

Study of fungal bioluminescence mechanisms generates development of a multicolor enzymatic chemiluminescence system.
Keywords: Neonothopanus, CIEEL, chemiluminescence, endoperoxide, hispidin, photochemistry, luciferase, luciferin, alpha-pyrone, enzyme promiscuity
Abstract
Bioluminescent fungi are spread throughout the globe, but details on their mechanism of light emission are still scarce. Usually, the process involves three key components: an oxidizable luciferin substrate, a luciferase enzyme, and a light emitter, typically oxidized luciferin, and called oxyluciferin. We report the structure of fungal oxyluciferin, investigate the mechanism of fungal bioluminescence, and describe the use of simple synthetic α-pyrones as luciferins to produce multicolor enzymatic chemiluminescence. A high-energy endoperoxide is proposed as an intermediate of the oxidation of the native luciferin to the oxyluciferin, which is a pyruvic acid adduct of caffeic acid. Luciferase promiscuity allows the use of simple α-pyrones as chemiluminescent substrates.
INTRODUCTION
Many living organisms emit light, which is a phenomenon named bioluminescence (1, 2). In most cases, light emission results from the chemical oxidation of a luciferin substrate catalyzed by a luciferase enzyme. The luciferin reacts with molecular oxygen, giving a high-energy intermediate (HEI) whose decomposition releases enough energy to produce the emitter oxyluciferin in the singlet electronically excited state. Fluorescence from this excited metabolite results in visible light emission used in nature for signaling and illumination (1). The luciferin/luciferase systems and the corresponding oxyluciferins of several bioluminescent insects, bacteria, and marine animals have been characterized (2). This knowledge has yielded major technological advances, such as genetically encoded labels using fluorescent proteins (3, 4), pyrosequencing (5), ultrasensitive enzyme quantification using triggered chemiluminescent substrates (6), and imaging via bioluminescence resonance energy transfer (7, 8).
Bioluminescent fungi are spread throughout the globe (9). The four known lineages of bioluminescent basidiomycete fungi contain around 80 different species (10). Recently, our group reported the chemical structure of fungal luciferin and its biosynthetic precursor (11). The enzymatic oxidation of the precursor (hispidin) by an NAD(P)H (nicotinamide adenine dinucleotide phosphate OR its reduced form)–dependent enzyme, hispidin-3-hydroxylase, produces 3-hydroxyhispidin (1), which acts as the fungal luciferin (Fig. 1A) (11). However, the molecular mechanism and reasoning behind fungal bioluminescence remained unclear (12–15). Here, we report the identification and characterization of fungal oxyluciferin.
Fig. 1. Characterization of the fungal oxyluciferin.
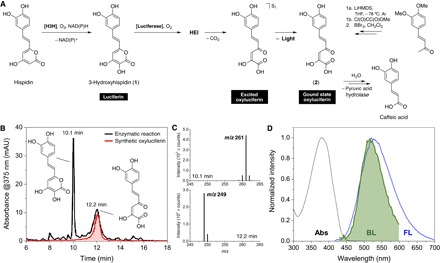
(A) General mechanism of the fungal bioluminescence and synthesis of the oxyluciferin (2). Hispidin is hydroxylated by a styrylpyrone hydroxylase [hispidin-3-hydroxylase (H3H)] in the presence of O2 and NAD(P)H, producing 3-hydroxyhispidin (1) (11), the fungal luciferin, which is enzymatically oxidized by O2, giving an HEI that decomposes in CO2 and the excited oxyluciferin. Fluorescence emission gives the ground-state oxyluciferin (2). The oxyluciferin was synthesized in two steps from 3,4-dimethoxybenzalacetone. LiHMDS, lithium bis(trimethylsilyl)amide; THF, tetrahydrofuran. (B) Comparison of HPLC-PDA-ESI-MS profiles of the enzymatic reaction (after 15 min) and the synthetic oxyluciferin. mAU, milliarbitrary unit. (C) Mass spectra of the luciferin 1 (Rt = 10.1 min, m/z = 261 [M − H]−) and compound 2 (Rt = 12.2 min, m/z = 249 [M − H]−). (D) Matching of the fungal bioluminescence (BL) spectrum and the fluorescence (FL) spectrum of 2 in acetone. The absorption spectrum of 2 is shown for reference (λmax = 380 nm).
RESULTS AND DISCUSSION
We analyzed the enzymatic oxidation of 1 by high-performance liquid chromatography (HPLC)–photodiode array (PDA)–electrospray ionization mass spectrometry (ESI-MS). After 15 min of the reaction of 1 with molecular oxygen in the presence of a luciferase-enriched protein fraction from the bioluminescent fungus Neonothopanus gardneri (16), two main chromatographic peaks corresponding to 1 [retention time (Rt) = 10.1 min, mass-to-charge ratio (m/z) = 261.04 [M − H]−] and to an unknown compound (Rt = 12.2 min, m/z = 249.04) are observed (Fig. 1B). The mass difference of 12 u suggests the loss of a single carbon atom, which is compatible with the addition of O2 and release of CO2 by 1, leading to compound 2 (Fig. 1, A and C).
Compound 2 was then synthesized in two steps from 3,4-dimethoxybenzalacetone (17), and its chromatographic profile and fluorescence spectrum were compared with those acquired during the study of the bioluminescent reaction. The retention time and absorption maxima matched those of the unknown product of the enzymatic oxidation of 1 (Fig. 1B). The fluorescence spectrum of 2 in acetone (ΦFL = 0.011; fig. S1) is in agreement with the bioluminescence spectrum of fungi containing 1, for example, Mycena chlorophos, N. gardneri, and N. nambi (Fig. 1D). The fact that 2 is not fluorescent in water [that is, phosphate buffer (pH 6 to 8)] suggests that the luciferase-oxyluciferin complex is important for light emission. This requirement also provides an explanation for the higher full width at half maximum of the bioluminescence spectrum, analogous to the data reported for coelenteramide, the oxyluciferin of Aequorea victoria that is fluorescent only when bound to aequorin (2). Chemiluminescence and singlet quantum yields (ΦCL and ΦS) of the native luciferin 1 increase almost threefold when pH is increased from 6 to 8 (figs. S2 to S4, and tables S1 and S2).
The kinetics of enzymatic oxidation of the luciferin 1 in the presence of a microsomal luciferase fraction of N. nambi were monitored for ca. 6 hours and revealed that the oxyluciferin 2 hydrolyzes enzymatically into caffeic acid (fig. S5, A and B). Accordingly, replacement of the native luciferin 1 by 3-hydroxybisnoryangonin (3) (11) yields the corresponding oxyluciferin 4 and 4-coumaric acid (fig. S5, C and D). The maximum wavelengths for the enzymatic chemiluminescence (λmaxCL) of 1 and 3 are 20 nm apart, indicating different light emitters (fig. S6). The chemiluminescence spectrum of 3 in acetone matches the fluorescence spectra of the oxyluciferin 4, confirming that the fungal oxyluciferin is the pyruvic acid adduct of caffeic acid (Fig. 1D and fig. S6).
The enzymatic oxidation of hispidin does not result in light emission. Therefore, the hydroxylation of hispidin is one of the key steps in fungal bioluminescence (11). The investigation of hispidin and 1 by cyclic voltammetry demonstrated a reversible oxidation potential at 428 mV (versus Ag/AgCl) for hispidin and a considerably lower potential for 1 (211 mV versus Ag/AgCl), showing that the oxidation of 1 is more favorable when compared to hispidin in water (Fig. 2A). Computational chemistry at the density functional theory (DFT) level revealed that spin density surfaces, which are generated from the optimized structures of the radical cations of both compounds, are similar (Fig. 2B). However, for 1+•, the deprotonation of the hydroxyl group at C-3 in the α-pyrone ring is 25 kJ mol−1 more thermodynamically favorable than the deprotonation of the hydroxyl group at C-4 in water. The resulting resonance-stabilized radical shows high spin density at C-6, which is diametrically opposed to C-3 in the pyrone ring.
Fig. 2. Experimental and theoretical study of the oxidation of 3-hydroxyhispidin.
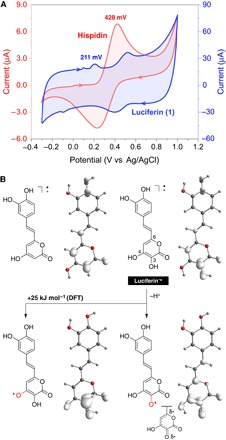
(A) Cyclic voltammetry of hispidin and the luciferin in aqueous KCl. (B) Spin density surface of the radical cations of hispidin and 3-hydroxyhispidin and the corresponding deprotonated radicals.
Endoperoxides have been proposed as HEIs in several chemiluminescent systems, including the notorious luminol reaction (18, 19). Despite the lack of examples of endoperoxides as HEIs in bioluminescent processes, previous studies (20–22) have shown that α-pyrone endoperoxides are chemiluminescent upon either thermal decarboxylation leading to 1,2-diacylethylenes or chemically initiated electron exchange luminescence. We then prepared the [16O]- and [18O]-labeled endoperoxides of the synthetic luciferin analog 3 by singlet oxygenation (Fig. 3A) (23). As expected, decomposition of the endoperoxide resulted in chemiluminescence (fig. S7). Unfortunately, the low intensity makes it impossible to acquire the chemiluminescence spectrum using conventional equipment. MS analyses of 3 (m/z = 245) before and after singlet oxygenation (given in Fig. 3B as normalized intensity difference) reveal the appearance of compounds with an m/z of 237 [(M + 4) − H]−. The main product ion upon collision-induced dissociation is found at an m/z of 163, indicating the loss of one 18O atom with the neutral fragment (that is, [(M + 2) − H]−; Fig. 3B). These results agree with the formation of a [4 + 2] cycloadduct between 3 and singlet oxygen (1O2), which, upon CO2 elimination, decomposes into the corresponding oxyluciferin. Carbon dioxide elimination should be a dominant dissociation channel in the negative mode for several carboxylic acids, for example, caffeic acid. Nevertheless, the formed oxyluciferins (2 and 4) are α-ketoacids (or α-hydroxyacids in their tautomeric form); thus, the loss of CO2 is readily followed by carbon monoxide elimination (fig. S8).
Fig. 3. Formation of the HEI via the oxidation of 3-hydroxyhispidin.
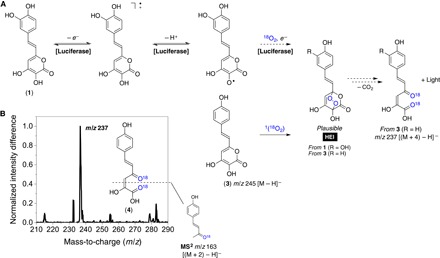
(A) General mechanistic proposal for the enzymatic generation of the HEI and the chemical preparation of the endoperoxide of 3 via methylene blue–sensitized photo-oxygenation with [18O]-labeled singlet oxygen [1(18O2)]. (B) Difference between the normalized mass spectra signal intensity before and after photo-oxygenation of 3 and the structure proposal for product ion at an m/z of 163. Molecular structures represent the neutral form of 1 and 3, not the anion detected by MS.
According to these results, the presence of the 3-OH group is key to the formation of the HEI in fungal bioluminescence. The exact mechanism for the generation of the HEI is yet to be investigated; however, our results suggest that the oxidation and deprotonation of the luciferin are related to the addition of two atoms of oxygen and that the decomposition of the resulting endoperoxide leads to the formation of the oxyluciferin in the electronically excited state. There is currently no evidence for the formation of a 1,2-dioxetanone as HEI in fungal bioluminescence, and the chemiluminescence efficiency of such peroxides is as low as that reported for endoperoxides (24–26).
The chemiluminescence arising from the enzymatic oxidation of both the native luciferin 1 and the analog 3 suggests that the fungal luciferase has some degree of promiscuity (27). To test this hypothesis, we synthesized five styryl-α-pyrones containing substituted aromatic moieties or aromatic heterocyclic rings (Fig. 4A) (28–31). The incubation of all compounds with the luciferase homogenate from N. gardneri in the presence of molecular oxygen resulted in chemiluminescence emission, except the thiophene derivative 5. The lack of chemiluminescence of the analog 5 could be explained by either its low affinity for the luciferase catalytic pocket, which can only be confirmed in further investigations, or by the low H-bond acceptor strength of the thiophene ring. The chemiluminescence quantum yields (ΦCL) and observed rate constants for the chemiluminescence decay (kobs) of luciferins 1, 3, and 6 to 9 were determined (Fig. 4, C and D, fig. S9, and tables S3 and S4). The values of ΦCL depend on the substituent, and the maximum efficiency was reached with the N,N-diethylaniline styryl-α-pyrone 7. These results suggest that light emission is observed, provided that the substrate contains the 3-hydroxy-α-pyrone ring. Moreover, the chemiluminescence wavelength maximum depends on the substitution pattern, supporting the idea that the oxyluciferin is the true emitter in fungal bioluminescence (Fig. 4B).
Fig. 4. Color modulation of fungal bioluminescence.
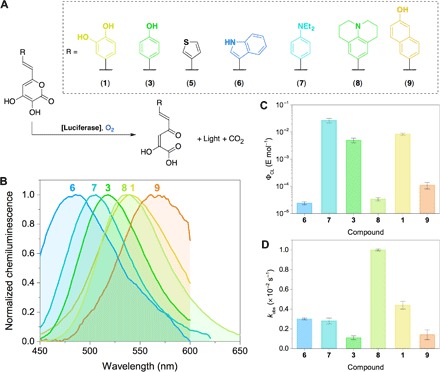
(A) Natural luciferin (1), synthetic analogs 3 and 5 to 9, and enzymatic chemiluminescent reaction in tris buffer (pH 7). (B) Chemiluminescence spectra of luciferins 1, 3, and 6 to 9. The Savitzky-Golay filter (20 points) was used to improve the signal-to-noise ratio due to very low light intensity. (C) Chemiluminescence quantum yields (ΦCL) and (D) the observed chemiluminescence decay rate constant for compounds 1, 3, and 6 to 9.
Our study provides insight into the mechanism of fungal bioluminescence by characterizing the oxyluciferin of 3-hydroxyhispidin and expanding the knowledge on how styryl-3-hydroxy-α-pyrones are chemiexcited in vivo. The enzyme-catalyzed hydrolysis of the oxyluciferin produces caffeic acid, which may be recycled in the biosynthesis of hispidin via the styrylpyrone pathway (32). These results suggest the usefulness of fungal bioluminescence as a model system for continuous conversion of chemical energy into light emission. The promiscuous nature of the fungal luciferase was confirmed by developing a group of substrates bearing substituents that conveniently fine-tune the color of chemiluminescence without requiring enzyme mutation. Future work on the isolation, characterization, and heterologous expression of the luciferase will stimulate the development of fungal bioluminescence–inspired applications.
MATERIALS AND METHODS
Materials and equipment
Fluorescence and bioluminescence spectra were recorded on a Cary Eclipse Fluorescence Spectrophotometer (Agilent Technologies) in Russia. In Brazil, luminescence data were recorded on a Sirius L tube luminometer (Berthold) and F4500 spectrofluorometer (Hitachi). Ultraviolet-visible (UV-vis) absorption spectra were recorded either on a Cary 100 Bio spectrophotometer (Varian) in Russia or on an NP80 NanoPhotometer (Implen) in Brazil. In Russia, chromatography was performed on a 1260 Infinity LC and high-resolution mass spectra were acquired on a 6224 time-of-flight (TOF) LC/MS System (both from Agilent Technologies). In Brazil, high-resolution mass analyses were accomplished in a micrOTOF-QII(Bruker Daltonics) coupled to a Prominence HPLC (Shimadzu). Scan and product ion spectra were acquired in a 6460 Triple Quadrupole coupled to a 1260 Infinity LC (Agilent Technologies). Nuclear magnetic resonance (NMR) spectra were recorded at 300 K on spectrometers Avance III 800 (with a 5-mm CPTXI cryoprobe), Avance III 700, Avance III 600, and Fourier 300 (all from Bruker) in DMSO-d6, CDCl3, CD3OD, or acetone-d6, using tetramethylsilane as internal standard.
All commercially available solvents and reagents were used without additional purification. Ultrapure water was generated in a Direct-Q8 system (Millipore). Kieselgel 60 (70-230 mesh, Merck) was used for column chromatography. Thin-layer chromatography was performed on silica gel 60 F254 glass-backed plates (Merck). UV light (254 or 312 nm) and staining with KMnO4 solution were used for visualization.
Cultures and preparation of enzymatic extracts
N. gardneri crude extract and luciferase-enriched fraction
Mycelium of the basidiomycete fungus N. gardneri (MycoBank MB519818) was obtained from mushrooms collected in the Brazilian Coconut Forest in Altos, Piauí, Brazil (16). The mycelium was inoculated on sterilized dialysis cellulose membrane squares (3 × 3 cm, Sigma-Aldrich) in 100-mm petri dishes over a medium composed of 1.0% (w/v) sugar cane molasses (82.2° Bx, Pol 56%) and 0.10% (w/v) yeast extract (Oxoid) in 2.0% (w/v) agar (Oxoid) (14). Cultures were maintained for 14 days in darkness inside a KMF 240 climatic chamber (Binder) at 25°C and ~80% humidity. Crude extracts were typically prepared by homogenization of 2 g of mycelium in 10 ml of 100 mM phosphate buffer (pH 7.5) containing 2 μl of 1 mM phenylmethylsulfonyl fluoride as protease inhibitor (Sigma-Aldrich) at 0°C (Potter-Elvehjem PTFE, Sigma-Aldrich). After centrifugation for 10 min at 10,000g and 4°C (5804R, rotor F34-6-38; Eppendorf) to clear debris, the resulting supernatant was further ultracentrifuged for 90 min at 200,000g and 4°C (RP50T-2, rotor P50AT2-716; Hitachi). The luciferase-enriched fraction was reconstituted in 1 ml of 100 mM phosphate buffer (pH 7.5) containing 2% glycine and 1 mM dithiothreitol (DTT).
N. nambi microsomal luciferase fraction
Mycelium of the fungus N. nambi was collected in the rainforest of southern Vietnam. The cultures were grown on 100-mm petri dishes covered with the liquid nutrient potato-sucrose medium (200 g of potatoes, 20 g of sucrose, and 1 liter of distilled water) at 26°C in darkness for 10 days. Before homogenization, the mycelial biomass was soaked in distilled water for 20 hours at 27°C. The mycelium (approximately 1 g of biomass from one petri dish) was homogenized in 30 ml of distilled water at 0°C and sonicated on ice for 1 min (ultrasonic disintegrator UD-20, Techpan). The homogenate was centrifuged for 20 min at 10,000g and 4°C in Avanti J-E centrifuge (Beckman Coulter). A 1 M CaCl2 solution was added to the resulting supernatant up to a final concentration of 10 mM, and the solution was incubated for 30 min at 4°C. After centrifugation for 20 min at 30,000g and 4°C, the microsomal fraction was reconstituted in 3 ml of 200 mM sodium phosphate buffer (pH 7.5) containing 0.1% Triton X-100, 1 mM DTT, and 75 μl of 2.5% lecithin solution in ethanol.
Identification of luciferin/luciferase reaction products
N. gardneri luciferase-enriched fraction assay
Synthetic luciferin (1) in acetone (200 μl, 3.8 μM) was added to a solution containing the luciferase-enriched fraction (300 μl) and 20 mM phosphate buffer (pH 7) (1500 μl) at 25°C. Three hundred microliters of the solution was immediately retrieved and equally divided for the chemiluminescence assay/spectrofluorimetry and HPLC-PDA-ESI-MS analyses. Additional samples (150 μl) were retrieved at 5, 10, 15, 20, and 30 min. Probes were prepared by the addition of formic acid (10 μl) and centrifugation for 5 min at 14,000g and 4°C. The resulting supernatants were concentrated and desalted online through a two-position, six-port valve containing a C18 disk (3 × 4 mm, 5 μm; Phenomenex). The samples (50 μl) were loaded using 0.1% aqueous formic acid (pump C) at 300 μl min−1, desalted for 2 min, and eluted in backflush mode at 300 μl min−1 with 90:10 (v/v) 0.1% aqueous formic acid (pump A) and acetonitrile (pump B). Chromatographic separation was achieved in a Luna C18(2) column (100 × 2.0 mm, 3 μm; Phenomenex) using the following gradients: 0 to 3 min, 10% B; 3 to 18 min, 10 to 45% B; 18 to 20 min, 45 to 95% B; 20 to 24 min, 95% B; 24 to 24.5 min, 95 to 10% B; and 24.5 to 30 min, 10% B. Eluting compounds were monitored by absorbance (200 to 700 nm; SPD-M20A, Shimadzu) and ESI in the negative mode (micrOTOF-Q II, Bruker Daltonics). Synthetic oxyluciferin (2) dissolved in acetone was cochromatographed under the same conditions.
N. nambi microsomal luciferase assay and dynamics of product formation
Synthetic luciferin (1) in DMSO (20 μl, 22 μg) was added to a solution of microsomal luciferase in 200 mM phosphate buffer (pH 7) (1000 μl) containing 0.05% Triton X-100 (hereafter referred to as microsomal luciferase solution). The reaction was incubated at 20°C, and samples (200 μl) were retrieved at 2, 25, 50, 100, 180, and 320 min. Probes were prepared by the addition of HCl (10 μl) and centrifugation for 5 min at 14,000g and 4°C. The resulting supernatants (100 μl) were immediately loaded onto a Zorbax Eclipse XDB-C18 column (150 × 3.0 mm, 5 μm; Agilent Technologies). Chromatographic separation was achieved at 0.8 ml min−1 using 0.1% aqueous formic acid (pump A) and acetonitrile (pump B) as mobile phase in a linear gradient from 5 to 40% B in 10 min. Eluting compounds were monitored by absorbance (200 to 700 nm; 1260 Infinity LC, Agilent Technologies). Aliquots of 100 μl were also retrieved from the reaction mixture for the chemiluminescence assay. Luciferin (1) in DMSO (5 μl, 10 μg) was also incubated at 20°C in 200 mM phosphate buffer (pH 7) (500 μl) containing 0.05% Triton X-100 as a control experiment (absence of the microsomal luciferase). Probes were prepared and analyzed as described above. In this case, the formation of oxyluciferin (2) was not observed. A supplementary control experiment was performed by the addition of oxyluciferin (2) in DMSO (15 μl, 25 μg) to the microsomal luciferase solution (1000 μl). After incubation at 20°C, probes were retrieved and analyzed as described above. In this case, the consumption of 2 was observed, suggesting that chromatographic peaks 2 to 4 (fig. S5A) are the degradation products of the oxyluciferin. These results may be explained by the insufficient degree of purification of the microsomal luciferase. It is likely that the microsomal fraction contains enzymes other than the luciferase. Nevertheless, chromatographic peak 3 was identified as caffeic acid through its spectral characteristics, mass-to-charge ratio, and cochromatography with an authentic standard.
Identification of compounds in chromatographic peaks 2 and 4
To isolate compounds corresponding to peaks 2 and 4, we incubated luciferin (1) in DMSO (100 μl, 85 μg) at 20°C with the microsomal luciferase solution (14 ml). After 2 hours, when the chemiluminescence of the reaction decreased to zero, concentrated HCl was added to adjust the pH to 2. The reaction mixture was centrifuged and concentrated in a 1-ml disposable C16 extraction cartridge (Diapack-C16, BioChemMak S&T) previously equilibrated with 10 mM HCl in 3% acetonitrile. The compounds were eluted with 75% acetonitrile and concentrated on a rotary evaporator. The residue was dissolved in DMSO (100 μl) and loaded onto a semipreparative HPLC column (Zorbax Eclipse XDB-C18, 250 × 9.4 mm, 5 μm; Agilent Technologies). Chromatographic separation was achieved at 3 ml min−1 using 0.1% aqueous formic acid (pump A) and acetonitrile (pump B) as mobile phase in a linear gradient from 5 to 40% B in 24 min. Eluting compounds were monitored by absorbance (PDA; 200 to 700 nm) and manually collected. Combined fractions containing the target peaks were concentrated on a rotary evaporator and identified by NMR (fig. S10), whose putative mechanism of formation is depicted in fig. S11.
Identification and dynamics of product formation for the reaction 3-hydroxybisnoryangonin/microsomal luciferase solution
3-Hydroxybisnoryangonin (3) in DMSO (50 μl, 85 μg) was added to the microsomal luciferase solution (2.9 ml). The reaction was incubated at 20°C, and samples (200 μl) were retrieved at 6, 60, 180, 440, and 1150 min. Probes were prepared and analyzed as described above (fig. S5C). The corresponding oxyluciferin (4) was synthesized and identified by cochromatography. 4-Coumaric acid was identified through its spectral characteristics, mass-to-charge ratio, and cochromatography with an authentic standard.
Mechanistic studies
Cyclic voltammetry
Commercial DropSens DS-110 screen-printed carbon electrodes were dried under N2 atmosphere and used throughout this work. A single drop (200 μl) of ~10−3 M solutions of the analyte in KCl (3 M) was added to the electrode. Cyclic voltammetry measurements were carried out in a μAutolab TYPE III potentiostat/galvanostat (scan rate, 10 mV s−1; potential range, −0.3 to 1.0 V). All the electrochemical experiments were performed at room temperature (25° ± 2°C).
Computational methods
The ground-state geometry of hispidin and the native luciferin (1) and their corresponding radical cations were fully optimized using the DFT at the B3LYP/6-31+g(d,p)/SMD level (restricted and unrestricted, respectively) (33, 34). Vibrational analyses revealed no imaginary frequencies, indicating that the optimized geometries were in a minimum of the potential energy surface. The geometry optimizations were performed with the aid of the Gaussian 09 rev. B01 package (35).
Photo-oxygenation of 3-hydroxybisnoryangonin in the presence of methylene blue under 18O2 atmosphere
3-Hydroxybisnoryangonin (400 μl, 0.4 mM) and methylene blue in acetone (40 μl, 3 mM) were added to a 1-ml septated clear chromatographic vial. Then, vacuum was made with a needle coupled to a vacuum pump, and 18O2 (99%; Isotec, Sigma-Aldrich) was bubbled for 5 s. The reaction mixture was irradiated with a red lamp (20 W, λmax = 610 nm) for 1.5 hours. Parallel control experiments (16O2, in darkness, and without methylene blue) were also performed. Reaction mixtures were filtered (Millex PVDF, 0.45 μm; Millipore) and analyzed by flow injection analysis with a 1260 Infinity LC coupled to a 6460 Triple Quadrupole mass spectrometer through an ESI source operated in the negative mode. Methanol and water [50:50 (v/v)] were used as carrying solvent at 200 μl min−1. Product ion spectra [tandem MS (MS/MS)] were acquired through collision-induced dissociation using nitrogen as collision gas.
Chemiluminescence from the reaction of 3-hydroxyhispidin in the presence of 1,4-dimethylnaphthalene endoperoxide (DMNO2)
To confirm the emission of light from the reaction of 3-hydroxyhispidin with singlet oxygen (1O2), we conducted the following experiments in a Berthold Sirius L luminometer. The luminometer test tube containing 150 μl of 290 mM DMNO2 in acetone was inserted into the instrument, and light emission was measured for 400 s. Next, the same experiment was repeated but with the addition of 150 μl of 0.45 mM luciferin to 150 μl of 290 mM DMNO2, with both solutions in acetone. These first two experiments were conducted at room temperature (ca. 25°C). Finally, the last assay was also repeated, but the solution (150 μl of 0.45 mM luciferin and 150 μl of 290 mM DMNO2) was heated in water at 55°C for 15 s before the insertion of the assay tube into the luminometer (see fig. S7). DMNO2 was prepared according to the procedure described in the literature (23, 36).
Photochemical properties of the luciferin, oxyluciferin, and analogs
Determination of the fluorescence quantum yield of the oxyluciferin (2)
UV-vis absorption spectra were recorded on Implen NP80 NanoPhotometer (in Brazil). Luminescence data were recorded on the Berthold Sirius L luminometer and Hitachi F4500 spectrofluorometer. The fluorescence quantum yield of the oxyluciferin 2 (ΦFL,OxyLn) was determined as described in the literature (37, 38). In summary, the absorbance of fluorescein (>99%, Sigma-Aldrich) solution (absorbance of 0.028 in aqueous 0.1 M NaOH at 460 nm) and oxyluciferin (absorbance of 0.045 in acetone at 460 nm) was acquired. The same solutions were excited at 460 nm to obtain fluorescence emission spectra (fig. S1). The area under each emission spectrum, fluorescein fluorescence quantum yield (0.89) (38), and refractive index of water (nwater = 1.33) and acetone (nacetone = 1.36) were used for determination of the oxyluciferin fluorescence quantum yield using the first equation in the Supplementary Materials, whose value is ΦFL,OxyLn = 0.011. Notably, oxyluciferin is not fluorescent in aqueous phosphate buffer (20 mM) at pH 6, 7, or 8.
Determination of the chemiluminescence and singlet quantum yields of the luciferin/luciferase reaction
The calibration of the Berthold Sirius L luminometer was adapted from the study by Stevani et al. (39). In summary, 100 μl of H2O2 (0.0003% or 0.1 mM) and 100 μl of hemin (Sigma-Aldrich) in phosphate buffer (absorbance of 0.1 at 414 nm) were added to a suitable test tube. A very intense but short burst of light was observed. When light decayed to baseline value (ca. 200 counts, 90 s), 300 μl of 1.0 nM luminol (ε = 7600 M−1 cm−1 at 347 nm) in phosphate buffer was added, and light was recorded (fig. S2). The chemiluminescence and singlet quantum yields of the reaction between 3-hydroxyhispidin (1) and luciferase fraction of N. gardneri were determined using luminol as standard. To a test tube containing 150 to 160 μl of 20 mM phosphate buffer (pH 6, 7, or 8) with bovine serum albumin (1 mg ml−1) (Sigma-Aldrich) and 30-μl crude extract of N. gardneri (total protein of 0.60 mg/ml determined by Bradford assay), we added 10 to 20 μl of luciferin in acetone [final concentrations of 76, 152, 304, 380, and 760 nM; 7.6 mM stock solution determined using hispidin molar absorptivity (ε = 13,200 M−1 cm−1) (40)] to trigger light emission. The final volume was 200 μl, and the reaction was performed at 25°C in duplicate for each concentration and pH and followed for 210 s (fig. S3 and table S1). To determine chemiluminescence quantum yields of luciferin/luciferase reaction, it was also necessary to correct the emission using the photomultiplier tube (PMT) sensitivity. Usually, PMTs are more sensitive in blue. Hence, there is no need to correct the light emission of luminol [445 nm (maximum); fPMT = 1.00], but different emissions must be corrected. The PMT sensitivity correction factor (fPMT) was determined by using the photocathode spectral response curve provided by Berthold Detection Systems (fig. S4). On the basis of the maximum emission of oxyluciferin (compound 2), the value of fPMT is 1.17.
Chemiluminescence, fluorescence, and absorption spectra of compounds 1 to 9
Fluorescence and chemiluminescence spectra were recorded on an Agilent 1260 Infinity LC spectrofluorometer, equipped with Agilent Cary Eclipse software in Russia. UV-vis absorption spectra were recorded on the Varian Cary 100 Bio spectrophotometer. Fluorescence emission spectra for compounds were obtained in acetone at λexcitation = 380 nm for compounds 1 to 3 and 5 to 7 and at λexcitation = 430 nm for compound 8 (figs. S6 and S9 and table S3).
Comparison of rate constants and chemiluminescence quantum yields of the luciferin 1 and analogs 3 and 5 to 9 in the presence of luciferase-enriched extract of N. gardneri
To a test tube containing 115 μl of tris buffer (50 mM, pH 7) and 25 μl of luciferase-enriched fraction of N. gardneri (in the same buffer), we added 10 μl of luciferin or analogs 3 and 5 to 9 (stock solution of ca. 0.1 μM and final concentration of ~7 nM) to trigger the chemiluminescent reaction. The analog 5 is not a substrate to the fungal luciferase. It is noteworthy to mention that the batch of luciferase-enriched fraction used here was different from the one used in previously described experiments; hence, chemiluminescence quantum yields are only comparable within this experiment. The light emission was monitored for 15 min using the Berthold Sirius L luminometer. The values of kobs and Q were determined as described above (here, the curves were extrapolated to zero counts, 5000 s). The singlet quantum yields (ΦS) were not determined in this case because the fluorescence quantum yields of each respective oxyluciferin are not known (table S4).
Synthetic procedures
Detailed synthetic procedures for the preparation of compounds 1 to 9, synthetic intermediates, building blocks, purification, and structure characterization are described in the Supplementary Materials (figs. S12 to S18).
Supplementary Material
Acknowledgments
We thank K. V. Antonov (Institute of Bioorganic Chemistry of the Russian Academy of Sciences, Moscow, Russia) for registration of the high-resolution mass spectra. Funding: This work was supported by the São Paulo Research Foundation [FAPESP grants 10/11578-5 (to A.G.O.), 13/16885-1 (to C.V.S.), 14/14866-2 (to E.L.B.), 13/07914-8 (to E.P. and F.A.D.), and 2012/12663-1 (to P.D.M.) and CEPID Redoxoma 2013/07937-8 (to P.D.M.)], the National Council for Scientific and Technological Development (CNPq) [301307/2013-0 (to P.D.M.)], NAP Redoxoma (PRPUSP) [2011.1.9352.1.8. (to P.D.M.)], the Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research (KAKENHI) [grant no. 16K07715 (to Y.O.)], Chubu University [grant AII28II M01 (to Y.O.)], and the Russian Science Foundation (grant 16-14-00052 to all Russian authors). Author contributions: Z.M.K. and A.S.T. synthesized all the compounds. V.N.P. and N.S.R. performed the incubation and HPLC experiments with luciferase and analogs. K.V.P. prepared the microsomal luciferase fractions. K.S.M. and Z.M.K. registered the NMR spectra. E.B.G., Y.S., and S.K. performed the HPLC experiments. Z.M.K., A.K., and N.S.B. registered the chemiluminescence and fluorescence spectra of the analogs. M.S.B., A.S.A., J.I.G., S.L., and Y.O. discussed and planned the experiments. E.P., F.A.D., E.L.B., P.D.M., C.V.S., T.A.P., R.P.C., A.G.O., and H.E.W. determined the quantum yields and conducted the photo-oxygenation and labeling experiments. E.L.B. determined the oxidation potentials and performed the theoretical calculations. I.V.Y., C.V.S., F.A.D., and E.L.B. designed the study. C.V.S., Z.M.K., F.A.D., E.L.B., and I.V.Y. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. All necessary data are included in this paper.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/4/e1602847/DC1
Synthetic procedures
fig. S1. Determination of fluorescence quantum yield of the oxyluciferin.
fig. S2. Determination of the chemiluminescence and singlet quantum yields of the luciferin/luciferase reaction.
fig. S3. Dependence of the chemiluminescence intensity from luciferin/luciferase reaction on the concentration of luciferin (76 to 760 nM) and phosphate buffer (pH 6 to 8).
fig. S4. Photocathode spectral response curve provided by Berthold Detection Systems (PMT type 9107).
fig. S5. Dynamics of the reaction between substrate and luciferase microsomal preparation of N. nambi.
fig. S6. Chemiluminescence spectra of 1 (orange curve) and 3 (green curve) and the fluorescence spectrum of 4 in acetone (dotted curve).
fig. S7. Chemiluminescence from the reaction of 1,4-dimethylnaphthalene endoperoxide (DMNO2) with 3-hydroxyhispidin (luciferin).
fig. S8. Collision-induced dissociation spectrum (ESI-MS/MS) of the labeled oxyluciferin—compound 4.
fig. S9. Absorption (blue) and fluorescence emission (red) spectra of compounds 2, 3, and 5 to 9.
fig. S10. Isolation and identification of compounds in peaks 2 and 4 of the luciferin 1/luciferase reaction.
fig. S11. Proposed mechanisms for oxyluciferin degradation.
fig. S12. Synthesis of the fungal luciferin 1.
fig. S13. Synthesis of 3,4-dihydroxy-6-methyl-2H-pyran-2-one 13.
fig. S14. Synthesis of compounds 14 to 16.
fig. S15. Synthesis of fungal luciferin analogs 3 and 5 to 9.
fig. S16. Synthesis of fungal oxyluciferins 2 and 4.
fig. S17. NMR spectra chemical shifts of compounds 18 to 23.
fig. S18. NMR spectra chemical shifts of luciferin analogs 3 and 5 to 9.
table S1. Observed decay rate constants (kobs), the total light emitted (Q) by the reaction of luciferin/luciferase reaction, and chemiluminescence and singlet quantum yields (ΦCL and ΦS) at different pH and substrate concentrations.
table S2. Dependence of chemiluminescence quantum yield (ΦCL) and singlet quantum yield (ΦS) on the pH of luciferin-luciferase reaction.
table S3. Spectral characteristics of compounds 1 to 3 and 5 to 9.
table S4. Observed decay rate constant (kobs), the total of light emission (Q), photomultiplier sensitivity correction factor (fPMT), and the chemiluminescence quantum yield (ΦCL) obtained from the reaction between the crude extract of N. gardneri and compounds 1, 3, and 5 to 9.
REFERENCES AND NOTES
- 1.T. Wilson, J. W. Hastings, Bioluminescence: Living Lights, Lights for Living (Harvard Univ. Press, 2013). [Google Scholar]
- 2.O. Shimomura, Bioluminescence: Chemical Principles and Methods (World Scientific, 2012). [Google Scholar]
- 3.Prasher D. C., Eckenrode V. K., Ward W. W., Prendergast F. G., Cormier M. J., Primary structure of the Aequorea victoria green-fluorescent protein. Gene 111, 229–233 (1992). [DOI] [PubMed] [Google Scholar]
- 4.Hastings J. W., Chemistries and colors of bioluminescent reactions: A review. Gene 173, 5–11 (1996). [DOI] [PubMed] [Google Scholar]
- 5.Ahmadian A., Ehn M., Hober S., Pyrosequencing: History, biochemistry and future. Clin. Chim. Acta 363, 83–94 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Adam W., Reinhardt D., Saha-Moller C. R., From the firefly bioluminescence to the dioxetane-based (AMPPD) chemiluminescence immunoassay: A retroanalysis. Analyst 121, 1527–1531 (1996). [Google Scholar]
- 7.Xia Z., Rao J., Biosensing and imaging based on bioluminescence resonance energy transfer. Curr. Opin. Biotechnol. 20, 37–44 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pfleger K. D. G., Eidne K. A., Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET). Nat. Methods 3, 165–174 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Oliveira A. G., Desjardin D. E., Perry B. A., Stevani C. V., Evidence that a single bioluminescent system is shared by all known bioluminescent fungal lineages. Photochem. Photobiol. Sci. 11, 848–852 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Chew A. L. C., Desjardin D. E., Tan Y.-S., Musa M. Y., Sabaratnam V., Bioluminescent fungi from Peninsular Malaysia—A taxonomic and phylogenetic overview. Fungal Divers. 70, 149–187 (2015). [Google Scholar]
- 11.Purtov K. V., Petushkov V. N., Baranov M. S., Mineev K. S., Rodionova N. S., Kaskova Z. M., Tsarkova A. S., Petunin A. I., Bondar V. S., Rodicheva E. K., Medvedeva S. E., Oba Y., Oba Y., Arseniev A. S., Lukyanov S., Gitelson J. I. and Yampolsky I. V., The chemical basis of fungal bioluminescence. Angew. Chem. Int. Ed. 54, 8124–8128 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Oliveira A. G., Carvalho R. P., Waldenmaier H. E., Stevani C. V., Fungi bioluminescence: Distribution, function and mechanism of light emission. Quim. Nova 36, 314–319 (2013). [Google Scholar]
- 13.Bechara E. J. H., Bioluminescence: A fungal nightlight with an internal timer. Curr. Biol. 25, R283–R285 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Oliveira A. G., Stevani C. V., Waldenmaier H. E., Viviani V., Emerson J. M., Loros J. J., Dunlap J. C., Circadian control sheds light on fungal bioluminescence. Curr. Biol. 25, 964–968 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Airth R. L., Foerster G. E., The isolation of catalytic components required for cell-free fungal bioluminescence. Arch. Biochem. Biophys. 97, 567–573 (1962). [DOI] [PubMed] [Google Scholar]
- 16.Capelari M., Desjardin D. E., Perry B. A., Asai T., Stevani C. V., Neonothopanus gardneri: A new combination for a bioluminescent agaric from Brazil. Mycologia 103, 1433–1440 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Crosby D. C., Lei X., Gibbs C. G., McDougall B. R., Robinson W. E. Jr, Reinecke M. G., Design, synthesis, and biological evaluation of novel hybrid dicaffeoyltartaric/diketo acid and tetrazole-substituted l-chicoric acid analogue inhibitors of human immunodeficiency virus type 1 integrase. J. Med. Chem. 53, 8161–8175 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Griesbeck A. G., Díaz-Miara Y., Fichtler R., von Wangelin A. J., Pérez-Ruiz R., Sampedro D., Steric enhancement of the chemiluminescence of luminols. Chem. Eur. J. 21, 9975–9979 (2015). [DOI] [PubMed] [Google Scholar]
- 19.W. J. Baader, C. V. Stevani, E. L. Bastos, Chemiluminescence of organic peroxides, in The Chemistry of Peroxides, Z. Rappoport, Ed. (Wiley, 2006), pp. 1211–1278. [Google Scholar]
- 20.Adam W., Erden I., ɑ-Pyrone endoperoxides. Synthesis, thermal decomposition, and chemiluminescence. J. Am. Chem. Soc. 101, 5692–5696 (1979). [Google Scholar]
- 21.Schuster G. B., Chemiluminescence of organic peroxides. Conversion of ground-state reactants to excited-state products by the chemically-initiated electron-exchange luminescence mechanism. Acc. Chem. Res. 12, 366–373 (1979). [Google Scholar]
- 22.Bastos E. L., da Silva S. M., Baader W. J., Solvent cage effects: Basis of a general mechanism for efficient chemiluminescence. J. Org. Chem. 78, 4432–4439 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Di Mascio P., Bechara E. J. H., Rubim J. C., Dioxygen Nir FT-emission (1Δg→3∑−g) and Raman spectra of 1,4-dimethylnaphthalene endoperoxide: A source of singlet molecular oxygen. Appl. Spectrosc. 46, 236–239 (1992). [Google Scholar]
- 24.Bartoloni F. H., de Oliveira M. A., Ciscato L. F. M. L., Augusto F. A., Bastos E. L., Baader W. J., Chemiluminescence efficiency of catalyzed 1,2-dioxetanone decomposition determined by steric effects. J. Org. Chem. 80, 3745–3751 (2015). [DOI] [PubMed] [Google Scholar]
- 25.de Oliveira M. A., Batoloni F. H., Augusto F. A., Ciscato L. F. M. L., Bastos E. L., Baader W. J., Revision of singlet quantum yields in the catalyzed decomposition of cyclic peroxides. J. Org. Chem. 77, 10537–10544 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Wilson T., Chemiluminescence from endoperoxide of 1,4-dimethoxy-9,10-diphenylanthracene. Photochem. Photobiol. 10, 441–444 (1969). [DOI] [PubMed] [Google Scholar]
- 27.Khersonsky O., Tawfik D. S., Enzyme promiscuity: A mechanistic and evolutionary perspective. Annu. Rev. Biochem. 79, 471–505 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Soldi C., Moro A. V., Pizzolatti M. G., Correia C. R. D., Heck-Matsuda arylation as a strategy to access kavalactones isolated from Polygala sabulosa, Piper methysticum, and analogues. Eur. J. Org. Chem. 2012, 3607–3616 (2012). [Google Scholar]
- 29.Adeva M., Sahagún H., Caballero E., Peláez-Lamamié de Clairac R., Medarde M., Tomé F., Open analogues of arcyriaflavin A. Synthesis through Diels-Alder reaction between maleimides and 1-aryl-3-tert-butyldimethylsiloxy-1,3-butadienes. J. Org. Chem. 65, 3387–3394 (2000). [DOI] [PubMed] [Google Scholar]
- 30.Bacardit R., Moreno-Mañas M., Pleixats R., Functionalization at C-5 and at the C-6 methyl group of 4-methoxy-6-methyl-2-pyrone. J. Heterocycl. Chem. 19, 157–160 (1982). [Google Scholar]
- 31.Hatzigrigoriou E., Varvoglis A., Bakola-Christianopoulou M., Preparation of [hydroxy(((+)-10-camphorsulfonyl)oxy)iodo]benzene and its reactivity toward carbonyl-compounds. J. Org. Chem. 55, 315–318 (1990). [Google Scholar]
- 32.Lee I.-K., Yun B.-S., Styrylpyrone-class compounds from medicinal fungi Phellinus and Inonotus spp., and their medicinal importance. J. Antibiot. 64, 349–359 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Marenich A. V., Cramer C. J., Truhlar D. G., Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Becke A. D., Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098–3100 (1988). [DOI] [PubMed] [Google Scholar]
- 35.M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09, Revision C.01 (Gaussian Inc., 2009). [Google Scholar]
- 36.Uemi M., Ronsein G. E., Miyamoto S., Medeiros M. H. G., Di Mascio P., Generation of cholesterol carboxyaldehyde by the reaction of singlet molecular oxygen [O2 (1Δg)] as well as ozone with cholesterol. Chem. Res. Toxicol. 22, 875–884 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Silva S. M., Wagner K., Weiss D., Beckert R., Stevani C. V., Baader W. J., Studies on the chemiexcitation step in peroxyoxalate chemiluminescence using steroid-substituted activators. Luminescence 17, 362–369 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Würth C., Grabolle M., Pauli J., Spieles M., Resch-Genger U., Relative and absolute determination of fluorescence quantum yields of transparent samples. Nat. Protoc. 8, 1535–1550 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Stevani C. V., Silva S. M., Baader W. J., Studies on the mechanism of the excitation step in peroxyoxalate chemiluminescence. Eur. J. Org. Chem. 2000, 4037–4046 (2000). [Google Scholar]
- 40.Khushbaktova Z. A., Yusupova S. M., Badal’yants K. L., Syrov V. N., Batirov É. K., Isolation of hispidin from a walnut-tree fungus and its antioxidant activity. Chem. Nat. Compd. 32, 27–29 (1996). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/4/e1602847/DC1
Synthetic procedures
fig. S1. Determination of fluorescence quantum yield of the oxyluciferin.
fig. S2. Determination of the chemiluminescence and singlet quantum yields of the luciferin/luciferase reaction.
fig. S3. Dependence of the chemiluminescence intensity from luciferin/luciferase reaction on the concentration of luciferin (76 to 760 nM) and phosphate buffer (pH 6 to 8).
fig. S4. Photocathode spectral response curve provided by Berthold Detection Systems (PMT type 9107).
fig. S5. Dynamics of the reaction between substrate and luciferase microsomal preparation of N. nambi.
fig. S6. Chemiluminescence spectra of 1 (orange curve) and 3 (green curve) and the fluorescence spectrum of 4 in acetone (dotted curve).
fig. S7. Chemiluminescence from the reaction of 1,4-dimethylnaphthalene endoperoxide (DMNO2) with 3-hydroxyhispidin (luciferin).
fig. S8. Collision-induced dissociation spectrum (ESI-MS/MS) of the labeled oxyluciferin—compound 4.
fig. S9. Absorption (blue) and fluorescence emission (red) spectra of compounds 2, 3, and 5 to 9.
fig. S10. Isolation and identification of compounds in peaks 2 and 4 of the luciferin 1/luciferase reaction.
fig. S11. Proposed mechanisms for oxyluciferin degradation.
fig. S12. Synthesis of the fungal luciferin 1.
fig. S13. Synthesis of 3,4-dihydroxy-6-methyl-2H-pyran-2-one 13.
fig. S14. Synthesis of compounds 14 to 16.
fig. S15. Synthesis of fungal luciferin analogs 3 and 5 to 9.
fig. S16. Synthesis of fungal oxyluciferins 2 and 4.
fig. S17. NMR spectra chemical shifts of compounds 18 to 23.
fig. S18. NMR spectra chemical shifts of luciferin analogs 3 and 5 to 9.
table S1. Observed decay rate constants (kobs), the total light emitted (Q) by the reaction of luciferin/luciferase reaction, and chemiluminescence and singlet quantum yields (ΦCL and ΦS) at different pH and substrate concentrations.
table S2. Dependence of chemiluminescence quantum yield (ΦCL) and singlet quantum yield (ΦS) on the pH of luciferin-luciferase reaction.
table S3. Spectral characteristics of compounds 1 to 3 and 5 to 9.
table S4. Observed decay rate constant (kobs), the total of light emission (Q), photomultiplier sensitivity correction factor (fPMT), and the chemiluminescence quantum yield (ΦCL) obtained from the reaction between the crude extract of N. gardneri and compounds 1, 3, and 5 to 9.