

ABSTRACT
While intratumor heterogeneity contributes to disease progression, metastasis, and resistance to chemotherapy, it also provides a route to understanding the evolution and drivers of disease. Defects in epigenetic landscapes are intimately linked to pathogenesis of a variety of human diseases, with epigenetic deregulation promoting tumorigenesis. Understanding epigenetic heterogeneity is crucial in hepatocellular carcinoma (HCC), where epigenetic alterations are frequent, early, and pathogenic events. We determined genome-wide DNA methylation and copy number variation leveraging the Infinium 450K in a series of regenerative nodules from within single patient livers. Bioinformatics strategies were used to ascertain within-patient heterogeneity, link epigenetic changes to clinical features, and determine their relevance to disease pathogenesis. Our data demonstrate that DNA methylation and copy number alterations evolve during the pre-neoplastic phase of HCC and independently segregate regenerative nodules into distinct clusters. Regenerative nodules with a high frequency of epigenetic changes have significantly lower copy number variation, suggesting that individual nodules have differential enrichment of epigenetic and genetic components, with both contributing to disease progression. Regenerative nodules were scored based on ‘epigenetic progression’ with higher scores associated with increased proliferation measured by Ki67 staining. Early events observed in epigenetically ‘aggressive’ nodules are enriched for genes involved in liver cancer. Our study demonstrates that marked epigenetic and genetic heterogeneity exists in early pre-neoplastic liver tissue within individual patients, emphasizing the potential contributions of each mechanism to driving liver disease progression, and it unveils strategies for identifying epigenetic drivers of hepatocellular carcinoma.
KEYWORDS: Copy number variation, DNA methylation, early changes, hepatocellular cancer, heterogeneity
Introduction
Hepatocellular carcinoma (HCC) is the 3rd leading cause of cancer-related death worldwide. HCC incidence has more than doubled in the United States during the last 20 years and is expected to continue increasing due in part to the growing number of patients with advanced hepatitis C virus (HCV) infection and metabolic syndrome.1 Based on current projections, HCC will surpass colorectal and breast cancer to become the 3rd leading cancer-killer in the United States by 2030.2 One and 3-year survival rates are a grim 29% and 8%, respectively, due in large part to the advanced stage at diagnosis and lack of curative therapies.1 Hepatitis and cirrhosis arising from chronic hepatitis B virus (HBV) and/or HCV infection of the liver are major predisposing factors to HCC.3
Most HCCs appear to develop through a progressive pathway from premalignant nodular lesions in the context of liver cirrhosis, with more than 80% of HCC patients having underlying cirrhosis.4 Within cirrhotic liver both regenerative (RN) and dysplastic (DN, low and high grade) nodules can be identified, typically at a ratio of ∼10:1. A recent study comparing large regenerative nodules (LRN, >5 mm) to dysplastic nodules revealed a higher frequency of nodule enlargement for DN (33% DN/16% LRN), a higher rate of transformation to HCC for DN (75% DN/32% LRN at 100 months follow-up), and more frequent progression of DN to HCC, suggesting LRN are low-risk and DN high-risk premalignant lesions.5 HCC also shows an evolution, with early/small HCCs (< 2 cm in diameter) generally containing one or more nodules of mixed grade and differentiation status, while later/large HCCs (> 2 cm in diameter) generally lack the differentiated cell component observed in early HCCs.4
Genetic and epigenetic mechanisms generally work in concert to deregulate key growth regulatory pathways.6 hTERT promoter mutations activating telomerase expression are among the most common alterations in HCC (59%) and pre-neoplastic liver nodules (25%).7,8 Copy number variation (CNV) also increased in frequency from regenerative, low- to high-grade dysplastic nodules, and to HCC.9 Deregulated DNA methylation (5mC) patterns, characterized by global hypomethylation and gene-specific hypermethylation, are a hallmark of tumor cells, including HCC.10-12 5mC targeted to promoters and enhancers is linked to transcriptional repression, while 5mC within gene bodies is linked positively to transcription. Methylation is regulated not only by the DNA methyltransferases (DNMTs) and their unique and overlapping specificities,13 but also by the Ten-eleven-translocation (TET)-driven oxidation of 5mC to 5-hydroxymethylcytosine (5hmC).14 DNA methylation signatures have been linked to grade, differentiation status, progression, and survival,11,15 although none have made their way into clinical use. Most changes during the pre-neoplastic cirrhotic phase appear to be epigenetic in nature.11,12,16 In HCC, DNA hypomethylation predominates globally, while hypermethylation events are centered on promoters, CpG islands (CGI), and CGI shores.12,15 While candidate gene-based studies have shown that RASSF1A, APC, and SOCS1 are hypermethylated in early RN and DN lesions, little is known about the full extent of 5mC changes in early premalignant nodular liver lesions, thus limiting our knowledge about when epigenetic changes occur and how they participate in disease progression.17-19
At the pathologic level, intratumoral phenotypic heterogeneity, typically assessed through immunohistochemical stains, has been recognized since the 1800s.20 Only recently has this phenotypic heterogeneity been linked to underlying genetic, epigenetic, and transcriptional alterations.21 Intratumoral heterogeneity is a defining characteristic of human tumors. Cells within a tumor possess discrete differences in proliferative and metastatic capacity, as well as response to treatment.22 Multiple mechanisms likely underlie this heterogeneity, including genetic and epigenetic determinants. For some tumors, the extent of heterogeneity correlates with invasion, poor outcome, metastatic progression, acquisition of drug resistance and likely also contributes to the difficulty in identifying or confirming reliable cancer biomarkers.22,23 Intratumoral heterogeneity is beginning to be examined at the genetic, and, to a lesser extent, the epigenetic levels, in several tumor types, including glioma, renal, and prostate cancer.24-29 In renal cancer, for example,25 phylogenetic reconstruction of intratumoral mutational patterns revealed that only ∼35% of mutations were found in all regions of the tumor.
We recently investigated the impact of two of the major environmental insults that contribute to HCC development on the liver epigenome, chronic HCV infection and alcohol abuse, revealing both etiology-specific and shared DNA methylation alterations occurring over large regions of the genome. Changes in DNA methylation were also temporally distinct, with HCV infected livers demonstrating more changes at the cirrhosis stage and chronic alcoholic livers showing more 5mC changes after HCC development.12 Effects of viral infection and alcohol abuse likely contribute to HCC heterogeneity at all levels. Major questions remain, however, including when during progression to HCC do epigenetic changes occur, what is the extent of epigenetic heterogeneity in liver disease, and does epigenetic heterogeneity contribute to disease progression? In the current study we begin to address these issues by defining genome-wide 5mC patterns in multiple spatially distinct early proliferative lesions (RN) in individual livers derived from patients undergoing transplant for end-stage liver disease. Phylogenetic reconstruction of DNA methylation and copy number data reveal extensive heterogeneity in 5mC patterns between individual nodules and allow us to build a measure of ‘epigenetic progression’ of premalignant nodular lesions toward HCC.
Results
Characterization of patient samples
Case 1 is a 56 year-old male with cirrhosis including hepatitis C infection and chronic alcoholism. Case 1 had two benign smooth-lined biliary cysts with clear serous fluid measuring less than 1.5 cm on the inferior right and left lobes, a reactive lymph node, no significant steatosis, and no intracytoplasmic inclusions based on PAS or PAS-D stains. Case 2 tissue originates from a 56 year-old male with diffusely nodular cirrhosis induced by HCV infection. Similar to case 1, a reactive lymph node was present with no cytoplasmic inclusions or significant steatosis. The patient presented with bile-ductular proliferation and moderate to severe iron deposition within hepatocytes. Case 3 is a 56 year-old male with moderately differentiated hepatocellular carcinoma (found incidentally, after pathologic examination) and background cirrhosis due to HCV infection. The HCC was multifocal with the largest tumor measuring 1.1 cm at the greatest dimension. Severe cholestasis and bile ductular proliferation were observed with increased deposition of iron into hepatocytes and Kuppfer cells. No intracytoplasmic inclusions were observed. Additional clinical features are listed in Table 1. Each liver was sectioned into 2.0 cm slices. Ten distinct spatially separated cirrhotic nodules were identified for sample collection (example shown in Fig. 1A). Hematoxylin and Eosin (H&E) and Ki67 staining was performed on each of the 10 nodules from all three cases as well as the tumor from case 3. A pathologist confirmed through H&E and gross pathology that all nodules were regenerative hepatic nodules. Histologically there was no difference among all of the nodules examined (representative examples are shown in Fig. 1B–D).
Table 1.
Summary of patient characteristics and molecular analyses. Each parameter is color coded from light to dark within each case based on increasing values (e.g. white/low to red/high for Ki67).
 |
Figure 1.
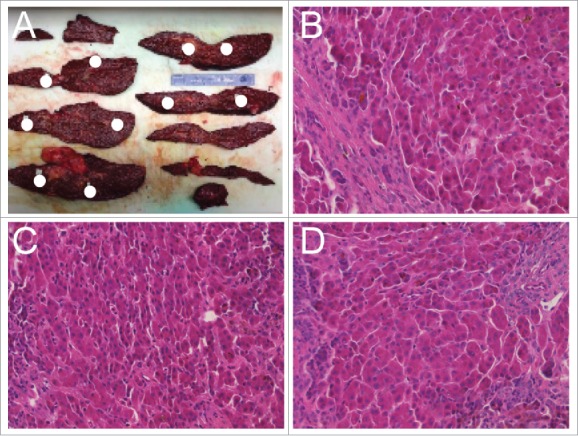
Tissue dissection and sample pathologic characterization. (A) Liver dissection with coronal sections shown from case 2. Positions of nodules isolated for analysis throughout the liver are indicated with white circles. (B-D) Representative H&E staining for three nodules demonstrating overall similarity of regenerative nodules.
Genetic and epigenetic heterogeneity in cirrhotic nodules
To assess the genetic and epigenetic heterogeneity in the cirrhotic nodules, we performed genome-wide DNA methylation profiling and copy-number analysis using the multiple samplings taken throughout the liver of three HCV-driven cirrhosis patients through application of the Infinium HumanMethylation450 Beadchip (450K array). This yielded genetic and epigenetic landscapes for 30 cirrhotic nodules (n = 10/patient) and one HCC (case 3 only). While all three patients were of the same gender, we focused on 5mC sites on autosomes for continuity, as methylation data from females is included in downstream analysis, and we excluded CpGs associated with single nucleotide polymorphisms, yielding 411,237 CpG sites for 5-methylcytosine and copy number assessment after initial filtering of data and quality control.
We first performed unsupervised hierarchical clustering of DNA methylation patterns from each case using the 5,000 most variable 5mC probes across nodules on the 450K array (Fig. 2). We also performed principal component analysis (PCA) for each case (Fig. S1). This analysis showed a surprising level of heterogeneity among nodules considering their similarity based on histologic analysis. Aberrant 5mC profiles from individual patients are shared among a subset of the cirrhotic nodules. Case 1 had a single outlier (nodule 6) that displayed a much higher frequency of DNA methylation changes compared with the other nine nodules, chiefly in the form of hypomethylation (Table 1, Fig. 2A). Case 2 showed two distinct clusters, with nodules 1, 2, 5, and 9 forming one distinct group that on average displayed more DNA methylation changes when compared with normal liver and in contrast to the remaining six nodules (P = 0.005 for total changes, P = 0.0005 for hypomethylation, Fig. 2B, orange on the dendrogram). The final sample set, case 3, consisted of 10 nodules and one HCC. Overall, the number of changes observed in this patient was relatively uniform but substantially higher in number than the other two cases (hypermethylation: P = 1.54E-8, hypomethylation: P = 0.002, total: P = 3.8E-6, Fig. 2C). While all samples from case 3 were more epigenetically divergent from normal liver, we still observed distinct clusters of nodules based on the most variable CpGs, with nodules 3, 4, 7, 8, and 9 demonstrating more aberrant events (P = 0.01), mimicking the tumor sample (Fig. 2C). The most variable CpGs within patients were spread throughout the genome, with a subset targeting key regulatory regions such as gene promoters and liver-specific enhancers in multiple samples (Fig. S2, S3).
Figure 2.

Unbiased analysis of DNA methylation and copy number heterogeneity in regenerative nodules. (A-C) Unsupervised hierarchical clustering of the 5,000 most variable CpGs for each case, where ‘outlier’ samples are color coded in orange and the remainder in green. The tumor for case 3 is labeled red. A color bar is shown to depict a range of methylation from low (blue, β = 0) to high (yellow, β = 1). (D-F) Frequency plots for copy number alterations in cases 1–3 where the relative proportion of samples with a gain (green) or loss (red) is shown for chromosomes 1–22 (chromosomes separated by dashed lines). Specific regions where green or red bars approach 1/-1 are conserved gains/losses across nodules. Case 3 includes the singular tumor.
An advantage of the 450K array is the ability to infer copy number changes.26,30 Therefore, we examined chromosomal abnormalities for all three cases (Fig. 2D–F) relative to a representative normal liver control (no cancer or cirrhosis). Case 1 had the fewest copy number changes, case 2 had a moderate increase in the number of CNVs, and case 3 had a relatively stable genome with the exception of one nodule (Table 1). Specifically, nodule 9 from case 3 had a much higher frequency of CNV relative to any other nodule. The tumor from case 3 was marked by chromosomal amplifications and relatively few losses. Very few common copy number alterations were observed between more than one nodule from each patient, suggesting marked genetic heterogeneity also exists in these nodules.
Evolution of DNA methylation changes in pre-neoplastic liver disease
Other groups have shown that phylogenetic trees provide insight into the evolution of DNA methylation changes during tumorigenesis and tumor recurrence.27-29 Therefore, to determine if these DNA methylation alterations have a shared evolution, we performed phylogenetic reconstruction from DNA methylation and copy number variation using the R package ‘ape’, similar to analyses reported previously examining the clonal evolution of prostate cancer.27,31 Phylogenetic trees from DNA methylation data (Fig. 3A–C) are similar to observations from unsupervised hierarchical clustering, highlighting the outlier nodule in case 1 (nodule 6) and the distinct clusters in cases 2 and 3. Interestingly, when we performed analysis using the chromosomal breakpoints identified from inference of CNV on the 450K array, we observed a strikingly similar structure for case 2, with the same four nodules forming a distinct group (Fig. 3E). We observe an outlier group from DNA methylation comprising nodules 1, 2, 5, and 9, with the same four nodules separating off in the CNV phylogenetic tree for case 2. However, closer examination reveals that DNA methylation changes are more pronounced in these outlier nodules (P = 0.005), while copy number changes are significantly fewer (P = 0.019). This trend is consistent for nodules from case 1 as well, with nodule 6 having the most aberrant methylation changes and the fewest copy number changes. Case 3 had a strong epigenetic component, but relatively few genetic changes, except for nodule 9, which had more CNVs than any other nodule (Figs. 3C, F, Table 1). Taken together, these data suggest that DNA methylation and CNV may, in some cases, evolve independently during the early phases of hepatocarcinogenesis, and poise specific nodules for eventual dysplasia and/or transformation through distinct mechanisms.
Figure 3.
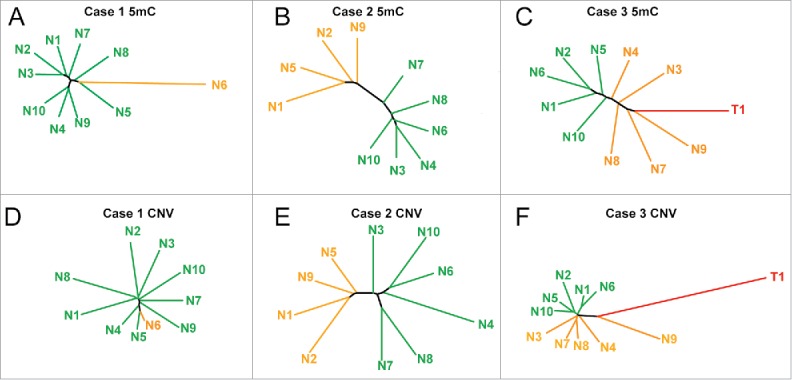
Phylogenetic trees based on DNA methylation landscapes and copy number variation. (A-C) Phylogenetic trees from the top 5,000 most variable 5mC sites from cases 1–3. (D-F) Phylogenetic trees from chromosomal aberrations identified in cases 1–3. Distinct groups and unique samples are labeled orange, with the remaining regenerative nodules in green, and the tumor for case 3 in red. Individual branches are color coded in the same manner as the dendrograms in Fig. 2.
Due to the distinct clustering that we observed in phylogenetic trees constructed from DNA methylation, we hypothesized that these epigenetic changes might reflect the potential for specific nodules to progress toward dysplasia and/or tumorigenesis. To test this, we incorporated DNA methylation landscapes from the 450K array from our previous publication 12 as well as publically available data from The Cancer Genome Atlas, into our analysis. In total, we compiled samples from normal (n = 30), cirrhosis (n = 30), and HCC (n = 46) livers, all obtained from patients with chronic HCV infection (the same etiology common to all nodules from cases 1–3). We examined the top 5,000 hyper- and hypo-methylation events in tumors relative to normal controls and plotted the phylogenetic tree (Fig. 4A). Single nucleotide polymorphisms were removed to ensure observed changes were due to DNA methylation. We observed that the resulting phylogenetic tree had three distinct clusters correlating with disease stage: normal liver (blue, no cirrhosis or HCC), cirrhosis (green), and HCC (red). Normal samples clustered closely together, while cirrhotic tissues were less similar, and tumors were the most divergent based on the Euclidean distance relative to the average of normal liver. The average Euclidean distance for normal (relative to the average of all normal tissues), cirrhotic, and HCC livers was 6.0, 8.1, and 35.8, respectively. Comparisons between the three groups were significantly different, indicating a stepwise increase in Euclidean distance from normal to cirrhotic to HCC liver tissues (P < 5E-9 for all comparisons). We took advantage of this phenomenon of increased Euclidean distance significantly correlating with disease stage to create a surrogate measure of nodule ‘aggressiveness’ or progression, which we term an ‘Epigenetic Progression’ (EP) score (Table 1, expanded upon further below).
Figure 4.
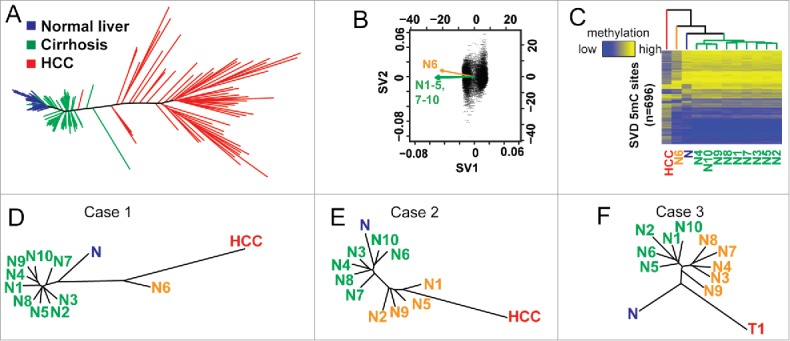
DNA methylation-based phylogenetic trees delineate disease stages. (A) A phylogenetic tree created using the R package ‘ape’ using normal (blue, n = 30), cirrhotic (green, n = 30), and HCC liver (red, n = 46). (B) Singular value decomposition based on CpGs from (A) performed on DNA methylation data from case 1. The original data set is filtered down to 696 CpGs by this method. (C) A heatmap of the 696 CpGs from (B, case 1) with the average of HCCs (red, n = 46), normal liver (blue, n = 30), the outlier nodule (N6, orange) and other nodules (N1–6; 7–10, green) indicated (D-F). Phylogenetic trees constructed after SVD analysis of CpGs identified from (A) for cases 1–3. The average of normal liver (n = 30) and HCC (n = 46) is shown for cases 1 and 2, while the tumor from case 3 (C3T1) is used in panel F.
We next examined these differentially methylated CpGs (hyper- and hypo-methylated) between normal liver and HCC to determine if these frequent targets of aberrant DNA methylation in hepatocarcinogenesis are also the ones targeted for epigenetic deregulation in the pre-neoplastic nodular lesions. As an added measure to increase the likelihood that individual 5mC sites are relevant, we performed singular value decomposition (SVD) on this subset of CpGs for each case to determine if they were capable of separating nodules into the previously observed clusters, thereby implying the clusters have relevance to HCC-specific epigenetic changes (Fig. 4B, Fig. S4). In principle, this method determines which events have the greatest impact on the structure of the tree, allowing one to identify events most likely to be relevant to pathogenesis. Plotting these CpGs (n = 696, 216, and 298 CpGs for cases 1–3, respectively, Fig. 4C, Fig. S5) shows that specific nodule clusters appear more similar to either the average of normal (n = 30) or tumors (n = 46). Indeed, these aberrant DNA methylation events segregated the outlier from case 1 from the remainder of the nodules with an EP score of 5.06 compared with an average of 2.93, with tumor having an EP score of 8.62 (Table 1, Fig. 4C–D). The same group of four sampled regions from case 2 (nodules 1, 2, 5, and 9) clustered independently with an average EP score of 0.82 higher than the other six regions, an increase of more than 50% (P<0.001, Fig. 4E). Importantly, the structure of the trees in cases 1 and 2 demonstrate that tumor associates more closely with the outliers. Case 3 is unique as we have the matching tumor from the same patient for comparison, and again we observe that the cirrhotic nodules form two clusters, one of which is more similar to the tumor (Fig. 4F). Our results therefore indicate that a set of DNA methylation changes that typify HCC are strongly reflected in the heterogeneity observed during early liver disease, which are also linked to the proliferative capacity of individual nodules.
Linking epigenetic phenotypes to the biology of liver disease
To investigate whether cirrhotic nodules that are less similar to normal liver are indeed more ‘aggressive’ and/or have a greater chance to progress to a more advanced state (e.g., RN to DN), we performed Ki67 immunohistochemistry on all of the samples analyzed for DNA methylation from cases 1–3. The outlier nodule from case 1 had the highest Ki67 score [8 cells per high powered field (HPF)] compared with an average of 2.4 for the other nodules (Fig. 4D, Table 1). Similarly, the four outlier nodules for case 2 averaged 9.5 cells/HPF relative to 4.0 for the other six nodules (P = 0.0036, Fig. 4E, Table 1). Case 3, which also showed the highest number of aberrant DNA methylation events, had relatively consistent Ki67 scores across nodules, with higher Ki67-positivity in the tumor (Fig. 4F, Table 1). All together, the 5mC changes identified here that drive nodule clustering into groups defined by Ki67 score suggest that individual cirrhotic nodules have already acquired epigenetic changes characteristic of hepatocellular cancers, which may predispose them toward transformation or sow the seeds for progression when combined with additional genetic and/or epigenetic lesions.
DNA methylation heterogeneity in cirrhosis converges on key liver cancer related pathways
We performed Ingenuity Pathway Analysis (IPA) on the genes associated with the most variable CpGs from unsupervised hierarchical clustering in Fig. 1, as well as the CpGs identified from SVD in Fig. 4. As case 2 exhibited two distinct groups, we focused on this patient as a proof-of-principle. Ingenuity pathway analysis using the 5,000 most variable CpGs from case 2 showed that the most enriched term was ‘Cancer’ (Fig. 5A). More in-depth analysis revealed specific links to liver disease and liver cancer genes with pathway terms that include ‘liver cancer’ (most significant category under ‘cancer’) and ‘gastrointestinal cancer’ (Fig. 5B). Similarly, IPA analysis of genes linked to singular value decomposition-selected CpGs included terms like ‘Cancer’ and ‘Hepatic System Disease’ (Fig. 5C). Other specific pathway enrichments include different cancers (skin, pancreas, liver), immortalization, and malignant tissue (Fig. 5D). Examination of specific loci reveals regions targeted for specific methylation gain/loss events in gene regulatory regions (Fig. 5E–F). Two such regions within the GSTO2 and UPB1 promoters demonstrate hyper- and hypo-methylation, respectively in the ‘aggressive’ nodules from case 2, suggesting that these changes are early initiators of disease progression. These data collectively implicate epigenetic changes as early events that may predispose select genes for deregulation, which in turn may facilitate progression of nodules to more advanced dysplasia or HCC.
Figure 5.
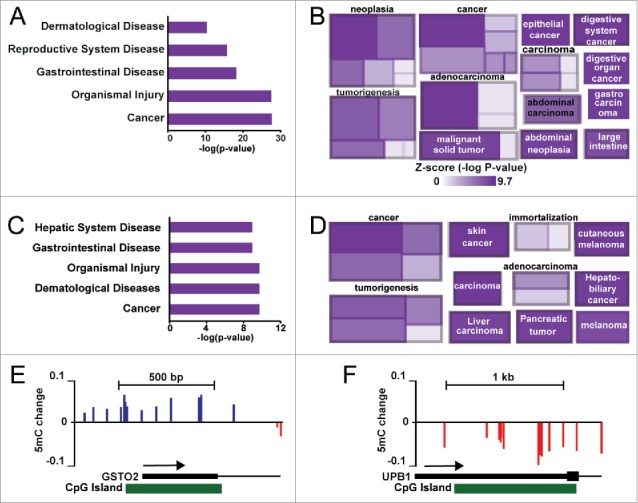
Gene ontology analysis of hypervariable CpG sites in case 2. (A) Ingenuity pathway analysis of diseases and disorders enriched in the top 5,000 most variable CpGs. (B) A cobblestone plot of processes associated with genes in (A). The color of the rectangle reflects the associated Z-score for the term, while the size represents the number of genes enriched. (C) Ingenuity pathway analysis based upon genes associated with CpGs (n = 216) identified from singular value decomposition of hyper- and hypo-methylated CpGs in HCC from case 2. (D) A cobblestone plot of processes associated with genes in (C). (E-F) UCSC genome browser views of the GSTO2 and UPB1 genes, depicting regions of hyper- and hypo-methylation in case 2 nodules 1, 2, 5, and 9 relative to the other nodules.
Discussion
In this study, we observed substantial epigenetic heterogeneity at the earliest stage of premalignant liver disease progression toward hepatocellular cancer. By constructing phylogenetic trees based on DNA methylation and CNV data, we inferred genetic and epigenetic heterogeneity that created distinct clusters of samples derived from individual patients despite their similarity at the histopathologic level. In one case, genetic- and epigenetic-based phylogenetic trees formed markedly similar structures, suggesting that genetic and epigenetic aberrations evolve in parallel in hepatocarcinogenesis. DNA methylation-based phylogenetic trees derived from all stages of disease show a linear progression from normal to cirrhotic to HCC liver states, indicating this method can link epigenetic patterns to tumorigenic potential. Indeed, through this method we showed that early aberrant epigenetic deregulation events were associated with increased proliferation based on Ki67 staining. Many of these early methylation changes target genes and pathways involved in hepatocarcinogenesis, implying they are functionally relevant. Overall, these results reveal that specific nodules may be epigenetically predisposed to advance to dysplasia and/or HCC and that the 5mC changes that delineate the more ‘progressed’ lesions might be particularly important for surveillance and/or the ability to predict liver disease progression.
We identified a subset of regenerative nodules with increased Ki67 staining that correlated with aberrant DNA methylation events and our epigenetic progression score. This phenomenon is particularly important as pathologists typically only refer to histologic analysis when determining whether liver nodules are regenerative or dysplastic. Combining H&E staining with epigenetic signatures such as those identified here could facilitate our ability to differentiate regenerative from dysplastic nodules and early HCC by identifying key molecular changes missed with histologic and morphologic criteria alone. Much like the importance of molecular subtypes in cancer, assigning pre-neoplastic lesions into groups that are more or less prone to progress could facilitate identification of the earliest cells responsible for disease progression based on inherent signatures, and permit identification of key drivers of disease initiation and progression. Epigenetic heterogeneity studies of topographically distinct sites in cancer reflect the history of the tumor's origins and should facilitate identification of aberrantly regulated genes.27 Indeed, analysis of epigenetic heterogeneity in glioma identified the TP73 gene as a potential driver.29 It is well established that HCC frequently displays heterogeneous growth patterns and/or cytological features within individual tumors,32 which may prove equally important early in disease based on evidence from our study. To our knowledge, our study is the first to integrate genetic and epigenetic heterogeneity in HCC at a genome-wide level within individual patients. A recent analysis of HBV-associated HCCs at the genetic level revealed marked intratumoral heterogeneity between patients (8–68% mutations in common between regions of the same tumor) with larger tumors having more heterogeneity.33 The potential importance of epigenetic events in progression to HCC is highlighted by studies showing that experimentally targeting DNA methylation defects to specific growth regulatory genes accelerates cell proliferation and drives spontaneous tumor formation.34,35
Our data, although derived from a limited number of samples, shows that while genetic and epigenetic alterations are prevalent in the early pre-neoplastic phases of HCC, one mechanism appears to predominate within an individual lesion. In all three cases examined, phylogenetic tree structure was similar based on DNA methylation- and copy number alteration-derived data, yet changes in the methylome occurred more frequently in nodules with fewer CNV events. As a parallel to this observation, epigenetic alterations in pediatric CNS tumors are the most frequent type of ‘mutation’, suggesting that pediatric ependymomas are almost exclusively driven by epigenetic defects; in contrast, other cancers have a very strong genetic component.36-38 There is robust evidence that changes in DNA methylation directly result in tumorigenesis, as demonstrated by Yu et al. with targeted hypermethylation of p16INK4A and subsequent induction of spontaneous tumor formation.34 In addition, tumor progression and metastasis is intimately linked to epigenetic modifications.39,40 The genetic and epigenetic contributions to carcinogenesis are difficult to separate since many tumors have cross-talk between genetic and epigenetic mechanisms, such as mutation of the IDH1 and SETD2 genes leading to distinct DNA methylation profiles that impact patient outcome,41,42 and epigenetic regulatory genes themselves (e.g., TET2 and DNMT3A) being targeted for mutation.43-45 While these findings link the genome to the epigenome bi-directionally, they also emphasize the importance of dissecting the individual contributions of each type of molecular event to carcinogenesis.
Since cirrhotic nodules implanted into mice do not form tumors, they cannot be tested directly for malignant potential in this conventional functional assay and therefore, surrogate methods must be used to infer this potential. To this end, heterogeneity of both genetic and epigenetic mutations lends insight into the history of the disease, allowing one to trace back to the initial events (drivers) promoting growth advantage and metastatic potential, and also yield information on the cell-of-origin, age, and other features of the patient and/or disease. While we observe distinct methylation patterns in subsets of nodules obtained from each patient, the normal liver also demonstrates epigenetic heterogeneity, especially in enhancer regions marked by H3K4me1.46 This suggests that the stochastic nature of the epigenome in the liver may inadvertently lead to DNA methylation landscapes predisposed to progression toward regenerative nodule formation and ultimately carcinogenesis, particularly when exposed to chronic environmental insults including hepatitis C viral infection. Our data suggest that early epigenetic changes could fuel progression to a more advanced proliferative state with increased propensity toward dysplasia and carcinogenesis when combined with additional lesions (epigenetic or genetic), or environmental insults that interfere with maintenance of cell-specific DNA methylation landscapes, such as inflammation.
We demonstrate here that pre-neoplastic lesions are enriched for DNA methylation changes associated with liver-specific cancer pathways in a distinct subset of nodules within the same patient. Further study of this process should not only facilitate stratification of early disease and permit better prediction of which nodules can/will advance to later disease stages, but also yield potential mechanisms underlying the poor response of HCC to chemotherapy. Frontline chemotherapy for HCC results in modest improvements in survival, with sorafenib conveying only a 3-month survival benefit on average in a phase III clinical trial.47 Currently, most clinical trials do not account for intratumoral heterogeneity.48 Epigenetic heterogeneity too may influence response to chemotherapy.49 In contrast to genetic and epigenetic heterogeneity, transcriptome level analyses revealed that premalignant liver lesions (DN) had minor changes in expression and were relatively homogeneous; extensive alteration of cancer-related pathways occurred late in HCC progression.50 This and other observations collectively highlight that: 1) epigenetic changes may precede and predispose to transcriptional changes; 2) epigenetic changes may serve as better prognostic/diagnostic markers since they occur before transcriptional and gross morphological changes; 3) use of early epigenetic changes could facilitate determination of whether a particular nodular lesion will progress to HCC; and 4) a more in-depth assessment of spatially separated tumor sections in a larger patient population is required to fully assess the role heterogeneity in HCC progression and response to treatment.
Materials and methods
Sample collection
Liver tissue from three 56-year-old male hepatitis C infected patients was collected as part of liver transplant surgery performed at the University of Florida Shands Hospital between March and September of 2014 (Table 1). An experienced liver pathologist sectioned the livers and micro-dissected 10 spatially separated cirrhotic nodules from all three cases; one case had an incidental finding of hepatocellular carcinoma after pathologic examination that had not been identified through imaging (Fig. 1). The freshly dissected liver tissues were stored at −80°C. A portion of the frozen tissue was used for DNA isolation, and a portion of tissue was fixed in formalin for routine histological examination. Protocols for tissue collection are approved by the Institutional Review Board and patient consent was obtained.
Genome-wide DNA methylation profiling
Genomic DNA was isolated using a standard phenol-chloroform protocol. Resultant DNA was quantified using the Quant-iT PicoGreen dsDNA Assay Kit (ThermoFisher) and checked for quality using a custom TaqMan probe. Five hundred micrograms of input DNA was used with the manufacturer's protocols to bisulfite treat and hybridize to the Infinium HumanMethylation450 BeadChip (450K; Illumina, San Diego, CA).
Data processing
Quality control of resultant 450K array data was performed using two methods: Genome Studio Methylation Module (Illumina, San Diego, CA) and the R bioconductor package ‘minfi’. Methylation data was normalized through subset within-array quantile normalization (SWAN) as described previously.12 Copy number variations were also obtained from the 450K array through the R package ‘ChAMP’, which infers copy number changes using intensity of individual and surrounding probes.30 Only events that reached a minimum threshold of |0.3| were included in downstream analysis. Heatmaps and phylogenetic trees were constructed using R with the ‘heatmap.3′ and ‘ape’ (Analyses of Phylogenetics and Evolution) packages. ‘Ape’ is a minimal evolution algorithm used to estimate trees with distance-based methods (Euclidean distance was used herein).31 We identified CpGs that have the largest impact on tree structure through singular value decomposition (SVD),29 which summarizes the main ways mean-centered data deviate from zero, allowing us to identify events that most influence the tree structure and thus are most likely to be important for disease pathogenesis. Gene ontology analysis was performed using Ingenuity Pathway Analysis (Qiagen, Redwood City, CA). Infinium 450K data are available in the NCBI GEO repository (GSE87056).
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This study was supported by an AASLD Clinical and Translational Research Award in Liver Diseases (RAH), R01 AA19976 (KDR and CL), the Mayo Clinic Center for Individualized Medicine (KDR), and the Mayo Clinic Cancer Center (KDR).
References
- 1.El-Serag HB. Hepatocellular carcinoma. N Engl J Med 2011; 365:1118-27; PMID:21992124; http://dx.doi.org/ 10.1056/NEJMra1001683 [DOI] [PubMed] [Google Scholar]
- 2.Njei B, Rotman Y, Ditah I, Lim JK. Emerging trends in hepatocellular carcinoma incidence and mortality. Hepatology 2015; 61:191-9; PMID:25142309; http://dx.doi.org/ 10.1002/hep.27388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology 2004; 127:S35-50; PMID:15508101; http://dx.doi.org/ 10.1053/j.gastro.2004.09.014 [DOI] [PubMed] [Google Scholar]
- 4.Kojiro M, Roskams T. Early hepatocellular carcinoma and dysplastic nodules. Semin Liver Dis 2005; 25:133-42; PMID:15918142; http://dx.doi.org/ 10.1055/s-2005-871193 [DOI] [PubMed] [Google Scholar]
- 5.Sato T, Kondo F, Ebara M, Sugiura N, Okabe S, Sunaga M, Yoshikawa M, Suzuki E, Ogasawara S, Shinozaki Y, et al.. Natural history of large regenerative nodules and dysplastic nodules in liver cirrhosis: 28-year follow-up study. Hepatol Int 2015; 9:330-6; PMID:25788204; http://dx.doi.org/ 10.1007/s12072-015-9620-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell 2012; 22:9-20; PMID:22789535; http://dx.doi.org/ 10.1016/j.ccr.2012.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, et al.. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet 2015; 47:505-11; PMID:25822088; http://dx.doi.org/ 10.1038/ng.3252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nault JC, Mallet M, Pilati C, Calderaro J, Bioulac-Sage P, Laurent C, Laurent A, Cherqui D, Balabaud C, Zucman-Rossi J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun 2013; 4:2218; PMID:23887712; http://dx.doi.org/ 10.1038/ncomms3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tornillo L, Carafa V, Sauter G, Moch H, Minola E, Gambacorta M, Vecchione R, Bianchi L, Terracciano LM. Chromosomal alterations in hepatocellular nodules by comparative genomic hybridization: high-grade dysplastic nodules represent early stages of hepatocellular carcinoma. Lab Invest 2002; 82:547-53; PMID:12003995; http://dx.doi.org/ 10.1038/labinvest.3780449 [DOI] [PubMed] [Google Scholar]
- 10.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3:415-28; PMID:12042769; http://dx.doi.org/ 10.1038/nrg816 [DOI] [PubMed] [Google Scholar]
- 11.Hernandez-Vargas H, Lambert MP, Le Calvez-Kelm F, Gouysse G, McKay-Chopin S, Tavtigian SV, Scoazec JY, Herceg Z. Hepatocellular carcinoma displays distinct DNA methylation signatures with potential as clinical predictors. PLoS One 2010; 5:e9749; PMID:20305825; http://dx.doi.org/ 10.1371/journal.pone.0009749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hlady RA, Tiedemann RL, Puszyk W, Zendejas I, Roberts LR, Choi JH, Liu C, Robertson KD. Epigenetic signatures of alcohol abuse and hepatitis infection during human hepatocarcinogenesis. Oncotarget 2014; 5:9425-43; PMID:25294808; http://dx.doi.org/ 10.18632/oncotarget.2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tiedemann RL, Putiri EL, Lee JH, Hlady RA, Kashiwagi K, Ordog T, Zhang Z, Liu C, Choi JH, Robertson KD. Acute depletion redefines the division of labor among DNA methyltransferaes in methylating the human genome. Cell Rep 2014; 9:1554-66; PMID:25453758; http://dx.doi.org/ 10.1016/j.celrep.2014.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev 2016; 30:733-50; PMID:27036965; http://dx.doi.org/ 10.1101/gad.276568.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villanueva A, Portela A, Sayols S, Battiston C, Hoshida Y, Mendez-Gonzalez J, Imbeaud S, Letouze E, Hernandez-Gea V, Cornella H, et al.. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology 2015; 61:1945-56; PMID:25645722; http://dx.doi.org/ 10.1002/hep.27732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet 2002; 31:339-46; PMID:12149612; http://dx.doi.org/ 10.1038/ng0802-339 [DOI] [PubMed] [Google Scholar]
- 17.Di Gioia S, Bianchi P, Destro A, Grizzi F, Malesci A, Laghi L, Levrero M, Morabito A, Roncalli M. Quantitative evaluation of RASSF1A methylation in the non-lesional, regenerative and neoplastic liver. BMC Cancer 2006; 6:89; PMID:16606445; http://dx.doi.org/ 10.1186/1471-2407-6-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee S, Lee HJ, Kim JH, Lee HS, Jang JJ, Kang GH. Aberrant CpG island hypermethylation along multistep hepatocarcinogenesis. Am J Pathol 2003; 163:1371-8; PMID:14507645; http://dx.doi.org/ 10.1016/S0002-9440(10)63495-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Um TH, Kim H, Oh BK, Kim MS, Kim KS, Jung G, Park YN. Aberrant CpG island hypermethylation in dysplastic nodules and early HCC of hepatitis B virus-related human multistep hepatocarcinogenesis. J Hepatol 2011; 54:939-47; PMID:21145824; http://dx.doi.org/ 10.1016/j.jhep.2010.08.021 [DOI] [PubMed] [Google Scholar]
- 20.Navin NE, Hicks J. Tracing the tumor lineage. Mol Oncol 2010; 4:267-83; PMID:20537601; http://dx.doi.org/ 10.1016/j.molonc.2010.04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Birkbak NJ, Andersen JB. Heterogeneity among liver cancer – a hurdle to optimizing therapy. Gastroenterology 2016; 150:818-21; PMID:26924088; http://dx.doi.org/ 10.1053/j.gastro.2016.02.063 [DOI] [PubMed] [Google Scholar]
- 22.Almendro V, Cheng YK, Randles A, Itzkovitz S, Marusyk A, Ametller E, Gonzalez-Farre X, Munoz M, Russnes HG, Helland A, et al.. Inference of tumor evolution during chemotherapy by computational modeling and in situ analysis of genetic and phenotypic cellular diversity. Cell Rep 2014; 6:514-27; PMID:24462293; http://dx.doi.org/ 10.1016/j.celrep.2013.12.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerlinger M, Swanton C. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br J Cancer 2010; 103:1139-43; PMID:20877357; http://dx.doi.org/ 10.1038/sj.bjc.6605912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al.. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012; 366:883-92; PMID:22397650; http://dx.doi.org/ 10.1056/NEJMoa1113205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR, et al.. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 2014; 46:225-33; PMID:24487277; http://dx.doi.org/ 10.1038/ng.2891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feber A, Guilhamon P, Lechner M, Fenton T, Wilson GA, Thirlwell C, Morris TJ, Flanagan AM, Teschendorff AE, Kelly JD, et al.. Using high-density DNA methylation arrays to profile copy number alterations. Genome Biol 2014; 15:R30; PMID:24490765; http://dx.doi.org/ 10.1186/gb-2014-15-2-r30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brocks D, Assenov Y, Minner S, Bogatyrova O, Simon R, Koop C, Oakes C, Zucknick M, Lipka DB, Weischenfeldt J, et al.. Intratumor DNA methylation heterogeneity reflects clonal evolution in aggressive prostate cancer. Cell Rep 2014; 8:798-806; PMID:25066126; http://dx.doi.org/ 10.1016/j.celrep.2014.06.053 [DOI] [PubMed] [Google Scholar]
- 28.Aryee MJ, Liu W, Engelmann JC, Nuhn P, Gurel M, Haffner MC, Esopi D, Irizarry RA, Getzenberg RH, Nelson WG, et al.. DNA methylation alterations exhibit intraindividual stability and interindividual heterogeneity in prostate cancer metastases. Sci Transl Med 2013; 5:169ra10; PMID:23345608; http://dx.doi.org/ 10.1126/scitranslmed.3005211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mazor T, Pankov A, Johnson BE, Hong C, Hamilton EG, Bell RJ, Smirnov IV, Reis GF, Phillips JJ, Barnes MJ, et al.. DNA methylation and somatic mutations converge on the cell cycle and define similar evolutionary histories in brain tumors. Cancer Cell 2015; 28:307-17; PMID:26373278; http://dx.doi.org/ 10.1016/j.ccell.2015.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morris TJ, Butcher LM, Feber A, Teschendorff AE, Chakravarthy AR, Wojdacz TK, Beck S. ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics 2014; 30:428-30; PMID:24336642; http://dx.doi.org/ 10.1093/bioinformatics/btt684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 2004; 20:289-90; PMID:14734327; https://doi.org/ 10.1093/bioinformatics/btg412 [DOI] [PubMed] [Google Scholar]
- 32.Friemel J, Rechsteiner M, Frick L, Bohm F, Struckmann K, Egger M, Moch H, Heikenwalder M, Weber A. Intratumor heterogeneity in hepatocellular carcinoma. Clin Cancer Res 2015; 21:1951-61; PMID:25248380; http://dx.doi.org/ 10.1158/1078-0432.CCR-14-0122 [DOI] [PubMed] [Google Scholar]
- 33.Xue R, Li R, Guo H, Guo L, Su Z, Ni X, Qi L, Zhang T, Li Q, Zhang Z, et al.. Variable intra-tumor genomic heterogeneity of multiple lesions in patients with hepatocellular carcinoma. Gastroenterology 2016; 150:998-1008; PMID:26752112; http://dx.doi.org/ 10.1053/j.gastro.2015.12.033 [DOI] [PubMed] [Google Scholar]
- 34.Yu DH, Waterland RA, Zhang P, Schady D, Chen MH, Guan Y, Gadkari M, Shen L. Targeted p16(Ink4a) epimutation causes tumorigenesis and reduces survival in mice. J Clin Invest 2014; 124:3708-12; PMID:25061879; http://dx.doi.org/ 10.1172/JCI76507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bernstein DL, Le Lay JE, Ruano EG, Kaestner KH. TALE-mediated epigenetic suppression of CDKN2A increases replication in human fibroblasts. J Clin Invest 2015; 125:1998-2006; PMID:25866970; http://dx.doi.org/ 10.1172/JCI77321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DT, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, et al.. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 2013; 24:660-72; PMID:24183680; http://dx.doi.org/ 10.1016/j.ccr.2013.10.006 [DOI] [PubMed] [Google Scholar]
- 37.Bjerke L, Mackay A, Nandhabalan M, Burford A, Jury A, Popov S, Bax DA, Carvalho D, Taylor KR, Vinci M, et al.. Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov 2013; 3:512-9; PMID:23539269; http://dx.doi.org/ 10.1158/2159-8290.CD-12-0426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., Kinzler KW. Cancer genome landscapes. Science 2013; 339:1546-58; PMID:23539594; http://dx.doi.org/ 10.1126/science.1235122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feinberg AP, Koldobskiy MA, Gondor A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet 2016; 17:284-99; PMID:26972587; http://dx.doi.org/ 10.1038/nrg.2016.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vanharanta S, Shu W, Brenet F, Hakimi AA, Heguy A, Viale A, Reuter VE, Hsieh JJ, Scandura JM, Massague J. Epigenetic expansion of VHL-HIF signal output drives multiorgan metastasis in renal cancer. Nat Med 2013; 19:50-6; PMID:23223005; http://dx.doi.org/ 10.1038/nm.3029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duncan CG, Barwick BG, Jin G, Rago C, Kapoor-Vazirani P, Powell DR, Chi JT, Bigner DD, Vertino PM, Yan H. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome Res 2012; 22:2339-55; PMID:22899282; http://dx.doi.org/ 10.1101/gr.132738.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang C, McKeithan TW, Gong Q, Zhang W, Bouska A, Rosenwald A, Gascoyne RD, Wu X, Wang J, Muhammad Z, et al.. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood 2015; 126:1741-52; PMID:26268241; http://dx.doi.org/ 10.1182/blood-2015-05-644591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, et al.. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet 2012; 44:760-4; PMID:22634756; http://dx.doi.org/ 10.1038/ng.2291 [DOI] [PubMed] [Google Scholar]
- 44.Plass C, Pfister SM, Lindroth AM, Bogatyrova O, Claus R, Lichter P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat Rev Genet 2013; 14:765-80; PMID:24105274; http://dx.doi.org/ 10.1038/nrg3554 [DOI] [PubMed] [Google Scholar]
- 45.Tiedemann RL, Hlady RA, Hanavan PD, Lake DF, Tibes R, Lee JH, Choi JH, Ho TH, Robertson KD. Dynamic reprogramming of DNA methylation in SETD2-deregulated renal cell carcinoma. Oncotarget 2016; 7:1927-46; PMID:26646321; http://dx.doi.org/ 10.18632/oncotarget.6481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gravina S, Dong X, Yu B, Vijg J. Single-cell genome-wide bisulfite sequencing uncovers extensive heterogeneity in the mouse liver methylome. Genome Biology 2016; 17:150; PMID:27380908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al.. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008; 359:378-90; PMID:18650514; http://dx.doi.org/ 10.1056/NEJMoa0708857 [DOI] [PubMed] [Google Scholar]
- 48.Mazor T, Pankov A, Song JS, Costello JF. Intratumoral heterogeneity of the epigenome. Cancer Cell 2016; 29:440-51; PMID:27070699; http://dx.doi.org/ 10.1016/j.ccell.2016.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, et al.. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010; 141:69-80; PMID:20371346; http://dx.doi.org/ 10.1016/j.cell.2010.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marquardt JU, Seo D, Andersen JB, Gillen MC, Kim MS, Conner EA, Galle PR, Factor VM, Park YN, Thorgeirsson SS. Sequential transcriptome analysis of human liver cancer indicates late stage acquisition of malignant traits. J Hepatol 2014; 60:346-53; PMID:24512821 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.