

Abstract
Objective
Limited efficacy of immune checkpoint inhibitors in pancreatic ductal adenocarcinoma (PDAC) has prompted investigation into combination therapy. We hypothesized that IL-6 blockade would modulate immunologic features of PDAC and enhance the efficacy of anti-PD-L1 checkpoint inhibitor therapy.
Design
Transcription profiles and IL-6 secretion from primary patient stellate cells (PSC) were determined via nanostring analysis and immunohistochemistry, respectively. In vivo efficacy and mechanistic studies were conducted with antibodies (Ab) targeting IL-6, PD-L1, CD4, or CD8 in subcutaneous or orthotopic models using Panc02, MT5, or KPC-luc cell lines; and the aggressive, genetically engineered PDAC model [KrasLSL-G12D, Trp53LSL-R270H, Pdx1.cre, Brca2F/F (KPC-Brca2 mice)]. Systemic and local changes in immunophenotype were measured by flow cytometry or IHC as appropriate.
Results
Patient-derived PSC (n=12) demonstrated prominent IL-6 expression, which was localized to stroma of tumors as confirmed via immunohistochemistry. Combined IL-6 and PD-L1 blockade elicited efficacy in in mice bearing subcutaneous MT5 (p<0.02) and Panc02 tumors (p=0.046) which was accompanied by increased intratumoral effector T lymphocytes (CD62L− CD44−). Administration of CD8-depleting, but not CD4-depleting Ab abrogated the efficacy of combined IL-6 and PD-L1 blockade in mice bearing Panc02 tumors (p=0.0016). This treatment combination also elicited significant anti-tumor activity in mice bearing orthotopic KPC-luc tumors and limited tumor progression in KPC-Brca2 mice (p<0.001). Histologic analysis revealed increased T cell infiltration and reduced α-SMA+ cells in tumors from multiple models. Finally, IL-6 and PD-L1 blockade increased overall survival in KPC-Brca2 mice compared to isotype controls (p=0.0012).
Conclusions
These pre-clinical results indicate that targeted inhibition of IL-6 may enhance the efficacy of anti-PD-L1 in PDAC.
Keywords: pancreatic cancer, IL-6, PD-L1
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is expected to surpass breast and colon cancer, becoming the second leading cause of cancer-related deaths by the year 2030.1 One of the major difficulties with PDAC is its clinical silence. Typically the disease only becomes apparent after the tumor invades surrounding tissues or metastasizes to distant organs.2 For many years, the current standard of care for most advanced PDAC patients has been gemcitabine-based regimens including combined therapy with nab-paclitaxel (Abraxane)3, or aggressive chemotherapy (e.g. FOLFIRNOX) as a strategy to de-bulk the tumor, and improve candidacy for surgery. Regardless, these advances may rightfully be classified as only incremental, and justify further research to identify novel strategies with potential for long term clinical responses and cures for this devastating malignancy.
Signaling downstream of IL-6 is important in PDAC genesis and progression.4, 5 This pleiotropic cytokine binds membrane receptor complexes containing the common signal transducing receptor chain GP130 (glycoprotein 130),6 thereby initiating a complex series of signaling events that include the JAK/STAT, MAPK and PI3K pathways.7, 8 In particular, STAT3 is activated via phosphorylation at Tyr705 in most human PDAC specimens, and cooperates with activated Kras to drive initiation and progression of PDAC in murine models.9, 10 The IL-6/STAT3 axis can simultaneously promote the expansion of immunosuppressive cells or alter the balance of T cell subsets. Among the most notable of these subsets are myeloid-derived suppressor cells (MDSCs) and T regulatory cells (T regs), given their prominent expansion and role as poor prognostic indicators in patients with advanced GI cancer.11–13 Interestingly, data from our group and others point to the pancreatic stroma as one likely source of IL–6. This cytokine is produced in abundance by components of the stroma including pancreatic stellate cells (PSC) and tumor associated myeloid cells.5, 14. In this manner, IL-6 can cooperate with other cytokines, either systemically or in the tumor microenvironment, to further amplify immune changes in patients. Recent studies using an inducible Kras-mediated PDAC murine model also showed that IL-6 was instrumental for PDAC progression.4, 15 In fact, lack of IL-6 completely ablated cancer progression, even in the presence of oncogenic Kras.4 In agreement with these data, recent results from our group emphasize the importance of systemic IL-6 in PDAC patients.16 We analyzed plasma from untreated patients with metastatic or non-resectable PDAC and found that IL-6, IL-10 and MCP-1 were significantly associated with overall survival, with IL-6 having a strong inverse relationship.16
There remains optimism that the dramatic efficacy of immunotherapy in other cancers may be realized in PDAC patients. Indeed, blockade of T cell checkpoint receptors with neutralizing antibodies (Ab) has emerged as a promising immunotherapeutic approach.17, 18 Programmed death-1-ligand 1 (PD-L1), also known as B7-H1, is a cell surface protein and one of two ligands for program death receptor 1 (PD-1), a costimulatory molecule that negatively regulates T cell responses.19, 20 Ligation of PD-L1 on cancer cells to PD-1 expressed on T cells suppresses T cell activation and proliferation, and can induce apoptosis. Intratumoral expression of PD-L1 correlates with poor prognosis in patients with PDAC.17, 21 Indeed, increased PD-L1 expression by cancer cells or the stroma is a fundamental escape mechanism from host immunity.
In theory, IL-6 blockade may modulate immunologic features of PDAC in both the systemic circulation and the tumor microenvironment, thereby enhancing antitumor activity of anti-PD-L1. IL-6 blocking Ab is an attractive component of combination therapy regimens for PDAC for a number of reasons. First, IL-6 targeted Ab are FDA-approved for other indications and could easily be re-purposed for oncology. Second, data from our group and others suggest that IL-6 is a major factor produced by multiple cellular compartments to promote immune suppression and PDAC progression.17, 22–24. Finally, blocking IL–6 may tune the phenotypic properties of both myeloid and T cell compartments of a tumor bearing host to be more amenable to efficacy from immunotherapy regimens.
This pre-clinical study tested the hypothesis that IL-6 blockade would enhance the efficacy of anti-PD-L1 checkpoint inhibitor therapy in preclinical models of PDAC. A significant benefit was observed from this therapy in in the subcutaneous MT-5 and Panc02 models, an orthotopic KPC luciferase model, and an aggressive genetically engineered mouse model (GEMM) of PDAC. Mechanistic studies revealed increased pancreatic effector T cell infiltration, and efficacy dependent on CD8+ T cells. These results indicate that targeting IL-6 may enhance the efficacy of Ab targeting PD-L1 in PDAC by improving the phenotypic properties and infiltration of T cells.
Experimental Methods
Cell lines and reagents
The murine pancreatic cancer cell line Panc02 was purchased from the ATCC and cultured in RPMI (Gibco) with 10% FBS, 10 mM L-glutamine, and antibiotics. Murine MT5 (KrasLSL-G12D, Trp53LSL-R270H, Pdx1-cre) pancreatic cells were a kind gift from Dr. Tuveson (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY) and cultured in RPMI (Gibco) with 10% FBS, 10 mM L-glutamine, and antibiotics. KPC-luc cells for orthotopic experiments were derived from KPC mice (KrasLSL-G12D, Trp53−/−, PDX-1-Cre) and transfected with enhanced firefly luciferase as previously described.25 Murine isotype controls (clone 3E5.2H12), anti-IL-6R (clone BP-5875), and anti-PD-L1 blocking Ab were obtained from either Genentech, Inc. (San Francisco, CA) for in vivo studies in the KPC-Brca2 murine model. Murine antibodies to IL-6 (Clone MP5-20F3), PD-L1 (Clone 10F.9G2), or isotype controls (Clones LTF-2 and HRPN) were purchased from BioXcell (West Lebanon, NH) for in vivo studies using the MT-5, Panc02, and KPC-luc cell lines.
Murine models of pancreatic cancer
KPC-Brca2 mice were generated by interbreeding Brca2flox2/flox2; KrasLSL-G12D/+ with Brca2flox2/flox2; Trp53LSL-R270H/+; Pdx1-cre animals.26 The mouse strains p53LSL-R270H (strain number 01XM3), KrasLSL-G12D (strain number 01XJ6), and Pdx1-cre (strain number 01XL5) were acquired from the National Cancer Institute (NCI) Frederick Mouse Repository. All transgenic mice generated in this study were maintained on a mixed 129/B6 genetic background. All studies involving MT5, Panc02, KPC-luc tumors utilized syngeneic, female C57BL/6 mice, 5–6 weeks of age.
In vivo efficacy studies
KPC-Brca2 mice (5 weeks of age) were treated with isotype controls, anti-IL-6R and/or anti-PD-L1 Ab (Genentech) at a dose of 20μg/mouse, 3 times each week (Monday, Wednesday, and Friday). Following 2 weeks of treatment, animals were euthanized via CO2 asphyxiation, followed by cardiac puncture. Plasma, splenocytes and tumor tissue were collected for further analysis. Pathology was assessed in H&E stained slides to determine the differentiation state of tissue as pancreatic intraepithelial neoplasia (PanIN) 1A, PanIN 1B, PanIN 2, or PDAC. For studies using MT5 and Panc02 tumors, 1×106 or 3×105 cells, respectively were injected subcutaneously in the flank of C57BL/6 mice 3 times each week with 20μg/mouse of isotype, anti-IL-6 or anti-PD-L1 Abs (BioXCell) Ab treatment starting once tumors reached 50–100mm3 volume. For orthotopic studies, C57BL/6 mice were injected with 1×106 KPC-luc (luciferase expressing) cells in Matrigel (BD Biosciences) in the tail of the pancreas. Tumor growth was analyzed once a week by bioluminescent imaging and end of study tumor weight was determined immediately post-mortem. Mice were treated 3 times each week with 200 μg/mouse of isotype, anti-IL-6 or anti-PD-L1 Abs (BioXCell). For T cell depletion studies, Ab to deplete CD4 (Clone GK1.5; BioXcell) or CD8 (Clone 2.43; BioXCell) were injected i.p. at 100 μg per mouse on days −2, −1, +1, +4, and every other 3 days afterwards until completion of the study as previously described.27 For survival studies, KPC-Brca2 mice were treated starting at 5 weeks of age with isotype control, IL-6 or PD-L1 Abs as single agents or in combination (200 μg/mouse each Ab, BioXCell) until mice were moribund as determined by IACUC protocol.
Pancreatic stellate cell isolation and Nanostring analysis
Tissue from human patient pancreatic tumors undergoing surgical resection at the James Cancer Hospital and Solove Research Institute (Columbus, OH) was obtained under an Institutional review board-approved protocol following informed consent. Tissue was dissected with a scalpel into 0.5–1mm3 pieces, then plated in 6-well 10 cm2 uncoated culture wells in DMEM with 10% FBS and antibiotics and incubated at 37°C. PSC typically grew out of the tissue in 2–3 weeks, and were characterized by morphology and histological analysis of alpha-smooth muscle actin (α-SMA+) staining. PSC were maintained in culture with fresh media added twice weekly for 3 passages and then RNA was collected by Trizol extraction. RNA was analyzed using the nCounter PanCancer Immune Profiling Panel (Nanostring Technologies, Seattle WA).
Immunohistochemical (IHC) analysis
Tumor tissue was prospectively obtained at surgery under an Institutional review board-approved protocol following informed consent and de-identified. Formalin fixed pancreatic tissue from human tissue specimens and in vivo experiments in mice were subjected to IHC analysis following staining with Ab against IL-6 (Abcam and eBioscience), pSTAT3 (Catalog 4904; Cell Signaling), α-SMA (Catalog M0851; Dako), PD-L1 (Catalog NBP1-76769) and CD3 (Catalog A0452; Dako). For α-SMA quantification, 20× magnification images of pancreata (8–10 images per mouse) were captured using PerkinElmer’s Vectra multispectral slide analysis system. inForm software tools were used to quantify positive SMA cells (Fast Red chromogen) within each image (Supplementary Figure 1). For analysis of CD3+ cells, blinded histological analysis of staining in the pancreas was counted at 40× magnification, with at least 10 fields counted per mouse.
Flow Cytometry
Immunophenotypic analyses of splenoctyes and single cell suspensions from tumors were assessed by flow cytometry. Antibodies to stain for MDSC were CD11b-APC (Clone M1/70; BD Biosciences), Ly6G-FITC (Clone 1A8; BD Biosciences), Ly6C-PE (Clone AL-21; BD Biosciences). For T cell activation markers, cells were stained with Ab specific for CD4-PE-Cy7 (Clone RM4-5; BD Biosciences), CD8-PE-Cy7 (Clone 53-6.7; BD Biosciences), CD62L-PE (Clone MEL-14; BD Biosciences), and CD44-FITC (Clone IM7; Biolegend). To determine Th1 and Th2 phenotypes, cells were stained using fluorochrome-conjugated Ab targeted CXCR3-PE-Cy7 (Clone CXCR13-173; Biolegend), CCR4-PE (Clone 2G12; Biolegend), and CCR6-APC (Clone CK4-L3; BD Biosciences). Cells were incubated on ice for 30 minutes, washed, and fixed in PBS containing 1% formalin for flow cytometric analysis on a LSRII flow cytometer (BD Biosciences).
Statistics
For data obtained by Nanostring (Nanostring Technologies, Seattle, WA), positive spike-in controls were first used to normalize assay efficiency. Negative controls were used to access background hybridization, and for filtering out low expression probes. Reference genes were used to normalize across biological samples. Linear mixed models were then used to detect differentially expressed RNAs. A variance smoothing method was used for improving variance estimates in testing.28 Significance was adjusted by controlling the mean number of false positives.29 Data obtained by flow cytometry and IHC as well as tumor volumes were log-transformed prior to analysis to meet model assumptions of normality and homoscedasticity. Poisson regression was used to model the number of PanIN3/tumor lesions in the KPC-Brca2 mice. Tumor volume was modeled over time using mixed-effects regression with fixed effects for group, time and the interaction of the two. Random intercepts and slopes by mouse were included with an unstructured covariance matrix for the random effects. For the T cell depletion experiments examining tumor growth, interaction contrasts were constructed to enable the direct testing of the hypotheses under consideration.30 For Kaplan-Meier survival curves a log-rank test to determine difference used. The other outcomes were compared using ANOVA. P-values less than 0.05 were considered significant and all analyses were conducted in SAS v9.4 (SAS Institute, Cary, NC) or R v3.2.3 (R Foundation for Statistical Computing, Vienna, Austria).
Results
The pancreatic tumor stroma is a major source of IL-6 in the tumor microenvironment
For these studies, an unsupervised assessment of gene expression was conducted to identify actionable immunologic targets in pancreatic stellate cells, a prominent component of PDAC stroma. Stellate cells derived from surgically resected pancreatic tissue of 10 different patients were isolated and cultured in vitro as previously described.14 After 3 passages, RNA was extracted and subjected to profiling of transcripts relevant to immunologic biomarkers using Nanostring technology (Nanostring PanCancer Immune Profiling Panel). RNA isolated from patient-derived PSC exhibited significantly increased expression of cytokines and chemokines including IL-6, and CCL11 as compared to RNA isolated from normal human pancreatic fibroblasts (Figure 1 and Supplementary Figure 2). Further investigation via IHC analysis confirmed a pattern of staining for IL-6 that localized predominantly in the desmoplastic, stromal regions of pancreatic tumors, with occasional staining in regions containing malignant pancreatic epithelial cells (Figure 2A). This trend was evident in tissue from several (n=13) representative patients (Figure 2B). These data were consistent with our analysis of transcripts in PSC, and suggested that the stromal compartment, is a major source of IL-6 in the pancreatic tumor microenvironment.
Figure 1. Profile of mRNA transcripts from patient-derived PSC.

RNA was isolated from 10 patient-derived pancreatic cancer stellate cells and analyzed utilizing the Nanostring nCounter PanCan Immune Profiling Panel. Data are expressed as the fold change in expression as compared to a normal human pancreatic fibroblast cell line and relative to several housekeeping genes. Genes are ranked from highest fold-change to lowest. Heat map with hierarchical clustering for genes with at least 2 fold change up or down with a p<0.01 cutoff. Significantly higher expression is shown in red and lower expression in green.
Figure 2. IL-6 expression is enriched in the stromal compartments of human PDAC tissue.
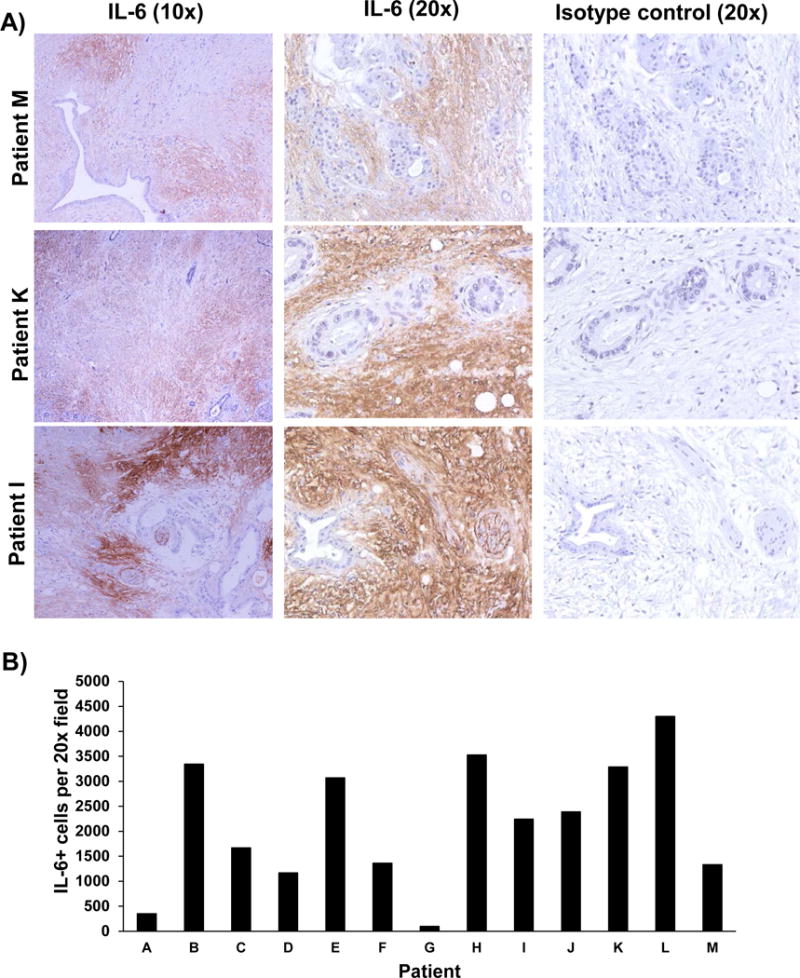
Human PDAC tumors (among n=13 patients) stained for IL-6 expression by immunohistochemistry. A) IL-6 (Brown Chromogen) IHC staining from three representative patient samples is displayed at 10× and 20× magnification. Tissue sections from each patient were stained with an isotype control Ab to account for background. B) Number of IL-6 positive cells per field was quantified.
Prominent expression of IL-6/JAK/STAT pathway components and PD-L1 in the pancreatic tumor microenvironment of mouse models of pancreatic cancer
Similar to human PDAC, histologic characterization of pancreata from PDAC orthotopic and genetically engineered mouse models (GEMM) revealed expression of IL-6 in both stroma, and more limited positive staining in foci of some tumors (Figure 3). Consistent with prior published studies, pancreata from GEMM (KPC and KPC-Brca2) recapitulated the histologic features of advanced PDAC observed in human disease. For example, KPC tumors displayed acinar to ductal metaplasia, while the KPC-Brca2 displayed desmoplastic stroma, PanIN, and ductal adenocarcinoma expression similar to observations in human tissue. Pancreata from both the orthotopic and genetic tumor models displayed a fibrotic stroma that both surrounded and intercalated within tumor foci (Figure 3; S, stroma; T, tumor tissue). Of note, robust phosphorylation of STAT3 was detectable in stroma and malignant epithelial cells in the pancreata from both KPC and KPC-Brca2 mice (Figure 3). Consistent with this immunosuppressive microenvironment, tumors expressed high levels of PD-L1, regardless of the animal model.
Figure 3. PDAC GEMM recapitulate IL-6 pathway activation and PD-L1 expression observed in human disease.
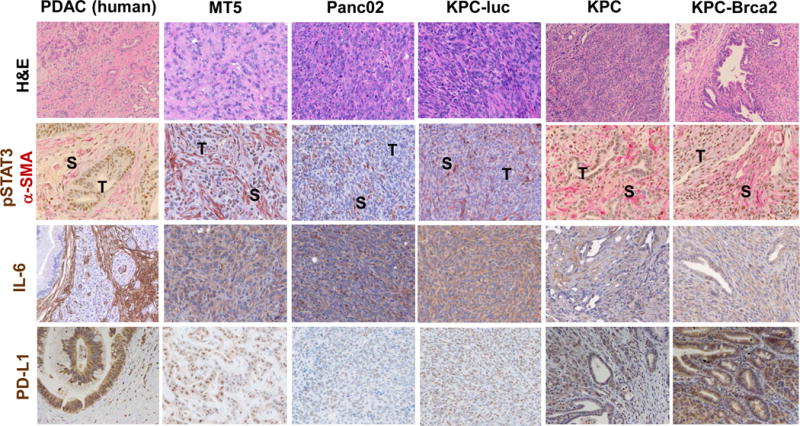
IHC analysis of representative H&E, pSTAT3 (brown)/α–SMA (pink), IL-6 (brown), and PD-L1 (brown) staining on tissue sections from human pancreatic cancer, orthotopic (KPC-luc), subcutaneous (MT5, Panc02), or pancreata from the following GEMM: KPC (KrasLSL-G12D, Trp53LSL-R270H, Pdx1-cre), and KPC-Brca2 (KrasLSL-G12D, Trp53LSL-R270H, Pdx1.cre, Brca2F/F). All images are presented at 40× magnification.
Combined blockade of IL-6 and PD-L1 inhibits murine pancreatic tumor growth and increases intratumoral effector T cells in vivo
We postulated that concurrent blockade of IL-6 and a potent immune-modulatory stimulus via PD-L1-targeting Ab would circumvent key mechanisms of resistance to T-cell mediated immunity to pancreatic cancer. We utilized murine MT5 tumor cells subcutaneously injected into C57BL/6 mice. This cell line was derived from KPC tumors and has both G12D mutated Kras and R172H Trp53.31 Once tumors were palpable, mice were treated with anti-IL-6, anti-PD-L1 or isotype controls until study endpoint. A significant inhibition of tumor growth was observed in response to the combination therapy as compared to isotype, anti-IL-6 or PD-L1 antibody therapy alone (Figure 4A; p<0.03). To carefully assess immune changes, and confirm our data in an additional PDAC model, the well-characterized, murine Panc02 model was also used, given its frequent use in published pre-clinical immunotherapy studies in PDAC. Similar to the MT5 cell line, Panc02 cells are also syngeneic to immune competent C57BL/6 mice.32, 33 Once tumors were palpable, mice received treatment until the study endpoint. A significant inhibition of tumor growth was observed in response to combined therapy with anti-IL-6 and anti-PD-L1 Ab as compared to isotype controls (Figure 4B; p=0.0457). Tumors were dissociated at the study endpoint and underwent phenotypic analysis by flow cytometry. These data confirmed a significantly greater percentage of infiltrating CD8+ T cells in the tumor tissue from mice receiving the combination (Figure 4C; p=0.0006). Further analysis indicated an increased proportion of cells with an effector CD62L−CD44+ phenotype in mice receiving combined blockade of IL-6 and PD-L1 (Figure 4D; p=0.0086 and E; p=0.0024). Further, we observed no difference in the level of other circulating or intratumoral immunosuppressive subsets (Granulocytic and Monocytic MDSC) between any of the treatment groups as compared to isotype controls (Supplementary Figure 3A–C). Importantly, treatment Ab blocking IL-6, PD-L1 or both Ab combined did not result in any overt signs of toxicity as evidenced by weight loss in all animal models tested (data not shown).
Figure 4. IL-6 and PD-L1 antibody blockade combination therapy decreases PDAC tumor progression and increases the percentage of intratumoral effector T cells.
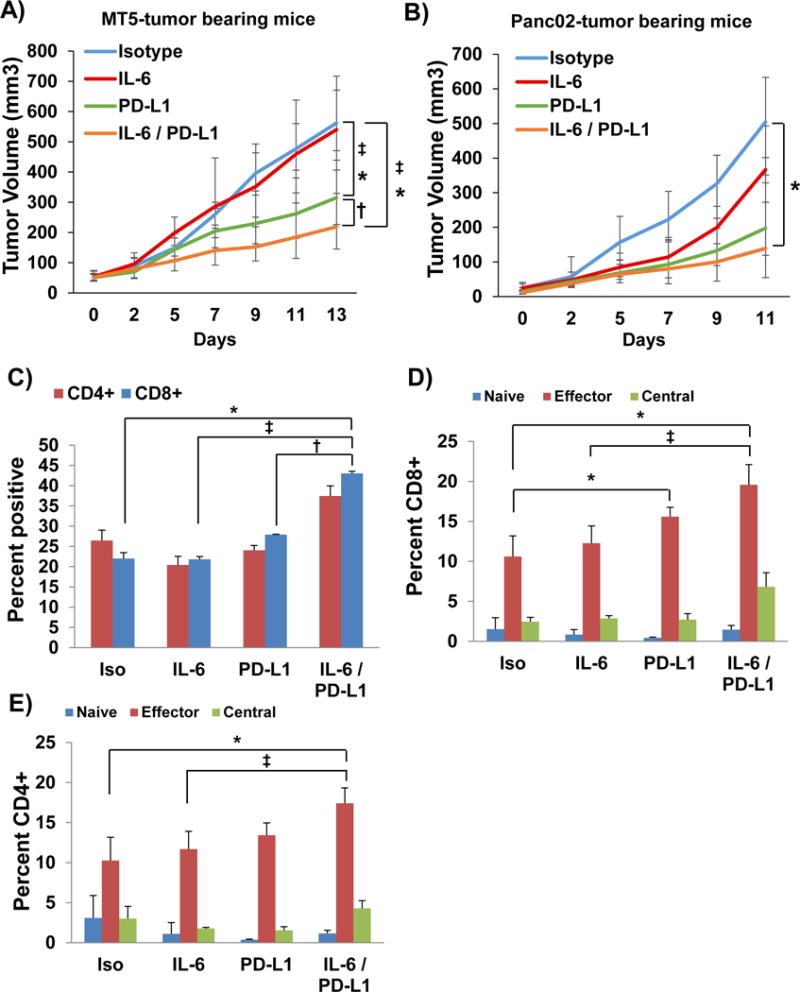
A) MT5 or B) Panc02 murine pancreatic tumor cells were subcutaneously injected into C57BL/6 mice with treatment beginning when tumors reached 50–100mm3. Mice were treated with 200μg (intraperitoneal injection 3 times/week) with isotype control, anti-IL-6 and/or anti-PD-L1 antibodies (n=5–6 mice/group) until mice met pre-specified IACUC-approved early removal criteria. Geometric means ± SD; *p<0.01 compared to Isotype; †p<0.03 compared to PD-L1; ‡p<0.05 compared to IL-6. Panc02 tumors were dissociated using Collagenase II and the Miltenyi Biotec gentleMACS dissociator to obtain a single cell suspension. Cells were stained and analyzed by flow cytometry for C) CD4+, CD8+ T cells and activation markers for different D) CD4+ and E) CD8+ T cell subsets (n=3/group). Naïve (CD62L+CD44−), Effector Memory (CD62L−CD44+), and Central Memory (CD62L+CD44+). Means ± SD; *p<0.002 compared to Isotype; †p<0.01 compared to PD-L1; ‡p<0.02 compared to IL-6.
Decreased tumor growth by combined blockade of IL-6 and PD-L1 is CD8+ T cell dependent
To determine whether the efficacy of combined IL-6 and PD-L1 blockade was T cell dependent, Panc02-bearing mice were administered CD4+ or CD8+ depleting Ab during the course of treatment. T cell depletion was confirmed by flow cytometry using splenocytes from mice at the study endpoint (Figure 5A and B). Administration of CD8 (p=0.0016), but not CD4 depleting Ab abrogated the efficacy of combined IL-6 and PD-L1 blockade in mice bearing Panc02 tumors (Figure 5C).
Figure 5. The anti-tumor response to combined IL-6 and PD-L1 blockade is CD8+ T cell dependent.
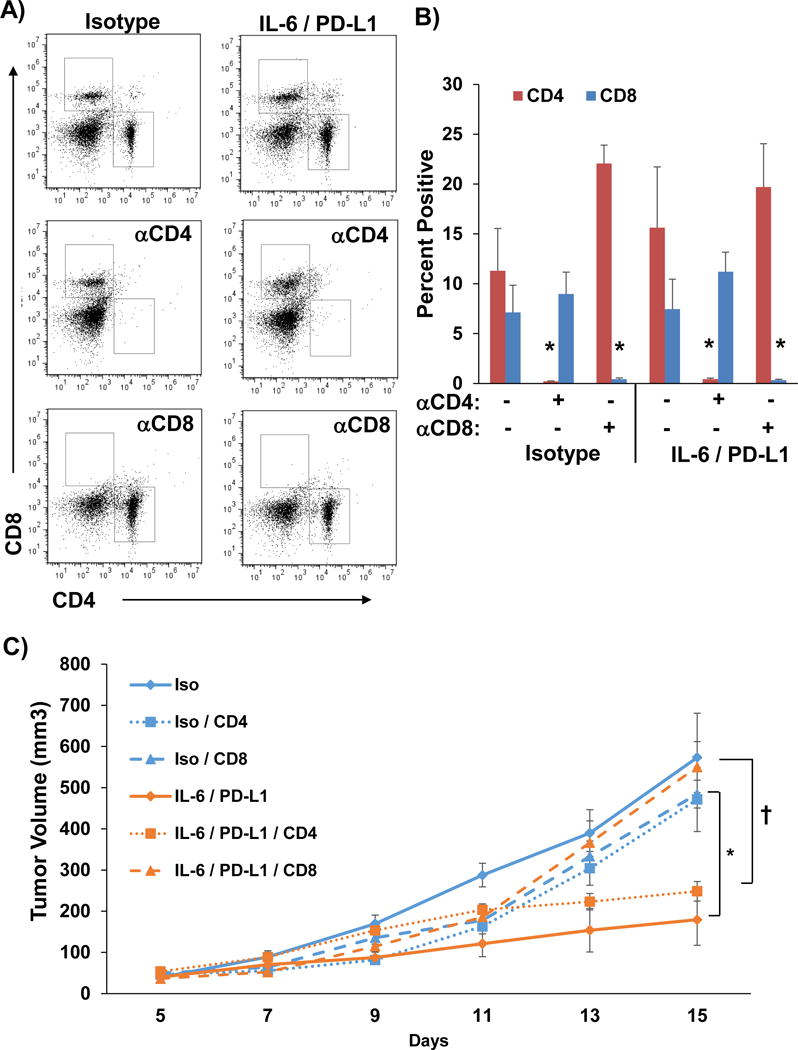
Panc02 murine pancreatic tumor cells were subcutaneously injected into C57BL/6 mice with treatment beginning when tumors reached 50–100mm3. Mice were also depleted of either CD4+ or CD8+ T cells by injecting depletion antibodies on days 5, 6, 8, 11, and 14. A) Representative flow cytometry and B) quantification of CD4 and CD8 staining of splenocytes to confirm depletion. Means ± SD; *p<0.001. C) On day 7, mice were treated with 200 μg (intraperitoneal injection 3 times/week) of isotype controls or anti-IL-6 and anti-PD-L1 antibodies combined (n=5 mice/group) until mice met pre-specified IACUC-approved early removal criteria. Means ± SD; *p<0.001 compared to isotype; †p<0.002 CD8 depletion vs CD4 depletion.
IL-6 and PD-L1 antibody blockade combination therapy reduces tumor progression in an orthotopic model of PDAC
To further investigate the efficacy of IL-6 blockade combined with PD-L1 inhibition, we utilized KPC-luc cells orthotopically injected into the tail of the pancreas of C57BL/6 mice. This luciferase-expressing cell line was derived from KPC tumors with G12D mutated Kras and R172H Trp53.25 Tumor growth was assessed by weekly bioluminescent imaging (Figure 6A and B). Once tumors were detectable by imaging the pancreas, mice were randomized and treated with anti-IL-6, anti-PD-L1 or isotype control Ab until the study endpoint. We observed a decreased final tumor weight in mice receiving combination therapy as compared to isotype control, anti-IL-6, or PD-L1 antibody therapy alone (Figure 6C; p=0.05530). A decrease in α-SMA+ stromal cells was also observed in the pancreata of mice treated with combination therapy as compared to isotype control Ab alone (p=0.0563). Furthermore, the number of CD3+ T cells was greater in the pancreata from mice treated with antibodies targeting both IL-6 and PD-L1 as compared to those treated with isotype control antibodies alone (p=0.0553).
Figure 6. IL-6 and PD-L1 antibody combination blockade reduces tumor growth in an orthotopic model of pancreatic cancer.
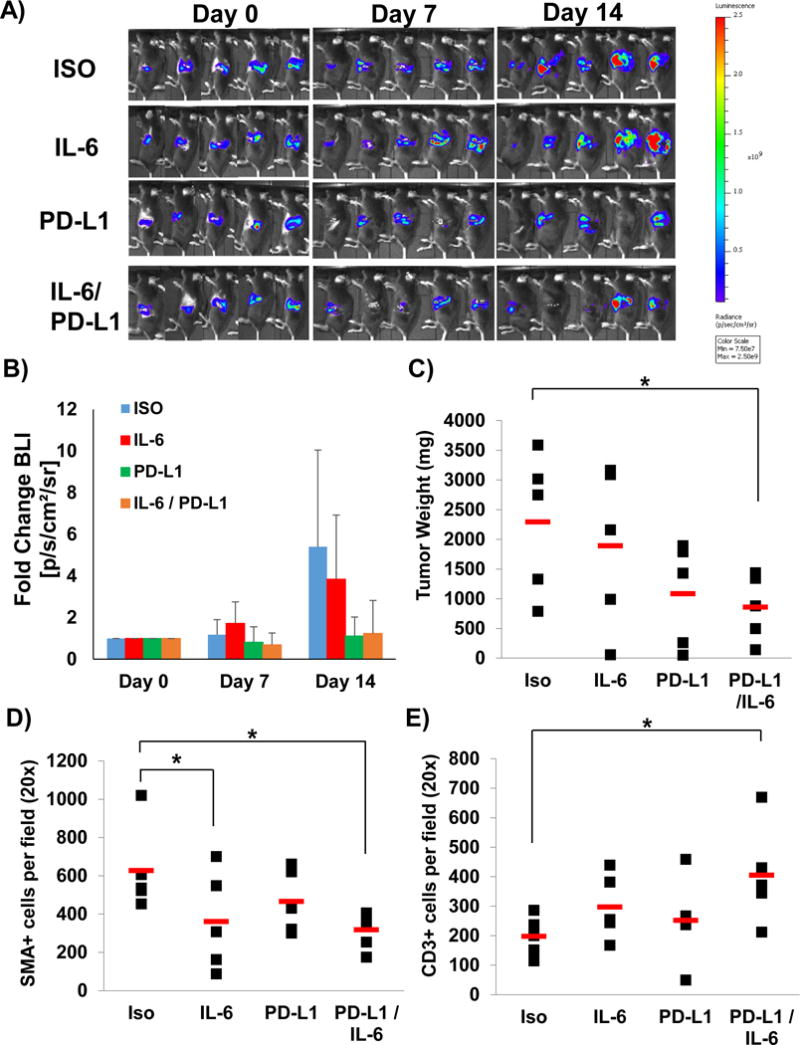
C57BL/6 mice were orthotopically injected with luciferase expressing KPC cells (KPC-luc) in matrigel and A) tumor growth assessed and B) quantified by bioluminescent imaging weekly. C) Tumor weights at completion of study. Means ± SD; *p=0.05530 compared to isotype. Pancreatic tissue was stained for D) α–SMA+ stromal cells (Means ± SD; *p=0.0563 compared to Isotype) and E) CD3+ T cells (Means ± SD; *p=0.0553 compared to Isotype) by IHC quantified at 20× magnification.
Antibodies targeting IL-6R and PD-L1 limit tumor progression in the aggressive KPC-Brca2 GEMM
To further confirm our data in a genetic model that better recapitulates many aspects of patient tumors, we used a highly aggressive, autochthonous model of spontaneously arising PDAC driven by mutant Kras, Trp53 and Brca2 (Figure 7A). Any efficacy in this oncogene driven tumor model would signify a promising approach to carry forward for further evaluation. At 5–6 weeks of age, KPC-Brca2 mice have PDAC with 100% penetrance (Figure 7A). Following two weeks of treatment with anti-IL-6R and/or anti-PD-L1, animals were euthanized and pancreata was isolated for histologic analysis. There was a statistically significant shift in the proportion of low grade PanIN lesions, with fewer PanIN3 or foci of adenocarcinoma in mice treated with antibodies targeting PD-L1 and IL-6R, as compared to animals treated with isotype control Ab (Figure 7A and B; p<0.001). Similar to the subcutaneous Panc02 model, depletion of CD8+ T cells limited the effect of combination therapy in the KPC-Brca2 GEMM. In fact, we observed a greater proportion of PanIN3/PDAC lesions in pancreata from CD8+ depleted mice treated with the combination when compared to mice treated with the combination alone (Figure 7B). Importantly, analysis revealed a significant decrease in the number of α-SMA+ stromal cells in pancreata of mice treated with Ab targeting both IL-6R and PD-L1 as compared to control mice (Figure 7C and D; p<0.05). Mice depleted of CD8+ T cells treated with combination therapy did not display the same decrease of α-SMA+ cells in the pancreas compared to mice treated with only the combination therapy (Figure 7D; p=0.0033). Further, treatment with Ab targeting IL-6, PD-L1 or both agents combined did not alter expression of IL-6 (Supplementary Figure 5A) or PD-L1 (Supplementary Figure 5B) in the pancreatic tumor microenvironment.
Figure 7. IL-6 and PD-L1 antibody blockade combination therapy decreases PDAC tumor progression and α-SMA+ cells in the pancreata from KPC-Brca2 mice.
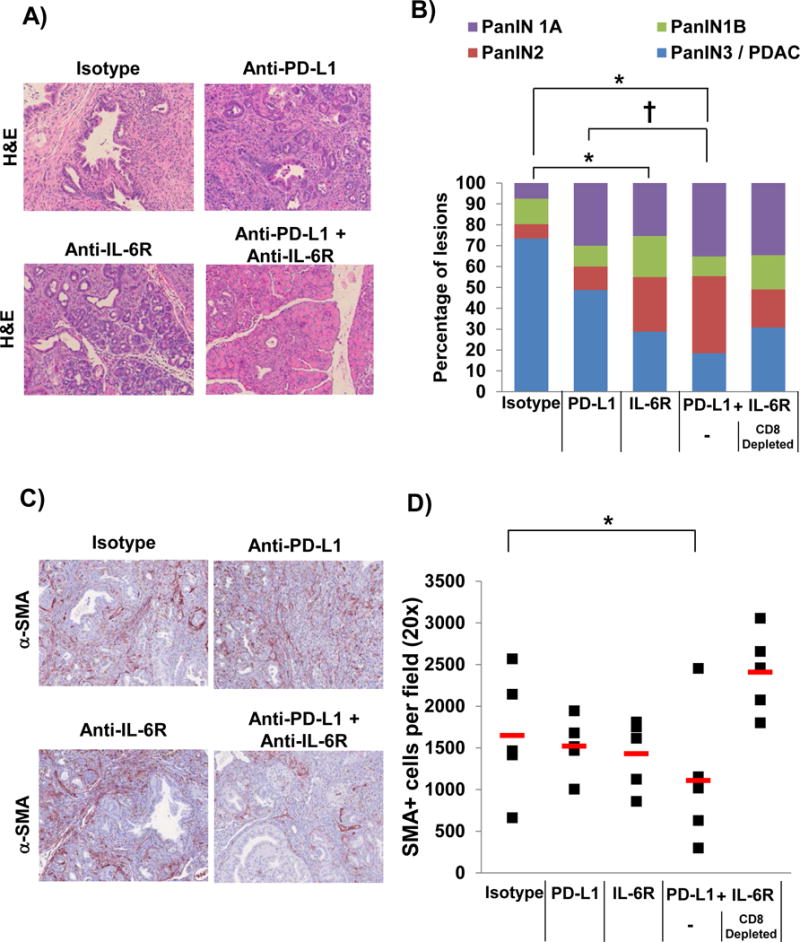
KPC-Brca2 mice were treated at 5–6 weeks of age with 200 μg (intraperitoneal injection 3 times/week) of isotype control, anti-IL-6R and/or anti-PD-L1 antibodies for 2 weeks (n=5 mice/group). A cohort of mice treated with anti-IL-6R and PD-L1 antibodies were also depleted for CD8+ T cells. A) Representative H&E staining of pancreata at 20× magnification and B) quantification of the pathology. Means ± SD; *p<0.005 compared to Isotype; †p=0.01) compared to PD-L1; C) Pancreatic tissue was stained for α–SMA+ stromal cells (red) by IHC and D) quantified at 20× magnification. Means ± SD; *p=0.0545 compared to Isotype.
Combined blockade of IL-6R and PD-L1 increases T cells with Th1 phenoytpic properties, and CD3+ cell infiltration into PDAC tumors in KPC-Brca2 mice
To better understand the mechanism by which combined IL-6 and PD-L1 blockade limited tumor progression in the aggressive KPC-Brca2 model, a series of immunophenotypic analyses were conducted. These data revealed a higher percentage of circulating CD4+ T cells with Th1 phenotypic properties (CXCR3+CCR4−CCR6−) in the spleens of mice treated with Ab targeting IL-6R and PD-L1 combined as compared to controls (Figure 8A; p<0.0001). No changes in CD4+ T cells with Th2 phenotypic properties were evident in any treatment group as compared to controls (Figure 8B). Importantly, assessment of the tumor microenvironment revealed significant increases in the infiltrating CD3+ T cells in the pancreata of combination treated mice (Figure 8C–D; p=0.0025). Further analysis revealed no difference in the level of other immunosuppressive subsets (MDSC and Tregs) between any of the treatment groups as compared to isotype controls (Supplementary Figure 4A–D). These data suggest that this treatment regimen may modulate phenotypic properties of T cells and their access to the tumor microenvironment.
Figure 8. Higher percentage of circulating T cells with Th1 phenotypic properties and intratumoral CD3+ cells in mice treated with IL-6 and PD-L1 antibody blockade.
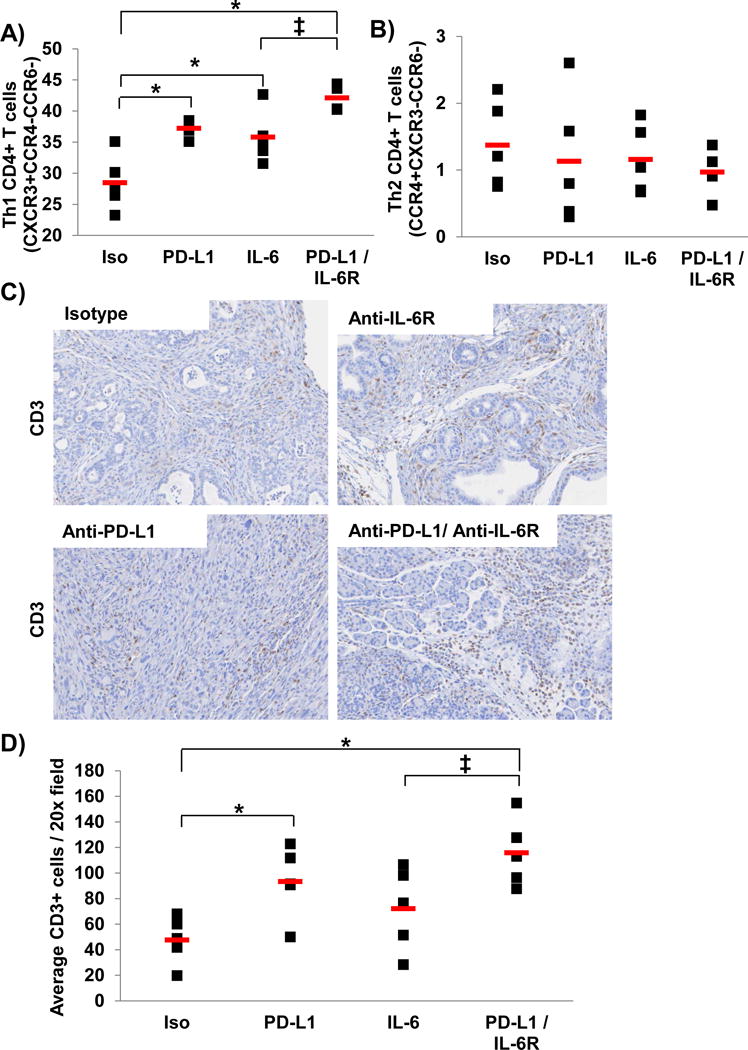
KPC-Brca2 mice were treated at 5–6 weeks of age with 200 μg (intraperitoneal injection 3 times/week) with isotype control, anti-IL-6R and/or anti-PD-L1 antibodies for 2 weeks (n=5 mice/group). Splenocytes were stained and analyzed by flow cytometry for A) T cells with Th1 phenotypic properties (CD4+CXCR4+CCR4−CCR6−) or B) T cells with Th2 phenotypic properties (CD4+CCR4+CXCR4−CCR6−). Means ± SD; *p<0.002 compared to Isotype; ‡p=0.028 compared to IL-6. Pancreatic tissue was stained for C) CD3+ T cells (brown) by IHC and D) quantified at 20× magnification. Means ± SD; *p<0.05 compared to Isotype; ‡p=0.052 compared to IL-6.
Combined blockade of IL-6 and PD-L1 extend overall survival in the KPC-Brca2 GEMM
The impact of this treatment combination on overall survival was evaluated in KPC-Brca2 mice. In this study, mice began long-term treatment at 5 weeks of age with antibodies targeting IL-6 and PD-L1 or isotype controls 3 times weekly until the animals were moribund. A significant prolongation in overall survival (35%; p=0.0012) was observed in mice treated with combined IL-6 and PD-L1 Abs, as compared to isotype control treated animals (Figure 9). Long term exposure to this treatment regimen did not lead to alterations in body weight of these animals, indicating its tolerability during a sustained period of administration (Supplementary Figure 6).
Figure 9. IL-6 and PD-L1 blockade increases overall survival of KPC-Brca2 mice.
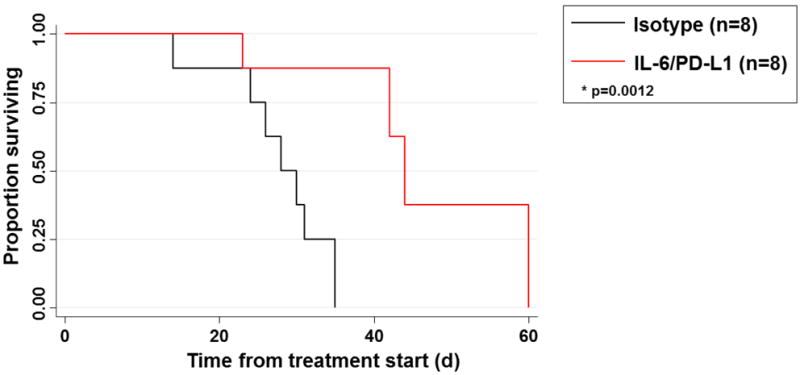
KPC-Brca2 beginning at 5 weeks of age mice were treated with isotype control antibodies or antibodies targeting IL-6 and PD-L1 (200μg/each) until mice were moribund and met pre-specified IACUC-approved early removal criteria. Kaplan-Meier survival curves with log-rank test for significance between isotype control and IL-6/PD-L1 antibodies (p=0.0012).
Discussion
In the present report we demonstrate that combined blockade of IL-6 and PD-L1 elicits efficacy and extends survival therapy in highly aggressive PDAC models. These data extend prior observations regarding IL-6 as a factor limiting the efficacy of immunotherapy in PDAC. Combination therapy skewed T cells to a Th1 phenotype, and increased the number of infiltrating effector cells into the tumor. This is significant, as limited T cell infiltration is considered a barrier for efficacy of immunotherapy in PDAC. Further, we showed that efficacy of combination therapy on tumor progression was dependent on CD8+ T cells in multiple models. Overall, these pre-clinical data support this combination as a rational, therapeutic approach to elicit improved T cell mediated immune responses to pancreatic cancer.
The next frontier of immunotherapy is discovering ways to enhance the efficacy of checkpoint inhibitors using combination approaches, especially in cancers where little effect is observed in the clinic. Before blindly moving forward with combination approaches, it will be important to prioritize therapies based on empirical data, and the relative importance of targeting each factor in the tumor-bearing host. IL-6 as a target is quite attractive for a number of reasons. For example, as recently reported by our group, higher baseline plasma IL-6 in treatment-naïve PDAC patients correlated with worse overall survival.16 IL-6 can also promote the differentiation of DCs, macrophages, and MDSC contributing to immunosuppression.19, 22 Further, IL-6 can lead to greater proliferation, invasiveness, and tumor progression in the pancreatic tumor microenvironment.16 This report represents an important series of pre-clinical studies to provide the first evidence of this treatment combination working in relevant animal models. The next logistical step for effective translation will be to understand how this drug combination works in the context of prior or concurrent chemotherapy, which will be most relevant in clinical trial scenarios.
IL-6 targeting agents have been utilized in pancreatic cancer but with limited success. This is potentially because IL-6 blockade has been attempted as a single agent, or in combination with chemotherapy in heavily pre-treated patients.34, 35 In all of these studies, there was no potent immunotherapeutic treatment that was given concurrently with blockade of IL-6. Others have shown that neutralization of IL-6 also suppresses signaling through the phosphoinositide 3-kinase/AKT pathway.36 A recent Phase-I study combining tocilizumab (anti-IL-6) and IFN-a2b in ovarian cancer showed feasibility/safety and a greater percentage of activated T cells.37 Thus, blockade of IL-6 could lead to potentially more efficacious results than targeting a single pathway (JAK/STAT, MEK, AKT) as it could simultaneously limit other pathways driven by aberrant IL-6 expression.
Our group and others have reported that tumors, myeloid cells, and stromal cells can secrete IL-6, fueling the tumor microenvironment and immunosuppression in PDAC. It is known that PDAC is profoundly immune suppressive, and clearly more data on clinical relevance of the stroma and its soluble factors, including IL-6 are needed. In a model of MCA205 cells transfected with OVA, Gr-1+ MDSC were the source of IL-6 in tumor bearing mice. Blockade of IL-6 led to increased differentiation of tumor-Ag specific effector T helper cells (Th1).24 In this report, IL-6 in stellate cells derived from PDAC stroma was a key factor that emerged in an unsupervised analysis of patient PSC at the RNA level and was confirmed via IHC. This was consistent with our prior in vitro data showing that patient-derived PSC secrete IL-6 that promotes the differentiation of MDSC.20 Other studies have clearly shown that IL-6 can arise from tumor derived macrophages, and may play a role via STAT3 in mediating immune suppression in the tumor microenvironment.5 These factors complement other cytokines such as GM-CSF, which is produced by tumor cells (and perhaps others) to fuel an immunosuppressive microenvironment.38 These data indicate that IL-6 represents an actionable therapeutic target that may modulate the immune architecture in PDAC tumors and can be manipulated to limit immune suppression.
It is likely that the PDAC stroma amplifies immunosuppression via IL-6-induced signaling, and limits the efficacy of immunotherapy. For instance, numerous reports suggest that a reciprocal relationship exists between tumor and stromal cells, leading to enhanced survival and growth of PDAC.39 Among these are studies demonstrating that fibroblast activating protein (FAP+) stromal cells maintain the stroma and suppress anti-tumor immunity.40–42 Other studies, using targeted disruption of sonic hedgehog signaling in the stroma led to accelerated PDAC progression.43, 44 These data provided insight into prior clinical studies using the hedgehog pathway inhibitor Saridegib which were halted early due to patients progressing faster than those on the control arm. Similarly, genetic ablation of SMA+ stromal cells led to accelerated metastasis and disease progression in a mutant KRas-driven model of PDAC.45 Despite these data, it was quite interesting to note that the response to immunotherapy with anti-CTLA4 Ab was enhanced in animals lacking SMA+ stroma cells. In contrast, these animals did not derive benefit from chemotherapy with gemcitabine.45
Subsequent studies should investigate further how IL-6 blockade enhances checkpoint immunotherapy. IL-6 could act to promote expansion of MDSC, although no alterations were observed in MDSC levels in tumor and/or spleen in either the genetic or subcutaneous murine models (Supplementary Figures 3 and 4). MDSC and macrophages are other potential sources of IL-6, and blockade could also render these cells less immunosuppressive, leading to enhanced efficacy of checkpoint inhibition. IL-6 could act at the level of antigen presenting cells, by down-regulating IL-12 production, and subsequently the Th1 immune response.24, 46 Another possibility is that IL-6 blockade could downregulate SOCS expression in T-cells, and restore the ability of IFN-gamma or other cytokines to act and promote Th1 differentiation.47 Although this is less likely given the redundancy in cytokines that might compensate and upregulate SOCS expression, it remains a possibility.
One final interesting and important observation was that anti-IL-6 and anti-PD-L1 combination increased T cell infiltration into tumors. T cell infiltration into tumors is a major issue that impacts the ability of a patient to respond to checkpoint inhibitors.48, 49 This regimen clearly adjusts the phenotypic properties of T cells that infiltrate tumors, and may make a subject more likely to respond to immunotherapy. Blockade of IL-6 in combination with checkpoint therapy could improve T cell trafficking, and change the profile of T cells in the tumor. We believe this combination may be a broadly applicable therapeutic strategy for multiple types of tumors that are driven by IL-6. Given the multiple facets of IL-6 in regulating pancreatic carcinogenesis and immune suppression, this represents a reasonable target with a rapid path to clinical translation since multiple IL-6 blocking Ab are already FDA approved for other indications.
Supplementary Material
Significance of this study.
What is already known on this subject?
Checkpoint blockade immunotherapy has limited efficacy in pancreatic cancer patients.
The pro-inflammatory cytokine, IL-6 is highly elevated in pancreatic cancer patients and is a prognostic factor for outcome.
What are the new findings?
Pancreatic stellate cells are a major source of IL-6 in the pancreatic tumor microenvironment.
Blockade of IL-6 and PD-L1 limits tumor progression and enhances overall survival in aggressive murine models of pancreatic cancer.
Combination therapy increased circulating cells with Th1 phenotypic characteristics and intratumoral effector T cells.
Combined blockade of IL-6 and PD-L1 immunotherapy elicits efficacy in a CD8+ T cell-dependent manner.
How might it impact on clinical practice in the foreseeable future?
These preclinical results elucidate a novel combination therapy that could be translated to the clinic.
Our results suggest that targeting IL-6 could improve pancreatic cancer patient responses to checkpoint blockade immunotherapy.
Acknowledgments
We thank the OSU Comprehensive Cancer Center Biostatistics, Genomics, Target Validation, Analytical Cytometry, and Comparative Pathology & Mouse Phenotyping Shared Resources supported in part by NCI grant P30 CA016058.
Grant Funding: NIH Grants 5T32CA009338-34 (Mace), 1R21AI124687-01 (Lesinski), 1R01CA208253-01 (Lesinski), P30CA016058-36 (Caligiuri), American-Italian Cancer Foundation Pancreatic Cancer Initiative grant (Bloomston), and Lustgarten Foundation grant (Zimmers). The project described was supported by Award Number UL1RR025755 from the National Center for Research Resources, funded by the Office of the Director, National Institutes of Health (OD) and supported by the NIH Roadmap for Medical Research. Supported by the William Hall Fund for pancreatic and liver research. The project described by Award Number Grant KL2TR001068 from the National Center for Advancing Translational Sciences. This work was also supported by the Pelotonia Fellowship Program. Any opinions, findings and conclusions expressed are those of the authors and do not necessarily reflect those of the Pelotonia Fellowship Program. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Advancing Translational Sciences, the National Center for Research Resources, or the National Institutes of Health.
Abbreviations
- PDAC
pancreatic ductal adenocarcinoma
- IL-6
interleukin-6
- PD-L1
Programmed death-1-ligand 1
- PSC
pancreatic stellate cell
- GEMM
genetically engineered mouse model
- JAK
Janus kinases
- STAT
signal transducer and activator of transcription
- PanIN
pancreatic intraepithelial lesions
- Treg
T regulatory
- MDSC
myeloid derived suppressor cell
References
- 1.Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–21. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 2.Vincent A, Herman J, Schulick R, et al. Pancreatic cancer. Lancet. 2011;378:607–20. doi: 10.1016/S0140-6736(10)62307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Hajeili M, Azmi AS, Choi M. Nab-paclitaxel: potential for the treatment of advanced pancreatic cancer. Onco Targets Ther. 2014;7:187–92. doi: 10.2147/OTT.S40705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y, Yan W, Collins A, et al. Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance. Cancer Res. 2013;73:6359–74. doi: 10.1158/0008-5472.CAN-13-1558-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lesina M, Kurkowski MU, Ludes K, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19:456–69. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 6.Rose-John S, Scheller J, Elson G, et al. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: role in inflammation and cancer. J Leukoc Biol. 2006;80:227–36. doi: 10.1189/jlb.1105674. [DOI] [PubMed] [Google Scholar]
- 7.Fisher DT, Appenheimer MM, Evans SS. The two faces of IL-6 in the tumor microenvironment. Semin Immunol. 2014;26:38–47. doi: 10.1016/j.smim.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scheller J, Chalaris A, Schmidt-Arras D, et al. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011;1813:878–88. doi: 10.1016/j.bbamcr.2011.01.034. [DOI] [PubMed] [Google Scholar]
- 9.Corcoran RB, Contino G, Deshpande V, et al. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011;71:5020–9. doi: 10.1158/0008-5472.CAN-11-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scholz A, Heinze S, Detjen KM, et al. Activated signal transducer and activator of transcription 3 (STAT3) supports the malignant phenotype of human pancreatic cancer. Gastroenterology. 2003;125:891–905. doi: 10.1016/s0016-5085(03)01064-3. [DOI] [PubMed] [Google Scholar]
- 11.Gabitass RF, Annels NE, Stocken DD, et al. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. 2011;60:1419–30. doi: 10.1007/s00262-011-1028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Markowitz J, Brooks TR, Duggan MC, et al. Patients with pancreatic adenocarcinoma exhibit elevated levels of myeloid-derived suppressor cells upon progression of disease. Cancer Immunol Immunother. 2015;64:149–59. doi: 10.1007/s00262-014-1618-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mundy-Bosse BL, Young GS, Bauer T, et al. Distinct myeloid suppressor cell subsets correlate with plasma IL-6 and IL-10 and reduced interferon-alpha signaling in CD4(+) T cells from patients with GI malignancy. Cancer Immunol Immunother. 2011;60:1269–79. doi: 10.1007/s00262-011-1029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mace TA, Bloomston M, Lesinski GB. Pancreatic cancer-associated stellate cells: A viable target for reducing immunosuppression in the tumor microenvironment. Oncoimmunology. 2013;2:e24891. doi: 10.4161/onci.24891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goumas FA, Holmer R, Egberts JH, et al. Inhibition of IL-6 signaling significantly reduces primary tumor growth and recurrencies in orthotopic xenograft models of pancreatic cancer. Int J Cancer. 2015 doi: 10.1002/ijc.29445. [DOI] [PubMed] [Google Scholar]
- 16.Farren MR, Mace T, Geyer S, et al. Systemic immune activity predicts overall survival in treatment naive patients with metastatic pancreatic cancer. Clin Cancer Res. 2015 doi: 10.1158/1078-0432.CCR-15-1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang X, Zhou J, Giobbie-Hurder A, et al. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res. 2013;19:598–609. doi: 10.1158/1078-0432.CCR-12-2731. [DOI] [PubMed] [Google Scholar]
- 18.Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015;125:3384–91. doi: 10.1172/JCI80011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–34. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldberg MV, Maris CH, Hipkiss EL, et al. Role of PD–1 and its ligand, B7–H1, in early fate decisions of CD8 T cells. Blood. 2007;110:186–92. doi: 10.1182/blood-2006-12-062422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24:207–12. doi: 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duluc D, Delneste Y, Tan F, et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood. 2007;110:4319–30. doi: 10.1182/blood-2007-02-072587. [DOI] [PubMed] [Google Scholar]
- 23.Marigo I, Bosio E, Solito S, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. 2010;32:790–802. doi: 10.1016/j.immuni.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 24.Tsukamoto H, Nishikata R, Senju S, et al. Myeloid-derived suppressor cells attenuate TH1 development through IL-6 production to promote tumor progression. Cancer Immunol Res. 2013;1:64–76. doi: 10.1158/2326-6066.CIR-13-0030. [DOI] [PubMed] [Google Scholar]
- 25.Ma Y, Hwang RF, Logsdon CD, et al. Dynamic mast cell-stromal cell interactions promote growth of pancreatic cancer. Cancer Res. 2013;73:3927–37. doi: 10.1158/0008-5472.CAN-12-4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ludwig T, Fisher P, Murty V, et al. Development of mammary adenocarcinomas by tissue-specific knockout of Brca2 in mice. Oncogene. 2001;20:3937–48. doi: 10.1038/sj.onc.1204512. [DOI] [PubMed] [Google Scholar]
- 27.Guenterberg KD, Lesinski GB, Mundy-Bosse BL, et al. Enhanced anti-tumor activity of interferon-alpha in SOCS1-deficient mice is mediated by CD4(+) and CD8(+) T cells. Cancer Immunol Immunother. 2011;60:1281–8. doi: 10.1007/s00262-011-1034-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sartor MA, Tomlinson CR, Wesselkamper SC, et al. Intensity-based hierarchical Bayes method improves testing for differentially expressed genes in microarray experiments. BMC Bioinformatics. 2006;7:538. doi: 10.1186/1471-2105-7-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gordon A, Glazko G, Qiu X, et al. Control of the Mean Number of False Discoveries, Bonferroni and Stability of Multiple Testing. Annals of Applied Statistics. 2007;1:179–190. [Google Scholar]
- 30.Slinker BK. The statistics of synergism. J Mol Cell Cardiol. 1998;30:723–31. doi: 10.1006/jmcc.1998.0655. [DOI] [PubMed] [Google Scholar]
- 31.Boj SF, Hwang CI, Baker LA, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324–38. doi: 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collignon A, Perles-Barbacaru AT, Robert S, et al. A pancreatic tumor-specific biomarker characterized in humans and mice as an immunogenic onco-glycoprotein is efficient in dendritic cell vaccination. Oncotarget. 2015;6:23462–79. doi: 10.18632/oncotarget.4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pilon-Thomas S, Nelson N, Vohra N, et al. Murine pancreatic adenocarcinoma dampens SHIP-1 expression and alters MDSC homeostasis and function. PLoS One. 2011;6:e27729. doi: 10.1371/journal.pone.0027729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki K, Ogura M, Abe Y, et al. Phase 1 study in Japan of siltuximab, an anti-IL-6 monoclonal antibody, in relapsed/refractory multiple myeloma. Int J Hematol. 2015;101:286–94. doi: 10.1007/s12185-015-1743-y. [DOI] [PubMed] [Google Scholar]
- 35.San-Miguel J, Blade J, Shpilberg O, et al. Phase 2 randomized study of bortezomib-melphalan-prednisone with or without siltuximab (anti-IL-6) in multiple myeloma. Blood. 2014;123:4136–42. doi: 10.1182/blood-2013-12-546374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hunsucker SA, Magarotto V, Kuhn DJ, et al. Blockade of interleukin-6 signalling with siltuximab enhances melphalan cytotoxicity in preclinical models of multiple myeloma. Br J Haematol. 2011;152:579–92. doi: 10.1111/j.1365-2141.2010.08533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dijkgraaf EM, Santegoets SJ, Reyners AK, et al. A phase I trial combining carboplatin/doxorubicin with tocilizumab, an anti-IL-6R monoclonal antibody, and interferon-alpha2b in patients with recurrent epithelial ovarian cancer. Ann Oncol. 2015;26:2141–9. doi: 10.1093/annonc/mdv309. [DOI] [PubMed] [Google Scholar]
- 38.Bayne LJ, Beatty GL, Jhala N, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21:822–35. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neesse A, Michl P, Frese KK, et al. Stromal biology and therapy in pancreatic cancer. Gut. 2011;60:861–8. doi: 10.1136/gut.2010.226092. [DOI] [PubMed] [Google Scholar]
- 40.Fearon DT. The carcinoma-associated fibroblast expressing fibroblast activation protein and escape from immune surveillance. Cancer Immunol Res. 2014;2:187–93. doi: 10.1158/2326-6066.CIR-14-0002. [DOI] [PubMed] [Google Scholar]
- 41.Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. 2013;110:20212–7. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lo A, Wang LC, Scholler J, et al. Tumor-Promoting Desmoplasia Is Disrupted by Depleting FAP-Expressing Stromal Cells. Cancer Res. 2015;75:2800–10. doi: 10.1158/0008-5472.CAN-14-3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rhim AD, Oberstein PE, Thomas DH, et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2014 doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee JJ, Perera RM, Wang H, et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci USA. 2014;111:E3091–100. doi: 10.1073/pnas.1411679111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ozdemir BC, Pentcheva-Hoang T, Carstens JL, et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell. 2014 doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dodge IL, Carr MW, Cernadas M, et al. IL-6 production by pulmonary dendritic cells impedes Th1 immune responses. J Immunol. 2003;170:4457–64. doi: 10.4049/jimmunol.170.9.4457. [DOI] [PubMed] [Google Scholar]
- 47.Wang Y, van Boxel-Dezaire AH, Cheon H, et al. STAT3 activation in response to IL-6 is prolonged by the binding of IL-6 receptor to EGF receptor. Proc Natl Acad Sci USA. 2013;110:16975–80. doi: 10.1073/pnas.1315862110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vassilakopoulou M, Avgeris M, Velcheti V, et al. Evaluation of PD-L1 Expression and Associated Tumor-Infiltrating Lymphocytes in Laryngeal Squamous Cell Carcinoma. Clin Cancer Res. 2015 doi: 10.1158/1078-0432.CCR-15-1543. [DOI] [PubMed] [Google Scholar]
- 49.Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–7. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.