

Abstract
The brain plays a key role in the controls of energy intake and expenditure and many genes associated with obesity are expressed in the central nervous system. Technological and conceptual advances in both basic and clinical neurosciences have expanded the traditional view of homeostatic regulation of body weight by mainly the hypothalamus to include hedonic controls of appetite by cortical and subcortical brain areas processing external sensory information, reward, cognition, and executive functions. Thus, hedonic controls interact with homeostatic controls to regulate body weight in a flexible and adaptive manner that takes environmental conditions into account. This new conceptual framework has several important implications for the treatment of obesity. Because much of this interactive neural processing is outside awareness, cognitive restraint in a world of plenty is made difficult and prevention and treatment of obesity should be more rationally directed to the complex and often redundant mechanisms underlying this interaction.
Keywords: appetite, physical activity, cognition, reward, self-control, hypothalamus, cortex, limbic system
The importance of consuming energy is reflected by the elaborate regulatory system that has evolved over millions of years, reaching its pinnacle in the high energy demand of homeotherms such as humans and birds1. It takes a truly remarkable regulatory system to manage the energy demands of a hummingbird which has a heart rate of up to 1,200 beats/min, a muscle oxygen consumption rate 10 times higher than that of an elite athlete, a reversible fat compartment allowing long migration flights, and the ability to enter a mode of reduced metabolism (torpor) to survive cold nights without food1. Similar to the hummingbird, our human ancestors also needed a regulatory system that could adaptively respond to periods of limited food supply and/or high physical activity, such as migrations and famines. In addition, the mechanisms for finding and selecting optimal food sources while avoiding toxic foods had to be strong enough to compete with other behaviors and had to be learned and passed to future generations.
Such a high energy throughput also requires a large gastrointestinal tract capable of efficiently digesting and absorbing necessary nutrients. This is particularly true when high physical activity is combined with low dietary energy density and a massive brain as in early hominids. At the highest level of physical activity, our ancestors might have eaten several kilograms of food and absorbed several thousand Kcals every day. It is thus not surprising that the gut brain axis is a crucial component of the control of food intake and regulation of energy balance (For an in-depth discussion of gut-brain communication please see chapter x in this issue by Monteiro and Batterham)
To be efficient, the regulatory system requires (1) complex and redundant nutrient sensing and monitoring mechanisms, (2) a flexible integrative mechanism that can learn from and adapt to changing external and internal conditions, and (3) powerful effector mechanisms for energy intake and metabolism (Fig. 1). The brain has inputs from and outputs to other organs just like a computer’s peripherals, but the crucial integrator is located in the brain. Indeed, research some 70 years ago identified the hypothalamus as indispensable for the regulation of food intake and body weight2–4, and the most recent genome wide association study found that an overwhelming majority of genes associated with body mass index are expressed in the central nervous system, many of them in the hypothalamus5.
Fig. 1.
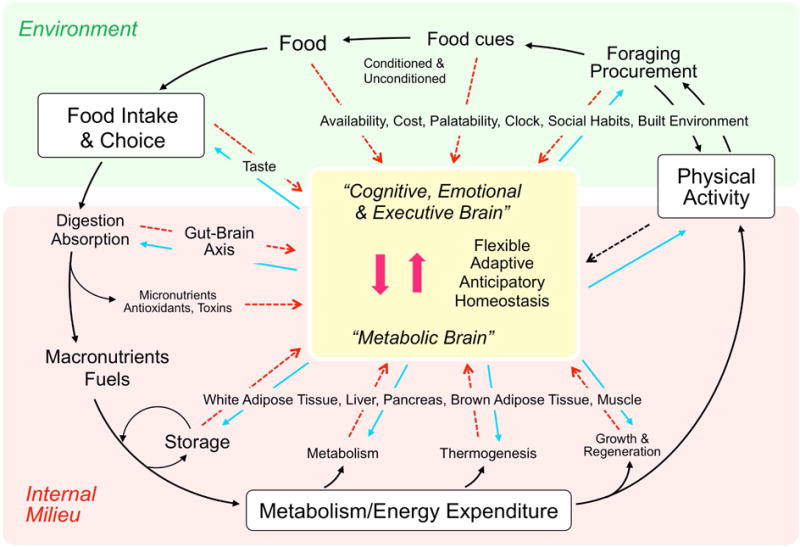
The brain uses nutritional information from both outside and inside the body before, during, and after ingestive behavior. During the initiation phase, attention may be shifted to ingestive behavior because of hunger or an opportunity to consume a highly rewarding food item in the absence of metabolic hunger. During the procurement phase, unconditioned and conditioned stimuli from food and food cues interact through the external senses with the cognitive, emotional, and executive brain. During the consumatory phase, interoceptive signals from taste and the gastrointestinal tract reaching the ‘metabolic’ brain guide the relatively stereotypical processes of digestion, transport, and absorption. Anticipatory reflexes shaped by prior experience thereby help keeping regulated parameters within optimal limits. Finally, during the postingestive or metabolic phase, signals from the gut, and organs that can store and/or metabolize energy such as adipose tissue and liver inform the brain about the metabolic consequences of the meal, providing reward from satisfaction and generating episodic memorial representations of the entire meal for future reference. By integrating external and internal information, the brain can regulate long-term body weight flexibly and adaptively, to accommodate special circumstances (allostasis).
In the following review we examine the mechanisms through which the nervous system detects both external and internal nutritional signals, integrates this information to control eating behavior and energy expenditure, and thereby ultimately regulates body weight. As several aspects of body weight regulation and obesity are addressed in other papers of this Special Issue and in earlier reviews, we will mainly focus on recent developments in the neurobiology of obesity. Given space limitations, we will often cite recent reviews summarizing specific aspects of this topic, rather than original papers.
Monitoring Nutrients
In order to make decisions about eating, an organism must have accurate information about available nutrients inside and outside the body. Internal sensors monitor the nutritional requirements of every cell, tissue, and organ, and signal impending deficits to the brain. For longer-term nutritional planning, an organism needs to know where to find specific kinds of nutrients and be able to balance the need for these nutrients with the time and effort necessary to procure them.
Sensing nutrients in the environment
All five classical senses are used to monitor nutrients in the environment. While humans, non-human primates, and birds rely quite heavily on the visual system, other species rely almost exclusively on smell and taste. We all know the power of pictures with appealing food items, even when we are not really hungry. The food industry exploits this power in advertisements6, and scientists have built an entire paradigm exploring brain function with visual food stimuli. Importantly, a visual stimulus does not have to show a food item to be salient; it can simply be a cue learned from prior experience with food6. Just like visual and other sensory stimuli can become conditioned cues for addictive behavior, they can signal availability of desired foods and drinks6.
Large areas of the brain are dedicated for processing sensory information through the visual, olfactory and auditory, as well as the oral taste systems. Final integration of sensory input takes place in polymodal association areas such as the orbitofrontal, prefrontal, and insular cortex7. The insular cortex is crucial as it also receives information related to oral taste and the internal milieu through the vagus nerve, while a specialized area of the human anterior insula has been implicated in the generation of self-awareness and consciousness8. Together with relevant spatial and temporal information, such multimodal sensory representations of foods are then laid down as “food memories” in a distributed network consisting of the hippocampal formation, prefrontal cortex, dorsal striatum, and amygdala9, 10. Memorial representations of prior experience with food are then used to guide future ingestive behavior. One manifestation of the involvement of learning and memory is anticipatory responding11
Sensing nutrient absorption and nutritional status
Each and every cell in the body has an evolutionarily preserved fuel sensing mechanism composed of nutrient sensors such as AMP kinase and mTOR12, 13. A fundamental question therefore is how the nutritional status of individual cells is communicated to other cells, tissues and organs, and most importantly, to the brain.
Once food passes into the gastrointestinal tract, it is processed and eventually absorbed into the blood or lymph within the alimentary canal. One remarkable discovery over the past decades is the fact that ‘taste’ doesn’t just occur within the mouth, but that the gut has a variety of taste receptors, as well as unique mechanosensors and chemoreceptors, which it uses to sense the volume and nutrient content of consumed food14. This information reaches the brain via several pathways, but two in particular are notable. First, the entire GI tract is densely innervated by vagal sensory nerves15, which are positioned to directly communicate nutritional information from the gut to brainstem. This vagal sensory information has been particularly linked to the process of satiety and meal termination, as disruption of vagal signaling leads to larger meal sizes and an impaired ability to modulate food intake in response to nutritional preloads16. A second communication mode is via the production of endocrine hormones in response to presence of food within the GI system. Unique hormones are produced in multiple sites along the GI tract, with these hormones not only acting locally to influence nutrient absorption and metabolism, but also acting directly in the brain to alter feeding behavior (reviewed in17). The effect of GI-derived hormones on feeding behavior and metabolism is a large and ever growing field, and as such is a specific emphasis of a separate chapter within this Special Issue.
Absorbed glucose, proteins and small lipids are first delivered to the liver via the hepatic portal vein before entering the general circulation, while larger lipids (chylomicrons) are absorbed via the intestinal lymph system, bypass the liver, and are delivered directly to the blood stream. Circulating nutrients are then utilized or stored by all tissues within the body, with the brain serving in some fashion to help coordinate this nutritional flux while also monitoring the nutritional status of the organism and potentially individual tissues. Over the past 20 years, there has been an explosion in our understanding of how nutritional information is transmitted to the brain. The primary driver of this expansion was the discovery of the hormone leptin, which is produced by adipose tissue in proportion to the adipose mass, and which acts in the brain to suppress food intake, stimulate energy expenditure, and also influence growth and reproduction (reviewed in18). In this manner, leptin effectively informs the brain as to the ‘status’ of energy stores throughout the body. It is now known that adipose tissue secretes a multitude of paracrine and endocrine hormones, often termed ‘adipokines’, which influences metabolism, immune function and other endpoints, and many of these signals are also detected by the brain19. Thus the view of adipose tissue has shifted from an ‘inert’ energy storage depot to a metabolically active and dynamic tissue which communicates directly with the brain. This view of adipose tissue as an endocrine organ has been extended to other tissues as well. For instance, muscle has been shown to secrete ‘myokines’ in response to exercise or metabolic stress, and some of these hormones appear to act on the brain (reviewed in20). Likewise, the liver has also been recently demonstrated to communicate to the brain via endocrine signals, with the metabolic hormone Fibroblast growth factor-21 (FGF21) being a notable example (reviewed in21). FGF21 acts on peripheral tissues and the brain to promote changes in energy expenditure, food intake and selection, glucose and lipid homeostasis and growth, and recent work suggests that FGF21 may be particularly important for coordinating metabolic responses to macronutrient imbalance and dietary protein restriction22.
Besides the role of primary (vagal and dorsal root) afferents in the communication of nutritional information from the periphery, many circulating nutrients, hormones, cytokines, and other factors can reach the brain directly at places with a weak or absent blood brain barrier or via transport mechanisms. Representatives of all of the macronutrients (glucose, amino acids, and fatty acids), gut hormones such as ghrelin and GLP-1, as well as many other hormones such as leptin, insulin, and FGF21, have relatively easy access to the brain and have been implicated in the control of food intake and the regulation of energy balance (reviewed in23).
In summary, the brain receives a great deal of external and internal nutritional information before, during, and after the ingestion of food. Integration of this information allows the brain to make metabolic adjustments, prioritize appropriate behavioral action, and optimize the nutritional value of ingested food, as discussed below.
Neural Control of Appetite and Food Intake
How does the brain use the many internal and external signals of nutrient availability discussed above to make the decision to eat and what to eat? Now classical studies conducted in the mid-1900s established the hypothalamus as the key brain area controlling food intake and regulating energy balance 3, 4, 24. Specifically, electrical stimulation of the lateral hypothalamus4 or lesions of the ventromedial hypothalamus3 resulted in voracious appetite or weight gain, leading to the labels “feeding center” and “satiety center”, respectively. While the massive research effort launched since then has undoubtedly both refined and expanded this original portrayal, at its core it remains the same. Thus, the hypothalamus is the only integrative brain area whose manipulation elicits profound eating and body weight phenotypes. For example, selective ablation or stimulation of only one cluster of chemically identified AGRP/NPY/GABA-expressing neurons in the basomedial hypothalamus (an estimated 20,000 neurons in the mouse) results in either starvation and death or dramatic overeating and obesity, respectively25, 26. The hypothalamus therefore remains a hotbed for research identifying the nuts and bolts of the molecular machinery leading to these profound changes in eating and body weight.
Perhaps the biggest addition to the original discovery is the realization that the hypothalamus does not act in isolation, but is intimately connected to representations of both the outside and inside world. It can thus be seen as the ultimate integrator of nutritional information from both the environment and the internal milieu27. Given the many discussions summarizing current knowledge of hypothalamic control of food intake and energy homeostasis (for example28, 29), the main purpose of this review is to highlight recent studies addressing a new conceptual framework that integrates both metabolic (generated by real or perceived nutrient needs) and hedonic (generated by other than nutrient needs) drives to eat.
Depletion of available nutrients, as signaled to the brain through changes in the numerous metabolites and hormones discussed above, is the main driver of ingestive behavior. The basomedial hypothalamic AGRP/NPY neurons are central for the drive to eat25. Recent evidence from selective optogenetic manipulation of these neurons suggests that their stimulation for as briefly as 1 minute triggers voracious food intake even if access to food is delayed by up to 30 minutes after cessation of stimulation30. Furthermore, mice were motivated to press a lever to obtain food reward, they learned to prefer a food that was eaten with optogenetic AGRP/NPY neuron stimulation over another food that was not paired with stimulation, and they learned to lever press for optogenetic AGRP/NPY neuron self-stimulation. In other words, these “hunger neurons drive feeding through a sustained positive reinforcement signal”30. Even though being hungry without access to food is associated with a negative emotional state31, the anticipation of, and the actual eating that follows are rewarding, suggesting that both negative and positive reinforcement mechanisms are operative during an ingestive bout.
Importantly, the downstream neural network driving feeding seems to consist of several redundant pathways, as selective stimulation of individual projections from the AGRP/NPY neurons to various brain areas is sufficient for the elicited behavior30. These observations are consistent with the idea that AGRP/NPY neurons translate hunger signals into sustained and complete appetitive and consumatory ingestive behavior by programming a downstream neural network that increases the incentive or reward value of particular foods. Therefore, AGRP/NPY neurons directly integrate classical homeostatic with reward and cognitive mechanisms (Fig. 2).
Fig. 2.
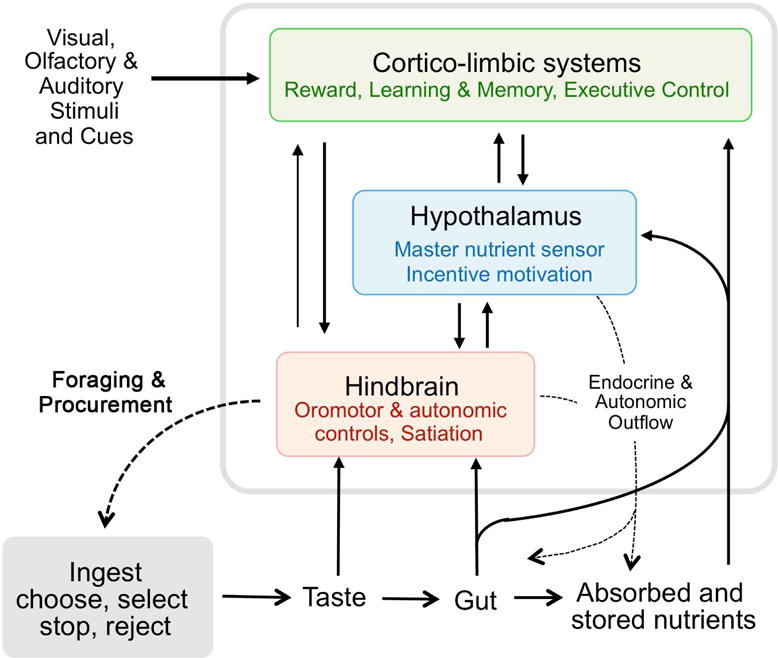
Schematic diagram showing the three heavily interconnected major brain areas constituting the core processor for the control of ingestive behavior and its relation to the gastrointestinal tract and other peripheral organs involved in energy storage and utilization. The hindbrain is mainly concerned with meal size control, as it possesses all the elements to detect sensory information mediated by vagal afferents and circulating factors, and generate motor output associated with the ingestion, digestion, and absorption of food. The cortico-limbic system, consisting of large cortical areas, basal ganglia, hippocampus, and amygdala, is intimately connected to the hypothalamus and brainstem and provides the emotional, cognitive, and executive support for ingestive behavior. The hypothalamus and its connections with the other areas is central for the drive to eat and can potently modulate peripheral organs by autonomic and endocrine outflow.
The hypothalamus, together with the corticolimbic system and the hindbrain can be seen as core processor in the control of appetite (Fig. 3). The hindbrain or brainstem is mainly concerned with meal size control, as it possesses all the elements to detect sensory information mediated by vagal afferents and circulating factors, and generate motor output associated with the ingestion, digestion, and absorption of food32. However, by itself, the brainstem alone cannot adjust food intake to external demands such as anticipation of a food shortage33.
Fig. 3.
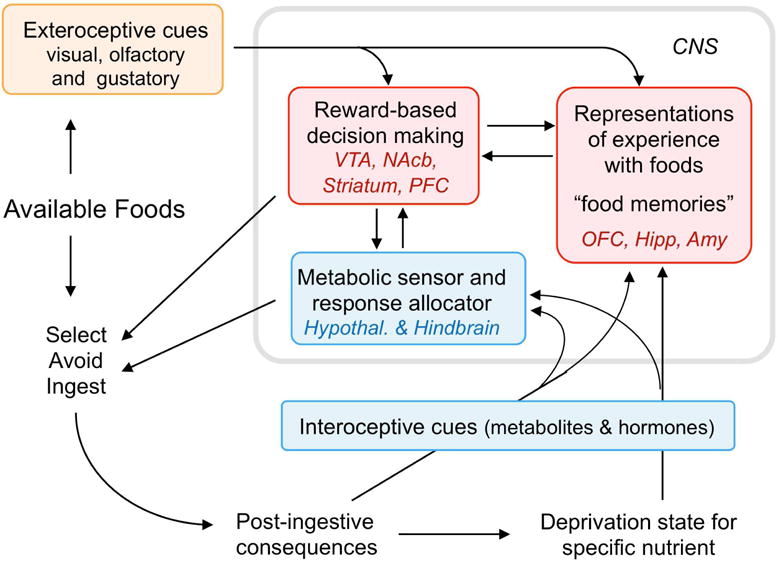
Schematic diagram showing the neural basis for the selection of specific foods. Before ingestion, available foods and their cues act through external sensory channels to retrieve representations with prior experience (‘food memories’) that have access to reward-based decision making circuits. If there is a specific need state for that food (nutrient), it is sampled. If the taste experience confirms the stored information retrieved from memory, ingestion follows, eliciting interoceptive signals such as metabolites and hormones which feed back to the hindbrain and hypothalamus to stop eating when satiated and update the memory. If tasting reveals any problem with the food (spoiled food or otherwise aversive), the food item is not ingested and the memory updated. Abbreviations: Amy, amygdala; Hipp, hippocampal complex; Nac, nucleus accumbens; OFC, orbitofrontal cortex; PFC, prefrontal cortex; VTA, ventral tegmental area.
The cortico-limbic system, consisting of large cortical areas, basal ganglia, hippocampus, and amygdala, is intimately connected to the hypothalamus and brainstem and provides the emotional, cognitive, and executive support for ingestive behavior34. The neurology of reward, economics, and decision making35 are increasingly recognized as important determinants of food intake36. Studies in rodents and humans have shown that when there is a choice between two foods, the cost/reward value of each food is compared before a choice is made37, 38 (Fig. 4). An intuitive and simple way to understand food reward is to parse it into two components, ‘liking’ and ‘wanting’39. Intrinsic liking is represented diffusely in both forebrain and hindbrain circuits with an important role for the mu-opioid receptor40, while conscious liking in humans is encoded in the prefrontal cortex41. Intrinsic wanting is encoded by the mesolimbic dopamine system mainly projecting to the nucleus accumbens in the ventral striatum42. While ‘liking’ is relatively independent of the prevailing nutritional state, ‘wanting’ is greatly amplified by hunger43. Thus, a particular food can be very much liked, but not necessarily wanted after just eating to full satiety. This latter example points to the process of devaluation. Eating a food to satiation devalues the salience of cues and sensory attributes of this food, but not necessarily to other types of food44, 45. The fact that most of us still readily eat a sweet dessert after being full on a savory meat dish is known as sensory-specific satiety46.
Fig. 4.
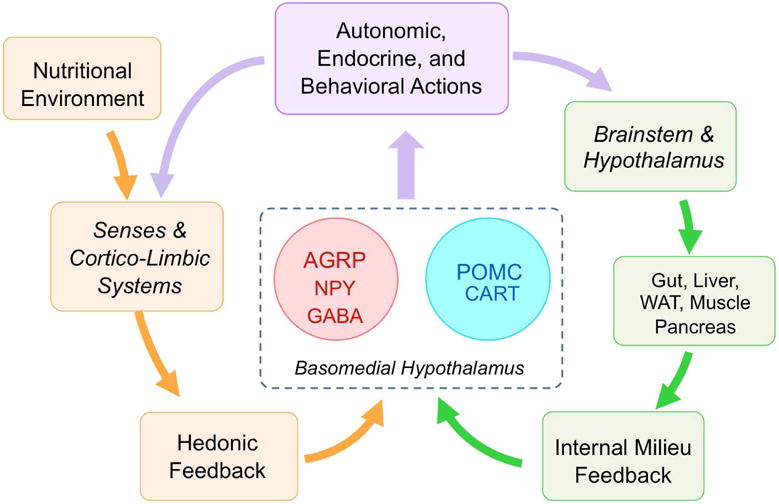
Basomedial hypothalamic neurons as master integrators of internal and external nutritional information to achieve allostasis. AGRP/NPY hunger neurons translate hunger signals from both the internal milieu and hedonic feedback into sustained and complete appetitive and consumatory ingestive behavior by programming a downstream neural network that increases the incentive or reward value of particular foods. While AGRP/NPY neurons could be thought of as the gas pedal, POMC/CART neurons represent the brake pedal, acting through parallel downstream pathways.
Through its access to multimodal sensory information and the skeletal motor system, the cortico-limbic system is the main interface with the environment. However, just like the other two nodes, components of the cortico-limbic system can also sense the availability of internal nutrients either directly through relevant nutrient and hormone receptors or indirectly through neural inputs from the hypothalamus and brainstem47. The hypothalamus, located strategically between the hindbrain and cortico-limbic system, is the key hub for detecting overall nutritional state, generating the drive to eat, prioritizing essential behaviors48, and regulating long-term energy balance by matching energy intake with expenditure (see below).
In summary, many brain areas are involved in a hierarchical fashion in the neural control of food intake. At the top of the hierarchy are the “hunger neurons” (AGRP/NPY) in the basomedial hypothalamus that directly translate low nutrient availability, for example after a fasting period, into motivated behavioral action. Other important nodes in the hypothalamus, brainstem, and cortico-limbic system are involved in the orchestration and execution of ingestive behavior vis-à-vis other behaviors. Most importantly, much of neural processing within this core processor occurs outside awareness, rendering it relatively inaccessible to conscious manipulation (for detailed discussion see27)
Neural Controls of Energy Expenditure
Besides energy intake, energy expenditure is the other component of energy balance regulation that can, at least partially, be controlled through the brain. Whole body energy expenditure can be divided into its individual components, such as basal metabolism, the thermic effect of food, and thermogenesis to maintain body temperature, and physical activity, each with its own neural controls and effector pathways. Whether surplus energy, for example after a larger than normal meal, is automatically expended through increased basal metabolism and thermogenesis, just like surplus water is excreted through the kidneys, has been a matter of intense debate.
Already some 35 years ago, intense research focused on thermogenesis in brown adipose tissue (BAT)49. While it was shown that the interscapular brown fat pad in rodents expresses very high levels of the uncoupling protein UCP1 and generates much of the necessary heat for maintenance of body temperature50, very little BAT was found in adult humans and the idea of using this tissue for body weight control remained dormant. However more recently, by using routine 18F-glucose imaging for the detection of neoplasms, considerable depots of BAT were discovered above the clavicular bones and near the cervical and thoracic vertebrae in at least some adult humans51. This discovery has stimulated renewed interest in harnessing this tissue for the fight against obesity52. How BAT, which is present in considerable amounts in newborns, can be preserved through adolescence and adulthood, and how it can be activated to burn energy is of great interest. Clearly, the brain exerts key control over BAT thermogenesis through the sympathetic nervous system (SNS)53, and much of the central neural circuitry has been deciphered in rodents (reviewed in54).
Discovery of brown fat-like adipocytes (also referred to as beige or brite adipocytes) within classical white fat storage depots has spurred even more excitement about the role of fat tissue in energy balance regulation and its exploitation for obesity prevention and treatment55. Researchers are feverishly studying the optimal conditions and signals for “beiging” of white fat depots in the hope of using them to “effortlessly” increase energy expenditure56. Until such a new “pill” is available, we will have to rely on the old-fashioned way of getting rid of excess energy intake, namely physical activity.
Before industrialization and automatization, physical activity was the major component of energy expenditure and could result in the utilization of several thousand calories per day. Modern lifestyles now involve sitting in a chair for most of the day, resulting in drastically decreased energy expenditure for many of us. It is thus imperative to develop strategies for increasing physical activity at the workplace and at home. Again, the brain is key to such strategies, as it controls not only skeletal muscle but also the motivation to engage in physical activity. Much clinical research is focusing on how children and adolescents can be motivated to engage in physical activity. Exergaming, which is taking advantage of the astonishing affinity of children for videos and electronic devices, has shown some promising results57, but much remains to be done58. Preclinical research is just beginning to study mechanisms and map the neural pathways underlying the drive for physical activity. For example, hypothalamic orexin-expressing neurons have been implicated in voluntary physical activity and the development of obesity59.
In summary, research trying to capitalize on the thermogenic capacity of brown and beige adipocytes to get rid of excess calories has exponentially increased in the last decade. However, success with this strategy may be complicated by compensatory changes in energy intake. Restoring adequate physical activity levels through exercise has been a longstanding strategy, although with only moderate success. There is no doubt that regular physical activity has many health benefits, but the realization that physical activity alone is not an efficient weight loss strategy60 has been a major roadblock. In addition, we do not understand how the brain regulates physical activity level and couples it with energy intake. Thus, a physical activity “mimetic” seems unlikely in the near future.
Neural Regulation of Body Weight
Set point theory
In a strict sense, energy intake and expenditure are controlled but not regulated processes, as the organism does not seek to maintain constant levels of either endpoint. In contrast, body weight (or perhaps adiposity) is regulated to maintain constant levels via the control of intake and expenditure, and this process is often termed energy homeostasis. Although the major attention is on eating behavior, obesity is primarily the result of defective regulation of energy balance and body weight, rather than defective eating controls. There is no doubt that excess food intake can lead to obesity, but only if the regulatory system fails to compensate for increased energy intake by either commensurately increasing energy expenditure or by reducing food intake at subsequent meals. A healthy regulatory system is able to defend body weight within narrow limits, often referred to as a set point. The principle of set point regulation has been demonstrated at the behavioral level in both rodents and humans. When body weight is artificially perturbed by periods of either under- or overfeeding, adaptive changes in food intake and energy expenditure promptly return body weight to pre-perturbation levels61.
Despite the evidence for the homeostatic regulation of body weight, a number of observations refute the notion of a specific set point. First, there is no fixed set point around which mammalian species regulate their body weight. This is best illustrated by the seasonably variable yet homeostatically defended body weight set point of hibernators, who spontaneously gain or lose weight in response to seasonal changes in photoperiod62. Interestingly, if these animals are artificially under- or overfed to induce weight loss or weight gain, they return to exactly the body weight corresponding to their current seasonal cycle upon removing the artificial feeding regimen. Hibernators are thus perfectly capable of cycling between leanness and obesity without any apparent metabolic complications while also defending this moving body weight target. These results are not consistent with the basic set point theory, and rather suggest a flexible or sliding set point.
Second, in non-hibernators, transition to obesity can change the defended body weight set point. This is certainly the case in the genetically obese, such as leptin-deficient rodents and humans, but has also been demonstrated in common obesity63, 64. For example, obese rodents that had been exposed to high-fat diet for an extended period of time will not fully return to lean levels upon cessation of high-fat diet feeding65, 66. In parallel, hypothalamic neural projections are permanently disrupted in genetic lines of diet-induced obesity-prone rats67. Another line of evidence suggests that overingestion of high-fat diets can rapidly produce hypothalamic inflammation and damage, causing the set point to change (as summarized in68. Thus, obese rodents and humans start defending their new, higher body weight rather than their pre-obesity lean body weight, again suggesting a sliding, rather than fixed set point (Fig. 5).
Fig. 5.
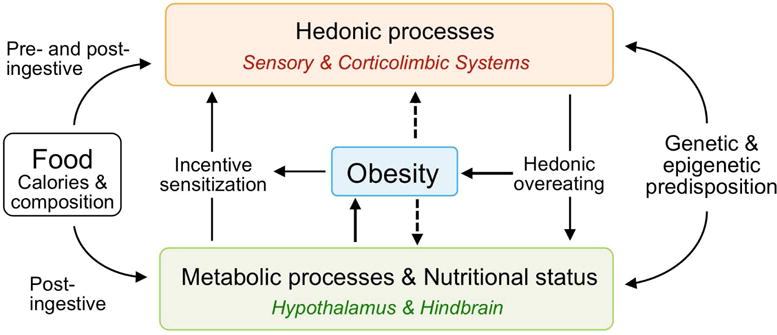
Schematic diagram showing interactions between metabolic and hedonic consequences of food and their relationship to obesity. Obesity can be caused by overstimulation of hedonic processes leading to hedonic overeating or by impaired metabolic processes leading to lack of compensation following overfeeding. Both processes are subject to an interaction with genetic and epigenetic predisposition. The obese state can also lead to a secondary impairment of both hedonic and metabolic processes, leading to a vicious circle and further progression of obesity, as well as causing metabolic disturbances in offspring.
Third, although the notion of set point receives considerable support from behavioral observations, we do not understand its neural and molecular basis. It likely involves the powerful basomedial hypothalamic circuitry consisting of AGRP/NPY and POMC/CART neurons, but obviously not in the rigid way purported by the original adipostat hypothesis69. An additional mechanism which allows the set point to slide according to seasonal or other variations in nutritional supply is necessary to explain the behavior of hibernators as well as humans. Given the novel mechanisms by which sensory, cognitive and emotional information can rapidly modulate activity of AGRP neurons (as discussed above), elucidation of these complex pathways and their molecular identity will be important for understanding this flexible homeostatic/allostatic regulator.
The brain and current hypotheses on the causes of obesity
New hypotheses about the cause of obesity spring up almost every day, and the purpose of this brief section is to highlight those that are clearly anchored in the CNS and have received the most attention.
Genetics and epigenetics
Genetic or epigenetic variation in genes that control energy homeostasis is a logical contributor to obesity (for a more detailed discussion see chapter xy in this issue). There are clear examples in humans where either leptin-deficiency or single-point mutations of the melanocortin-4 receptor cause severe obesity, and preclinical research has identified signaling through leptin and MC4 receptors in the hypothalamus as most critical70. However, common obesity is not generally caused by mutations in any single gene, and obesity is more likely induced by the accrual of multiple genes that more subtly predispose an individual to obesity. A recent genome wide association study (GWAS) in over 300,000 subjects found that a majority of genes within BMI-associated loci are preferentially and significantly enriched in the CNS5. Most of these genes are involved in functional and structural aspects of neurotransmission that could affect any component of the neural control of energy intake, energy expenditure, and regulation of body weight and any of the mechanisms discussed in this review and below. However, because only about 20% of BMI variation can be accounted for by common genetic variation5, other factors and mechanisms must contribute to obesity. Epigenetic modifications of gene expression during early life and even later in life is also increasingly recognized as an important contributor to obesity, and preclinical studies have identified several epigenetically modified genes in the hypothalamus associated with obesity71.
Increased availability of palatable energy-dense foods
The relatively recent rise in obesity prevalence has been the main argument against a major role of inheritance and a more prominent role of environment and lifestyle. Among the many potential environmental factors causing obesity, reaching from viruses72 through toxins73 to the use of artificial light74, changes in nutrition and food hedonics have attracted the most attention. Particularly the shift away from relatively low-calorie, high-fiber diets towards energy-dense, palatable diets is considered an important contributor to the increased prevalence of obesity. This effect can be clearly demonstrated in laboratory animals64 and pets75. Although similar well-controlled, randomized long-term studies in humans have not been conducted, studies on populations with voluntary or involuntary long-term calorie-restriction or over-feeding are generally supportive of the overall beneficial metabolic effects of reducing the availability of energy dense, palatable foods76, 77.
Increased exposure to food cues
In most industrialized countries and urbanized populations of the modern world, there is an onslaught of advertisements for enticing foods (often referred to as “food porn”) in the public space as well as at home through the media, with the sole purpose to sell more food. Because such signals are processed by the brain largely outside awareness, they are quite resistant to conscious inhibitory control27. As a result, cognitive reasoning, impulsivity, and executive self-control are challenged on a daily basis6. In addition, a considerable portion of total food intake is in restaurants and other food-selling businesses. Much of the low-cost food available at such places is salty, greasy and/or sweet and thus relatively palatable, particularly if paired with large amounts of sweetened beverages. Exposure to these food cues constitutes a constant challenge for homeostasis28, 39. One key neural mechanism for disordered eating is the transition from normal ‘liking’ and ‘wanting’ to addictive behavior. The debate about sugary foods goes on, and while there is no strong direct evidence for sugar addiction or sugar-induced obesity, it has been demonstrated that rats readily chose sweet over cocaine or nicotine reward and have a greater attraction for sweet versus drug cues78, 79.
Stress-induced overeating
The modern environment, hectic lifestyle, and socio-economic inequalities have considerably increased the psychological stress burden starting at childhood80. Stress-induced overeating can be seen as another disorder of reward mechanisms. Reward from comfort food is considered an attempt at self-medication to relieve the negative emotion and depressive state associated with chronic psychological stress81, 82. Thus stress management through a variety of techniques is an important component in obesity care83.
Mindless and habit-driven food intake
For many, eating has become completely detached from procuring and preparing food, and is often done rapidly and in the presence of a TV or other significant distraction. In short, eating has become less mindful but more automatic and habitual, and this change poses a challenge to homeostatic regulation. Distracted eating caused by watching television, text messaging, or playing computer games impairs the memory of the meal and increases later snack intake, suggesting that the attentive and mindful experience of a meal is necessary for adequate satiation mechanisms and proper inhibitory controls83, 84. Because automatic, habitual eating takes place outside awareness, conscious cognitive influences such as response inhibition by prefrontal cortical activity is largely ineffective. In addition, mechanisms by which satiety signals devalue incentive motivation appear to be desensitized in habitual food intake and may lead to overeating and obesity85.
What does the neural control of food intake and energy balance tell us about strategies for the treatment or prevention of obesity and metabolic disease?
If body weight were indeed regulated by a sliding set point mechanism and we understood its neural and molecular basis, all we would have to do is change the setting, just like we change the setting of our living room thermostat. In the meantime, obesity treatment remains symptomatic, using almost “anything that works”. However, given from what we know, some approaches seem to work better than others. Because the regulatory system varies both energy intake and expenditure to maintain a given steady state, energy intake will change to compensate when expenditure is challenged (exercise), while energy expenditure compensates when intake is challenged (calorie restriction). Successful treatment will require uncoupling of these compensatory mechanisms. As long as the exact molecular mechanisms responsible for coupling are not understood, two-prong approaches attacking both energy intake and expenditure will likely yield superior results. The validity of this approach is supported by the observation that physically active rats lose more weight during three weeks of 50% calorie-restriction compared with physical inactive rats86, and analysis of weight loss in a large cohort from the 2009–2012 National Health and Nutrition Examination Surveys showed that long-term weight losers consumed fewer calories and reported more vigorous leisure activity than did overweight or obese individuals87. However, only randomized controlled intervention studies will provide the necessary rigor to more conclusively illuminate this important issue.
In addition, because the neural pathways controlling intake and expenditure are complex and involve redundant mechanisms controlled by a multitude of genes, successful treatment will likely require combination approaches (Supplementary Fig. S1). Combinations, for example a drug and bariatric surgery, which manipulate a single pathway/mechanism may also have relatively small effects. Instead, successful combinations are likely to target multiple mechanisms in order to produce effects that are fully additive or even synergistic. Most of the pertinent literature has been discussed in a recent review88.
The superior efficacy of combination drug treatments has already been demonstrated in preclinical studies89, with many of them currently in phase 1 or phase 2 clinical trials88. A better understanding of the complexity and redundancy of neural and non-neural pathways underlying body weight regulation should lead to more rationally designed combinations.
Conclusions and Outlook
The worldwide increase in the prevalence of obesity is best explained by gene-environment interactions whereby rapid changes in food supply and built environment trigger overeating and/or sedentary behavior in individuals with genetic predisposition. Many of the genes underlying this predisposition are expressed in the brain, the final arbitrator of ingestive and locomotor behavior and regulator of body weight. Although the hypothalamus has long been known to be a key player in the control of food intake and regulation of body weight, technological advances in manipulating and listening to the brain of both rodents and humans have significantly expanded this view by demonstrating hypothalamic connections and functional interactions with other major brain areas such as the corticolimbic system and the hindbrain. Thus, the traditional dichotomy between homeostatic and non-homeostatic/hedonic systems should be replaced by a much larger, highly interactive system that unifies homeostasis with reward, cognition and emotion. This new conceptual framework has several important implications for the treatment of obesity. First, obesity-disposing genes may exert their influence not only on classical homeostatic pathways, but also on the large brain areas involved in the hedonic aspects of food intake. Second, because hedonic processes are intricately interacting with homeostatic hypothalamic processes, which operate completely outside awareness, hedonic processes are also less under conscious control. It is therefore unlikely that obese individuals can simply will themselves to weight loss. Third, the complexity and redundancy of neural pathways controlling ingestive behavior and regulation of body weight suggests combination therapies that attack more than one mechanism to be the most efficient in combating obesity. Finally, deciphering the molecular underpinnings of flexible set point regulation should be a high priority goal for basic research.
Supplementary Material
Acknowledgments
The original research supporting some of the conclusions reached in this review was supported by National Institutes of Health grants DK 047348 (HRB), DK105032 and DK081563 (CDM), and DK092587 (HM).
Biographies

Hans-Rudolf Berthoud

Christopher D. Morrison

Heike Münzberg
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hargrove JL. Adipose energy stores, physical work, and the metabolic syndrome: lessons from hummingbirds. Nutr J. 2005;4:36. doi: 10.1186/1475-2891-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hess W. Das Zwischenhirn: Syndrome, Localisationen, Funktionen. Basel: Schwabe; 1949. [Google Scholar]
- 3.Brobeck JR. Mechanism of the development of obesity in animals with hypothalamic lesions. Physiol Rev. 1946;26:541–59. doi: 10.1152/physrev.1946.26.4.541. [DOI] [PubMed] [Google Scholar]
- 4.Bruegger M. Fresstrieb als hypothalamisches Syndrom. Helvetica Physiologica Acta. 1943;1:183–198. [Google Scholar]
- 5.Locke AE, Kahali B, Berndt SI, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spence C, Okajima K, Cheok AD, et al. Eating with our eyes: From visual hunger to digital satiation. Brain and Cognition. 2015 doi: 10.1016/j.bandc.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 7.Verhagen JV. The neurocognitive bases of human multimodal food perception: Consciousness. Brain Res Brain Res Rev. 2006 doi: 10.1016/j.brainresrev.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Craig AD. How do you feel–now? The anterior insula and human awareness. Nat Rev Neurosci. 2009;10:59–70. doi: 10.1038/nrn2555. [DOI] [PubMed] [Google Scholar]
- 9.Petrovich GD. Forebrain networks and the control of feeding by environmental learned cues. Physiology and Behavior. 2013;121:10–8. doi: 10.1016/j.physbeh.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen J, Papies EK, Barsalou LW. A core eating network and its modulations underlie diverse eating phenomena. Brain and Cognition. 2016 doi: 10.1016/j.bandc.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 11.Ramsay DS, Woods SC. Physiological Regulation: How It Really Works. Cell Metab. 2016;24:361–4. doi: 10.1016/j.cmet.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hardie DG. AMPK–sensing energy while talking to other signaling pathways. Cell Metab. 2014;20:939–52. doi: 10.1016/j.cmet.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Psichas A, Reimann F, Gribble FM. Gut chemosensing mechanisms. Journal of Clinical Investigation. 2015;125:908–17. doi: 10.1172/JCI76309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berthoud HR, Neuhuber WL. Distribution and morphology of vagal afferents supplying the digestive system. Boca Raton: CRC Press; 1994. [Google Scholar]
- 16.Schwartz GJ. The role of gastrointestinal vagal afferents in the control of food intake: current prospects. Nutrition. 2000;16:866–73. doi: 10.1016/s0899-9007(00)00464-0. [DOI] [PubMed] [Google Scholar]
- 17.Hussain SS, Bloom SR. The regulation of food intake by the gut-brain axis: implications for obesity. Int J Obes (Lond) 2013;37:625–33. doi: 10.1038/ijo.2012.93. [DOI] [PubMed] [Google Scholar]
- 18.Munzberg H, Morrison CD. Structure, production and signaling of leptin. Metabolism: Clinical and Experimental. 2015;64:13–23. doi: 10.1016/j.metabol.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stern JH, Rutkowski JM, Scherer PE. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis through Adipose Tissue Crosstalk. Cell Metab. 2016;23:770–84. doi: 10.1016/j.cmet.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pedersen BK, Febbraio MA. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol. 2012;8:457–65. doi: 10.1038/nrendo.2012.49. [DOI] [PubMed] [Google Scholar]
- 21.Owen BM, Mangelsdorf DJ, Kliewer SA. Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol Metab. 2015;26:22–9. doi: 10.1016/j.tem.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laeger T, Henagan TM, Albarado DC, et al. FGF21 is an endocrine signal of protein restriction. Journal of Clinical Investigation. 2014;124:3913–22. doi: 10.1172/JCI74915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berthoud HR, Morrison C. The brain, appetite, and obesity. Annu Rev Psychol. 2008;59:55–92. doi: 10.1146/annurev.psych.59.103006.093551. [DOI] [PubMed] [Google Scholar]
- 24.Hoebel BG, Teitelbaum P. Hypothalamic control of feeding and self-stimulation. Science. 1962;135:375–7. doi: 10.1126/science.135.3501.375. [DOI] [PubMed] [Google Scholar]
- 25.Luquet S, Perez FA, Hnasko TS, et al. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005;310:683–5. doi: 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- 26.Krashes MJ, Koda S, Ye C, et al. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest. 2011 doi: 10.1172/JCI46229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Munzberg H, Qualls-Creekmore E, Yu S, et al. Hedonics Act in Unison with the Homeostatic System to Unconsciously Control Body Weight. Front Nutr. 2016;3:6. doi: 10.3389/fnut.2016.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berthoud HR. Metabolic and hedonic drives in the neural control of appetite: who is the boss? Current Opinion in Neurobiology. 2011;21:888–96. doi: 10.1016/j.conb.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berthoud HR. Mind versus metabolism in the control of food intake and energy balance. Physiol Behav. 2004;81:781–93. doi: 10.1016/j.physbeh.2004.04.034. [DOI] [PubMed] [Google Scholar]
- 30.Chen Y, Lin YC, Zimmerman CA, et al. Hunger neurons drive feeding through a sustained, positive reinforcement signal. Elife. 2016;5 doi: 10.7554/eLife.18640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Betley JN, Xu S, Cao ZF, et al. Neurons for hunger and thirst transmit a negative-valence teaching signal. Nature. 2015;521:180–5. doi: 10.1038/nature14416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berthoud HR. The caudal brainstem and the control of food intake and energy balance. In: Stricker EM, Woods SC, editors. Handbook of Behavioral Neurobiology. Vol. 14. New York: Plenum; 2004. pp. 195–240. [Google Scholar]
- 33.Kaplan JM, Seeley RJ, Grill HJ. Daily caloric intake in intact and chronic decerebrate rats. Behav Neurosci. 1993;107:876–81. [PubMed] [Google Scholar]
- 34.Kelley AE, Baldo BA, Pratt WE, et al. Corticostriatal-hypothalamic circuitry and food motivation: integration of energy, action and reward. Physiology and Behavior. 2005;86:773–95. doi: 10.1016/j.physbeh.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 35.Hosokawa T, Kennerley SW, Sloan J, et al. Single-neuron mechanisms underlying cost-benefit analysis in frontal cortex. Journal of Neuroscience. 2013;33:17385–97. doi: 10.1523/JNEUROSCI.2221-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murray EA, Rudebeck PH. The drive to strive: goal generation based on current needs. Front Neurosci. 2013;7:112. doi: 10.3389/fnins.2013.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morris RW, Dezfouli A, Griffiths KR, et al. Action-value comparisons in the dorsolateral prefrontal cortex control choice between goal-directed actions. Nat Commun. 2014;5:4390. doi: 10.1038/ncomms5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Atalayer D, Robertson KL, Haskell-Luevano C, et al. Food demand and meal size in mice with single or combined disruption of melanocortin type 3 and 4 receptors. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1667–74. doi: 10.1152/ajpregu.00562.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berridge KC, Ho CY, Richard JM, et al. The tempted brain eats: Pleasure and desire circuits in obesity and eating disorders. Brain Res. 2010;1350:43–64. doi: 10.1016/j.brainres.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berridge KC, Robinson TE. Parsing reward. Trends Neurosci. 2003;26:507–13. doi: 10.1016/S0166-2236(03)00233-9. [DOI] [PubMed] [Google Scholar]
- 41.Kringelbach ML. Food for thought: hedonic experience beyond homeostasis in the human brain. Neuroscience. 2004;126:807–19. doi: 10.1016/j.neuroscience.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 42.Kelley AE, Berridge KC. The neuroscience of natural rewards: relevance to addictive drugs. J Neurosci. 2002;22:3306–11. doi: 10.1523/JNEUROSCI.22-09-03306.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robinson MJ, Fischer AM, Ahuja A, et al. Roles of “Wanting” and “Liking” in Motivating Behavior: Gambling, Food, and Drug Addictions. Curr Top Behav Neurosci. 2016;27:105–36. doi: 10.1007/7854_2015_387. [DOI] [PubMed] [Google Scholar]
- 44.Galarce EM, Crombag HS, Holland PC. Reinforcer-specificity of appetitive and consummatory behavior of rats after Pavlovian conditioning with food reinforcers. Physiology and Behavior. 2007;91:95–105. doi: 10.1016/j.physbeh.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.West EA, DesJardin JT, Gale K, et al. Transient inactivation of orbitofrontal cortex blocks reinforcer devaluation in macaques. Journal of Neuroscience. 2011;31:15128–35. doi: 10.1523/JNEUROSCI.3295-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rolls BJ, Rolls ET, Rowe EA, et al. Sensory specific satiety in man. Physiol Behav. 1981;27:137–42. doi: 10.1016/0031-9384(81)90310-3. [DOI] [PubMed] [Google Scholar]
- 47.Williams DL. Neural integration of satiation and food reward: role of GLP-1 and orexin pathways. Physiology and Behavior. 2014;136:194–9. doi: 10.1016/j.physbeh.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paxinos G, Bindra D. Hypothalamic and midbrain neural pathways involved in eating, drinking, irritability, aggression, and copulation in rats. Journal of Comparative and Physiological Psychology. 1973;82:1–14. doi: 10.1037/h0033799. [DOI] [PubMed] [Google Scholar]
- 49.Rothwell NJ, Stock MJ. Luxuskonsumption, diet-induced thermogenesis and brown fat: the case in favour. Clin Sci (Colch) 1983;64:19–23. doi: 10.1042/cs0640019. [DOI] [PubMed] [Google Scholar]
- 50.Feldmann HM, Golozoubova V, Cannon B, et al. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 2009;9:203–9. doi: 10.1016/j.cmet.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 51.Cohade C, Osman M, Pannu HK, et al. Uptake in supraclavicular area fat (“USA-Fat”): description on 18F-FDG PET/CT. Journal of Nuclear Medicine. 2003;44:170–6. [PubMed] [Google Scholar]
- 52.Mukherjee J, Baranwal A, Schade KN. Classification of Therapeutic and Experimental Drugs for Brown Adipose Tissue Activation: Potential Treatment Strategies for Diabetes and Obesity. Curr Diabetes Rev. 2016 doi: 10.2174/1573399812666160517115450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tupone D, Madden CJ, Morrison SF. Autonomic regulation of brown adipose tissue thermogenesis in health and disease: potential clinical applications for altering BAT thermogenesis. Front Neurosci. 2014;8:14. doi: 10.3389/fnins.2014.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Munzberg H, Qualls-Creekmore E, Berthoud HR, et al. Neural Control of Energy Expenditure. Handb Exp Pharmacol. 2016;233:173–94. doi: 10.1007/164_2015_33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tharp KM, Stahl A. Bioengineering Beige Adipose Tissue Therapeutics. Front Endocrinol (Lausanne) 2015;6:164. doi: 10.3389/fendo.2015.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fan W, Evans R. METABOLISM. The quest to burn fat, effortlessly and safely. Science. 2016;353:749–50. doi: 10.1126/science.aah6189. [DOI] [PubMed] [Google Scholar]
- 57.Staiano AE, Marker AM, Beyl RA, et al. A randomized controlled trial of dance exergaming for exercise training in overweight and obese adolescent girls. Pediatr Obes. 2016 doi: 10.1111/ijpo.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hochsmann C, Schupbach M, Schmidt-Trucksass A. Effects of Exergaming on Physical Activity in Overweight Individuals. Sports Medicine. 2016;46:845–60. doi: 10.1007/s40279-015-0455-z. [DOI] [PubMed] [Google Scholar]
- 59.Perez-Leighton CE, Boland K, Billington CJ, et al. High and low activity rats: elevated intrinsic physical activity drives resistance to diet-induced obesity in non-bred rats. Obesity (Silver Spring) 2013;21:353–60. doi: 10.1002/oby.20045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johannsen DL, Redman LM, Ravussin E. The role of physical activity in maintaining a reduced weight. Curr Atheroscler Rep. 2007;9:463–71. doi: 10.1007/s11883-007-0062-z. [DOI] [PubMed] [Google Scholar]
- 61.Harris RB, Kasser TR, Martin RJ. Dynamics of recovery of body composition after overfeeding, food restriction or starvation of mature female rats. Journal of Nutrition. 1986;116:2536–46. doi: 10.1093/jn/116.12.2536. [DOI] [PubMed] [Google Scholar]
- 62.Morgan PJ, Ross AW, Mercer JG, et al. Photoperiodic programming of body weight through the neuroendocrine hypothalamus. Journal of Endocrinology. 2003;177:27–34. doi: 10.1677/joe.0.1770027. [DOI] [PubMed] [Google Scholar]
- 63.Levin BE, Dunn-Meynell AA. Defense of body weight depends on dietary composition and palatability in rats with diet-induced obesity. Am J Physiol Regul Integr Comp Physiol. 2002;282:R46–54. doi: 10.1152/ajpregu.2002.282.1.R46. [DOI] [PubMed] [Google Scholar]
- 64.Sclafani A, Springer D. Dietary obesity in adult rats: similarities to hypothalamic and human obesity syndromes. Physiol Behav. 1976;17:461–71. doi: 10.1016/0031-9384(76)90109-8. [DOI] [PubMed] [Google Scholar]
- 65.Ravussin Y, Leibel RL, Ferrante AW., Jr A missing link in body weight homeostasis: the catabolic signal of the overfed state. Cell Metab. 2014;20:565–72. doi: 10.1016/j.cmet.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ferrannini E, Rosenbaum M, Leibel RL. The threshold shift paradigm of obesity: evidence from surgically induced weight loss. American Journal of Clinical Nutrition. 2014;100:996–1002. doi: 10.3945/ajcn.114.090167. [DOI] [PubMed] [Google Scholar]
- 67.Bouret SG, Gorski JN, Patterson CM, et al. Hypothalamic neural projections are permanently disrupted in diet-induced obese rats. Cell Metab. 2008;7:179–85. doi: 10.1016/j.cmet.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thaler JP, Guyenet SJ, Dorfman MD, et al. Hypothalamic inflammation: marker or mechanism of obesity pathogenesis? Diabetes. 2013;62:2629–34. doi: 10.2337/db12-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cowley MA, Pronchuk N, Fan W, et al. Integration of NPY, AGRP, and melanocortin signals in the hypothalamic paraventricular nucleus: evidence of a cellular basis for the adipostat. Neuron. 1999;24:155–63. doi: 10.1016/s0896-6273(00)80829-6. [DOI] [PubMed] [Google Scholar]
- 70.Farooqi IS, Keogh JM, Yeo GS, et al. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085–95. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- 71.Plagemann A, Harder T, Brunn M, et al. Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: an epigenetic model of obesity and the metabolic syndrome. J Physiol. 2009;587:4963–76. doi: 10.1113/jphysiol.2009.176156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rogers PM, Fusinski KA, Rathod MA, et al. Human adenovirus Ad-36 induces adipogenesis via its E4 orf-1 gene. Int J Obes (Lond) 2008;32:397–406. doi: 10.1038/sj.ijo.0803748. [DOI] [PubMed] [Google Scholar]
- 73.Rutkowska AZ, Diamanti-Kandarakis E. Polycystic ovary syndrome and environmental toxins. Fertility and Sterility. 2016;106:948–58. doi: 10.1016/j.fertnstert.2016.08.031. [DOI] [PubMed] [Google Scholar]
- 74.Versteeg RI, Stenvers DJ, Kalsbeek A, et al. Nutrition in the spotlight: metabolic effects of environmental light. Proceedings of the Nutrition Society. 2016:1–13. doi: 10.1017/S0029665116000707. [DOI] [PubMed] [Google Scholar]
- 75.Zoran DL. Obesity in dogs and cats: a metabolic and endocrine disorder. Veterinary Clinics of North America. Small Animal Practice. 2010;40:221–39. doi: 10.1016/j.cvsm.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 76.Kitada M, Kume S, Takeda-Watanabe A, et al. Calorie restriction in overweight males ameliorates obesity-related metabolic alterations and cellular adaptations through anti-aging effects, possibly including AMPK and SIRT1 activation. Biochimica et Biophysica Acta. 2013;1830:4820–7. doi: 10.1016/j.bbagen.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 77.Weiss EP, Fontana L. Caloric restriction: powerful protection for the aging heart and vasculature. Am J Physiol Heart Circ Physiol. 2011;301:H1205–19. doi: 10.1152/ajpheart.00685.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lenoir M, Serre F, Cantin L, et al. Intense sweetness surpasses cocaine reward. PLoS ONE. 2007;2:e698. doi: 10.1371/journal.pone.0000698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Madsen HB, Ahmed SH. Drug versus sweet reward: greater attraction to and preference for sweet versus drug cues. Addict Biol. 2015;20:433–44. doi: 10.1111/adb.12134. [DOI] [PubMed] [Google Scholar]
- 80.Hemmingsson E. A new model of the role of psychological and emotional distress in promoting obesity: conceptual review with implications for treatment and prevention. Obes Rev. 2014;15:769–79. doi: 10.1111/obr.12197. [DOI] [PubMed] [Google Scholar]
- 81.Dallman MF, Pecoraro NC, la Fleur SE. Chronic stress and comfort foods: self-medication and abdominal obesity. Brain Behav Immun. 2005;19:275–80. doi: 10.1016/j.bbi.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 82.Morris MJ, Beilharz JE, Maniam J, et al. Why is obesity such a problem in the 21st century? The intersection of palatable food, cues and reward pathways, stress, and cognition. Neuroscience and Biobehavioral Reviews. 2015;58:36–45. doi: 10.1016/j.neubiorev.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 83.O’Reilly GA, Cook L, Spruijt-Metz D, et al. Mindfulness-based interventions for obesity-related eating behaviours: a literature review. Obes Rev. 2014;15:453–61. doi: 10.1111/obr.12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Robinson E, Kersbergen I, Higgs S. Eating ‘attentively’ reduces later energy consumption in overweight and obese females. British Journal of Nutrition. 2014;112:657–61. doi: 10.1017/S000711451400141X. [DOI] [PubMed] [Google Scholar]
- 85.Horstmann A, Dietrich A, Mathar D, et al. Slave to habit? Obesity is associated with decreased behavioural sensitivity to reward devaluation. Appetite. 2015;87:175–83. doi: 10.1016/j.appet.2014.12.212. [DOI] [PubMed] [Google Scholar]
- 86.Smyers ME, Bachir KZ, Britton SL, et al. Physically active rats lose more weight during calorie restriction. Physiology and Behavior. 2015;139:303–13. doi: 10.1016/j.physbeh.2014.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wilson P. Physical Activity and Dietary Determinants of Weight Loss Success in the US General Population. American Journal of Public Health. 2016;106:321–6. doi: 10.2105/AJPH.2015.302956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tschop MH, Finan B, Clemmensen C, et al. Unimolecular Polypharmacy for Treatment of Diabetes and Obesity. Cell Metab. 2016;24:51–62. doi: 10.1016/j.cmet.2016.06.021. [DOI] [PubMed] [Google Scholar]
- 89.Finan B, Yang B, Ottaway N, et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nature Medicine. 2015;21:27–36. doi: 10.1038/nm.3761. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.