients achieved CHR versus 5% in BAT arm. [Passamonti et al. 2017]. The RESPONSE 2 study was necessary to address some of the shortcomings of the RESPONSE trial, such as the effectiveness of ruxolitinib in the absence of splenomegaly. However, the trial continues to compare ruxolitinib with BAT in a difficult cohort of patients, small numbers were recruited and no predetermined endpoints for thrombosis or transformation to MF or AML.
The phase III Randomized Switch Study from HC to Ruxolitinib for RELIEF of PPV symptoms trial [ClinicalTrials.gov identifier: NCT0632904] evaluated the efficacy and safety of ruxolitinib versus HC for the control of disease-related symptoms (cytokine total symptom score ; TSS-C) in patients who have controlled PV haematological parameters but continue to report symptoms on HU. At 16 weeks, 43% in the ruxolitinib arm and 29.6% in the HU arm achieved a ⩾50% reduction from baseline in TSS-C. Specifically, on evaluating relieve of pruritus and fatigue, the proportion of patients in the ruxolitinib versus HC arms achieving a ⩾50% reduction symptom score were 40% versus 26% (p > 0.05) and 54% versus 32% (p > 0.05), respectively. The trial did not demonstrate a significant difference in symptom control between ruxolitinib and HC; however, it did show that ruxolitinib was well tolerated. The commonest nonhaematological side effects included fatigue (20% ruxolitinib versus 11% HC), headache (17% versus 5%), and dizziness (13% versus 9%). There was no grade 3 or 4 anaemia or thrombocytopaenia reported in the ruxolitinib arm [Mesa et al. 2017].
The outcomes of the outlined studies suggest ruxolitinib is generally well tolerated, and it appears that the side effect profile in PV mirrors that reported previously reported in MF. The success of targeted therapy in other myeloproliferative neoplasms such as chronic myeloid leukaemia with tyrosine kinase inhibitors, was evidenced by the reduction of the burden of disease-causing mutation to achieve haematological and molecular remission. In the long-term follow up of 236 JAK2 V617F-positive MF patients treated with ruxolitinib, Deininger and colleagues reported a 50% reduction in JAK2 V617F allele burden in 28% of patients; six patients (2.5%) had values below quantifiable limits [Deininger et al. 2015]. JAK2V617F mutant clone reduction was also demonstrated in the RESPONSE study, with reductions of 12% at week 32, 22% at week 80, and 40% at week 208 [Vannucchi et al. 2015]. Although monitoring of allele burden remains investigational and has not been integrated into any of the risk stratification algorithms, there is increasing speculation that high JAK2 V617F allele burden correlates with disease progression and constitutional symptoms [Zhao et al. 2016].
There is also interest in employing histone deacetylase (HDAC) inhibitors in PV, such an agent was givinostat which resulted in improved symptom control and reduction in spleen size in combination with HC in patients resistant or intolerant to HC [Finazzi et al. 2013]. There is ongoing interest in targeted therapy with these agents. More sophisticated is the quest for combination therapy, targeting the haematological and constitutional symptom control achieved by ruxolitinib in combination with IFN which has demonstrated impressive reductions in JAK2 V617F allele burden [Bjorn et al. 2015]. The Danish COMBI-Trial further reported 75% of patients receiving the combination therapy achieved CHR within 3 months. However, there needs to be long-term assessment of the benefits and drawbacks of such a combination, in particular with regards to tolerability and long-term complications.
Clinical scenarios
Many clinicians will encounter a challenging patient with PV; the following two clinical cases are real scenarios we have encountered in managing with resistance or intolerance to first-line therapies. The two cases illustrate our experience in using ruxolitinib and considerations that need to be taken in the event of adverse effects.
Case 1
A 50-year-old female, with no significant past medical history, presented with acute abdominal discomfort, fatigue and facial flushing. On presentation, her full blood parameters were: total white cell count (wcc) 9.6 × 109/l, haemoglobin (Hb) 171 g/l, HCT 52%, platelets 1322 × 109/l and neutrophil count of 7.6 × 109/l. Ultrasound imaging demonstrated an enlarged spleen measuring 16 cm. On review there was no history of weight loss or night sweats, the peripheral blood smear was normal, JAK2 V617F was positive, erythropoietin level was reduced and the bone marrow was compatible with a diagnosis of PV.
The patient began a phlebotomy programme and low-dose aspirin (75 mg daily). Unfortunately, her symptoms continued unabated, and she needed to attend for phlebotomy every 6 weeks. Altogether these issues resulted in the loss of her job, and an inability to care for her family. Thus, in view of the ongoing need for phlebotomy, the elevated platelet count and profound constitutional symptom burden she was commenced on HC (patient preference). As depicted in Figure 3, despite escalation of the daily dose to 2 g daily, the HCT and platelet count remained elevated, and her symptoms persisted. She was referred to our centre, HC was stopped and PEGylated IFN at a dose of 45 mcg weekly was started. However, despite an IFN dose of 225 µg weekly she remained symptomatic with worsening splenomegaly and ongoing requirements for phlebotomy. As she fulfilled the modified ELN criteria for resistance (Table 2) to both HC and failed to respond to PEGylated IFN, she commenced on ruxolitinib 10 mg daily; her constitutional symptom burden improved, with resolution of splenomegaly and normalization of her haematological parameters.
Figure 3.

Treatment responses to hydroxycarbamide (HC), interferon (IFN) and ruxolitinib (Rux) in a patient intolerant of HC.
HC, hydroxycarbamide; HCT, haematocrit; IFN, interferon; Rux, ruxolitinib.
Unfortunately, 12 weeks into her treatment with ruxolitinib, she developed a Herpes zoster infection which was treated effectively with the oral antiviral drug aciclovir. She has otherwise remained well and independent of phlebotomy, and takes prophylactic aciclovir.
Case 2
A 57-year-old male, with a diagnosis of JAK2 V617F positive PV complicated by a hepatic vein thrombosis diagnosed 12 months previously, was referred for a second opinion. His full blood count was: Hb 165 g/l, wcc 8.4 × 109 /l, platelets 319 × 109/l, HCT 48%. He had been anticoagulated and phlebotomy was started. As he had a history of severe depression, he commenced on HC rather than IFN (Figure 4). Unfortunately, with a HC dose escalation to 500 mg twice daily, he began to develop oral ulcers; this was followed by lower limb ulceration when the dose of HC was escalated to 1000 mg twice a day. A dose reduction in HC was made, however, the ulcers did not improve, and he began to require increasing numbers of phlebotomies. He developed iron deficiency anaemia with worsening fatigue. We therefore commenced him on ruxolitinib 10 mg twice daily, and escalated to 20 mg twice daily within 6 weeks. This last dose achieved good control of HCT, and eliminated the need for regular phlebotomy. The patient tolerated the ruxolitinib, however, he gained over 14 pounds in body weight. He then was referred to a dietician, although he modified his diet, he continues to be above his baseline weight and has required initiation of cholesterol-lowering drugs. Later he became thrombocytopaenic, with a platelet count of 100 × 109 /l and the dose of ruxolitinib was subsequently reduced to 15 mg twice daily. Unfortunately, this failed to control his HCT and consequently the higher dose was reinstated. We continue to monitor the platelet count regularly.
Figure 4.

Treatment responses to hydroxycarbamide (HC) and ruxolitinib (Rux) in a patient intolerant of HC. HC, hydroxycarbamide; HCT, haematocrit; IFN, interferon; Rux, ruxolitinib.
The two clinical cases demonstrated real life examples encountered in managing patients with PV. Ruxolitinib has provided an option for symptomatic and high-risk patients who are intolerant to first-line therapeutic agents. Both patients highlighted in the cases became phlebotomy-independent with obvious improvement in their quality of life, however both had significant adverse effects that required intervention. Herpes zoster infection was noted in over 5% of patients in the RESPONSE trial compared with none in the BAT, confirming the immune vulnerability of patients on JAK inhibitors [Vannucchi, 2015]. We treat promptly with antiviral medications, and if we experience recurrences we may opt to commence patients on prophylaxis treatment [Galli et al. 2014]. In our experience, the most frequently reported adverse effects are that of headaches and dizziness, which are short lived and tolerated by patients which may respond to a dose reduction [Galli et al. 2014]. Like our patient, in the RESPONSE study, haematological toxicity was primarily of grade 1 or 2; however, almost 4% had grade 3 or 4 anaemia and thrombocytopaenia. Weight gain is a common adverse event which we encounter, and we always offer a review by a dietician.
On commencing ruxolitinib we counsel all our patients with regards to the risk of cytopaenia, weight gain, immunosuppression and also long-term risk of nonmelanoma skin neoplasms [Verstovsek, 2013; Vannucchi et al. 2015]. We perform baseline virology in our patients including hepatitis B, C and HIV. If we diagnose a new case of hepatitis or HIV we co-manage their case with our colleagues from the infectious diseases department. We have successfully treated a number of patients who have the hepatitis B virus. Depending on symptoms and haematological values, on commencing ruxolitinib it is our practice to initiate treatment at a dose of 10 mg twice daily and escalate depending on tolerance and response. Finally, we review haematological and clinical response regularly.
Currently, ruxolitinib is the only JAK inhibitor available for patients with PV, and has demonstrated superior outcomes in clinical trials improving constitutional symptoms, HCT control and reducing spleen size [Vannucchi, 2015]. However, it is a relatively costly therapeutic option in the face of a financially challenged healthcare system. It is important to note that not all trials demonstrate superiority of ruxolitinib over BAT; for example, the phase II RELIEF [ClinicalTrials.gov identifier: NCT01632904] study, evaluated ruxolitinib versus HC in the absence of resistance or intolerance and did not report any significant difference in outcomes between the two arms (Mesa et al. 2017). To date, we remain unaware of the impact of JAK inhibitors in reducing the risk of thromboembolic events or frequency of transformation to MF or AML in patients with PV. The multicentre randomized phase II MAJIC study (EudraCT.201100527918) is evaluating the efficacy and safety of ruxolitinib versus BAT in patients with high-risk PV and essential thrombocythaemia who are resistant or intolerant to HC. This trial will be informative in guiding our understanding of the rates of thromboembolic events in ruxolitinib-treated patients and rate of transformations and molecular response in patients managed with ruxolitinib.
In summary, our current practice in low-risk patients remains aspirin and phlebotomy as the standard of care. In patients who fall into the high-risk category and require cytoreduction, both HC and IFN are effective agents tolerated by the majority of patients, in our experience. In those who develop resistance or intolerance, we recognize that ruxolitinib will have an integral role. For the PV patient who has intolerance or resistance to first or second-line therapies, ruxolitinib has become an invaluable option in an otherwise limited pharmacotherapy platform for high-risk patients. Figure 5 summaries our current approach to managing patients with PV.
Figure 5.
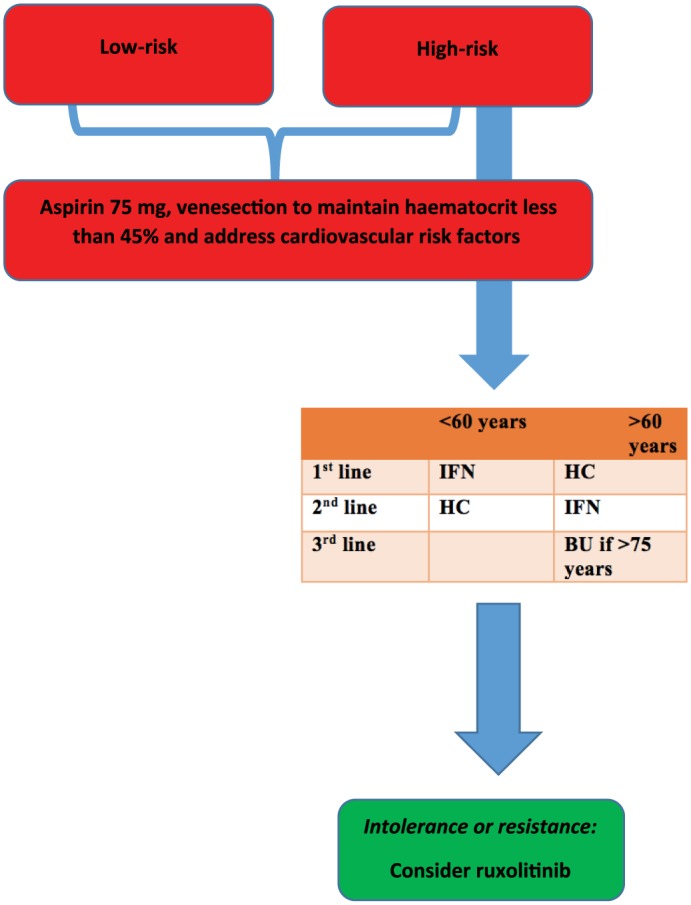
Our local approach to managing patients with polycythaemia vera.
BU, busulfan; HC, hydroxycarbamide; IFN, interferon.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement: The authors declare that there is no conflict of interest.
Contributor Information
Samah Alimam, Guy’s and St Thomas’s NHS Foundation Trust, London, UK.
Claire Harrison, Guy’s and St Thomas’s NHS Foundation Trust, London, UK.
References
- Alvarez-Larrán A., Martínez-Aviles L., Hernández-Boluda J., Ferrer-Marín F., Antelo M., Burgaleta C., et al. (2014) Busulfan in patients with polycythemia vera or essential thrombocythemia refractory or intolerant to hydroxyurea. Ann Hematol 93: 2037–2043. [DOI] [PubMed] [Google Scholar]
- Alvarez-Larrán A., Pereira A., Cervantes F., Arellano-Rodrigo E., Hernández-Boluda J., Ferrer-Marín F., et al. (2012) Assessment and prognostic value of the European leukaemia criteria for clinic hematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood 119: 1363–1369. [DOI] [PubMed] [Google Scholar]
- Alvarez-Larrán A., Pérez-Encinas M., Ferrer-Marín F., Hernández-Boluda J., Ramírez M, Martínez J., et al. (2016) Prevention of thrombosis in patients with polycythemia vera treated with hydroxuyurea plus phlebotomies or hydroxyurea alone. EHA Meeting Abstract P301. [Google Scholar]
- Arber D., Orazi A., Hasserjian R., Thiele J., Borowitz M., Le Beau M., et al. (2016) The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127: 2391–2405. [DOI] [PubMed] [Google Scholar]
- Barosi G., Birgegard G., Finazzi G., Griesshammer M., Harrison C., Hasselbalch H., et al. (2010) A unified definition of clinical resistance and intolerance to hydroxycarbamide in polycythaemia vera and primary myelofibrosis: results of a European Leukemianet (Eln) Consensus Process. Br J Haematol 148: 961–963. [DOI] [PubMed] [Google Scholar]
- Bjorn M., De Stricker K., Kjaer L., Ellemann K., Hasselbalch H. (2015) Corrigendum to “Combination therapy with interferon and JAK1-2 inhibitor is feasible. Proof of concept with rapid reduction in JAK2V617F-allele burden in polycythemia vera” [Leuk. Res. Rep. 3 (2) (2014) 73–75]. Leuk Res Rep 4: 31.26101742 [Google Scholar]
- Cervantes F., Vannucchi A., Kiladjian J., Al-Ali H., Sirulnik A., Stalbovskaya V., et al. (2013) Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood 122: 4047–4053. [DOI] [PubMed] [Google Scholar]
- Deacon B., Abramowitz J. (2006) Fear of needles and vasovagal reactions among phlebotomy patients. J Anxiety Disord 20: 946–960. [DOI] [PubMed] [Google Scholar]
- Deininger M., Radich J., Burn T., Huber R., Paranagama D., Verstovsek S. (2015) The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood 126: 1551–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finazzi G., Barbui T. (2008) Evidence and expertise in the management of polycythemia vera and essential thrombocythemia. Leukemia 22: 1494–1502. [DOI] [PubMed] [Google Scholar]
- Finazzi G., Caruso V., Marchioli R., Capnist G., Chisesi T., Finelli C., et al. (2005) Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood 105: 2664–2670. [DOI] [PubMed] [Google Scholar]
- Finazzi G., Vannucchi A., Martinelli V., Ruggeri M., Nobile F., Specchia G., et al. (2013) A phase II study of givinostat in combination with hydroxycarbamide in patients with polycythaemia vera unresponsive to hydroxycarbamide monotherapy. Br J Haematol 161: 688–694. [DOI] [PubMed] [Google Scholar]
- Fruchtman S., Mack K., Kaplan M., Peterson P., Berk P., Wasserman L. (1997) From efficacy to safety: a polycythemia vera study group report on hydroxyurea in patients with polycythemia vera. Semin Hematol 34: 17–23. [PubMed] [Google Scholar]
- Galli S., Mclornan D., Harrison C. (2014) Safety evaluation of ruxolitinib for treating myelofibrosis. Expert Opin Drug Saf 13: 967–976. [DOI] [PubMed] [Google Scholar]
- Geyer H., Scherber R., Kosiorek H., Dueck A., Kiladjian J., Xiao Z., et al. (2016) Symptomatic profiles of patients with polycythemia vera: implications of inadequately controlled disease. J Clin Oncol 34: 151–159. [DOI] [PubMed] [Google Scholar]
- Gisslinger H., Klade C., Georgiev P., Skotnicki A., Gercheva-Kyuchukova L., Egyed M., et al. (2016) Final results from PROUD-PV a randomized controlled phase 3 trial comparing ropeginterferon alfa-2b to hydroxyurea in polycythemia vera patients. ASH Annual Meeting Session 634: Abstract 475. [Google Scholar]
- Gruppo Italiano Studio P. (1995) Polycythemia vera: the natural history of 1213 patients followed for 20 years. Ann Intern Med 123: 656–664. [DOI] [PubMed] [Google Scholar]
- Hasselbalch H., Bjorn M. (2015) MPNs as inflammatory diseases: the evidence, consequences, and perspectives. Mediators Inflamm 2015: 102476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiladjian J., Cassinat B., Chevret S., Turlure P., Cambier N., Roussel M., et al. (2008) Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 112: 3065–3072. [DOI] [PubMed] [Google Scholar]
- Kiladjian J., Chevret S., Dosquet C., Chomienne C., Rain J. (2011) Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncol 29: 3907–3913. [DOI] [PubMed] [Google Scholar]
- Kumar C., Purandare A., Lee F., Lorenzi M. (2009) Kinase drug discovery approaches in chronic myeloproliferative disorders. Oncogene 28: 2305–2313. [DOI] [PubMed] [Google Scholar]
- Landolfi R., Marchioli R., Kutti J., Gisslinger H., Tognoni G., Patrono C., et al. (2004) Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med 350: 114–124. [DOI] [PubMed] [Google Scholar]
- Louis J. (1958) Treatment of polycythemia vera with busulfan (myleran). J Am Med Assoc 168: 1880–1882. [DOI] [PubMed] [Google Scholar]
- Marchioli R., Finazzi G., Landolfi R., Kutti J., Gisslinger H., Patrono C., et al. (2005) Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol 23: 2224–2232. [DOI] [PubMed] [Google Scholar]
- Marchioli R., Finazzi G., Specchia G., Cacciola R., Cavazzina R., Cilloni D., et al. (2013) Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med 368: 22–33. [DOI] [PubMed] [Google Scholar]
- Marchioli R., Finazzi G., Specchia G., Masciulli A., Mennitto M., Barbui T. (2011) The CYTO-PV: a large-scale trial testing the intensity of CYTOreductive therapy to prevent cardiovascular events in patients with polycythemia vera. Thrombosis 2011: 794240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullin M., Bareford D., Campbell P., Green A., Harrison C., Hunt B., et al. (2005) Guidelines for the diagnosis, investigation and management of polycythaemia/erythrocytosis. Br J Haematol 130: 174–195. [DOI] [PubMed] [Google Scholar]
- McMullin M., Reilly J., Campbell P., Bareford D., Green A., Harrison C., et al. (2007) Amendment to the guideline for diagnosis and investigation of polycythaemia/erythrocytosis. Br J Haematol 138: 821–822. [DOI] [PubMed] [Google Scholar]
- Mcmullin M.F., Wilkins B.S., Harrison C.N. (2016) Management of polycythaemia vera: a critical review of current data. Br J Haematol 172: 337–349. [DOI] [PubMed] [Google Scholar]
- Mehta J., Wang H., Iqbal S., Mesa R. (2014) Epidemiology of myeloproliferative neoplasms in the United States. Leuk Lymphoma 55: 595–600. [DOI] [PubMed] [Google Scholar]
- Mesa R., Kiladjian J., Verstovsek S., Al-Ali H., Gotlib J., Gisslinger H., et al. (2014) Comparison of placebo and best available therapy for the treatment of myelofibrosis in the phase 3 comfort studies. Haematologica 99: 292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesa R., Niblack J., Wadleigh M., Verstovsek S., Camoriano J., Barnes S., et al. (2007) The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international internet-based survey of 1179 MPD patients. Cancer 109: 68–76. [DOI] [PubMed] [Google Scholar]
- Mesa R., Vannucchi A.M., Yacoub A., Zachee P., Garg M., Lyons R., et al. (2017) The efficacy and safety of continued hydroxycarbamide therapy versus switching to ruxolitinib in patients with polycythaemia vera: a randomized, double-blind, double-dummy, symptom study (RELIEF). Br J Haematol 176: 76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osler W. (1908) A clinical lecture on erythraemia (polycythaemia with cyanosis, maladie de vaquez). Lancet 1: 143–146. [Google Scholar]
- Passamonti F., Griesshammer M., Palandri F., Egyed M., Benevolo G., Devos T., et al. (2017) Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (Response-2): a randomised, open-label, Phase 3b study. Lancet Oncol 18: 88–99. [DOI] [PubMed] [Google Scholar]
- Passamonti F., Rumi E., Pietra D., Elena C., Boveri E., Arcaini L., et al. (2010) A prospective study of 338 patients with polycythemia vera: the impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia 24: 1574–1579. [DOI] [PubMed] [Google Scholar]
- Passamonti F., Saydam G., Lim L., Khan M., Mounedji N., Griesshammer M. (2014) RESPONSE 2: a phase 3b study evaluating the efficacy and safety of ruxolitinib in patients with hydroxyurea-resistant/intolerant polycythemia vera vs best available therapy. J Clin Oncol Abstract 32: TPS7128^. [Google Scholar]
- Prchal J., Gordeuk V. (2013) Treatment target in polycythemia vera. N Engl J Med 368: 1555–1556. [DOI] [PubMed] [Google Scholar]
- Quintas-Cardama A., Abdel-Wahab O., Manshouri T., Kilpivaara O., Cortes J., Roupie A.L., et al. (2013) Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon alpha-2a. Blood 122: 893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintas-Cardama A., Kantarjian H., Manshouri T., Luthra R., Estrov Z., Pierce S., et al. (2009) Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol 27: 5418–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sever M., Newberry K., Verstovsek S. (2014) Therapeutic options for patients with polycythemia vera and essential thrombocythemia refractory/resistant to hydroxyurea. Leuk Lymphoma 55: 2685–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver R. (1988) Recombinant interferon-alpha for treatment of polycythaemia vera. Lancet 2: 403. [DOI] [PubMed] [Google Scholar]
- Silver R., Kiladjian J., Hasselbalch H. (2013) Interferon and the treatment of polycythemia vera, essential thrombocythemia and myelofibrosis. Expert Rev Hematol 6: 49–58. [DOI] [PubMed] [Google Scholar]
- Spivak J. (2002) Polycythemia vera: myths, mechanisms, and management. Blood 100: 4272–4290. [DOI] [PubMed] [Google Scholar]
- Stein B., Tiu R. (2013) Biological rationale and clinical use of interferon in the classical BCR-ABL-negative myeloproliferative neoplasms. J Interferon Cytokine Res 33: 145–153. [DOI] [PubMed] [Google Scholar]
- Stein B., Moliterno A., Tiu R. (2014) Polycythemia vera disease burden: contributing factors, impact on quality of life, and emerging treatment options. Ann Hematol 93: 1965–1976. [DOI] [PubMed] [Google Scholar]
- Tefferi A., Vardiman J. (2008) Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia 22: 14–22. [DOI] [PubMed] [Google Scholar]
- Tefferi A., Rumi E., Finazzi G., Gisslinger H., Vannucchi A., Rodeghiero F., et al. (2013) Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia 27: 1874–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tefferi A., Barbui T. (2015) Polycythemia vera and essential thrombocythemia: 2015 update on diagnosis, risk-stratification and management. Am J Hematol 90: 162–173. [DOI] [PubMed] [Google Scholar]
- Them N., Bagienski K., Berg T., Gisslinger B., Schalling M., Chen D., et al. (2015) Molecular responses and chromosomal aberrations in patients with polycythemia vera treated with peg-proline-interferon alpha-2b. Am J Hematol 90: 288–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibes R., Mesa R. (2013) Emerging drugs for polycythemia vera. Expert Opin Emerg Drugs 18: 393–404. [DOI] [PubMed] [Google Scholar]
- Vannucchi A. (2014) How I treat polycythemia vera. Blood 124: 3212–3220. [DOI] [PubMed] [Google Scholar]
- Vannucchi A. (2015) Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med 372: 1670–1671. [DOI] [PubMed] [Google Scholar]
- Vannucchi A., Kiladjian J., Griesshammer M., Masszi T., Durrant S., Passamonti F., et al. (2015) Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med 372: 426–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstovsek S. (2013) Ruxolitinib: an oral Janus kinase 1 and Janus kinase 2 inhibitor in the management of myelofibrosis. Postgrad Med 125: 128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstovsek S., Passamonti F., Rambaldi A., Barosi G., Rosen P., Rumi E., et al. (2014) A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer 120: 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstovsek S., Vannucchi A., Griesshammer M., Masszi T., Durrant S., Passamonti F., et al. (2016) Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica 101: 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S., Zhang X., Xu Y., Feng Y., Sheng W., Cen J., et al. (2016) Impact of JAK2V617F mutation burden on disease phenotype in Chinese patients with JAK2V617F-positive polycythemia vera (PV) and essential thrombocythemia (ET). Int J Med Sci 13: 85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]