

Abstract
Oxidative cyclizations are important transformations that occur widely during natural product biosynthesis. The transformations from acyclic precursors to cyclized products can afford morphed scaffolds, structural rigidity and biological activities. Some of the most dramatic structural alterations in natural product biosynthesis occur through oxidative cyclization. In this review, we examine the different strategies used by Nature to create new intra-(inter-)-molecular bonds via redox chemistry. The review will cover both oxidation- and reduction-enabled cyclization mechanisms, with an emphasis on the former. Radical cyclizations catalyzed by P450, nonheme iron, α-KG dependent oxygenases and radical SAM enzymes are discussed to illustrate the use of molecular oxygen and S-adenosylmethionine to forge new bonds at unactivated sites via one-electron manifolds. Nonradical cyclizations catalyzed by flavin-dependent monooxygenases and NAD(P)H-dependent reductases are covered to show the use of two-electron manifolds in initiating cyclization reactions. The oxidative installation of epoxides and halogens into acyclic scaffolds to drive subsequent cyclizations are separately discussed as examples of “disappearing” reactive handles. Lastly, oxidative rearrangement of rings systems, including contractions and expansions will be covered.
Graphical Abstract
1. INTRODUCTION
The importance of natural products to human health is unquestioned. Many of the natural products and their semisynthetic derivatives have served as front line therapeutics to treat a wide variety of diseases, ranging from microbial infections, cancer, cardiovascular conditions, and as immunosuppressants.1–3 The vast collection of bioactivities displayed by natural products is a direct result of the immense structural diversity introduced during the biosynthetic assembly.4 Several major families of natural products have been classified during the decades of isolation and structural characterization since the golden age of antibiotics. Based on biosynthetic origin and fundamental building blocks, the major families of natural products include polyketides (PKs), nonribosomal peptides (NRPs), ribosomally synthesized and post-translationally modified peptides (RiPPs), isoprenoids, alkaloids including phenylpropanoids and indole alkaloids, aminoglycosides, nucleosides, etc.5
For the major families of natural products including PKs, NRPs, RiPPs, terpenes, etc., nature builds structural complexity in two major phases. The first phase involves the polymerization of building blocks, such as acetate for PKs, amino acids for NRPs and RiPPs, and isoprene units for terpenes. The polymerization process builds the carbon backbone of the molecules, and in most cases, cyclizes the molecule in a regioselective fashion to yield a basic scaffold of the final natural product. This scaffold, which can be called the aglycone in some cases, is then subjected to a barrage of post-assembly modifications that serve a variety of purposes, such as rigidifying the molecule and fixing the three-dimensional conformation; introducing polar groups to increase the water solubility; reveal structural motifs and reactive sites as warheads for target inhibition; and appending oligosaccharides that bind to specific DNA sequences. It is during the post-assembly steps that the natural products can undergo significant structural morphing to result in the final bioactive compounds. Obtaining a complete understanding of these enzymatic transformations is therefore important to not only allow chemists to appreciate Nature’s chemical strategy to forge complex small molecules, but also increases the toolbox of biocatalysts that can used to generate structural diversity and biological activities into both biological and synthetic molecules.
The types of enzyme modifications that will be reviewed here are cyclization reactions through redox alteration of acyclic substrates. We define oxidative (or reductive) cyclization as 1) formation of new rings as a result of oxidative modification, such as in the case of P450-catalyzed formation of phenyl ether rings during vancomycin biosynthesis; 2) a tandem redox/cyclization combination, in which a discrete redox modification occurs to set up the subsequent enzyme-catalyzed/spontaneous step that forges a new ring system. This is exemplified in the epoxide-mediated cyclization of terpenes and polyethers, as well as the cryptic oxidative halogenation steps that lead to cyclopropanation; and 3) oxidative morphing of existing ring systems to either contract, expand or rearrange to a new ring system. All cyclization reactions covered in this review must involve a net redox state change in the starting substrates, i.e. addition or removal of electrons to drive the formation of new intramolecular covalent bonds, although in many cases the overall reaction can be redox neutral. Nonredox-mediated cyclization mechanisms are not covered except a brief discussion of [4+2] cycloadditions that are setup by prior redox steps. Nonredox cyclizations include acyltransferase/thioesterase catalyzed macrocyclization of PKs and NRPs,6 intramolecular Aldol and Claisen condensation reactions in aromatic PK biosynthesis,7 pyran synthase in many PK pathways,8 etc.
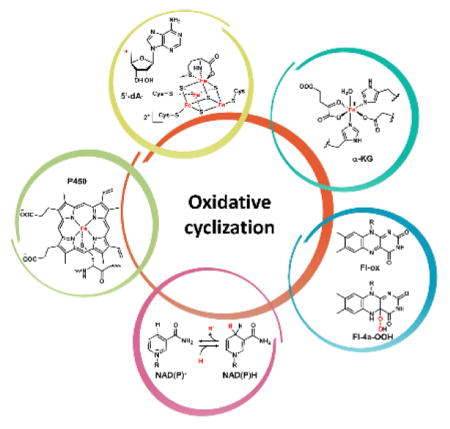
Oxidative cyclization can introduce some of the most dramatic structural changes in a natural product biosynthetic pathway. The use of high-valent iron-containing enzymes such as P450 and nonheme iron α-ketoglurarate (α-KG, also known as 2-oxoglutarate) dependent oxygenases can result in the formation of radicals through homolysis of different types of bonds in a molecules, including unactivated C-H bonds, phenolic O-H bonds, indole N-H bonds and thiol S-H bonds.9,10 The resulting radical can then be the starting point to regioselective forging of new bonds that are otherwise difficult to construct. Section 2 will cover this type of oxidative cyclization sorted by both enzymes and the types of bonds that are formed. In addition to the one-electron manifold oxidation of substrates, Nature also uses two-electron redox manifolds facilitated by the flavin and β-nicotinamide adenine dinucleotide (phosphate), reduced form (NAD(P)H) cofactors. While enzymes use these cofactors to catalyze oxidation/reduction of more reactive sites in the molecules, the flexibility of the flavin cofactor, serving either as an electron sink in desaturation reactions or as an electrophilic [OH+] equivalent, propels this family of enzymes into central importance during oxidative cyclizations.11 Section 3 will discuss prominent examples of nonradical-mediated oxidative cyclizations, including both flavin and NAD(P)H mediated mechanisms. Several examples of [4+2] cycloaddition will also be discussed, although we use these to highlight the importance of precise redox tuning of the acyclic substrates that set up such dramatic transformations. Section 4 and 5 will discuss the use of reactive handles installed by oxidative enzymes to initiate a cyclization sequence, ultimately forging new ring systems while masking the use of the handles in the final molecules. Section 4 will discuss the use of “disappearing” epoxides in promoting ring closing steps in a variety of natural product pathways. Regioselective epoxidation of a double bond in the substrate can trigger a cascade of cyclization steps that rigidify an otherwise linear precursor, as in the example of lanosterol biosynthesis. Section 5 will discuss the oxidative halogenation of natural products as a strategy to initiate cyclization. Both the vanadium haloperoxidases (VHPOs) and α-KG dependent families of oxidative halogenases will be discussed. In Section 6, oxidative ring rearrangements catalyze by all major families of oxygenases will be discussed. Finally in Section 7, a unifying discussion of the examples presented here will conclude the review.
Types of Redox Enzyme in Nature
Before we discuss the specific examples of oxidative cyclizations, a general introduction to the types of enzymes that participated in these reaction will be briefly summarized here. The mechanisms of the reactions have been well-worked out and have been reviewed in many excellent articles and books that are cited throughout.
Iron and Heme-Dependent Cytochrome P450 Monooxygenases
P450s are ubiquitous oxidative enzymes that span across all organisms, from bacteria to humans. P450s are heme-binding enzymes and share a highly conserved protein fold.12 These oxygenases form iron-porphyrin complexes to oxidize a vast multitude of different substrates using molecular oxygen. Despite their substantial range of substrate diversity, P450 oxidations can be highly stereoselective and regioselective. P450 enzymes in natural product biosynthesis use a single-electron manifold to produce radical intermediates via a high-valent oxoiron cationic radical (or FeIV=O, ferryl) (Scheme 1).13,14 This Compound I can readily abstract hydrogen from C-H, N-H, O-H and S-H bonds to yield a reactive radical species.10,12 These reactive radical intermediates can lead to a variety of modifications including hydroxyl rebound, epoxide formation, dehydrogenation, radical addition, diradical combination, etc.10,12 In typical hydroxylation mechanisms, rebound of the [OH•] from the resulting FeIV-OH leads to insertion of the oxygen atom which is derived from molecular oxygen. Alternatively, the radical species can perform intramolecular addition (for example, with an olefin) or recombination with another radical to forge new bonds in the product scaffold. In this case, the net reaction is the four-electron reduction of molecular oxygen without insertion of any oxygen atom into the final product. The use of these powerful oxidative enzymes in natural product cyclization will be discussed in Section 2.2.
Scheme 1.

Catalytic Cycle of Cytochrome P450
Nonheme Iron Dependent Oxygenases
In addition to heme-dependent oxygenases, nonheme iron-dependent oxygenases are also widely found in biosynthetic pathways.15,16 In particular, mononuclear nonheme Fe(II)- α-KG-dependent oxygenases comprise a large family of oxidative enzymes that are widely distributed in Nature.17,18 Enzymes in this family utilizes an FeIV-oxoiron intermediate to initiate otherwise challenging oxidative transformations at unactivated sites, such as hydroxylation, desaturation and halogenation.19 Mechanistic and structural studies of this family of enzymes have clarified the catalytic cycle.9,20 As shown in Scheme 2 using a classical hydroxylation reaction as an example, the resting FeII is coordinated by two His residues and the carboxylate from an Asp residue to form the 2-His-1-carboxylate facial triad. This can be coordinated to three water molecules until binding of α-KG to the FeII center displaces two of the three metal-bound water molecules. Binding of substrate (R-H) to the enzyme active site displaces the remaining water molecule and vacates a site for binding to the triplet O2 molecule to generate the FeIII-superoxo intermediate. The distal oxygen atom of the FeIII-superoxo species can attack C2 of α-KG to yield a peroxohemiketal bicyclic intermediate, which collapses via oxidative decarboxylation to release CO2. This step also sets up a high valence FeIV=O (ferryl) intermediate and a bound succinate. It is this ferryl species (functionally analogous to compound I in the P450 cycle) that can abstract a hydrogen atom from the substrate (R-H) to reduce the iron to the FeIII-OH state with concomitant formation of the substrate radical (R•). To complete the catalytic cycle, hydroxyl radical rebound onto the substrate radical yields the hydroxylated product (R-OH), returning the iron back to the FeII state with product and succinate release. Examples of using this family of enzymes in natural product cyclization will be covered in Section 2.3. The use of nonheme, noncofactor-dependent oxygeneases to perform oxidative cyclization involving unactivated C-H bonds will be covered in Section 2.4. One notable example is the Rieske oxygenase discussed in section 2.4.3. Similar to the α-KG-dependent oxygenases, the iron within the Rieske nonheme iron-dependent oxygenases is also coordinated by the 2-His-1-carboxylate facial triad.21 The key difference is that duing the catalytic process, instead of using α-KG directly as the reductant, the Rieske oxygenases use a [2Fe-2S] cluster to deliver the necessary electrons from an external reductant.21
Scheme 2.

Catalytic Cycle of Nonheme Iron α-KG-Dependent Oxygenase
Radical SAM Superfamily
The radical S -adenosylmethionine (SAM) enzymes is a relatively new enzyme superfamily discovered to catalyze radical reactions such as H-atom abstraction from an unactivated C–H bond, mostly under anaerobic conditions in the bacterial world.22 This family is rarely found in eukaryotic organisms such as fungi and plants, but are vastly powerful biocatalysts in both primary and secondary metabolisms of bacteria.22 As shown in Scheme 3, these enzymes use a [4Fe-4S] cluster to transfer an electron from an external source (such as flavodoxin shown) to SAM, which is homolytically cleaved to methionine and the reactive 5′-deoxyadenosyl radical intermediate (5′dA•).23,24 This 5′dA• radical is able to abstract a proton and an electron from unactivated substrate (R-H) to form 5′dA and generate a radical (R•) that can participate in downstream oxidation and cyclization reactions. This superfamily of enzymes, which has over one hundred thousand homologs in the database of which mostly of unknown function, greatly expands Nature’s ability to use Fe-S clusters in oxidative catalysis beyond the textbook examples of electron transport.25 Some examples will be covered in Section 2.4.
Scheme 3.

Catalytic Cycle of Radical SAM Enzyme
Copper-Dependent Tyrosinase
Copper is a relatively rarely used metal cofactor in enzymes catalysis. Three notable examples that have relevance to metabolism are cytochrome c oxidase,26 laccase,27 and tyrosinase28. The most well-studied example of tyrosinase is the hydroxylation of tyrosine to yield L-3,4-dihydroxyphenylalanine (DOPA) as shown in Scheme 4.29–31 In the active site of tyrosinase, six histidine residues coordinate to a pair of copper ions (CuII) and one oxygen molecule to give the oxy starting complex. The substrate monophenol (M) binds to one of the copper metals and forms the oxy-M intermediate. This weakens the O-O bond, resulting in cleavage and rearrangement of original trigonal bipyramidal active site and forming the diphenolate (D) intermediate (met-D). The product is then oxidized to the quinone through the transfer of two electrons to the coppers, with the active site in the reduced di-CuI form (oxy-red) to be reoxidized by molecular oxygen for a second round of catalysis.
Scheme 4.
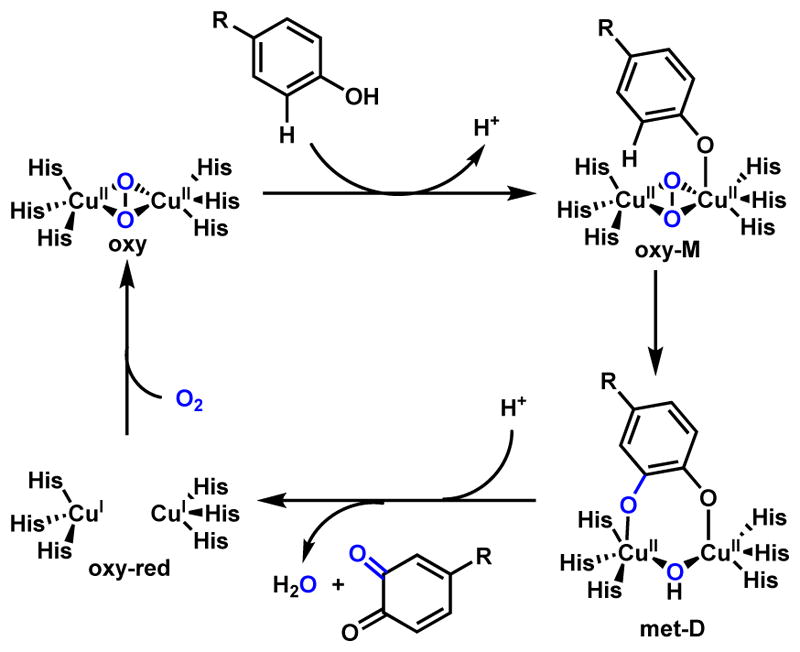
Catalytic Cycle of Copper-Dependent Tyrosinase
Flavin-Dependent Monooxygenase
Flavin-dependent monooxygenases (FMOs) are widespread enzymes that catalyze a large variety of substrate oxidations such as dehydrogenation, hydroxylations, epoxidations, Baeyer-Villiger oxidations, and sulfoxidations.32,33 FMOs use a flavin cofactor such as flavin adenine dinucleotide (FAD) or flavin mononucleotide (FMN), to generate reactive peroxyl species that serve as nucleophiles (peroxyflavin, Fl-4a-OO−) or electrophiles (hydroperoxyflavin, Fl-4a-OOH) (Scheme 5A).11 After each round of catalysis, the flavin cofactor can be reduced in the presence of NAD(P)H to repeat the catalytic cycle.
Scheme 5.

Catalytic Cycle of Flavin-Dependent Monooxygenase
The oxidized flavin can also serve as electron sink in oxidases that catalyze dehydrogenation reactions such as the berberine bridge enzyme family (Scheme 5B). Here the flavin is often covalently attached to the active site through histidine and cysteine residues.34,35 During the net two-electron reduction of molecular oxygen, a corresponding oxidation of substrate takes place to generate a degree of unsaturation that result in an electrophilic carbon (C=N, C=O, etc). This carbon is then subject to intramolecular attack by a nucleophile to forge a new bond and a cyclized structure as will be shown in Section 3. The reduced flavin is oxidized back to the Fl-ox form with release of hydrogen peroxide. As this species can be reactive and toxic to the cell, an accompanying catalase is often found in the gene cluster for detoxification.
NAD(P)H Dependent Reductases/Dehydrogenases
NAD(P)H-dependent enzymes catalyze reversible redox reactions including reduction and dehydrogenation as shown in Scheme 6.36,37 The reduced form of the cofactor NAD(P)H is employed in substrate reduction, while the oxidized form NAD(P)+ are used in oxidative dehydrogenation. During substrate reduction such as ketone/aldehyde to alcohols, NAD(P)H is a hydride-donating cofactor. Delivery of a hydride from the dihydropyridine ring to substrate in a stereospecific manner is coupled with oxidation of NAD(P)H to NAD(P)+. In the reverse reaction of dehydrogenation, such as from alcohols to ketone/aldehydes, two hydrogen atoms are removed from the substrate with one of them transferred as a hydride to reduce NAD(P)+ to NAD(P)H while the other as a proton to captures by the aqueous solution. While NAD(P)H is typically a non-covalent cofactor in these enzymes, Erb and coworkers observed a covalent ene intermediate between NADPH and α,β-unsaturated carbonyl substrate during catalysis of crotonyl-CoA carboxylase/reductase.38,39 One notable use of NAD(P)H as a reducing cofactor in oxidative cyclization is in the reductive release of PK and NRP products that are bound as thioesters to the thiolation domains.40 Releasing the products as aldehydes can trigger intramolecular cyclization reactions that are widely found in many natural product biosynthetic pathways (Section 3.2).
Scheme 6.
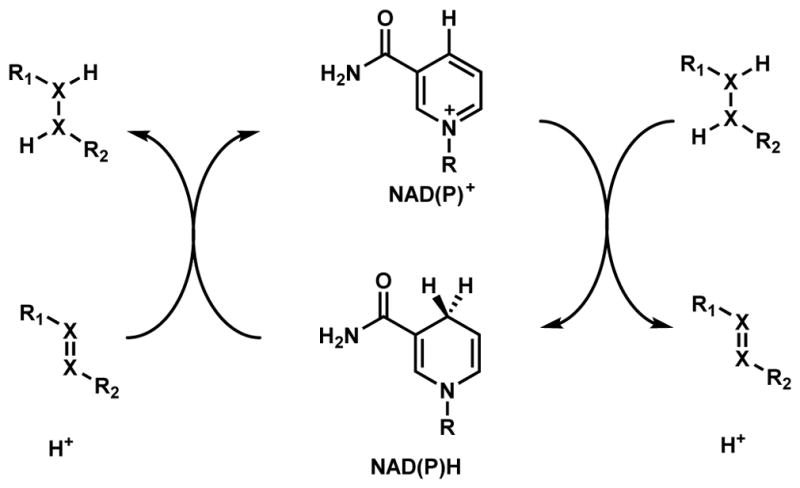
NAD(P)H-Dependent Reductases and Dehydrogenases
2. RADICAL CYCLIZATION MECHANISMS
2.1 Introduction
This section will describe examples of oxidative cyclization in which radical intermediates are formed through one electron oxidation of carbon or heteroatoms. The main enzyme families involved are those use a high-valent ferryl oxoiron reactive species, including P450s and nonheme iron α-KG-dependent oxygenases. Other enzyme families such as copper-dependent tyrosinases and radical SAM-dependent enzymes are also used for this purpose. Structural morphing catalyzed by these enzymes are typically the most dramatic and chemically challenging. Several recurring cases of radical mechanisms are summarized in Scheme 7. In Scheme 7A, an otherwise unactivated C-H bond is subjected to homolytic cleavage to yield a carbon radical, which can be further oxidized into carbocation or alcohol through [OH•] rebound, and subject to intramolecular nucleophilic attack. This mechanism is observed in the synthesis of nitrogen heterocycles, as in the formation of methlyproline from either L-leucine or L-isoleucine (Scheme 46). In Scheme 7B, an X-H bond is oxidized to the corresponding X•, which can intramolecularly add to an electron-rich double bond to form a cyclized intermediate and form a new C-radical. This is followed by an additional one electron transfer to the iron center to yield the doubly oxidized product. There are increasing evidences to support this mechanism in oxidative cyclization, such as during spirocycle formation during griseofulvin biosynthesis (Scheme 25). In Scheme 7C, two successive oxidations at two different sites in the same molecules result in formation of diradicals that can be combined to form the cyclized product. For many reactions listed in this section, both mechanisms B and C can be proposed, such as for the two C-C bond forming steps during rebeccamycin synthesis (Section 2.2.2). Differentiation between the two mechanisms has come from crystal structures, computational analysis or both. The remaining section will be grouped by enzyme families that catalyze these reactions.
Scheme 7.

Models of Radical Cyclization in Natural Product Biosynthesis
Scheme 46.

Mechanisms of Methylproline Formation in NRP Natural Products
Scheme 25.

Possible Mechanisms of GsfF catalyzed C-O Coupling in Griseofulvin Biosynthesis
2.2. Cyclization Catalyzed by Heme-dependent Enzymes
This section will discuss oxidative cyclization catalyzed by heme-dependent enzymes. In each example, the cyclization process starts with abstraction of one hydrogen and generation of a radical intermediate. In most examples, the oxygenation half of the reaction is not completed as in the P450 catalytic cycle (no water rebound), and instead the radical is combined internally with an electron rich site (Scheme 7B), or with another radical to forge the new cyclizing bond (Scheme 7C). Mechanisms can be proposed using either scheme for many of the cyclizing reactions. We will first discuss the P450 catalyzed cyclization during the biosynthesis of vancomycin and rebeccamycin families, each featuring multiple C-C and C-O coupling steps. This will be followed by examples of cyclization categorized by the different types of atoms coupled to carbon.
2.2.1. P450-Catalyzed Phenyl Crosslinking in Glycopeptide Biosynthesis
The members of glycopeptide family compounds are a class of antibiotics of microbial origin, exemplified by the anti-infective antibiotics vancomycin (1) and teicoplanin (2) (Scheme 8A).41,42 Compounds in this family are considered to be the last line of defense against antibiotic resistant bacteria, especially methicillin resistant Staphylococcus aureus.41 The heptapeptide backbones for both are derived from NRPS assembly lines, incorporating a number of unnatural amino acids that have dedicated biosynthetic enzymes in the gene clusters.41,42 Using 2 as an example, seven nonproteinogenic aromatic amino acids are introduced, including three 4-OH-phenylglycine (rings B, E and G), two 3-chloro-tyrosine (rings A and C), and two 3,5-dihydroxyphenylglycin (rings D and F). Extensive phenyl-coupling reactions take place after completion of linear peptide synthesis to effectively crosslink the side chains and introduce architectural rigidity that is crucial for the biological properties. The five phenol-couplings are catalyzed by five dedicated P450 monooxygenases. In a related compound complestatin (3), there is also a C-C coupling between the phenyl ring of 4-OH-phenylglycine and the phenyl ring of the tryptophan.43,44 In the biosynthesis of arylomycin A2 (4), a lipopeptide anti-infective agent, similar P450 catalyzed coupling reaction was proposed for C-C coupling between two phenyl side chains.45–47
Scheme 8.

P450-Catalyzed Phenol Coupling Reactions in Glycopeptide Biosynthesis
Two different types of P450-catalyzed phenol coupling reactions take place between the different phenol rings: ortho-ortho C-C coupling and ortho C-O coupling. Both coupling regioselectivity will be seen in many other examples in this section of the review and is therefore useful to examine the possible mechanisms (Scheme 9). Based on structural and mechanistic studies of these P450s,48–55 diradical coupling mechanisms were proposed for these reactions. In a single round of iron reduction from compound I (FeIV=O) to FeIII-OH2, two sequential hydrogen abstraction steps from two phenolic hydroxyl groups of 11 yields the diradical 12. In the C-O coupling case, delocalization of one phenoxyl radical to the ortho carbon to give 13 is followed by diradical combination to yield the new C-O bond in 14 which can rearomatize into the diphenyl 15 as seen with three of the four coupled rings. If resonance delocalization of both radicals in 12 takes place to the ortho position as in 16a, diradical combination forges the new C-C bond in 17 and can similarly rearomatize to 18 as in the case of D and E rings. Single-electron transfer and nucleophilic addition via 16b can also be proposed for the C-C bond forming step. Current state of research favors the diradical mechanism over the radical addition mechanism, although more data are needed to fully distinguish the two. The radical addition mechanism will be presented for other coupling pathways in this section, some of which are favored over diradical coupling.
Scheme 9.

Mechanistic Proposal of C-C and C-O Phenol Coupling Catalyzed by P450
The order of these P450 reactions during maturation of 2 is shown in Scheme 8B. All the coupling steps take place after the heptapeptide is assembled and while still attached to the thiolation (T) domain as a thioester 5. The new C-C and C-O bonds are introduced in a stepwise fashion: i) OxyB catalyzed C-O coupling of A/B phenyl rings to yield 6; OxyE catalyzed C-O of the F/G phenyl rings to yield 7; iii) OxyA catalyzed C-O coupling of B/C phenyl rings to yield 8; and iv) OxyC catalyzed C-C coupling of D/E phenyl rings to yield 9. The rigidified aglycone 10 is then released from the NRPS assembly through hydrolysis by the terminal thioesterase domain, which can be further glycosylated to yield the mature glycopeptide 2. The crystal structures of all four P450s in the pathway have been solved, giving insights into the mechanistic proposal in Scheme 9 and their differences in substrate specificity.48–50,55 In addition, the structural basis of the interaction between the P450s and the NRPS enzyme was recently elucidated by Cryle and coworkers.56 They showed that there is an X domain present in the last module of the teicoplanin NRPSs. While the X domain is structurally similar to the Condensation (C) domains used by NRPS for peptide bond formation, it is catalytically inactive due to mutations in the active site. Extensive protein-protein interactions are present between the X domain and the suite of P450s as shown by crystal structure and biophysical characterization.56,57 The working model is that together with the adjacent T domain, the X domain is crucial in the recruitment of P450 enzymes to the NRPS for on-assembly line oxidative cyclization.
2.2.2. Oxidative Coupling in Indolocarbazole Biosynthesis
Rebeccamycin (19), together with staurosporine (20), and K-252a (21) (Scheme 10A), are examples of the indolocarbazole superfamily of natural alkaloids produced by actinomycete strains.58 Members of this family have been found to exhibit a broad spectrum of antitumor activities.58 For example, 20 and 21 are potent inhibitors of protein kinase C, and 19 is an inhibitor of topoisomerase I.59–61 Biosynthetic studies revealed that the aglycones of these compounds are derived from two molecules of L-tryptophan via a series of oxidative coupling transformations.62–68 The pathway to the aglycones follow a unified route. Variations in biosynthetic routes of this family include the chlorination step in the pathway of 19 and the different oxidation state of the pyrrole-derived five-membered ring.
Scheme 10.

Oxidative Coupling in Indolocarbazole Biosynthesis
During the biosynthesis of the aglycone, the first oxidative cyclization is the coupling of two L-tryptophan derived indole-3-pyruvate imine (IPAI, 22) to form the key biosynthetic intermediate chromopyrrolic acid (CPA, 27), catalyzed by the heme-dependent oxidase RebD and its homologs (Scheme 10B).62,66 The cytochrome b protein RebD first generates two IPAI radicals 23 and catalyzes the formation of the new C-C bond to yield the adduct 24. Tautomerization of one of the imines to an enamine forms a free amine group in 25, which can attack the remaining imine group to form the dihydropyrrole 26. Finally, the pyrrole ring in 27 is formed by removing the ammonia. Recent mechanistic studies revealed that during the C-C oxidative coupling step, RebD can use both peroxidase and peroxygenase chemistry as shown in Scheme 10C.69
Although current studies of RebD provide evidence for the diradical coupling mechanism to form 27, the radical addition route is still not ruled out. As shown in Scheme 11 the first step in this mechanism is the tautomerization of the first molecule of 22 to yield a free amine 28. This can serve as the nucleophile to attack the imine group of a second molecule of 22 to form an intermolecular C-N bond and 29. This intermediate can lose ammonia to form the imine 30. RebD can then catalyze the hydrogen abstraction at the allylic position to generate the C-radical 31, which can lead to radical addition to the C=C double bond in the left half of the molecule to form the new C-C bond in 32 that can also form pyrrole radical 33. Abstraction of a second hydrogen following radical migration yields 34 which can followed by tautomerization to yield the pyrrole ring and complete the biosynthesis of 27.
Scheme 11.

Mechanistic Proposal for Chromopyrrolic Acid Formation
The next biosynthetic step in the biosynthetic pathway is the oxidative C2-C2 coupling of the two indole rings in 27 to form the indolocarbazole aglycone 41. This reaction is catalyzed by a second P450, StaP in the staurosporine pathway or RebP in the rebeccamycin pathway which works on the 7,7-dichloro-CPA.68 Recent X-ray crystal structural studies suggested a catalytic mechanism for StaP as shown in Scheme 12.65 The first step is the one electron oxidation of the one indole NH by StaP to afford 35 or 36, which can undergo hydrogen abstraction to form the N-radical 37. From here both diradical combination and radical addition mechanisms can be proposed. In the diradical route a, a second hydrogen abstraction of the other indole yields the second nitrogen radical 38. Following migration of both radicals to the C2 carbons and C-C coupling to give 39, tautomerization of the adduct forms the indolocarbazole ring and 41. In the radical addition route b, migration of the single radical of 37 to C2 can lead to addition to the other indole ring and generate a C3 radical intermediate 40, which can be followed by electron transfer to the heme to form the final product 41. In both mechanisms, a single round of iron reduction from the ferryl state to the FeIII state is sufficient to perform the C2-C2 coupling. In the Scheme 15, a P450-catalyzed indole couple with C3-C3′ regioselectivity is discussed.
Scheme 12.

Proposed Catalytic Mechanism of Indole Coupling in Indolocarbazole Biosynthesis
Scheme 15.

Formation of the Communesin Core by the P450 CnsC
Shaik and coworkers performed both computational and experimental studies with StaP to propose a new mechanism (Scheme 13).70 The first step is again hydrogen abstraction from one of the indole NH to yield a N-radical 37. This is followed by radical addition to form the C2-C2′ bond together with a single electron transfer from the NH of the recipient indole 42 to heme-iron to form the iminium 43 and the FeIII-hydroxide species. Abstraction of a proton gives 44 and returns the heme to the FeIII-OH state followed by indole tautomerization of 45 yields the indolocarbazole ring system.
Scheme 13.

Alternative Proposal for the Mechanism of StaP
In the structures of 20 and 21, one additional ring is formed via two C-N bonds between the indolocarbazole aglycone and the deoxysugar 2,3,6-trideoxy-3-aminoaldohexose. Biosynthetic studies revealed that this ring formation is catalyzed by the P450 StaN in the pathway to 20.71 The first C-N bond is formed by StaG, an N-glycosyltransferase using the TDP-activated form of the deoxysugar and K252c (46) as substrates. This yields holyrine A 47 which then undergoes intramolecular oxidative cyclization by StaN to yield staurosporine 20. A proposed mechanism is shown in Scheme 14. The first step is the hydroxylation on the C5 through the rebound mechanism shown in Scheme 2.1A to form a hemiketal 48a, which undergoes dehydration to form the oxonium ion 48b. The amine from the indole ring can then act as a nucleophile to attack the oxonium ion to form the new C-N bond in 49, which is further transformed to 20 by both O- and N-methylations. Therefore, in the remarkably complex biosynthetic pathway of 20, a total of three P450 enzymes are recruited to form two C-C bonds and one C-N bond.
Scheme 14.

Proposed Catalytic Mechanism for the Last C-N Coupling Step in Staurosporine Biosynthesis
2.2.3. C-C Coupling
2.2.3.1. Fungal P450 Catalyzed Cyclization
Communesins (65–67) are fungal indole alkaloids produced by various Penicillium species and have been noted to have strong insecticidal properties.72–76 There are ~ 10 members of this family isolated, each containing a heptacyclic core that is among the most complex observed in fungal alkaloids. The seven interconnected rings consist of two indole containing fragments fused through a vicinal quaternary C-C bond and two aminal bonds. Four contiguous stereocenters run through the junction of two fragments. While numerous total syntheses of communesin have been reported,77–87 the biosynthetic pathway was only recently uncovered from P. expansum and was shown to be highly efficient (Scheme 15).88,89 The two indole-containing building blocks are tryptamine (50) derived from decarboxylation of L-tryptophan, and (−)-aurantioclavine (53) derived from the decarboxylative cyclization of 4-dimethylallyltryptophan. A P450 enzyme (CnsC) was found to be solely responsible in combining the two fragments to generate the core structure 63, which is one N-methylation step away from the stable communesin K (65).89 65 is then further elaborated through epoxidation and N-acylation to yield communesin A (66) and communesin B (67).
Characterization of the P450 using either yeast based-biotransformation and/or in vitro microsomal assay showed formation of the three bonds between 50 and 53 is regioselective as no other products are formed.89 Formation of the two aminal bonds (N1-C2-N1′ and N10-C2′-N10′) in 63 requires ring opening of the pyrroloindole 60 to generate the aniline species 62. The N1 hydrogens on the indole nitrogens of both 50 and 53 were found to be required for coupling, as N-methyl substituents were not reactive. The P450 displayed broad substrate promiscuity towards C3-substituted indoles. When N10-methyl tryptamine (51) was used in place of 50, a coupled “isocommunesin” product 59 with alternative aminal bond regioselectivity was the major product (4:1 ratio between isocommunesin 59 and communesin scaffold 64). The isocommunesin scaffold of 59 is not naturally observed and is derived from the direct N1-C2-N10′ and N1′-C2′-N10 aminal bonds formation after C3-C3′ coupling.
Density functional theory (DFT) calculation was performed to assess whether a radical addition mechanism, involving a tryptaminyl radical addition to the indole of 53, takes place.89 Computation showed the radical addition has a 105 preference for the formation of C3-C2′ adduct, therefore excluding this mechanism and favoring the diradical coupling between 52 and 54 as shown in Scheme 15. Other mechanisms such as radical cation addition, electrophilic aromatic addition were also ruled out based on C-C coupling regioselectivity. Further calculation on the aminal formation steps showed that formation of the isocommunesin scaffold 59 is energetically favorable and is predicted to take place spontaneously from 56 via 57 or 58, while formation of the communesin scaffold 63 from 55 is energetically uphill in the pyrroloindole ring opening step (60 or 61 to 62). Hence, it was proposed that CnsC is responsible for the formation of all three bonds: first catalyzing the C-C coupling to yield 55, followed by promoting the regioselective aminal bond forming steps (while suppressing isocommunesin formation).
In parallel with the biosynthetic studies, Movassaghi and coworkers developed an efficient total synthesis of 65 using a strategy that mimics the diradical coupling strategy shown in Scheme 16.87 In their strategy, the two indole fragments were first synthesized as azepine 68 and cyclotryptamine 69. Coupling of the two fragments can yield the sulfamide 70 on a gram scale. Chemoselective oxidation in the presence of N-chloro-N-methylbenzamide and 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine yields the diazene 71, which can be subjected to photoexcitation and release of N2 to yield the diradical 72 that can be coupled to forge the quaternary C-C bond in 73. This reaction yields a single diastereomer, which can be reacted in three more steps to yield 65. Such later stage coupling strategy can be applied to the synthesis of other homo- and heterodimeric indole alkaloid natural products.
Scheme 16.

Biomimetic Synthesis of Communesin K by Movassaghi
Mycocyclosin (82) is a cyclized diketopiperazine (DKP) produced by Mycobacterium tuberculosis (Scheme 17).90 Two genes are responsible for the biosynthesis. The first is Rv2275, which is a cyclodipeptide synthase (CDPS) that synthesizes cyclo-Tyr-Tyr (cYY, 75) using two molecules of tyrosyl-tRNATyr (74).91 The second is a P450 CYP121 that was shown to perform the C-C coupling of 75 to yield 82.90 Interestingly, CYP121 is essential for M. tuberculosis viability, as knockout of the gene requires plasmid complement of cyp121 to survive.92 The activity of CYP121 was confirmed in vitro, and the structure in complex with 75 was solved.90 It was shown that the DPK ring of 75 is perpendicular to and offset from the center with respect to the heme, with one phenol pointing at the iron and the other pointing away. A diradical combination mechanism was proposed in route b, in which the first radical is formed on the phenolic oxygen in the presence of Compound I, followed by delocalization of the radical in the benzene ring. This is followed by rotation around the DKP core and placing the other phenol at the iron center to undergo second hydrogen abstraction by Compound II to yield the diradical 80.90 Computation modeling of the reaction steps and energetics led to a later proposal that the second radical formation may be due to proton-coupled electron transfer (PCET) instead of substrate rotation.93 Diradical combination of the C-diradical 81 that yields 79 likely takes place in bulk solution and is followed by aromatization to yield the rigidified 82. An alternative mechanism involving radical addition can also be proposed in route a, in which the first radical 77 directly adds to the ortho position of the second phenyl ring to forge the C-C bond and yield 78. This can be followed by a second step of hydrogen abstraction and formation of the diketone 79 which can aromatize into 82. CYP121 was shown to be highly specific to 75, as alternative DKP substrates were not recognized. The physiological role of 75 and 82 in M. tuberculosis virulence are not clearly understood, however, the indispensable role of CYP121 in M. tuberculosis viability makes it an intriguing therapeutic target.
Scheme 17.

Possible Mechanisms of CYP121 catalyzed C-C coupling in Mycocyclosin Pathway
A similar P450-catalyzed intramolecular coupling of phenols can be seen in herquline A (93) and herquline B (91), which are platelet aggregation inhibitors produced by Penicillium strains.94–97 93 has also been shown to inhibit replication of the influenza virus without notable toxicity to humans.94 In contrast to the DKP core of 82, 91 contains a reduced piperazine core, likely derived from a multitude of reductions on the L-Tyr-L-Tyr dipeptide. Both radical addition (route a) and diradical combination (route b) mechanisms can be proposed starting from diphenylpiperazine precursor 83 and the phenoxy radical 84 as shown in Scheme 18. Whereas the C-C coupled product 89 can undergo aromatization in mycocyclosin pathway, here 89 is proposed to be further reduced to yield the fused cyclohexanone structure 90. While the stereochemistry of the coupling reaction is not known (and is also masked in mycocyclosin), it is expected that the two cyclohexanone rings will be oriented in anti-fashion to minimize steric and electronic clashes. Selective methylation of one of the nitrogen yields 91, which may undergo isomerization to yield the α-β unsaturated 92 that be subjected to intramolecular 1,4-addition to yield 93. The extensive number of reduction steps involved in herquline biosynthesis thus provide stark contrast to that of mycocyclosin.
Scheme 18.

Possible Mechanisms of P450-catalyzed C-C Coupling in Herquline Pathway
Usnic acid (98) is a polyketide-derived substance widely found in lichens.98–100 The biosynthetic proposal is shown in Scheme 19 in which two molecules of methylphloroacetophenone (94) are coupled regioselectively via radical intermediates 95 to form the dienone 96. Again both radical addition and diradical combination (shown) can be proposed for this coupling step. As in the herquline pathway and will be seen repeatedly in following examples, 1,4-addition of the phenoxy oxygen to the dienone establishes the 6-5-6 ring system in hydrated usnic acid 97, which can readily dehydrate to form 98.
Scheme 19.

Proposed Pathway for C-C Coupling in Usnic Acid
Viridicatumtoxin (105) is a meroterpenoid (polyketide-terpene hybrid) synthesized by the fungus Penicillium aethiopicum (Scheme 20).101,102 The spirocycle-containing compound shows strong antibacterial properties especially towards methicillin resistant Staphylococcus aureus (MRSA).103 The entire biosynthetic pathway of 105 has been mapped from genome sequencing and biochemically studied.104,105 The anhydrotetracycline-like aglycone is synthesized by a polyketide synthase using malonamyl-CoA as a starter unit. A prenyltransferase VrtC was shown to perform Friedel-Craft like alkylation of the C ring with geranylgeranyl diphosphate as the electrophile to yield 99.106 The linear prenyl chain then must undergo two cyclization steps and form two new C-C bonds to yield 105, a reaction that was surprisingly shown to be catalyzed by a single P450 VrtK.104 The proposed mechanism, which is supported by DFT calculations, is shown in Scheme 20. The allylic C17 position is first proposed to undergo hydrogen abstraction to yield the radical 100. A second electron transfer to the heme yields the allylic cation 101. [OH•] rebound at the allylic radical site follow by loss of water may also lead to the cation 101. From there, it was suggested that a C15-C19 cyclization step can yield the C20 tertiary carbocation 102, which can undergo concerted 1,2-alkyl shift and 1,3-hydride shift to yield a new C15 tertiary carbocation 103. The origin of the two hydrogen atoms on C19 (shown in blue) is consistent with prior labeling studies using C3-2H-labeled mevalonate. From 103, quenching of the carbocation by the aromatic D-ring (C7) forges the second C-C bond (C7-C15) in 104 and rearomatization yields 105. VrtK represents the first example of a P450 capable of catalyzing terpene cyclization, although its exact catalytic role in the C-C bond forming steps are not known.
Scheme 20.

Computationally Predicted Oxidative Cyclization Mechanism of VrtK
The last example in P450 catalyzed C-C cyclization in fungi is that of brefeldin A (111), a potent inhibitor for blockade of secretory cargo moving from the endoplasmic reticulum to the Golgi organelles.107–109 111 is a bicyclic polyketide containing a cyclopentane fused to a 13-membered macrolide. Biosynthetic reasoning suggests the cyclopentane ring is formed via an intramolecular C-C coupling within the 16-membered macrolactone. The likely bref biosynthetic cluster was found in Eupenicillium brefeldianum.110 Reconstitution of the bref-PKS and an associated hydrolase in yeast led to the formation of the triene acid 106, which is of the same length (16 carbons) and expected functionalization required for formation of 111.110 The gene cluster encodes four P450 enzymes, one of which is proposed to be responsible for a radical mediated cyclization shown in Scheme 21. The activities of the P450s have not been reconstituted, thereby leaving timing of the cyclopentane formation in question. The inability of the reconstituted PKS to form the macrocycle provides some evidence for cyclopentane formation to proceed macrocyclization, with the former possibly rigidifying the hydrocarbon backbone for macrolactone formation. The P450 catalyzed C-C coupling can either take place on the PKS-bound intermediate 107 or the isolated linear acid 106. Abstraction of the C9 hydrogen to give 108 followed by radical addition to C5 yields the allylic radical 109, which can undergo [OH•] rebound to yield 110. Further modification including C7 hydroxylation and macrocyclization would yield 111. A detailed study of the roles of the P450s is needed to complete the biosynthetic pathway.
Scheme 21.

Proposed Radical Mediated Cyclization in Brefeldin A Biosynthesis
2.2.3.2. Plant P450 Catalyzed Cyclization
P450-catalyzed C-C couplings are widespread in the plant kingdom, especially during alkaloid and phenylpropanoid biosynthesis.111–113 Recently, comparative transcriptome analysis has enabled the identification of many plant biosynthetic pathways in their entirety and has revealed the key P450 steps that have been elusive for many years.114–116 A case in point is the biosynthetic pathway of morphine (117), which has been referred to as the king of alkaloids. A key step in forging the morphine scaffold is the C-C coupling of R-reticuline (112) to yield salutaridine (116) (Scheme 22A). In the 1950s, Barton and Cohen proposed that such a connection can be formed via phenolic coupling.117 A P450 activity that catalyzes this reaction was first observed in the microsomal fractions of Papaver somniferum capsule.118,119 Zenk et al observed that both NADPH and O2 are required for the reaction, indicative of heme chemistry. Kutchan and coworkers identified CYP719B1 is the responsible P450, by first comparing P450 transcriptome profile to non-producing P. somniferum strains, followed by expression in insect cells and activity confirmation.120 The enzyme exhibited very strict substrate specificity towards 112 and has essentially no activity towards all other structurally related isoquinoline alkaloids. Both radical addition via 114 (route a) and diradical coupling via 115 (route b) have been proposed for this reaction, with either one starting with abstraction of one phenolic hydrogen to generate the phenolate radical 113. Under either mechanism, a single cycle of iron redox is sufficient to turn over the coupled product 116. At nearly the same time, Sato and coworkers discovered a comparable P450 CYP80G2 in the biosynthetic pathway of magnoflorine (120) in Coptis japonica, which can perform phenolic coupling on S-reticuline (118) to yield S-corytuberine (119) (Scheme 22B).121 Both CYP719B1 and CYP80G2 are close sequence homologs to berbamunine synthase (CYP80), which was discovered to catalyze C-O phenolic coupling during biosynthesis of bisbenzylisoquinoline alkaloids in higher plants.122
Scheme 22.

P450 Catalyzed C-C Coupling in Salutaridine and Magnoflorine Biosynthesis
The Amaryllidaceae alkaloids which include galantamine (125), marithidine (128) and lycorine (131) display a variety of biological activities (Scheme 23).123 125 inhibits acetylcholine esterase and binds to nicotinic receptor, and has been used in the treatment of Alzheimer’s disease.124 These related alkaloids are all multicyclic compounds derived from L-Phe and L-Tyr, and go through a common intermediate norbelladine.125 Following O-methylation by a specific methyltransferase to give 4-O′-methylnorbelladine (121)126, a P450 is suggested to initiate phenolic coupling by hydrogen abstraction from the B ring. Delocalization of the radical on the ortho and para positions of the ring can lead to different coupling regioselectivity as shown in Scheme 23. Both radical addition and diradical combination mechanisms can be written for the coupling steps. Formation of different dienone intermediates are proposed depending on the coupling regioselectivity, each can undergo spontaneous 1,4-addition by either the phenolic oxygen in ring B or the secondary amine nitrogen to yield tetracyclic products. The paraA-paraB connection, which occurs most readily under chemical conditions, leads to dienone 126. Intramolecular 1,4-addition forms noroxomaritidine (127) (C-N bond) which can be reduced and methylated to yield 128. The P450 enzyme CYP96T1 responsible for the coupling was identified from transcriptome analysis of producing plants.127 The paraA-orthoB coupling product 123 can undergo 1,4-addition to yield the dibenzofuran 124, which can be N-methylated to yield 125.127 The orthoA-paraB coupling product 129 can be converted to noroxopluviine 130, which is the precursor to 131.127 The tandem P450-coupling/1,4-addition steps for Amaryllidaceae alkaloids parallel closely that of herquline A biosynthesis (Scheme 18), although the former is from the plant world while the latter is found in fungi, demonstrating converging biosynthetic strategy to construct the constrained alkaloids.
Scheme 23.

P450 Catalyzed Phenol Coupling in Amaryllidaceae Alkaloid Biosynthesis
The homo-coupling of monolignol (E)-coniferyl alcohol (132) to yield (+)-pinoresinol (136) is an essential step in the biosynthesis of plant lignans (Scheme 24).128 One electron oxidation of 132 can readily generate the phenoxy radical 133. Delocalization of the radical in the phenyl ring through resonance can lead to 134 and coupled products with different regioselectivity and stereoselectivity, in a process that at first appears random. Lewis and coworkers discovered a noncatalytic dirigent protein from Forsythia intermedia that is responsible for controlling the coupling step between the allylic coniferyl alcohol radical 134 to yield exclusively (+)-C8-C8′ linked product 135, which can undergo addition by the hydroxyl group to yield the furanofuran 136.128 Mechanistic studies showed the dirigent proteins bind to the radical 134 with an apparent KM of 10 nM, ~40,000-fold lower than that of the weak binding 132, suggesting the radical species is the substrate and the dirigent protein spatially controls the diradical combination outcome.129 Schaller and coworkers subsequently purified an enantiocomplementary dirigent protein from Arabidopsis thaliana that mediates the exclusive formation of (−)-pinoresinol (137).130 A P450 CYP81Q1 catalyzes methylenedioxy bridge formation in 136 to yield sesamin (138),131 a cyclization mode that will be discussed in Scheme 33 (Section 2.2.4). In addition to serving as a lignan build block, 136 is also the biosynthetic precursor to the antitumor podophyllotoxin as shown in Scheme 39 (Section 2.3.1).
Scheme 24.

Diradical Combination in (+)-Pinoresinol Biosynthesis
Scheme 33.

P450 Catalyzed Formation of Methylenedioxy Bridge in Natural Products
Scheme 39.

Biosynthetic Pathway of (−)-4-Desmethylepipodophyllotoxin in Mayapple
2.2.4. C-O Coupling
In addition to catalyzing C-C bond formation and cyclization, P450s are also widely involved in the formation of C-O bonds. We have already seen an example of this in the formation of aryl ethers during vancomycin biosynthesis (Scheme 9, Section 2.2.1). This section will provide more examples of oxidative cyclization via C-O bonds catalyzed by P450s. As in the previous section, both radical addition or diradical combination mechanisms can be written for many of the reactions described here.
The first example is the formation of the grisan spirocycle in griseofulvin (146) biosynthesis (Scheme 25). Griseofulvin in a “classical” fungal polyketide that helped established the polyketide hypothesis. It was originally used as an antifungal drug, but recently has been shown to disrupt mitotic spindles and as a potential antitumor compound.132–135 146 is produced by various Aspergillus and Penicillium species, and the biosynthetic pathway from P. aethiopicum was elucidated first.105,136 The carbon backbone is synthesized from a nonreducing PKS and undergoes one aldol and one Claisen cyclization to yield the benzophenone scaffold seen in griseophenone. Methylation and chlorination of the nascent cyclized product afford griseophenone B (139), which is the substrate of P450-catalyzed conversion to the grisan product desmethyl-dehydrogriseofulvin A (145). An additional O-methylation and enoylreduction yield the final product 146.
The P450 GsfF catalyzes the oxidative transformation of benzophenone 139 to grisan structure 145 and formation of a oxa-spiro core. The activity was verified using yeast microsomes containing overexpressed enzyme. Two mechanisms (B and C) were initially proposed (Scheme 25). Mechanism B involves the formation and recombination of a diradical intermediate 142 through two successive phenolic hydrogen abstraction steps. In mechanism C, the enzyme catalyzes epoxidation of A ring to 143 followed by nucleophilic opening of the epoxide by the B ring phenol to yield the hemiacetal 144 that can be aromatized to yields 145. The C-O coupling step was studied computationally by Grandner et al, in which they concluded a third, mechanism A, is the most likely and energetically favorable.137 This is the radical addition mechanism in which the enzyme performs the O-H abstraction on B ring to give 140, but avoids diradical formation. Direct attack of the phenoxy radical on A ring leads to ring closure and intermediate 141, subsequent phenolic O-H abstraction by the heme affords 145.
An interesting contrast to the use of GsfF in forming the spirocycle is found in the (+)-geodin (150) and trypacidin (154) pathways, which contain a similar 6-5-6 spiro grisan structure derived from benzophenone precursors (Scheme 26). 150 is a common fungal metabolite produced by many strains, most notably A. terreus. After formation of dihydrogeodin (147) by the polyketide synthase and accessory enzymes, a multi-copper protein GedJ was found to be responsible for transformation into geodin.138,139 The enzyme, dihydrogeodin oxidase (DHGO) shows significant sequence homology to other copper-containing oxidases, including laccase and ascorbate oxidase. Based on the known mechanisms of laccases,27 formation of a phenoxy radical intermediate 148 and diradical coupling of 149 have been proposed (Scheme 26). The closely related 154 isolated from A. fumigatus was found to be a potential virulence factor and has a protective function against phagocytosis during invasion.140,141 The gene clusters of 150 and 154 are highly homologous and a side by side comparison suggests TynJ is the responsible copper oxidase for C-O bond formation.141
Scheme 26.

Multi-copper Protein Catalyzed C-O Coupling to from Grisan-Containing Compounds
Radical recombination is also proposed in the biosynthesis of tubocurarine (160), which is a monocationic, dimeric benzylisoquinoline alkaloid isolated from the South American plant Chondrodendron tormentosum.142 The compound is an acetylcholine agonist and has been used as skeletal muscle relaxant in African arrow poison.143,144 The biosynthesis involves a heterodimeric radical coupling between the enantiomers (R)-N-methyl-coclaurine (155) and (S)-N-methyl-coclaurine (157) (Scheme 27). Both building blocks are derived from dopamine and 4-hydroxyphenylacetaldehyde via Pictet-Spengler reaction (Section 3.2.2). A P450-catalyzed dimerization mechanism has been proposed in which diradical species of both enantiomers are formed. Two O-radicals are formed on the (S)-isomer in 156, while two C-radicals are generated on the (R)-isomer via O-radical delocalization in 158. Two regioselective C-O couplings between the diradical species forge two new C-O bonds in the dimer 159, which can be methylated at one of the tertiary amine to yield 160.
Scheme 27.

Heterodimeric Radical Coupling in Tubocurarine Biosynthesis
Another pathway where the diradical combination mechanism is likely at play is during the synthesis of aureothin (165), which is an antitumor, antifungal, and insecticidal compound isolated from Streptomyces thioluteus (Scheme 28).145,146 The notable structural features include a nitroaryl moiety and an exo-methylene tetrahydrofuran (THF) ring. Biosynthetic studies revealed that the carbon backbone of aureothin is assembled by a modular PKS using p-nitrobenzoic acid as the starter unit.147 Starting from 161, formation of the exo-methylene THF moiety is catalyzed by a multifunctional P450 AurH, using a stepwise mechanism.148–150 First, AurH catalyzes the hydroxylation of C7 position to yield 162 using standard hydrogen abstraction and [OH•] rebound mechanism. Two possible routes are likely to transform 162 into 165. Route a requires a second P450 catalyzed hydroxylation at the C9a position to yield 163, followed by cyclization and dehydration to form the THF ring. Route b involves abstraction of two hydrogens from C9a and the C7 hydroxyl group during a single P450 catalytic cycle to generate the allylic C-radical and the O-radical in 164, respectively. Diradical combination then furnishes the THF ring and the 165.
Scheme 28.

Proposed Mechanisms of P450-catalyzed Formation of Tetrahydrofuran in Aureothin
Like the previous example, two mechanisms can also be proposed for cyclic ether bond formation during penitrem D (171) biosynthesis. Penitrems are tremorgenic mycotoxins isolated from several Penicillium species and are the most complex indole diterpenes isolated to date.151–154 These compounds contain a paxilline backbone fused with a 6-4-8 tricyclic ring system. Formation of the six-membered ring involves a ring-expansion step that will be discussed in Scheme 135 (Section 6.2.6). Reconstitution of the biosynthetic pathway of penitrem A in A. oryzae revealed that the P450 PtmU is responsible for the construction of the eight-membered cyclized ether ring in 171 using secopenitrem D (166) as a substrate (Scheme 29).155 The mechanism of the cyclization step is unresolved but is expected to be analogous to that of the THF ring in 165. Two routes are proposed in which route a is diradical 168 formation and recombination; while route b is via hydroxylation of the cyclopentane ring in the paxilline core to give 169, followed by dehydration to 170 and nucleophilic addition to arrive at 171.
Scheme 135.

P450 Catalyzed Ring Expansion in Penitrem A Biosynthesis
Scheme 29.

Proposed mechanisms of P450 Catalyzed Oxidative Cyclization in Penitrem D Pathway
Pyrrocidine B (173)156 and related compounds, such as hirsutellone B (172)157, GKK1032A2 (174)158 and embellicine A (175)159 (Scheme 30A), are multicyclic hybrid PK-NRP compounds isolated from different fungal species with various biological activities.160 These compounds share several common structural features: a fused 6-5-6 decahydrofluorene core, a 13-membered and strained paracyclophane, and a 5-hydroxypyrrolidinone moiety derived from reductive cyclization catalyzed by PKS-NRPS (see section 3.2).160 Early isotopic labeling studies of 174 carried out by the Oikawa group established the biosynthetic origin of the carbon backbone of the molecule.161 Recently, Nay and coworkers performed feeding studies of 173 by using doubly labeled 18O,13C-L-tyrosine 176.162 The feeding studies confirmed that both of the labeled atoms were incorporated into 173, suggesting that oxygen atom of cyclophane ether is derived from the nucleophilic phenolic oxygen of tyrosine. Several P450-catalyzed cyclization mechanisms can be proposed for morphing the acyclic PK-NRP product 177 into the paracyclophane as shown in Scheme 30B. In route a, cyclization is initiated through the allylic hydroxylation of the terminal C16 methyl to give 178, which can trigger olefin rearrangement to yield a C4 carbocation that can be quenched by the phenolate oxygen to form paracyclophane 182. This common intermediate for all three mechanisms can undergo intramolecular Diels-Alder cyclization to yield the 6-5-6 ring system and 183. In route b, epoxidation of the acyclic precursor to 179 can set up a general base/general acid catalyzed epoxide opening to form the cyclohexane ring 181. Attack by the phenolate oxygen at C4 is followed by dehydration to afford the Diels-Alderase substrate 182. Finally in route c, P450-catalyzed hydrogen abstraction by the high-valent oxoiron yields a phenoxy radical 180 that can add to the triene system with the FeIV-OH as the second electron recipient to directly setup the diene and dienophile system in 182. Following Diels-Alder cyclization, several tailoring steps complete the biosynthetic pathway to 173. Regardless of which exact mechanism is used, it is worth noting the rapid construction of all the rings systems of this family of compounds from a PK-NRP product via oxidation.
Scheme 30.

Proposed Oxidative Cyclization Mechanisms in Pyrrocidine Biosynthesis
Salinamides are members of a family of unusual bicyclic depsipeptides with antibacterial and anti-inflammatory bioactivities isolated from the marine actinomycete Streptomyces sp. CNB-091 (Scheme 31A).163–165 The backbone of salinamides is assembled by a NRPS and PKS hybrid biosynthetic pathway. A type I thioesterase performs an intermolecular transesterification of the salinamide aglycone with a (4-methylhexa-2,4-dienoyl)glycine handle to yield the monocyclic desmethylsalinamide C (184).166 Forging an intramolecular C-O bond between the phenyl ring of the depsipeptide and the olefin of newly added handle is followed by epoxidation to yield salinamide A (185). This reaction was shown to be catalyzed by the P450 Sln10, but the exact mechanism is not known. A phenoxy radical addition mechanism can be proposed as shown in Scheme 31B. Following one hydrogen abstraction to yield the phenoxy radical 188, addition of the radical to the terminal double bond in the ester handle yields a new radical species 189 that is stabilized by the conjugated α-β double bond. Abstraction of a second hydrogen by the FeIV=O heme center affords the exo-methylene intermediate 190. Epoxidation of this compound by the same P450 installs the epoxide in 185. This epoxide can be subjected SN2 ring opening by either Cl− or OH− to yield salinamide B (186) or salinamide F (187), respectively.
Scheme 31.

Proposed Mechanism of P450 Catalyzed Phenoxy Radical Addition in Salinamide Pathway
Allene oxides (193 and 196) are unstable vinyl epoxides and are key biosynthetic intermediates to a series of plant signaling molecules.167 The epoxides are formed by the enzymatic dehydration and cyclization of lipoxygenase-catalyzed peroxidation of polyunsaturated fatty acids, such as linolenic acid (191) and arachidonic acid (194) (Scheme 32). The responsible enzyme for this transformation is the P450 allene oxide synthase (CYP74A). A proposed mechanism is shown in Scheme 32 in which the first step is the homolytic cleavage of the O-O bond of the peroxide (either 192 or 196) to generate an O-radical 197. This is proposed to be catalyzed by the FeIII state of the heme center. The O-radical can add to the adjacent C=C double bond to form the epoxide ring and an allylic C-radical 198. One electron oxidation of this intermediate by the FeIV=O heme center yields the allylic carbocation 199, which can form the vinyl epoxide by proton abstraction. Recently another type of allene oxide synthase was identified from corals, which is a catalase-like hemoprotein.168 The allene epoxide sets up significant morphing of the linear fatty acid into jasmonic acid and clavulone I as described in Scheme 112 (Section 4.5).
Scheme 32.

Mechanism of Reactions Catalyzed by Allene Oxide Synthase (CYP74A)
Scheme 112.

Cyclopentenone Formation in Jasmonic acid and Clavulone I Biosynthesis
The cyclized 1,3-benzodioxole functional group is a common structural unit found in many plant and microbial natural products. A list of representative compounds (200–210) is shown in Scheme 33A, in addition to the compounds already discussed in this review such as lycorine (131)169 and sesamin (138)170. This ring system is formed via a methylenedioxy O-C-O bridge between phenylic oxygen and an ortho methoxy carbon, catalyzed by P450 enzymes as shown in Scheme 33B.171–174 P450 catalyzes hydroxylation of the methoxy group 211 via a hydrogen abstraction and [OH•] rebound to form the hemiacetal 212. After dehydration to 213, the ortho hydroxyl group serve as nucleophile to attack the oxonium cation to form the new C-O bond in 214.
2.2.5. C-N Coupling
Two examples of P450-catalyzed C-N coupling and intramolecular cyclization will be included here. The first is during biosynthesis of fumitremorgin C (219), which is a L-Trp-L-Pro diketopiperazine mycotoxin produced by A. fumigatus that has been well-characterized as a breast cancer resistance protein inhibitor (Scheme 34).175–177 219 contains a fused 6-5-6-6-5 rigid ring system that is formed after oxidative coupling between the amide nitrogen in the DKP ring and a C5 prenyl chain grafted onto the indole ring. The biosynthetic gene cluster of 219 was recently identified by Osada and coworkers and studies showed the transformation from tryprostatin A (215) to 219 is catalyzed by the P450 FtmE.178 Two oxidative mechanisms can be proposed for this reaction. The first step is hydrogen abstraction in the dimethylallyl unit to generate 216 of which the radical is stabilized by both the indole ring and the Δ2-olefin. Subsequently in route a, a second hydrogen abstraction from the amide nitrogen generates a diradical intermediate 217 that can undergo recombination to yield 219. Alternatively in route b, a second electron transfer from the allylic radical to the heme center yields the carbocation 218, which can be quenched by the amide nitrogen to afford the rigidified ring system. It is interesting to note in many indole alkaloid pathways, the amide nitrogen originally derived from tryptophan acts as a nucleophile to attack the C2 position of the indole ring to form a 6-5-5-6 ring system. In these cases, the C-N bond formation is triggered by either epoxidation of the indole ring as in the notoamide example (Scheme 106, Section 4.4.2), or alkylation of the C3 position (such as prenylation179 or methylation180). In the case of 219, the oxidative cyclization occurs exo of the indole ring. 219 can still undergo indole epoxidation and further structural morphing to yield the spirocyclic spirotryprostatin as shown in Scheme 107 (Section 4.4.2).
Scheme 34.

Possible Mechanisms of P450 Catalyzed C-N Coupling in Fumitremorgin C Pathway
Scheme 106.

Indole Epoxidation in Notoamide Biosynthesis
Scheme 107.

Indole Epoxidation Sets Up Spirocycle Formation in Spirotryprostatin
The indole alkaloids teleocidin B (220) and lyngbyatoxin A (221) are bacterial natural products that share the same cyclic indole backbone (the indolactam V core, 222) (Scheme 35). These compounds are potent activator of various protein kinase C isozymes,181 making them potentially valuable reagents in pharmaceutical research. Biosynthetic studies of these compounds showed that 222 is cyclized from N-methyl-L-valyl-tryptophanol (223), which is synthesized by a bimodular NRPS that releases product as a free alcohol.182–184 Cyclization of 223 into the 9-membered ring indolactam V structure is catalyzed by a P450, such as LtxB from the biosynthetic pathway of lyngbyatoxin A, via C-N coupling. Three different routes have been proposed for the mechanism of ring construction. Route a is via epoxidation on the phenyl ring of the indole moiety to give 224, followed nucleophilic ring opening upon attack by the amine group to yield 225. Rearomatizing the phenyl ring completes the cyclization to 222. Route b is via a diradical coupling between the C-radical on the phenyl ring and the N-radical from the amine group in 227, presumably catalyzed by two consecutive hydrogen abstraction (via 226) by the heme center. Route c is via formation of the N-radical 226, followed by radical addition to the phenyl ring and hydrogen abstraction that affords 228 and rearomatization to 222. Alkylation of 222 by geranylgeranyl diphosphate leads to 221.
Scheme 35.

Possible Mechanisms of P450-Catalyzed C-N Coupling in Indolactam Pathway
2.2.6. C-S Coupling
Griseoviridin (229), belonging to the streptogramin family, is a cyclic polyunsaturated macrolactam produced by S. griseoviridis and exhibits broad-spectrum antibacterial activity (Scheme 36).185 229 contains a unique 9-membered lactone with a thioene connection (S-C2 linkage). The macrolactam backbone of 229 is biosynthesized by a hybrid PKS-NRPS assemble line featuring one oxazole derived from cyclization and dehydration of serine (Scheme 68, Section 3.3.3).186 Gene inactivation experiments identified that a P450 monooxygenase, SgvP, is responsible for forging the thio-ene linkage starting with 232.187 Both epoxidation and radical mechanisms can be proposed for the C-S coupling catalyzed by SgvP, analogous to those outlined for 220. Route a initiates with epoxidation of the α-β C2-C3 double bond to 233, followed by nucleophilic attack of the thiol to open the epoxide ring and forms the C-S bond in 234, and subsequent dehydration leads to 229. Route b is the radical addition mechanism in which extraction of the thiol hydrogen leads to a S-radical in 235 that can add to the C2-C3 double bond in 236. Abstraction the C2 hydrogen results in the thio-ene moiety. Two other examples of cyclic natural products containing the thioether linkage, α-amanitin (230) and phallacidin (231) have been isolated from the mushroom Amanita bisporigera.188 The biosynthetic origin and the enzymatic basis for these examples of C-S coupling remains unexplored.
Scheme 36.

Possible Mechanisms of P450 Catalyzed C-S Coupling in Griseoviridin Pathway
Scheme 68.

Mechanisms of Oxazole and Thiozole Formation
2.3. Cyclization Catalyzed by Nonheme Iron α-KG-Dependent Oxygenases
In section 2.2, we have reviewed P450-catalyzed oxidative cyclization during natural product biosynthesis. The use of a high-valent oxoiron heme center to abstract hydrogen atoms from different scaffolds and heteroatoms is also catalyzed by nonheme iron α-KG-dependent oxygenases (Scheme 2). As in the previous section, different mechanisms of the transformations can also be proposed, including diradical recombination, radical addition, and cyclization mediated through [OH•] rebound. The examples in this section will also be grouped by the type of atom that is coupled to the carbon atom to forge the new cyclic structures.
2.3.1. C-C Coupling
Cycloclavine (245) is an ergot indole alkaloid produced by the fungus A. japonicas (Scheme 37).189 The most notable structural feature of 245 is a cyclopropyl moiety fused to the ergot core. O’Connor and coworkers recently identified and heterologously reconstituted the pathway in S. cerevisiae.190 The biosynthetic pathway, as with other ergot alkaloids, is derived from the oxidative cyclization of 4-dimethylallyl-L-tryptophan (237) through oxidation of the prenyl chain. Chanoclavine-I aldehyde (238) was shown to be a precursor of 245, and is first proposed to undergo ene-reduction by the Old Yellow Enzyme (OYE) EasA to 239 and cyclize to yield the imine 240 (Scheme 37A). Tautomerization of the imine to enamine 241 then sets up the oxidative transformation by the nonheme iron α-KG-dependent enzyme EasH as shown in Scheme 37B. Two likely mechanisms of intramolecular cyclopropanation are shown. Route a involves the hydroxylation of the enamine 241 to 242, followed by water elimination to 244. Although chlorination of the same carbon is also possible using the mechanism discussed in Section 5.1, EasH lacks the active site motif that is signature of an oxidative chlorinase. Alternatively, route b involves hydrogen abstraction at the same position and radical migration to yield the intermediate 243, which can undergo a second hydrogen abstraction by EasH to afford the iminium 244, which can be reduced by EasG to yield 245. In section 3.5.2, we discuss an alternative fate of 238 to yield lysergic acid.
Scheme 37.

Possible Mechansims of the EasH Catalyzed Cyclopropane Formation in Cycloclavine
Nogalamycin (250) is an anthracycline antibiotic produced by the soil-dwelling S. nogalater (Scheme 38).191 Unlike other anthracyclines, 250 is derived from a diglycosylated anthracycline scaffold 246. The C7 position is glycosylated with D-olivose, while the unusual bicyclic ring systems attached to C1 and C2 are derived from rhodosamine. The O1- rhodosamine deoxysugar is further coupled to the C2 position via a new C-C bond, but is also epimerized at the C4′ position to give nogalamine in the final product. Prior genetic studies showed that the glycosyltransferase SnoD and SnoE are glycosyltransferases that establish the classic O-glycosidic bonds at C1 and C7 respectively in 246.192 Recently, two α-KG dependent iron enzymes from the gene cluster were characterized.193 SnoK was shown to catalyze the C-C bond formation of 248. The radical mechanism is shown in Scheme 38, in which abstraction of C5′ hydrogen to 247 and subsequently the C2-hydrogen by the iron center can lead to radical coupling for formation of the new C-C bond in 248. The other α-KG dependent iron enzyme SnoN was found to be the C4′ epimerase that affords nogalamycin R (249) via a diradical mechanism. It is interesting that SnoK and SnoN share 38% sequence identity and have very similar active sites as determined by X-ray crystallography, yet catalyze very different reactions in the late stage of this pathway.
Scheme 38.

Mechanism of SnoK Catalyzed C-C Coupling in Nogalamycin Biosynthesis
In Scheme 24, we showed (+)-pinoresinol (136) is derived from the regioselective and stereoselective coupling of coniferyl alcohol radical in the presence of a dirigent protein. In mayapple, 136 is further modified into the antitumor natural product podophyllotoxin 262 as shown in Scheme 39.114 The biosynthetic pathway of 262 is of considerable interest since the semisynthetic etoposide (261) is a front-line anticancer drug.194,195 The transformation of 136 to (−)-pluviatolide (253) was known for some time, while the remaining pathway was recently mapped by Satterly and coworkers using tobacco plant as a heterologous host.114 Successive introduction of candidate genes identified from transcriptome analysis enabled the heterologous production of (−)-4-desmethylepipodophyllotoxin (260), which is the precursor of the desired stereochemistry for 261 semisynthesis.196 Of note in the pathway are several cyclization reactions, including dehydrogenation of (−)-secoisolariciresinol (251) to (−)-matairesinol (252); P450-catalyzed formation of methylenedioxy bridge in 253, and the key C-C coupling in (−)-yatein (254) to forge the nonaromatic, 6-membered central ring of the 5-6-6-5 tetracyclic framework in (−)-deoxypodophyllotoxin (258). The responsible enzyme 2-ODD is a nonheme iron α-KG-dependent enzyme of which the activity was confirmed using purified enzyme. The proposed mechanism of 2-ODD is first the radical mediated benzylic hydroxylation to yield 255, which can lose water to form the intermediate 256. Aromatic addition to the enenone furnishes the C-C bond in 257 and rearomatization gives 258. Identification and reconstitution of the entire set of enzymes required to transform pinoresinol to 260 sets the stage for a more scalable and cost-effective pathway to 261.
2.3.2. C-O Coupling
Returning to ergot alkaloids briefly, the ergopeptine are well-documented toxins that can cause intoxications in mammals.197 Dihydroergotamine (266) is a known precursor to some of the ergopeptine natural products. Biosynthesis of the diketopiperazine-derived core in 266 starts with two NRPSs, LPS1 and LPS2, that use D-lysergic acid as a starter unit and adds L-alanine, L-phenylalanine and L-proline to yield a tetrapeptide.198 Intramolecular cyclization releases L,L-dihydroergotamam (263) as the product of the NRPS enzymes (Scheme 40). Cloning and characterization of cpEasH from the ergot fungus Claviceps purpurae demonstrated this nonheme iron α-KG-dependent enzyme is responsible for the formation of the oxazolidinone ring found in 266.198 The reaction mechanism is proposed to be first hydrogen abstraction at the α-carbon of the alanine unit in the DKP substrate to give 264, followed by [OH•] rebound to yield the transient hydroxylated intermediate 265. Spontaneous attack of the newly introduced hydroxyl group on the prolyl amide carbonyl forms the oxazolidinone ring in 266. Structural characterization of cpEasH was performed and showed this enzyme is highly similar to phytanoyl-CoA hydroxylase PhyH from humans.
Scheme 40.

cpEasH Catalyzed C-O coupling in Dihydroergotamine Biosynthesis
Orthosomycins are oligosaccharide antibiotics that contain at least one interglycosidic spirocyclic ortho-δ-lactone linkages between a pair of carbohydrates.199 This family of natural products are exemplified by avilamycin A (267), everninomicin D (268), and hygromycin B (269) (Scheme 41A), which are broad spectrum antibiotics.199 The orthoether linkages are essential for the biological activities of these natural products. In both 267 and 268, methylenedioxy bridges are also present. The biosynthetic gene clusters of 267 and 268 are large and encode up to fifty enzymes, many of them with unknown biosynthetic function.200,201 Recently, Bachmann and Iverson and coworkers identified a group of conserved nonheme iron α-KG-dependent enzymes from the biosynthetic gene clusters and solved their X-ray crystal structures.202 Although direct biochemical evidence is still lacking, the presence of long substrate channels in these structures, and co-crystal structure of HygX with 269 provide initial evidence that the orthoether linkages are forged by these enzymes. Genetic inactivation abolished the biosynthesis, further demonstrating their essential roles. The proposed mechanism for the orthoether linkage formation is shown in Scheme 41B. After formation of the high-valent FeIV=O ferryl oxo species, abstraction of hydrogen from the α-carbon of one sugar unit 270 forms the C-radical 271 and FeIII-OH. A second hydrogen abstraction from the hydroxyl group forms the O-radical that can be combined with the C-radical to form the spirocyclic structure 272 and return the iron to the resting state. Although no direct evidence is available, the same enzyme may also catalyze the methylenedioxy bridge 275 starting with 273 with the same mechanism as proposed for P450 enzymes in Scheme 33 (Section 2.2.4), using a different prosthetic iron center.
Scheme 41.

Oxidative Cyclization in Orthosomycin Biosynthesis
Clavulanic acid (286) is a commercially important β-lactam that is produced by S. clavuligerus (Scheme 42).203 It has a bicyclic ring system, consists of one 4-membered β-lactam and one 5-membered oxazolidine ring. Like penicillin and cephalosporinβ-lactams, the multicyclic ring system is derived from oxidative cyclization of a linear tripeptide 276 (Scheme 42A).204 Following β-lactam formation to yield deoxyguanidinoproclavaminic acid (277), a nonheme iron α-KG-dependent enzyme clavaminate synthase (CAS) plays key roles in the remaining pathway.205–212 CAS first catalyzes the stereoselective C3 hydroxylation to yield guanidinoclavaminic acid (278), which can be hydrolyzed to proclavaminic acid (279). CAS then performs oxidative cyclization between the C3 hydroxyl group and the C4′ position in the β-lactam to yield dihydroclavaminic acid (281). CAS next performs a third oxidation reaction to dehydrogenatively form clavaminic acid (285), which can be eventually converted to 286. Scheme 42B shows two of the proposed mechanisms for CAS in the cyclization step. Route a is similar to the mechanism discussed in the formation of the orthoether linkage in Scheme 41. The hydrogen on C4′ can be first abstracted by the FeIV=O ferryl species to form the C-radical 280. A second hydrogen abstraction on the C3 hydroxyl group and accompanying C-O coupling forms the oxazolidine ring and 281. In route b, the first hydrogen abstraction is on the hydroxyl group to form O-radical 282, followed by a retro-aldol-like decomposition to form an aminoaldehyde and a C2 radical 283. After another hydrogen abstraction from C4′ to yield the azomethine ylide 284, a 1,3-dipolar cycloaddition between the ylide and the aldehyde forms the 281. Regardless of the mechanism, CAS is a remarkable enzyme that catalyzes three different oxidation reactions in one biosynthetic pathway: hydroxylation, C-O oxidative cyclization and dehydrogenation. The crystal structure of CAS showed an unusual jellyroll fold uncharacteristic of this family of enzymes.213,214
Scheme 42.

Mechanism of Oxidative Cyclization During Clavaminic Acid Formation
Another iterative nonheme iron α-KG-dependent oxygenase is hyoscyamine 6β-hydroxylase (H6H) found in the biosynthetic pathway of the classic plant alkaloid scopolamine (290, also known as S-hyoscine) (Scheme 43A).215–217 290 is a tropane alkaloid that is used in the treatment of motion sickness and postoperative nausea. The tropane portion is derived from putrescine (287), which originated from L-ornithine. The phenyl portion is derived from L-phenylalanine via oxidative rearrangements catalyzed by P450. Conversion hyoscyamine (288) to 290 is catalyzed by H6H via two successive oxidation steps (Scheme 43B). In the first step, H6H catalyzes the hydroxylation at the C6 position via hydrogen abstraction and [OH•] rebound to yield 6β-hydroxyl-hyoscyamine 289. The second step catalyzed by H6H is the oxidative cyclization to form the epoxide. The likely mechanism is first abstraction of hydrogen from C7 to generate a C-radical (291), followed by abstraction of hydrogen from the freshly introduced C6-OH to facilitate radical C-O coupling.
Scheme 43.

Proposed Mechanism of Epoxide Formation in Scopolamine by Hyoscyamine 6β-Hydroxylase
Verruculogen (292) is a tremorgenic mycotoxin produced by various Aspergillus and Penicillium strains (Scheme 44A).218 The most unusual feature of 292 and its prenylated derivative fumitremorgin A (293) is the eight membered endoperoxide ring bridging the two middle rings of the 6-5-6-6-6 system. We have previously discussed the P450 catalyzed C-N bond formation that gives rise to the unusual 6-5-6-6-6 system in fumitremorgin C (219) (Scheme 34, section 2.2.5). Further modification in A. fumigatus converts 219 to fumitremorgin B (294), which is N-prenylated in the indole ring and hydroxylated in the middle ring (at C13). FtmOx1 is a nonheme iron α-KG-dependent oxygenase that connects the two linear prenyl chains in 294 with molecular oxygen to yield the endoperoxide-containing ring of 292.219 The crystal structure of FtmOx1 was solved and detailed mechanistic studies were carried out by Zhang and coworkers.220 The results from the assays with different ratios of α-KG/FtmOx1/substrate suggested one equivalent of α-KG and two equivalents of O2 were consumed during each turnover. The C13 ketone version of 292, 295, is the dominant product under the assay conditions. One important finding is that a tyrosine residue (Y224) was confirmed to participate in the catalytic cycle of FtmOx1 during endoperoxide formation.
Scheme 44.

Proposed Mechanism of FtmOx1 Catalyzed Endoperoxide Formation
A radical mechanism was proposed for the endoperoxide formation as shown in Scheme 44B. The first molecule of oxygen is activated to form the active species FeIV=O. Then a tyrosine (Y224) O-radical was formed through a hydrogen abstraction step. This phenoxy radical then abstracts a hydrogen from the N-linked prenyl group to yield 296 which is a C-radical that can be resonance stabilized. The C-radical then adds to a second molecule of oxygen to yield a peroxyl radical 297, which can undergo radical addition to the olefin present in the prenyl chain of the neighboring ring to forge the endoperoxide ring and the C-radical 298. Y224 is then re-oxidized to a tyrosine O-radical and affords 292. Starting from here, two pathways are possible: in the first pathway, this tyrosine O-radical can restart the catalytic cycle to turnover additional endoperoxide 292 from 294. In the second and more dominant pathway observed under assay conditions, a two electron oxidation of the C13 hydroxyl group in 292 to 295 reduces the iron in FtmOx1 back to the resting state.
2.3.3. C-N Coupling
The examples in this section are not direct radical coupling between carbon and nitrogen atoms. Instead, these example illustrate a sequence in which a nonheme iron α-KG-dependent enzyme catalyzes oxidation of unactivated C-H bond leading to an electrophilic carbon that can be subjected to nucleophilic attack by a nitrogen to form a new C-N bond and a cyclic structure. We previously illustrated this example in the P450-catalyzed conversion of holyrine (47) to staurosporine (20) (Scheme 14, Section 2.2.2). In both examples here, formation of unnatural, cyclized amino acids that are incorporated into NRP assembly lines are illustrated.
The tuberactinomycin family natural products, such as viomycin (299) and capreomycin IIA (300) (Scheme 45) are pentacyclic NRPs produced by different actinomycetes and display anti-tuberculosis activity.221 The common pharmacophore of this family has a number of nitrogen-containing unnatural amino acids, including the cyclic guanidine 2S,3R-capreomycidine (301), β-lysine and β-ureidodehydroalanine. The cyclic guanidine is essential for the activity of the tuberactinomycin compounds and has been found to be derived from L-arginine (302). Two enzymes are required for the cyclic C-N bond formation.222,223 The first enzyme (VioC) is a nonheme iron α-KG-dependent oxygenase that hydroxylates the β-position of 302 to yield 3S-OH-L-arginine 303. The second enzyme (VioD) is a pyridoxal phosphate (PLP) dependent aminotransferase that reacts with 3S-OH-L-arginine to form an PLP-imine 304. Elimination across α-β carbons affords (E)-2,3-dehydroarginine/PLP adduct 305, which can undergo cyclization by nucleophilic attack of the guanidine amine to give 306 and subsequent product release to afford 301.
Scheme 45.

Mechanism of C-N Coupling in Capreomycidine Formation
Methylproline residues are found as building blocks in numerous NRP natural products, such as echinocandin B (307)224, nostopeptolide A1 (308)225, polyoxypeptin B (309)226, griselimycin (310)227, perthamide C (311)228, and monamycin D (312)229 (Scheme 46A). Different combinations of methyl substitution regioselectivity and stereochemistry on the prolyl ring have been observed, including 2S,4R-4-methylproline (317), 2S,4S-4-methylproline (321), and 2S,3S-3-methylproline (326). The dedicated biosynthetic machinery for synthesizing 317 and 321 have been found, all originating from the acyclic aliphatic amino acid L-leucine (313) (Scheme 46B). In the echinocandin pathway, formation of 317 requires EcdK, a nonheme iron α-KG-dependent oxygenase.230 Enzyme assays support the mechanism of consecutive hydroxylation of the terminal methyl to yield 4R-5-OH-leucine first then the gem diol product 314, which can dehydrate to form the γ-methyl-glutamic acid-γ-semialdehyde (315) that can exist in equilibrium with the cyclic imine 3R-methyl-Δ1-pyrroline-5-carboxylic acid (316). Subsequent reduction in an NAD(P)H-dependent fashion can afford 317. Although no such reductase was found in the ecd pathway, it was proposed that the pyrroline-5-carboxylate reductase from the proline biosynthetic pathway could be shared. In the biosynthesis of 321 for nostopeptolide assembly in Nostoc sp. GSV224.6, Moore and coworkers showed following the first hydroxylation to yield 4S-5-OH-leucine (318), an NAD+ dependent alcohol dehydrogenase NosE catalyzes the dehydrogenation of the hydroxyl group to form the aldehyde 319.231,232 The cyclized imine 320 can be reduced by NosF, a dedicated reductase to yield the building block 321. Ogawa subsequently found an α-KG dependent leucine 5-hydroxylase in Nostoc punctiforme that can hydroxylate 313 to 318, suggesting this enzyme is widely present in other Nostoc species, including the nostopeptolide producer.233 Hence the two examples above provide contrasts in biosynthetic strategy to generate the 4-methylproline piece. In fungi, a radical mechanism via α-KG oxygenase is employed all the way to the aldehyde product, while in bacteria the first hydroxylation is radical dependent but the second oxidation is NAD+ dependent, a route that will be discussed more in section 3.5. Although the exact enzymes in biosynthesis of 326 have not been identified, it is expected that similar transient oxidation of the C5 position in L-isoleucine (322) followed by cyclization and reduction are involved (Scheme 46B).
2.4. Other Fe-Dependent Oxygenases and Oxygen-Independent Radical Cyclases
Besides P450 and nonheme iron α-KG-dependent oxygenases, nature also uses an arsenal of other iron-dependent oxygenases in oxidative cyclization. These include i) mononuclear, nonheme dependent oxygenase, such as HppE involved in epoxide formation in fosfomycin biosynthesis, and isopenicillin N synthase (IPNS) in penicillin biosynthesis; and ii) Rieske-type oxygenases that are non-heme iron-dependent oxygenases using a [2Fe-2S] cluster as one electron mediators.
In addition there is an extensive set of molecular oxygen–independent cyclases that work by radical mechanisms. These are the radical SAM enzymes that use bound 4Fe-4S clusters to cleave SAM into methionine and 5′dA•, the latter serving as the radical initiator. This section will cover notable examples of cyclization catalyzed by enzymes in each of these subfamilies.
2.4.1. Noncofactor Fe-Dependent Peroxidase: Fosfomycin Biosynthesis
Fosfomycin (332) is a representative member of the phosphonate family of natural products, all of which contain a C-P bond that is rarely observed among primary metabolites (Scheme 47A).234,235 332 is a clinically useful antibiotic produced by various Streptomyces and Pseudomonas species.236 The warhead of 332 is the epoxide ring and its biological target is uridine diphosphate (UDP)-N-acetylglucosamine enolpyruvyl transferase (MurA), which catalyzes the first committed step in bacterial cell wall peptidoglycan assembly.237 Biosynthetic studies had shown that the biosynthesis of all phosphonates start with the action of PEP mutase, which isomerizes phosphoenolpyruvate (327, PEP) into phosphonopyruvate (328). Following decarboxylation to 329, aldehyde reduction to HEP (330), and C-methylation by a radical SAM-and cobalt-dependent enzyme (Fom3), the immediate precursor to 332, (S)-2-hydroxypropylphosphonate (331, S-2-HPP) is formed.238 The last step is the oxidative cyclization of 331 to form the epoxide ring and 332, which is catalyzed by a mononuclear non-heme iron dependent enzyme HppE.239,240 HppE has a cupin fold that is associated with other noncofactor dependent oxygenases.241 The mechanism of HppE remained elusive, especially with regard to the nature of the oxidant. Recent studies by Liu and coworkers confirmed that HppE is actually a non-heme iron peroxidase.242
Scheme 47.

Biosynthesis of Fosfomycin and Proposed Mechanism of HppE
The mechanism for the H2O2-driven epoxide ring formation is shown in Scheme 47B: the substrate 331 is first coordinated to the active site FeII through the C2 oxygen and one of the phosphate oxygen atoms (in addition to two His and one Asp residue from HppE) to give 333. Reduction of H2O2 to H2O leads to the formation of the high-valent FeIV=O reactive species 334, which abstract a hydrogen from the C1 methylene to form 335 and is followed by an additional one electron oxidation to form a C1 carbocation 336 that can be stabilized by the adjacent phosphonate. Quenching of the carbocation 336 by the C2 oxygen forms a new C-O bond and completes the biosynthesis of the epoxide ring in 332. The notable feature of the overall reaction catalyzed by HppE is that the oxygen atom in the epoxide ring originates from the C2-hydroxyl group of 331, not from H2O2 or O2. During the mechanistic studies of HppE, this enzyme was shown to catalyze two interesting reactions when alternative substrates are used (Scheme 47C): R-2-HPP (337) is converted to the ketone 338, while S-1-HPP (339) is converted to the aldehyde 340 via oxidative 1,2-phosphono migration reaction.243,244
2.4.2. Noncofactor Fe-Dependent Oxidase: Penicillin and Cephalosporin Biosynthesis
Penicillins and cephalosporins are well known β-lactam antibiotics for the treatment of bacterial infections. The β-lactam antibiotics irreversibly inhibit the function of transpeptidase that are essential to the biosynthesis of the bacterial cell wall peptidoglycan.245 The shared precursor to penicillins and cephalosporins is isopenicillin N (345), which is synthesized in two steps (Scheme 48A).246,247 In the first step, a trimodular NRPS ACV synthetase assembles the tripeptide precursor δ-(L-α-aminoadipoyl)-L-cysteinyl-D-valine (ACV) (344). The linear tripeptide 344 is then oxidatively cyclized into the fused 4,5-ring system in 345 by the non-heme iron dependent oxidase, isopenicillin N synthase (IPNS). In the biosynthetic pathway to cephalosporins, 345 will be epimerized to penicillin N, followed by ring expansion of the 4,5-fused ring system into the 4,6-fused ring system by deacetoxycephalosporin C synthetase, another non-heme iron dependent enzyme, which will be discussed in Scheme 131 (Section 6.2.2).
Scheme 48.

Mechanism of Isopenicillin N Synthase
Scheme 131.

Oxidative Ring Expansion in Desacetoxycephalosporin C Biosynthesis
Mechanistic studies of the IPNS have been performed for decades and we will only briefly summarize here as other reviews can provide more detailed analysis. As shown in Scheme 48B, the reaction proceeds in two stages, each resulting in a two electron oxidative cyclization via C-H bond cleavage. The net result in terms of oxygen consumption is that one O2 is reduced by four electrons into two water molecules, therefore the cofactor α-KG is not needed. In the first stage, the substrate 344 and molecule oxygen bind to the active site iron center, forming the peroxide radical 347, which can abstract a hydrogen from the β-carbon of the cysteine residue to form the C-radical 348 (first C-H cleavage). After this, a C=S double bond and 349 is formed with one electron reduction of the FeIII to FeII. Nucleophilic attack of the valinyl amide nitrogen on the thioaldehyde forms the β-lactam ring in 350, this is accompanied by cleavage of the peroxo bond to generate FeIV=O and loss of water. In the second stage, a valinyl radical 351 is generated via hydrogen abstraction by the ferryl-oxoiron (second C-H cleavage). Attack of the valinyl radical on the sulfur yields the new C-S bond and thereby the thiazolidine ring in 352 and completes the penicillin core structure that is released as 345. Recent transient-state kinetics and spectroscopic methods with labeled substrates led to the identification of both C-H cleavage intermediates (348 and 351), thus validating the proposed mechanism.248 We also note the comparison to the 4,5-lactam-oxazolidine ring system formed during clavulanic acid (286) biosynthesis in Scheme 42 (Section 2.3.2). There the β-lactam ring is formed nonoxidatively and α-KG is used as cofactor by CAS to generate the high valent ferryl oxoiron required for the oxazolidine ring formation.
2.4.3. Rieske Oxygenase: Prodiginine Biosynthesis
Prodiginines, including metacycloprodigiosin (353)249, streptorubin B (354)250, and marineosin A (355)251, are a family of red-pigmented compounds containing the tri-pyrrole moiety (Scheme 49A). These 10- and 12-membered carbocycles have different bioactivities, such as immunosuppressive and anticancer activities, and are produced by both Gram-positive and Gram-negative bacteria.252,253 The most interesting connectivities in the structures of members in this family is the C-C bond between the pyrrole ring and the unreactive alkyl side chain. Oxidative cyclizations of the carbocycle from the acyclic precursors in 353 and 354 are catalyzed by the Rieske non-heme iron-dependent oxygenases McpG and RedG, respectively.254 The substrate of these enzymes is the linear undecylprodigiosin (356), which is derived from the condensation of 4-methoxy-2,2′-bipyrrole-5-carbaldehyde and 2-undecylpyrrole.255 The differences in regioselectivity and stereoselectivity of the C-C bond forming step lead to different products as seen in Scheme 49A.
Scheme 49.

Proposed Catalytic Cycle of Rieske Oxygenases in Prodiginine Biosynthesis
Rieske oxygenases are non-heme iron-dependent oxygenases with a N-terminal [2Fe-2S] cluster and a C-terminal nonheme iron center. Electron transfer from the [2Fe-2S] cluster to the iron center is facilitated by a hydrogen bond network between them. The proposed catalytic cycle of RedG is shown in Scheme 49B: after the substrate 356 and oxygen sequentially bind to the nonheme iron center, and with the assistant of one electron transfer from the [2Fe-2S] cluster, the FeIII-OOH complex abstracts the pro-R hydrogen from the alkyl chain of 356 and generates the FeIV=O species.256 The C-radical in the 357 then adds to the pyrrole ring stereoselectively to form the new C-C bond and the (S)-configured carbocycle 358. Another hydrogen abstraction from the hydropyrrole ring by the ferryl oxoiron, followed by rearomatization forms the pyrrole ring and the product 354. Reduction of the active iron center to the resting state releases the product.
In the biosynthetic pathway of 355, the enzyme responsible for the oxidative cyclization is MarG, which is a homolog to RedG and McpG.257 The C-H cleavage and C-C bond forming steps of MarG are similar to RedG starting from the acyclic 359 and the radical intermediates 360 and 361. A subsequent nucleophile attack of the terminal hydroxyl group on the double bond of 362 forms an additional 6-membered ring fused to the carbocycle (Scheme 49C).
2.4.4. Radical SAM Enzyme: C-C Coupling
The mechanism of radical SAM enzymes is shown in Scheme 3. Although radical SAM-mediated oxidative cyclization is not as widespread as the other iron-dependent enzymes shown above, several notable examples warrant discussion here. Because of the autoxidizable 4Fe/4S cluster that is the donor of one electron to the bound SAM substrate, the radical SAM enzyme family not only does not use O2 as cosubstrate, this enzyme family is inactivated by O2 so these reactions run in anaerobic microenvironments.
Streptide (363) is a ribosomally biosynthesized peptide (RiPP) that produced by Streptococcus thermophiles, a non-pathogenic streptococcal model strain used in the fermentation of dairy products.258 Previous studies by Monnet and co-workers demonstrated that 363 is a nonapeptide processed from a 30-mer ribosomally translated precursor 364.259–261 Recently, Seyedsayamdost and coworkers solved the structure of this compound and elucidated the biosynthetic pathway (Scheme 50).258 A macrocycle is generated by forming a C-C bond between the Cβ of the terminal lysine residue and C7 of the phenyl ring of a tryptophan residue. This key oxidative cyclization step is catalyzed by a radical SAM enzyme StrB acting on the 30-mer precursor peptide 364. A mechanistic model for the oxidative coupling of the otherwise unreactive carbons is shown in Scheme 50. The first step is to form the radical reagent, 5′-dA•, from SAM and iron-sulfur clusters in the active site of StrB. The 5′-dA• initiates the radical mediated cyclization by first abstracting a hydrogen from the Cβ of the Lys residue to form a new C-radical 365. This is followed by radical addition to the phenyl ring of the Trp residue, creating the Lys-to-Trp crosslink and an indolyl radical 366. Reduction of the auxiliary iron–sulfur cluster together with deprotonation and rearomatization complete the biosynthesis of the cross-linked peptide 367, which can be further processed by a protease to give 363.
Scheme 50.

Proposed Mechanism of C-C coupling in Streptide Biosynthesis
Menaquinone (374), also known as vitamin K2, is a membrane-soluble molecule that delivers electrons to protein complexes in the respiratory chain. The classic biosynthetic pathway of 374 involves eight steps starting from chorismate.262 Dairi and coworkers discovered an alternative chorismate-derived pathway to menaquinone biosynthesis, named the futalosine pathway (Scheme 51).263 Futalosine (368) is converted to dehypoxanthine futalosine (DHFL, 369) by the hydrolase MqnB, followed by a C-C coupling step that affords cyclic-DHFL (CDHFL, 372) by the radical SAM enzyme MqnC. The spirocyclic 372 is further converted to 1,4-dihydroxy-6-naphthoic acid (373), the precursor to 374. The proposed mechanism of the oxidative cyclization catalyzed by MqnC264 is a radical addition mechanism that parallels that proposed for the cyclization step of streptide shown above. After 5′-dA• mediated generation of the C-radical 370, radical addition to the phenyl ring forges the new C-C bond in 371. Deprotonation and one electron oxidation yields 372.
Scheme 51.

Proposed mechanism of C-C Coupling in Menaquinone Biosynthesis
Jawsamycin (FR900848, 378) is a potent antifungal natural product produced by Streptoverticillium fervens HP-891 (Scheme 52A).265 It is one of only two natural products isolated to date that contain the highly unusual polycyclopropanated fatty acyl chain, which has resemblance to the jaws of sharks that accounts for the compound name. In 378, the polycyclopropanyl chain is amidated with 5′-amino-5′-deoxy-5,6-dihydrouridine. Feeding studies suggested the fatty acyl chain is derived from a polyketide pathway, while the methylene groups of the cyclopropane units are derived from L-methionine.266,267 Recently, Oikawa and coworkers identified and heterologously reconstituted the biosynthetic pathway of 378.268 A notable finding is that a set of three enzymes are responsible for forming the polycyclopropanated polyketide, they are the iterative PKS Jaw4 (KS-AT-DH-ACP), the associated ketoreductase Jaw6 and the radical SAM enzyme Jaw5. Based on the lack of polyketide production in the absence of Jaw5, and the incorporation of the cyclopropanated acyl fragments into the final product, it is proposed that Jaw5 functions on the α–β unsaturated polyketide acyl chain after each iteration of chain extension, ketoreduction and dehydration.
Scheme 52.

Proposed Mechanism of Radical-SAM Enzyme Catalyzed Cyclopropanation in Jawsamycin
Two possible mechanisms are proposed for the Jaw5-catalyzed cyclopropanation, using the first polyketide intermediate crotonyl-ACP 375 as an example (Scheme 52B). The two mechanisms use reactive species of SAM generated by 5′-dA•, hence two SAM per catalytic cycle are consumed as in other radical SAM methyltransferases.269 The first hydrogen abstraction facilitated by 5′-dA• yields the SAM methyl radical 383, which may receive an electron from the [4Fe-4S] cluster to become the SAM ylide 384 (Scheme 52C). In route a, SAM ylide 384 performs a 1,4-addition on 375 to generate the adduct 379, which can eliminate SAH to yield the methylcyclopropyl thioester 376. In route b, direct attack of the SAM radical 383 on the double bond in 375 yields the adduct 380 that can afford 376 through the elimination of the SAM radical carbocation, which can be further reduced to SAH by receiving one electron from the [4Fe-4S] cluster. The cyclopropanated polyketide intermediate 376 is then proposed to undergo additional chain extension to assemble the full length polyketide product that will be ligated to 5′-amino-5′-deoxy-5,6-dihydrouridine. Effectively, Jaw5 becomes part of the PKS programming cycle to stereoselectively convert the α–β unsaturation into the R-cyclopropane. Interestingly, the timing of Jaw5 is stringently controlled as three of the unsaturations are retained in the final structure, suggestive of close coordination of the three enzymes during construction of the remarkable compound.
2.4.5. Radical SAM Enzyme: C-S Coupling
Biotin (389) is used as an enzyme cofactor in many enzyme-catalyzed reactions involving CO2, such as carboxylation, decarboxylation, and trans-carboxylation (Scheme 53). The structure of 389 contains a ureido (tetrahydroimidizalone) ring fused to a thiophane ring (5,5-ring system). The last step in biotin biosynthesis is the insertion of sulfur into dethiobiotin (385) to form the thiophane ring, and is catalyzed by a radical SAM enzyme named biotin synthase. Mechanistic studies confirmed that the [4Fe-4S] cluster is responsible for the 5′-dA• formation, while a separate [2Fe-2S] cluster is responsible for sulfur insertion and cyclization.270–274 A brief mechanism is shown in Scheme 53: following homolytic cleavage of the C-H bond in the methyl group by 5′-dA•, attack of the resulting C-radical 386 on the μ-sulfide within the [2Fe-2S] cluster forms the first C-S bond and 9-mercaptodethiobiotin 387. Subsequently, a new 5′-dA• radical cleaves a second C-H bond in the methylene group. The new C-radical 388 attacks the same sulfur atom completes thiophane ring formation and releases 389 from biotin synthase.
Scheme 53.

Mechanism of C-S Coupling Steps in Biotin Biosynthesis
The 35-residue head-to-tail cyclized subtilosin A (390) is a ribosomally synthesized antibiotic produced by soil bacterium Bacillus subtilis (Scheme 54A).275,276 It belongs to the sactipeptide family that features unusual sulfur to Cα thioether bridges that rigidify the macrocycle at multiple positions. In 390, there are three thioether bridges of which two are between cysteine thiol and Cα-Phe and one is between cysteine thiol and Cα-Thr. Recent biosynthetic studies showed that the formation of all three thioether bridges are catalyzed by a radical SAM enzyme AlbA that acts on the linear 43-mer precursor peptide.276,277 Similar to streptide biosynthesis (Scheme 50), completion of oxidative cyclization is followed by leader peptide cleavage and macrocyclization to yield 390, which is exported to the extracellular space by an ABC transporter.
Scheme 54.

Proposed Mechanisms of C-S Coupling Steps in Subtilosin Biosynthesis
A C-S radical coupling mechanism was first proposed to account for the reaction catalyzed by AlbA.276 As shown in Scheme 54B, the reaction is initialed by the removal of a proton from the thiol group to form the iron-sulfur coordinate bond in 391, with the iron coming from the [4Fe-4S] cluster. Then cleavage of SAM generates a 5′-dA• radical that can cleave the Cα-H bond in either the Phe or Thr residue. The resultant C-radical 392 is then coupled to the cysteine thiol to release from the [4Fe-4S] cluster and form one thioether bridge. More recently, Berteau and coworkers performed mutagenesis studies on AlbA and proposed a different nucleophilic addition mechanism (Scheme 54C).277 The first step is proton abstraction from the cysteine thiol but no Fe coordination. The second step remains the 5′-dA• mediated cleavage of Cα-H bond to form the Cα radical 393. However, instead of radical coupling as in Scheme 54B, the Cα radical migrates to become an O-radical 394 through resonance, and is further transformed to an imine 395 by one electron oxidation. Finally, the C-S thioether bond is formed through the nucleophile addition of deprotonated thiol to imine.
3. NONRADICAL CYCLIZATION MECHANISMS
3.1. Introduction
The previous section dealt with a set of cyclizations initiated by oxidative reactions proceeding through carbon radical intermediates, many of them driven by enzymatic reductive activation of molecular oxygen. Analogously, the next section in this review deals with oxygenases in the form of epoxide-mediated cyclizations.
In this section we consider ring formations attendant with or subsequent to two electron redox pathways. These can, in a few cases involve enzymatic reductions that set up cyclizations, as in nostocyclopeptide, saframycin and ikarugamycin assembly, but are largely cyclizations enabled by oxidation of substrates. The oxidations can involve hydride transfers from substrates as in the berberine bridge and related flavoenzymes, or can yield carbonyl groups that undergo intramolecular capture, as in spirocycle formation in reveromycin. Internal capture of transient imines occurs in scaffolds as simple as the pipecolic acid building blocks and as complex as in fumiquinazolines C and D and saframycin.
We also take up a set of biosynthetic transformations that are formal [4+2] cyclizations, including solanapyrone, lovastatin, spinosyn, abyssomycin and pyrroindomycins. In some of these enzyme cases evidence for Diels-Alder concerted mechanisms is gradually accruing. The cyclizations themselves, whether stepwise or concerted, are nonoxidative and will be covered in a review by Liu et al in this issue. However, the prior enzymatic steps, setting up the diene and dienophile combinations are oxidative. Viewed in this context we look at the two step enzymatic logic for such biosynthetic cyclopentyl and cyclohexyl ring constructions.
Scheme 55 depicts four general possibilities for the reactions in this section. Scheme 55A involves reduction of a carboxylic acid derivative (typically a thioester) to the aldehyde, followed by internal capture by an amine nucleophile. Scheme 55B schematizes intramolecular attack by a nucleophile concomitant with, or after, hydride transfer out of a substrate. Scheme 55C reflects oxidation of a substrate alcohol or amine group to the carbonyl or imine followed by internal capture by a nucleophile. Often subsequent reduction occurs as in pipecolate formation. Scheme 55D suggests assembly-line redox transformation to set up the diene or dienophile for subsequent [4+2] (or perhaps [6+4]) ring-forming reaction.
Scheme 55.

Models of Nonradical Cyclization in Natural Product Biosynthesis
3.2. Reduction-Enabled Cyclization
In the biosynthesis of polyketide and nonribosomal peptide frameworks by PKS and NRPS assembly lines, the polyketide or peptidyl chains grow as a series of elongated thioesters, tethered to carrier protein domains.278 The canonical mechanism of product release is through the redox-neutral mechanisms of hydrolysis, esterification and amide formation.40,279 This is the basis for forming most macrocycle natural products. There are also increasing examples of NAD(P)H-dependent reduction as a release mechanism. The mechanism involves first the reduction of the thioester 396 to the thiohemiacetal 397 by hydride transfer, and this tetrahedral adduct decomposes spontaneously to release the aldehyde product 398 (Scheme 56A). Sometimes the aldehyde can persist, but in many cases the aldehyde is captured by intramolecular nucleophilic attack to form a cyclized product. Attack by alcohol can yield hemiacetal, while attack by amine can lead to imino formation. Alternatively, in PKS-NRPS assembly lines, the aldehyde can be attacked by the Cα carbon of the 1,3-diketo group of the polyketide fragment to yield tetramate or pyrrolidone structures. These examples are abundant, here only a few will be highlighted.
Scheme 56.

Reduction-Enabled Cyclization in Nostocyclopeptide A2 Biosynthesis
3.2.1. Nostocyclopeptide: Reductive Release from NRPS
Nostocyclopeptides, such as nostocyclopeptide A2 (401), are cyclic peptides produced by terrestrial cyanobacterium Nostoc sp. ATCC53789 (Scheme 56B).280 Several features of 401 are worth noting, including the incorporation of the unnatural amino acid such as 4-methylproline of which the formation was discussed in Scheme 46 (Section 2.3.3). The imino linkage between the α-amino group of Tyr1 and the carbonyl of Phe7 suggests reductive release of the nascent linear heptapeptide 399. This was reflected in the last module of the NPRS in which a Reductive domain (R) was found.281 The formation of an aldehyde intermediate 400 in an NADH fashion and the subsequent capture by amino of Tyr1 was confirmed in vitro.282 The carbinolamine adduct loses water and the cyclic imine is stable as found in the accumulating product.
3.2.2. Saframycin: Iterative Pictet-Spengler Condensations
Saframycins including saframycin A (413) are NRP-fatty acid hybrid compounds produced by Streptomyces and Pseudomonas species, and are members of the tetrahydroisoquinoline natural product.283 This family displays strong antitumor properties and is best represented by the saframycin analog ecteinascidin 743 (ET-743) which is approved for treating soft-tissue sarcoma.284,285 The central pentacyclic core of 413 from S. lavendulae and ET-743 are the same, thereby attracting interests to understand saframycin biosynthesis towards rational engineering the pathway to afford ET743, of which a likely biosynthetic gene cluster was identified through metagenomic approach.286,287
Oikawa and coworkers showed a reductive NRPS assembly line release mechanism is in play (Scheme 57).288 Three peptidyl aldehydes (405, 409 and 411) are formed transiently via hydride-mediated release from the carrier protein thiol arm of the NRPS assembly line. The first is the N-acyl-Ala-Gly dipeptidyl aldehyde 405 derived from thioester 404. It is condensed with the amine of the tyrosyl-derived methyl DOPA-S-carrier protein intermediate 403. The initial imine 406 is then captured intramolecularly (Scheme 57A) by C2 of the catecholic ring, acting as an accessible carbanion by virtue of the adjacent phenol oxygen. This adduct 408 then undergoes a second cycle of NADPH mediated reductive release to yield 409. It then is captured by the identical aminoacyl-S-carrier protein 403 for a second Pictet Spengler reaction, creating the second bicyclic scaffold 410. This peptidyl thioester is now subjected to a third NADPH-mediated reductive release cycle to give 411. When 411 is captured internally by the indicated secondary amine to yield 412, a stable cyclic carbinolamine ring is formed and accumulates (Scheme 57B). The iterative strategy of three reductive release steps on increasingly complex peptidyl thioesters builds the three core six member rings in the saframycin pentacyclic scaffold.
Scheme 57.

Pictet-Spengler Reactions in Saframycin A Biosynthesis
3.2.3. Leporin: Inverse Electron-Demand Diels-Alder
The 2-pyridone leporin B (419) is synthesized by a PKS-NRPS hybrid enzyme from A. flavus (Scheme 58A).289 The biosynthetic gene cluster was identified from overexpression of a transcription factor in the cluster which led to the overproduction of the metabolite.289 419 was shown to form a trimeric complex with iron, which was attributed to its antiinsectan and antifeedant properties.289 The biosynthesis of 419 involves a number of initially unobvious biosynthetic transformations.289 The initial expansion of the five member tetramate product 414 from PKS-NRPS to the 6-membered 2-pyridone 415 was shown for aspyridone290 was described in Scheme 134 (Section 6.2.5). It is proposed that the subsequent steps begin with enzymatic reduction of the exocyclic ketone 415 to the alcohol 416. Its subsequent elimination as H2O creates the exocyclic double bond in 417. Although mechanistic details are not yet available, one formulation of the next step is an inverse electron-demand Diels-Alder reaction creating two rings in 418 simultaneously, involving the pyridone carbonyl as the surrogate diene. N-hydroxylation completes the assembly of tetracyclic 419.
Scheme 58.

Reduction-Enabled Hetero Diel-Alder Cyclization in Leporin Biosynthesis
Scheme 134.

P450 Catalyzed Tetramic Acid to Pyridone in Aspyridone Biosynthesis
The hetero Diels-Alder reaction was investigated using synthetic strategies as shown Scheme 58B.291 Knoevenagel condensation of 4-hydroxy-5-phenyl-2-pyridone (420) with 2-methyl-6E,8E-decadienal (421) forms the unstable O-quinone methide intermediate 417 that is also proposed for the biosynthetic transformation. A one pot experiment with 420 and 421 in ethanol and trimethylamine at 160°C for 20 hours afforded the desired inverse electron-demand Diels-Alder product 418 with 35% yield. A majority of products were the adduct 422 that displayed alternative stereochemistry as well as the normal Diels-Alder cyclization spiro-adduct 423. The latter also be formed from the Claisen rearrangement of 422 (but not 418). Therefore, the synthetic studies suggest enzymatic control is required during leporin biosynthesis to ensure formation of the required leporin intermediate. In the unrelated marine compounds dibenzo-p-dioxins, Moore and coworkers proposed a P450-catalyzed quinone formation leads to a hetero Diels Alder cycloaddition.292
3.2.4. Ikarugamycin Cyclization: Hydride-Mediated Coupling
Ikarugamycin (428) is isolated from Streptomyces sp. and displays a variety of potent biological activities.293 It is a member of the polycyclic tetramate macrolactams (PTMs) family that also includes clifednaminde (429)294, dihydromaltophilin (HSAF, 430)295 and frontalamide A (431)296. The PTMs are PK-NRP hybrid metabolites with a tetramic acid moiety and most notably 5,6,5 or 5,5,6 fused tricyclic element itself embedded in a 16-membered macrolactam.297 Later in this section of the review we will note the spinosyn system where a [4+2] enzymatic reaction has been shown for the comparable 5,6,5 tricyclic system (Scheme 83, Section 3.10.4). The logic of assembly of this structural element in ikarugamycin (and presumably its congeners) is completely distinct (Scheme 59).
Scheme 83.

Cyclization Steps in Spinosyn A Biosynthesis
Scheme 59.

Hydride-Mediated Cyclization in Ikarugamycin Biosynthesis
Zhang and coworkers elucidated the enzymatic basis for forming 428, which involves only three enzymes.298 The PKS-NRPS IkaA synthesizes the acyclic tetramic acid 424 that consists of two polyenyl fragments (pentaene and tetraene). IkaB has sequence homology to phytoene desaturase family enzymes, and has been proposed to be responsible for forming the first two rings (5,6 system), although the mechanism is not understood. Formation of the third ring is catalyzed by IkaC, an enzyme with sequence homology to alcohol dehydrogenase.298 The strategy to form this five-membered ring was shown to be reductive, driven by a hydride donation from NADPH on the terminus of the conjugated 1,6-dienone moiety in 425 (Scheme 59). The incipient carbanion (can be written with charge density all the way out on the oxygen) can mount a transannular attack on the terminus of the conjugated dienyl amide building the indicated cyclopentane ring in 426. Following IkaC-assisted isomerization (to yield 427) and ketonization, the natural product 428 is formed. The IkaABC cassette therefore represents an exceptionally concise enzymatic pathway; unsurprisingly, these three genes are well-conserved in the gene clusters of all PTMs family of products.296,298–300
3.2.5. Iridoid and Related Plant Metabolites
The 5,6-fused oxazepine system is found in iridoid plant metabolites as noted in Scheme 60, including nepetalactone (432)301, aucubin (433)302 and catapol (434)302.303 The iridoid intermediate nepetalactol (439) is an on-pathway metabolite to secologanin and strictosidine, the latter of which is the branch point for medicinally important monoindole alkaloids such as camptothecin and vinblastine.304 The bicyclic framework of iridoid arises ultimately from the common monoterpene building block geraniol (435). The C1-OH undergoes oxidation to the aldehyde while one of the terminal methyl groups is oxygenated twice in succession to yield the acyclic C1-C8 dialdehyde 8-oxogeranial (436). The enzyme iridoid synthase (ISY) responsible for the cyclization was identified using comparative transcriptome analysis from Catharanthus roseus.305 ISY has sequence homology to progesterone 5β-reductase (P5βR)306, a short-chain NADPH-dependent dehydrogenase. ISY mediates transfer of a hydride equivalent from NADPH to the C4 olefinic terminus of the enone 436 and generates an enolate anion 437. This sets up intramolecular cyclization to create the fused 5,6-bicyclic ring system in 439. This could happen in a stepwise fashion, by a Michael addition to form the cyclopentane 438a and then the oxazepine 439. Alternatively, a hetero Diels-Alder reaction, vide supra, as shown with 438b would construct both rings simultaneously as shown in the scheme. The product cis-trans 439 are in equilibrium with the ring opened iridodial (440).
Scheme 60.

Reductive Cyclization of Iridoid by Iridoid Synthase in Plants
The O’Connor group subsequently obtained the crystal structure of ISY to demonstrate the most likely pathway is the stepwise enolization and 1,4-addition.307 The backbone amide of Ile145 and the phenol of Tyr178 form an oxyanion anchor that stabilizes the enolate anion 437. The orientation of the active site and cocrystalized NADP+ is also consistent with the stereochemistry of the product. The enzyme is also proposed to undergo conformation change after enolate formation accommodate folding of the substrate and subsequent cyclization.
3.3. Oxidation Enabled Cyclization
The subsequent examples of ring formations during natural product assembly in this section proceed by oxidative not reductive mechanisms as depicted in Scheme 55B. The first set (Section 3.3) involves O-C, N-C, or C-C bond formation at the site where a hydride is ejected from the substrate and passed to FAD in the active site of the relevant enzyme. The second set (Section 3.5) involve oxidation of alcohol or amine groups to the corresponding carbonyl which are then captured by internal nucleophiles. A somewhat distinct mechanistic category that epitomizes oxidative ring construction are disulfide bond formations between two sulfhydryl groups in a metabolite.
3.3.1. Berberine Bridge Enzyme: Nucleophilic Attack with Hydride Transfer to FAD
The best characterized enzyme in this mechanistic class is the berberine bridge enzyme (BBE), involved in plant alkaloid biosynthesis.34,35,308–315 The enzyme converts (S)-reticuline (118) derived from L-tyrosine, to (S)-scoulerine (443) which is on the way to more complex benzophenanthridine alkaloids including berberine (205). BBE constructs a new C-C bond between the N-methyl and the catechol ring in 443 as shown in Scheme 61. The puzzling element in the reaction had been how the unactivated N-methyl group in 118 is converted to the bridging CH2 in 443 in a low energy path. Interestingly, there is no known synthetic counterpart to this reaction.
Scheme 61.

The Reaction Catalyzed by Berberine Bridge Enzyme in S-Scoulerine Biosynthesis
As shown in Scheme 61A, the BBE enzyme continues the theme of hydride transfer to or from substrates during biosynthetic ring constructions elaborated in Section 3.2. In this case it is FAD that is the hydride acceptor from substrate rather than NADPH the donor to substrate that sets off the reaction flux. The X-ray crystal structure of Eschscholzia californica BBE in complex with 118 showed a glutamic acid E417 is the catalytic base in deprotonating the phenol, and site-directed mutagenesis of this residue had significant effect on catalysis.311 This mechanistic investigation suggested hydride transfer and SN2 carbanion attack to form the C-C bond in 441 occur in a concerted fashion, facilitated by the deprotonation of the phenol (resonance contributor to the phenolate anion in a single transition state). More recently, a series of deuterium kinetic isotope effect studies were conducted with both the wild type BBE and the E417Q mutant, suggesting that deprotonation does not occur before or during C–H bond cleavage.310 Therefore, a step-wise mechanism for BBE enzyme was proposed (Scheme 61B), in which hydride transfer from the N-methyl to the bivalently attached flavin cofactor leads to formation of the iminium cation 442, followed by attack of the ortho phenolate anion to forge the C-C bond to generate 441, which can aromatize to yield 443.
3.3.2. Mechanistic Analogies to the Berberine Bridge Enzyme Transformation
A mechanistically comparable pathway to the plant BBE transformation was found through genome mining from A. fumigatus. Transcriptional factor overexpression accompanied with comparative metabolomics found the fsq cluster is able to synthesize the tricyclic isoquinoline fumoisoquins A (449) and B (450) and the related benzoquinone fumisoquins C (451).316 Formation of the berberine-like C-C bond is proposed to arise from a modified dipeptidyl-S-enzyme intermediate 444 on a two module NRPS assembly line (Scheme 62). On-assembly line N-methylation and catechol formation by dissociated enzyme partners generate the identical arrangement 445 for a BBE-like oxidative cyclization catalyzed by the flavin-containing enzyme FsqB to give 448. The reaction was diagrammed as a two-step process, hydride transfer to 446 followed by cyclization to 447 in the original publication (Scheme 62A), but has not been investigated for timing and could happen in one transition state as proposed above for the BBE, with hydride extrusion and C-C bond formation occurring synchronously to give 447 before rearomatization (Scheme 62B).
Scheme 62.

Proposed Role of BBE in Fumoisoquins Biosynthesis
The last steps in tetrahydrocannabinol (455) assembly involves cyclization of the geranyl side chain in 452 onto the electron rich aromatic polyketide moiety to form the tricyclic framework of the cannabinols.317 Scheme 63 illustrates a comparable chemical logic as BBE for ring construction catalyzed by THCA synthase. X-ray structure of THCA synthases showed a bivalently attached flavin cofactor, identical to that in the BBE enzyme.317 Phenol-assisted ejection of a hydride ion from the benzylic position in 452 to enzyme-FAD generates (perhaps in a single transition state) the conjugated dienone 453 that acts as electrophile for the π electrons of the terminal double bond. The pyran ring in 454 may be formed in the same transition state as suggested in Scheme 63. The immediate result is formation of the fused tricyclic platform of tetrahydrocannibinol is C-C and C-O bond formation. Tetrahydrocannabinolic acid (454) is readily decarboxylated to for 455, the active ingredient of marijuana.
Scheme 63.

Proposed Mechanism of Tetrahydrocannabinol Biosynthesis
Lankacidins, including Lankacidin C (459) produced from S. rochei are macrocyclic antibiotics that display significant antibiotic activities towards many bacteria, and are used in agriculture to treat porcine infections.318–323 The acyclic substrate LC-KA05 (456) is formed by a hybrid NRPS-PKS assembly line with γ-lactone formation as the termination and product release step.324 Concerted desaturation of the secondary amine to the product iminium (457) and capture by the enol carbon to create the new C-N bond that forms the 17 atom macrocycle lankacidinol A (458) is catalyzed by the enzyme LkcE (Scheme 64). Subsequent oxidation of α-OH and deacetylation affords 459. It is of note here that the macrocyclic framework of 458 has an embedded lactone but its formation is not the route to the macrocycle.
Scheme 64.

Desaturative Macrocyclization in Lankacidin Biosynthesis
An additional example of hydride removal to create an electrophilic center captured in ring formation is provided during biosynthesis of the mycotoxin cyclopiazonic acid (464) (Scheme 65). The pentacyclic metabolite, fashioned from polyketide, amino acid, and isoprenoid building blocks in a short, three enzyme pathway325,326 and is a nanomolar inhibitor of Ca2+-ATPase327 (Scheme 65). CpaS is a two module PKS/NRPS hybrid assembly line, notable for a Dieckmann type cyclization as release mechanism to generate the tetramic acid cycloacetoacetyl-tryptophan (461).328 CpaD is a prenyltransferase specifically installing the prenyl group at C4 of the indole ring to give 462.329 The third enzyme is the redox catalyst. CpaO contains covalently bound FAD and is proposed to utilize the indole NH as neighboring group to expel a hydride ion from the indicated methylene center to FAD.330–332 The conjugated indole iminium 463 is an electrophile capturable by the π electrons of the Δ3-olefin of the prenyl group. Simultaneous participation of the amide nitrogen from the tetramic acid moiety (analogous to the –OH participation in tetrahydrocannabinol construction just above) would again create two rings at once and give the angular pentacyclic framework of the potent mycotoxin 464.
Scheme 65.

Desaturative Cyclization in Cyclopiazonic acid Biosynthesis
Chlorizidine A (467) provides the sixth and final example in this subsection of a hydride expulsion/ring forming logic, in this case to make a new N-C bond rather than the O-C and C-C bonds noted thus far (Scheme 66).333 467 is a tetrachlorinated metabolite isolated from a marine Streptomyces, and contains an unprecedented pyrrolo-isoindolone ring system.334 The precursor molecule 465 shown in the scheme is derived a polyketide assembly line in which two molecules of dichloropyrrole are used as building blocks. Following pyrrole nitrogen mediated chain release and cyclization, phenolate-assisted expulsion of a CH2-derived hydride ion to FAD yields the conjugated enone 466 that is capturable by the dichloroimidazole nitrogen atom to produce the 5,5 bicyclic system.
Scheme 66.

Mechanism of Chlorizidine A Cyclization
These examples provided in this section highlight the ability of the two most abundant cellular redox coenzymes, NAD(P)H and FAD, to engage in hydride transfers, as donor and acceptor respectively, in the biosynthetic directions. This hydride transfer capability undergirds the central enabling chemical logic for this structurally and functionally diverse array of biosynthetic reductive and oxidative cyclizations in plants, bacteria and fungi. These are two electron pathways that lead to a variety of fused ring systems, in contrast to the manifold one electron pathways in the prior Section 2 which also enable great diversification of natural product frameworks.
3.3.3. Peptidyl Oxazoles and Thiazoles
A number of microbially generated nascent proteins can be morphed into small molecule stable frameworks containing five membered oxazole and thiazole rings.335–337 These include members of the cyanobactin family such as patellamides A (468) and C (469)338, telomestatin (470)339, and the thiocillin class of antibiotics (471) with a trithiazolylpyridine tetracyclic core340 (Scheme 67).
Scheme 67.

Selected Natural Products that Contain Oxazole and Thiazole Rings
The oxazole (477) and methyloxazole (478) rings arise from cyclization of Ser (472) and Thr (473) residues to the corresponding oxazolines, respectively.341 The thiazole ring (479) arises from Cys residues (474) via comparable thiazolidine intermediates.341 The cyclization reaction manifolds begin by attack of the Ser/Thr/Cys oxygen or thiolate side chain in the upstream carbonyl (Scheme 68). The resultant tetrahedral adducts 475 are enzymatically phosphorylated on oxygen by a cyclodehydratase that uses ATP as cosubstrate.342,343 Breakdown of the phosphorylated adduct in the front direction involves elimination of the elements of phosphoric acid to create the oxazoline and thiazoline rings (476).342,343 These are hydrolytically labile.
The redox step happens next. An FMN-containing dehydrogenase enzyme catalyzes proton abstraction from C4 and hydride transfer from C5 of the dihydroheterocycles 476 to create the 2,3-olefin that completes heteroaromatization to the oxazole and thiazole rings.341 These are stable and represent a dramatic morphing of the peptide backbone of the protein precursors into these protease-resistant heterocycles. The patellamides and telomestatin have heterocycles embedded within the macrocycle where some of the oxazolines have not undergone desaturative aromatization.338,339 The thiocillin biosynthetic pathway creates the three central thiazoles first with pyridine ring construction simultaneous with 26 atom macrocycle formation as the last biosynthetic step.344,345
3.4. S-S Bond Formation from Dithiols
The formation of disulfides from dithiols is featured as a common posttranslational modification in proteins excreted from cells into oxidizing environments. The dithiol-disulfide redox equilibrium is also central to primary metabolism to keep intracellular protein thiols essentially fully reduced.346 The agent for this function is typically the tripeptide glutathione, or its relatives in a variety of prokaryotes and single cell eukaryotes. Reduced glutathione is accumulated in a 10/1 ration over the glutathione disulfide by the enzyme glutathione reductase which uses hydride equivalents from NADPH, run through the bound FAD in the enzyme (Scheme 69A).347
Scheme 69.

Disulfide Bond Formation from Dithiols in Natural Products Biosynthesis
There are a relatively small number of natural products which utilize disulfide bridges as conformationally rigidifying and activity-conferring structural elements. Among them are the histone deacetylase (HDAC) inhibitor spiruchostatins of which FK228 (Romidepsin, 481) has been approved as a therapeutic agent,348,349 holomycins (485)350, and the epidithio/disulfide forms of the fungal DKP toxins, of which gliotoxin (483)351 is the best known (Scheme 69B). All of these metabolite sets are made by the producing microbes as the dithiol forms (480, 482 and 484) and in the last or penultimate biosynthetic steps, then oxidized to the disulfides by flavoenzymes in the glutathione reductase superfamily.352–356 The disulfide forms impart specific conformations to the pathway end products and may confer the toxigenic properties since they can react with thiols of cellular constituents (e.g. proteins) to form covalent mixed disulfide adducts.357
Two disulfide variants in natural products are worth mention. One is the disulfide monoxide link found in the DNA damaging macrocyclic leinamycin (488) produced from S. atroolivaceus (Scheme 70A).358,359 Enzymatic oxygenation of the disulfide 487 fromed from dithiol 486 is the likely origin of the disulfide monoxide group. The higher oxidation state of the sulfoxide destabilizes the S-S linkage and makes the sulfoxide more likely to depart in the presence of a nucleophile. Thus, one can consider this to be an activated form of a typical disulfide electrophile.
Scheme 70.

Examples of Disulfide Variants in Leinamycin and Echinomycin
The second intriguing variant is found in the dimeric echinomycins (492) (Scheme 70B). These are formed from the N-acylated-Ser-Ala-N-MeCys-N-MeVal tetrapeptidyl-S-enzyme species by head to tail dimerization as the release mechanism from the biosynthetic NRPS assembly line.360 The bislactones 489 have the two Cys-SH side chains in proximity across the macrocyclic ring and are subject to oxidation to the disulfide 490 as shown. This could be nonenzymatic via a sulfenic acid, now electrophilic for capture by the transannular -SH partner. Of special note is that the bridging disulfide can undergo a catalyzed rearrangement to the thiomethyl thioether in 492 in the presence of S-adenosylmethionine and via intermediate 491.361 Unlike the disulfide which is labile to reduction, the thioether bridge formation should be irreversible and may provide a locked in conformation that is relevant to its bioactivity.
3.5. Capture of Transient Carbonyls
Perhaps the most straightforward two electron route to oxidatively enabled ring formations during natural product biosynthesis is the use of NAD (or less often FAD) as hydride acceptor during transient oxidation of a substrate alcohol or amine to the corresponding ketone or imine. Capture by an intramolecular nucleophile could generate a stable ring system on its own (Scheme 55C, Section 3.1). If not, coupled re-reduction by the just generated NAD(P)H would yield the stable cyclic product.
3.5.1. Pipecolic Acid: Hidden Redox Mechanism
The enzymatic conversion of the proteinogenic amino acid L-lysine (497) to the six-member heterocyclic pipecolic acid (501) is a simple example of this ring-forming logic (Scheme 71).362,363 Enzymatic oxidation at the C2 amine yields the acyclic imine 498 and NADH. Capture by the C6 amine gives 499, and loss of NH3 from the tetrahedral adduct yields the Δ1 -cyclic imine 500. Now transfer of the hydride back from NADH to C2 yields the cyclic 501. The redox process is cryptic, does not show up in the reaction stoichiometry. 501 is the next higher homolog of the amino acid proline. It is incorporated into several PK-NRP hybrid natural products, among them rapamycin (493)364, FK506 (494)365, meridamycin (495)366 and antascomicin B (496)367 (Scheme 71). 493 and 494 are macrocycles that have been approved as therapeutic immunosuppressive agents. In these pathways, 501 is activated by the terminal NRPS-module and appended to the growing chain as an amide thioester, which is macrocyclized through an internal hydroxyl group to yield the macrolactone/lactam. 368–372
Scheme 71.

Formation of L-Pipecolic Acid in Natural Products
3.5.2. Lysergic Acid: Formation of the Fourth Ring
Lysergic acid (505) and its biosynthetic offsprings are members of the ergot alkaloids from the spores of the fungus Claviceps purpurea.373 These alkaloids can elicit a number of digestive, circulatory and neurological responses in humans. 505 is derived from the tricyclic intermediate chanoclavine-I (502), which is derived from the oxidative cyclization of 4-dimethylallyltryptophan (237). A comparable cyclization strategy to 501 is in play in the conversion of 502 to 505 (Scheme 72).373 NAD-linked dehydrogenation of the primary alcohol in 505 yields chanoclavine-I aldehyde(238) and NADH. The aldehyde can be attacked by the exocyclic N-methyl amine to give the carbinolamine, then the iminium ion 503 as shown. Addition of hydride back from NADH yields the stable cyclic amine in agroclavine (504). This metabolite is three oxygenations away from 505, all involving successive oxygen transfers to the methyl group. The last olefin isomerization to be in conjugation with the indole is thought to be spontaneous. The reversible oxidation of alcohol 502 to aldehyde 238 and back reduction of iminium 503 to secondary cyclic amine 504 hides the redox process from cursory inspection of reaction stoichiometry. 505 can then serve as a highly unusual starter unit for the LPS NRPS, which can ultimately lead to the formation of ergotamine peptide 266 that is discussed in Scheme 40 (Section 2.3.2).
Scheme 72.

Cyclization of Chanoclavine-I to D-Lysergic Acid
3.5.3. Reveromycin: 6,6-Spirocycle
The spiroacetal reveromycin A (515) from Streptomyces sp. SN-593 is a 35-carbon branched polyketide metabolite that can inhibit bone resorption by induction of apoptosis in osteoclast.374,375 The nascent polyketide RM-A2a (506) released from the polyketide synthase machinery undergoes a truncation and spiroacetalization tailoring by RevG and RevJ (Scheme 73A).376 A P450 mediated hydroxylation at C18 in 513 then introduces an OH group in 514 that is the site of subsequent hemisuccinylation in 515.
Scheme 73.

Oxidative Formation of 6,6-Spiroacetal as Shown in Reveromycin
The distinguishing 6,6-spiroacetal feature of reveromycin A is installed by action of the short-chain dehydrogenase RevG for NAD-dependent dehydrogenation of the C15-OH in 506 to the ketone in the acyclic precursor 507. Intramolecular hemiacetal formation creates the first six membered ring 508. Elimination of water by action of RevJ and transient formation of the oxonium ion 510 sets up stereospecific attack by the C11-OH to complete formation of the stable 15S acetal RM-A3a (512) while creating the spiro arrangement. In the absence of RevJ, the cyclization cascade can also occur, but affords a 3:2 ratio of the 15S to 15R isomers. Comparable 6,6-spiroacetal moieties are found in tautomycin (516)377, avermectin B1a (517),378 and spirangien A (518)379 which may utilize the same dihydroxyketone functional group array for spiroacetalization (Scheme 73B). The spiroketal formation in avermectin biosynthesis was characterized by Liu and coworkers in vitro378. On the other hand, the 5,6-spiroacetal linkage in monensin rises via epoxyketone cyclization as described in Section 4.2.1.
3.5.4. Fumiquinazoline C and D: Hemiacetal and Hemiaminal
A. fumigatus strains make fumiquinazoline peptidyl alkaloids constitutively.380 We note the formation of fumiquinazoline A and related tryptoquialanine in the Section 4 on epoxidative cyclizations at the 2,3-position of indole rings. In Scheme 74 the further enzymatic processing of fumiquinazoline A (519) to fumiquinazoline C (521) and D (522) by the FAD-dependent enzyme Af12070 is depicted.381 The enzyme oxidizes the secondary amine in the C ring of the quinazolinedione tricyclic moiety to the imine 520. This can be captured intramolecularly by the C3-OH of the imidazoindolone moiety to give 521 as the kinetic product. This spiroheptacyclic structure has a hemiacetal linkage. It can revert back to and equilibrate with the imine 520. The thermodynamic product that accumulates, nonenzymatically, at later time is the corresponding hemiaminal in 522, resulting from imine capture instead by the -NH of the imidazoindolone group, yielding an altered heptacyclic connectivity scaffold.
Scheme 74.

Oxidative Scaffold Maturation in Fungal Indole Alkaloid Biosynthesis
During tryptoquialanine biosynthesis, oxidation of 15-methyl-2-epi-fumiquinazoline A (523) by TqaG yields the analogous imine product 524 (Scheme 74B).382 The imine is then hydrolyzed by water to yield the acid 525, which can be captured by the C3-OH to form the five-membered lactone deoxytryptoquialanone (526), which is three steps (ketone reduction and acetylation, imidazo-nitrogen hydroxylation) from tryptoquialanine.
3.6. Transient Oxidation of Catechols to Set up Cyclization
Two examples of oxidative cyclization of catechols are shown in Scheme 75. The first is the conversion of L-DOPA (528) to cyclo- L-DOPA (531), revealing the logic of oxidative conversion of catechol 528 to orthoquinone 529 to function as electrophile in intramolecular 1,4-addition by the amine group (Scheme 75A).383 The tyrosinase is a copper-containing oxidase functioning with a binuclear copper mechanism as described in Scheme 4. A related biosynthetic logic applies in the conversion of the indicated chalcone 532 to aurone (536) by aureosidine synthase.384–386 Aurones are responsible for the yellow color in flowers such as yellow snapdragon flower, from which the first aureosidine synthase was cloned from. Enzymatic oxygenation of the phenol 532 to the catechol 533 and then to the orthoquinone 534 creates the electrophile that determines the regiospecificity of attack of the phenol oxygen on the neighboring double bond which gives 535. Aromatization creates the conjugated aurone system in 536 and the yellow color (Scheme 75B).
Scheme 75.

Transient Oxidation of Catechols to Set up Cyclization
A third example of catechol oxidation is found in the maturation of the phenalenone tricyclic framework, fused tricyclic hydroxyperinaphthenones.387 Dozens of these aromatic tricyclic metabolites are known in plant and microbial metabolism. The fungal product phenalenone (540) and related compounds are synthesized by the strain P. herquei.387 Earlier labeling studies suggested the molecules are synthesized from polyketide pathways, which was confirmed with studies using an identified phn biosynthetic pathway.388–390 Yeast reconstitution studies showed that only PhnA and PhnB are required to synthesize 540. A nonreducing PKS PhnA was shown to synthesize the angular, hemiketal-containing naphtho-γ-pyrone prephenalenone (537) (Scheme 76). A FMO PhnB then catalyzes the hydroxylation of 537 to the trihydroxyphenol product 538 through abstraction of the γ-pyrone hydroxyl proton. The hydroxylated product 538 was then proposed to be more prone to C3-proton abstraction by general base, which can drive the formation of the C-C bond from C8 of the catechol onto the C13 ketone to afford 539. Subsequent loss of water gives the stable, aromatic perinaphtheneone framework in 540. This core structure can then be subjective to a variety of modifications, including prenylation, methylation, and dimerization yield a large assortment of more complex products.
Scheme 76.

Oxidative Formation of Phenalenone
3.7. Enterocin: Favorskii Rearrangement
S. maritimus produces the polyketide enterocin (547) with an unusual tricyclic core that has undergone a skeletal rearrangement from the linear nascent product 541 released from aromatic polyketide synthase using benzoic acid as a starting unit.391,392 In particular, the C2-C3 bond has been cleaved while a new C2-C4 connectivity has been established along with a new lactone (C7-oxygen to C3 ketone) (Scheme 77A). The connectivity alterations are mediated by an FAD dependent enzyme EncM that functions as a C4 oxygenase, yielding the transient C3, C4, C5 triketone 543 (via the C4-OH 542).393,394 It is proposed that this undergoes a Favorskii type rearrangement, through a cyclopropanone 544 formation and subsequent opening to 545 that is the connectivity altering step. The direction of cyclopropanone opening is controlled by the participation of the C7-OH in lactone formation. The remaining two rings of the tricyclic core are then proposed to be established by intramolecular aldol condensations (C5-C10, C2-C9) from 545: a short path to the unique and compact tricyclic core of 547. Mechanistic and structural examination of the key EncM catalyst revealed the first example of an N5-oxide of FAD (548) as an on pathway oxygen transfer agent (Scheme 77B).395,396
Scheme 77.

Flavoenzyme Catalyzed Favorskii Rearrangement in Enterocin Biosynthesis
3.8. Piperazic Acid: N-N Bond Formation
We noted the enzymatic cyclization of lysine to pipecolic-acid (501) in Scheme 71 (Section 3.5.1) above, creating the proline homolog that gets inserted into rapamycin (493), FK506 (494) and related NRP-PK cyclic scaffolds. A related building block homolog to pipecolic acid is piperazic acid (549), with an unusual N-N (hydrazo) bond. Piperazate residues are found in several nonribosomal peptide frameworks including sanglifehrin A (550)397, kutzneride 1 (551)398, polyoxypeptins (309)399, dentigerumycin (552)400 and azinothricin (553)401 (Scheme 78A). The mechanisms of N-N bond formation is not fully understood but the first step is enzymatic N5-hydroxylation of L-ornithine (554) by a flavin-dependent oxygenase (KtzI from kutzneride pathway).402 Whether the -OH group in N5-OH-Orn (555) is further functionalized (top pathway in Scheme 78B) to a leaving group 556 for N-N bond formation or some other oxidative cyclization route to the hydrazo linkage in 557 occurs is yet to be determined.
Scheme 78.

Cyclization of L-Ornithine to Piperazic Acid in NRP Natural Products
3.9. Aurafuron
Aurafuron (562) is a 5-alkenyl-3 3(2H) furanone metabolite from myxobacterium Stigmatella aurantiaca DW4/3-1.403 It is included in this review because of an oxygenative cleavage of the nascent polyketide chain 558 that simultaneously creates the five ring furanone scaffold (Scheme 79).404 The cleavage is also proposed to be the release step of the truncated chain 561 from its thioester tether 558 on the polyketide synthase AufG. As shown, “on assembly line” oxygenation at C2 by a cytochrome P450 to yield 559 is followed by action of a flavoenzyme Baeyer-Villigerase AufJ to create the tetrahedral adduct/oxoester 560 which can decompose to the released triketone. Cyclization of the 6-OH of the enol 561 onto the C2 ketone yields the product 562.
Scheme 79.

Proposed Formation of Furanone Scaffold in Aurafuron A Biosynthesis
3.10. [4+2] Cyclizations Enabled by Prior Oxidations
As noted in the introduction of this section, there are a set of biosynthetic transformations creating 5 and 6 membered carbocycles that can be written as formal [4+2] cyclizations of diene and dienophile.405,406 Those ring-forming reactions do not involve formal redox chemistry. However, many of them occur in pathways immediately after an oxidative step, often one creating the third olefin in the substrate that then undergoes the ring-forming reaction in the next step. We examine those pairs of steps in six examples below.
3.10.1. Sordarin
Sordarin (569) is a glycoside antibiotic with a tetracyclic diterpene aglycone produced by the mold Sordaria araneosa Cain (Scheme 80).407 It is an antifungal agent that targets elongation factor 2.408,409 Labeling studies indicated that the diterpene cycloaraneosene (563) is a biosynthetic precursor of 569.410 Recently, the sordarin biosynthetic gene cluster was identified and the diterpene cyclase responsible for the biosynthesis of 563 was biochemically characterized.411 It was proposed the flavin-dependent monooxygenase SdnN is a multifunctional enzyme that converts the diol 564 to 569 by catalyzing dehydrogenation of 564 to the α-hydroxyl ketone 565, Baeyer-Villiger oxygenation to generate the ester 566, and hydrolysis and dehydration to yield the aldehyde 567 that can undergo [4+2] cycloaddition to furnish the ring system in sordaricin (568). The exact mechanism of SdnN and the oxidative cascades are not known. Further glycosylation and methylation complete the biosynthetic pathway to 569.
Scheme 80.

Proposed Diels-Alder Reaction in Sordarin Biosynthesis
3.10.2. Solanapyrone
Solanapyrones are a phytotoxin isolated from the plant pathogen Alternaria solani.412 The solanapyrone case is somewhat distinct from the next four example that follow in that the three olefinic components are present in the precursor alcohol metabolite 570, which does not engage in the putative Diels-Alder type cyclization. Instead the next enzyme, an FAD dependent dehydrogenase converts the exocyclic alcohol to the aldehyde 571 (Scheme 81).413,414 At this point the exo-and endo- derived product solanapyrones A (572) and D (574) are generated, putatively from the indicated transition state geometries. The argument has been made that the conjugated aldehyde in 571 affects the orbitals of the olefin adjacent to and in conjugation with the pyrone to lower the energy barrier for the cis-decalin formation. Interestingly, the biosynthetic pathways continue through reduction of the aldehyde back to the alcohol to yield solanapyrone B (573) and E (575), masking the introduction of the aldehyde during the cycloaddition step.
Scheme 81.

Oxidation Mediated [4+2] Cyclizations in Solanapyrone Biosynthesis
3.10.3. Lovastatin
Lovastatin (581), the prototypic statin that spawned the synthesis of a range of approved statin drugs, has been extensively examined for biosynthesis over the past two decades (Scheme 82).415 The compound is biosynthesized from a polyketide pathway in which the nonaketide synthase LovB has been intensively studied.416–419 LovB together with the enoylreductase LovC catalyze eight chain elongation cycles and reductive tailoring to yield the decalin containing intermediate dihydromonacolin L (580).417 Formation of the endo decalin core is proposed to take place during the elongation step at the hexaketide stage using the acyclic intermediate 578. The electron deficient diene (α-β unsaturated carbonyl) and the dienophile are set up by the precise reductive tailoring of the iterative LovB together with LovC. LovC selectively skips three enoylreduction steps at the diketide 576, triketide 577 and hexaketide 578 stages to yield the enzyme-tethered 579 (Scheme 82). The exact domain responsible for the cycloaddition has not been uncovered, but the activity must reside in the multidomain LovB as in vitro reconstitution with LovB and LovC alone were sufficient to generate dihydromonacolin L.417 While the synchronicity or lack thereof of decalin construction is still debated in the lov system, the presence of the decalin ring system in natural products has become a default suggestion of [4+2] enzymatic cyclization chemistry.417
Scheme 82.

On-Assembly Line Diels-Alder Reaction During Lovastatin Biosynthesis
3.10.4. Spinosyn
The polyketide spinosyn A (588) produced from Saccharopolyspora spinose is the active ingredient of highly effective insecticides use today.420 588 is a structurally complex polyketide natural product containing a perhydro-as-indacene moiety fused to a 12-membered macrolactone. Formation of the 5,6,5 tricyclic system embedded within the polyketide macrolactone framework has been intensively studied for the timing and mechanism of cyclization.421 The SpnF enzyme has been purified to homogeneity, characterized for enzyme activity and X-ray structure determination.422 It accelerates a nonenzymatic reaction from the macrocyclic precursor 584 to the 5,6-bicyclic system 585 (Scheme 83) by 50-fold. This modest but real acceleration indicates that many of the diene/dienophile substructures are poised to react nonenzymatically and the enzymes may be lowering energy barriers but not changing the intrinsic reactivity profiles of their substrates. While a transannular [4+2] mechanism can be written to produce the 6,5 fused bicyclic 585 in the SpnF reaction, calculations of the energy barriers423 have also suggested that a [6+4] electrocyclization could proceed to first form 586, followed by a rapid Cope rearrangement to get to the observed product 586. Glycosylation of 585 by the glycosyltransferase SpnG affords 587.
The steps immediately antecedent to SpnF action are catalyzed by SpnJ and SpnM to create a ketone from an alcohol 582 and then dehydration to the γ-hydroxy α,β-enone 583 from which dehydration unveils the diene 584 required for SpnF action. Thus the three enzymes SpnJ, SpnM and SpnF can be viewed as an oxidative generation of the diene that can then undergo the cyclization reaction.
Completion of the tricyclic core in 588 from 587 is achieved by the next enzyme SpnL, a homolog of SAM-dependent methyltransferases, to form five-membered cyclopentene C ring (Scheme 83). A Rauhut-Currier reaction mechanism421, has been proposed but not validated. The Rauhut-Currier reaction “involves the coupling of one active alkene/latent enolate to a second Michael acceptor, creating a new C–C bond between the α-position of one activated alkene and the β-position of a second alkene under the influence of a nucleophilic catalyst”.424 This reaction was also proposed for nepetalactol (439) formation from 8-oxogeranial (436) (Scheme 60, Section 3.2.5), but was ruled out after structural studies of ISY.307
It is worth comparing the chemical logic and enzymatic machinery to create the 5,6,5 tricyclic core in 588 with the related 5,6,5 core in ikarugamycin (428) in Scheme 59 (Section 3.2.4). In the ikarugamycin case the last cyclization was initiated by a hydride transfer from NADH rather than the proposed Rauhut-Currier route, while formation of the first two 5,6 ring system is not completely understood.
3.10.5. Abyssomicin
The abyssomicins, including abyssomicin C (593) are polyketide metabolites isolated from marine Streptomycete collected from the abyss in the Sea of Japan.425 They act as irreversible inhibitors of para-aminobenzoate synthase in bacterial metabolism.425 Recent studies on the enzymes AbyA4, AbyA5 and AbyU have provided evidence for a [4+2] cyclization catalyzed by AbyU426 (Scheme 84) on the acyclic 591 to create the indicated two ring 5,6-spirocycle 592 at the core of the 593 framework. The Aby4 and Aby5 enzymes act on the precursor tetronate ring 589 to acetylate (to give 590) and then eliminate the -OH group to produce the exo methylene double bond in 591. This oxidation process creates the dienophile that then participates in the AbyU cyclization reaction.
Scheme 84.

[4+2] Cyclization in Formation of 5,6-Spirocycle in Abyssomicin C
3.10.6. Pyrroindomycins
The pyrroindomycin B (597) biosynthetic pathway is noted here not so much for how the substrates are manipulated by biosynthetic enzymes to create the diene and dienophile partners but rather because two enzymatic [4+2] transformations occur in consecutive steps in the maturation pathway (Scheme 85). PyrE3 is known to produce the characteristic monounsaturated cis-decalin moiety 595 from 594.427 It is argued to arise from a [4+2] process as shown. Then the next enzyme PyrI4 reprises the AbyU logic, this time on an exo methylene tetramate rather than an exo methylene tetronate to produce the spiro 6,5-bicyclic moiety 596. In two enzymatic steps remarkable complexity has been built into the aglycon of the pyrroindomycin framework. An enzyme catalyzing the analogous cycloaddition as PyrI4 to yield the spirocycle in versipelostatin.428
Scheme 85.

[4+2] Cyclizations in Formation of cis-Decalin and 5,6-Spirocycle in Pyrroindomycin B
4. CYCLIZATION VIA EPOXIDATION
4.1. Introduction
Olefin epoxidation is a widely employed functional group transformation in both synthetic organic chemistry, and in natural product biosynthesis across the major structural classes of secondary metabolites.429–435 Conversion of the olefin via reaction of its π electrons to the three membered oxygen ring (epoxide = oxirane), creates a potential electrophile. The epoxide group can and does persist in many natural product scaffolds. These can subsequently react with nucleophilic groups (Nu) on cellular targets and lead to covalent adducts that can be sources of problematic toxicity.436
The susceptibility of epoxides to ring opening reactions by covalent capture by nucleophiles is a key strategic element of the biosynthetic logic in a wide range of natural product scaffold maturations, as detailed in this section. Scheme 86 shows five schematic contexts in which epoxides are generated and then used as key functional groups for ring-forming reactions. The two steps of epoxide formation and then nucleophilic opening to larger ring systems constitute a general strategy for oxidative cyclizations. When the two steps are taken together, the epoxide is a “disappearing” functional group, not persisting in the maturing scaffold, but essential for ring creations.
Scheme 86.

Models of Cyclization via Epoxidation in Natural Product Biosynthesis
In Scheme 86A, intramolecular attack on either the proximal or distal carbon of an epoxide by a nucleophile (Nu) represents as a common pattern will be exemplified in lasalocid and aurovertin production, among others. Scheme 86B represents intramolecular capture of the epoxide by the oxygen atom of a tetrahedral adduct arising from (prior/simultaneous) nucleophilic attack in a carbonyl group. This is a common route to polycyclic ethers in monensin and nanchangmycin assembly. Scheme 86C shows a prototypic bis-epoxidation of a pair of olefins with water attack as the external nucleophile, leading to cyclic ether formation, as found in glabrescol and solamin assembly. Scheme 86D involves regiospecific epoxidation of one among multiple olefins that sets off a cascade of carbocycle formation as the other olefins react as nucleophiles via their π electrons. This is the classic oxidosqualene cyclase cascade, among others, that turns a linear substrate into a rigidified polycyclic product. Scheme 86E is a variant of D in which a terminal intramolecular nucleophile initiates the olefin cascade on the epoxide and is seen in meroterpenoid assembly.
Routes to Epoxide Formation
Before taking up the specific examples of epoxide-mediated cyclizations in this section we note in Scheme 87 that the two major classes of monooxygenases, flavin-based and iron-based- can each deliver one oxygen atom from O2 to olefinic cosubstrates. The schematic mechanism for the hemeprotein P450 oxygenases can be formulated as a one electron pathway with donation of one electron from the olefin to the high-valent oxoiron species and then collapse of the FeIV-O-carbon radical to the coordinated epoxide by an effective [OH•] transfer (Scheme 87A). As shown in Scheme 87B, the proposed oxygen transfer route in the flavin-based epoxygenases (such as squalene-2,3-epoxidase) is from the flavin-4a-OOH and delivery of an [OH+] equivalent to the π electrons of the olefin as nucleophile, result in olefin epoxidation and oxidized flavin. A NAD(P)H-dependent two-electron-reduction of the flavin completes the catalytic cycle of epoxygenases. In all the pathways noted in the subsequent subsections the first step of the two step epoxide formation/epoxide opening with attendant ring formation is always catalyzed by one of the above forms of epoxygenases. Epoxidation can also be catalyzed by nonheme iron α-KG dependent oxygenase and is shown in an example of ring contraction in Section 6.1.1.
Scheme 87.

P450 and Flavoenzyme Catalyzed Formation of Epoxides
4.2. Formation of Cyclic Polyethers via Epoxide Intermediates
4.2.1. Polyether Formation
The formation of the large class of polyethers has been intensively investigated for decades and has borne out seminal hypotheses that the placement and stereochemistry of cyclic ether rings in the isoprene and polyketide-derived class can be explained by conversion of nascent polyolefinic frameworks to epoxides which could then be opened in intramolecular cascade reactions.437 Scheme 88A shows such a transformation of the acyclic hexaene 598 into glabrescol (600) with five furan rings via hexa-epoxide intermediate 599.438 Six of the seven oxygens in 600 derive from molecular oxygen, the seventh from the water molecule that initiates the epoxide opening furan-forming cascade. Analogously, the paired furan and pyran rings in the ionophore lasalocid A (604) arise by action of Lsd19 on bisepoxyprelasalocid (602) as shown in Scheme 88B. The flavin-dependent epoxidase Lsd18 first oxidizes the two E-olefins in prelasalocid A (601) into epoxides to give 602. Water initiated cyclization into 604 occurs in two tandem epoxide opening steps, catalyzed by a didomain epoxide hydrolase Lsd19.439–442 The N-terminal domain Lsd19A catalyzes the 5-exo-tet cyclization of 602 to form the tetrahydrofuran ring in 603, while the C-terminal domain Lsd19B catalyzes an anti-Baldwin 6-endo-tet cyclization to form the pyran ring and give 604.443 The X-ray crystal structure of Lsd19 showed that Lsd19A and Lsd19B are arranged in a head-to-tail manner. The substrate binding pocket of Lsd19A is unoccupied while Lsd19B contains the product 604.444 Comparison of lasalocid A-Lsd19B and product analogue-Lsd19B complexes shows that the change of conformation of Lsd19B active site residues as well as precise three-dimensional arrangement of the substrate are needed to overcome the thermodynamically disfavored 6-endo cyclization (while suppressing 5-exo-tet cyclization) under enzymatic catalysis.444 Computation modeling of the crystal structure of Lsd19B supports the mechanistic proposal for favoring 6-endo-tet under enzymatic catalysis conditions.443,444
Scheme 88.

Cyclic Polyethers formation via Epoxide Intermediates
Scheme 89 and 90 depict analogous strategies of epoxidation during monensin A (611) and nanchangmycin (615) assembly, respectively. The formation of the initial hydroxypyran 606 from the linear substrate 605 in monensin is an example of Scheme 86B, where a tetrahedral adduct oxygen is the initiating nucleophile in the epoxide opening cascade. Interestingly, although the MonBI/MonBII pair are functional parallels of Lsd19A/Lsd19B, MonBI/MonBII have been shown to catalyze three ring opening reactions (via 607, 608 and 609) to afford the pentacyclic structure 610.445,446 The X-ray crystal structure of MonBI dimers suggested that a KSD motif is important in MonBI/MonBII interactions.447 Likewise, in formation of the cyclized intermediate 614 of nanchangmycin (615), following formation of the bisepoxide 613 from the PKS-bound 612, one of the spiro pyran/furan pair arises by Scheme 86B while the second pair exemplifies the mechanism Scheme 86C.448
Scheme 89.

Mechanism of Polyether Formation in Monensin Biosynthesis
The family of heronapyrroles A-D (620–623), which contains an unusual nitropyrrole moiety attached with an oxidized farnesyl chain, represents alternative fates of epoxide ring opening from two bis-epoxide regioisomers (617 and 618) vs the tris-epoxide metabolite (619) (Scheme 91).449 In heronapyrroles A (621) and B (620), the two isolated epoxides of 617 are opened hydrolytically without neighboring group participation in ring formations; whereas in heronapyrroles D (622), neighboring group participation in 618 generates the tetrahydrofuran ring. By comparison, the tris-epoxide (619) can be opened with intramolecular formation of the indicated bis furan grouping in heronapyrrole C (623).450
Scheme 91.

Proposed mechanism of Polyether Formation in Heronapyrrole Biosynthesis
4.2.2. Formation of Other Cyclic Ethers
The conversion of muricadienin (624) into solamin (626) is another example of the “disappearing” epoxide biosynthetic logic as shown in Scheme 92A.451 Double epoxidation of two Z-olefins in 624 yields the bis-epoxide 625 that is the progenitor to the indicated dihydroxy furan functionality in 626. The stereochemistry of the epoxidation step dictates the outcome of the two hydroxyl groups to be either trans (shown) or cis.452 Aurovertins are polyketide metabolites from fungal strains that have potent cytotoxic activities and are noncompetitive inhibitors of ATP synthases.453,454 Biosynthesis of the simplest member, aurovertin E (632) was studied in the endophytic fungus Calcarisporum arbuscula.455 Formation of 632 from its hexaene-pyrone polyketide precursor 627 reflects three epoxidative steps by the only one FMO AurC (Scheme 92B).455 The first two epoxidations of the (E, Z) terminal diene 628 yield the indicated bis-epoxide 629 which gets converted in a water-addition/epoxide opening cascade to the now anticipated dihydroxyfuran 630 by the hydrolase AurD. A subsequent epoxidation of the exocyclic double bond (the third overall from AurC action) to 631 sets up an intramolecular anti-Baldwin 6-endo-tet cyclization under the catalytic aegis of AurD again to generate 2,6-dioxabicyclo [3.2.1]-octane ring scaffold of 632. Overall only four enzymes are required to assemble aurovertin E, representing a remarkably efficient biosynthetic pathway.
Scheme 92.

Cyclic Ethers formation via Epoxide Intermediates in Solamin and Aurovertin
4.3. Cyclizations via Epoxides on Terpene Scaffolds
4.3.1. 2,3-Oxidosqualene Cyclase and Triterpenes
The most famous of the terpene cyclizations that proceed via an epoxy substrate are those carried out by oxidosqualene cyclases.456–458 In this tandem epoxidation-cyclization reaction, the head-to-head coupled C30 hexaenoic hydrocarbon squalene (598) is morphed into polycyclic six- and five ring fused triterpenes that account for all of the steroids in plants (cycloartenol, 642),459 animal and fungi (lanosterol, 647) (Scheme 93).460 While 598 itself can be cyclized by the squalene-hopene cyclase (SHC) into hopene (652) and hopanol (653) (Scheme 94),461,462 for the context of this review, the majority of discussion will be devoted to the epoxidative route.
Scheme 93.

Triterpene Cyclization via Epoxide and Cation Intermediates
Scheme 94.

Engineering of Squalene-Hopene Cyclase as a Protonase
The relevant substrate arises by the action of the FAD-containing squalene epoxidase which selects one of the terminal double bonds in 598 to form 3S-2,3-oxidosqualene (633) via the general mechanism of Scheme 93.457 The effect of this action is to polarize the squalene scaffold and results in epoxide at the end serving as an electrophilic functional group. Oxidosqualene cyclases then catalyze the cascade of cyclization steps, starting from a pre-chair-boat-chair configuration, to form the proposed protosterol cation intermediate (640). The epoxide opening step is facilitated through the concerted protonation of an active site Brønsted acid. The implications of which will be discussed in subsequent section. Following a series of 1,2-hydride and methyl shifts, abstraction of a proton leads to the internal double bond and yield 647, as catalyzed by lanosterol synthase. Under different cyclases from different organisms, the cation 640 can also undergo rearrangement to form the C11 cation 641, which can be quenched by either a) new C-C bond formation catalyzed by cycloartenol synthase in biosynthesis of 642 that results in cyclopropane ring formation.463 This is the starting point of all steroids in plants;464 b) migration of the cation from C11 to C6 in 648 followed by quenching via proton removal yields the internal olefin placement in cucurbitadienol (650) by cucurbitadienol synthase;465 c) C12 proton elimination to yield parkeol (649).466 This is just gliding over the surface of the many variants that can leak out of squalene oxidocyclase active sites. Baruol synthase from Arabidopsis thaliana can leak up to 22 minor products, reflecting cations quenched at eleven carbon sites in the tetracyclic sterol framework.458
A different cyclization of 633 by a cascade of four internal olefins yields the classic tetracyclic scaffold of the sterol natural product family, as the dammarenyl cation (643).467 This is the path directed by the Lup1 variant of the oxidocyclase plant family. Rearrangement of the 643 via the remaining olefin yields the lupenyl cation (644), which can be quenched by loss of a proton to afford the pentacyclic lupeol (645), which has a variety of biological activities.468 Further rearrangement of the 644 yields the ring expanded oleanyl cation, which can be quenched by proton abstraction to yield β-amyrin (646),469 the most abundant cyclic triterpenes in most higher plants.
In some triterpene cyclization pathways, the squalene epoxidase can further epoxidize the other terminal olefin in 633 to yield bis-epoxide intermediate 634. This can be cyclized into 24,25-oxido-lanosterol (636) by lanosterol synthase, the derailed product preonocerin (635) or the epoxydammarenyl cation (637) in the presence of Lup1.467 The latter cation 637 can be captured as pentacyclic scaffolds with a pyran or furan to yield the epoxydammerane products 638 and 639. This pathway shows that the epoxidase can actually act also on the other terminal olefin to form 634 which can be carried through all the way to 636 by the lanosterol synthase member of the squalene oxidocyclase family.
The reaction catalyzed by oxidosqualene cyclase exemplifies the role of the epoxide in facilitating the electrophilic cyclizations observed in many other families of natural products, including indole diterpenes and meroterpenoids. The concerted protonation and anchimeric addition of the C6-C7 double bond is facilitated by the presence of a Brønsted acid residue in the terminal end of the active site. Crystal structure of the human lanosterol synthase showed a conserved Asp residue donates an syn proton that is hydrogen-bonded to two Cys residues.456 In SHC (Scheme 94), protonation of the terminal double bond of 598 is more challenging and requires the presence of a stronger Brønsted acid.461 In turn, crystal structure showed the SHC performs protonation with the anti-oriented Asp acid proton.461 The acidity of the proton is further increased through a specialized amino acid network that includes a protonated histidine side chain that polarizes the Asp acid carbonyl, and hydrogen bonded water molecule that bridges the carboxylated oxygen and a tyrosine side chain. In turn to take advantage of this strong active site Brønsted acid, the SHC was rationally engineered into a protonation based biocatalyst (protonase).470 Structural based mutagenesis to enable better binding of smaller substrates led to the isolation of the Y420W or Y420W/G600F mutant that can stereoselectively cyclize S-(6,7)-epoxygeraniol (654) to hydroxylcyclogeraniol (656) via 655.470
A very different cyclization pattern is observed in yardenone (659) and abudinol (662 and 663), which are triterpenes isolated from marine sponges Ptilocaulis spiculifer.471 Multiple epoxidations on different olefins of squalene 598 are proposed to initiate the cyclization cascades (Scheme 95). The formation of the pentacyclic framework of 659 requires formation of the tris-epoxide 657. An epoxide-opening cascade on the right half of the precursor substrate is accompanied by an analogous capture of a proton as stand-in electrophile, on the left hand side of substrate. These cyclizations build the 7/5 and 7/6 bicyclic rings in 658 respectively, on the route to the final 659 (with an additional furan moiety).472 The formation of abudinol A (662) and B (663) is proposed to start from the tetra-epoxidized substrate 660.473–475 Once the epoxide-opening cascade on the left half of 661 is completed, abstraction of H14 could induce the tandem cyclization on the right half of the molecule to generate 662 and 663 through different epoxide opening regioselectivity.
Scheme 95.

Alternative Triterpene Cyclization in Yardenone and Abudinol Biosynthesis
4.3.2. Indole Terpenes
Three examples of cascade reactions on the terpene side chains in three indole terpene biosynthetic pathways illustrate the applicability of the “disappearing” epoxide logic in this hybrid class of natural products. One is the conversion of 3-farnesyl indole (664) into xiamycin (667) (Scheme 96). XiaO catalyze the epoxidation on the terminal olefin of the C15 664 to form the 3-(epoxyfarnesyl)-indole (665). Like a terpene cyclase, XiaH sets up the enzyme-mediated cyclization cascade to form the 6,6 bicyclic system in preindosespene (666).476 Further, epoxidation of the indole ring sets the fifth ring, bridging six membered ring and a ring expansion to the seven membered cyclic ether in ring D of 667 (see section 4.4.2).
Scheme 96.

Epoxide Mediated Cyclization in Xiamycin Biosynthesis
The second case is the related processing of fungal indole diterpene family compounds, such as paxilline (675) and emindoles (679 and 680) (Scheme 97). This is a large family of natural product frequently found in fungal sclerotia and exhibit potent insecticidal properties.477,478 Construction of these molecules starts with the C3-alkylation of indole-3-glycerol phosphate to yield geranylgeranyl-indole 668. In biosynthesis of 675, regiospecific epoxidation is effected at the third of the four olefins in the C20 side chain by PaxM to 669, as preamble to the intramolecular epoxide ring opening by PaxB through cations 670 and 671, yielding a pentacyclic framework of 672 with one remaining olefin.479 A second epoxidation by PaxM on the residual olefin yield 673 and capture by the indicated -OH group creates the pyran ring in 674 as the sixth ring in the paxilline scaffold. The final enzymes in the pathway are the P450s PaxP and PaxQ which are involved in creation of the enone functionality and hydroxylation in the end product 675. The emindole framework represents an alternative capture and diversion of the indicated cationic intermediate 671 by Ats5B1 to create another tetracyclic framework 676 with one remaining olefin. Subsequent epoxidation of the residual olefin by AtmM to yield 677 and pyran formation in 678 create the connectivity and architecture found in the prenylated emindole PA (679) and emindole PB (680).480
Scheme 97.

Epoxide Mediated Cyclization in Paxilline and Emindoles Biosynthesis
Scheme 98 is an expansion of the enzymatic processing of one of the cation intermediates (670) in the pathway of the previous Scheme 97. Unlike PaxB, AtS2B or AfB cyclases can catalyze three additional rearrangements of cation 670 with i) quenching by proton removal of 682 yields the anominine (686) scaffold, which is a proposed precursor of tubingensin A/B (690 and 691) catalyzed by an unidentified FMO or P450, following an indole 2,3-oxidation induced cyclization (for 690) and a further series of cation shifts induced rearrangement (for 691, like xiamycin biosynthesis, Scheme 108) ; ii) instead of quenching 682, two additional intervening cation rearrangements via cations including 683 and 684 yield the olefins in the context of the highly rearranged congeners of aflavinine (687) with endocyclic (689) and exocyclic (685) olefin regioisomers. Interestingly, the anominine and aflavinine cyclases (AtS2B and AfB) were found to be separated from the enzymes that produce 686, suggesting the likelihood of siphoning this substrate from other indole diterpene pathways.480 The unclustered cyclases was found using a genome-guided, heterologous reconstitution approach in yeast. The activities of these compounds as strong insecticidal natural products found in the sclerotia of fungi hints likely differential regulation of these unclustered genes. Overall, the ability of cationic intermediates to drive skeletal rearrangements in these reactions and the promiscuity of epoxidases allows evolution of new skeletal frameworks in natural products.
Scheme 98.

Epoxide Mediated Cyclization in Anominine, Aflavinine and Tubingensin Biosynthesis
Scheme 108.

Indole Epoxidation Mediated Carbazole Formation in Xiamycin Biosynthesis
4.3.3. Fungal Meroterpenoids
Meroterpenoids constitute a large family of molecules of mixed terpene-polyketide origin, exhibit diverse chemical structures and a wide range of biological activities (Scheme 99).481 Compounds in this family share a modular assembly pathway and can diverge in structure during the oxidative cyclization and rearrangement steps (Scheme 100).482 The typical sequence of biosynthetic steps is i) synthesis of an aromatic polyketide unit such as methyl-orsellinic acid; ii) Friedel-Craft alkylation of the polyketide with either farnesyl-PP or geranylgeranyl-PP; iii) epoxidation of the terminal olefin in the prenyl chain to set up the cascade of cyclization steps; iv) protonation-assisted cyclization of the prenyl portion; and v) oxidative rearrangement of the scaffolds. The tandem epoxidation/cyclization steps therefore serve as the key steps in defining the cyclized skeleton of the final products.
Scheme 99.

Epoxide Mediated Cyclization in Pyripyopene Biosynthesis
Scheme 100.

Biosynthetic Pathway of Fungal Meroterpenoids
The first completely elucidated pathway for compounds in this family is that of the pyridine-containing pyripyropene A (695), followed by related compounds oxalicine (696 and 697) and decaturins (698–703) (Scheme 99).483–487 695 produced from A. fumigatus is one of the strongest acyl-coenzyme A: cholesterol acyltransferase (ACAT) inhibitor, and is a potential antihypercholesterolemia drug.484 The polyketide building block, 4-hydroxy-6-(3-pyridinyl)-2H-pyran-2-one (HPPO) is synthesized by a nonreducing polyketide synthase, using nicotinyl-CoA as the start unit.488 Attachment of farnesyl group to the C3 position of the pyrone yields the linear precursor farnesyl-HPPO (692). The flavin-dependent enzyme Pyr5 installs the epoxide at the terminal olefin to yield 693. Pyr4 was discovered as the first membrane-bound cyclase that can perform the proton-initiated cyclization of 693 to yield the cyclized deacetyl-pyripyropene E (694).488 The cyclization cascade can be written as in Scheme 99 to initiate from the pyrone 4-hydroxyl group, an example of the mechanism shown in Scheme 86E. The result of the cyclization step is a pentacyclic product 694, which contains a fused pyrone-pyran-two carbocyclc core. Further hydroxylations and acetylations yield the natural product 695. In the oxalicine and decaturin pathways, more oxidative rearrangements are responsible for the spirocycle formation.487
Additional complexity/versatility in meroterpenoid biosynthesis continues in Scheme 100, which represents alternative fates of one common polyketide precursor 3,5-dimethylorsellinic acid (704) to different complex meroterpenoid scaffolds through regio- and stereo-selective prenylation and epoxidation.482 For example, tropolactone D (708) arises from the S-farnesyl adduct 705 in an epoxide opening cascade (via 706) to provide tetracyclic intermediate (707) exemplifying Scheme 86B. This is followed by oxidation of the cyclohexanol of 707 to the ketone, hydroxylation and a Baeyer-Villiger enzymatic conversion to the lactone that gives tropolactone D.489
Alternatively, action of the R-specific C15 prenylation (AdrG/AusN/Trt2) to 709 leads ultimately to more complex andrastin (713)/austinol (715)/terretonin (717) and novofumigatonin (721) scaffolds.490–493 Unlike biosynthesis of 708, the S-configuration epoxidation opening cascade starting from 718 leads to the formation of a cationic intermediate 719 instead of tetracyclic intermediate 707 in 708 biosynthesis.494–496 Quenching via H-shift results in the formation of spirocyclic intermediate 720, which is proposed to be the precursor of 721.493 Another set of epoxidases (AdrH/AusM/Trt8) with different product selectivity yield R-configuration epoxidation intermediate 710. When the initiating nucleophile in the epoxide-opening cascade is not the pyrone-OH or the tetrahedral adduct oxygen, but instead the olefin to form the cation intermediate 711, the frameworks of 713, 715 and 717 are generated through loss of different protons in 711. One lesson is that the pyrone moiety can employ three distinct nucleophiles in the intramolecular cascade of epoxide opening to enter into three distinct kinds of scaffold connectivity and complexity.
One more pathway for anditomin (726) biosynthesis requires conversion of 704 to phthalide (722), a transformation catalyzed by a chimeric protein (AndK) consists of P450 monooxygenase and hydrolase domains.497,498 Although R-specific C15 prenylation is installed on 722 by AndD to yield 723, the epoxide opening cascade follows that of the S-configured 724 via 723, and afford a tetracyclic intermediate preandiloid A (725) that can be further morphed into 726 with extensive oxygenation and rearrangement.498
4.3.4. Quinolone and Quinoline Alkaloids
Two insecticidal quinolone alkaloids, penigequinolone A/B (737 and 738) (from P. thymicola) and aspoquinolone A/B (742 and 743) (from A. nidulans) feature the same quinolone alkaloid core (as in quinolinone B, 729) attached with differently cyclized C10 terpenoid pieces (Scheme 101).499–501 729 is derived from 4′-methoxyviridicatin (728), which is rearranged from the cyclic dipeptide 4′-methoxycyclopeptide (727) by the PhyH-like dioxygenase AsqJ through an epoxidation reaction (see Section 6.1.1).502 Following formation of 729, two prenyltransferases PenI and PenG sequentially transfer two DMAPP molecules, together with the action of a FAD-dependent dehydrogenase PenH, to form the hydroxylated, acyclic precursor 730. The most striking result of using three enzymes to install the ten-carbon chain in 730, instead of a direct geranylation, is the installment of a C3 hydroxyl group that is critical for the subsequent epoxide rearrangement reactions.503
Scheme 101.

Enzyme-Catalyzed Cationic Epoxide Rearrangements in Quinolone Alkaloid Biosynthesis
In both pathways, a flavin-dependent monooxygenase (PenE and AsqG) is responsible for epoxidizing the terminal olefin in 730 to yield the epoxide 731, which can readily undergo the canonical 5-exo-tet cyclization to yield the tetrahydrofuran containing yaequinolone C (732).504,505 732 was initially thought of as an intermediate towards the formation of penigequinolone and aspoquinolone. Subsequent feeding and biochemical studies, however, demonstrated this is a shunt and co-metabolite.506 A dedicated epoxide hydrolase is encoded in each pathway to further accelerate the formation of 732 as a cometabolite. In the penigequinolone pathway, a carotenoid hydratase-like enzyme, PenF, catalyzes a novel cationic epoxide rearrangement to yield the aldehyde 734, which can spontaneously cyclize to form the hemiacetal yaequinolone D (735).506 This compound can be dehydrated and reduced to form the pyran-containing 737 and 738. PenF is proposed to not only suppress the favorable 5-exo-tet cyclization, but more impressively, to promote a discrete epoxide cleavage step to yield the carbocation 733, which can undergo a Meinwald-like rearrangement to afford 734 and the quaternary C8. Such reaction requires strong acid in organic synthesis, but occurs under physiological condition in the active site of PenF which was proposed to have a strong Brønsted acid as found in SHC (Scheme 94). In the aspoquinolone pathway, a dehydratase (AsqC) catalyzes the loss of water from epoxide 731 to form the dienyl epoxide 739. The ability of PenF to catalyze cationic epoxide rearrangement was further demonstrated through converting 739 to the aldehyde 740. A related Brønsted acid enzyme AsqO was found in the aspoquinolone pathway to catalyze the 3-exo-tet cyclization of 739 followed by intramolecular quenching of the carbocation 741 to yield the cyclopropyl furan containing 742 and 743. Hence, the dienyl epoxide 739 can be directed towards two distinct epoxide opening pathways dependent on exposure to either PenF or AsqO.506
Aurachins are secondary metabolites produced by the myxobacterium Stigmatella aurantiaca Sg a15 (Scheme 102).507 The gene cluster was found by Müller and coworkers to be localized on two separate regions in the genome.508 The bicyclic quinolone aurachin D (744) is converted to the tricyclic ring system of aurachin A (751) under the auspices of two oxygenases. AurG is proposed to transiently epoxidize the quinolone double bond of aurachin C (745), the N-hydroxylation product of 744 to 746, which is then opened intramolecularly with assistance by the N-OH functionality of 745. 1,2-Migration of the farnesyl side chain of epoxide-opened 747 through a proposed semi-pinacol rearrangement that gives 748 is followed by reduction of ketone to hydroxyl group by AurH to give aurachin B (749). Yet another epoxidation of the proximal olefin of the farnesyl side chain in 749 to 750 is followed by capture by the adjacent -OH group via a 5-exo-tet cyclization. This forms the fused pyran ether and the side chain -OH in 751.
Scheme 102.

Proposed Cyclization Mechanism during Aurachin A Biosynthesis
4.3.5. Oxetane Formation During Taxol Maturation
During the late stages of taxol (756) biosynthesis, the tricyclic C20 hydrocarbon taxadiene core is oxygenated at multiple sites around the periphery and acylated with acyl groups of differing complexity.509 The most striking oxygen functionality in mature taxol is the four membered oxetane ring with an acetoxy side chain. The formation of the oxetane is postulated to arise as shown in Scheme 103. Epoxidation of the exocyclic olefin 752 to 753 would lead to subsequent internal opening by the oxy anion of a tetrahedral adduct of the acetoxy side chain to yield the adduct 754. The oxy anion from the opened epoxide could attack 754, becoming the four membered Oxetane 755 as the acetoxy group is reconstituted, now with a net 1,2 shift.
Scheme 103.

Oxetane Formation via an Epoxide Intermediate During Paclitaxel Maturation
4.4. Arene Oxides as Biosynthetic Intermediates
4.4.1. Phenyl Ring Epoxidations
Among the most notable proposed arene oxides in biosynthesis of natural products are those in the pathways to epidithiodioxopiperazines, such as acetylaranotin (760) and the notorious fungal metabolite gliotoxin (483).510,511 As depicted in Scheme 104, 760 assembly from the epidisulfide adduct of the Phe-Phe diketopiperazine 757 is proposed to occur via action of a cytochrome P450 oxygenase (AtaF) that acts on both phenyl rings to form a proposed bis-epoxide intermediate 758. While the arene oxides could rearrange to the phenols, a typical outcome, they are instead captured intramolecularly by the amide nitrogens of the diketopiperazine ring. This action forms the two C-N bonds and generates the cyclohexadienol forms of the pentacyclic precursor 759 that can be converted to 760.510 Action of a cognate arene epoxide enzyme (GliC or GliF) on 761 during 483 assembly yields the dithiol form 482 of gliotoxin, thought to oxidize to the disulfide 483 as shown in Section 3.4 as the producing A. fumigatus cells export the metabolite512. Ring expansion oxygen insertion (discussed in Section 6.2.4) occurs in the cyclohexadienol rings of 760 but not in 483.
Scheme 104.

Proposed Epoxide Intermediates in Acetylaranotin and Gliotoxin Biosynthesis
4.4.2. Indole 2,3-Epoxidations
The heterocyclic pyrrole ring in the bicyclic indole system has recently been found to undergo epoxidation by a class of flavin-dependent oxygenases involved in fungal natural product biogenesis, exampled as fungal alkaloids fumiquinazoline A (519), tryptoquialanine A (767), notoamide D (778) and spirotryprostatin A (781).513 Scheme 105 shows the conversion of the indole moiety of fumiquinazoline F (763)514 in the presence of O2, a single module NRPS (used for activation of L-alanine), and an epoxidase (AF12060 or TqaH) into the annulated imidazoneindole tricyclic ring structure of 519.382,515 The proposed 2,3-epoxyindole 764 can be in equilibrium with the indicated hydroxyiminium form that can be captured by the amine group of the NRPS-bound alanine or aminoisobutyrate cosubstrate 765 to yield 766; and then the indoline NH can complete the annulation by capture of the aminoacyl thioester carbonyl and yield 519. Further elaboration of the fumiquinazoline scaffold gives both fumiquinazoline D (522) (see Scheme 74) or 767. Scheme 105 also depicts additional fungal peptidyl alkaloids 768–772 that arise via comparable indole ring epoxidation.516–520
Scheme 105.

Indole Epoxidation as a Key Step in Fungal Indole Alkaloid Biosynthesis
Another example in the indole epoxidation manifold is provided for the biosyntheses of notoamide C (776) and D (778) from notoamide E (773) (Scheme 106). When the proposed 2,3-epoxyindole 774 catalyzed by NotB is formed, two pathway of the epoxidation intermediate could be occurred. Like fumiquinazoline scaffold biosynthesis, 778 is pictured as arising by internal capture of the hydroxyiminium tautomer 777 by the diketopiperazine amide nitrogen, giving rise to the complex 6,6,5,5,6,5 hexacyclic framework of 778.521 Alternatively, a comparable participation of the para-oxygen lone pair on the substituted phenol 774 yields 775, followed by ketonization of the carbinolamine and a migration of a C-C single bond yields the isomerized framework of 776, which result in shift of the reverse prenyl group from C2 to C3 in indole ring.
The spirotryprostatins produced by A. fumigatus are fungal peptidyl alkaloids with antimitotic properties.522 The key feature is the quaternary carbon at the nexus of a 5,5-spiro center. Scheme 107 shows the third example for indole epoxidative rearrangement of fumitremorgin C (219) into spirotryprostatin A (781). In this case the lone pair of electrons on the methoxyphenol moiety of 779 is suggested as the internal initiating nucleophile. The tetrahedral carbinolamine adduct 780 rearranges with C-C bond migration leading to produce the spiro system in 781.523 A different oxidative rearrangement mechanism that affords spirotryprostatin G is shown in Section 6.1.5.
The indole epoxidation mechanism is proposed to be involved in bacterial alkaloid xiamycin (667) biosynthesis.524 The indole epoxidation “disappears” (shown in Scheme 108) as an early step in creation of the carbazole framework in 667.524 The proposed 2,3-epoxyindole 783 can be in equilibrium with the indicated hydroxyiminium form 784 that can be captured by the adjacent double bond to form a new C-C bond and generate a cation intermediate 785. The product after cation quenching and water elimination is the pentacyclic cyclohexene intermediate 786, which can be ring-expanded to form 667.
Except FMOs, the indole 2,3-epoxidation can also be catalyzed by P450 enzymes. Two distinct plant cytochrome P450s convert the plant secondary metabolite brassinin (787) into spriobrassinol (789) or cyclobrassinin (790) as depicted in Scheme 109.115 Internal opening of the putative indole epoxide 788 at either carbon center by the thioimidate yields the 5 or 6-membered nitrogen and sulfur heterocycles.
Scheme 109.

Indole Epoxidation Mediated Cyclization in Plant Metabolite Biosynthesis
4.5. Additional Epoxide-Mediated Cyclizations
The hybrid polyketide/peptide amides, known as heronamides, from Heron Island in Australia feature two kinds of cyclizations in proceeding from heronamides C (791) and F (792) to heronamide A (797) and B (798), respectively. (Scheme 110). The completed natural products contain a 5,5 ring system embedded within the larger polyene macrocycle.525 The first cyclization follows the O2-mediated regioselective epoxidation of one of the seven double bonds in substrate to yield 795. The exact trigger of this epoxidation is unclear and was proposed to be possible without enzyme catalysis. Spontaneous internal capture by the amide nitrogen builds the first five membered ring in 796.526 The subsequent conversion to the 5,5- bicyclic 797 and 798 is thought to be either a [4+2] or a direct [6+4] (followed by a facile Cope rearrangement) cyclization route.527 The starting macrocyclic compounds 791 and 792 can also be cyclized into heronamide B (793) and heronamide E (794) when exposed to UV light.
Scheme 110.

Epoxidation Mediated Cyclization in Heronamides Biosynthesis
Pseurotin A (804) produced by A. fumigatus arises from a short, hybrid nonribosomal peptide/polyketide assembly line (Scheme 111).528 Chain release from the tethered thioester enzyme intermediate 799 is mediated by reduction to the thiohemiacetal and dissociation of the free aldehyde 800 as shown in Scheme 56A. Tetramate ring formation in 801 is proposed to occur by attack of the acetoacetyl enolate on the aldehyde 800. An extensive list of oxidative tailoring steps ensues to afford the oxo-spirocyclic framework in 804. An interesting enzyme encoded in the gene cluster is an unusual didomain PsoF, which contains a N-terminal methyltransferase domain and a C-terminal FMO domain.529,530 The MT domain was shown to be essential for introducing the α-methyl group in the polyketide portion of the 799.531 The FMO domain was shown to be required for the formation of the oxo-spiro structure of 803.531 The proposed mechanism of PsoF is the epoxidation of the endocyclic olefin in the pyrrolidone portion of 801 to yield 802, which sets up an internal opening/capture by the enol to produce 803. Recent studies by Watanabe and coworkers show that trans to cis isomerization of the diene portion requires a glutathione S-transferase PsoE.532 PsoF was shown to be responsible for an additional epoxidation step to introduce an epoxide which can be hydrolyzed to yield the trans diol found in 804.530
Scheme 111.

Epoxidation Mediated Oxo-Spiro Ring Formation in Pseurotin A Biosynthesis
Jasmonic acid (808), the important regulator in plant responses to biotic and abiotic stresses as well as in development, is a long chain fatty acid-derived plant hormone biosynthesized from linolenic acid (191).530 As shown in Scheme 112A, S-hydroperoxide 192, installed by lipoxygenase, is the internal source of the resultant epoxide oxygen in the allene oxide 193 (see Scheme 32). One electron opening by allene oxide cyclase yields a delocalized radical pair (805 and 806) that can cyclize and unravel to the cyclopentenone core intermediate 807.533 Further chain shortening through β-oxidation as well as reduction leads to the 808. On the other hand as shown in Scheme 112B, the R-hydroperoxide 195 from arachidonic acid (194) is the precursor of clavulone I (809), which possibly carries a similar pathway of 808.534 Additionally, instead of β-oxidation and reduction, the post-modification steps including hydroxylation, acetylation and O-methylation are required for biosynthesis of 809 following opening of the allene oxide 196.
5. CYCLIZATION VIA HALOGENATION
5.1. Introduction: Haloperoxidases and Oxygen-Consuming Halogenases
In synthetic organic transformations, the displacement of halogen substituents by nucleophiles can constitute efficient substitution reactions. In natural product biosyntheses two of the four major classes of biosynthetic halogenases can participate in oxidative cyclization processes. The two halogenase classes that can carry out the generalized transformations noted in Scheme 113 are hydrogen peroxide-dependent vanadium halogenases, known as vanadium haloperoxidases (VHPOs), and mononuclear nonheme iron oxygen-dependent halogenases. The VHPOs can engage directly in concomitant halogenation and cyclization as exemplified in Scheme 113A and 113B. The iron-dependent halogenases are part of a two-step process (Scheme 113C), where a second enzyme carries out intramolecular displacement of the halide.
Scheme 113.

Models of Cyclization via Halogenation in Natural Product Biosynthesis
Scheme 114A shows the catalytic cycle for VHPOs in which H2O2 is the exogenous source of oxidizing equivalent for the halide ions. HOOH replaces a coordinated water molecule as a vanadium ligand. Binding of a halide, typically chloride or bromide ion (in seawater microenvironments) leads to generation of a hypochlorite or hypobromite equivalent ligated to the vanadium. The -OX species acts as an [X+] equivalent: the polarity of chloride and bromide ions has been reversed from X− to X+ and can be captured with nucleophiles. Scheme 114B depicts the ligand sphere around the nonheme FeII with chloride as a first sphere ligand. Reaction of O2 with the iron halogenases yields a high-valent FeIV=O. In a typical oxygenase mode it would cleave a bound C-H to yield a carbon centered radical and transfer an [OH•] equivalent in a rebound step to effect substrate hydroxylation. In halogenase mode, instead a [Cl•] equivalent is transferred to a carbon radical to produce the halogenated product.
Scheme 114.

Catalytic Cycles of Vanadium Haloperoxidases and α-KG Dependent Halogenase
5.2. Vanadium Haloperoxidases as Cyclization Catalysts
The biosynthetic capability of VHPOs as a cyclization catalyst is demonstrated in the merochlorin biosynthetic pathways (Scheme 115). Tetrahyrdoxynaphthalene (THN, 810) is a prototypic aromatic polyketide arising from utilization of five molecules of malonyl-CoA.535,536 Installation of a rearranged farnesyl moiety by Mcl23 creates the substrate 811 for haloperoxidase-mediated chlorinations.537,538 Mlc24, the vanadium-dependent chloroperoxidase, catalyzes a site-selective naphthol chlorination to 812a and initiates oxidative dearomatization to afford cation intermediate 812b. This triggers terpene cyclization to create the possible cation 813. Two possible cyclization routes complete the stereochemically complex carbon framework of merochlorin A (814) and merochlorin A (815) in one more step.
Scheme 115.

VHPO Catalyzed Formation of Merochlorins
A third variation in route leads to merochlorin C (820). As in the other two cases above, chlorination can occur on a nucleophilic carbon ortho to a phenol in 817 to yield merochlorin D (818). Then generation of the cyclic chloronium ion at the terminal olefin in 819 can be followed by attack of the indicated OH to install a second chlorine substituent as the macrocyclic ether 820 forms.539
Scheme 116 shows a route from the same starting substrate 810, with ortho chlorination and also double prenylation, with a C5 and a C10 unit to yield SF2415B1 (821). Again, attack of the terminal olefin on V-OCl in the haloperoxidase active site by NpaH1 leads to a species shown as a cyclic chloronium ion intermediate 822, which is captured by the indicated OH to create the pyran ring fused to the original naphthalene core to form SF2415B3 (824).540,541 In the case of bromine activation, formation of the bromonium 823 lead to the bromo-SF2415B3 (825) as shown. In some organisms, 824 can be elaborated to a further chlorinated product A80915C (826) by a comparable olefin attack on a vanadium-coordinated [Cl+] equivalent.
Scheme 116.

Halogenation as a Key Step in SF2415B3 Biosynthesis
Two examples of brominated isoprenoids induced cyclization are displayed in Scheme 117. In Scheme 117A the laurencin (828) and prelaureatin (830) are generated by attack on the V-OBr by the olefins in laurediols (827 and 829), respectively. The source of the [Br+] is from KBr shown in the scheme.542 Cyclic brominated eight-membered ethers are the core frameworks in the products. In Scheme 117B, the origin of the three α-,β-and γ-snyderols (834–836) is the isoprene nerolidol (831). The nascent cation 833 from attack of the olefin on the bromonium ion 832 can be quenched by loss of a proton from the endocyclic (α-snyderol, 834 and γ-snyderol, 836), or exocyclic (β-snyderol, 835) position.543
Scheme 117.

Bromination Triggered Cyclization in Laurencin, Laureatin and Snyderol Biosynthesis
5.3. Iron Halogenases and Partner Cyclases
Three examples of two step enzymatic processes to convert an unactivated carbon center into a cyclopropyl group are shown in this subsection. In all three cases the halogenases act on a precursor metabolite tethered in thioester linkage to an acyl carrier protein domain/subunit that is part of either a PKS (curacin, 841) or a NRPS (coronatine, 847 and kutzneride, 851) assembly line. The nonheme iron halogenases as noted in Scheme 114B reductively activate and cleave O2 while generating a high-valent FeIV=O active site reagent that can cleave an unactivated C-H bond in a bound substrate. The resultant carbon radical is thought to capture the [Cl•] in a rebound step.
As shown in Scheme 118 the CurA halogenase has effected a radical chlorination of the unactivated terminal methyl group of the Δ3-prenyl-acyl carrier protein 837.544–546 Subsequent conjugate addition of a hydride equivalent from NADH by the enolyl reductase (ER) domain component of the PKS assembly machinery yields the C2 thioester enolate anion 839. This carbanion equivalent can now carry out an intramolecular SN2 displacement on the chlorine substituent. In this ring-forming reaction the cyclopropane 840 has been created on its thioester, ready to be incorporated as a building block in the assembly line to 841.
Scheme 118.

Chlorination Mediated Cyclopropane Formation During Curacin A Biosynthesis
Coronatine 847 is a hybrid PK/NRP produced by phytopathic bacteria as a mimic of the jasmonic acid hormone noted in Scheme 112. It causes opening of the stomata in plant leaves allowing the pathogenic Pseudomonas syringae producers to gain access to the internal nutrients of the leaf.547,548 The aminocarboxycyclopropane building block is generated from L-allo-isoleucine (842) by a comparable strategy to the curacin case (Scheme 119).549 842 is selected, activated and tethered on its peptidyl carrier protein CmaD by the CmaA enzyme to yield 843. CmaB is an O2-requiring, nonheme mononuclear iron halogenase that regiospecifically chlorinates the unactivated Cγ methyl group to afford 844. CmaC can remove the activated Cα-H as a proton, generating the thioester enolate 845. That has carbanion character at Cα and can react in an intramolecular SN2 displacement of the chlorine under the catalytic agency of the enzyme CmaC. The result is the cyclopropane-containing aminocarboxy building block known as coronamic acid (846) that gets incorporated into the hormone agonist 847.
Scheme 119.

Chlorination Mediated Cyclopropane Formation During Coronatine Biosynthesis
The third example is embedded in the antifungal hexapeptidolactone kutzneride (851) (Scheme 120). All six amino acid constituents are nonproteinogenic, signaling its nonribosomal peptide synthetase assembly line origin.550 The 2-(1-methylcyclopropyl)-D-glycine is the most unusual residue. The details of mechanism in this case are not clearly worked out but KtzD is again an oxygen-reducing mononuclear iron halogenase so a cryptic chlorination mechanism for cyclopropane 850 formation starting with the KtzC-tethered isoleucine 848 is anticipated.551 Despite the fact that a corresponding Cα carbanion equivalent derived from the chlorinated 849 will not suffice for formation of the methylcyclopropyl glycine scaffold (but can form 850).
Scheme 120.

Chlorination Mediated Cyclopropane Formation During Kutzneride Biosynthesis
6. OXIDATIVE RING REARRANGEMENT
Ring sizes in natural product scaffolds can expand or, less often, contract during enzyme-mediated transformations, many of these occur during iron-dependent oxygenase catalysis.552 As noted in Section 2 the hallmark of iron-based oxygenases are the involvement of carbon-centered radicals at different stages in the catalytic cycles and those can drive scaffold rearrangements, both to larger and smaller rings systems.
6.1. Ring Contractions
6.1.1. Viridicatin: Diazepinone Contraction
4′-methoxyviridicatin (728), the precursor of penigequinolone and aspoquinolone discussed in Section 4.3.4, contains a 6,6 quinolone scaffold. This core structure is derived from cyclic dipeptide 852 built from cyclization of anthranilate and O-methyl-tyrosine by a two module NRPS assembly line.502 The biosynthesis of 728 from cyclic dipeptide substrate 852 was confirmed through in vitro biochemical analysis of a dioxygenase enzyme AsqJ.502,553,554 The experiments revealed two key roles of AsqJ in driving scaffold rearrangement and expulsion of methylisocyanate (857). As noted in Scheme 121, the first transformation effected by AsqJ is a net desaturation to generate an exocyclic olefin 854 on the diazepinedione moiety. It is shown as a radical process in which the carbon centered radical 853 arising from the initial C-H homolysis step transfers a second electron to the FeIII-OH rather than experiencing an [OH•] rebound. This exocyclic olefin 854 is then substrate for a second catalytic cycle by the AsqJ enzyme, this time resulting in a more typical epoxide product 855. This epoxide is spontaneously opened by neighboring group participation of the π electrons of the anthranilyl aromatic ring, creating a tetracyclic framework 856. This can break down with expulsion of 857 and yield the phenyl substituted dioxoquinolone 858 framework in viridicatin.
Scheme 121.

Oxidative Ring Contraction to Form Viridicatin
The net result is a ring contraction of the seven member diazepinedione of the starting cyclic dipeptide 852 to the six member dioxopyridine ring in the quinolone product 858. The one carbon bridge between the phenyldiazepinedione and the methoxyphenol ring in starting substrate is the carbon atom that gets extruded as 857, along with one of the two nitrogens in the diazepine ring.
6.1.2. Gibberellic Acid 12: Cyclohexanol Contraction
The gibberellins are cyclic diterpenes that have a variety of hormonal and developmental effects in plants.555 Gibberellic acid 12 (GA12, 864) is generated from ent-7α-hydroxykaurenoic acid (859) by the steps catalyzed by P450 CYP88A, involving two oxygen transfer events (Scheme 122).556,557 The first step is an exceptional one. Homolytic cleavage of the indicated C-H bond by the high-valent oxoiron in the CYP88A active site yields a radical 860 that rearranges rather than undergoing immediate hydroxylation. The net result is conversion of the B ring of the initial substrate radical from a hydroxycyclohexane to a cyclopentane with an exocyclic •CHOH functional group as in 861. At this juncture the [OH•] rebound occurs to give the product as an aldehyde hydrate 862, which is in equilibrium with the aldehyde 863. A second CYP88A mediated oxygenation of the aldehyde 863 to carboxylic acid functionality creates 864. The net result of CYP88A action is to trap out the ring-contracted gibberellic acid framework in the form of the aldehyde intermediate 863.
Scheme 122.

P450 Catalyzed Ring Contraction in Gibberellic Acid 12 Biosynthesis
6.1.3. Saliniketal: Macrocycle Opening and Dioxabicyclo[3.2.1]octane Formation
Saliniketals (872 and 873) from Salinospora arenicola block transcription of the gene encoding ornithine decarboxylase, the gatekeeper enzyme in polyamine biosynthesis.558 The ansa bridge is reminiscent of rifamycin framework. Moore and colleagues have shown that rifamycins and saliniketal share common biosynthetic intermediates and then diverge at the stage of 34a-deoxyrifamycin W (865) (Scheme 123).559 The cytochrome P450 CYPSare1259 is proposed to oxidize the C34 methyl group up to the carboxylate (866) (via three successive oxygen transfer events). At this juncture loss of the carboxylate as CO2 is enabled by the vinylogous keto group to stabilize the product enol 867. Dioxygenative cleavage of that 11, 12-double bond would generate the acyclic keto acid 868, breaking up the macrocycle. Subsequent reductive saturation of the 28, 29-double bond gives a conformationally mobile chain 869 for hemiketal 870 formation. The oxyanion of the hemiketal is postulated to act as trans-annular nucleophile for displacement of the original 27-OH group. This generates the dioxabicyclo-[3.2.1]octane ring system 871, the precursor of 872 and 873 prior to C-N bond cleavage. In a formal sense the large macrocyclic bridge of the rifamycin precursor has been replaced by the much more compact bicyclooctane in 871.
Scheme 123.

Dioxabicyclo[3.2.1]octane Formation in Saliniketals Biosynthesis
6.1.4. Griseorhodin A: Ring Contraction in a 6,6- to a 6,5- Spiroketal System
Griseorhodin A (881) is a member of the rubromycin family of spiroketal-containing polyketides, assembled by oxidative cleavage and rearrangement of the angular tridecaketide collinone (874) (Scheme 124).560,561 From gene deletion and heterologous expression studies, Piel and colleagues showed that the 6,6-spiroketal 875 is further along the biosynthetic pathway and need to lose one more carbon atom to form 881. An FAD-dependent oxygenase (GrhO6) was identified as the catalyst likely to contract the 6,6-spiroketal framework into the 6,5-spiroketal framework in the rubromycin family.562 One of two possible routes proposed was the Baeyer-Villiger pathway (route a) to the carbonate ester 876 followed by water attack to the hydroxy acid (877). Decarboxylation attendant to ring closure would generate the contracted 5,6-spiroketal 880. In route b, the transformation would start with hydroxylation at the B/C ring juncture to yield 878. Loss of CO2 would be driven by rearomatization that generates a transient cyclic oxonium ion 879 that gets captured in 6,5 spiroketal 880 formation.
Scheme 124.

Oxidative Ring Contraction in Griseorhodin A Biosynthesis
6.1.5. Spirotryprostatin: Ring Contraction in a 6,5- to a 5,5- Spiroketal System
We noted the formation of spirotryprostatin A (781) in Scheme 107 via a proposed 2,3-indole epoxidation route. Spirotryprostatin G (887) differs from 781 by an additional desaturation in the C ring. Scheme 125 shows the proposed route from fumitremorgin C (219), involving both spirocycle formation and ring contraction of the C ring of the substrate. FqzG is the responsible P450 oxygenase.523 First it inserts an -OH group at the C/D ring juncture via the C-radical 882 to yield 883. In its second catalytic cycle FqzG starts the reaction by homolysis of a specific C-H bond in the substrate to give a new C-radical 884. In this instance it is in resonance with radical density at the lower B/C ring juncture and gives the indolyl radical 885. That is the site of delivery of [OH•] in the rebound step to give the diol 886. The diol is proposed to be the transient species which rearranges, with ring contraction, to the spirocycle framework of 887 as the initial –OH group is expelled from the C/D bridgehead position and the second -OH group becomes the indolone carbonyl.
Scheme 125.

Oxidative Ring Contraction in Spirotryprostatin G Biosynthesis
6.1.6. Xenovulene A: Cyclopentenone Formation via a Ring Expansion-Contraction Mechanism
Active against human GABA receptor, xenovulene A (899) is a meroterpenoid consist of a sesquiterpene fused with 2,4-dihydroxyl-3,6-dimethyl-benzoic acid (888).563 The sesquiterpene portion in 899 is reminiscent of the humulene scaffolding. Its biosynthesis is remarkable for a net contraction of a fused methyl phenol in substrate to a fused cyclopentenone ring in 899.564 Most remarkably, the starting phenol 889 first undergoes catalyzed ring expansion to a tropolone 891 (Scheme 126) before two successive ring contractions by extrusion of formyl groups and their subsequent hydrolytic elimination as formic acid. The rearrangements are set off by three oxygenation steps that produce the 5,6-hydroxy formyl intermediate 890 which rearranges to the 5,7-trisubstituted tropolone 891. In turn that can ring contract to an alternate aldehyde 893 from which loss of formic acid regenerates a fused 5,6-ring system in 894 with the six-membered ring now a catechol. One more oxygen transfer yields the trihydroxybenzene metabolite 895 which can undergo a second round of ring contraction and formyl group extrusion to give 897. Repeat of the hydrolytic loss of formate yields the tautomer 898 of the final 5,5-hydroxycyclopentenone ring of 899.
Scheme 126.

Cyclopentenone Ring Formation in Xenovulene A Biosynthesis
6.2. Ring Expansion Processes
6.2.1. Baeyer-Villigerases: Cyclic Ketones to Lactones
A predominant route to ring expansions in cyclic ketones is the oxygen insertion adjacent to the carbonyl group carried out by oxygenases that act as Baeyer-Villigerases.565–568 Most of those enzymes are flavoproteins, using the flavin-4a-OO− as the initiating nucleophile on the substrate carbonyl group to generate a covalent Criegee intermediate (Scheme 5A). We note below one example in the hemeprotein cytochrome P450 family during maturation of the brassinosteroid plant hormones.
Pentalenolactone (906) is a Streptomyces sesquiterpenoid antibiotic with good activity against both Gram-positive and Gram-negative bacteria as well as a variety of fungi and protozoa.569,570 It features a unique 5-5-6-3 fused cyclic structure within the lactone ring proposed to be synthesized from the expansion of intermediate 901, the oxidative product of pentalenene (900). Scheme 127 shows the expansion of the methylcyclopentanone ring in 901 to the ring-expanded isomeric lactones to yield pentalenolactone D (904) or neopentenolactone D (903) that can arise from the migration of either neighboring carbon after formation of the Criegee intermediate 902.571–573 It is notable that 904, the precursor of 906 is not the favored product based on migratory aptitude (2° vs. 3°) by Baeyer-Villiger reactions. Further dehydrogenation by an nonheme α-KG iron dependent oxygenase yields an exo olefin that can be epoxidized by the same enzyme to yield pentalenolactone F (905), which is one additional oxidation step away from the final product 906.573
Scheme 127.

Baeyer-Villiger Oxidation in Pentalenolactone Biosynthesis
Baeyer-Villigerases catalyzed ring expansion is also widely found during fungal meroterpene biosynthesis, such as the transformation of preaustinoid A (907) to preaustinoid A1 (909) (Scheme 128). The FMO AusC inserts one oxygen atom between C2 and C3 to yield 909 forms the 7-membere lactone.574 The Criegee intermediate 908 is shown and in this case the preferred migratory aptitude is followed in the ester formation.
Scheme 128.

Baeyer-Villiger Oxidation in Preaustinoid A1 Biosynthesis
A novel Baeyer-Villigerase that can catalyze transformation of a ketone to a carbonate functionality was confirmed in the biosynthetic pathway of cytochalasin E, a PK/NRP hybrid angiogenesis inhibitor produced by A. clavatus.575,576 Through in vitro biochemical characterization, the FMO CcsB was confirmed to catalyze the transformation of cytochalasin Z16 (918) from ketochalasin (910) via two successive oxygen insertion reactions (Scheme 129).577 The first step converts the 11-membered macrocyclic bis-ketone 910 to the 12-membered ketolactone 912 via the Criegee intermediate 911, notable for the size of the cyclic ketone expanded. The second oxygen insertion creates a cyclic carbonate 918 with a 13-membered macrocyclic ring. While a similar Criegee intermediate can be written for the second oxygen insertion, Scheme 129 depicts a route via epoxy intermediate that might be in play for generation of the unique cyclic carbonate functional group.577 The mechanism starts with the 1,4-addition of the flavin-peroxo anion onto 912 to yield the adduct 913. This leads to formation of the epoxide 914 that can ring open to give the α-alkoxide 915. Subsequent attack of the alkoxide yields the tetrahedral intermediate 916 that can collapse to yield the conjugated carbonate 917. Ketonization of 917 then affords 918 that is two more oxidation steps away from cytochalasin E. This mechanism depends on the 1,5-vinylogous diketone functionality located on the southern portion of the starting 910, with the distal ketone serving as an electron sink during the transformation. The discovery of CcsB expands the repertoire of activities of Baeyer-Villigerase and provides a possible synthetic strategy for transformation of ketones into carbonates.
Scheme 129.

Baeyer-Villiger Oxidation in Cytochalasin Biosynthesis
The biosynthesis of the brassinosteroid plant hormones (924) involves a cytochrome P450-mediated Baeyer-Villiger expansion of the B-ring ketone in castasterone (922) as shown in Scheme 130.578 CYP85A3 in tomato plants can convert 6-deoxycastasterone (919) to 922, by double hydroxylation at C6 (first to the alcohol 920, then to the ketone hydrate 921, in equilibrium with the ketone 922).579 922 is then the substrate for a third oxygen transfer via CYP85A3, this time as a Baeyer-Villigerase ketone to lactone expandase to yield 924. The FeIII-OOH is the presumed species attacking the 6-keto group of 922 to yield a Criegee intermediate 923.
Scheme 130.

P450 Catalyzed Baeyer-Villiger Ring Expansion in Brassinosteroid Biosynthesis
6.2.2. Penicillin N to Desacetoxycephalosporin C: β-Lactam Expandase
Perhaps the most famous of the enzymes with expandase activity in natural product biosynthesis is the desacetoxycephalosporin synthase (DAOCS), catalyzing the conversion of the penicillin class of β-lactam antibiotics into the cephalosporin class.580 The penam to cephem conversion involves expansion of the 4,5- bicyclic scaffold penicillin N (925) to the 4,6-scaffold in desacetoxy-cephalosporin C (929) with an endocyclic double bond in the six-membered ring. As schematized in Scheme 131, DAOCS is a Fe(II)/α-KG-dependent dioxygenase, reducing O2 with no oxygen insertion into product, while generating substrate radicals 926 and 928 via C-H bond homolytic cleavages.581,582 The rearrangement of the initial methyl radical species 926 to the episulfide radical 927 and its opening to the 4,6 bicyclic system is depicted. Quenching of the product radical 928 by H• transfer to iron, creates the endocyclic cephem double bond in 929 and returns the active site iron to its starting FeII oxidation state. Semisynthetic versions of the cephem antibiotics, in which the 4,6 bicyclic warhead is conserved and synthetic changes to the aminoadipate and 3-methyl substituent implemented, are among the bestselling antibiotics globally.
6.2.3. Stipitatic Acid: Tropolone Ring Formation
A variety of natural products contain the 7-membered hydroxy cycloheptatrienone ring system known trivially as tropolone.583 A short fungal pathway converts 3-methylorcinaldehyde (930) (from action of polyketide synthase TropA) to the 5,7-scaffold of stipitatic acid (935) by way of the tropolone intermediate stipitaldehyde (934) (Scheme 132).584 TropB is a flavoenzyme oxygenase that hydroxylates ortho to the phenolic oxygen, producing a dearomatized cyclohexenone scaffold 931. Nonheme iron α-KG-dependent oxygenase TropC is proposed to oxygenate the adjacent methyl group in 931 to the primary alcohol 933 via a rebound mechanism, which sets off the ring expansion to the 934 as shown.585 We noted an analogous ring expansion to a tropolone in xenovulene biosynthesis in Scheme 126 before an iterated set of ring contractions that takes troplone down to a cyclopentenone. A different mechanism for tropolone ring formation via an epoxide intermediate was confirmed in the degradation of phenylacetyl-CoA,586 and was proposed in the biosynthesis of omega-cycloheptyl fatty acids. 587
Scheme 132.

Tropolone Ring Formation in Stipitatic Acid Biosynthesis
6.2.4. Acetylaranotin: 4,5–Dihydrooxepine Ring Formation
We have noted an aspect of acetylaranotin (760) biosynthesis in Scheme 104 (Section 4.4), where the focus was on enzymatic formation of the aromatic epoxide and its internal capture by the amide nitrogen of the diketopiperazine neighboring group to construct the C-N bonds from each of the DKP nitrogens as the pentacyclic scaffold is created. We also noted the likely route to epidisulfide formation in Scheme 69 (Section 3.4).510 A brief return to acetylaranotin maturation here is on the conversion of each of the cyclohexadienol esters in 936 to the ring expanded 4,5-dihydrooxepines in 760, catalyzed by the cytochrome P450 monooxygenase AtaY (Scheme 133). The likely route is via a transient epoxide with ring opening to the seven-membered oxepine 937 followed by a second round of ring expansion to 760, but the exact mechanism of this transformation remains unresolved.
Scheme 133.

Oxidative Ring Expansion in Acetylaranotin Biosynthesis
6.2.5. Aspyridone: Tetramic Acid to Pyridone
Aspyridone (945) was first isolated from A. nidulans following targeted overexpression of the transcription factor that controls the transcription of the apd biosynthetic pathway.588 Subsequent expression of the hybrid PKS-NRPS ApdA bimodular enzyme in the heterologous host S. cerevisiae results in release of the tetramic acid preaspyridone (939) shown in Scheme 134. This is the starting compound for expansion of the five member tetramic acid ring to the 2-pyridone ring found in 945, tenellin, and related metabolites.589 The P450 enzyme ApdE (and related ones in other pathways) is proposed to carry out homolytic C-H cleavage as the initiation step at the benzylic CH2 group to yield 940.290 While direct [OH•] rebound can yield the off-pathway hydroxy metabolite 941, a competing internal rearrangement to the cyclopropanol radical 942 can lead to the ring expansion as shown in Scheme 134. Capture of that ring-expanded carbon radical 943 by [OH•] rebound yields the hydroxy precursor 944 to aspyridones. The delivery of an OH radical equivalent after the substrate radical has rearranged is reminiscent of the spirotryprostatin G biosynthetic sequence in Scheme 125.
6.2.6. Ring Expansion During Penitrem E Biosynthesis
Penitrem A (950) and the deschloro-version penitrem E (949) are the most complex indole diterpenes isolated from filamentous fungal species to date.153 The fully tailored natural product features a contiguous decacyclic ring system including a 4,6-bicycle fused to the indole phenyl ring. Formation of the nine-membered macrocyclic ether ring in penitrem D (171) from secopenitrem D (166) was covered in Scheme 29 (Section 2.2). Formation of 166 itself involves an unusual oxidative ring expansion step with unresolved mechanism. The prenyltransferase PtmE was shown to catalyze the prenylation-initiated cationic cyclization between DMAPP and the prenylated paxilline derivative 946 to yield PC-M4 (947). The reaction starting from 947 catalyzed by the P450 PtmK has been reconstituted in the heterologous host A. oryzae (Scheme 135).590 A possible mechanism is shown where consecutive C-H bond homolytic cleavage of the two methyl group, first in 947 followed by 948, leads to expansion of the cyclopentane to a cyclohexane ring and exo methylene formation in 166.591
6.2.7. Ring Expansion During Anditomin Biosynthesis
A dazzling arrays of oxidative modifications catalyzed by two nonheme iron α-KG-dependent oxygenases AndA and AndF are proposed for the transformation of preandiloid C (951) to anditomin (726).574 The epoxide-mediated formation of the meroterpenoid scaffold was discussed previously in Scheme 100 (Section 4.3). Starting with 951, generation of the bicyclo[2.2.2]octane ring system in andiconin (956) is catalyzed by AndA (Scheme 136). Methyl C-H bond homolytic cleavage in 951 yields the substrate radical 952, which can initiate the C-O bond cleavage to form fused 1,3-cyclohexanedione-furanone radical intermediate 953. The radical then adds to the newly exposed exo methylene to form a new seven-membered ring radical intermediate 954. This radical adds to the furanone double bond to yield a new C-C bond and eventual quenching of the newly formed intermediate 955 arrives at 956. After an identical Baeyer-Villiger ring expansion as depicted for preaustinoid A1 (909) in Scheme 128 to afford andilesin C (957), AndF receives the oxidative relay baton and cleaves the C-H bond in the middle cyclopentane ring to form 958. Following an [OH•] rebound that gives 959, a ring expansion step takes place to expel water and form 726. The sequence of [OH•] rebound followed by elimination to drive rearrangement is observed once more in this example (See Scheme 134 and others). In total the dramatic morphing from 951 to 726 forges three new carbon-carbon bonds and represents one of the most complicated sequence of rearrangements characterized to date.
Scheme 136.

Ring Modifications in Anditomin Biosynthesis
6.3. Additional Oxidative Ring Rearrangements
6.3.1. Fumagillin: Multifunctional P450-Catalyzed Morphing
The A. fumigatus metabolite fumagillin (970) has a meroterpenoid structure, reflecting convergence of an isoprenoid and polyketide biosynthetic pathways.592 970 displays anti-angiogenic activities and has been considered as an antitumor lead.593 The key structural feature is the heavily oxidized cyclohexanone ring, including the bis-epoxide moiety. The epoxide groups are sufficiently reactive as electrophiles to target the methionine aminopeptidase in protein biosynthesis.594–596 Identification and reconstitution of the fumagillin biosynthetic pathway revealed several interesting enzymes. First the sesquiterpene cyclase (Af520) that synthesizes β-trans-bergamotene (960) from farnesyl-diphosphate was the first membrane-bound Class I-type terpene cyclase discovered.592 Af520 converts farnesyl-PP to the corresponding nerolidyl-PP and then cyclization via a bisabolyl cation to the indicated 4,6-bicyclic hydrocarbon product.
Second, the P450 Af510 was shown to be a powerful, multifunctional oxidase/oxygenase that converts 960 into the bis-epoxide ketone intermediate 968 (Scheme 137).51 This remarkable transformation by Af510 corresponds to a net eight-electron oxidation of the substrate. The first oxygen-transfer to the bridgehead alcohol to yield the monohydroxylated 961. From there several mechanistic proposals have been presented, two of which are shown here.597 In route a, C-H bond cleavage in the prenyl portion that affords 962 is followed by a second one electron oxidation to yield the carbocation intermediate 963. This species is then attacked by the Iron-peroxo anion to form the heme-bound peroxide 964, which can collapse to the epoxy-ketone 967 in a concerted series of electron transfer from the bridgehead hydroxyl to the iron center. An alternative mechanism is shown in route b that excludes formation of the secondary carbocation 963. Here Af510 installs another alcohol group in the prenyl side chain to yield a doubly hydroxylated bergamotene product 965. The third catalytic cycle of Af510 results in fragmentation of the bicyclic framework to yield the indicated cyclohexanone radical 966. A fourth P450 cycle converts the side chain alcohol to the epoxide 967 before the exo methylene is itself epoxidized (five Af510 catalytic cycles) to create the bisepoxide framework 968. To finish the fumagillin pathway, 968 is further oxidized at the ortho position, methylated and finally reduced at the ketone to yield fumagillol (969). 969 is then acylated with a pentaene polyketide product to yield prefumagillin, of which the terminal olefin can be oxidative cleaved to yield 970.
Scheme 137.

Proposed Mechanisms of Carbon Scaffold Rearrangement during Fumagillin Biosynthesis
6.3.2. Roquefortine to Meleagrin: Reconfiguring Connectivity in a Tetracyclic Framework
Roquefortines, natural products produced by fungi that are named for their cheese associations, are simple C3-prenylated diketopiperazine metabolites.598,599 Roquefortine D (971) arises from a tryptophanyl-histidine NRPS bimodular assembly line by internal cyclizing release as the DKP forms.600,601 Subsequent prenylation at C3 of the indole is attended by amide nitrogen attack on the incipient indoline at C2 to create the tetracyclic core of 971 (Scheme 138).602 RoqR in analogy to AsqJ in viridicatin biosynthesis (Section 6.1) effects a homolytic desaturation of the bridge between the DKP core and the pendant imidazole to produce roquefortine C (972).602 Gene-knock out results indicate that two oxidative enzymes (P450 RoqM and FMO RoqO) are responsible for transformation of 972 to glandicoline B (976). First, N-hydroxylation by RoqM generates an electrophilic imino N-oxide roquefortine L (973). The second hydroxylation catalyzed by RoqO is proposed to occur at the C/D ring juncture to yield a carbinolamine 974, which reverts to the carbonyl, mediating a transannular disconnection and produce a nine-member ring intermediate 975. Now another amide nitrogen from the L-histidine portion of DKP core can attack at C2 of the indole-N-oxide, which creates the crowded tetracyclic scaffold of 976 and then meleagrin (977) after O-methylation. The connectivity of the tetracyclic framework between 971 and 977 has been distinctly altered, from a 6,5,5,6 starting array to a 6,5,6,5 order in the product.
Scheme 138.

Tri-aminal Formation during Meleagrin Biosynthesis
6.3.3. Xantholipin: Quinone to Xanthone Ring Conversion
The angular heptacyclic antitumor antibiotic xantholipin (986) produced by S. flavogriseus is assembled by a type II polyketide synthase.524,603 The conversion of the quinone D ring of the pentatcyclic polyketide intermediate 978 to the xanthone D ring in 985 on the way to 986 is initiated by a FMO XanO4 (Scheme 139).604 Two successive oxygenations are proposed for the C and D rings of the substrate 978. The first oxygenation via the adduct 979 yields 980, which is an arene oxide in the C ring. The second adduct 981 leads to transient ring expansion to the oxazepine 982 that undergoes hydrolysis to liberate the OH group in 983 that attacks the adjacent C ring epoxide and afford 984. The system is now set up for decarboxylation (and expulsion of methanol), effectively carving out the carbon atom that had been at the top of the D ring. Simultaneously, the oxygen atom from the second Baeyer-Villiger oxygenation reaction has been inserted as the xanthone forms in ring D to arrive at 985. Subsequent P450-mediated formation of the methylenedioxy bridge and pyridone ring completes the complex heptacyclic scaffold of 986.
Scheme 139.

Xanthone Formation via a Cryptic Demethoxylation in Xantholipin Biosynthesis
6.3.4. Chartarin: Linear to Angular Polyaromatic Framework
The DNA-binding antitumor agent chartreusin (993) produced by S. chartreusis is a disaccharide of a pentacyclic aromatic polyketide aglycone chartarin (992).605 As shown in Scheme 140, 992 is derived from a linear tetracyclic anthracycline framework 987. The conversion to the rearranged connectivity in 992 is initiated by the oxygenase ChaZ which cleaves the B ring quinone as indicated, yielding the acyclic hydroxy ketone 988.606 Rotation around the C-C single bond precedes reclosure with C-C bond formation as indicated to yield the angular framework 989. The quinone is restored but the connectivity between the B and C rings has been flipped. This twist places the carbomethoxy substituent close to the indicated -OH group in 990 which engages in lactone formation to yield 991. A cupin-like dioxygenase ChaP is proposed to mediate the last step: an oxygenative removal of one of the carbonyl carbons in the cyclohexadienone 991 to 992 by successive possible reactions including hydroxylation, ring opening and lactonization as CO2 is released. The oxidative carving out of the one carbon unit as CO2 has analogies to the xantholipin logic above.
Scheme 140.

Angular Polyaromatic Framework Formation in Chartreusin Biosynthesis
6.3.5. Jadomycin: Quinone to Oxazolidinone
Jadomycin A (1001) is a type-II angucycline polyketide produced by Streptomyces sp. with antitumor activity.607,608 As shown in Scheme 141, the benzo[d]naphthopyrone framework of 1001 derives from benz[a]anthracene precursors such as dehydrorabelomycin (994), the starting substrate, via a series of substantial rearrangements in ring construction, especially ring C cleavage.609–611 An FMO JadG and its reductase JadY partner work together to hydroxylate the C ring of 994, as well as to carry out a Baeyer-Villiger type oxygen insertion to create the seven-membered hydroxylactone intermediate 996. Hydrolytic opening of 996 yields the aldehyde acid 997. Next, incorporation of L-isoleucine can lead to formation of 1001. Two possible mechanisms are proposed for this step. In route a, a Schiff base intermediate 998 is formed via the reaction of isoleucine amine group with aldehyde in 997, this is followed by attack on the quinone ring to form the oxazolidinone ring in 1001. Alternatively in route b, attack of cosubstrate isoleucine amine group in a 1,4-Michael addition to the B ring quinone takes place first to yield 999. The acid group initially present in the B-ring quinone can now decarboxylate in a low energy pathway since it is a β-ketoacid, and forms 1000. Attack of the α-NH on the aldehyde in 1000 creates a tetrahedral adduct whose oxyanion can react at the isoleucine carboxylate to create the oxazolidinone ring in the end product 1001. Combined with the examples of xantholipin and chartarin, this is the third example of an oxidative strategy to carve out one carbon unit from a polycyclic framework during biosynthetic morphing.
Scheme 141.

Proposed pathways of Oxazolidinone Formation in Jadomycin Biosynthesis
6.3.6. PD116198: Anthracycline to Angucycline
The benz[a]anthraquinone antibiotic PD116198 (1011) has also undergone skeletal rearrangement from the anticipated octaketonyl-S-PKS enzyme intermediate.612,613 As suggested in Scheme 142, the initial oxidative tetracyclic metabolite 1003 derived from the released product 1002 from the PKS enzyme undergoes oxygenative Baeyer-Villiger type expansion of the D-ring ketone to the lactone 1004, catalyzed by BexE.614 Hydrolysis to the hydroxy acid 1005 allows rotation and reformation of an angular ketone 1008, thereby altering the connectivity from the linear tetracyclic starting substrate. A second Baeyer-Villiger insertion by the BexM enzyme to afford 1009 is proposed to be followed by a ring contraction back to the indicated hydroxy cyclohexanone 1010. One more hydroxylation by BexK yields the end metabolite 1011.614
Scheme 142.

Anthracycline to Angucycline Rearrangement in PD116198 Biosynthesis
6.3.7. Aflatoxin Structural Rearrangement from Anthraquinone
The most famous fungal toxin aflatoxin B1 (1034) is produced by many filamentous fungi. The biosynthesis of 1034 involves significant structural morphing from an aromatic polyketide product to the final product that is hardly recognizable to be derived from polyketide origin.615 Many of the oxidative rearrangement cascades observed in the previous examples, including Baeyer-Villiger ring expansion, lactone cleavage and CO2 expulsion will again be in play in this pathway.616,617 As shown in Scheme 143, the biosynthetic pathway of 1034 involves at least 21 steps starting from the polyketide product norsolorinate anthrone (1012), which can undergo oxidation into the anthraquinone 1013, and reduction of the benzylic ketone to yield norsolorinic acid (1014).506,618–620 From there the structural morphing can be artificially divided into four stages.
Scheme 143.

Oxidative Ring Arrangements During Aflatoxin Maturation
The first stage results in formation of the hydroxyl-tetrahydrofuran ring in 1′-hydroxyversicolorone (1018). C5′-hydroxylation of 1014 by the P450 AvnA to (1′S, 5′S)-5′-hydroxyaverantin (1015) is followed by dehydrogenation of the newly introduced hydroxyl group yields the 5′-ketone intermediate (5′-oxoaverantin, OVAN, 1016).621 This can be converted to (1′S, 5′S)-averufin (1017) by the OAVN cyclase via intramolecular acetal formation.622 Subsequently, based on in vivo knockout results in the sterigmatocystin pathway, the P450 StcB and FMO StcW are responsible for converting 1017 to 1018.623 Based on the isotope labeling together with feeding of synthetically prepared analogs, either a cation- or radical-dependent mechanism can be proposed for this unusual oxidative rearrangement.624–626
The second stage includes four reactions to construct the pentacyclic intermediate versicolorin A (1022) which contains the bisfuran moiety. 1018 is first converted to versiconal hemiacetal acetate (1019) via a Baeyer-Villiger reaction, during which the aliphatic ketone is converted to an ester.623,627,628 Hydrolysis of the acetate reveals the terminal hydroxyl group as in versiconal (1020), which can be cyclized into versicolorin B (1021) by versiconal cyclase.629,630 The terminal tetrahydrofuran ring can be dehydrogenated to form 1022.
The third stage converts 1022 to demethylsterigmatocystin (1030), which converts the linearly fused anthraquinone to the angular xanthone, both pentacyclic compounds. Two enzymes, a P450 AflN and a probable NADPH-dependent oxidoreductase AflM are involved in this transformation.631 First, P450-mediated aryl epoxidation by AflN affords the arene oxide 1023. Oxirane opening converts ring A in 1023 into the hydroxylated dienone in 1024. AflM mediated reduction of the dienone followed by dehydration yield the triketone intermediate 1026. Next, as observed in the xantholipin biosynthesis in Scheme 139, the quinone ring is converted to xanthone through Baeyer-Villiger oxygen insertion to 1027, hydrolytic cleavage to 1028, xanthone formation to 1029, and expulsion of carbon dioxide and water to yield demethylsterigmatocystin (1030).
The fourth stage starts with methylation of 1030 to yield O-methylsterigmatocystin (1031), which is morphed into 1034 in a net ring contraction sequence.632,633 The exact order of reactions and the corresponding enzymes have not been elucidated. It is proposed that a P450-catalyzed hydroxylation to give 1032, an additional Baeyer-Villiger oxidation to give 1033 are required for the ring expansion, rearrangement and contraction steps.634–636
6.3.8. Betalamic Acid: Catechol to Tetrahydropyridine Dicarboxylate
Betalamic acid(1036) (Scheme 144) is the chromophore in a variety of plant pigments, including betacyanins (1037 and 1038) and betaxanthins (1039), which are red and yellow pigments respectively.637,638 It forms spontaneously from 4,5-seco DOPA (1035), which arises from action of regiospecific plant dioxygenases on the abundant metabolite L-DOPA (528).639 The enzyme is a member of the extradiol dioxygenase family, cleaving adjacent to one of the catechol-OH groups.640 The classic product pattern is conversion of the phenol carbon to the carboxylate and the adjacent carbon to the aldehyde, and that is the structure of 1035. The enol isomerizes to the C5 ketone that is then captured by the amine. Loss of water generates the 1036 with its dicarboxy tetrahydropyridine framework. The resulting aldehyde can engage in imine formation with several amines in plant cells to form the chromophoric betalins.
Scheme 144.

Oxidative Rearrangements in Betalamic Acid Biosynthesis
7. CONCLUDING THOUGHTS
Ubiquity of cyclic structures in natural products
The preponderance of the >500,000 natural products contain one or more carbocyclic or heterocyclic rings and most of those have multiple fused rings in their scaffolds. Five and Six membered rings predominate although we have delved in depth into the great biosynthetic versatility of the three atom epoxide rings. Seven member diazepinediones and tropolone, and eight member cyclooctanes are also found. In the polyketide family of macrolides, macrolactones from 12–22 atoms are generated by producer microorganisms.
The structural and functional group diversity of cyclic natural product frameworks is nothing short of awesome and have inspired generations of synthetic and medicinal chemists. Many of these scaffolds have still not yielded to total synthesis and the complex ones that have been prepared are typically not suitable for industrial scale production compared to the biological sources. Complexity generation in the conversion of simple, primary metabolic building blocks to polycyclic product architectures can occur by short efficient enzymatic pathways, as noted several times in the several sections of this review.
Several hypotheses have been advanced to comment on the utility of cyclic frameworks to producer organisms. The enzymatic conversion of acyclic precursors into cyclic natural products pays the energy costs upfront for building in entropic constraints. The resulting end products have order of magnitude higher affinities for particular biologic targets (DNA, RNA, Proteins, bacterial lipid II, peptidoglycan) than the linear precursors. This argument for cyclic architectures has the built-in assumption that these natural products are generated by producers for such specific recognition/ligation. The putative functions of the small molecule secondary metabolites span a spectrum of signaling, antibiotic activity, plant defense against predators (phytoalexins, phytoanticipins), development, and pheromone functions.641
Many of the natural products that have been active in human pharmacology assays are unlikely to have been the intended targets by the microbial or plant producers. They may reflect shapes, polarities and functional group arrays that have universal chemical properties for interaction with proteins (e.g the immunosuppressive fungal molecules FK506 and rapamycin) or DNA (bleomycin), just to pick two types of human targets.
Nonoxidative cyclizations: outside the scope of this review
In this review we have considered the chemical logic and enzymatic machinery for only a subset of cyclizations of natural products, those that are enabled by or directly involve one or more redox steps. Three examples of large families of nonoxidative cyclizations include macrolactonizing release of polyketides of the erythromycin, fidaxomycin class40 and the aromatization of polyketonic intermediates into tri-and tetracyclic aromatic ring frameworks (e.g. tetracenomycin)7,642,643. (By contrast, prior reduction of the C9 ketone to the alcohol reflects a redox-controlled cyclization to pretetramid, the precursor to tetracycline). The third major non-redox cyclization route is exemplified by the plethora of cation-driven rearrangements of mono-, sesqui-, di-, and triterpenes into many thousands of mono, bi-, tri-, and tetracyclic product scaffolds.644–648 The bacterial squalene-hopene cyclases run in parallel to the oxidosqualene cyclases (Section 4.3), reflecting the alternate (and probably earlier evolutionary route) of initiating tetracyclic triterpene formations by either H+ (non-oxidative) or epoxide (oxidative from the previous enzymatic step) as initiating electrophiles.649–651
Redox-Driven Cyclizations in Natural Product Assembly
The biosynthetic cyclizations that are covered in this review encompass C-C, C-O, C-N, C-S, N-N, and S-S bond forming steps in ring formations and occur in all the major classes of natural products. We have noted two distinct logics in play for the multitude of C-C and C-O bond formations: one electron and two electron reaction manifolds.
One Electron Routes
While radical chemistry is relatively rare in primary metabolism, it is a signature route for the hundreds to thousands of iron-based oxygenases that are sprinkled liberally through so many biosynthetic maturation pathways. The iron-based oxygenases generate high-valent oxoiron species as powerful active site-contained oxidants that can cleave unactivated C-H bonds homolytically in bound substrates. That sets off two possible downstream reaction manifolds. One is [OH•] rebound leading to oxygen transfer. The second is a competing intramolecular reaction/rearrangement of the carbon centered radical (often with electron transfer from internal olefins) that results in ring formations without any net transfer of oxygen atoms to product. The broad reach of these alternate product-determining routes are the subject of Section 2. In many of the examples evidence has not yet accrued to distinguish between formation of a radical pair that combine vs pathways that involve migration of the initial radical with bond formations before transfer of a second electron to the oxoiron cofactor in the enzyme active sites.
The enzymatic generation of the penicillin and cephalosporin classes of β-lactam antibiotics,246,247 historically the most widely used antibiotics globally over the past forty years, illustrates the separation of O2-reductive activation, carbon-centered substrate radicals, and the formation of carbacylic and heterocyclic fused ring systems (Section 2.4). The separation of O2-induced substrate carbon radicals from [OH•] rebound is also emphasized in the O2-dependent halogenases noted in Section 5. In those cases chlorination, like epoxidation, can be a transient fate on the way in those cases to intramolecular SN2 eliminations to cyclopropane rings.
Among the various examples of radical-mediated coupling and ring formations the biosynthesis of the glycosylated indolocarbazole staurosporine stands out for having three radical steps during assembly (Section 2.2.2).64,65,68,71 The first is in construction of the C-C bond in chromopyrrolic acid, the second in the C-C coupling of the two pyrrole rings of the indole moieties to create the hexacyclic indolocarbazole platform. The third is the second N-C bond-creating step in bicyclic attachment of the sugar to create the final octacyclic staurosporine metabolite.
Other prominent examples of biosynthetic generation of radical species on electron rich aromatic rings of pathway intermediates include the oxidative coupling of five side chains in vancomycin and all seven in the heptapeptide framework of the teicoplanin family of antibiotics (Section 2.2.1).41,42 These one electron manifolds for C-C bond formation also occur in a key step in morphine biosynthesis, (S)-reticuline to salutaridine (Section 2.2.3).120
At the other end of the oxygen availability spectrum are a set of S-adenosylmethionine (SAM)-dependent radical reactions that require anaerobicity because of attendant auto-oxidizable iron/sulfur clusters as electron donors. More than a hundred thousand predicted radical SAM open reading frames are in protein data bases, suggesting that an enormous range of anaerobic radical chemistry, most of it in secondary metabolism, awaits discovery.22,23,25 SAM is progenitor to the 5′-deoxyradical as initiator of C-H bond homolytic cleavages to set one electron reaction manifolds in motion (Section 2.4).23 It is likely that the radical SAM logic and enzymatic machinery evolved early on in anaerobic/microaerophilic organisms before O2 rose to current levels of 20% in the atmosphere and aerobic enzymatic machinery came into widespread distribution in secondary metabolic pathways.
The biosynthetically most useful feature of oxygenase chemistry is olefin epoxidation (Section 4). As in synthetic organic chemistry, the epoxides are a useful staging point for further complexity generation.430,652,653 The three member oxirane ring provides either of two carbon atoms for attack by nucleophiles, intramolecular in all the cases discussed in Section 4. Further, the epoxide oxygen as it is opened by nucleophilic attack at the adjacent carbon center can function as nucleophile, most clearly illustrated in the two step biosynthetic conversion of olefins into furan- and pyran-containing cyclic polyether metabolites.
The most celebrated pathway for two step creation then ring-opening of an epoxy intermediate is in the conversion of squalene to thousands of cyclic triterpene sterol-like frameworks, via 2,3-oxidosqualene. Epoxide-induced cyclizations are also prevalent in indole terpene biosynthesis648 and in the tri-epoxide skeletal morphing in aurovertin E biosynthesis.455 In addition to aliphatic epoxide creation and opening are several examples of arene and heteroarene epoxidative intermediates, exemplified in gliotoxin biosynthesis512 as well as spirotryprostatin spirocycle formations.523
Two Electron Routes
A second major route to biosynthetic ring formations during the assembly of natural products involves two electron pathways. These involve heterolytic mechanisms for C-C bond formations, with carbanion equivalents capturing electrophilic partners. Not surprisingly the enzymes that set up these cyclization routes use the two most readily available redox coenzymes of cellular metabolism, NAD(P)H and FAD.
NAD(P)H is the prototypic diffusible reduction currency in cells. In its reduced form it is a hydride donor; in its oxidized form, NAD(P)+ is a hydride acceptor. FAD by contrast is almost always tightly bound to its partner redox enzymes because in its dihydro form it is rapidly autoxidized by O2. This is a crucially useful feature for FAD-enzymes to function as oxygenases, as noted through examples in Sections 3.6–3.10. But for the reactions in Section 3.3 it is the ability of FAD to accept hydride ions from substrates undergoing oxidation that is determinative.
Hydride Transfers to Drive Ring Formations
Both reductive and oxidative two electron pathways can be utilized to drive ring formations. The ikarugamycin case emphasizes how a hydride ion transfer from NADPH to an olefin sets off a transannular 1,6 carbonyl addition that builds a cyclopentane ring (Section 3.2.4).298 In a different context NADH-mediated reductive cleavage of peptidyl thioesters from an NRPS assembly line yields the nascent peptidyl aldehyde that can be cyclized to the imine and then captured in an intramolecular Pictet-Spengler reaction, twice, on the way to the antitumor scaffold of saframycins (Section 3.2.2).288
The oxidative transformations that feature hydride transfer to enzyme-bound FAD during cyclization are illustrated by the berberine bridge enzyme in alkaloid assembly (Section 3.3).654 Comparable hydride transfer logic to Enzyme-bound FMN converts peptidyl thiazoline and oxazoline rings to the stable heteroaromatic thiazole and oxazoles in such metabolites as patellamides and telomestatin (Scheme 67 and 68).337 Likewise, hydride transfer to flavoenzymes sets the cyclization cascade in tetrahydrocannabinol (Scheme 63) and in cyclopiazonate (Scheme 65) framework maturations.317,330–332
Transient Formation of Carbonyls as Cyclization Electrophiles
An alternative logic for two electron reaction manifolds to generate electrophiles employs enzymatic oxidation of alcohols to aldehydes or ketones and of amines to the corresponding imines. Capture of such transient carbonyls is at work in pipecolic acid formation for FK506 and rapamycin (Scheme 71)363, chanoclavine-I to lysergic acid (Scheme 72)373, and flavones to the yellow pigment aurones (Scheme 75)384,386. Notably this hydroxy ketone route is the path to assembly of the 6,6-spirocycle in reveromycin (Scheme 73).376 (In passing we note this oxygen-independent route to spirocycles differs from the O2-dependent routes, e.g. to griseofulvin (Scheme 25)136,137, reflecting independent routes to this geometry-imposing bicyclic unit.).
Oxidations that Set up [4+2] Cyclizations
We have dealt briefly in this review with examples of [4+2] cyclizations that have differential accrued evidence for concerted (Diels-Alder) vs stepwise mechanisms (Section 3.10). Although the five and six-member ring-forming steps themselves do not involve oxidation or reduction, the enzymes catalyzing the prior step(s) do carry out oxidative transformations to set up the diene/dienophile reactive array. This can be manifested differently in solanapyrone (Scheme 81) where a conjugated aldehyde lowers energy barriers413,414, versus the lovastatin (Scheme 82)417, spinosyn (Scheme 83)421, and abyssomicin (Scheme 84)426 systems where the prior enzymes create either the dienophilic olefin or the diene array. The pyrroindomycin (Scheme 85)427 case illustrates the two observed types of [4+2] cyclizations in two successive steps of one biosynthetic pathway.
The hirsutellone maturation logic is worth note at this juncture (Scheme 30). There are three distinct routes for C-C bond formation: (1) epoxidation, (2) radical coupling with phenolate, (3) an apparent Diels-Alder to create the 6-5-6 tricyclic scaffold element.162
Redox Dependent Morphing of Existing Ring Structures
Sections 2–5 deal with transformations that create new ring systems in the maturing natural products. Section 6 examines a distinct biosynthetic logic: the redox-dependent interconversion of one ring type to another. In a formal sense the conversion of epoxides to furans and pyrans in the polyethers by the epoxide hydrolases, considered on their own, would also fall into this category. The conversion of the 4,5-fused β-lactam in penicillin N to the fused 4,6-β-lactam in desacetoxycephalosporin is a notable additional example (Scheme 131), by one electron, O2-dependent enzyme chemistry.580–582
The rearrangements can be divided into ring contractions and ring expansions. Notable among the oxidative ring contractions are the 7-member diazepinedione to 2,3-dioxoquinolone system in viridicatin assembly (Scheme 121)502 and also the cyclohexanol ring contraction in 7-hydroxy-ent-kaurenic acid to gibberellic acid-12 (Scheme 122)557. As a third route to a spirocyclic system the pathway from collinone to griseorhodin carves out one carbon from a fused 6,6-bicyclic element to the 5,6-spirocycle in the end product metabolite (Scheme 124)562. Perhaps the most convoluted molecular traverse is in xenovulene assembly (Scheme 126)564: a formyl catechol is first expanded to a tropolone then successively contracted to a formyl cyclohexanedione and, via loss of formate, generates ultimately a formyl cyclopentenone. Loss of a second formate results in oxidative carving out of two one carbon units to create the final hydroxycyclopentanone ring.
Ring expansion as a route to interconvert an existing ring into a larger lactone is a common strategy employed by Baeyer-Villigerases. Notable among the cadre of flavoenzyme Baeyer-Villigerases is the double insertion of oxygen to convert a ketone macrocycle to a cyclic carbonate ester in cytochalasin scaffold diversification (Scheme 129)577. One identified hemeprotein cytochrome P450 with Baeyer-Villiger lactonizing activity occurs at a late stage in maturation of the brassinosteroid plant hormones (Scheme 130).579
Enzymatic ring expansion of a tetramate five ring into the six ring pyridone in aspyridone assembly is proposed to proceed via an oxycyclopropyl radical intermediate (Scheme 134).290,589 Reconfiguring connectivity and/or ring identity within fused polycyclic scaffolds also occurs in a number of biosynthetic pathways. One such is the conversion of the linear tetracyclic connectivity in roquefortine D to the angular framework of meleagrin, driven by a transient hydroxylation step (Scheme 138).601,602 Another example is in the formation of the benz[a]anthraquinone polyketide antibiotic PD116198 (Scheme 142).614
The interchange of an internal benzoquinone to a xanthone in xantholipin (Scheme 139)604 and aflatoxin (Scheme 143)631 formation, and of a comparable internal quinone to an oxazolidinone in jadomycin maturation (Scheme 141)609,610 are three examples of remarkable interchanges of oxidized carbacycles to heterocycles with dramatic changes in functional group properties in the products. An additional remarkable rearrangement is the formation of the pyridazine ring in the phthalazinone meroterpenoid azamerone.655
In sum, natural product biosynthesis pathways construct compact polycyclic metabolites whose functional group arrays, connectivities, shapes, and polarities depend both on the initial building blocks and the associated tailoring enzymes. Creation of the active, mature scaffolds employs the two most available two electron redox coenzymes, NAD(P)H and FAD and the one electron radical generators O2 and SAM to drive an enormous range of carbacycle and heterocycle architectures.
Scheme 90.

Mechanism of Polyether Formation in Nanchangmycin Biosynthesis
Acknowledgments
Research activities in related areas in YT lab are supported by the by NIH 1R35GM118056 and 1U01GM110706, and the David and Lucile Packard Fellowship in Science and Engineering. K.W. thanks Japan Society for the Promotion of Science (JSPS) Program for Advancing Strategic International Networks to Accelerate the Circulation of Talented Researchers (No. G2604).
ABBREVIATIONS
- ACAT
acyl-coenzyme A:cholesterol acyltransferase
- ACP
acyl carrier protein
- ACV
δ-(L-α-aminoadipoyl)-L-cysteinyl-D-valine
- AT
acyltransferase
- ATP
adenosine 5′-triphosphate
- BBE
berberine bridge enzyme
- C domain
condensation domain
- CAS
clavaminate synthase
- CDHFL
cyclic- dehypoxanthine futalosine
- CDPS
cyclodipeptide synthase
- CPA
chromopyrrolic acid
- cYY
cyclo-Tyr-Tyr
- 5′dA•
5′-deoxyadenosyl radical
- DAOCS
desacetoxycephalosporin synthase
- DFT
density functional theory
- DH
dehydratase
- DHFL
dehypoxanthine futalosine
- DHGO
dihydrogeodin oxidase
- DKP
diketopiperazine
- DMAPP
dimethylallyl pyrophosphate
- DOPA
L-3,4-dihydroxyphenylalanine
- ER
enolylreductase
- FAD
flavin adenine dinucleotide
- Fl-OO−
peroxyflavin
- Fl-OOH
hydroperoxyflavin
- FMO
flavin dependent monooxygenase
- FMN
flavin mononucleotide
- GABA
γ-aminobutyric acid
- GA12
gibberellic acid 12
- H6H
hyoscyamine 6β-hydroxylase
- HDAC
histone deacetylase
- 2-HPP
2-hydroxypropylphosphonate
- HPPO
4-hydroxy-6-(3-pyridinyl)-2H-pyran-2-one
- IPAI
indole-3-pyruvate imine
- IPNS
isopenicillin N synthase
- ISY
iridoid synthase
- α-KG
α-KG
- KR
ketoreductase
- KS
ketosynthase
- MRSA
methicillin resistant Staphylococcus aureus
- MT
methyltransferase
- NAD(P)+
β-nicotinamide adenine dinucleotide (phosphate)
- NAD(P)H
β-nicotinamide adenine dinucleotide (phosphate), reduced form
- nM
nanomolar
- NRP
nonribosomal peptide
- NRPS
nonribosomal peptide synthetase
- Nu
nucleophile
- OYE
old yellow enzyme
- OVAN
5′-oxoaverantin
- P5βR
progesterone 5β-reductase
- PCET
proton-coupled electron transfer
- PEP
phosphoenolpyruvate
- PK
polyketide
- PKS
polyketide synthase
- PLP
pyridoxal phosphate
- PTM
polycyclic tetramate macrolactam
- R domain
reductase domain
- RiPP
ribosomally synthesized and post-translationally modified peptide
- SAH
S-adenosyl-L-homocysteine
- SAM
S-adenosyl-L-methionine
- SHC
squalene-hopene cyclase
- T domain
thiolation domain
- TDP
thymidine diphosphate
- THF
tetrahydrofuran
- THN
tetrahyrdoxynaphthalene
- UDP
uridine diphosphate
- VHPO
vanadium haloperoxidase
Biographies
Man-Cheng Tang was born in 1982 in Hunan, China. He received his B. Sc. in Chemistry from Wuhan University in 2003 and his Ph. D. in Organic Chemistry from Shanghai Institute of Organic Chemistry (SIOC), CAS, in 2010 under the supervision of Prof. Gong-Li Tang. He then worked as a research associate in Prof. Gong-Li Tang’s group at SIOC and was appointed as an Associate Professor at SIOC in 2013. In 2014, he joined Prof. Yi Tang’s lab at University of California, Los Angeles as a postdoctoral scholar. His is interested in understanding the enzymology of natural products biosynthesis.
Yi Zou was born in 1983 in Guizhou, China. He received his B. Sc. degree in 2006 and MS degree in 2009 in Pharmaceutical Engineering from Southwest University, China. He received his Ph.D. in Microbiology in 2013 from Shanghai JiaoTong Univeristy, China, under the guidance of Prof. Shuangjun Lin. During his doctoral work, he worked on identifying the gene clusters and novel enzymes involved in polyketides and alkaloids biosynthesis from Streptomyces. In 2014, He joined Prof. Yi Tang’s lab at University of California, Los Angeles as a postdoctoral scholar. His current research interests focus on the biosynthesis of fungal natural products.
Kenji Watanabe, born in 1969, received his Ph.D. in 2000 in bioorganic chemistry from Hokkaido University, Japan. His postdoctoral fellowships included a year at University of Wisconsin-Madison and a two-year appointment at Stanford University. In 2003, he returned to Hokkaido University as an assistant professor. In 2004, he joined the department of pharmaceutical sciences at University of Southern California as a research assistant professor. In 2008, he received a tenure-track position at Okayama University. He was promoted to associate professor in 2009 and professor in 2016 at University of Shizuoka. His research interest has been focused on developing a heterologous production system for natural products from PKS, NRPS and other important molecules of interest. Also, he has placed much effort into the mechanistic and structural characterization of enzymes involved in natural product biosynthesis.
Christopher T. Walsh, born in 1944, studied biology at Harvard and completed his PhD in biochemistry under Fritz Lipmann at The Rockefeller University. From 1972 to 1987 he held a faculty position at MIT and later moved on to continue his work at Harvard Medical School. He served as Chair of the Department of Chemistry at MIT from 1978 to 1982 and of the Department of Biological Chemistry & Molecular Pharmacology at Harvard Medical School from 1987 to 1995. In 2014, he was appointed a Consulting Professor of Chemistry at Stanford University and a member of the Stanford Institute for Chemistry, Engineering, and Medicine for Human Health (ChEM-H). His research interests focus on the molecular logic and enzymatic machinery of peptide-based natural product biosynthesis.
Yi Tang, born in 1976, received his undergraduate degree in Chemical Engineering and Material Science from Penn State University in 1997. He received his Ph.D. in Chemical Engineering from California Institute of Technology under the guidance of Prof. David A. Tirrell in 2002. After NIH postdoctoral training in Chemical Biology from Prof. Chaitan Khosla at Stanford University, he started his independent career at University of California Los Angeles in 2004. He is currently Professor in the Department of Chemical and Biomolecular Engineering at UCLA, and holds joint appointments in the Department of Chemistry and Biochemistry; and Department of Bioengineering. His lab is interested in identifying new enzymes from the biosynthetic pathways of polyketides, nonribosomal peptides, terpenoids, alkaloids and hybrid compounds.
References
- 1.Newman DJ, Cragg GM. Natural Products as Sources of New Drugs from 1981 to 2014. J Nat Prod. 2016;79(3):629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 2.Cragg GM, Newman DJ. Natural Products: A Continuing Source of Novel Drug Leads. Biochim Biophys Acta. 2013;1830(6):3670–3695. doi: 10.1016/j.bbagen.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mishra BB, Tiwari VK. Natural Products: An Evolving Role in Future Drug Discovery. Eur J Med Chem. 2011;46(10):4769–4807. doi: 10.1016/j.ejmech.2011.07.057. [DOI] [PubMed] [Google Scholar]
- 4.Walsh CT. A Chemocentric View of the Natural Product Inventory. Nat Chem Biol. 2015;11(9):620–624. doi: 10.1038/nchembio.1894. [DOI] [PubMed] [Google Scholar]
- 5.Walsh CT, Fischbach MA. Natural Products Version 2.0: Connecting Genes to Molecules. J Am Chem Soc. 2010;132(8):2469–2493. doi: 10.1021/ja909118a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kopp F, Marahiel MA. Macrocyclization Strategies in Polyketide and Nonribosomal Peptide Biosynthesis. Nat Prod Rep. 2007;24(4):735–749. doi: 10.1039/b613652b. [DOI] [PubMed] [Google Scholar]
- 7.Zhou H, Li Y, Tang Y. Cyclization of Aromatic Polyketides from Bacteria and Fungi. Nat Prod Rep. 2010;27(6):839–868. doi: 10.1039/b911518h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Helfrich EJ, Piel J. Biosynthesis of Polyketides by trans-AT Polyketide Synthases. Nat Prod Rep. 2016;33(2):231–316. doi: 10.1039/c5np00125k. [DOI] [PubMed] [Google Scholar]
- 9.Martinez S, Hausinger RP. Catalytic Mechanisms of Fe(II)- and 2-Oxoglutarate-dependent Oxygenases. J Biol Chem. 2015;290(34):20702–20711. doi: 10.1074/jbc.R115.648691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meunier B, de Visser SP, Shaik S. Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem Rev. 2004;104(9):3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 11.Walsh CT, Wencewicz TA. Flavoenzymes: Versatile Catalysts in Biosynthetic Pathways. Nat Prod Rep. 2013;30(1):175–200. doi: 10.1039/c2np20069d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and Chemistry of Cytochrome P450. Chem Rev. 2005;105(6):2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 13.Jung C. The Mystery of Cytochrome P450 Compound I: A Mini-Review Dedicated to Klaus Ruckpaul. Biochim Biophys Acta. 2011;1814(1):46–57. doi: 10.1016/j.bbapap.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 14.Jung C, de Vries S, Schunemann V. Spectroscopic Characterization of Cytochrome P450 Compound I. Arch Biochem Biophys. 2011;507(1):44–55. doi: 10.1016/j.abb.2010.12.029. [DOI] [PubMed] [Google Scholar]
- 15.Costas M, Mehn MP, Jensen MP, Que L. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem Rev. 2004;104(2):939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 16.Abu-Omar MM, Loaiza A, Hontzeas N. Reaction Mechanisms of Mononuclear Non-Heme Iron Oxygenases. Chem Rev. 2005;105(6):2227–2252. doi: 10.1021/cr040653o. [DOI] [PubMed] [Google Scholar]
- 17.Kovaleva EG, Lipscomb JD. Versatility of Biological Non-Heme Fe(II) Centers in Oxygen Activation Reactions. Nat Chem Biol. 2008;4(3):186–193. doi: 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu LF, Meng S, Tang GL. Ferrous Iron and Alpha-Ketoglutarate-Dependent Dioxygenases in the Biosynthesis of Microbial Natural Products. Biochim Biophys Acta. 2016;1864(5):453–470. doi: 10.1016/j.bbapap.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 19.Krebs C, Galonic Fujimori D, Walsh CT, Bollinger JM., Jr Non-Heme Fe(IV)-Oxo Intermediates. Acc Chem Res. 2007;40(7):484–492. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schofield CJ, Zhang Z. Structural and Mechanistic Studies on 2-Oxoglutarate-Dependent Oxygenases and Related Enzymes. Curr Opin Struct Biol. 1999;9(6):722–731. doi: 10.1016/s0959-440x(99)00036-6. [DOI] [PubMed] [Google Scholar]
- 21.Barry SM, Challis GL. Mechanism and Catalytic Diversity of Rieske Non-Heme Iron-Dependent Oxygenases. ACS Catal. 2013;3(10):2362–2370. doi: 10.1021/cs400087p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Broderick JB, Duffus BR, Duschene KS, Shepard EM. Radical S-Adenosylmethionine Enzymes. Chem Rev. 2014;114(8):4229–4317. doi: 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Challand MR, Driesener RC, Roach PL. Radical S-Adenosylmethionine Enzymes: Mechanism, Control and Function. Nat Prod Rep. 2011;28(10):1696–1721. doi: 10.1039/c1np00036e. [DOI] [PubMed] [Google Scholar]
- 24.Frey PA, Booker SJ. Radical Mechanisms of S-Adenosylmethionine-Dependent Enzymes. Adv Protein Chem. 2001;58:1–45. doi: 10.1016/s0065-3233(01)58001-8. [DOI] [PubMed] [Google Scholar]
- 25.Frey PA, Hegeman AD, Ruzicka FJ. The Radical SAM Superfamily. Crit Rev Biochem Mol Biol. 2008;43(1):63–88. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- 26.Richter OM, Ludwig B. Cytochrome c Oxidase--Structure, Function, and Physiology of a Redox-Driven Molecular Machine. Rev Physiol Biochem Pharmacol. 2003;147:47–74. doi: 10.1007/s10254-003-0006-0. [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez Couto S, Toca Herrera JL. Industrial and Biotechnological Applications of Laccases: A Review. Biotechnol Adv. 2006;24(5):500–513. doi: 10.1016/j.biotechadv.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 28.Ramsden CA, Riley PA. Tyrosinase: the Four Oxidation States of the Active Site and Their Relevance to Enzymatic Activation, Oxidation and Inactivation. Bioorg Med Chem. 2014;22(8):2388–2395. doi: 10.1016/j.bmc.2014.02.048. [DOI] [PubMed] [Google Scholar]
- 29.Decker H, Tuczek F. Tyrosinase/Catecholoxidase Activity of Hemocyanins: Structural Basis and Molecular Mechanism. Trends Biochem Sci. 2000;25(8):392–397. doi: 10.1016/s0968-0004(00)01602-9. [DOI] [PubMed] [Google Scholar]
- 30.Sanchez-Ferrer A, Rodriguez-Lopez JN, Garcia-Canovas F, Garcia-Carmona F. Tyrosinase: A Comprehensive Review of Its Mechanism. Biochim Biophys Acta. 1995;1247(1):1–11. doi: 10.1016/0167-4838(94)00204-t. [DOI] [PubMed] [Google Scholar]
- 31.Rolff M, Schottenheim J, Decker H, Tuczek F. Copper-O2 Reactivity of Tyrosinase Models Towards External Monophenolic Substrates: Molecular Mechanism and Comparison with the Enzyme. Chem Soc Rev. 2011;40(7):4077–4098. doi: 10.1039/c0cs00202j. [DOI] [PubMed] [Google Scholar]
- 32.Huijbers MM, Montersino S, Westphal AH, Tischler D, van Berkel WJ. Flavin Dependent Monooxygenases. Arch Biochem Biophys. 2014;544:2–17. doi: 10.1016/j.abb.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 33.van Berkel WJ, Kamerbeek NM, Fraaije MW. Flavoprotein Monooxygenases, a Diverse Class of Oxidative Biocatalysts. J Biotechnol. 2006;124(4):670–689. doi: 10.1016/j.jbiotec.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 34.Winkler A, Motz K, Riedl S, Puhl M, Macheroux P, Gruber K. Structural and Mechanistic Studies Reveal the Functional Role of Bicovalent Flavinylation in Berberine Bridge Enzyme. J Biol Chem. 2009;284(30):19993–20001. doi: 10.1074/jbc.M109.015727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winkler A, Hartner F, Kutchan TM, Glieder A, Macheroux P. Biochemical Evidence that Berberine Bridge Enzyme Belongs to a Novel Family of Flavoproteins Containing a Bi-Covalently Attached FAD Cofactor. J Biol Chem. 2006;281(30):21276–21285. doi: 10.1074/jbc.M603267200. [DOI] [PubMed] [Google Scholar]
- 36.Kavanagh KL, Jörnvall H, Persson B, Oppermann U. Medium- and Short-Chain Dehydrogenase/Reductase Gene and Protein Families. Cell Mol Life Sci. 2008;65(24):3895–3906. doi: 10.1007/s00018-008-8588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oppermann U, Filling C, Hult M, Shafqat N, Wu XQ, Lindh M, Shafqat J, Nordling E, Kallberg Y, Persson B, et al. Short-Chain Dehydrogenases/Reductases (SDR): the 2002 Update. Chem Biol Interact. 2003;143–144:247–253. doi: 10.1016/s0009-2797(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 38.Rosenthal RG, Ebert MO, Kiefer P, Peter DM, Vorholt JA, Erb TJ. Direct Evidence for a Covalent Ene Adduct Intermediate in NAD(P)H-Dependent Enzymes. Nat Chem Biol. 2014;10(1):50–55. doi: 10.1038/nchembio.1385. [DOI] [PubMed] [Google Scholar]
- 39.Rosenthal RG, Vogeli B, Quade N, Capitani G, Kiefer P, Vorholt JA, Ebert M-O, Erb TJ. The Use of Ene Adducts to Study and Engineer Enoyl-Thioester Reductases. Nat Chem Biol. 2015;11(6):398–400. doi: 10.1038/nchembio.1794. [DOI] [PubMed] [Google Scholar]
- 40.Du L, Lou L. PKS and NRPS Release Mechanisms. Nat Prod Rep. 2010;27(2):255–278. doi: 10.1039/b912037h. [DOI] [PubMed] [Google Scholar]
- 41.Kahne D, Leimkuhler C, Lu W, Walsh C. Glycopeptide and Lipoglycopeptide Antibiotics. Chem Rev. 2005;105(2):425–448. doi: 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- 42.Yim G, Thaker MN, Koteva K, Wright G. Glycopeptide Antibiotic Biosynthesis. J Antibiot (Tokyo) 2014;67(1):31–41. doi: 10.1038/ja.2013.117. [DOI] [PubMed] [Google Scholar]
- 43.Park OK, Choi HY, Kim GW, Kim WG. Generation of New Complestatin Analogs by Heterologous Expression of the Complestatin Biosynthetic Gene Cluster fromStreptomyces Chartreusis AN1542. Chembiochem. 2016;17(18):1725–1731. doi: 10.1002/cbic.201600241. [DOI] [PubMed] [Google Scholar]
- 44.Chiu HT, Hubbard BK, Shah AN, Eide J, Fredenburg RA, Walsh CT, Khosla C. Molecular Cloning and Sequence Analysis of the Complestatin Biosynthetic Gene Cluster. Proc Natl Acad Sci U S A. 2001;98(15):8548–8553. doi: 10.1073/pnas.151246498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holtzel A, Schmid DG, Nicholson GJ, Stevanovic S, Schimana J, Gebhardt K, Fiedler HP, Jung G. Arylomycins A and B, New Biaryl-Bridged Lipopeptide Antibiotics Produced by Streptomyces sp. Tü 6075. II. Structure Elucidation. J Antibiot (Tokyo) 2002;55(6):571–577. doi: 10.7164/antibiotics.55.571. [DOI] [PubMed] [Google Scholar]
- 46.Liu WT, Kersten RD, Yang YL, Moore BS, Dorrestein PC. Imaging Mass Spectrometry and Genome Mining Via Short Sequence Tagging Identified the Anti-Infective Agent Arylomycin in Streptomyces Roseosporus. J Am Chem Soc. 2011;133(45):18010–18013. doi: 10.1021/ja2040877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schimana J, Gebhardt K, Höltzel A, Schmid DG, Süssmuth R, Müller J, Pukall R, Fiedler HP. Arylomycins A and B, New Biaryl-Bridged Lipopeptide Antibiotics Produced by Streptomyces sp. Tü 6075. I. Taxonomy, Fermentation, Isolation and Biological Activities. J Antibiot (Tokyo) 2002;55(6):565–570. doi: 10.7164/antibiotics.55.565. [DOI] [PubMed] [Google Scholar]
- 48.Zerbe K, Pylypenko O, Vitali F, Zhang W, Rouset S, Heck M, Vrijbloed JW, Bischoff D, Bister B, Süssmuth RD, et al. Crystal Structure of OxyB, a Cytochrome P450 Implicated in an Oxidative Phenol Coupling Reaction During Vancomycin Biosynthesis. J Biol Chem. 2002;277(49):47476–47485. doi: 10.1074/jbc.M206342200. [DOI] [PubMed] [Google Scholar]
- 49.Li Z, Rupasinghe SG, Schuler MA, Nair SK. Crystal Structure of a Phenol-Coupling P450 Monooxygenase Involved in Teicoplanin Biosynthesis. Proteins. 2011;79(6):1728–1738. doi: 10.1002/prot.22996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pylypenko O, Vitali F, Zerbe K, Robinson JA, Schlichting I. Crystal Structure of OxyC, a Cytochrome P450 Implicated in an Oxidative C-C Coupling Reaction during Vancomycin Biosynthesis. J Biol Chem. 2003;278(47):46727–46733. doi: 10.1074/jbc.M306486200. [DOI] [PubMed] [Google Scholar]
- 51.Haslinger K, Maximowitsch E, Brieke C, Koch A, Cryle MJ. Cytochrome P450 OxyBtei Catalyzes the First Phenolic Coupling Step in Teicoplanin Biosynthesis. ChemBioChem. 2014;15(18):2719–2728. doi: 10.1002/cbic.201402441. [DOI] [PubMed] [Google Scholar]
- 52.Geib N, Woithe K, Zerbe K, Li DB, Robinson JA. New Insights into the First Oxidative Phenol Coupling Reaction During Vancomycin Biosynthesis. Bioorg Med Chem Lett. 2008;18(10):3081–3084. doi: 10.1016/j.bmcl.2007.11.093. [DOI] [PubMed] [Google Scholar]
- 53.Zerbe K, Woithe K, Li DB, Vitali F, Bigler L, Robinson JA. An Oxidative Phenol Coupling Reaction Catalyzed by OxyB, a Cytochrome P450 from the Vancomycin-Producing Microorganism. Angew Chem Int Ed Engl. 2004;43(48):6709–6713. doi: 10.1002/anie.200461278. [DOI] [PubMed] [Google Scholar]
- 54.Woithe K, Geib N, Zerbe K, Li DB, Heck M, Fournier-Rousset S, Meyer O, Vitali F, Matoba N, Abou-Hadeed K, et al. Oxidative Phenol Coupling Reactions Catalyzed by OxyB: A Cytochrome P450 from the Vancomycin Producing Organism. Implications for Vancomycin Biosynthesis. J Am Chem Soc. 2007;129(21):6887–6895. doi: 10.1021/ja071038f. [DOI] [PubMed] [Google Scholar]
- 55.Haslinger K, Cryle MJ. Structure of OxyAtei: Completing Our Picture of the Glycopeptide Antibiotic Producing Cytochrome P450 Cascade. FEBS Lett. 2016;590(4):571–581. doi: 10.1002/1873-3468.12081. [DOI] [PubMed] [Google Scholar]
- 56.Haslinger K, Peschke M, Brieke C, Maximowitsch E, Cryle MJ. X-Domain of Peptide Synthetases Recruits Oxygenases Crucial for Glycopeptide Biosynthesis. Nature. 2015;521(7550):105–109. doi: 10.1038/nature14141. [DOI] [PubMed] [Google Scholar]
- 57.Brieke C, Peschke M, Haslinger K, Cryle MJ. Sequential in Vitro Cyclization by Cytochrome P450 Enzymes of Glycopeptide Antibiotic Precursors Bearing the X-Domain from Nonribosomal Peptide Biosynthesis. Angew Chem Int Ed Engl. 2015;54(52):15715–15719. doi: 10.1002/anie.201507533. [DOI] [PubMed] [Google Scholar]
- 58.Sanchez C, Mendez C, Salas JA. Indolocarbazole Natural Products: Occurrence, Biosynthesis, and Biological Activity. Nat Prod Rep. 2006;23(6):1007–1045. doi: 10.1039/b601930g. [DOI] [PubMed] [Google Scholar]
- 59.Bailly C, Riou JF, Colson P, Houssier C, Rodrigues-Pereira E, Prudhomme M. DNA Cleavage by Topoisomerase I in the Presence of Indolocarbazole Derivatives of Rebeccamycin. Biochemistry. 1997;36(13):3917–3929. doi: 10.1021/bi9624898. [DOI] [PubMed] [Google Scholar]
- 60.Kase H, Iwahashi K, Nakanishi S, Matsuda Y, Yamada K, Takahashi M, Murakata C, Sato A, Kaneko M. K-252 Compounds, Novel and Potent Inhibitors of Protein Kinase C and Cyclic Nucleotide-Dependent Protein Kinases. Biochem Biophys Res Commun. 1987;142(2):436–440. doi: 10.1016/0006-291x(87)90293-2. [DOI] [PubMed] [Google Scholar]
- 61.Ward NE, O’Brian CA. Kinetic Analysis of Protein Kinase C Inhibition by Staurosporine: Evidence that Inhibition Entails Inhibitor Binding at a Conserved Region of the Catalytic Domain but Not Competition with Substrates. Mol Pharmacol. 1992;41(2):387–392. [PubMed] [Google Scholar]
- 62.Howard-Jones AR, Walsh CT. Enzymatic Generation of the Chromopyrrolic Acid Scaffold of Rebeccamycin by the Tandem Action of RebO and RebD. Biochemistry. 2005;44(48):15652–15663. doi: 10.1021/bi051706e. [DOI] [PubMed] [Google Scholar]
- 63.Sanchez C, Butovich IA, Brana AF, Rohr J, Mendez C, Salas JA. The Biosynthetic Gene Cluster for the Antitumor Rebeccamycin: Characterization and Generation of Indolocarbazole Derivatives. Chem Biol. 2002;9(4):519–531. doi: 10.1016/s1074-5521(02)00126-6. [DOI] [PubMed] [Google Scholar]
- 64.Onaka H, Taniguchi S, Igarashi Y, Furumai T. Cloning of the Staurosporine Biosynthetic Gene Cluster from Streptomyces sp. TP-A0274 and Its Heterologous Expression in Streptomyces Lividans. J Antibiot (Tokyo) 2002;55(12):1063–1071. doi: 10.7164/antibiotics.55.1063. [DOI] [PubMed] [Google Scholar]
- 65.Makino M, Sugimoto H, Shiro Y, Asamizu S, Onaka H, Nagano S. Crystal Structures and Catalytic Mechanism of Cytochrome P450 StaP that Produces the Indolocarbazole Skeleton. Proc Natl Acad Sci U S A. 2007;104(28):11591–11596. doi: 10.1073/pnas.0702946104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nishizawa T, Gruschow S, Jayamaha DH, Nishizawa-Harada C, Sherman DH. Enzymatic Assembly of the Bis-Indole Core of Rebeccamycin. J Am Chem Soc. 2006;128(3):724–725. doi: 10.1021/ja056749x. [DOI] [PubMed] [Google Scholar]
- 67.Chiu H-T, Chen Y-L, Chen C-Y, Jin C, Lee M-N, Lin Y-C. Molecular Cloning, Sequence Analysis and Functional Characterization of the Gene Cluster for Biosynthesis of K-252a and Its Analogs. Mol Biosyst. 2009;5(10):1180–1191. doi: 10.1039/b905293c. [DOI] [PubMed] [Google Scholar]
- 68.Howard-Jones AR, Walsh CT. Staurosporine and Rebeccamycin Aglycones Are Assembled by the Oxidative Action of StaP, StaC, and RebC on Chromopyrrolic Acid. J Am Chem Soc. 2006;128(37):12289–12298. doi: 10.1021/ja063898m. [DOI] [PubMed] [Google Scholar]
- 69.Spolitak T, Ballou DP. Evidence for Catalytic Intermediates Involved in Generating the Chromopyrrolic Acid Scaffold of Rebeccamycin by RebO and RebD. Arch Biochem Biophys. 2015;573:111–119. doi: 10.1016/j.abb.2015.03.020. [DOI] [PubMed] [Google Scholar]
- 70.Wang Y, Chen H, Makino M, Shiro Y, Nagano S, Asamizu S, Onaka H, Shaik S. Theoretical and Experimental Studies of the Conversion of Chromopyrrolic Acid to an Antitumor Derivative by Cytochrome P450 StaP: The Catalytic Role of Water Molecules. J Am Chem Soc. 2009;131(19):6748–6762. doi: 10.1021/ja9003365. [DOI] [PubMed] [Google Scholar]
- 71.Onaka H, Asamizu S, Igarashi Y, Yoshida R, Furumai T. Cytochrome P450 Homolog Is Responsible for C-N Bond Formation Between Aglycone and Deoxysugar in the Staurosporine Biosynthesis ofStreptomyces sp. TP-A0274. Biosci Biotechnol Biochem. 2005;69(9):1753–1759. doi: 10.1271/bbb.69.1753. [DOI] [PubMed] [Google Scholar]
- 72.Dalsgaard PW, Blunt JW, Munro MH, Frisvad JC, Christophersen C. Communesins G and H, New Alkaloids from the Psychrotolerant Fungus Penicillium Rivulum. J Nat Prod. 2005;68(2):258–261. doi: 10.1021/np049646l. [DOI] [PubMed] [Google Scholar]
- 73.Numata A, Takahashi C, Ito Y, Takada T, Kawai K, Usami Y, Matsumura E, Imachi M, Ito T, Hasegawa T. Communesins, Cytotoxic Metabolites of a Fungus Isolated from a Marine Alga. Tetrahedron Lett. 1993;34(14):2355–2358. [Google Scholar]
- 74.Jadulco R, Edrada RA, Ebel R, Berg A, Schaumann K, Wray V, Steube K, Proksch P. New Communesin Derivatives from the Fungus Penicillium sp. Derived from the Mediterranean Sponge Axinella Verrucosa. J Nat Prod. 2004;67(1):78–81. doi: 10.1021/np030271y. [DOI] [PubMed] [Google Scholar]
- 75.Hayashi H, Matsumoto H, Akiyama K. New Insecticidal Compounds, Communesins C, D and E, fromPenicillium Expansum Link MK-57. Biosci Biotechnol Biochem. 2004;68(3):753–756. doi: 10.1271/bbb.68.753. [DOI] [PubMed] [Google Scholar]
- 76.Andersen B, Smedsgaard J, Frisvad JC. Penicillium Expansum: Consistent Production of Patulin, Chaetoglobosins, and Other Secondary Metabolites in Culture and Their Natural Occurrence in Fruit Products. J Agric Food Chem. 2004;52(8):2421–2428. doi: 10.1021/jf035406k. [DOI] [PubMed] [Google Scholar]
- 77.May JA, Zeidan RK, Stoltz BM. Biomimetic Approach to Communesin B (a.k.a. Nomofungin) Tetrahedron Lett. 2003;44(6):1203–1205. [Google Scholar]
- 78.Han SJ, Vogt F, Krishnan S, May JA, Gatti M, Virgil SC, Stoltz BM. A Diastereodivergent Synthetic Strategy for the Syntheses of Communesin F and Perophoramidine. Org Lett. 2014;16(12):3316–3319. doi: 10.1021/ol5013263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zuo ZW, Ma DW. Enantioselective Total Syntheses of Communesins A and B. Angew Chem Int Ed Engl. 2011;50(50):12008–12011. doi: 10.1002/anie.201106205. [DOI] [PubMed] [Google Scholar]
- 80.Seo JH, Liu P, Weinreb SM. Evolution of a Strategy for Total Synthesis of the Marine Fungal Alkaloid (+/−)-Communesin F. J Org Chem. 2010;75(8):2667–2680. doi: 10.1021/jo100339k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Han SJ, Vogt F, May JA, Krishnan S, Gatti M, Virgil SC, Stoltz BM. Evolution of a Unified, Stereodivergent Approach to the Synthesis of Communesin F and Perophoramidine. J Org Chem. 2015;80(1):528–547. doi: 10.1021/jo502534g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Crawley SL, Funk RL. A Synthetic Approach to Nomofungin/Communesin B. Org Lett. 2003;5(18):3169–3171. doi: 10.1021/ol034407v. [DOI] [PubMed] [Google Scholar]
- 83.Zuo ZW, Xie WQ, Ma DW. Total Synthesis and Absolute Stereochemical Assignment of (-)-Communesin F. J Am Chem Soc. 2010;132(38):13226–13228. doi: 10.1021/ja106739g. [DOI] [PubMed] [Google Scholar]
- 84.Yang J, Wu HX, Shen LQ, Qin Y. Total Synthesis of (+/−)-Communesin F. J Am Chem Soc. 2007;129(45):13794–13795. doi: 10.1021/ja075705g. [DOI] [PubMed] [Google Scholar]
- 85.Belmar J, Funk RL. Total Synthesis of (+/−)-Communesin F via a Cycloaddition with Indol-2-One. J Am Chem Soc. 2012;134(41):16941–16943. doi: 10.1021/ja307277w. [DOI] [PubMed] [Google Scholar]
- 86.Liu P, Seo JH, Weinreb SM. Total Synthesis of the Polycyclic Fungal Metabolite (+/−)-Communesin F. Angew Chem Int Ed Engl. 2010;49(11):2000–2003. doi: 10.1002/anie.200906818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lathrop SP, Pompeo M, Chang WT, Movassaghi M. Convergent and Biomimetic Enantioselective Total Synthesis of (-)-Communesin F. J Am Chem Soc. 2016;138(24):7763–7769. doi: 10.1021/jacs.6b04072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lin H-C, Chiou G, Chooi Y-H, McMahon TC, Xu W, Garg NK, Tang Y. Elucidation of the Concise Biosynthetic Pathway of the Communesin Indole Alkaloids. Angew Chem Int Ed Engl. 2015;54(10):3004–3007. doi: 10.1002/anie.201411297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lin H-C, McMahon TC, Patel A, Corsello M, Simon A, Xu W, Zhao M, Houk KN, Garg NK, Tang Y. P450-Mediated Coupling of Indole Fragments to Forge Communesin and Unnatural Isomers. J Am Chem Soc. 2016;138(12):4002–4005. doi: 10.1021/jacs.6b01413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Belin P, Le Du MH, Fielding A, Lequin O, Jacquet M, Charbonnier JB, Lecoq A, Thai R, Courçon M, Masson C, et al. Identification and Structural Basis of the Reaction Catalyzed by CYP121, an Essential Cytochrome P450 in Mycobacterium Tuberculosis. Proc Natl Acad Sci U S A. 2009;106(18):7426–7431. doi: 10.1073/pnas.0812191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gondry M, Sauguet L, Belin P, Thai R, Amouroux R, Tellier C, Tuphile K, Jacquet M, Braud S, Courcon M, et al. Cyclodipeptide Synthases Are a Family of tRNA-Dependent Peptide Bond-Forming Enzymes. Nat Chem Biol. 2009;5(6):414–420. doi: 10.1038/nchembio.175. [DOI] [PubMed] [Google Scholar]
- 92.McLean KJ, Carroll P, Lewis DG, Dunford AJ, Seward HE, Neeli R, Cheesman MR, Marsollier L, Douglas P, Smith WE, et al. Characterization of Active Site Structure in CYP121. A Cytochrome P450 Essential for Viability of Mycobacterium Tuberculosis H37Rv. J Biol Chem. 2008;283(48):33406–33416. doi: 10.1074/jbc.M802115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dumas VG, Defelipe LA, Petruk AA, Turjanski AG, Marti MA. QM/MM Study of the C-C Coupling Reaction Mechanism of CYP121, an Essential Cytochrome P450 of Mycobacterium Tuberculosis. Proteins. 2014;82(6):1004–1021. doi: 10.1002/prot.24474. [DOI] [PubMed] [Google Scholar]
- 94.Chiba T, Asami Y, Suga T, Watanabe Y, Nagai T, Momose F, Nonaka K, Iwatsuki M, Yamada H, Omura S, et al. Herquline A, Produced by Penicillium Herquei FKI-7215, Exhibits Anti-Influenza Virus Properties. Biosci Biotechnol Biochem. 2016:1–4. doi: 10.1080/09168451.09162016.01162084. [DOI] [PubMed] [Google Scholar]
- 95.Enomoto Y, Shiomi K, Hayashi M, Masuma R, Kawakubo T, Tomosawa K, Iwai Y, Omura S. Herquline B, a New Platelet Aggregation Inhibitor Produced by Penicillium Herquei Fg-372. J Antibiot (Tokyo) 1996;49(1):50–53. doi: 10.7164/antibiotics.49.50. [DOI] [PubMed] [Google Scholar]
- 96.Omura S, Hirano A, Iwai Y, Masuma R. Herquline, a New Alkaloid Produced by Penicillium Herquei. Fermentation, Isolation and Properties. J Antibiot (Tokyo) 1979;32(8):786–790. doi: 10.7164/antibiotics.32.786. [DOI] [PubMed] [Google Scholar]
- 97.Yu X, Liu F, Zou Y, Tang M-C, Hang L, Houk KN, Tang Y. Biosynthesis of Strained Piperazine Alkaloids - Uncovering the Concise Pathway of Herquline A. J Am Chem Soc. 2016;138(41):13529–13532. doi: 10.1021/jacs.6b09464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Taguchi H, Sankawa U, Shibata S. Biosynthesis of Natural ProductsVI. Biosynthesis of Usnic Acid in Lichens. A General Scheme of Biosynthesis of Usnic Acid. Chem Pharm Bull (Tokyo) 1969;17(10):2054–2060. doi: 10.1248/cpb.17.2054. [DOI] [PubMed] [Google Scholar]
- 99.Taguchi H, Sankawa U, Shibata S. Biosynthesis of Natural Products. VII. Biosynthesis of Usnic Acid in Lichens. Seasonal Variation Observed in Usnic Acid Biosynthesis. Chem Pharm Bull (Tokyo) 1969;17(10):2061–2064. doi: 10.1248/cpb.17.2061. [DOI] [PubMed] [Google Scholar]
- 100.Taguchi H, Sankawa U, Shibata S. Biosynthesis of Usnic Acid in Lichens. Tetrahedron Lett. 1966;(42):5211–5214. doi: 10.1016/s0040-4039(01)99879-x. [DOI] [PubMed] [Google Scholar]
- 101.Hutchison RD, Steyn PS, Van Rensburg SJ. Viridicatumtoxin, a New Mycotoxin from Penicillium Viridicatum Westling. Toxicol Appl Pharmacol. 1973;24(3):507–509. doi: 10.1016/0041-008x(73)90057-4. [DOI] [PubMed] [Google Scholar]
- 102.Kabuto C, Silverton JV, Akiyama T, Sankawa U, Hutchison RD, Steyn PS, Vleggaar R. X-Ray Structure of Viridicatumtoxin: A New Class of Mycotoxin from Penicillium Viridicatum Westling. J Chem Soc, Chem Commun. 1976;(18):728–729. [Google Scholar]
- 103.Zheng CJ, Yu HE, Kim EH, Kim WG. Viridicatumtoxin B, a New Anti-MRSA Agent from Penicillium sp. FR11. J Antibiot (Tokyo) 2008;61(10):633–637. doi: 10.1038/ja.2008.84. [DOI] [PubMed] [Google Scholar]
- 104.Chooi Y-H, Hong YJ, Cacho RA, Tantillo DJ, Tang Y. A Cytochrome P450 Serves as an Unexpected Terpene Cyclase During Fungal Meroterpenoid Biosynthesis. J Am Chem Soc. 2013;135(45):16805–16808. doi: 10.1021/ja408966t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chooi Y-H, Cacho R, Tang Y. Identification of the Viridicatumtoxin and Griseofulvin Gene Clusters from Penicillium Aethiopicum. Chem Biol. 2010;17(5):483–494. doi: 10.1016/j.chembiol.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chooi Y-H, Wang P, Fang J, Li Y, Wu K, Wang P, Tang Y. Discovery and Characterization of a Group of Fungal Polycyclic Polyketide Prenyltransferases. J Am Chem Soc. 2012;134(22):9428–9437. doi: 10.1021/ja3028636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Oda K, Hirose S, Takami N, Misumi Y, Takatsuki A, Ikehara Y. Brefeldin A Arrests the Intracellular Transport of a Precursor of Complement C3 Before its Conversion Site in Rat Hepatocytes. FEBS Lett. 1987;214(1):135–138. doi: 10.1016/0014-5793(87)80028-5. [DOI] [PubMed] [Google Scholar]
- 108.Kato S, Ito S, Noguchi T, Naito H. Effects of Brefeldin A on the Synthesis and Secretion of Egg White Proteins in Primary Cultured Oviduct Cells of Laying Japanese Quail (Coturnix Coturnix Japonica) Biochim Biophys Acta. 1989;991(1):36–43. doi: 10.1016/0304-4165(89)90025-1. [DOI] [PubMed] [Google Scholar]
- 109.Misumi Y, Misumi Y, Miki K, Takatsuki A, Tamura G, Ikehara Y. Novel Blockade by Brefeldin A of Intracellular Transport of Secretory Proteins in Cultured Rat Hepatocytes. J Biol Chem. 1986;261(24):11398–11403. [PubMed] [Google Scholar]
- 110.Zabala AO, Chooi Y-H, Choi MS, Lin H-C, Tang Y. Fungal Polyketide Synthase Product Chain-Length Control by Partnering Thiohydrolase. ACS Chem Biol. 2014;9(7):1576–1586. doi: 10.1021/cb500284t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mizutani M, Ohta D. Diversification of P450 Genes During Land Plant Evolution. Annu Rev Plant Biol. 2010;61:291–315. doi: 10.1146/annurev-arplant-042809-112305. [DOI] [PubMed] [Google Scholar]
- 112.Mizutani M. Impacts of Diversification of Cytochrome P450 on Plant Metabolism. Biol Pharm Bull. 2012;35(6):824–832. doi: 10.1248/bpb.35.824. [DOI] [PubMed] [Google Scholar]
- 113.Nelson D, Werck-Reichhart D. A P450-Centric View of Plant Evolution. Plant J. 2011;66(1):194–211. doi: 10.1111/j.1365-313X.2011.04529.x. [DOI] [PubMed] [Google Scholar]
- 114.Lau W, Sattely ES. Six Enzymes from Mayapple that Complete the Biosynthetic Pathway to the Etoposide Aglycone. Science. 2015;349(6253):1224–1228. doi: 10.1126/science.aac7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Klein AP, Sattely ES. Two Cytochromes P450 Catalyze S-Heterocyclizations in Cabbage Phytoalexin Biosynthesis. Nat Chem Biol. 2015;11(11):837–839. doi: 10.1038/nchembio.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hagel JM, Facchini PJ. Dioxygenases Catalyze the O-Demethylation Steps of Morphine Biosynthesis in. Opium Poppy Nat Chem Biol. 2010;6(4):273–275. doi: 10.1038/nchembio.317. [DOI] [PubMed] [Google Scholar]
- 117.Barton DHR, Cohen T. Festschrift Arthur Stall. Birkhauser; Basel: 1957. pp. 117–144. [Google Scholar]
- 118.Zenk MH, Gerardy R, Stadler R. Phenol Oxidative Coupling of Benzylisoquinoline Alkaloids is Catalysed by Regio- and Stereo-Selective Cytochrome P-450 Linked Plant Enzymes: Salutaridine and Berbamunine. J Chem Soc, Chem Commun. 1989;(22):1725–1727. [Google Scholar]
- 119.Gerardy R, Zenk MH. Formation of Salutaridine from (R)-Reticuline by a Membrane-Bound Cytochrome P-450 Enzyme from Papaver Somniferum. Phytochemistry. 1993;32(1):79–86. [Google Scholar]
- 120.Gesell A, Rolf M, Ziegler J, Díaz Chávez ML, Huang FC, Kutchan TM. CYP719B1 Is Salutaridine Synthase, the C-C Phenol-coupling Enzyme of Morphine Biosynthesis in Opium Poppy. J Biol Chem. 2009;284(36):24432–24442. doi: 10.1074/jbc.M109.033373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ikezawa N, Iwasa K, Sato F. Molecular Cloning and Characterization of CYP80G2, a Cytochrome P450 that Catalyzes an Intramolecular C-C Phenol Coupling of (S)-Reticuline in Magnoflorine Biosynthesis, from Cultured Coptis Japonica Cells. J Biol Chem. 2008;283(14):8810–8821. doi: 10.1074/jbc.M705082200. [DOI] [PubMed] [Google Scholar]
- 122.Kraus PF, Kutchan TM. Molecular Cloning and Heterologous Expression of a cDNA Encoding Berbamunine Synthase, a C-O Phenol-Coupling Cytochrome P450 from the Higher Plant Berberis Stolonifera. Proc Natl Acad Sci U S A. 1995;92(6):2071–2075. doi: 10.1073/pnas.92.6.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jin Z. Amaryllidaceae and Sceletium Alkaloids. Nat Prod Rep. 2013;30(6):849–868. doi: 10.1039/c3np70005d. [DOI] [PubMed] [Google Scholar]
- 124.Marco L, do Carmo Carreiras M. Galanthamine, a Natural Product for the Treatment of Alzheimer’s Disease. Recent Pat CNS Drug Discov. 2006;1(1):105–111. doi: 10.2174/157488906775245246. [DOI] [PubMed] [Google Scholar]
- 125.Kilgore MB, Kutchan TM. The Amaryllidaceae Alkaloids: Biosynthesis and Methods for Enzyme Discovery. Phytochem Rev. 2016;15(3):317–337. doi: 10.1007/s11101-015-9451-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kilgore MB, Augustin MM, Starks CM, O’Neil-Johnson M, May GD, Crow JA, Kutchan TM. Cloning and Characterization of a Norbelladine 4′-O-Methyltransferase Involved in the Biosynthesis of the Alzheimer’s Drug Galanthamine in Narcissus sp. aff. Pseudonarcissus. PLoS ONE. 2014;9(7):e103223. doi: 10.1371/journal.pone.0103223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kilgore MB, Augustin MM, May GD, Crow JA, Kutchan TM. CYP96T1 of Narcissus sp. aff. Pseudonarcissus Catalyzes Formation of the Para-Para’ C-C Phenol Couple in the Amaryllidaceae Alkaloids. Front Plant Sci. 2016;7:225. doi: 10.3389/fpls.2016.00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Davin LB, Wang HB, Crowell AL, Bedgar DL, Martin DM, Sarkanen S, Lewis NG. Stereoselective Bimolecular Phenoxy Radical Coupling by an Auxiliary (Dirigent) Protein without an Active Center. Science. 1997;275(5298):362–366. doi: 10.1126/science.275.5298.362. [DOI] [PubMed] [Google Scholar]
- 129.Halls SC, Davin LB, Kramer DM, Lewis NG. Kinetic Study of Coniferyl Alcohol Radical Binding to the (+)-Pinoresinol Forming Dirigent Protein. Biochemistry. 2004;43(9):2587–2595. doi: 10.1021/bi035959o. [DOI] [PubMed] [Google Scholar]
- 130.Pickel B, Constantin MA, Pfannstiel J, Conrad J, Beifuss U, Schaller A. An Enantiocomplementary Dirigent Protein for the Enantioselective Lacease-Catalyzed Oxidative Coupling of Phenols. Angew Chem Int Ed Engl. 2010;49(1):202–204. doi: 10.1002/anie.200904622. [DOI] [PubMed] [Google Scholar]
- 131.Ono E, Nakai M, Fukui Y, Tomimori N, Fukuchi-Mizutani M, Saito M, Satake H, Tanaka T, Katsuta M, Umezawa T, et al. Formation of Two Methylenedioxy Bridges by a Sesamum CYP81Q Protein Yielding a Furofuran Lignan, (+)-Sesamin. Proc Natl Acad Sci U S A. 2006;103(26):10116–10121. doi: 10.1073/pnas.0603865103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gentles JC. Experimental Ringworm in Guinea Pigs: Oral Treatment with Griseofulvin. Nature. 1958;182(4633):476–477. doi: 10.1038/182476a0. [DOI] [PubMed] [Google Scholar]
- 133.Gull K, Trinci AP. Griseofulvin Inhibits Fungal Mitosis. Nature. 1973;244(5414):292–294. doi: 10.1038/244292a0. [DOI] [PubMed] [Google Scholar]
- 134.Rebacz B, Larsen TO, Clausen MH, Ronnest MH, Loffler H, Ho AD, Kramer A. Identification of Griseofulvin as an Inhibitor of Centrosomal Clustering in a Phenotype-Based Screen. Cancer Res. 2007;67(13):6342–6350. doi: 10.1158/0008-5472.CAN-07-0663. [DOI] [PubMed] [Google Scholar]
- 135.Panda D, Rathinasamy K, Santra MK, Wilson L. Kinetic Suppression of Microtubule Dynamic Instability by Griseofulvin: Implications for Its Possible Use in the Treatment of Cancer. Proc Natl Acad Sci U S A. 2005;102(28):9878–9883. doi: 10.1073/pnas.0501821102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cacho RA, Chooi Y-H, Zhou H, Tang Y. Complexity Generation in Fungal Polyketide Biosynthesis: A Spirocycle-Forming P450 in the Concise Pathway to the Antifungal Drug Griseofulvin. ACS Chem Biol. 2013;8(10):2322–2330. doi: 10.1021/cb400541z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Grandner JM, Cacho RA, Tang Y, Houk KN. Mechanism of the P450-Catalyzed Oxidative Cyclization in the Biosynthesis of Griseofulvin. ACS Catal. 2016;6(7):4506–4511. doi: 10.1021/acscatal.6b01068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nielsen MT, Nielsen JB, Anyaogu DC, Holm DK, Nielsen KF, Larsen TO, Mortensen UH. Heterologous Reconstitution of the Intact Geodin Gene Cluster in Aspergillus Nidulans through a Simple and Versatile PCR Based Approach. PLoS ONE. 2013;8:e72871. doi: 10.1371/journal.pone.0072871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Huang KX, Fujii I, Ebizuka Y, Gomi K, Sankawa U. Molecular Cloning and Heterologous Expression of the Gene Encoding Dihydrogeodin Oxidase, a Multicopper Blue Enzyme from Aspergillus Terreus. J Biol Chem. 1995;270(37):21495–21502. doi: 10.1074/jbc.270.37.21495. [DOI] [PubMed] [Google Scholar]
- 140.Gauthier T, Wang X, Sifuentes Dos Santos J, Fysikopoulos A, Tadrist S, Canlet C, Artigot MP, Loiseau N, Oswald IP, Puel O. Trypacidin, a Spore-Borne Toxin from Aspergillus Fumigatus, Is Cytotoxic to Lung Cells. PLoS ONE. 2012;7(2):e29906. doi: 10.1371/journal.pone.0029906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mattern DJ, Schoeler H, Weber J, Novohradská S, Kraibooj K, Dahse HM, Hillmann F, Valiante V, Figge MT, Brakhage AA. Identification of the Antiphagocytic Trypacidin Gene Cluster in the Human-Pathogenic Fungus Aspergillus Fumigatus. Appl Microbiol Biotechnol. 2015;99(23):10151–10161. doi: 10.1007/s00253-015-6898-1. [DOI] [PubMed] [Google Scholar]
- 142.Wintersteiner O, Dutcher JD. Curare Alkaloids from Chondodendron Tomentosum. Science. 1943;97(2525):467–470. doi: 10.1126/science.97.2525.467. [DOI] [PubMed] [Google Scholar]
- 143.Steinbach JH, Chen Q. Antagonist and Partial Agonist Actions of d-Tubocurarine at Mammalian Muscle Acetylcholine Receptors. J Neurosci. 1995;15(1 Pt 1):230–240. doi: 10.1523/JNEUROSCI.15-01-00230.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nasiripourdori A, Taly V, Grutter T, Taly A. From Toxins Targeting Ligand Gated Ion Channels to Therapeutic Molecules. Toxins (Basel) 2011;3(3):260–293. doi: 10.3390/toxins3030260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Washizu F, Umezawa H, Sugiyama N. Chemical Studies on a Toxic Product of Streptomyces Thioluteus, Aureothin. J Antibiot (Tokyo) 1954;7(2):60. [PubMed] [Google Scholar]
- 146.Schwartz JL, Tishler M, Arison BH, Shafer HM, Omura S. Identification of Mycolutein and Pulvomycin as Aureothin and Labilomycin Respectively. J Antibiot (Tokyo) 1976;29(3):236–241. doi: 10.7164/antibiotics.29.236. [DOI] [PubMed] [Google Scholar]
- 147.He J, Hertweck C. Iteration as Programmed Event During Polyketide Assembly: Molecular Analysis of the Aureothin Biosynthesis Gene Cluster. Chem Biol. 2003;10(12):1225–1232. doi: 10.1016/j.chembiol.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 148.He J, Müller M, Hertweck C. Formation of the Aureothin Tetrahydrofuran Ring by a Bifunctional Cytochrome P450 Monooxygenase. J Am Chem Soc. 2004;126(51):16742–16743. doi: 10.1021/ja046104h. [DOI] [PubMed] [Google Scholar]
- 149.Richter ME, Traitcheva N, Knüpfer U, Hertweck C. Sequential Asymmetric Polyketide Heterocyclization Catalyzed by a Single Cytochrome P450 Monooxygenase (AurH) Angew Chem Int Ed Engl. 2008;47(46):8872–8875. doi: 10.1002/anie.200803714. [DOI] [PubMed] [Google Scholar]
- 150.Zocher G, Richter ME, Mueller U, Hertweck C. Structural Fine-Tuning of a Multifunctional Cytochrome P450 Monooxygenase. J Am Chem Soc. 2011;133(7):2292–2302. doi: 10.1021/ja110146z. [DOI] [PubMed] [Google Scholar]
- 151.Kyriakidis N, Waight ES, Day JB, Mantle PG. Novel Metabolites from Penicillium Crustosum, Including Penitrem E, a Tremorgenic Mycotoxin. Appl Environ Microbiol. 1981;42(1):61–62. doi: 10.1128/aem.42.1.61-62.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wagener RE, Davis ND, Diener UL. Penitrem A and Roquefortine Production by Penicillium Commune. Appl Environ Microbiol. 1980;39(4):882–887. doi: 10.1128/aem.39.4.882-887.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.de Jesus AE, Steyn PS, van Heerden FR, Vleggaar R, Wessels PL, Hull WE. Tremorgenic Mycotoxins from Penicillium Crustosum: Isolation of Penitrems A-F and the Structure Elucidation and Absolute Configuration of Penitrem A. J Chem Soc, Perkin Trans 1. 1983:1847–1856. [Google Scholar]
- 154.Wilson BJ, Wilson CH, Hayes AW. Tremorgenic Toxin from Penicillium Cyclopium Grown on Food Materials. Nature. 1968;220(5162):77–78. doi: 10.1038/220077b0. [DOI] [PubMed] [Google Scholar]
- 155.Liu C, Tagami K, Minami A, Matsumoto T, Frisvad JC, Suzuki H, Ishikawa J, Gomi K, Oikawa H. Reconstitution of Biosynthetic Machinery for the Synthesis of the Highly Elaborated Indole Diterpene Penitrem. Angew Chem Int Ed Engl. 2015;54(19):5748–5752. doi: 10.1002/anie.201501072. [DOI] [PubMed] [Google Scholar]
- 156.He HY, Yang HY, Bigelis R, Solum EH, Greenstein M, Carter GT. Pyrrocidines A and B, New Antibiotics Produced by a Filamentous Fungus. Tetrahedron Lett. 2002;43(9):1633–1636. [Google Scholar]
- 157.Isaka M, Rugseree N, Maithip P, Kongsaeree P, Prabpai S, Thebtaranonth Y. Hirsutellones A-E, Antimycobacterial Alkaloids from the Insect Pathogenic Fungus Hirsutella Nivea BCC 2594. Tetrahedron. 2005;61(23):5577–5583. [Google Scholar]
- 158.Koizumi F, Hasegawa K, Ando K, Ogawa T, Hara A. JP 2001147574 A2001147572 2001200109 Jpn Kokai Tokkyo Koho. 2001
- 159.Ebrahim W, Aly AH, Wray V, Mandi A, Teiten MH, Gaascht F, Orlikova B, Kassack MU, Lin W, Diederich M, et al. Embellicines A and B: Absolute Configuration and NF-kappaB Transcriptional Inhibitory Activity. J Med Chem. 2013;56(7):2991–2999. doi: 10.1021/jm400034b. [DOI] [PubMed] [Google Scholar]
- 160.Li XW, Ear A, Nay B. Hirsutellones and Beyond: Figuring Out the Biological and Synthetic Logics Toward Chemical Complexity in Fungal PKS-NRPS Compounds. Nat Prod Rep. 2013;30(6):765–782. doi: 10.1039/c3np70016j. [DOI] [PubMed] [Google Scholar]
- 161.Oikawa H. Biosynthesis of Structurally Unique Fungal Metabolite GKK1032A2: Indication of Novel Carbocyclic Formation Mechanism in Polyketide Biosynthesis. J Org Chem. 2003;68(9):3552–3557. doi: 10.1021/jo0267596. [DOI] [PubMed] [Google Scholar]
- 162.Ear A, Amand S, Blanchard F, Blond A, Dubost L, Buisson D, Nay B. Direct Biosynthetic Cyclization of a Distorted Paracyclophane Highlighted by Double Isotopic Labelling of L-Tyrosine. Org Biomol Chem. 2015;13(12):3662–3666. doi: 10.1039/c5ob00114e. [DOI] [PubMed] [Google Scholar]
- 163.Miao SC, Anstee MR, LaMarco K, Matthew J, Huang LHT, Brasseur MM. Inhibition of Bacterial RNA Polymerases. Peptide Metabolites from the Cultures of Streptomyces sp. J Nat Prod. 1997;60(8):858–861. [Google Scholar]
- 164.Trischman JA, Tapiolas DM, Jensen PR, Dwight R, Fenical W, Mckee TC, Ireland CM, Stout TJ, Clardy J. Salinamides A and B: Anti-inflammatory Depsipeptides from a Marine Streptomycete. J Am Chem Soc. 1994;116(2):757–758. [Google Scholar]
- 165.Degen D, Feng Y, Zhang Y, Ebright KY, Ebright YW, Gigliotti M, Vahedian-Movahed H, Mandal S, Talaue M, Connell N, et al. Transcription Inhibition by the Depsipeptide Antibiotic Salinamide A. Elife. 2014;3:e02451. doi: 10.7554/eLife.02451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ray L, Yamanaka K, Moore BS. A Peptidyl-Transesterifying Type I Thioesterase in Salinamide Biosynthesis. Angew Chem Int Ed Engl. 2016;55(1):364–367. doi: 10.1002/anie.201508576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tijet N, Brash AR. Allene Oxide Synthases and Allene Oxides. Prostaglandins Other Lipid Mediat. 2002;68–69:423–431. doi: 10.1016/s0090-6980(02)00046-1. [DOI] [PubMed] [Google Scholar]
- 168.Teder T, Lohelaid H, Boeglin WE, Calcutt WM, Brash AR, Samel N. A Catalase-Related Hemoprotein in Coral Is Specialized for Synthesis of Short-chain Aldehydes: DISCOVERY OF P450-TYPE HYDROPEROXIDE LYASE ACTIVITY IN A CATALASE. J Biol Chem. 2015;290(32):19823–19832. doi: 10.1074/jbc.M115.660282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wildman WC, Kaufman CJ. Isolation of Tazettine and Lycorine from Certain Hymenocallis Species. J Am Chem Soc. 1954;76(22):5815–5816. [Google Scholar]
- 170.Doskotch RW, El-Feraly FS. Isolation and Characterization of (+)-Sesamin and Beta-Cyclopyrethrosin from Pyrethrum Flowers. Can J Chem. 1969;47(7):1139–1142. [Google Scholar]
- 171.Ikezawa N, Iwasa K, Sato F. CYP719A Subfamily of Cytochrome P450 Oxygenases and Isoquinoline Alkaloid Biosynthesis in Eschscholzia Californica. Plant Cell Rep. 2009;28(1):123–133. doi: 10.1007/s00299-008-0624-8. [DOI] [PubMed] [Google Scholar]
- 172.Ikezawa N, Tanaka M, Nagayoshi M, Shinkyo R, Sakaki T, Inouye K, Sato F. Molecular Cloning and Characterization of CYP719, a Methylenedioxy Bridge-Forming Enzyme that Belongs to a Novel P450 Family, from Cultured Coptis Japonica Cells. J Biol Chem. 2003;278(40):38557–38565. doi: 10.1074/jbc.M302470200. [DOI] [PubMed] [Google Scholar]
- 173.Ikezawa N, Iwasa K, Sato F. Molecular Cloning and Characterization of Methylenedioxy Bridge-Forming Enzymes Involved in Stylopine Biosynthesis in Eschscholzia Californica. FEBS J. 2007;274(4):1019–1035. doi: 10.1111/j.1742-4658.2007.05652.x. [DOI] [PubMed] [Google Scholar]
- 174.Diaz Chavez ML, Rolf M, Gesell A, Kutchan TM. Characterization of Two Methylenedioxy Bridge-Forming Cytochrome P450-Dependent Enzymes of Alkaloid Formation in the Mexican Prickly Poppy Argemone Mexicana. Arch Biochem Biophys. 2011;507(1):186–193. doi: 10.1016/j.abb.2010.11.016. [DOI] [PubMed] [Google Scholar]
- 175.Rabindran SK, Ross DD, Doyle LA, Yang W, Greenberger LM. Fumitremorgin C Reverses Multidrug Resistance in Cells Transfected with the Breast Cancer Resistance Protein. Cancer Res. 2000;60(1):47–50. [PubMed] [Google Scholar]
- 176.Cui CB, Kakeya H, Okada G, Onose R, Osada H. Novel Mammalian Cell Cycle Inhibitors, Tryprostatins A, B and Other Diketopiperazines Produced by Aspergillus Fumigatus. I. Taxonomy, Fermentation, Isolation and Biological Properties. J Antibiot (Tokyo) 1996;49(6):527–533. doi: 10.7164/antibiotics.49.527. [DOI] [PubMed] [Google Scholar]
- 177.Cui CB, Kakeya H, Osada H. Novel Mammalian Cell Cycle Inhibitors, Tryprostatins A, B and Other Diketopiperazines Produced by Aspergillus Fumigatus. II. Physico-Chemical Properties and Structures. J Antibiot (Tokyo) 1996;49(6):534–540. doi: 10.7164/antibiotics.49.534. [DOI] [PubMed] [Google Scholar]
- 178.Kato N, Suzuki H, Takagi H, Asami Y, Kakeya H, Uramoto M, Usui T, Takahashi S, Sugimoto Y, Osada H. Identification of Cytochrome P450s Required for Fumitremorgin Biosynthesis in Aspergillus Fumigatus. ChemBioChem. 2009;10(5):920–928. doi: 10.1002/cbic.200800787. [DOI] [PubMed] [Google Scholar]
- 179.Yin W-B, Grundmann A, Cheng J, Li S-M. Acetylaszonalenin Biosynthesis in Neosartorya Fischeri. Identification of the Biosynthetic Gene Cluster by Genomic Mining and Functional Proof of the Genes by Biochemical Investigation. J Biol Chem. 2009;284(1):100–109. doi: 10.1074/jbc.M807606200. [DOI] [PubMed] [Google Scholar]
- 180.Liu J, Ng T, Rui Z, Ad O, Zhang W. Unusual Acetylation-Dependent Reaction Cascade in the Biosynthesis of the Pyrroloindole Drug Physostigmine. Angew Chem Int Ed Engl. 2014;53(1):136–139. doi: 10.1002/anie.201308069. [DOI] [PubMed] [Google Scholar]
- 181.Ma D. Recent Advances in the Discovery of Protein Kinase C Modulators Based on the Structures of Natural Protein Kinase C Activators. Curr Med Chem. 2001;8(2):191–202. doi: 10.2174/0929867013373868. [DOI] [PubMed] [Google Scholar]
- 182.Huynh MU, Elston MC, Hernandez NM, Ball DB, Kajiyama S, Irie K, Gerwick WH, Edwards DJ. Enzymatic Production of (-)-Indolactam V by LtxB, a Cytochrome P450 Monooxygenase. J Nat Prod. 2010;73(1):71–74. doi: 10.1021/np900481a. [DOI] [PubMed] [Google Scholar]
- 183.Edwards DJ, Gerwick WH. Lyngbyatoxin Biosynthesis: Sequence of Biosynthetic Gene Cluster and Identification of a Novel Aromatic Prenyltransferase. J Am Chem Soc. 2004;126(37):11432–11433. doi: 10.1021/ja047876g. [DOI] [PubMed] [Google Scholar]
- 184.Awakawa T, Zhang L, Wakimoto T, Hoshino S, Mori T, Ito T, Ishikawa J, Tanner ME, Abe I. A Methyltransferase Initiates Terpene Cyclization in Teleocidin B Biosynthesis. J Am Chem Soc. 2014;136:9910–9913. doi: 10.1021/ja505224r. [DOI] [PubMed] [Google Scholar]
- 185.Bartz QR, John E, Penner KM, Smith RM. 3023204. US Pat. 1962
- 186.Xie Y, Wang B, Liu J, Zhou J, Ma J, Huang H, Ju J. Identification of the Biosynthetic Gene Cluster and Regulatory Cascade for the Synergistic Antibacterial Antibiotics Griseoviridin and Viridogrisein in Streptomyces Griseoviridis. Chembiochem. 2012;13(18):2745–2757. doi: 10.1002/cbic.201200584. [DOI] [PubMed] [Google Scholar]
- 187.Xie Y, Li Q, Song Y, Ma J, Ju J. Involvement of SgvP in Carbon-Sulfur Bond Formation During Griseoviridin Biosynthesis. ChemBioChem. 2014;15(8):1183–1189. doi: 10.1002/cbic.201400062. [DOI] [PubMed] [Google Scholar]
- 188.Hallen HE, Luo H, Scott-Craig JS, Walton JD. Gene Family Encoding the Major Toxins of Lethal Amanita Mushrooms. Proc Natl Acad Sci U S A. 2007;104(48):19097–19101. doi: 10.1073/pnas.0707340104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Furuta T, Koike M, Abe M. Isolation of Cycloclavine from the Culture Broth of Aspergillus Japonicus SAITO. Agric Biol Chem. 1982;46(7):1921–1922. [Google Scholar]
- 190.Jakubczyk D, Caputi L, Hatsch A, Nielsen CA, Diefenbacher M, Klein J, Molt A, Schröder H, Cheng JZ, Naesby M, et al. Discovery and Reconstitution of the Cycloclavine Biosynthetic Pathway-Enzymatic Formation of a Cyclopropyl Group. Angew Chem Int Ed Engl. 2015;54(17):5117–5121. doi: 10.1002/anie.201410002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Klanova K, Blumauerova M, Vanek Z. Spontaneous and Induced Variability in Streptomyces Nogalater Producing Nogalamycin. Folia Microbiol (Praha) 1977;22(4):286–294. doi: 10.1007/BF02877658. [DOI] [PubMed] [Google Scholar]
- 192.Siitonen V, Claesson M, Patrikainen P, Aromaa M, Mantsala P, Schneider G, Metsa-Ketela M. Identification of Late-Stage Glycosylation Steps in the Biosynthetic Pathway of the Anthracycline Nogalamycin. Chembiochem. 2012;13(1):120–128. doi: 10.1002/cbic.201100637. [DOI] [PubMed] [Google Scholar]
- 193.Siitonen V, Selvaraj B, Niiranen L, Lindqvist Y, Schneider G, Metsä-Ketelä M. Divergent Non-Heme Iron Enzymes in the Nogalamycin Biosynthetic Pathway. Proc Natl Acad Sci U S A. 2016;113(19):5251–5256. doi: 10.1073/pnas.1525034113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Hande KR. Etoposide: Four Decades of Development of a Topoisomerase II Inhibitor. Eur J Cancer. 1998;34(10):1514–1521. doi: 10.1016/s0959-8049(98)00228-7. [DOI] [PubMed] [Google Scholar]
- 195.Nitiss JL. Targeting DNA Topoisomerase II in Cancer Chemotherapy. Nat Rev Cancer. 2009;9(5):338–350. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Sinkule JA. Etoposide: A Semisynthetic Epipodophyllotoxin. Chemistry, Pharmacology, Pharmacokinetics, Adverse Effects and Use as an Antineoplastic Agent. Pharmacotherapy. 1984;4(2):61–73. doi: 10.1002/j.1875-9114.1984.tb03318.x. [DOI] [PubMed] [Google Scholar]
- 197.Guerre P. Ergot Alkaloids Produced by Endophytic Fungi of the Genus Epichloë. Toxins (Basel) 2015;7(3):773–790. doi: 10.3390/toxins7030773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Havemann J, Vogel D, Loll B, Keller U. Cyclolization of D-Lysergic Acid Alkaloid Peptides. Chem Biol. 2014;21(1):146–155. doi: 10.1016/j.chembiol.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 199.McCranie EK, Bachmann BO. Bioactive Oligosaccharide Natural Products. Nat Prod Rep. 2014;31(8):1026–1042. doi: 10.1039/c3np70128j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Hosted TJ, Wang TX, Alexander DC, Horan AC. Characterization of the Biosynthetic Gene Cluster for the Oligosaccharide Antibiotic, Evernimicin, in Micromonospora Carbonacea var. Africana ATCC39149. J Ind Microbiol Biotechnol. 2001;27(6):386–392. doi: 10.1038/sj.jim.7000189. [DOI] [PubMed] [Google Scholar]
- 201.Gaisser S, Trefzer A, Stockert S, Kirschning A, Bechthold A. Cloning of an Avilamycin Biosynthetic Gene Cluster from Streptomyces Viridochromogenes Tü57. J Bacteriol. 1997;179(20):6271–6278. doi: 10.1128/jb.179.20.6271-6278.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.McCulloch KM, McCranie EK, Smith JA, Sarwar M, Mathieu JL, Gitschlag BL, Du Y, Bachmann BO, Iverson TM. Oxidative Cyclizations in Orthosomycin Biosynthesis Expand the Known Chemistry of an Oxygenase Superfamily. Proc Natl Acad Sci U S A. 2015;112(37):11547–11552. doi: 10.1073/pnas.1500964112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Reading C, Cole M. Clavulanic Acid: A Beta-Lactamase-Inhiting Beta-Lactam from Streptomyces Clavuligerus. Antimicrob Agents Chemother. 1977;11(5):852–857. doi: 10.1128/aac.11.5.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Song JY, Jensen SE, Lee KJ. Clavulanic Acid Biosynthesis and Genetic Manipulation for Its Overproduction. Appl Microbiol Biotechnol. 2010;88(3):659–669. doi: 10.1007/s00253-010-2801-2. [DOI] [PubMed] [Google Scholar]
- 205.Salowe SP, Krol WJ, Iwata-Reuyl D, Townsend CA. Elucidation of the Order of Oxidations and Identification of an Intermediate in the Multistep Clavaminate Synthase Reaction. Biochemistry. 1991;30:2281–2292. doi: 10.1021/bi00222a034. [DOI] [PubMed] [Google Scholar]
- 206.Borowski T, De Marothy S, Broclawik E, Schofield CJ, Siegbahn PE. Mechanism for Cyclization Reaction by Clavaminic Acid Synthase. Insights from Modeling Studies. Biochemistry. 2007;46(12):3682–3691. doi: 10.1021/bi602458m. [DOI] [PubMed] [Google Scholar]
- 207.Salowe SP, Marsh EN, Townsend CA. Purification and Characterization of Clavaminate Synthase from Streptomyces Clavuligerus: An Unusual Oxidative Enzyme in Natural Product Biosynthesis. Biochemistry. 1990;29:6499–6508. doi: 10.1021/bi00479a023. [DOI] [PubMed] [Google Scholar]
- 208.Janc JW, Egan LA, Townsend CA. Purification and Characterization of Clavaminate Synthase from Streptomyces Antibioticus. A Multifunctional Enzyme of Clavam Biosynthesis. J Biol Chem. 1995;270(10):5399–5404. doi: 10.1074/jbc.270.10.5399. [DOI] [PubMed] [Google Scholar]
- 209.Busby RW, Townsend CA. A Single Monomeric Iron Center in Clavaminate Synthase Catalyzes Three Nonsuccessive Oxidative Transformations. Bioorg Med Chem. 1996;4(7):1059–1064. doi: 10.1016/0968-0896(96)00088-0. [DOI] [PubMed] [Google Scholar]
- 210.Townsend CA. The Stereochemical Fate of Chiral-Methyl Valines in Cephalosporin C Biosynthesis. J Nat Prod. 1985;48(5):708–724. doi: 10.1021/np50041a002. [DOI] [PubMed] [Google Scholar]
- 211.Marsh EN, Chang MD, Townsend CA. Two Isozymes of Clavaminate Synthase Central to Clavulanic Acid Formation: Cloning and Sequencing of Both Genes from Streptomyces Clavuligerus. Biochemistry. 1992;31(50):12648–12657. doi: 10.1021/bi00165a015. [DOI] [PubMed] [Google Scholar]
- 212.Khaleeli N, Li RF, Townsend CA. Origin of the Beta-Lactam Carbons in Clavulanic Acid from an Unusual Thiamine Pyrophosphate-Mediated Reaction. J Am Chem Soc. 1999;121(39):9223–9224. [Google Scholar]
- 213.Zhang Z, Ren J-S, Harlos K, Mckinnon CH, Clifton IJ, Scho CJ. Crystal Structure of a Clavaminate Synthase-Fe(II)-2-Oxoglutarate-Substrate-NO Complex: Evidence for Metal Centered Rearrangements. FEBS Lett. 2002;517(1–3):7–12. doi: 10.1016/s0014-5793(02)02520-6. [DOI] [PubMed] [Google Scholar]
- 214.Zhang Z, Ren J, Stammers DK, Baldwin JE, Harlos K, Schofield CJ. Structural Origins of the Selectivity of the Trifunctional Oxygenase Clavaminic Acid Synthase. Nat Struct Biol. 2000;7(2):127–133. doi: 10.1038/72398. [DOI] [PubMed] [Google Scholar]
- 215.Hashimoto T, Yamada Y. Hyoscyamine 6Beta-Hydroxylase, a 2-Oxoglutarate-Dependent Dioxygenase, in Alkaloid-Producing Root Cultures. Plant Physiol. 1986;81(2):619–625. doi: 10.1104/pp.81.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Matsuda J, Okabe S, Hashimoto T, Yamada Y. Molecular Cloning of Hyoscyamine 6 Beta-Hydroxylase, a 2-Oxoglutarate-Dependent Dioxygenase, from Cultured Roots of Hyoscyamus Niger. J Biol Chem. 1991;266(15):9460–9464. [PubMed] [Google Scholar]
- 217.Zhang L, Ding R, Chai Y, Bonfill M, Moyano E, Oksman-Caldentey KM, Xu T, Pi Y, Wang Z, Zhang H, et al. Engineering Tropane Biosynthetic Pathway in Hyoscyamus Niger Hairy Root Cultures. Proc Natl Acad Sci U S A. 2004;101(17):6786–6791. doi: 10.1073/pnas.0401391101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Patterson DS, Shreeve BJ, Roberts BA, MacDonald SM. Verruculogen Produced by Soil Fungi in England and Wales. Appl Environ Microbiol. 1981;42(5):916–917. doi: 10.1128/aem.42.5.916-917.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Steffan N, Grundmann A, Afiyatullov S, Ruan H, Li S-M. FtmOx1, a Non-Heme Fe(II) and Alpha-Ketoglutarate-Dependent Dioxygenase, Catalyses the Endoperoxide Formation of Verruculogen in Aspergillus Fumigatus. Org Biomol Chem. 2009;7(19):4082–4087. doi: 10.1039/b908392h. [DOI] [PubMed] [Google Scholar]
- 220.Yan W, Song H, Song F, Guo Y, Wu C-H, Sae Her A, Pu Y, Wang S, Naowarojna N, Weitz A, et al. Endoperoxide Formation by an α-Ketoglutarate-Dependent Mononuclear Non-Haem Iron Enzyme. Nature. 2015;527(7579):539–543. doi: 10.1038/nature15519. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 221.Wank H, Rogers J, Davies J, Schroeder R. Peptide Antibiotics of the Tuberactinomycin Family as Inhibitors of Group I Intron RNA Splicing. J Mol Biol. 1994;236(4):1001–1010. doi: 10.1016/0022-2836(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 222.Ju J, Ozanick SG, Shen B, Thomas MG. Conversion of (2S)-Arginine to (2S,3R)-Capreomycidine by VioC and VioD from the Viomycin Biosynthetic Pathway of Streptomyces sp. Strain ATCC11861. ChemBioChem. 2004;5(9):1281–1285. doi: 10.1002/cbic.200400136. [DOI] [PubMed] [Google Scholar]
- 223.Yin X, McPhail KL, Kim KJ, Zabriskie TM. Formation of the Nonproteinogenic Amino Acid 2S,3R-Capreomycidine by VioD from the Viomycin Biosynthesis Pathway. ChemBioChem. 2004;5(9):1278–1281. doi: 10.1002/cbic.200400187. [DOI] [PubMed] [Google Scholar]
- 224.Nyfeler R, Keller-Schierlein W. Metabolites of Microorganisms. 143. Echinocandin B, a Novel Polypeptide-Antibiotic from Aspergillus Nidulans var. Echinulatus: Isolation and Structural Components. Helv Chim Acta. 1974;57(8):2459–2477. doi: 10.1002/hlca.19740570818. [DOI] [PubMed] [Google Scholar]
- 225.Golakoti T, Yoshida WY, Chaganty S, Moore RE. Isolation and Structures of Nostopeptolides A1, A2 and A3 from the Cyanobacterium Nostoc sp. GSV224. Tetrahedron. 2000;56(46):9093–9102. [Google Scholar]
- 226.Umezawa K, Nakazawa K, Ikeda Y, Naganawa H, Kondo S. Polyoxypeptins A and B Produced by Streptomyces: Apoptosis-Inducing Cyclic Depsipeptides Containing the Novel Amino Acid (2S,3R)-3-Hydroxy-3-Methylproline. J Org Chem. 1999;64(9):3034–3038. doi: 10.1021/jo981512n. [DOI] [PubMed] [Google Scholar]
- 227.Terlain B, Thomas JP. Structure of Griselimycin, Polypeptide Antibiotic Extracted from Cultures of Streptomyces. II. Structure of Griselimycin. Bull Soc Chim Fr. 1971;(6):2357–2362. [PubMed] [Google Scholar]
- 228.Festa C, De Marino S, Sepe V, Monti MC, Luciano P, D’Auria MV, Debitus C, Bucci M, Vellecco V, Zampella A. Perthamides C and D, Two New Potent Anti-Inflammatory Cyclopeptides from a Solomon Lithistid Sponge Theonella Swinhoei. Tetrahedron. 2009;65(50):10424–10429. [Google Scholar]
- 229.Bevan K, Davies JS, Hall MJ, Hassall CH, Morton RB, Phillips DA, Ogihara Y, Thomas WA. The Monamycins, a New Family of Cyclodepsipeptide Antibiotics. Experientia. 1970;26(2):122–123. doi: 10.1007/BF01895528. [DOI] [PubMed] [Google Scholar]
- 230.Jiang W, Cacho RA, Chiou G, Garg NK, Tang Y, Walsh CT. EcdGHK Are Three Tailoring Iron Oxygenases for Amino Acid Building Blocks of the Echinocandin Scaffold. J Am Chem Soc. 2013;135(11):4457–4466. doi: 10.1021/ja312572v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Luesch H, Hoffmann D, Hevel JM, Becker JE, Golakoti T, Moore RE. Biosynthesis of 4-Methylproline in Cyanobacteria: Cloning of NosE and NosF Genes and Biochemical Characterization of the Encoded Dehydrogenase and Reductase Activities. J Org Chem. 2003;68(1):83–91. doi: 10.1021/jo026479q. [DOI] [PubMed] [Google Scholar]
- 232.Hoffmann D, Hevel JM, Moore RE, Moore BS. Sequence Analysis and Biochemical Characterization of the Nostopeptolide A Biosynthetic Gene Cluster from Nostoc sp. GSV224 Gene. 2003;311:171–180. doi: 10.1016/s0378-1119(03)00587-0. [DOI] [PubMed] [Google Scholar]
- 233.Hibi M, Kawashima T, Sokolov PM, Smirnov SV, Kodera T, Sugiyama M, Shimizu S, Yokozeki K, Ogawa J. L-Leucine 5-Hydroxylase of Nostoc punctiforme Is a Novel Type of Fe(II)/Alpha-Ketoglutarate-Dependent Dioxygenase that Is Useful as a Biocatalyst. Appl Microbiol Biotechnol. 2013;97(6):2467–2472. doi: 10.1007/s00253-012-4136-7. [DOI] [PubMed] [Google Scholar]
- 234.Hendlin D, Stapley EO, Jackson M, Wallick H, Miller AK, Wolf FJ, Miller TW, Chaiet L, Kahan FM, Foltz EL, et al. Phosphonomycin, a New Antibiotic Produced by Strains of Streptomyces. Science. 1969;166(3901):122–123. doi: 10.1126/science.166.3901.122. [DOI] [PubMed] [Google Scholar]
- 235.Christensen BG, Leanza WJ, Beattie TR, Patchett AA, Arison BH, Ormond RE, Kuehl FA, Jr, Albers-Schonberg G, Jardetzky O. Phosphonomycin: Structure and Synthesis. Science. 1969;166(3901):123–125. doi: 10.1126/science.166.3901.123. [DOI] [PubMed] [Google Scholar]
- 236.Kim SY, Ju KS, Metcalf WW, Evans BS, Kuzuyama T, van der Donk WA. Different Biosynthetic Pathways to Fosfomycin in Pseudomonas Syringae and Streptomyces Species. Antimicrob Agents Chemother. 2012;56(8):4175–4183. doi: 10.1128/AAC.06478-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kahan FM, Kahan JS, Cassidy PJ, Kropp H. The Mechanism of Action of Fosfomycin (Phosphonomycin) Ann N Y Acad Sci. 1974;235(0):364–386. doi: 10.1111/j.1749-6632.1974.tb43277.x. [DOI] [PubMed] [Google Scholar]
- 238.Hidaka T, Goda M, Kuzuyama T, Takei N, Hidaka M, Seto H. Cloning and Nucleotide Sequence of Fosfomycin Biosynthetic Genes of Streptomyces Wedmorensis. Mol Gen Genet. 1995;249(3):274–280. doi: 10.1007/BF00290527. [DOI] [PubMed] [Google Scholar]
- 239.Liu P, Murakami K, Seki T, He X, Yeung S, Kuzuyama T, Seto H, Liu H-W. Protein Purification and Function Assignment of the Epoxidase Catalyzing the Formation of Fosfomycin. J Am Chem Soc. 2001;123(19):4619–4620. doi: 10.1021/ja004153y. [DOI] [PubMed] [Google Scholar]
- 240.Kuzuyama T, Seki T, Kobayashi S, Hidaka T, Seto H. Cloning and Expression in Escherichia coli of 2-Hydroxypropylphosphonic Acid Epoxidase from the Fosfomycin Producing Organism, Pseudomonas Syringae PB-5123. Biosci Biotechnol Biochem. 1999;63(12):2222–2224. doi: 10.1271/bbb.63.2222. [DOI] [PubMed] [Google Scholar]
- 241.McLuskey K, Cameron S, Hammerschmidt F, Hunter WN. Structure and Reactivity of Hydroxypropylphosphonic Acid Epoxidase in Fosfomycin Biosynthesis by a Cation- and Flavin-Dependent Mechanism. Proc Natl Acad Sci U S A. 2005;102(40):14221–14226. doi: 10.1073/pnas.0504314102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Wang C, Chang W-C, Guo Y, Huang H, Peck SC, Pandelia ME, Lin G-M, Liu H-W, Krebs C, Bollinger JMJ. Evidence that the Fosfomycin-Producing Epoxidase, HppE, Is a Non–Heme-Iron Peroxidase. Science. 2013;342(6161):991–995. doi: 10.1126/science.1240373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Chang W-C, Dey M, Liu P, Mansoorabadi SO, Moon SJ, Zhao Z-K, Drennan CL, Liu H-W. Mechanistic Studies of an Unprecedented Enzyme-Catalysed 1,2-Phosphono-Migration Reaction. Nature. 2013;496(7443):114–118. doi: 10.1038/nature11998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Zhao Z, Liu P, Murakami K, Kuzuyama T, Seto H, Liu H-W. Mechanistic Studies of HPP Epoxidase: Configuration of the Substrate Governs Its Enzymatic Fate. Angew Chem Int Ed Engl. 2002;41(23):4529–4532. doi: 10.1002/1521-3773(20021202)41:23<4529::AID-ANIE4529>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 245.Waxman DJ, Strominger JL. Penicillin-Binding Proteins and the Mechanism of Action of Beta-Lactam Antibiotics. Annu Rev Biochem. 1983;52:825–869. doi: 10.1146/annurev.bi.52.070183.004141. [DOI] [PubMed] [Google Scholar]
- 246.Hamed RB, Gomez-Castellanos JR, Henry L, Ducho C, McDonough MA, Schofield CJ. The Enzymes of Beta-Lactam Biosynthesis. Nat Prod Rep. 2013;30(1):21–107. doi: 10.1039/c2np20065a. [DOI] [PubMed] [Google Scholar]
- 247.Ozcengiz G, Demain AL. Recent Advances in the Biosynthesis of Penicillins, Cephalosporins and Clavams and Its Regulation. Biotechnol Adv. 2013;31(2):287–311. doi: 10.1016/j.biotechadv.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 248.Tamanaha E, Zhang B, Guo Y, Chang W-C, Barr EW, Xing G, St Clair J, Ye S, Neese F, Bollinger JMJ, et al. Spectroscopic Evidence for the Two C–H-Cleaving Intermediates of Aspergillus Nidulans Isopenicillin N Synthase. J Am Chem Soc. 2016;138(28):8862–8874. doi: 10.1021/jacs.6b04065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Wasserman HH, Rodgers GC, Keith DD. Metacycloprodigiosin, a Tripyrrole Pigment from Streptomyces Longisporus Ruber. J Am Chem Soc. 1969;91(5):1263–1264. doi: 10.1021/ja01033a065. [DOI] [PubMed] [Google Scholar]
- 250.Gerber NN, Mcinnes AG, Smith DG, Walter JA, Wright JLC, Vining LC. Biosynthesis of Prodiginines. C-13 Resonance Assignments and Enrichment Patterns in Nonyl-, Cyclononyl-, Methylcyclodecyl-, and Butylcycloheptylprodiginine Produced by Actinomycete Cultures Supplemented with C-13-Labeled Acetate and N-15-Labeled Nitrate. Can J Chem. 1978;56(9):1155–1163. [Google Scholar]
- 251.Boonlarppradab C, Kauffman CA, Jensen PR, Fenical W. Marineosins A and B, Cytotoxic Spiroaminals from a Marine-Derived Actinomycete. Org Lett. 2008;10(24):5505–5508. doi: 10.1021/ol8020644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Perez-Tomas R, Vinas M. New Insights on the Antitumoral Properties of Prodiginines. Curr Med Chem. 2010;17(21):2222–2231. doi: 10.2174/092986710791331103. [DOI] [PubMed] [Google Scholar]
- 253.Stankovic N, Senerovic L, Ilic-Tomic T, Vasiljevic B, Nikodinovic-Runic J. Properties and Applications of Undecylprodigiosin and Other Bacterial Prodigiosins. Appl Microbiol Biotechnol. 2014;98(9):3841–3858. doi: 10.1007/s00253-014-5590-1. [DOI] [PubMed] [Google Scholar]
- 254.Sydor PK, Barry SM, Odulate OM, Barona-Gomez F, Haynes SW, Corre C, Song L, Challis GL. Regio- and Stereodivergent Antibiotic Oxidative Carbocyclizations Catalysed by Rieske Oxygenase-Like Enzymes. Nat Chem. 2011;3(5):388–392. doi: 10.1038/nchem.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Haynes SW, Sydor PK, Stanley AE, Song L, Challis GL. Role and Substrate Specificity of the Streptomyces Coelicolor RedH Enzyme in Undecylprodiginine Biosynthesis. Chem Commun (Camb) 2008;(16):1865–1867. doi: 10.1039/b801677a. [DOI] [PubMed] [Google Scholar]
- 256.Withall DM, Haynes SW, Challis GL. Stereochemistry and Mechanism of Undecylprodigiosin Oxidative Carbocyclization to Streptorubin B by the Rieske Oxygenase RedG. J Am Chem Soc. 2015;137(24):7889–7897. doi: 10.1021/jacs.5b03994. [DOI] [PubMed] [Google Scholar]
- 257.Salem SM, Kancharla P, Florova G, Gupta S, Lu W, Reynolds KA. Elucidation of Final Steps of the Marineosins Biosynthetic Pathway Through Identification and Characterization of the Corresponding Gene Cluster. J Am Chem Soc. 2014;136(12):4565–4574. doi: 10.1021/ja411544w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Schramma KR, Bushin LB, Seyedsayamdost MR. Structure and Biosynthesis of a Macrocyclic Peptide Containing an Unprecedented Lysine-to-TryptophanCrosslink. Nat Chem. 2015;7(5):431–437. doi: 10.1038/nchem.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Ibrahim M, Guillot A, Wessner F, Algaron F, Besset C, Courtin P, Gardan R, Monnet V. Control of the Transcription of a Short Gene Encoding a Cyclic Peptide in Streptococcus Thermophilus: A New Quorum-Sensing System? J Bacteriol. 2007;189(24):8844–8854. doi: 10.1128/JB.01057-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Gardan R, Besset C, Guillot A, Gitton C, Monnet V. The Oligopeptide Transport System Is Essential for the Development of Natural Competence in Streptococcus Thermophilus Strain LMD-9. J Bacteriol. 2009;191(14):4647–4655. doi: 10.1128/JB.00257-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Fleuchot B, Gitton C, Guillot A, Vidic J, Nicolas P, Besset C, Fontaine L, Hols P, Leblond-Bourget N, Monnet V, et al. Rgg Proteins Associated with Internalized Small Hydrophobic Peptides: A New Quorum-Sensing Mechanism in Streptococci. Mol Microbiol. 2011;80(4):1102–1119. doi: 10.1111/j.1365-2958.2011.07633.x. [DOI] [PubMed] [Google Scholar]
- 262.Dairi T, Kuzuyama T, Nishiyama M, Fujii I. Convergent Strategies in Biosynthesis. Nat Prod Rep. 2011;28(6):1054–1086. doi: 10.1039/c0np00047g. [DOI] [PubMed] [Google Scholar]
- 263.Hiratsuka T, Furihata K, Ishikawa J, Yamashita H, Itoh N, Seto H, Dairi T. An Alternative Menaquinone Biosynthetic Pathway Operating in Microorganisms. Science. 2008;321(5896):1670–1673. doi: 10.1126/science.1160446. [DOI] [PubMed] [Google Scholar]
- 264.Cooper LE, Fedoseyenko D, Abdelwahed SH, Kim SH, Dairi T, Begley TP. In Vitro Reconstitution of the Radical S-Adenosylmethionine Enzyme MqnC Involved in the Biosynthesis of Futalosine-Derived Menaquinone. Biochemistry. 2013;52(27):4592–4594. doi: 10.1021/bi400498d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Yoshida M, Ezaki M, Hashimoto M, Yamashita M, Shigematsu N, Okuhara M, Kohsaka M, Horikoshi K. A Novel Antifungal Antibiotic, Fr-900848. I. Production, Isolation, Physico-Chemical and Biological Properties. J Antibiot (Tokyo) 1990;43(7):748–754. doi: 10.7164/antibiotics.43.748. [DOI] [PubMed] [Google Scholar]
- 266.Watanabe H, Tokiwano T, Oikawa H. Biosynthetic Study of FR-900848: Origin of the Aminodeoxynucleoside Part. J Antibiot (Tokyo) 2006;59(9):607–610. doi: 10.1038/ja.2006.82. [DOI] [PubMed] [Google Scholar]
- 267.Watanabe H, Tokiwano T, Oikawa H. Biosynthetic Study of FR-900848: Unusual Observation on Polyketide Biosynthesis that Did Not Accept Acetate as Origin of Acetyl-CoA. Tetrahedron Lett. 2006;47(9):1399–1402. [Google Scholar]
- 268.Hiratsuka T, Suzuki H, Kariya R, Seo T, Minami A, Oikawa H. Biosynthesis of the Structurally Unique Polycyclopropanated Polyketide-Nucleoside Hybrid Jawsamycin (FR-900848) Angew Chem Int Ed Engl. 2014;53(21):5423–5426. doi: 10.1002/anie.201402623. [DOI] [PubMed] [Google Scholar]
- 269.Bauerle MR, Schwalm EL, Booker SJ. Mechanistic Diversity of Radical S-Adenosylmethionine (SAM)-Dependent Methylation. J Biol Chem. 2015;290(7):3995–4002. doi: 10.1074/jbc.R114.607044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Lotierzo M, Tse Sum Bui B, Florentin D, Escalettes F, Marquet A. Biotin Synthase Mechanism: An Overview. Biochem Soc Trans. 2005;33(Pt 4):820–823. doi: 10.1042/BST0330820. [DOI] [PubMed] [Google Scholar]
- 271.Lotierzo M, Raux E, Tse Sum Bui B, Goasdoue N, Libot F, Florentin D, Warren MJ, Marquet A. Biotin Synthase Mechanism: Mutagenesis of the YNHNLD Conserved Motif. Biochemistry. 2006;45(40):12274–12281. doi: 10.1021/bi060662m. [DOI] [PubMed] [Google Scholar]
- 272.Bui BT, Florentin D, Fournier F, Ploux O, Mejean A, Marquet A. Biotin Synthase Mechanism: on the Origin of Sulphur. FEBS Lett. 1998;440(1–2):226–230. doi: 10.1016/s0014-5793(98)01464-1. [DOI] [PubMed] [Google Scholar]
- 273.Rana A, Dey S, Agrawal A, Dey A. Density Functional Theory Calculations on the Active Site of Biotin Synthase: Mechanism of S Transfer from the Fe2S2 Cluster and the Role of 1st and 2nd Sphere Residues. J Biol Inorg Chem. 2015;20(7):1147–1162. doi: 10.1007/s00775-015-1296-9. [DOI] [PubMed] [Google Scholar]
- 274.Taylor AM, Stoll S, Britt RD, Jarrett JT. Reduction of the [2Fe-2S] Cluster Accompanies Formation of the Intermediate 9-Mercaptodethiobiotin in Escherichia coli Biotin Synthase. Biochemistry. 2011;50(37):7953–7963. doi: 10.1021/bi201042r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Babasaki K, Takao T, Shimonishi Y, Kurahashi K. Subtilosin A, a New Antibiotic Peptide Produced by Bacillus Subtilis 168: Isolation, Structural Analysis, and Biogenesis. J Biochem. 1985;98(3):585–603. doi: 10.1093/oxfordjournals.jbchem.a135315. [DOI] [PubMed] [Google Scholar]
- 276.Flühe L, Knappe TA, Gattner MJ, Schäfer A, Burghaus O, Linne U, Marahiel MA. The Radical SAM Enzyme AlbA Catalyzes Thioether Bond Formation in Subtilosin A. Nat Chem Biol. 2012;8(4):737–737. doi: 10.1038/nchembio.798. [DOI] [PubMed] [Google Scholar]
- 277.Benjdia A, Guillot A, Lefranc B, Vaudry H, Leprince J, Berteau O. Thioether Bond Formation by SPASM Domain Radical SAM Enzymes: Cα H-Atom Abstraction in Subtilosin A Biosynthesis. Chem Commun (Camb) 2016;52(37):6249–6252. doi: 10.1039/c6cc01317a. [DOI] [PubMed] [Google Scholar]
- 278.Meier JL, Burkart MD. The Chemical Biology of Modular Biosynthetic Enzymes. Chem Soc Rev. 2009;38(7):2012–2045. doi: 10.1039/b805115c. [DOI] [PubMed] [Google Scholar]
- 279.Akey DL, Kittendorf JD, Giraldes JW, Fecik RA, Sherman DH, Smith JL. Structural Basis for Macrolactonization by the Pikromycin Thioesterase. Nat Chem Biol. 2006;2(10):537–542. doi: 10.1038/nchembio824. [DOI] [PubMed] [Google Scholar]
- 280.Golakoti T, Yoshida WY, Chaganty S, Moore RE. Isolation and Structure Determination of Nostocyclopeptides A1 and A2 from the Terrestrial Cyanobacterium Nostoc sp. ATCC53789. J Nat Prod. 2001;64(1):54–59. doi: 10.1021/np000316k. [DOI] [PubMed] [Google Scholar]
- 281.Becker JE, Moore RE, Moore BS. Cloning, Sequencing, and Biochemical Characterization of the Nostocyclopeptide Biosynthetic Gene Cluster: Molecular Basis for Imine Macrocyclization. Gene. 2004;325:35–42. doi: 10.1016/j.gene.2003.09.034. [DOI] [PubMed] [Google Scholar]
- 282.Kopp F, Mahlert C, Grünewald J, Marahiel MA. Peptide Macrocyclization: The Reductase of the Nostocyclopeptide Synthetase Triggers the Self-Assembly of a Macrocyclic Imine. J Am Chem Soc. 2006;128(51):16478–16479. doi: 10.1021/ja0667458. [DOI] [PubMed] [Google Scholar]
- 283.Arai T, Takahashi K, Kubo A. New Antibiotics Saframycins A, B, C, D and E. J Antibiot (Tokyo) 1977;30(11):1015–1018. doi: 10.7164/antibiotics.30.1015. [DOI] [PubMed] [Google Scholar]
- 284.Cuevas C, Francesch A. Development of Yondelis (Trabectedin, ET-743). A Semisynthetic Process Solves the Supply Problem. Nat Prod Rep. 2009;26(3):322–337. doi: 10.1039/b808331m. [DOI] [PubMed] [Google Scholar]
- 285.Molinski TF, Dalisay DS, Lievens SL, Saludes JP. Drug Development from Marine Natural Products. Nat Rev Drug Discov. 2009;8(1):69–85. doi: 10.1038/nrd2487. [DOI] [PubMed] [Google Scholar]
- 286.Rath CM, Janto B, Earl J, Ahmed A, Hu FZ, Hiller L, Dahlgren M, Kreft R, Yu F, Wolff JJ, et al. Meta-Omic Characterization of the Marine Invertebrate Microbial Consortium That Produces the Chemotherapeutic Natural Product ET-743. ACS Chem Biol. 2011;6(11):1244–1256. doi: 10.1021/cb200244t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Schofield MM, Jain S, Porat D, Dick GJ, Sherman DH. Identification and Analysis of the Bacterial Endosymbiont Specialized for Production of the Chemotherapeutic Natural Product ET-743. Environ Microbiol. 2015;17(10):3964–3975. doi: 10.1111/1462-2920.12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Koketsu K, Watanabe K, Suda H, Oguri H, Oikawa H. Reconstruction of the Saframycin Core Scaffold Defines Dual Pictet-Spengler Mechanisms. Nat Chem Biol. 2010;6(6):408–410. doi: 10.1038/nchembio.365. [DOI] [PubMed] [Google Scholar]
- 289.Cary JW, Uka V, Han Z, Buyst D, Harris-Coward PY, Ehrlich KC, Wei Q, Bhatnagar D, Dowd PF, Martens SL, et al. An Aspergillus Flavus Secondary Metabolic Gene Cluster Containing a Hybrid PKS-NRPS Is Necessary for Synthesis of the 2-Pyridones, Leporins. Fungal Genet Biol. 2015;81:88–97. doi: 10.1016/j.fgb.2015.05.010. [DOI] [PubMed] [Google Scholar]
- 290.Wasil Z, Pahirulzaman KAK, Butts C, Simpson TJ, Lazarus CM, Cox RJ. One Pathway, Many Compounds: Heterologous Expression of a Fungal Biosynthetic Pathway Reveals Its Intrinsic Potential for Diversity. Chem Sci. 2013;4(10):3845–3856. [Google Scholar]
- 291.Snider BB, Lu Q. Total Synthesis of (+/−)-Leporin A. J Org Chem. 1996;61(8):2839–2844. doi: 10.1021/jo952053i. [DOI] [PubMed] [Google Scholar]
- 292.Agarwal V, Moore BS. Enzymatic Synthesis of Polybrominated Dioxins from the Marine Environment. ACS Chem Biol. 2014;9(9):1980–1984. doi: 10.1021/cb5004338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Jomon K, Kuroda Y, Ajisaka M, Sakai H. A New Antibiotic, Ikarugamycin. J Antibiot (Tokyo) 1972;25(5):271–280. doi: 10.7164/antibiotics.25.271. [DOI] [PubMed] [Google Scholar]
- 294.Cao S, Blodgett JA, Clardy J. Targeted Discovery of Polycyclic Tetramate Macrolactams from an Environmental Streptomyces Strain. Org Lett. 2010;12(20):4652–4654. doi: 10.1021/ol1020064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Graupner PR, Thornburgh S, Mathieson JT, Chapin EL, Kemmitt GM, Brown JM, Snipes CE. Dihydromaltophilin; a Novel Fungicidal Tetramic Acid Containing Metabolite from Streptomyces sp. J Antibiot (Tokyo) 1997;50(12):1014–1019. doi: 10.7164/antibiotics.50.1014. [DOI] [PubMed] [Google Scholar]
- 296.Blodgett JA, Oh DC, Cao S, Currie CR, Kolter R, Clardy J. Common Biosynthetic Origins for Polycyclic Tetramate Macrolactams from Phylogenetically Diverse Bacteria. Proc Natl Acad Sci U S A. 2010;107(26):11692–11697. doi: 10.1073/pnas.1001513107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Zhang G, Zhang W, Saha S, Zhang C. Recent Advances in Discovery, Biosynthesis and Genome Mining of Medicinally Relevant Polycyclic Tetramate Macrolactams. Curr Top Med Chem. 2016;16(15):1727–1739. doi: 10.2174/1568026616666151012112818. [DOI] [PubMed] [Google Scholar]
- 298.Zhang G, Zhang W, Zhang Q, Shi T, Ma L, Zhu Y, Li S, Zhang H, Zhao YL, Shi R, et al. Mechanistic Insights into Polycycle Formation by Reductive Cyclization in Ikarugamycin Biosynthesis. Angew Chem Int Ed Engl. 2014;53(19):4840–4844. doi: 10.1002/anie.201402078. [DOI] [PubMed] [Google Scholar]
- 299.Lou L, Qian G, Xie Y, Hang J, Chen H, Zaleta-Rivera K, Li Y, Shen Y, Dussault PH, Liu F, et al. Biosynthesis of HSAF, a Tetramic Acid-Containing Macrolactam from Lysobacter Enzymogenes. J Am Chem Soc. 2011;133(4):643–645. doi: 10.1021/ja105732c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Antosch J, Schaefers F, Gulder TA. Heterologous Reconstitution of Ikarugamycin Biosynthesis in. E coli Angew Chem Int Ed Engl. 2014;53(11):3011–3014. doi: 10.1002/anie.201310641. [DOI] [PubMed] [Google Scholar]
- 301.Liblikas I, Santangelo EM, Sandell J, Baeckstrom P, Svensson M, Jacobsson U, Unelius CR. Simplified Isolation Procedure and Interconversion of the Diastereomers of Nepetalactone and Nepetalactol. J Nat Prod. 2005;68(6):886–890. doi: 10.1021/np049647d. [DOI] [PubMed] [Google Scholar]
- 302.Sticher O, Meier B. Quantitative Analysis and Isolation of Aucubin and Catalpol from Folium Plantaginis by HPLC. Planta Med. 1978;33(3):295–296. [Google Scholar]
- 303.Dinda B, Debnath S. In: Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes. Ramawat GK, Mérillon J-M, editors. Springer Berlin Heidelberg; Berlin, Heidelberg: 2013. [DOI] [Google Scholar]
- 304.Kries H, O’Connor SE. Biocatalysts from Alkaloid Producing Plants. Curr Opin Chem Biol. 2016;31:22–30. doi: 10.1016/j.cbpa.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 305.Geu-Flores F, Sherden NH, Courdavault V, Burlat V, Glenn WS, Wu C, Nims E, Cui Y, O’Connor SE. An Alternative Route to Cyclic Terpenes by Reductive Cyclization in Iridoid Biosynthesis. Nature. 2012;492(7427):138–142. doi: 10.1038/nature11692. [DOI] [PubMed] [Google Scholar]
- 306.Munkert J, Costa C, Budeanu O, Petersen J, Bertolucci S, Fischer G, Müller-Uri F, Kreis W. Progesterone 5Beta-Reductase Genes of the Brassicaceae Family as Function-Associated Molecular Markers. Plant Biol J. 2015;17(6):1113–1122. doi: 10.1111/plb.12361. [DOI] [PubMed] [Google Scholar]
- 307.Kries H, Caputi L, Stevenson CE, Kamileen MO, Sherden NH, Geu-Flores F, Lawson DM, O’Connor SE. Structural Determinants of Reductive Terpene Cyclization in Iridoid Biosynthesis. Nat Chem Biol. 2016;12(1):6–8. doi: 10.1038/nchembio.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Dittrich H, Kutchan TM. Molecular Cloning, Expression, and Induction of Berberine Bridge Enzyme, an Enzyme Essential to the Formation of Benzophenanthridine Alkaloids in the Response of Plants to Pathogenic Attack. Proc Natl Acad Sci U S A. 1991;88:9969–9973. doi: 10.1073/pnas.88.22.9969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Facchini PJ, Penzes C, Johnson AG, Bull D. Molecular Characterization of Berberine Bridge Enzyme Genes from. Opium Poppy Plant Physiol. 1996;112(4):1669–1677. doi: 10.1104/pp.112.4.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Gaweska HM, Roberts KM, Fitzpatrick PF. Isotope Effects Suggest a Stepwise Mechanism for Berberine Bridge Enzyme. Biochemistry. 2012;51(37):7342–7347. doi: 10.1021/bi300887m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Winkler A, Łyskowski A, Riedl S, Puhl M, Kutchan TM, Macheroux P, Gruber K. A Concerted Mechanism for Berberine Bridge Enzyme. Nat Chem Biol. 2008;4(12):739–741. doi: 10.1038/nchembio.123. [DOI] [PubMed] [Google Scholar]
- 312.Kutchan TM, Dirtrich H. Characterization and Mechanism of the Berberine Bridge Enzyme, a Covalently Flavinylated Oxidase of Benzophenanthridine Alkaloid Biosynthesis in Plants. J Biol Chem. 1995;270(41):24475–24481. doi: 10.1074/jbc.270.41.24475. [DOI] [PubMed] [Google Scholar]
- 313.Wallner S, Winkler A, Riedl S, Dully C, Horvath S, Gruber K, MacHeroux P. Catalytic and Structural Role of a Conserved Active Site Histidine in Berberine Bridge Enzyme. Biochemistry. 2012;51(31):6139–6147. doi: 10.1021/bi300411n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Winkler A, Puhl M, Weber HP, Kutchan TM, Gruber K, Macheroux P. Berberine Bridge Enzyme Catalyzes the Six Electron Oxidation of (S)-Reticuline to Dehydroscoulerine. Phytochemistry. 2009;70(9):1092–1097. doi: 10.1016/j.phytochem.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 315.Winkler A, Kutchan TM, Macheroux P. 6-S-Cysteinylation of Bi-Covalently Attached FAD in Berberine Bridge Enzyme Tunes the Redox Potential for Optimal Activity. J Biol Chem. 2007;282(33):24437–24443. doi: 10.1074/jbc.M703642200. [DOI] [PubMed] [Google Scholar]
- 316.Baccile JA, Spraker JE, Le HH, Brandenburger E, Gomez C, Bok JW, Macheleidt J, Brakhage AA, Hoffmeister D, Keller NP, et al. Plant-Like Biosynthesis of Isoquinoline Alkaloids in Aspergillus Fumigatus. Nat Chem Biol. 2016;12(6):419–424. doi: 10.1038/nchembio.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Shoyama Y, Tamada T, Kurihara K, Takeuchi A, Taura F, Arai S, Blaber M, Shoyama Y, Morimoto S, Kuroki R. Structure and Function of Δ1-Tetrahydrocannabinolic Acid (THCA) Synthase, the Enzyme Controlling the Psychoactivity of Cannabis Sativa. J Mol Biol. 2012;423(1):96–105. doi: 10.1016/j.jmb.2012.06.030. [DOI] [PubMed] [Google Scholar]
- 318.Tsuchiya K, Yamazaki T, Takeuchi Y, Oishi T. Studies on T-2636 Antibiotics. IV. In Vitro and in Vivo Antibacterial Activity of T-2636 Antibiotics. J Antibiot (Tokyo) 1971;24(1):29–41. doi: 10.7164/antibiotics.24.29. [DOI] [PubMed] [Google Scholar]
- 319.Harada S. Studies on Lankacidin-Group (T-2636) Antibiotics. VI. Chemical Structures of Lankacidin-Group Antibiotics. II. Chem Pharm Bull (Tokyo) 1975;23(10):2201–2210. doi: 10.1248/cpb.23.2201. [DOI] [PubMed] [Google Scholar]
- 320.Harada S, Kishi T. Studies on Lankacidin-Group (T-2636) Antibiotics. V. Chemical Structures of Lankacidin-Group Antibiotics. 1. Chem Pharm Bull (Tokyo) 1974;22(1):99–108. doi: 10.1248/cpb.22.99. [DOI] [PubMed] [Google Scholar]
- 321.Uramoto M, Otake N, Ogawa Y, Yonehara H. The Structures of Bundlin A (Lankacidin) and Bundlin B. Tetrahedron Lett. 1969;(27):2249–2254. doi: 10.1016/s0040-4039(01)88133-8. [DOI] [PubMed] [Google Scholar]
- 322.Harada S, Higashide E, Fugono T, Kishi T. Isolation and Structures of T-2636 Antibiotics. Tetrahedron Lett. 1969;(27):2239–2244. doi: 10.1016/s0040-4039(01)88131-4. [DOI] [PubMed] [Google Scholar]
- 323.Hayashi T, Suenaga I, Narukawa N, Yamazaki T. In Vitro and in Vivo Activities of Sedecamycin Against Treponema Hyodysenteriae. Antimicrob Agents Chemother. 1988;32(4):458–461. doi: 10.1128/aac.32.4.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Arakawa K, Sugino F, Kodama K, Ishii T, Kinashi H. Cyclization Mechanism for the Synthesis of Macrocyclic Antibiotic Lankacidin in Streptomyces Rochei. Chem Biol. 2005;12(2):249–256. doi: 10.1016/j.chembiol.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 325.Tokuoka M, Seshime Y, Fujii I, Kitamoto K, Takahashi T, Koyama Y. Identification of a Novel Polyketide Synthase-Nonribosomal Peptide Synthetase (PKS-NRPS) Gene Required for the Biosynthesis of Cyclopiazonic Acid in Aspergillus Oryzae. Fungal Genet Biol. 2008;45(12):1608–1615. doi: 10.1016/j.fgb.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 326.Chang PK, Horn BW, Dorner JW. Clustered Genes Involved in Cyclopiazonic Acid Production Are Next to the Aflatoxin Biosynthesis Gene Cluster in Aspergillus Flavus. Fungal Genet Biol. 2009;46(2):176–182. doi: 10.1016/j.fgb.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 327.Seidler NW, Jona I, Vegh M, Martonosi A. Cyclopiazonic Acid is a Specific Inhibitor of the Ca2+-ATPase of Sarcoplasmic Reticulum. J Biol Chem. 1989;264(30):17816–17823. [PubMed] [Google Scholar]
- 328.Liu X, Walsh CT. Cyclopiazonic Acid Biosynthesis in Aspergillus sp.: Characterization of a Reductase-Like R* Domain in Cyclopiazonate Synthetase that Forms and Releases Cyclo-Acetoacetyl-L-Tryptophan. Biochemistry. 2009;48(36):8746–8757. doi: 10.1021/bi901123r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Liu X, Walsh CT. Characterization of Cyclo-Acetoacetyl-L-Tryptophan Dimethylallyltransferase in Cyclopiazonic Acid Biosynthesis: Substrate Promiscuity and Site Directed Mutagenesis Studies. Biochemistry. 2009;48(46):11032–11044. doi: 10.1021/bi901597j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Kenney WC, Edmondson DE, Singer TP, Steenkamp DJ, Schabort JC. Identification and Properties of the Covalently Bound Flavin of Beta-Cyclopiazonate Oxidocyclase. Biochemistry. 1976;15(22):4931–4935. doi: 10.1021/bi00667a028. [DOI] [PubMed] [Google Scholar]
- 331.Schabort JC, Wilkens DC, Holzapfel CW, Potgieter DJJ, Neitz AW. Beta-Cyclopiazonate Oxidocyclase from Penicillium Cyclopium. 1. Assay Methods, Isolation and Purification. Biochim Biophys Acta. 1971;250(2):311–328. doi: 10.1016/0005-2744(71)90188-4. [DOI] [PubMed] [Google Scholar]
- 332.Steenkamp DJ, Schabort JC, Ferreira NP. Beta-Cyclopiazonate Oxidocyclase from Penicillium Cyclopium. 3. Preliminary Studies on the Mechanism of Action. Biochim Biophys Acta. 1973;309(2):440–456. doi: 10.1016/0005-2744(73)90042-9. [DOI] [PubMed] [Google Scholar]
- 333.Mantovani SM, Moore BS. Flavin-Linked Oxidase Catalyzes Pyrrolizine Formation of Dichloropyrrole-Containing Polyketide Extender Unit in Chlorizidine A. J Am Chem Soc. 2013;135(48):18032–18035. doi: 10.1021/ja409520v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Alvarez-Mico X, Jensen PR, Fenical W, Hughes CC. Chlorizidine, a Cytotoxic 5H-Pyrrolo[2,1-a]Isoindol-5-One-Containing Alkaloid from a Marine Streptomyces sp. Org Lett. 2013;15(5):988–991. doi: 10.1021/ol303374e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Oman TJ, van der Donk WA. Follow the Leader: the Use of Leader Peptides to Guide Natural Product Biosynthesis. Nat Chem Biol. 2010;6(1):9–18. doi: 10.1038/nchembio.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Donia MS, Ravel J, Schmidt EW. A Global Assembly Line for Cyanobactins. Nat Chem Biol. 2008;4(6):341–343. doi: 10.1038/nchembio.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Melby JO, Nard NJ, Mitchell DA. Thiazole/Oxazole-Modified Microcins: Complex Natural Products from Ribosomal Templates. Curr Opin Chem Biol. 2011;15(3):369–378. doi: 10.1016/j.cbpa.2011.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Ireland CM, Durso AR, Newman RA, Hacker MP. Antineoplastic Cyclic Peptides from the Marine Tunicate Lissoclinum Patella. J Org Chem. 1982;47(10):1807–1811. [Google Scholar]
- 339.Kim MY, Vankayalapati H, Shin-Ya K, Wierzba K, Hurley LH. Telomestatin, a Potent Telomerase Inhibitor that Interacts Quite Specifically with the Human Telomeric Intramolecular G-Quadruplex. J Am Chem Soc. 2002;124(10):2098–2099. doi: 10.1021/ja017308q. [DOI] [PubMed] [Google Scholar]
- 340.Bagley MC, Dale JW, Merritt EA, Xiong X. Thiopeptide Antibiotics. Chem Rev. 2005;105(2):685–714. doi: 10.1021/cr0300441. [DOI] [PubMed] [Google Scholar]
- 341.Dunbar KL, Mitchell DA. Revealing Nature’s Synthetic Potential Through the Study of Ribosomal Natural Product Biosynthesis. ACS Chem Biol. 2013;8(3):473–487. doi: 10.1021/cb3005325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Dunbar KL, Chekan JR, Cox CL, Burkhart BJ, Nair SK, Mitchell DA. Discovery of a New ATP-Binding Motif Involved in Peptidic Azoline Biosynthesis. Nat Chem Biol. 2014;10(10):823–829. doi: 10.1038/nchembio.1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Dunbar KL, Melby JO, Mitchell DA. YcaO Domains Use ATP to Activate Amide Backbones During Peptide Cyclodehydrations. Nat Chem Biol. 2012;8(6):569–575. doi: 10.1038/nchembio.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Wever WJ, Bogart JW, Baccile JA, Chan AN, Schroeder FC, Bowers AA. Chemoenzymatic Synthesis of Thiazolyl Peptide Natural Products Featuring an Enzyme-Catalyzed Formal [4 + 2] Cycloaddition. J Am Chem Soc. 2015;137(10):3494–3497. doi: 10.1021/jacs.5b00940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Wieland Brown LC, Acker MG, Clardy J, Walsh CT, Fischbach MA. Thirteen Posttranslational Modifications Convert a 14-Residue Peptide into the Antibiotic Thiocillin. Proc Natl Acad Sci U S A. 2009;106(8):2549–2553. doi: 10.1073/pnas.0900008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Kemp M, Go YM, Jones DP. Nonequilibrium Thermodynamics of Thiol/Disulfide Redox Systems: A Perspective on Redox Systems Biology. Free Radic Biol Med. 2008;44(6):921–937. doi: 10.1016/j.freeradbiomed.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Rahman I, Kode A, Biswas SK. Assay for Quantitative Determination of Glutathione and Glutathione Disulfide Levels Using Enzymatic Recycling Method. Nat Protoc. 2006;1(6):3159–3165. doi: 10.1038/nprot.2006.378. [DOI] [PubMed] [Google Scholar]
- 348.Grant C, Rahman F, Piekarz R, Peer C, Frye R, Robey RW, Gardner ER, Figg WD, Batest SE. Romidepsin: A New Therapy for Cutaneous T-Cell Lymphoma and a Potential Therapy for Solid Tumors. Expert Rev Anticancer Ther. 2010;10(7):997–1008. doi: 10.1586/era.10.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-cell Lymphoma. Oncologist. 2007;12(10):1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 350.Kenig M, Reading C. Holomycin and an Antibiotic (MM 19290) Related to Tunicamycin, Metabolites of Streptomyces Clavuligerus. J Antibiot (Tokyo) 1979;32(6):549–554. doi: 10.7164/antibiotics.32.549. [DOI] [PubMed] [Google Scholar]
- 351.Johnson JR, Bruce WF, Dutcher JD. Gliotoxin, the Antibiotic Principle of Gliocladium Fimbriatum. I. Production, Physical and Biological Properties. J Am Chem Soc. 1943;65:2005–2009. [Google Scholar]
- 352.Gardiner DM, Waring P, Howlett BJ. The Epipolythiodioxopiperazine (ETP) Class of Fungal Toxins: Distribution, Mode of Action, Functions and Biosynthesis. Microbiology. 2005;151(Pt 4):1021–1032. doi: 10.1099/mic.0.27847-0. [DOI] [PubMed] [Google Scholar]
- 353.Scharf DH, Groll M, Habel A, Heinekamp T, Hertweck C, Brakhage AA, Huber EM. Flavoenzyme-Catalyzed Formation of Disulfide Bonds in Natural Products. Angew Chem Int Ed Engl. 2014;53(8):2221–2224. doi: 10.1002/anie.201309302. [DOI] [PubMed] [Google Scholar]
- 354.Wang C, Wesener SR, Zhang H, Cheng YQ. An FAD-Dependent Pyridine Nucleotide-Disulfide Oxidoreductase Is Involved in Disulfide Bond Formation in FK228 Anticancer Depsipeptide. Chem Biol. 2009;16(6):585–593. doi: 10.1016/j.chembiol.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 355.Li B, Walsh CT. Streptomyces Clavuligerus HlmI Is an Intramolecular Disulfide-Forming Dithiol Oxidase in Holomycin Biosynthesis. Biochemistry. 2011;50(21):4615–4622. doi: 10.1021/bi200321c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Scharf DH, Remme N, Heinekamp T, Hortschansky P, Brakhage AA, Hertweck C. Transannular Disulfide Formation in Gliotoxin Biosynthesis and Its Role in Self-Resistance of the Human Pathogen Aspergillus Fumigatus. J Am Chem Soc. 2010;132(29):10136–10141. doi: 10.1021/ja103262m. [DOI] [PubMed] [Google Scholar]
- 357.Giessen TW, Marahiel MA. Rational and Combinatorial Tailoring of Bioactive Cyclic Dipeptides. Front Microbiol. 2015;6:785. doi: 10.3389/fmicb.2015.00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Hara M, Asano K, Kawamoto I, Takiguchi T, Katsumata S, Takahashi K, Nakano H. Leinamycin, a New Antitumor Antibiotic from Streptomyces: Producing Organism, Fermentation and Isolation. J Antibiot (Tokyo) 1989;42(12):1768–1774. doi: 10.7164/antibiotics.42.1768. [DOI] [PubMed] [Google Scholar]
- 359.Tang GL, Cheng YQ, Shen B. Leinamycin Biosynthesis Revealing Unprecedented Architectural Complexity for a Hybrid Polyketide Synthase and Nonribosomal Peptide Synthetase. Chem Biol. 2004;11(1):33–45. doi: 10.1016/j.chembiol.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 360.Watanabe K, Hotta K, Praseuth AP, Koketsu K, Migita A, Boddy CN, Wang CC, Oguri H, Oikawa H. Total Biosynthesis of Antitumor Nonribosomal Peptides in Escherichia coli. Nat Chem Biol. 2006;2(8):423–428. doi: 10.1038/nchembio803. [DOI] [PubMed] [Google Scholar]
- 361.Hotta K, Keegan RM, Ranganathan S, Fang M, Bibby J, Winn MD, Sato M, Lian M, Watanabe K, Rigden DJ, et al. Conversion of a Disulfide Bond into a Thioacetal Group During Echinomycin Biosynthesis. Angew Chem Int Ed Engl. 2014;53(3):824–828. doi: 10.1002/anie.201307404. [DOI] [PubMed] [Google Scholar]
- 362.Jiang H, Haltli B, Feng X, Cai P, Summers M, Lotvin J, He M. Investigation of the Biosynthesis of the Pipecolate Moiety of Neuroprotective Polyketide Meridamycin. J Antibiot (Tokyo) 2011;64(8):533–538. doi: 10.1038/ja.2011.45. [DOI] [PubMed] [Google Scholar]
- 363.Gatto GJ, Boyne MT, Kelleher NL, Walsh CT. Biosynthesis of Pipecolic Acid by RapL, a Lysine Cyclodeaminase Encoded in the Rapamycin Gene Cluster. J Am Chem Soc. 2006;128(11):3838–3847. doi: 10.1021/ja0587603. [DOI] [PubMed] [Google Scholar]
- 364.Sehgal SN, Baker H, Vezina C. Rapamycin (AY-22,989), a New Antifungal Antibiotic. II. Fermentation, Isolation and Characterization. J Antibiot (Tokyo) 1975;28(10):727–732. doi: 10.7164/antibiotics.28.727. [DOI] [PubMed] [Google Scholar]
- 365.Tanaka H, Kuroda A, Marusawa H, Hatanaka H, Kino T, Goto T, Hashimoto M, Taga T. Structure of FK506, a Novel Immunosuppressant Isolated from Streptomyces. J Am Chem Soc. 1987;109(16):5031–5033. [Google Scholar]
- 366.Salituro GM, Zink DL, Dahl A, Nielsen J, Wu E, Huang LY, Kastner C, Dumont FJ. Meridamycin: A Novel Nonimmunosuppressive FKBP12 Ligand from Streptomyces Hygroscopicus. Tetrahedron Lett. 1995;36(7):997–1000. [Google Scholar]
- 367.Fehr T, Sanglier JJ, Schuler W, Gschwind L, Ponelle M, Schilling W, Wioland C. Antascomicins A, B, C, D and E. Novel FKBP12 Binding Compounds from a Micromonospora Strain. J Antibiot (Tokyo) 1996;49(3):230–233. doi: 10.7164/antibiotics.49.230. [DOI] [PubMed] [Google Scholar]
- 368.Sun Y, Hong H, Samboskyy M, Mironenko T, Leadlay PF, Haydock SF. Organization of the Biosynthetic Gene Cluster in Streptomyces sp. DSM 4137 for the Novel Neuroprotectant Polyketide Meridamycin. Microbiology. 2006;152(Pt12):3507–3515. doi: 10.1099/mic.0.29176-0. [DOI] [PubMed] [Google Scholar]
- 369.Molnár I, Aparicio JF, Haydock SF, Khaw LE, Schwecke T, König A, Staunton J, Leadlay PF. Organisation of the Biosynthetic Gene Cluster for Rapamycin in Streptomyces Hygroscopicus: Analysis of Genes Flanking the Polyketide Synthase. Gene. 1996;169(1):1–7. doi: 10.1016/0378-1119(95)00799-7. [DOI] [PubMed] [Google Scholar]
- 370.He M, Haltli B, Summers M, Feng X, Hucul J. Isolation and Characterization of Meridamycin Biosynthetic Gene Cluster from Streptomyces sp. NRRL 30748 Gene. 2006;377:109–118. doi: 10.1016/j.gene.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 371.Schwecke T, Aparicio JF, Molnár I, König A, Khaw LE, Haydock SF, Oliynyk M, Caffrey P, Cortés J, Lester JB, et al. The Biosynthetic Gene Cluster for the Polyketide Immunosuppressant Rapamycin. Proc Natl Acad Sci U S A. 1995;92(17):7839–7843. doi: 10.1073/pnas.92.17.7839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Motamedi H, Shafiee A. The Biosynthetic Gene Cluster for the Macrolactone Ring of the Immunosuppressant FK506. Eur J Biochem. 1998;256(3):528–534. doi: 10.1046/j.1432-1327.1998.2560528.x. [DOI] [PubMed] [Google Scholar]
- 373.Schardl CL, Panaccione DG, Tudzynski P. Ergot Alkaloids - Biology and Molecular Biology. Alkaloids Chem Biol. 2006;63:45–86. doi: 10.1016/s1099-4831(06)63002-2. [DOI] [PubMed] [Google Scholar]
- 374.Mead B, Morgan H, Mann-Knowlton A, Tedeschi L, Sloan C, Lang S, Hines C, Gragg M, Stofer J, Riemann K, et al. Reveromycin A-Induced Apoptosis in Osteoclasts Is Not Accompanied by Necrosis. J Cell Biochem. 2015;116(8):1646–1657. doi: 10.1002/jcb.25125. [DOI] [PubMed] [Google Scholar]
- 375.Osada H, Koshino H, Isono K, Takahashi H, Kawanishi G. Reveromycin A, a New Antibiotic Which Inhibits the Mitogenic Activity of Epidermal Growth Factor. J Antibiot (Tokyo) 1991;44(2):259–261. doi: 10.7164/antibiotics.44.259. [DOI] [PubMed] [Google Scholar]
- 376.Takahashi S, Toyoda A, Sekiyama Y, Takagi H, Nogawa T, Uramoto M, Suzuki R, Koshino H, Kumano T, Panthee S, et al. Reveromycin A Biosynthesis Uses RevG and RevJ for Stereospecific Spiroacetal Formation. Nat Chem Biol. 2011;7(7):461–468. doi: 10.1038/nchembio.583. [DOI] [PubMed] [Google Scholar]
- 377.Li W, Ju J, Rajski SR, Osada H, Shen B. Characterization of the Tautomycin Biosynthetic Gene Cluster from Streptomyces Spiroverticillatus Unveiling New Insights into Dialkylmaleic Anhydride and Polyketide Biosynthesis. J Biol Chem. 2008;283(42):28607–28617. doi: 10.1074/jbc.M804279200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Sun P, Zhao Q, Yu F, Zhang H, Wu Z, Wang Y, Wang Y, Zhang Q, Liu W. Spiroketal Formation and Modification in Avermectin Biosynthesis Involves a Dual Activity of AveC. J Am Chem Soc. 2013;135(4):1540–1548. doi: 10.1021/ja311339u. [DOI] [PubMed] [Google Scholar]
- 379.Frank B, Knauber J, Steinmetz H, Scharfe M, Blöcker H, Beyer S, Müller R. Spiroketal Polyketide Formation in Sorangium: Identification and Analysis of the Biosynthetic Gene Cluster for the Highly Cytotoxic Spirangienes. Chem Biol. 2007;14(2):221–233. doi: 10.1016/j.chembiol.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 380.Frisvad JC, Rank C, Nielsen KF, Larsen TO. Metabolomics of Aspergillus Fumigatus. Med Mycol. 2009;47:S53–S71. doi: 10.1080/13693780802307720. [DOI] [PubMed] [Google Scholar]
- 381.Ames BD, Haynes SW, Gao X, Evans BS, Kelleher NL, Tang Y, Walsh CT. Complexity Generation in Fungal Peptidyl Alkaloid Biosynthesis: Oxidation of Fumiquinazoline A to the Heptacyclic Hemiaminal Fumiquinazoline C by the Flavoenzyme Af12070 from Aspergillus Fumigatus. Biochemistry. 2011;50(40):8756–8769. doi: 10.1021/bi201302w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Gao X, Chooi YH, Ames BD, Wang P, Walsh CT, Tang Y. Fungal Indole Alkaloid Biosynthesis: Genetic and Biochemical Investigation of the Tryptoquialanine Pathway in Penicillium Aethiopicum. J Am Chem Soc. 2011;133(8):2729–2741. doi: 10.1021/ja1101085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Strack D, Vogt T, Schliemann W. Recent Advances in Betalain Research. Phytochemistry. 2003;62(3):247–269. doi: 10.1016/s0031-9422(02)00564-2. [DOI] [PubMed] [Google Scholar]
- 384.Molitor C, Mauracher SG, Pargan S, Mayer RL, Halbwirth H, Rompel A. Latent and Active Aurone Synthase from Petals of C. Grandiflora: A Polyphenol Oxidase with Unique Characteristics. Planta. 2015;242(3):519–537. doi: 10.1007/s00425-015-2261-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Molitor C, Mauracher SG, Rompel A. Crystallization and Preliminary Crystallographic Analysis of Latent, Active and Recombinantly Expressed Aurone Synthase, a Polyphenol Oxidase, from Coreopsis Grandiflora. Acta Crystallogr F Struct Biol Commun. 2015;71(Pt6):746–751. doi: 10.1107/S2053230X15007542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Kaintz C, Molitor C, Thill J, Kampatsikas I, Michael C, Halbwirth H, Rompel A. Cloning and Functional Expression in E. coli of a Polyphenol Oxidase Transcript from Coreopsis Grandiflora Involved in Aurone Formation. FEBS lett. 2014;588(18):3417–3426. doi: 10.1016/j.febslet.2014.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Elsebai MF, Saleem M, Tejesvi MV, Kajula M, Mattila S, Mehiri M, Turpeinen A, Pirttila AM. Fungal Phenalenones: Chemistry, Biology, Biosynthesis and Phylogeny. Nat Prod Rep. 2014;31(5):628–645. doi: 10.1039/c3np70088g. [DOI] [PubMed] [Google Scholar]
- 388.Gao S-S, Duan A, Xu W, Yu P, Hang L, Houk KN, Tang Y. Phenalenone Polyketide Cyclization Catalyzed by Fungal Polyketide Synthase and Flavin-Dependent Monooxygenase. J Am Chem Soc. 2016;138(12):4249–4259. doi: 10.1021/jacs.6b01528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Simpson TJ. C-13 Nuclear Magnetic Resonance Structural and Biosynthetic Studies on Deoxyherqueinone and Herqueichrysin, Phenalenone Metabolites of Penicillium Herquei. J Chem Soc, Perkin Trans 1. 1979;(5):1233–1238. [Google Scholar]
- 390.Simpson TJ. Biosynthesis of Deoxyherqueinone in Penicillium Herquei from [C-13]Acetate and [C-13]Malonate-Assembly Pattern of Acetate into Phenalenone Ring System. J Chem Soc, Chem Commun. 1976;(7):258–260. [Google Scholar]
- 391.Piel J, Hertweck C, Shipley PR, Hunt DM, Newman MS, Moore BS. Cloning, Sequencing and Analysis of the Enterocin Biosynthesis Gene Cluster from the Marine Isolate ‘Streptomyces Maritimus’: Evidence for the Derailment of an Aromatic Polyketide Synthase. Chem Biol. 2000;7(12):943–955. doi: 10.1016/s1074-5521(00)00044-2. [DOI] [PubMed] [Google Scholar]
- 392.Cheng Q, Xiang L, Izumikawa M, Meluzzi D, Moore BS. Enzymatic Total Synthesis of Enterocin Polyketides. Nat Chem Biol. 2007;3(9):557–558. doi: 10.1038/nchembio.2007.22. [DOI] [PubMed] [Google Scholar]
- 393.Xiang L, Kalaitzis JA, Nilsen G, Chen L, Moore BS. Mutational Analysis of the Enterocin Favorskii Biosynthetic Rearrangement. Org Lett. 2002;4(6):957–960. doi: 10.1021/ol0255155. [DOI] [PubMed] [Google Scholar]
- 394.Xiang L, Kalaitzis JA, Moore BS. EncM, a Versatile Enterocin Biosynthetic Enzyme Involved in Favorskii Oxidative Rearrangement, Aldol Condensation, and Heterocycle Forming Reactions. Proc Natl Acad Sci U S A. 2004;101(44):15609–15614. doi: 10.1073/pnas.0405508101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Teufel R, Miyanaga A, Michaudel Q, Stull F, Louie G, Noel JP, Baran PS, Palfey B, Moore BS. Flavin-Mediated Dual Oxidation Controls an Enzymatic Favorskii-Type Rearrangement. Nature. 2013;503(7477):552–556. doi: 10.1038/nature12643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Teufel R, Stull F, Meehan MJ, Michaudel Q, Dorrestein PC, Palfey B, Moore BS. Biochemical Establishment and Characterization of EncM’s Flavin-N5-Oxide Cofactor. J Am Chem Soc. 2015;137(25):8078–8085. doi: 10.1021/jacs.5b03983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Fehr T, Kallen J, Oberer L, Sanglier JJ, Schilling W. Sanglifehrins A, B, C and D, Novel Cyclophilin-Binding Compounds Isolated from Streptomyces sp. A92-308110. II. Structure Elucidation, Stereochemistry and Physico-Chemical Properties. J Antibiot (Tokyo) 1999;52(5):474–479. doi: 10.7164/antibiotics.52.474. [DOI] [PubMed] [Google Scholar]
- 398.Broberg A, Menkis A, Vasiliauskas R. Kutznerides 1–4, Depsipeptides from the Actinomycete Kutzneria sp. 744 Inhabiting Mycorrhizal Roots of Picea Abies Seedlings. J Nat Prod. 2006;69(1):97–102. doi: 10.1021/np050378g. [DOI] [PubMed] [Google Scholar]
- 399.Umezawa K, Nakazawa K, Uemura T, Ikeda Y, Kondo S, Naganawa H, Kinoshita N, Hashizume H, Hamada M, Takeuchi T, et al. Polyoxypeptin Isolated from Streptomyces: A Bioactive Cyclic Depsipeptide Containing the Novel Amino Acid 3-Hydroxy-3-Methylproline. Tetrahedron Lett. 1998;39(11):1389–1392. [Google Scholar]
- 400.Oh DC, Poulsen M, Currie CR, Clardy J. Dentigerumycin: A Bacterial Mediator of an Ant-Fungus Symbiosis. Nat Chem Biol. 2009;5(6):391–393. doi: 10.1038/nchembio.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Maehr H, Liu CM, Palleroni NJ, Smallheer J, Todaro L, Williams TH, Blount JF. Microbial Products. VIII. Azinothricin, a Novel Hexadepsipeptide Antibiotic. J Antibiot (Tokyo) 1986;39(1):17–25. doi: 10.7164/antibiotics.39.17. [DOI] [PubMed] [Google Scholar]
- 402.Neumann CS, Jiang W, Heemstra JR, Gontang EA, Kolter R, Walsh CT. Biosynthesis of Piperazic Acid Via N5-Hydroxy-Ornithine in Kutzneria spp. 744. ChemBioChem. 2012;13(7):972–976. doi: 10.1002/cbic.201200054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Kunze B, Reichenbach H, Müller R, Hofle G. Aurafuron A and B, New Bioactive Polyketides from Stigmatella Aurantiaca and Archangium Gephyra (Myxobacteria). Fermentation, Isolation, Physico-Chemical Properties, Structure and Biological Activity. J Antibiot (Tokyo) 2005;58(4):244–251. doi: 10.1038/ja.2005.28. [DOI] [PubMed] [Google Scholar]
- 404.Frank B, Wenzel SC, Bode HB, Scharfe M, Blöcker H, Müller R. From Genetic Diversity to Metabolic Unity: Studies on the Biosynthesis of Aurafurones and Aurafuron-Like Structures in Myxobacteria and Streptomycetes. J Mol Biol. 2007;374(1):24–38. doi: 10.1016/j.jmb.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 405.Minami A, Oikawa H. Recent Advances of Diels-Alderases Involved in Natural Product Biosynthesis. J Antibiot (Tokyo) 2016;69(7):500–506. doi: 10.1038/ja.2016.67. [DOI] [PubMed] [Google Scholar]
- 406.Klas K, Tsukamoto S, Sherman DH, Williams RM. Natural Diels-Alderases: Elusive and Irresistable. J Org Chem. 2015;80(23):11672–11685. doi: 10.1021/acs.joc.5b01951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Hauser D, Sigg HP. Isolation and Decomposition of Sordarin. Helv Chim Acta. 1971;54(4):1178–1190. doi: 10.1002/hlca.19710540427. [DOI] [PubMed] [Google Scholar]
- 408.Dominguez JM, Kelly VA, Kinsman OS, Marriott MS, Gomez de las Heras F, Martin JJ. Sordarins: A New Class of Antifungals with Selective Inhibition of the Protein Synthesis Elongation Cycle in Yeasts. Antimicrob Agents Chemother. 1998;42(9):2274–2278. doi: 10.1128/aac.42.9.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Soe R, Mosley RT, Justice M, Nielsen-Kahn J, Shastry M, Merrill AR, Andersen GR. Sordarin Derivatives Induce a Novel Conformation of the Yeast Ribosome Translocation Factor eEF2. J Biol Chem. 2007;282(1):657–666. doi: 10.1074/jbc.M607830200. [DOI] [PubMed] [Google Scholar]
- 410.Borschberg H-J. Juris-Druck und -Verl. 1975. Zur Biogenese des Sordarins. [Google Scholar]
- 411.Kudo F, Matsuura Y, Hayashi T, Fukushima M, Eguchi T. Genome Mining of the Sordarin Biosynthetic Gene Cluster from Sordaria Araneosa Cain ATCC 36386: Characterization of Cycloaraneosene Synthase and GDP-6-Deoxyaltrose Transferase. J Antibiot (Tokyo) 2016;69(7):541–548. doi: 10.1038/ja.2016.40. [DOI] [PubMed] [Google Scholar]
- 412.Ichihara A, Tazaki H, Sakamura S. Solanapyrones A, B and C, Phytotoxic Metabolites from the Fungus Alternaria Solani. Tetrahedron Lett. 1983;24(48):5373–5376. [Google Scholar]
- 413.Kasahara K, Miyamoto T, Fujimoto T, Oguri H, Tokiwano T, Oikawa H, Ebizuka Y, Fujii I. Solanapyrone Synthase, a Possible Diels-Alderase and Iterative Type I Polyketide Dynthase Encoded in a Biosynthetic Gene Cluster from Alternaria Solani. ChemBioChem. 2010;11(9):1245–1252. doi: 10.1002/cbic.201000173. [DOI] [PubMed] [Google Scholar]
- 414.Katayama K, Kobayashi T, Oikawa H, Honma M, Ichihara A. Enzymatic Activity and Partial Purification of Solanapyrone Synthase: First Enzyme Catalyzing Diels-Alder Reaction. Biochim Biophys Acta. 1998;1384(2):387–395. doi: 10.1016/s0167-4838(98)00040-5. [DOI] [PubMed] [Google Scholar]
- 415.Mulder KC, Mulinari F, Franco OL, Soares MS, Magalhaes BS, Parachin NS. Lovastatin Production: From Molecular Basis to Industrial Process Optimization. Biotechnol Adv. 2015;33(6 Pt 1):648–665. doi: 10.1016/j.biotechadv.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 416.Ma SM, Tang Y. Biochemical Characterization of the Minimal Polyketide Synthase Domains in the Lovastatin Nonaketide Synthase LovB. FEBS J. 2007;274(11):2854–2864. doi: 10.1111/j.1742-4658.2007.05818.x. [DOI] [PubMed] [Google Scholar]
- 417.Ma SM, Li JW, Choi JW, Zhou H, Lee KK, Moorthie VA, Xie X, Kealey JT, Da Silva NA, Vederas JC, et al. Complete Reconstitution of a Highly Reducing Iterative Polyketide Synthase. Science. 2009;326(5952):589–592. doi: 10.1126/science.1175602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Kennedy J, Auclair K, Kendrew SG, Park C, Vederas JC, Hutchinson CR. Modulation of Polyketide Synthase Activity by Accessory Proteins During Lovastatin Biosynthesis. Science. 1999;284(5418):1368–1372. doi: 10.1126/science.284.5418.1368. [DOI] [PubMed] [Google Scholar]
- 419.Cacho RA, Thuss J, Xu W, Sanichar R, Gao Z, Nguyen A, Vederas JC, Tang Y. Understanding Programming of Fungal Iterative Polyketide Synthases: The Biochemical Basis for Regioselectivity by the Methyltransferase Domain in the Lovastatin Megasynthase. J Am Chem Soc. 2015;137(50):15688–15691. doi: 10.1021/jacs.5b11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Galm U, Sparks TC. Natural Product Derived Insecticides: Discovery and Development of Spinetoram. J Ind Microbiol Biotechnol. 2016;43(2–3):185–193. doi: 10.1007/s10295-015-1710-x. [DOI] [PubMed] [Google Scholar]
- 421.Kim HJ, Ruszczycky MW, Choi SH, Liu YN, Liu HW. Enzyme-Catalysed [4+2] Cycloaddition Is a Key Step in the Biosynthesis of Spinosyn A. Nature. 2011;473:109–112. doi: 10.1038/nature09981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Fage CD, Isiorho EA, Liu Y, Wagner DT, Liu H-W, Keatinge-Clay AT. The Structure of SpnF, a Standalone Enzyme that Catalyzes [4 + 2] Cycloaddition. Nat Chem Biol. 2015;11(4):256–258. doi: 10.1038/nchembio.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Patel A, Chen Z, Yang Z, Gutiérrez O, Liu H-W, Houk KN, Singleton DA. Dynamically Complex [6+4] and [4+2] Cycloadditions in the Biosynthesis of Spinosyn A. J Am Chem Soc. 2016;138(11):3631–3634. doi: 10.1021/jacs.6b00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Aroyan CE, Dermenci A, Miller SJ. The Rauhut-Currier Reaction: A History and Its Synthetic Application. Tetrahedron. 2009;65(21):4069–4084. [Google Scholar]
- 425.Bister B, Bischoff D, Ströbele M, Riedlinger J, Reicke A, Wolter F, Bull AT, Zahner H, Fiedler HP, Süssmuth RD. Abyssomicin C-A Polycyclic Antibiotic from a Marine Verrucosispora Strain as an Inhibitor of the p-Aminobenzoic Acid/Tetrahydrofolate Biosynthesis Pathway. Angew Chem Int Ed Engl. 2004;43(19):2574–2576. doi: 10.1002/anie.200353160. [DOI] [PubMed] [Google Scholar]
- 426.Byrne MJ, Lees NR, Han L-C, van der Kamp MW, Mulholland AJ, Stach JE, Willis CL, Race PR. The Catalytic Mechanism of a Natural Diels–Alderase Revealed in Molecular Detail. J Am Chem Soc. 2016;138(19):6095–6098. doi: 10.1021/jacs.6b00232. [DOI] [PubMed] [Google Scholar]
- 427.Tian Z, Sun P, Yan Y, Wu Z, Zheng Q, Zhou S, Zhang H, Yu F, Jia X, Chen D, et al. An Enzymatic [4+2] Cyclization Cascade Creates the Pentacyclic Core of Pyrroindomycins. Nat Chem Biol. 2015;11(4):259–265. doi: 10.1038/nchembio.1769. [DOI] [PubMed] [Google Scholar]
- 428.Hashimoto T, Hashimoto J, Teruya K, Hirano T, Shin-ya K, Ikeda H, Liu HW, Nishiyama M, Kuzuyama T. Biosynthesis of Versipelostatin: Identification of an Enzyme-Catalyzed [4+2]-Cycloaddition Required for Macrocyclization of Spirotetronate-Containing Polyketides. J Am Chem Soc. 2015;137(2):572–575. doi: 10.1021/ja510711x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Meninno S, Lattanzi A. Organocatalytic Asymmetric Reactions of Epoxides: Recent Progress. Chemistry. 2016;22(11):3632–3642. doi: 10.1002/chem.201504226. [DOI] [PubMed] [Google Scholar]
- 430.He J, Ling J, Chiu P. Vinyl Epoxides in Organic Synthesis. Chem Rev. 2014;114(16):8037–8128. doi: 10.1021/cr400709j. [DOI] [PubMed] [Google Scholar]
- 431.Thibodeaux CJ, Chang WC, Liu HW. Enzymatic Chemistry of Cyclopropane, Epoxide, and Aziridine Biosynthesis. Chem Rev. 2012;112(3):1681–1709. doi: 10.1021/cr200073d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.de Vries EJ, Janssen DB. Biocatalytic Conversion of Epoxides. Curr Opin Biotechnol. 2003;14(4):414–420. doi: 10.1016/s0958-1669(03)00102-2. [DOI] [PubMed] [Google Scholar]
- 433.Taylor SK. Reactions of Epoxides with Ester, Ketone and Amide Enolates. Tetrahedron. 2000;56(9):1149–1163. [Google Scholar]
- 434.Sienel G, Rieth R, Rowbottom KT. Ullmann’s Encyclopedia of Industrial Chemistry. Wiley-VCH Verlag GmbH & Co. KGaA; 2000. [DOI] [Google Scholar]
- 435.Jacobsen EN. Asymmetric Catalysis of Epoxide Ring-Opening Reactions. Acc Chem Res. 2000;33(6):421–431. doi: 10.1021/ar960061v. [DOI] [PubMed] [Google Scholar]
- 436.Krake SH, Bergmeier SC. Inter- and Intramolecular Reactions of Epoxides and Aziridines with Pi-Nucleophiles. Tetrahedron. 2010;66(37):7337–7360. [Google Scholar]
- 437.Dominguez de Maria P, van Gemert RW, Straathof AJ, Hanefeld U. Biosynthesis of Ethers: Unusual or Common Natural Events? Nat Prod Rep. 2010;27(3):370–392. doi: 10.1039/b809416k. [DOI] [PubMed] [Google Scholar]
- 438.Morimoto Y, Iwai T, Kinoshita T. Revised Structure of Squalene-Derived PentaTHF Polyether, Glabrescol, through Its Enantioselective Total Synthesis: Biogenetically Intriguing Cs vs C2 Symmetric Relationships. J Am Chem Soc. 2000;122(29):7124–7125. [Google Scholar]
- 439.Matsuura Y, Shichijo Y, Minami A, Migita A, Oguri H, Watanabe M, Tokiwano T, Watanabe K, Oikawa H. Intriguing Substrate Tolerance of Epoxide Hydrolase Lsd19 Involved in Biosynthesis of the Ionophore Antibiotic Lasalocid A. Org Lett. 2010;12(10):2226–2229. doi: 10.1021/ol100541e. [DOI] [PubMed] [Google Scholar]
- 440.Minami A, Migita A, Inada D, Hotta K, Watanabe K, Oguri H, Oikawa H. Enzymatic Epoxide-Opening Cascades Catalyzed by a Pair of Epoxide Hydrolases in the Ionophore Polyether Biosynthesis. Org Lett. 2011;13(7):1638–1641. doi: 10.1021/ol200100e. [DOI] [PubMed] [Google Scholar]
- 441.Tosin M, Smith L, Leadlay PF. Insights into Lasalocid A Ring Formation by Chemical Chain Termination in Vivo. Angew Chem Int Ed Engl. 2011;50(50):11930–11933. doi: 10.1002/anie.201106323. [DOI] [PubMed] [Google Scholar]
- 442.Minami A, Shimaya M, Suzuki G, Migita A, Shinde SS, Sato K, Watanabe K, Tamura T, Oguri H, Oikawa H. Sequential Enzymatic Epoxidation Involved in Polyether Lasalocid Biosynthesis. J Am Chem Soc. 2012;134(17):7246–7249. doi: 10.1021/ja301386g. [DOI] [PubMed] [Google Scholar]
- 443.Hotta K, Chen X, Paton RS, Minami A, Li H, Swaminathan K, Mathews II, Watanabe K, Oikawa H, Houk KN, et al. Enzymatic Catalysis of Anti-Baldwin Ring Closure in Polyether Biosynthesis. Nature. 2012;483(7389):355–358. doi: 10.1038/nature10865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Wong FT, Hotta K, Chen X, Fang M, Watanabe K, Kim C-Y. Epoxide Hydrolase–Lasalocid A Structure Provides Mechanistic Insight into Polyether Natural Product Biosynthesis. J Am Chem Soc. 2015;137(1):86–89. doi: 10.1021/ja511374k. [DOI] [PubMed] [Google Scholar]
- 445.Gallimore AR, Stark CB, Bhatt A, Harvey BM, Demydchuk Y, Bolanos-Garcia V, Fowler DJ, Staunton J, Leadlay PF, Spencer JB. Evidence for the Role of the MonB Genes in Polyether Ring Formation During Monensin Biosynthesis. Chem Biol. 2006;13(4):453–460. doi: 10.1016/j.chembiol.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 446.Leadlay PF, Staunton J, Oliynyk M, Bisang C, Cortes J, Frost E, Hughes-Thomas ZA, Jones MA, Kendrew SG, Lester JB, et al. Engineering of Complex Polyketide Biosynthesis--Insights from Sequencing of the Monensin Biosynthetic Gene Cluster. J Ind Microbiol Biotechnol. 2001;27(6):360–367. doi: 10.1038/sj.jim.7000204. [DOI] [PubMed] [Google Scholar]
- 447.Minami A, Ose T, Sato K, Oikawa A, Kuroki K, Maenaka K, Oguri H, Oikawa H. Allosteric Regulation of Epoxide Opening Cascades by a Pair of Epoxide Hydrolases in Monensin Biosynthesis. ACS Chem Biol. 2014;9(2):562–569. doi: 10.1021/cb4006485. [DOI] [PubMed] [Google Scholar]
- 448.Sun Y, Zhou X, Dong H, Tu G, Wang M, Wang B, Deng Z. A Complete Gene Cluster from Streptomyces Nanchangensis NS3226 Encoding Biosynthesis of the Polyether Ionophore Nanchangmycin. Chem Biol. 2003;10(5):431–441. doi: 10.1016/s1074-5521(03)00092-9. [DOI] [PubMed] [Google Scholar]
- 449.Raju R, Piggott AM, Barrientos Diaz LX, Khalil Z, Capon RJ. Heronapyrroles A–C: Farnesylated 2-Nitropyrroles from an Australian Marine-Derived Streptomyces sp. Org Lett. 2010;12(22):5158–5161. doi: 10.1021/ol102162d. [DOI] [PubMed] [Google Scholar]
- 450.Schmidt J, Khalil Z, Capon RJ, Stark CB. Heronapyrrole D: A Case of Co-Inspiration of Natural Product Biosynthesis, Total Synthesis and Biodiscovery. Beilstein J Org Chem. 2014;10:1228–1232. doi: 10.3762/bjoc.10.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Gleye C, Raynaud S, Hocquemiller R, Laurens A, Fourneau C, Serani L, Laprévote O, Roblot F, Leboeuf M, Fournet A, et al. Muricadienin, Muridienins and Chatenaytrienins, the Early Precursors of Annonaceous Acetogenins. Phytochemistry. 1998;47(5):749–754. [Google Scholar]
- 452.Hu Y, Cecil ARL, Frank X, Gleye C, Figadere B, Brown RCD. Natural cis-Solamin Is a Mixture of Two Tetra-Epimeric Diastereoisomers: Biosynthetic Implications for Annonaceous Acetogenins. Org Biomol Chem. 2006;4(7):1217–1219. doi: 10.1039/b601943a. [DOI] [PubMed] [Google Scholar]
- 453.Johnson KM, Swenson L, Opipari AW, Reuter R, Zarrabi N, Fierke CA, Börsch M, Glick GD. Mechanistic Basis for Differential Inhibition of the F1Fo-ATPase by Aurovertin. Biopolymers. 2009;91(10):830–840. doi: 10.1002/bip.21262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Huang T-C, Chang H-Y, Hsu C-H, Kuo W-H, Chang K-J, Juan H-F. Targeting Therapy for Breast Carcinoma by ATP Synthase Inhibitor Aurovertin B. J Proteome Res. 2008;7(4):1433–1444. doi: 10.1021/pr700742h. [DOI] [PubMed] [Google Scholar]
- 455.Mao XM, Zhan ZJ, Grayson MN, Tang MC, Xu W, Li YQ, Yin WB, Lin HC, Chooi YH, Houk KN, et al. Efficient Biosynthesis of Fungal Polyketides Containing the Dioxabicyclo-Octane Ring System. J Am Chem Soc. 2015;137(37):11904–11907. doi: 10.1021/jacs.5b07816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Thoma R, Schulz-Gasch T, D’Arcy B, Benz J, Aebi J, Dehmlow H, Hennig M, Stihle M, Ruf A. Insight into Steroid Scaffold Formation from the Structure of Human Oxidosqualene Cyclase. Nature. 2004;432(7013):118–122. doi: 10.1038/nature02993. [DOI] [PubMed] [Google Scholar]
- 457.Chen N, Wang S, Smentek L, Hess BA, Jr, Wu R. Biosynthetic Mechanism of Lanosterol: Cyclization. Angew Chem Int Ed Engl. 2015;54(30):8693–8696. doi: 10.1002/anie.201501986. [DOI] [PubMed] [Google Scholar]
- 458.Nes WD. Biosynthesis of Cholesterol and Other Sterols. Chem Rev. 2011;111(10):6423–6451. doi: 10.1021/cr200021m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Rees HH, Goad LJ, Goodwin TW. 2,3-Oxidosqualene Cycloartenol Cyclase from Ochromonas Malhamensis. Biochim Biophys Acta. 1969;176(4):892–894. [PubMed] [Google Scholar]
- 460.Dean PDG, DemontelPr Bloch K, Corey EJ. A Soluble 2,3,Oxidosqualene Sterol Cyclase. J Biol Chem. 1967;242(12):3014–3015. [PubMed] [Google Scholar]
- 461.Wendt KU, Poralla K, Schulz GE. Structure and Function of a Squalene Cyclase. Science. 1997;277(5333):1811–1815. doi: 10.1126/science.277.5333.1811. [DOI] [PubMed] [Google Scholar]
- 462.Hoshino T, Sato T. Squalene-Hopene Cyclase: Catalytic Mechanism and Substrate Recognition. Chem Commun (camb) 2002;(4):291–301. doi: 10.1039/b108995c. [DOI] [PubMed] [Google Scholar]
- 463.Gas-Pascual E, Berna A, Bach TJ, Schaller H. Plant Oxidosqualene Metabolism: Cycloartenol Synthase-Dependent Sterol Biosynthesis in Nicotiana Benthamiana. PloS ONE. 2014;9(10):e109156. doi: 10.1371/journal.pone.0109156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Ohyama K, Suzuki M, Kikuchi J, Saito K, Muranaka T. Dual Biosynthetic Pathways to Phytosterol Via Cycloartenol and Lanosterol in Arabidopsis. Proc Natl Acad Sci U S A. 2009;106(3):725–730. doi: 10.1073/pnas.0807675106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Shibuya M, Adachi S, Ebizuka Y. Cucurbitadienol Synthase, the First Committed Enzyme for Cucurbitacin Biosynthesis, Is a Distinct Enzyme from Cycloartenol Synthase for Phytosterol Biosynthesis. Tetrahedron. 2004;60(33):6995–7003. [Google Scholar]
- 466.Takase S, Saga Y, Kurihara N, Naraki S, Kuze K, Nakata G, Araki T, Kushiro T. Control of the 1,2-Rearrangement Process by Oxidosqualene Cyclases During Triterpene Biosynthesis. Org Biomol Chem. 2015;13(26):7331–7336. doi: 10.1039/c5ob00714c. [DOI] [PubMed] [Google Scholar]
- 467.Shan H, Segura MJR, Wilson WK, Lodeiro S, Matsuda SPT. Enzymatic Cyclization of Dioxidosqualene to Heterocyclic Triterpenes. J Am Chem Soc. 2005;127(51):18008–18009. doi: 10.1021/ja055822g. [DOI] [PubMed] [Google Scholar]
- 468.Saleem M. Lupeol, a Novel Anti-Inflammatory and Anti-Cancer Dietary Triterpene. Cancer Lett. 2009;285(2):109–115. doi: 10.1016/j.canlet.2009.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Hoshino T, Miyahara Y, Hanaoka M, Takahashi K, Kaneko I. β-Amyrin Biosynthesis: The Methyl-30 Group of (3S)-2,3-Oxidosqualene Is More Critical to Its Correct Folding To Generate the Pentacyclic Scaffold than the Methyl-24 Group. Chemistry. 2015;21(44):15769–15784. doi: 10.1002/chem.201502389. [DOI] [PubMed] [Google Scholar]
- 470.Hammer SC, Marjanovic A, Dominicus JM, Nestl BM, Hauer B. Squalene Hopene Cyclases Are Protonases for Stereoselective Brønsted Acid Catalysis. Nat Chem Biol. 2015;11(2):121–126. doi: 10.1038/nchembio.1719. [DOI] [PubMed] [Google Scholar]
- 471.Rudi A, Stein Z, Goldberg I, Yosief T, Kashman Y, Schleyer M. Yardenone and Abudinol Two New Triterpenes from the Marine Sponge Ptilocaulis Spiculifer. Tetrahedron Lett. 1998;39(11):1445–1448. [Google Scholar]
- 472.Domingo V, Arteaga JF, Quilez del Moral JF, Barrero AF. Unusually Cyclized Triterpenes: Occurrence, Biosynthesis and Chemical Synthesis. Nat Prod Rep. 2009;26(1):115–134. doi: 10.1039/b801470c. [DOI] [PubMed] [Google Scholar]
- 473.Boone MA, Tong R, McDonald FE, Lense S, Cao R, Hardcastle KI. Biomimetic Syntheses from Squalene-Like Precursors: Synthesis of Ent-Abudinol B and Reassessment of the Structure of Muzitone. J Am Chem Soc. 2010;132(14):5300–5308. doi: 10.1021/ja1006806. [DOI] [PubMed] [Google Scholar]
- 474.Tong R, McDonald FE. Mimicking Biosynthesis: Total Synthesis of the Triterpene Natural Product Abudinol B from a Squalene-Like Precursor. Angew Chem Int Ed Engl. 2008;47(23):4377–4379. doi: 10.1002/anie.200800749. [DOI] [PubMed] [Google Scholar]
- 475.Tong R, Valentine JC, McDonald FE, Cao R, Fang X, Hardcastle KI. Total Syntheses of Durgamone, Nakorone, and Abudinol B Via Biomimetic Oxa- and Carbacyclizations. J Am Chem Soc. 2007;129(5):1050–1051. doi: 10.1021/ja068826+. [DOI] [PubMed] [Google Scholar]
- 476.Li H, Sun Y, Zhang Q, Zhu Y, Li SM, Li A, Zhang C. Elucidating the Cyclization Cascades in Xiamycin Biosynthesis by Substrate Synthesis and Enzyme Characterizations. Org Lett. 2015;17(2):306–309. doi: 10.1021/ol503399b. [DOI] [PubMed] [Google Scholar]
- 477.Nozawa K, Nakajima S, Kawai K, Udagawa S. Isolation and Structures of Indoloditerpenes, Possible Biosynthetic Intermediates to the Tremorgenic Mycotoxin, Paxilline, from Emericella Striata. J Chem Soc, Perkin Trans 1. 1988;(9):2607–2610. [Google Scholar]
- 478.Hosoe T, Itabashi T, Kobayashi N, Udagawa S, Kawai K. Three New Types of Indoloditerpenes, Emindole PA-PC, from Emericella Purpurea. Revision of the Structure of Emindole PA. Chem Pharm Bull (Tokyo) 2006;54(2):185–187. doi: 10.1248/cpb.54.185. [DOI] [PubMed] [Google Scholar]
- 479.Tagami K, Liu C, Minami A, Noike M, Isaka T, Fueki S, Shichijo Y, Toshima H, Gomi K, Dairi T, et al. Reconstitution of Biosynthetic Machinery for Indole-Diterpene Paxilline in Aspergillus Oryzae. J Am Chem Soc. 2013;135(4):1260–1263. doi: 10.1021/ja3116636. [DOI] [PubMed] [Google Scholar]
- 480.Tang M-C, Lin H-C, Li D, Zou Y, Li J, Xu W, Cacho RA, Hillenmeyer ME, Garg NK, Tang Y. Discovery of Unclustered Fungal Indole Diterpene Biosynthetic Pathways through Combinatorial Pathway Reassembly in Engineered Yeast. J Am Chem Soc. 2015;137(43):13724–13727. doi: 10.1021/jacs.5b06108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Geris R, Simpson TJ. Meroterpenoids Produced by Fungi. Nat Prod Rep. 2009;26(8):1063–1094. doi: 10.1039/b820413f. [DOI] [PubMed] [Google Scholar]
- 482.Matsuda Y, Abe I. Biosynthesis of Fungal Meroterpenoids. Nat Prod Rep. 2016;33(1):26–53. doi: 10.1039/c5np00090d. [DOI] [PubMed] [Google Scholar]
- 483.Zhang Y, Li C, Swenson DC, Gloer JB, Wicklow DT, Dowd PF. Novel Antiinsectan Oxalicine Alkaloids from Two Undescribed Fungicolous Penicillium spp. Org Lett. 2003;5(5):773–776. doi: 10.1021/ol0340686. [DOI] [PubMed] [Google Scholar]
- 484.Hayashi A, Arai M, Fujita M, Kobayashi M. Pyripyropenes, Fungal Sesquiterpenes Conjugated with Alpha-Pyrone and Pyridine Moieties, Exhibits Anti-Angiogenic Activity Against Human Umbilical Vein Endothelial Cells. Biol Pharm Bull. 2009;32(7):1261–1265. doi: 10.1248/bpb.32.1261. [DOI] [PubMed] [Google Scholar]
- 485.Ubillas R, Barnes CL, Gracz H, Rottinghaus GE, Tempesta MS. X-Ray Crystal Structure of Oxalicine A, a Novel Alkaloid from Penicillium Oxalicum. J Chem Soc, Chem Commun. 1989;(21):1618–1619. [Google Scholar]
- 486.Abe M, Imai T, Ishii N, Usui M, Okuda T, Oki T. Isolation of an Insecticidal Compound Oxalicine B from Penicillium sp. TAMA 71 and Confirmation of Its Chemical Structure by X-Ray Crystallographic Analysis. J Pest Sci. 2007;32(2):124–127. [Google Scholar]
- 487.Yaegashi J, Romsdahl J, Chiang Y-M, Wang CC. Genome Mining and Molecular Characterization of the Biosynthetic Gene Cluster of a Diterpenic Meroterpenoid, 15-Deoxyoxalicine B, in Penicillium Canescens. Chem Sci. 2015;6(11):6537–6544. doi: 10.1039/c5sc01965f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Itoh T, Tokunaga K, Matsuda Y, Fujii I, Abe I, Ebizuka Y, Kushiro T. Reconstitution of a Fungal Meroterpenoid Biosynthesis Reveals the Involvement of a Novel Family of Terpene Cyclases. Nat Chem. 2010;2(10):858–864. doi: 10.1038/nchem.764. [DOI] [PubMed] [Google Scholar]
- 489.Cueto M, MacMillan JB, Jensen PR, Fenical W. Tropolactones A-D, Four Meroterpenoids from a Marine-Derived Fungus of the Genus Aspergillus. Phytochemistry. 2006;67(16):1826–1831. doi: 10.1016/j.phytochem.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 490.Nielsen KF, Dalsgaard PW, Smedsgaard J, Larsen TO. Andrastins A-D, Penicillium Roqueforti Metabolites Consistently Produced in Blue-Mold-Ripened Cheese. J Agric Food Chem. 2005;53(8):2908–2913. doi: 10.1021/jf047983u. [DOI] [PubMed] [Google Scholar]
- 491.Simpson TJ, Stenzel DJ, Bartlett AJ, O’Brien E, Holker JSE. Studies on Fungal Metabolites. Part 3. 13C N.M.R. Spectral and Structural Studies on Austin and New Related Meroterpenoids from Aspergillus Ustus, Aspergillus Variecolor, and Penicillium Diversum. J Chem Soc, Perkin Trans 1. 1982;(0):2687–2692. [Google Scholar]
- 492.Springer JP, Dorner JW, Cole RJ, Cox RH. Terretonin, a Toxic Compound from Aspergillus Terreus. J Org Chem. 1979;44(26):4852–4854. [Google Scholar]
- 493.Rank C, Phipps RK, Harris P, Fristrup P, Larsen TO, Gotfredsen CH. Novofumigatonin, a New Orthoester Meroterpenoid from Aspergillus Novofumigatus. Org Lett. 2008;10(3):401–404. doi: 10.1021/ol7026834. [DOI] [PubMed] [Google Scholar]
- 494.Matsuda Y, Awakawa T, Abe I. Reconstituted Biosynthesis of Fungal Meroterpenoid Andrastin A. Tetrahedron. 2013;69(38):8199–8204. [Google Scholar]
- 495.Lo HC, Entwistle R, Guo CJ, Ahuja M, Szewczyk E, Hung JH, Chiang YM, Oakley BR, Wang CC. Two Separate Gene Clusters Encode the Biosynthetic Pathway for the Meroterpenoids Austinol and Dehydroaustinol in Aspergillus Nidulans. J Am Chem Soc. 2012;134(10):4709–4720. doi: 10.1021/ja209809t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Guo CJ, Knox BP, Chiang YM, Lo HC, Sanchez JF, Lee KH, Oakley BR, Bruno KS, Wang CC. Molecular Genetic Characterization of a Cluster in A. Terreus for Biosynthesis of the Meroterpenoid Terretonin. Org Lett. 2012;14(22):5684–5687. doi: 10.1021/ol302682z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Simpson TJ, Walkinshaw MD. Anditomin, a New C25 Metabolite from Aspergillus Variecolor. J Chem Soc, Chem Commun. 1981;(17):914–915. [Google Scholar]
- 498.Matsuda Y, Wakimoto T, Mori T, Awakawa T, Abe I. Complete Biosynthetic Pathway of Anditomin: Nature’s Sophisticated Synthetic Route to a Complex Fungal Meroterpenoid. J Am Chem Soc. 2014;136(43):15326–15336. doi: 10.1021/ja508127q. [DOI] [PubMed] [Google Scholar]
- 499.Kusano M, Koshino H, Uzawa J, Fujioka S, Kawano T, Kimura Y. Nematicidal Alkaloids and Related Compounds Produced by the Fungus Penicillium cf. Simplicissimum. Biosci Biotechnol Biochem. 2000;64(12):2559–2568. doi: 10.1271/bbb.64.2559. [DOI] [PubMed] [Google Scholar]
- 500.Kimura Y, Kusano M, Koshino H, Uzawa J, Fujioka S, Tani K. Penigequinolones A and B, Pollen-Growth Inhibitors Produced by Penicilium sp., No. 410. Tetrahedron Lett. 1996;37(28):4961–4964. [Google Scholar]
- 501.Scherlach K, Hertweck C. Discovery of Aspoquinolones A-D, Prenylated Quinoline-2-One Alkaloids from Aspergillus Nidulans, Motivated by Genome Mining. Org Biomol Chem. 2006;4(18):3517–3520. doi: 10.1039/b607011f. [DOI] [PubMed] [Google Scholar]
- 502.Ishikawa N, Tanaka H, Koyama F, Noguchi H, Wang CC, Hotta K, Watanabe K. Non-heme Dioxygenase Catalyzes Atypical Oxidations of 6,7-Bicyclic Systems to Form the 6,6-Quinolone Core of Viridicatin-Type Fungal Alkaloids. Angew Chem Int Ed Engl. 2014;53(47):12880–12884. doi: 10.1002/anie.201407920. [DOI] [PubMed] [Google Scholar]
- 503.Zou Y, Zhan ZJ, Li D, Tang M, Cacho RA, Watanabe K, Tang Y. Tandem Prenyltransferases Catalyze Isoprenoid Elongation and Complexity Generation in Biosynthesis of Quinolone Alkaloids. J Am Chem Soc. 2015;137(15):4980–4983. doi: 10.1021/jacs.5b03022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Uchida R, Imasato R, Yamaguchi Y, Masuma R, Shiomi K, Tomoda H, Omura S. Yaequinolones, New Insecticidal Antibiotics Produced by Penicillium sp. FKI-2140. I. Taxonomy, Fermentation, Isolation and Biological Activity. J Antibiot (Tokyo) 2006;59(10):646–651. doi: 10.1038/ja.2006.86. [DOI] [PubMed] [Google Scholar]
- 505.Uchida R, Imasato R, Tomoda H, Omura S. Yaequinolones, New Insecticidal Antibiotics Produced by Penicillium sp. FKI-2140. II. Structural Elucidation. J Antibiot (Tokyo) 2006;59(10):652–658. doi: 10.1038/ja.2006.87. [DOI] [PubMed] [Google Scholar]
- 506.Zou Y, Garcia-Borràs M, Tang M-C, Hirayama Y, Li D-H, Li L, Watanabe K, Houk KN, Tang Y. Enzyme-Catalyzed Cationic Epoxide Rearrangements in Quinolone Alkaloid Biosynthesis. Nat Chem Biol. 2016 doi: 10.1038/nchembio.2283. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Kunze B, Hofle G, Reichenbach H. The Aurachins, New Quinoline Antibiotics from Myxobacteria: Production, Physico-Chemical and Biological Properties. J Antibiot (Tokyo) 1987;40(3):258–265. doi: 10.7164/antibiotics.40.258. [DOI] [PubMed] [Google Scholar]
- 508.Pistorius D, Li Y, Sandmann A, Müller R. Completing the Puzzle of Aurachin Biosynthesis in Stigmatella Aurantiaca Sg a15. Mol Biosyst. 2011;7(12):3308–3315. doi: 10.1039/c1mb05328k. [DOI] [PubMed] [Google Scholar]
- 509.Willenbring D, Tantillo DJ. Mechanistic Possibilities for Oxetane Formation in the Biosynthesis of Taxol’s D Ring. Russ J Gen Chem. 2008;78(4):723–731. [Google Scholar]
- 510.Guo CJ, Yeh HH, Chiang YM, Sanchez JF, Chang SL, Bruno KS, Wang CC. Biosynthetic Pathway for the Epipolythiodioxopiperazine Acetylaranotin in Aspergillus Terreus Revealed by Genome-Based Deletion Analysis. J Am Chem Soc. 2013;135(19):7205–7213. doi: 10.1021/ja3123653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Gardiner DM, Howlett BJ. Bioinformatic and Expression Analysis of the Putative Gliotoxin Biosynthetic Gene Cluster of Aspergillus Fumigatus. FEMS Microbiol Lett. 2005;248(2):241–248. doi: 10.1016/j.femsle.2005.05.046. [DOI] [PubMed] [Google Scholar]
- 512.Scharf DH, Heinekamp T, Remme N, Hortschansky P, Brakhage AA, Hertweck C. Biosynthesis and Function of Gliotoxin in Aspergillus Fumigatus. Appl Microbiol Biotechnol. 2012;93(2):467–472. doi: 10.1007/s00253-011-3689-1. [DOI] [PubMed] [Google Scholar]
- 513.Walsh CT, Haynes SW, Ames BD, Gao X, Tang Y. Short Pathways to Complexity Generation: Fungal Peptidyl Alkaloid Multicyclic Scaffolds from Anthranilate Building Blocks. ACS Chem Biol. 2013;8(7):1366–1382. doi: 10.1021/cb4001684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Gao X, Haynes SW, Ames BD, Wang P, Vien LP, Walsh CT, Tang Y. Cyclization of Fungal Nonribosomal Peptides by a Terminal Condensation-Like Domain. Nat Chem Biol. 2012;8(10):823–830. doi: 10.1038/nchembio.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Ames BD, Liu X, Walsh CT. Enzymatic Processing of Fumiquinazoline F: A Tandem Oxidative-Acylation Strategy for the Generation of Multicyclic Scaffolds in Fungal Indole Alkaloid Biosynthesis. Biochemistry. 2010;49(39):8564–8576. doi: 10.1021/bi1012029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Larsen TO, Petersen BO, Duus JØ. Lumpidin, a Novel Biomarker of Some Ochratoxin A Producing Penicillia. J Agric Food Chem. 2001;49(10):5081–5084. doi: 10.1021/jf010345g. [DOI] [PubMed] [Google Scholar]
- 517.Wong SM, Musza LL, Kydd GC, Kullnig R, Gillum AM, Cooper R. Fiscalins: New Substance P Inhibitors Produced by the Fungus Neosartorya Fischeri. Taxonomy, Fermentation, Structures, and Biological Properties. J Antibiot (Tokyo) 1993;46(4):545–553. doi: 10.7164/antibiotics.46.545. [DOI] [PubMed] [Google Scholar]
- 518.Fremlin LJ, Piggott AM, Lacey E, Capon RJ. Cottoquinazoline A and Cotteslosins A and B, Metabolites from an Australian Marine-Derived Strain of Aspergillus Versicolor. J Nat Prod. 2009;72(4):666–670. doi: 10.1021/np800777f. [DOI] [PubMed] [Google Scholar]
- 519.Jiao RH, Xu S, Liu JY, Ge HM, Ding H, Xu C, Zhu HL, Tan RX. Chaetominine, a Cytotoxic Alkaloid Produced by Endophytic Chaetomium sp. IFB-E015. Org Lett. 2006;8(25):5709–5712. doi: 10.1021/ol062257t. [DOI] [PubMed] [Google Scholar]
- 520.Goetz MA, Lopez M, Monaghan RL, Chang RS, Lotti VJ, Chen TB. Asperlicin, a Novel Non-Peptidal Cholecystokinin Antagonist from Aspergillus Alliaceus. Fermentation, Isolation and Biological Properties. J Antibiot (Tokyo) 1985;38(12):1633–1637. doi: 10.7164/antibiotics.38.1633. [DOI] [PubMed] [Google Scholar]
- 521.Li S, Finefield JM, Sunderhaus JD, McAfoos TJ, Williams RM, Sherman DH. Biochemical Characterization of NotB as an FAD-Dependent Oxidase in the Biosynthesis of Notoamide Indole Alkaloids. J Am Chem Soc. 2012;134(2):788–791. doi: 10.1021/ja2093212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Cui CB, Kakeya H, Osada H. Spirotryprostatin B, a Novel Mammalian Cell Cycle Inhibitor Produced by Aspergillus Fumigatus. J Antibiot (Tokyo) 1996;49(8):832–835. doi: 10.7164/antibiotics.49.832. [DOI] [PubMed] [Google Scholar]
- 523.Tsunematsu Y, Ishikawa N, Wakana D, Goda Y, Noguchi H, Moriya H, Hotta K, Watanabe K. Distinct Mechanisms for Spiro-Carbon Formation Reveal Biosynthetic Pathway Crosstalk. Nat Chem Biol. 2013;9(12):818–825. doi: 10.1038/nchembio.1366. [DOI] [PubMed] [Google Scholar]
- 524.Li H, Zhang Q, Li S, Zhu Y, Zhang G, Zhang H, Tian X, Zhang S, Ju J, Zhang C. Identification and Characterization of Xiamycin A and Oxiamycin Gene Cluster Reveals an Oxidative Cyclization Strategy Tailoring Indolosesquiterpene Biosynthesis. J Am Chem Soc. 2012;134(21):8996–9005. doi: 10.1021/ja303004g. [DOI] [PubMed] [Google Scholar]
- 525.Raju R, Piggott AM, Conte MM, Capon RJ. Heronamides A-C, New Polyketide Macrolactams from an Australian Marine-Derived Streptomyces sp. A Biosynthetic Case for Synchronized Tandem Electrocyclization. Org Biomol Chem. 2010;8(20):4682–4689. doi: 10.1039/c0ob00267d. [DOI] [PubMed] [Google Scholar]
- 526.Booth TJ, Alt S, Capon RJ, Wilkinson B. Synchronous Intramolecular Cycloadditions of the Polyene Macrolactam Polyketide Heronamide C. Chem Commun (Camb) 2016;52(38):6383–6386. doi: 10.1039/c6cc01930g. [DOI] [PubMed] [Google Scholar]
- 527.Yu P, Patel A, Houk KN. Transannular [6 + 4] and Ambimodal Cycloaddition in the Biosynthesis of Heronamide A. J Am Chem Soc. 2015;137(42):13518–13523. doi: 10.1021/jacs.5b06656. [DOI] [PubMed] [Google Scholar]
- 528.Bloch P, Tamm C, Bollinger P, Petcher TJ, Weber HP. Pseurotin, a New Metabolite of Pseudeurotium Ovalis STOLk Having an Unusual Hetero-Spirocyclic System. Helv Chim Acta. 1976;59(1):133–137. doi: 10.1002/hlca.19760590114. [DOI] [PubMed] [Google Scholar]
- 529.Maiya S, Grundmann A, Li X, Li S-M, Turner G. Identification of a Hybrid PKS/NRPS Required for Pseurotin A Biosynthesis in the Human Pathogen Aspergillus Fumigatus. ChemBioChem. 2007;8(14):1736–1743. doi: 10.1002/cbic.200700202. [DOI] [PubMed] [Google Scholar]
- 530.Tsunematsu Y, Fukutomi M, Saruwatari T, Noguchi H, Hotta K, Tang Y, Watanabe K. Elucidation of Pseurotin Biosynthetic Pathway Points to trans-Acting C-Methyltransferase: Generation of Chemical Diversity. Angew Chem Int Ed Engl. 2014;53(32):8475–8479. doi: 10.1002/anie.201404804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Zou Y, Xu W, Tsunematsu Y, Tang M, Watanabe K, Tang Y. Methylation-Dependent Acyl Transfer between Polyketide Synthase and Nonribosomal Peptide Synthetase Modules in Fungal Natural Product Biosynthesis. Org Lett. 2014;16(24):6390–6393. doi: 10.1021/ol503179v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Yamamoto T, Tsunematsu Y, Hara K, Suzuki T, Kishimoto S, Kawagishi H, Noguchi H, Hashimoto H, Tang Y, Hotta K, et al. Oxidative trans to cis Isomerization of Olefins in Polyketide Biosynthesis. Angew Chem Int Ed Engl. 2016;55(21):6207–6210. doi: 10.1002/anie.201600940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Brash AR, Boeglin WE, Stec DF, Voehler M, Schneider C, Cha JK. Isolation and Characterization of Two Geometric Allene Oxide Isomers Synthesized from 9S-Hydroperoxylinoleic Acid by Cytochrome P450 CYP74C3: STEREOCHEMICAL ASSIGNMENT OF NATURAL FATTY ACID ALLENE OXIDES. J Biol Chem. 2013;288(29):20797–20806. doi: 10.1074/jbc.M113.482521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Oldham ML, Brash AR, Newcomer ME. The Structure of Coral Allene Oxide Synthase Reveals a Catalase Adapted for Metabolism of a Fatty Acid Hydroperoxide. Proc Natl Acad Sci U S A. 2005;102(2):297–302. doi: 10.1073/pnas.0406352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Fujii I, Mori Y, Watanabe A, Kubo Y, Tsuji G, Ebizuka Y. Enzymatic Synthesis of 1,3,6,8-Tetrahydroxynaphthalene Solely from Malonyl Coenzyme A by a Fungal Iterative Type I Polyketide Synthase PKS1. Biochemistry. 2000;39(30):8853–8858. doi: 10.1021/bi000644j. [DOI] [PubMed] [Google Scholar]
- 536.Funa N, Ohnishi Y, Fujii I, Shibuya M, Ebizuka Y, Horinouchi S. A New Pathway for Polyketide Synthesis in Microorganisms. Nature. 1999;400(6747):897–899. doi: 10.1038/23748. [DOI] [PubMed] [Google Scholar]
- 537.Kaysser L, Bernhardt P, Nam SJ, Loesgen S, Ruby JG, Skewes-Cox P, Jensen PR, Fenical W, Moore BS. Merochlorins A-D, Cyclic Meroterpenoid Antibiotics Biosynthesized in Divergent Pathways with Vanadium-Dependent Chloroperoxidases. J Am Chem Soc. 2012;134(29):11988–11991. doi: 10.1021/ja305665f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Teufel R, Kaysser L, Villaume MT, Diethelm S, Carbullido MK, Baran PS, Moore BS. One-Pot Enzymatic Synthesis of Merochlorin A and B. Angew Chem Int Ed Engl. 2014;53(41):11019–11022. doi: 10.1002/anie.201405694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Diethelm S, Teufel R, Kaysser L, Moore BS. A Multitasking Vanadium-Dependent Chloroperoxidase as an Inspiration for the Chemical Synthesis of the Merochlorins. Angew Chem Int Ed Engl. 2014;53(41):11023–11026. doi: 10.1002/anie.201405696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Bernhardt P, Okino T, Winter JM, Miyanaga A, Moore BS. A Stereoselective Vanadium-Dependent Chloroperoxidase in Bacterial Antibiotic Biosynthesis. J Am Chem Soc. 2011;133(12):4268–4270. doi: 10.1021/ja201088k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Winter JM, Moffitt MC, Zazopoulos E, McAlpine JB, Dorrestein PC, Moore BS. Molecular Basis for Chloronium-Mediated Meroterpene Cyclization: Cloning, Sequencing, and Heterologous Expression of the Napyradiomycin Biosynthetic Gene Cluster. J Biol Chem. 2007;282(22):16362–16368. doi: 10.1074/jbc.M611046200. [DOI] [PubMed] [Google Scholar]
- 542.Ishihara J, Kanoh N, Murai A. Enzymatic Reaction of (3E,6S,7S)-Laurediol and the Molecular Modeling Studies on the Cyclization of Laurediols. Tetrahedron Lett. 1995;36(5):737–740. [Google Scholar]
- 543.Carter-Franklin JN, Butler A. Vanadium Bromoperoxidase-Catalyzed Biosynthesis of Halogenated Marine Natural Products. J Am Chem Soc. 2004;126(46):15060–15066. doi: 10.1021/ja047925p. [DOI] [PubMed] [Google Scholar]
- 544.Chang ZX, Sitachitta N, Rossi JV, Roberts MA, Flatt PM, Jia JY, Sherman DH, Gerwick WH. Biosynthetic Pathway and Gene Cluster Analysis of Curacin A, an Antitubulin Natural Product from the Tropical Marine Cyanobacterium Lyngbya Majuscula. J Nat Prod. 2004;67(8):1356–1367. doi: 10.1021/np0499261. [DOI] [PubMed] [Google Scholar]
- 545.Gu L, Jia J, Liu H, Hakansson K, Gerwick WH, Sherman DH. Metabolic Coupling of Dehydration and Decarboxylation in the Curacin A Pathway: Functional Identification of a Mechanistically Diverse Enzyme Pair. J Am Chem Soc. 2006;128(28):9014–9015. doi: 10.1021/ja0626382. [DOI] [PubMed] [Google Scholar]
- 546.Gu L, Wang B, Kulkarni A, Geders TW, Grindberg RV, Gerwick L, Hakansson K, Wipf P, Smith JL, Gerwick WH, et al. Metamorphic Enzyme Assembly in Polyketide Diversification. Nature. 2009;459(7247):731–735. doi: 10.1038/nature07870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547.Geng XQ, Cheng JY, Gangadharan A, Mackey D. The Coronatine Toxin of Pseudomonas Syringae Is a Multifunctional Suppressor of Arabidopsis Defense. Plant Cell. 2012;24(11):4763–4774. doi: 10.1105/tpc.112.105312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548.Geng XQ, Jin L, Shimada M, Kim MG, Mackey D. The Phytotoxin Coronatine Is a Multifunctional Component of the Virulence Armament of Pseudomonas Syringae. Planta. 2014;240(6):1149–1165. doi: 10.1007/s00425-014-2151-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Vaillancourt FH, Yeh E, Vosburg DA, O’Connor SE, Walsh CT. Cryptic Chlorination by a Non-Haem Iron Enzyme During Cyclopropyl Amino Acid Biosynthesis. Nature. 2005;436(7054):1191–1194. doi: 10.1038/nature03797. [DOI] [PubMed] [Google Scholar]
- 550.Fujimori DG, Hrvatin S, Neumann CS, Strieker M, Marahiel MA, Walsh CT. Cloning and Characterization of the Biosynthetic Gene Cluster for Kutznerides. Proc Natl Acad Sci U S A. 2007;104(42):16498–16503. doi: 10.1073/pnas.0708242104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Neumann CS, Walsh CT. Biosynthesis of (–)-(1S,2R)-Allocoronamic Acyl Thioester by an FeII-Dependent Halogenase and a Cyclopropane-Forming Flavoprotein. J Am Chem Soc. 2008;130(43):14022–14023. doi: 10.1021/ja8064667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Cox R. Oxidative Rearrangements During Fungal Biosynthesis. Nat Prod Rep. 2014;31(10):1405–1424. doi: 10.1039/c4np00059e. [DOI] [PubMed] [Google Scholar]
- 553.Bräuer A, Beck P, Hintermann L, Groll M. Structure of the Dioxygenase AsqJ: Mechanistic Insights into a One-Pot Multistep Quinolone Antibiotic Biosynthesis. Angew Chem Int Ed Engl. 2016;55(1):422–426. doi: 10.1002/anie.201507835. [DOI] [PubMed] [Google Scholar]
- 554.Chang WC, Li J, Lee JL, Cronican AA, Guo Y. Mechanistic Investigation of a Non-Heme Iron Enzyme Catalyzed Epoxidation in (-)-4′-Methoxycyclopenin Biosynthesis. J Am Chem Soc. 2016;138(33):10390–10393. doi: 10.1021/jacs.6b05400. [DOI] [PubMed] [Google Scholar]
- 555.Brian PW. Effects of Gibberellins on Plant Growth and Development. Biol Rev. 1959;34(1):37–77. [Google Scholar]
- 556.Helliwell CA, Poole A, Peacock WJ, Dennis ES. Arabidopsis Ent-Kaurene Oxidase Catalyzes Three Steps of Gibberellin Biosynthesis. Plant Physiol. 1999;119(2):507–510. doi: 10.1104/pp.119.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Helliwell CA, Chandler PM, Poole A, Dennis ES, Peacock WJ. The CYP88A Cytochrome P450, Ent-Kaurenoic Acid Oxidase, Catalyzes Three Steps of the Gibberellin Biosynthesis Pathway. Proc Natl Acad Sci U S A. 2001;98(4):2065–2070. doi: 10.1073/pnas.041588998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558.Williams PG, Asolkar RN, Kondratyuk T, Pezzuto JM, Jensen PR, Fenical W. Saliniketals A and B, Bicyclic Polyketides from the Marine Actinomycete Salinispora Arenicola. J Nat Prod. 2007;70(1):83–88. doi: 10.1021/np0604580. [DOI] [PubMed] [Google Scholar]
- 559.Wilson MC, Gulder TAM, Mahmud T, Moore BS. Shared Biosynthesis of the Saliniketals and Rifamycins in Salinispora Arenicola is Controlled by the Sare1259-Encoded Cytochrome P450. J Am Chem Soc. 2010;132(36):12757–12765. doi: 10.1021/ja105891a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Stroshane RM, Chan JA, Rubalcaba EA, Garretson AL, Aszalos AA, Roller PP. Isolation and Structure Elucidation of a Novel Griseorhodin. J Antibiot (Tokyo) 1979;32(3):197–204. doi: 10.7164/antibiotics.32.197. [DOI] [PubMed] [Google Scholar]
- 561.Li A, Piel J. A Gene Cluster from a Marine Streptomyces Encoding the Biosynthesis of the Aromatic Spiroketal Polyketide Griseorhodin A. Chem Biol. 2002;9(9):1017–1026. doi: 10.1016/s1074-5521(02)00223-5. [DOI] [PubMed] [Google Scholar]
- 562.Yunt Z, Reinhardt K, Li A, Engeser M, Dahse HM, Gutschow M, Bruhn T, Bringmann G, Piel J. Cleavage of Four Carbon-Carbon Bonds During Biosynthesis of the Griseorhodin a Spiroketal Pharmacophore. J Am Chem Soc. 2009;131(6):2297–2305. doi: 10.1021/ja807827k. [DOI] [PubMed] [Google Scholar]
- 563.Ainsworth AM, Chicarelli-Robinson MI, Copp BR, Fauth U, Hylands PJ, Holloway JA, Latif M, O’Beirne GB, Porter N, Renno DV, et al. Xenovulene A, a Novel GABA-Benzodiazepine Receptor Binding Compound Produced by Acremonium Strictum. J Antibiot (Tokyo) 1995;48(7):568–573. doi: 10.7164/antibiotics.48.568. [DOI] [PubMed] [Google Scholar]
- 564.Raggatt ME, Simpson TJ, Raggatt ME, Chicarelli-Robinson MI. Biosynthesis of Xenovulene A®: Formation of a Cyclopentenone Via a Unique Ring Expansion-Ring Contraction Mechanism. Chem Commun (camb) 1997;(22):2245–2247. [Google Scholar]
- 565.Kamerbeek NM, Janssen DB, van Berkel WJH, Fraaije MW. Baeyer-Villiger Monooxygenases, an Emerging Family of Flavin-Dependent Biocatalysts. Adv Syn Catal. 2003;345(6–7):667–678. [Google Scholar]
- 566.Leisch H, Morley K, Lau PCK. Baeyer-Villiger Monooxygenases: More Than Just Green Chemistry. Chem Rev. 2011;111(7):4165–4222. doi: 10.1021/cr1003437. [DOI] [PubMed] [Google Scholar]
- 567.Pazmino DET, Dudek HM, Fraaije MW. Baeyer-Villiger Monooxygenases: Recent Advances and Future Challenges. Curr Opin Chem Biol. 2010;14(2):138–144. doi: 10.1016/j.cbpa.2009.11.017. [DOI] [PubMed] [Google Scholar]
- 568.Mascotti ML, Lapadula WJ, Ayub MJ. The Origin and Evolution of Baeyer-Villiger Monooxygenases (BVMOs): An Ancestral Family of Flavin Monooxygenases. PloS ONE. 2015;10(7):e0132689. doi: 10.1371/journal.pone.0132689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569.Cane DE, Rossi T. The Isolation and Structural Elucidation of Pentalenolactone E. Tetrahedron Lett. 1979;20(32):2973–2974. [Google Scholar]
- 570.Tillman AM, Cane DE. Pentalenolactone F, a New Metabolite Isolated from Streptomyces. Isolation and Structure Elucidation. J Antibiot (Tokyo) 1983;36(2):170–172. doi: 10.7164/antibiotics.36.170. [DOI] [PubMed] [Google Scholar]
- 571.Cane DE, Oliver JS, Harrison PHM, Abell C, Hubbard BR, Kane CT, Lattman R. Biosynthesis of Pentalenene and Pentalenolactone. J Am Chem Soc. 1990;112(11):4513–4524. [Google Scholar]
- 572.Tetzlaff CN, You Z, Cane DE, Takamatsu S, Omura S, Ikeda H. A Gene Cluster for Biosynthesis of the Sesquiterpenoid Antibiotic Pentalenolactone in Streptomyces Avermitilis. Biochemistry. 2006;45(19):6179–6186. doi: 10.1021/bi060419n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573.Seo M-J, Zhu D, Endo S, Ikeda H, Cane DE. Genome Mining in Streptomyces. Elucidation of the Role of Baeyer–Villiger Monooxygenases and Non-Heme Iron-Dependent Dehydrogenase/Oxygenases in the Final Steps of the Biosynthesis of Pentalenolactone and Neopentalenolactone. Biochemistry. 2011;50(10):1739–1754. doi: 10.1021/bi1019786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574.Matsuda Y, Awakawa T, Wakimoto T, Abe I. Spiro-Ring Formation is Catalyzed by a Multifunctional Dioxygenase in Austinol Biosynthesis. J Am Chem Soc. 2013;135(30):10962–10965. doi: 10.1021/ja405518u. [DOI] [PubMed] [Google Scholar]
- 575.Scherlach K, Boettger D, Remme N, Hertweck C. The Chemistry and Biology of Cytochalasans. Nat Prod Rep. 2010;27(6):869–886. doi: 10.1039/b903913a. [DOI] [PubMed] [Google Scholar]
- 576.Qiao KJ, Chooi YH, Tang Y. Identification and Engineering of the Cytochalasin Gene Cluster from Aspergillus Clavatus NRRL 1. Metab Eng. 2011;13(6):723–732. doi: 10.1016/j.ymben.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Hu YC, Dietrich D, Xu W, Patel A, Thuss JAJ, Wang JJ, Yin WB, Qiao KJ, Houk KN, Vederas JC, et al. A Carbonate-Forming Baeyer-Villiger Monooxygenase. Nat Chem Biol. 2014;10(7):552–554. doi: 10.1038/nchembio.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578.Jager CE, Symons GM, Nomura T, Yamada Y, Smith JJ, Yamaguchi S, Kamiya Y, Weller JL, Yokota T, Reid JB. Characterization of Two Brassinosteroid C-6 Oxidase Genes in Pea. Plant Physiol. 2007;143(4):1894–1904. doi: 10.1104/pp.106.093088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.Nomura T, Kushiro T, Yokota T, Kamiya Y, Bishop GJ, Yamaguchi S. The Last Reaction Producing Brassinolide Is Catalyzed by Cytochrome P-450s, CYP85A3 in Tomato and CYP85A2 in Arabidopsis. J Biol Chem. 2005;280(18):17873–17879. doi: 10.1074/jbc.M414592200. [DOI] [PubMed] [Google Scholar]
- 580.Valegard K, van Scheltinga AC, Lloyd MD, Hara T, Ramaswamy S, Perrakis A, Thompson A, Lee HJ, Baldwin JE, Schofield CJ, et al. Structure of a Cephalosporin Synthase. Nature. 1998;394(6695):805–809. doi: 10.1038/29575. [DOI] [PubMed] [Google Scholar]
- 581.Dotzlaf JE, Yeh WK. Purification and Properties of Deacetoxycephalosporin C Synthase from Recombinant Escherichia coli and its Comparison with the Native Enzyme Purified from Streptomyces Clavuligerus. J Biol Chem. 1989;264(17):10219–10227. [PubMed] [Google Scholar]
- 582.Lee HJ, Lloyd MD, Harlos K, Clifton IJ, Baldwin JE, Schofield CJ. Kinetic and Crystallographic Studies on Deacetoxycephalosporin C Synthase (DAOCS) J Mol Biol. 2001;308(5):937–948. doi: 10.1006/jmbi.2001.4649. [DOI] [PubMed] [Google Scholar]
- 583.Bentley R. Aromatic Synthesis in Molds: Formation of the Tropolone, Stipitatic Acid. Biochim Biophys Acta. 1958;29(3):666–667. doi: 10.1016/0006-3002(58)90040-4. [DOI] [PubMed] [Google Scholar]
- 584.Davison J, al Fahad A, Cai M, Song Z, Yehia SY, Lazarus CM, Bailey AM, Simpson TJ, Cox RJ. Genetic, Molecular, and Biochemical Basis of Fungal Tropolone Biosynthesis. Proc Natl Acad Sci U S A. 2012;109(20):7642–7647. doi: 10.1073/pnas.1201469109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585.Cox RJ, Al-Fahad A. Chemical Mechanisms Involved During the Biosynthesis of Tropolones. Curr Opin Chem Biol. 2013;17(4):532–536. doi: 10.1016/j.cbpa.2013.06.029. [DOI] [PubMed] [Google Scholar]
- 586.Teufel R, Friedrich T, Fuchs G. An Oxygenase that Forms and Deoxygenates Toxic Epoxide. Nature. 2012;483(7389):359–362. doi: 10.1038/nature10862. [DOI] [PubMed] [Google Scholar]
- 587.Moore BS, Walker K, Tornus I, Handa S, Poralla K, Floss HG. Biosynthetic Studies of ω-Cycloheptyl Fatty Acids in Alicyclobacillus Cycloheptanicus. Formation of Cycloheptanecarboxylic Acid from Phenylacetic Acid. J Org Chem. 1997;62(7):2173–2185. doi: 10.1021/jo962402o. [DOI] [PubMed] [Google Scholar]
- 588.Bergmann S, Schumann J, Scherlach K, Lange C, Brakhage AA, Hertweck C. Genomics-Driven Discovery of PKS-NRPS Hybrid Metabolites from Aspergillus Nidulans. Nat Chem Biol. 2007;3(4):213–217. doi: 10.1038/nchembio869. [DOI] [PubMed] [Google Scholar]
- 589.Xu W, Cai X, Jung ME, Tang Y. Analysis of Intact and Dissected Fungal Polyketide Synthase-Nonribosomal Peptide Synthetase in Vitro and in Saccharomyces Cerevisiae. J Am Chem Soc. 2010;132(39):13604–13607. doi: 10.1021/ja107084d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590.Liu C, Tagami K, Minami A, Matsumoto T, Frisvad JC, Suzuki H, Ishikawa J, Gomi K, Oikawa H. Reconstitution of biosynthetic machinery for the synthesis of the highly elaborated indole diterpene penitrem. Angew Chem Int Ed Engl. 2015;54(19):5748–5752. doi: 10.1002/anie.201501072. [DOI] [PubMed] [Google Scholar]
- 591.Minami A, Liu C, Oikawa H. Total Biosynthesis of Fungal Indole Diterpenes Using Cell Factories. Heterocycles. 2016;92(3):397–421. [Google Scholar]
- 592.Lin H-C, Chooi YH, Dhingra S, Xu W, Calvo AM, Tang Y. The Fumagillin Biosynthetic Gene Cluster in Aspergillus Fumigatus Encodes a Cryptic Terpene Cyclase Involved in the Formation of β-trans-Bergamotene. J Am Chem Soc. 2013;135(12):4616–4619. doi: 10.1021/ja312503y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Ingber D, Fujita T, Kishimoto S, Sudo K, Kanamaru T, Brem H, Folkman J. Synthetic Analogs of Fumagillin That Inhibit Angiogenesis and Suppress Tumor Growth. Nature. 1990;348(6301):555–557. doi: 10.1038/348555a0. [DOI] [PubMed] [Google Scholar]
- 594.Sin N, Meng L, Wang MQ, Wen JJ, Bornmann WG, Crews CM. The Anti-Angiogenic Agent Fumagillin Covalently Binds and Inhibits the Methionine Aminopeptidase, MetAP-2. Proc Natl Acad Sci U S A. 1997;94(12):6099–6103. doi: 10.1073/pnas.94.12.6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595.Griffith EC, Su Z, Niwayama S, Ramsay CA, Chang YH, Liu JO. Molecular Recognition of Angiogenesis Inhibitors Fumagillin and Ovalicin by Methionine Aminopeptidase 2. Proc Natl Acad Sci U S A. 1998;95(26):15183–15188. doi: 10.1073/pnas.95.26.15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596.Liu SP, Widom J, Kemp CW, Crews CM, Clardy J. Structure of Human Methionine Aminopeptidase-2 Complexed with Fumagillin. Science. 1998;282(5392):1324–1327. doi: 10.1126/science.282.5392.1324. [DOI] [PubMed] [Google Scholar]
- 597.Ortiz de Montellano PRE. Cytochrome P450-Structure, Mechanism, and Biochemistry. Springer; New York: 2015. [Google Scholar]
- 598.Ohmomo S, Oguma K, Ohashi T, Abe M. Isolation of a New Indole Alkaloid, Roquefortine D, from the Cultures of Penicillium Roqueforti. Agric Biol Chem. 1978;42(12):2387–2389. [Google Scholar]
- 599.Sumarah MW, Miller JD, Blackwell BA. Isolation and Metabolite Production by Penicillium Roqueforti, P. Paneum and P. Crustosum Isolated in Canada. Mycopathologia. 2005;159(4):571–577. doi: 10.1007/s11046-005-5257-7. [DOI] [PubMed] [Google Scholar]
- 600.Bhat B, Harrison DM, Lamont HM. The Biosynthesis of the Mould Metabolites Roquefortine and Aszonalenin from L-[2,4,5,6,7-2H5]Tryptophan. Tetrahedron. 1993;49(46):10663–10668. [Google Scholar]
- 601.Garcia-Estrada C, Ullan RV, Albillos SM, Fernandez-Bodega MA, Durek P, von Dohren H, Martin JF. A Single Cluster of Coregulated Genes Encodes the Biosynthesis of the Mycotoxins Roquefortine C and Meleagrin in Penicillium Chrysogenum. Chem Biol. 2011;18(11):1499–1512. doi: 10.1016/j.chembiol.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 602.Ali H, Ries MI, Nijland JG, Lankhorst PP, Hankemeier T, Bovenberg RAL, Vreeken RJ, Driessen AJM. A Branched Biosynthetic Pathway Is Involved in Production of Roquefortine and Related Compounds in Penicillium Chrysogenum. PloS ONE. 2013;8(6):e65328. doi: 10.1371/journal.pone.0065328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 603.Terui Y, Chu YW, Li JY, Ando T, Yamamoto H, Kawamura Y, Tomishima Y, Uchida S, Okazaki T, Munetomo E, et al. Xantholipin, a Novel Inhibitor of HSP47 Gene Expression Produced by Streptomyces sp. Tetrahedron Lett. 2003;44(29):5427–5430. [Google Scholar]
- 604.Kong L, Zhang W, Chooi YH, Wang L, Cao B, Deng Z, Chu Y, You D. A Multifunctional Monooxygenase XanO4 Catalyzes Xanthone Formation in Xantholipin Biosynthesis via a Cryptic Demethoxylation. Cell Chem Biol. 2016;23(4):508–516. doi: 10.1016/j.chembiol.2016.03.013. [DOI] [PubMed] [Google Scholar]
- 605.Leach BE, Calhoun KM, Johnson LE, Teeters CM, Jackson WG. Chartreusin, a New Antibiotic Produced by Streptomyces Chartreusis, a New Species. J Am Chem Soc. 1953;75(16):4011–4012. [Google Scholar]
- 606.Xu Z, Jakobi K, Welzel K, Hertweck C. Biosynthesis of the Antitumor Agent Chartreusin Involves the Oxidative Rearrangement of an Anthracyclic Polyketide. Chem Biol. 2005;12(5):579–588. doi: 10.1016/j.chembiol.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 607.Ayer SW, Mcinnes AG, Thibault P, Walter JA, Doull JL, Parnell T, Vining LC. Jadomycin, a Novel 8H-Benz[b]Oxazolo[3,2-f]Phenanthridine Antibiotic from Streptomyces Venezuelae ISP5230. Tetrahedron Lett. 1991;32(44):6301–6304. [Google Scholar]
- 608.Doull JL, Ayer SW, Singh AK, Thibault P. Production of a Novel Polyketide Antibiotic, Jadomycin B, by Streptomyces Venezuelae Following Heat Shock. J Antibiot (Tokyo) 1993;46(5):869–871. doi: 10.7164/antibiotics.46.869. [DOI] [PubMed] [Google Scholar]
- 609.Tibrewal N, Pahari P, Wang GJ, Kharel MK, Morris C, Downey T, Hou YP, Bugni TS, Rohr J. Baeyer-Villiger C-C Bond Cleavage Reaction in Gilvocarcin and Jadomycin Biosynthesis. J Am Chem Soc. 2012;134(44):18181–18184. doi: 10.1021/ja3081154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 610.Fan KQ, Pan GH, Peng XJ, Zheng JT, Gao WB, Wang J, Wang WS, Li Y, Yang KQ. Identification of JadG as the B Ring Opening Oxygenase Catalyzing the Oxidative C-C Bond Cleavage Reaction in Jadomycin Biosynthesis. Chem Biol. 2012;19(11):1381–1390. doi: 10.1016/j.chembiol.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 611.Han L, Yang KQ, Ramalingam E, Mosher RH, Vining LC. Cloning and Characterization of Polyketide Synthase Genes for Jadomycin B Biosynthesis in Streptomyces Venezuelae ISP5230. Microbiology. 1994;140:3379–3389. doi: 10.1099/13500872-140-12-3379. [DOI] [PubMed] [Google Scholar]
- 612.Gould SJ, Cheng X-C. Biosynthesis of the Benz[α]Anthraquinone Antibiotic PD 116198. Tetrahedron. 1993;49(48):11135–11144. [Google Scholar]
- 613.Gould SJ, Halley KA. Biosynthesis of the Benz[a]Anthraquinone Antibiotic PD 116198: Evidence for a Rearranged Skeleton. J Am Chem Soc. 1991;113(13):5092–5093. [Google Scholar]
- 614.Sasaki E, Ogasawara Y, Liu HW. A Biosynthetic Pathway for BE-7585A, a 2-Thiosugar-Containing Angucycline-Type Natural Product. J Am Chem Soc. 2010;132(21):7405–7417. doi: 10.1021/ja1014037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 615.Yu J, Chang PK, Ehrlich KC, Cary JW, Bhatnagar D, Cleveland TE, Payne GA, Linz JE, Woloshuk CP, Bennett JW. Clustered Pathway Genes in Aflatoxin Biosynthesis. Appl Environ Microbiol. 2004;70(3):1253–1262. doi: 10.1128/AEM.70.3.1253-1262.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616.Townsend CA. Aflatoxin and Deconstruction of Type I, Iterative Polyketide Synthase Function. Nat Prod Rep. 2014;31(10):1260–1265. doi: 10.1039/c4np00092g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 617.Yabe K, Nakajima H. Enzyme Reactions and Genes in Aflatoxin Biosynthesis. Appl Microbiol Biotechnol. 2004;64(6):745–755. doi: 10.1007/s00253-004-1566-x. [DOI] [PubMed] [Google Scholar]
- 618.McKeown DSJ, McNicholas C, Simpson TJ, Willett NJ. Biosynthesis of Norsolorinic Acid and Averufin: Substrate Specificity of Norsolorinic Acid Synthase. Chem Commun (camb) 1996;(3):301–302. [Google Scholar]
- 619.Crawford JM, Korman TP, Labonte JW, Vagstad AL, Hill EA, Kamari-Bidkorpeh O, Tsai SC, Townsend CA. Structural Basis for Biosynthetic Programming of Fungal Aromatic Polyketide Cyclization. Nature. 2009;461(7267):1139–1143. doi: 10.1038/nature08475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 620.Ehrlich KC, Scharfenstein LL, Chang PK, Li P, Bhatnagar D. Characterization of the Anthrone Oxidase in Aflatoxin Biosynthesis. FASEB J. 2009;23(1 Supplement):535.532. [Google Scholar]
- 621.Yu JJ, Chang PK, Cary JW, Bhatnagar D, Cleveland TE. AvnA, a Gene Encoding a Cytochrome P-450 Monooxygenase, Is Involved in the Conversion of Averantin to Averufin in Aflatoxin Biosynthesis in Aspergillus Parasiticus. Appl Environ Microbiol. 1997;63(4):1349–1356. doi: 10.1128/aem.63.4.1349-1356.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622.Sakuno E, Wen Y, Hatabayashi H, Arai H, Aoki C, Yabe K, Nakajima H. Aspergillus Parasiticus Cyclase Catalyzes Two Dehydration Steps in Aflatoxin Biosynthesis. Appl Environ Microbiol. 2005;71(6):2999–3006. doi: 10.1128/AEM.71.6.2999-3006.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 623.Keller NP, Watanabe CMH, Kelkar HS, Adams TH, Townsend CA. Requirement of Monooxygenase-Mediated Steps for Sterigmatocystin Biosynthesis by Aspergillus Nidulans. Appl Environ Microbiol. 2000;66(1):359–362. doi: 10.1128/aem.66.1.359-362.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624.Townsend CA, Christensen SB. Concerning the Role of Nidurufin in Aflatoxin Biosynthesis. J Am Chem Soc. 1985;107(1):270–271. [Google Scholar]
- 625.Townsend CA, Davis SG, Koreeda M, Hulin B. A Cationic Model of the Chain-Branching Step in Aflatoxin Biosynthesis. J Org Chem. 1985;50(25):5428–5430. [Google Scholar]
- 626.Townsend CA, Isomura Y, Davis SG, Hodge JA. Reaction Models of the Oxidative Rearrangement of Averufin to 1′-Hydroxyversicolorone: The First Step in Dihydrobisfuran Formation in Aflatoxin Biosynthesis. Tetrahedron. 1989;45(8):2263–2276. [Google Scholar]
- 627.Yabe K, Chihaya N, Hamamatsu S, Sakuno E, Hamasaki T, Nakajima H, Bennett JW. Enzymatic Conversion of Averufin to Hydroxyversicolorone and Elucidation of a Novel Metabolic Grid Involved in Aflatoxin Biosynthesis. Appl Environ Microbiol. 2003;69(1):66–73. doi: 10.1128/AEM.69.1.66-73.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628.Yu J, Chang PK, Bhatnagar D, Cleveland TE. Genes Encoding Cytochrome P450 and Monooxygenase Enzymes Define One End of the Aflatoxin Pathway Gene Cluster in Aspergillus Parasiticus. Appl Microbiol Biotechnol. 2000;53(5):583–590. doi: 10.1007/s002530051660. [DOI] [PubMed] [Google Scholar]
- 629.Lin BK, Anderson JA. Purification and Properties of Versiconal Cyclase from Aspergillus Parasiticus. Arch Biochem Biophys. 1992;293(1):67–70. doi: 10.1016/0003-9861(92)90366-5. [DOI] [PubMed] [Google Scholar]
- 630.Yabe K, Hamasaki T. Stereochemistry During Aflatoxin Biosynthesis: Cyclase Reaction in the Conversion of Versiconal to Versicolorin B and Racemization of Versiconal Hemiacetal Acetate. Appl Environ Microbiol. 1993;59(8):2493–2500. doi: 10.1128/aem.59.8.2493-2500.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Henry KM, Townsend CA. Ordering the Reductive and Cytochrome P450 Oxidative Steps in Demethylsterigmatocystin Formation Yields General Insights into the Biosynthesis of Aflatoxin and Related Fungal Metabolites. J Am Chem Soc. 2005;127(11):3724–3733. doi: 10.1021/ja0455188. [DOI] [PubMed] [Google Scholar]
- 632.Yu J, Cary JW, Bhatnagar D, Cleveland TE, Keller NP, Chu FS. Cloning and Characterization of a cDNA from Aspergillus Parasiticus Encoding an O-Methyltransferase Involved in Aflatoxin Biosynthesis. Appl Environ Microbiol. 1993;59(11):3564–3571. doi: 10.1128/aem.59.11.3564-3571.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633.Lee LW, Chiou CH, Linz JE. Function of Native OmtA in Vivo and Expression and Distribution of this Protein in Colonies of Aspergillus Parasiticus. Appl Environ Microbiol. 2002;68(11):5718–5727. doi: 10.1128/AEM.68.11.5718-5727.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 634.Yabe K, Nakamura M, Hamasaki T. Enzymatic Formation of G-Group Aflatoxins and Biosynthetic Relationship Between G- and B-Group Aflatoxins. Appl Environ Microbiol. 1999;65(9):3867–3872. doi: 10.1128/aem.65.9.3867-3872.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 635.Yu J, Chang PK, Ehrlich KC, Cary JW, Montalbano B, Dyer JM, Bhatnagar D, Cleveland TE. Characterization of the Critical Amino Acids of an Aspergillus Parasiticus Cytochrome P-450 Monooxygenase Encoded by OrdA that Is Involved in the Biosynthesis of Aflatoxins B1, G1, B2, and G2. Appl Environ Microbiol. 1998;64(12):4834–4841. doi: 10.1128/aem.64.12.4834-4841.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 636.Prieto R, Woloshuk CP. Ord1, an Oxidoreductase Gene Responsible for Conversion of O-Methylsterigmatocystin to Aflatoxin in Aspergillus Flavus. Appl Environ Microbiol. 1997;63(5):1661–1666. doi: 10.1128/aem.63.5.1661-1666.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637.Kimler L, Larson RA, Messenger L, Moore JB, Mabry TJ. Betalamic Acid, a New Naturally Occurring Pigment. J Chem Soc D. 1971;(21):1329–1330. [Google Scholar]
- 638.Gandia-Herrero F, Escribano J, Garcia-Carmona F. Purification and Antiradical Properties of the Structural Unit of Betalains. J Nat Prod. 2012;75(6):1030–1036. doi: 10.1021/np200950n. [DOI] [PubMed] [Google Scholar]
- 639.Gandia-Herrero F, Garcia-Carmona F. Characterization of Recombinant Beta Vulgaris 4,5-DOPA-Extradiol-Dioxygenase Active in the Biosynthesis of Betalains. Planta. 2012;236(1):91–100. doi: 10.1007/s00425-012-1593-2. [DOI] [PubMed] [Google Scholar]
- 640.Lipscomb JD. Mechanism of Extradiol Aromatic Ring-Cleaving Dioxygenases. Curr Opin Struct Biol. 2008;18(6):644–649. doi: 10.1016/j.sbi.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 641.Demain AL, Fang A. The Natural Functions of Secondary Metabolites. Adv Biochem Eng Biotechnol. 2000;69:1–39. doi: 10.1007/3-540-44964-7_1. [DOI] [PubMed] [Google Scholar]
- 642.Crawford JM, Townsend CA. New Insights into the Formation of Fungal Aromatic Polyketides. Nat Rev Microbiol. 2010;8(12):879–889. doi: 10.1038/nrmicro2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643.Hertweck C, Luzhetskyy A, Rebets Y, Bechthold A. Type II Polyketide Synthases: Gaining a Deeper Insight into Enzymatic Teamwork. Nat Prod Rep. 2007;24(1):162–190. doi: 10.1039/b507395m. [DOI] [PubMed] [Google Scholar]
- 644.Dickschat JS. Bacterial Terpene Cyclases. Nat Prod Rep. 2016;33(1):87–110. doi: 10.1039/c5np00102a. [DOI] [PubMed] [Google Scholar]
- 645.Ashour M, Wink M, Gershenzon J. Biochemistry of Terpenoids: Monoterpenes Sesquiterpenes and Diterpenes. Annu Plant Rev. 2010;40:258–303. [Google Scholar]
- 646.Tholl D. Biosynthesis and Biological Functions of Terpenoids in Plants. Adv Biochem Eng Biotechnol. 2015;148:63–106. doi: 10.1007/10_2014_295. [DOI] [PubMed] [Google Scholar]
- 647.Abe I. Enzymatic Synthesis of Cyclic Triterpenes. Nat Prod Rep. 2007;24(6):1311–1331. doi: 10.1039/b616857b. [DOI] [PubMed] [Google Scholar]
- 648.Baunach M, Franke J, Hertweck C. Terpenoid Biosynthesis Off the Beaten Track: Unconventional Cyclases and Their Impact on Biomimetic Synthesis. Angew Chem Int Ed Engl. 2015;54(9):2604–2626. doi: 10.1002/anie.201407883. [DOI] [PubMed] [Google Scholar]
- 649.Hammer SC, Syren PO, Seitz M, Nestl BM, Hauer B. Squalene Hopene Cyclases: Highly Promiscuous and Evolvable Catalysts for Stereoselective C-C and C-X Bond Formation. Curr Opin Chem Biol. 2013;17(2):293–300. doi: 10.1016/j.cbpa.2013.01.016. [DOI] [PubMed] [Google Scholar]
- 650.Siedenburg G, Jendrossek D. Squalene-Hopene Cyclases. Appl Environ Microbiol. 2011;77(12):3905–3915. doi: 10.1128/AEM.00300-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 651.Syren PO, Henche S, Eichler A, Nestl BM, Hauer B. Squalene-Hopene Cyclases-Evolution, Dynamics and Catalytic Scope. Curr Opin Struct Biol. 2016;41:73–82. doi: 10.1016/j.sbi.2016.05.019. [DOI] [PubMed] [Google Scholar]
- 652.Crotti P, Pineschi M. In: Aziridines and Epoxides in Organic Synthesis. Yudin AK, editor. chap. 8. Wiley-VCH; Weinheim: 2006. pp. 271–313. [Google Scholar]
- 653.Olofsson B, Somfai P. In: Aziridines and Epoxides in Organic Synthesis. Yudi AK, editor. chap. 9. Wiley-VCH; Weinheim: 2006. pp. 315–347. [Google Scholar]
- 654.Hagel JM, Facchini PJ. Benzylisoquinoline Alkaloid Metabolism: A Century of Discovery and a Brave New World. Plant Cell Physiol. 2013;54(5):647–672. doi: 10.1093/pcp/pct020. [DOI] [PubMed] [Google Scholar]
- 655.Winter JM, Jansma AL, Handel TM, Moore BS. Formation of the Pyridazine Natural Product Azamerone by Biosynthetic Rearrangement of an Aryl Diazoketone. Angew Chem Int Ed Engl. 2009;48(4):767–770. doi: 10.1002/anie.200805140. [DOI] [PMC free article] [PubMed] [Google Scholar]