

Abstract
The peptide hormone relaxin regulates the essential maternal haemodynamic adaptations in early pregnancy through direct actions on the renal and systemic vasculature. These vascular actions of relaxin occur mainly through endothelium‐derived NO‐mediated vasodilator pathways and improvements in arterial compliance in small resistance‐size arteries. This work catalysed a plethora of studies which revealed quite heterogeneous responses across the different regions of the vasculature, and also uncovered NO‐independent mechanisms of relaxin action. In this review, we first describe the role of endogenous relaxin in maintaining normal vascular function, largely referring to work in pregnant and male relaxin‐deficient animals. We then discuss the diversity of mechanisms mediating relaxin action in different vascular beds, including the involvement of prostanoids, VEGF, endothelium‐derived hyperpolarisation and antioxidant activity in addition to the classic NO‐mediated vasodilatory pathway. We conclude the review with current perspectives on the vascular remodelling capabilities of relaxin.
Linked Articles
This article is part of a themed section on Recent Progress in the Understanding of Relaxin Family Peptides and their Receptors. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.10/issuetoc
Abbreviations
- Ang II
angiotensin II
- BK
bradykinin
- EDH
endothelium‐derived hyperpolarisation
- eNOS
endothelial NOS
- ET1
endothelin‐1
- ECs
endothelial cells
- IKCa
intermediate‐conductance calcium‐activated K+ channel
- PAs
parenchymal arterioles
- PlGF
placental growth factor
- Rln−/−
relaxin‐deficient mice
- RXFP1
relaxin/insulin‐like family peptide receptor 1
- SHRs
spontaneously hypertensive rats
- VSMC
vascular smooth muscle cell
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Enzymes c |
| AT2 receptors | Adenylate cyclase |
| B2 receptors | Akt (PKB) |
| ETB receptors | COX |
| RXFP1 receptors | eNOS |
| Voltage‐gated ion channels b | iNOS |
| IKCa (KCa3.1) | PI3K |
| SKCa |
| LIGANDS | |
|---|---|
| ACh | MMP2 |
| Ang II | NO |
| BK, bradykinin | PGI2 |
| cGMP | PlGF, placental growth factor |
| cAMP | Relaxin |
| ET‐1 | TNF‐α |
| Indomethacin | VEGF |
| L‐NAME |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a,b,c).
Introduction
The 6 kDa peptide relaxin is considered a hormone of pregnancy mainly because the highest circulating relaxin concentrations are measured during pregnancy in animals and women. It is now known to contribute to important maternal cardiovascular adaptations of pregnancy (Conrad, 2011a,b). These beneficial effects of relaxin are associated with haemodynamic changes in the maternal systemic and renal vasculature (Conrad, 2010, 2011a,b). They include increased GFR, effective renal plasma flow and global arterial compliance, and reduced systemic vascular resistance, and are explained by the vasodilatory actions of relaxin on small arteries. The negative consequences on the vasculature of a lack of circulating endogenous relaxin are best illustrated in relaxin‐deficient (Rln −/−) rodents during pregnancy (Novak et al., 2001; Vodstrcil et al., 2012; Gooi et al., 2013; Marshall et al., 2016). However, relaxin is also produced in arteries of non‐pregnant female and male rodents (Novak et al., 2006), and Rln −/− male mice have vascular phenotypes that affect normal blood vessel function (Leo et al., 2014a; Ng et al., 2015). This review considers the current literature and provides some new perspectives on the role of endogenous relaxin in maintaining normal vascular function.
The concept that relaxin is a vasodilator is strongly supported by immunohistochemical data that localized the relaxin receptor, RXFP1, in both endothelial and smooth muscle cells in a broad range of arteries and veins (Novak et al., 2006; Jelinic et al., 2014; Ng et al., 2015). The unexpected finding was the differential expression of RXFP1 receptors across the vasculature that now seems to contribute to the diversity of vascular actions of the ligand. In small resistance‐size arteries, vascular tone is mediated by endothelium‐derived factors, including NO, prostanoids and endothelium‐derived hyperpolarisation (EDH). The pioneering studies of Novak and Conrad (Novak et al., 2002) on small renal arteries in rodents demonstrated that recombinant human relaxin (rhRLX) acts predominantly through endothelium‐dependent NO‐mediated vasodilator pathways involving MMPs, endothelial NOS (eNOS), PI3K activation and Akt phosphorylation (Conrad, 2010). More recent work has identified several alternative vasodilatory pathways that are activated by rhRLX and do not entirely involve NO (Leo et al., 2014b; Leo et al., 2016a,b). This review describes the heterogeneous actions of relaxin in different vascular beds and highlights the diversity of mechanisms involved, in addition to the classic NO‐mediated vasodilatory pathway. Although much of the research has focused on modulation of vascular tone, relaxin is also capable of remodelling the extracellular matrix within the vessel wall, thus influencing distensibility and/or stiffness of blood vessels (Conrad and Shroff, 2011). Therefore, we conclude the review with current perspectives on the vascular remodelling capabilities of relaxin.
Relaxin regulates vascular homeostasis in pregnant females
The vascular actions of relaxin were first reported almost 30 years ago when Massicotte and colleagues demonstrated reduced vasoconstrictor responses to noradrenaline and arginine vasopressin in perfused rat mesenteric arteries after in vivo infusion with rat relaxin for 2 days (Massicotte et al., 1989). Evidence that porcine relaxin is a vasodilator and alters coronary blood flow through a NO‐dependent pathway in rat and guinea pig hearts originated from the work of Bani and co‐workers (Bani‐Sacchi et al., 1995). These studies were pivotal in catalysing research on relaxin's role as a regulator of vascular function in pregnancy. Such research is epitomized by numerous studies from the Conrad group and others in which non‐pregnant rats were treated exogenously with purified porcine relaxin or rhRLX to mimic circulating concentrations of relaxin in mid‐pregnancy (20–40 ng·mL−1). Vascular responses, for example reduced myogenic reactivity, were demonstrated in rat small renal and mesenteric arteries after in vivo rhRLX treatment (Novak et al., 2002; van Drongelen et al., 2011; van Drongelen et al., 2012), whereas ex vivo rhRLX application induced vasodilation in isolated rodent small renal and mesenteric arteries (Li et al., 2005; McGuane et al., 2011b) and human small gluteal and subcutaneous arteries but not in pulmonary resistance arteries (Fisher et al., 2002; McGuane et al., 2011b). Although the direct vasodilator effects of rhRLX in isolated blood vessels have not been observed by all investigators (Leo et al., 2016a), these studies established important vascular functions and underlying mechanisms of relaxin action in renal and mesenteric arteries (discussed in more detail later).
Early maternal vascular adaptations to pregnancy are essential to maternal health and the survival and normal development of the fetus. During pregnancy, the maternal vasculature becomes refractory to the actions of vasoconstrictors such as the thromboxane A2 mimetic U46619 (Kim et al., 1994) and angiotensin II (Ang II) (Novak et al., 1997; Hermsteiner et al., 2001). There is also enhanced endothelium‐dependent vasodilation (Kim et al., 1994; Cooke and Davidge, 2003) involving NO, prostacyclin (PGI2) and EDH (Luksha et al., 2010; Majed and Khalil, 2012; Johal et al., 2014). The maternal uteroplacental vasculature also undergoes modifications to increase uterine blood flow and delivery of nutrients to the growing fetus. Specifically, there is dramatic remodelling of the uterine artery wall (Osol and Mandala, 2009). Initially, there is outward hypertrophic remodelling (increase in lumen diameter and wall cross‐sectional area) caused by vascular smooth muscle cell (VSMC) de‐differentiation. The lumen diameter then widens, but without a large expansion of vessel wall thickness. Uterine and radial arteries also become longer and more distensible in both axial and radial directions (Cipolla and Osol, 1994). Maximum remodelling of the main uterine artery occurs during late pregnancy and is associated with VSMC hypertrophy and hyperplasia (Cipolla and Osol, 1994; van der Heijden et al., 2005; Osol and Mandala, 2009).
Endogenous relaxin is now a leading candidate molecule for regulating these essential vascular adaptations in early pregnancy. In a small pilot study of women with ovarian failure who become pregnant through egg donation, in vitro fertilization and embryo transfer (with no detectable circulating relaxin), the first trimester increase in GFR is blunted significantly. They also have increased plasma osmolality relative to women with normal ovarian function (Smith et al., 2006). Serum relaxin concentrations are first detected 6–7 days after the lutenizing hormone peak in non‐conceptive and conceptive cycles and reach highest circulating concentrations (1–2 ng·mL−1) in the first trimester (Stewart et al., 1990). Correlated with this first trimester increase in relaxin are changes in the uterine artery (Anumba et al., 2009), suggesting that relaxin mediates modifications to the uteroplacental vasculature. Analysis of serum relaxin levels in pregnancies complicated by preeclampsia or chronic kidney disease revealed no differences between normal pregnancies and those complicated by disease (Lafayette et al., 2011; Bramham et al., 2016). Nor was there a correlation between relaxin levels and GFR, mean arterial pressure, renal blood flow or renal vascular resistance (Lafayette et al., 2011). However, these studies measured relaxin late in pregnancy, prior to delivery. Sampling of serum in the first trimester revealed that women were at increased risk of developing late‐onset preeclampsia in combination with a small for gestational age newborn if they had lower than normal relaxin concentrations (Uiterweer et al., 2014). Thus, a deficiency or complete lack of circulating relaxin, specifically in early pregnancy, may compromise the maternal vascular adaptations to pregnancy and affect both renal and placental function (Conrad, 2016).
Animal models of relaxin deficiency confirm the hypothesis that a lack of relaxin during pregnancy can have significant consequences on the vasculature (Table 1). Depletion of circulating relaxin with a neutralizing monoclonal antibody in midterm pregnant rats prevented the normal pregnancy‐induced increase in GFR and effective renal plasma flow, and the normal reduction in renal vascular resistance, plasma osmolality and myogenic reactivity of small renal arteries (Novak et al., 2001). These relaxin‐neutralizing antibodies also blocked the gestational elevation in cardiac output and global arterial compliance, and decrease in systemic vascular resistance (Debrah et al., 2006). In late pregnant rats, relaxin‐neutralizing antibody treatment reduced uterine artery inner and outer diameters, and increased circumferential passive wall stiffness (Vodstrcil et al., 2012), but there was no significant change in wall thickness or wall‐to‐lumen ratio. There was also no measureable change in collagen expression and composition, or expression of MMP2 and MMP9 (Vodstrcil et al., 2012), so as yet, the precise mechanistic pathways underlying these structural modifications are unclear. Pregnant Rln −/− mice also have stiffer uterine arteries, with reduced elastin, MMP2, MMP10 and MMP14 gene expression in the vessel wall (Gooi et al., 2013). The less profound changes in the arteries of the pregnant Rln −/− rats are explained by the relatively short antibody treatment time (they received the relaxin‐neutralizing antibodies for 3 days), whereas the Rln −/− mice lacked relaxin for the duration of their pregnancy (19 days). This longer period of relaxin deficiency might be necessary to cause changes in the structural composition of the vascular wall in the absence of relaxin.
Table 1.
Vascular phenotypes in relaxin‐deficient rodents
| Animal model | Artery | Vascular phenotype |
|---|---|---|
| MCA1 treated Long‐Evans rats – mid pregnant (Novak et al., 2001) | small renal artery | ↑ myogenic constriction |
| Rln −/− mice – male and female (Novak et al., 2006; Debrah et al., 2011) | small renal artery | ↑ myogenic constriction inward geometric remodelling, ↓ passive compliance |
| Rln −/− mice – late pregnant, 5 months old (Gooi et al., 2013) | uterine artery | ↑ passive circumferential wall stiffness |
| Rln −/− mice – late pregnant (Marshall et al., 2016) | mesenteric artery | ↑ sensitivity to Ang II (endothelium‐independent) |
| Rln −/− mice – male (Leo et al., 2014a) | mesenteric artery | ↑ sensitivity to the α1‐adrenoceptor agonist phenylephrine and the thromboxane A2 mimetic U46619 |
| – | ↓ endothelium‐dependent relaxation to ACh ↓ volume compliance but no change in circumferential wall stiffness | |
| Rln −/− mice – male (Ng et al., 2015) | aorta | ↑ superoxide production
↓ total eNOS protein and basal NOS activity ↑ eNOS phosphorylation at Ser1177 ↓ sensitivity to the PGI2 analogue, iloprost no reduction in endothelium‐dependent relaxation to ACh |
| Rln −/− mice – male (Debrah et al., 2011; Jelinic et al., 2015) | femoral artery; external iliac artery | no effect |
MCA1 = monoclonal antibody to rat relaxin.
A novel vascular smooth muscle phenotype was recently identified in the mesenteric arteries of late pregnant female Rln −/− mice. Unlike their wild‐type counterparts, responses to Ang II were not reduced, indicating that normal vascular adaptation to pregnancy is compromised in Rln −/− mice (Marshall et al., 2016). Surprisingly, the hyper‐responsiveness to Ang II was endothelium‐independent and appeared to be associated with a reduction in vasodilator prostanoids derived from the vascular smooth muscle (Marshall et al., 2016). Despite these vascular phenotypes, Rln −/− mice are not hypertensive during pregnancy (O'Sullivan et al., 2016), although pups born to Rln −/− dams are growth restricted (Gooi et al., 2013). It is tempting to speculate that a deficiency or complete lack of relaxin in the first trimester of pregnancy may predispose women to hypertensive disorders, including preeclampsia, because the renal and mesenteric vasculature fail to adapt sufficiently to pregnancy. However, there is no direct evidence to support this hypothesis in pregnant women.
Vascular actions of endogenous relaxin in males?
Despite being primarily classified as a pregnancy hormone, relaxin and RXFP1 receptors are detected in the small renal arteries, mesenteric arteries and aorta of male rodents (Novak et al., 2006; Jelinic et al., 2014; Ng et al., 2015). This suggests that relaxin may act as an autocrine or paracrine factor to mediate vascular function. Studies in Rln −/− male mice demonstrated that both vascular function and structure were impaired in small renal and mesenteric arteries, and the aorta (Novak et al., 2006; Leo et al., 2014a; Ng et al., 2015). Interestingly, the degree of vascular impairment is vessel‐specific, highlighting the heterogeneity of vascular dysfunction associated with a deficiency of relaxin (Table 1).
Relaxin phenotypes in small renal and mesenteric arteries
In small renal arteries of Rln −/− mice, myogenic constriction was increased relative to wild‐type mice (Novak et al., 2006), a finding uncovered by incubating arteries in L‐arginine or D‐arginine. Although the exact mechanism of action for this increased myogenic reactivity in small renal arteries is not yet known (Novak et al., 2006), it is likely to involve impaired NO signal transduction pathways. In addition, when compared with wild‐type mice, the Rln −/− mice demonstrated relatively hypotrophic geometric remodelling associated with reduced VSMC density and unstressed wall area, as well as increased collagen‐to‐total protein ratio, leading to reduced passive compliance (Novak et al., 2006; Debrah et al., 2011). Therefore, it is possible that in Rln −/− mice, there is impaired vascular remodelling and compromised endothelium‐derived NO function, resulting in a more ‘constricted’ and ‘stiffer’ small renal artery phenotype compared with that in wild‐type mice.
Small mesenteric arteries (150–200 μm) of Rln −/− mice exhibit super‐sensitivity to the α1‐adrenoceptor agonist, phenylephrine and the thromboxane A2 mimetic, U46619 (Leo et al., 2014a). The hyper‐sensitivity to these vasoconstrictors is endothelium‐dependent because these changes were absent in endothelium‐denuded mesenteric arteries. Further analysis using pharmacological blockers revealed that the enhanced responses to these vasoconstrictors were underpinned by an impairment of NO and vasodilator prostanoid pathways (Leo et al., 2014a). In further support of these findings, mesenteric arteries from Rln −/− mice were less responsive to the endothelium‐dependent agonist, ACh, indicating endothelial dysfunction (Leo et al., 2014a). In contrast to the small renal arteries (Novak et al., 2006), endothelial dysfunction of the mesenteric arteries was not due to impaired NO or EDH. Instead, it was associated with an increased contribution of vasoconstrictor prostanoids, independent of changes in thromboxane receptors or COX enzymes (Leo et al., 2014a).
Mesenteric arteries from Rln −/− mice also have reduced volume compliance but no changes in passive circumferential wall stiffness (Leo et al., 2014a). A recent study that assessed the impact of ageing on mesenteric arteries demonstrated age‐dependent increases in circumferential wall stiffness, but not volume compliance in wild‐type mice with no additional detrimental effects of relaxin deficiency (Jelinic et al., 2015). However, vascular stiffness was exacerbated in mesenteric arteries of younger Rln −/− mice compared with Rln +/+ of equivalent age, highlighting the importance of endogenous relaxin in regulating vascular remodelling of these arteries in younger animals. However, the negative consequences of relaxin deficiency are surpassed by other influences during the ageing process.
Relaxin phenotypes in large arteries (>500 μm)
In contrast to small renal and mesenteric arteries, the vascular phenotypes in the aorta of Rln −/− mice are less pronounced. Superoxide production was increased but was independent of changes in SOD or NADPH oxidase protein expression (Ng et al., 2015). Furthermore, eNOS protein expression and basal NOS activity were reduced in Rln −/− mice. Despite reduced NO bioavailability and increased basal superoxide levels in the aorta, endothelium‐dependent and ‐independent vasorelaxation were unaffected. The NO contribution to endothelium‐dependent relaxation was impaired, but this was only revealed following blockade of COX activity (Ng et al., 2015). There was also a compensatory increase in eNOS phosphorylation at Ser1177 to maintain endothelial function under conditions of increased oxidative stress (Ng et al., 2015). In addition to NO, the aortae of Rln −/− mice also had modifications in the prostanoid pathway. Specifically, there was reduced smooth muscle sensitivity to the PGI2 analogue, iloprost, which was partly explained by decreased expression of PGI2 receptors. Expression of the PG synthase enzymes Cox1, Cox2 and Ptgis were not affected (Ng et al., 2015). In contrast, in the aorta of aged Rln −/− mice (16 months), Cox1 was increased whereas Cox2 was decreased (Ng et al., 2015). Despite these changes in COX gene expression in the older mice, there was no overt evidence of vascular dysfunction.
There has been no analysis of the passive mechanical wall properties in the aorta, and there were no effects of relaxin deficiency on other larger arteries, for example femoral and external iliac (Debrah et al., 2011; Jelinic et al., 2015). To summarize, endogenous relaxin plays an important role in the maintenance of endothelial function and vascular remodelling in both males and females, particularly in small resistance‐size arteries. Impaired vascular function in Rln −/− mice is artery‐specific and is mediated by compromised NO and prostanoid production. The consequences of relaxin deficiency on cardiovascular function are unclear because mean arterial pressure, heart rate and left ventricular dimensions are not compromised in Rln −/− mice (Du et al., 2003). Moreover, the lifespan of Rln −/− mice is not different from Rln +/+ mice, which age up to 24 months (Jelinic et al., 2015). However, old male Rln −/− mice have impeded left ventricular diastolic filling and increased atrial weights, most likely due to increased ventricular collagen and chamber stiffness (Du et al., 2003). Although Rln −/− mice display no overt cardiovascular phenotype, the underlying impairments in vascular function may worsen the progression of cardiovascular disease when animals are exposed to a secondary ‘insult’.
Heterogeneous vascular actions of relaxin
The vascular endothelium regulates the tone of the underlying smooth muscle via the production of various endothelium‐derived relaxing and contracting factors. Different vascular beds exhibit a marked heterogeneity in the relative contribution of these factors to the regulation of tone (Edwards et al., 2010; Zhao et al., 2015). In the aorta, endothelium‐dependent relaxation is entirely mediated by NO whereas in smaller resistance‐size vessels, there is a pronounced contribution of EDH (Luksha et al., 2009; Sandow et al., 2012). We now know that RXFP1 receptors are present in a variety of blood vessels, with quite different localization patterns in the endothelium and underlying VSMCs (Vodstrcil et al., 2012; Jelinic et al., 2014). The differential localization pattern of RXFP1 receptors in various vessel and cell types also supports the hypothesis that relaxin treatment will produce differential responses between the different vessel beds. To date, expression of vascular RXFP1 receptors has not been compared between males and females, and the possible involvement of sex steroids in regulating vascular RFXP1 receptors is unknown. Neither age, obesity nor hypertension altered Rxfp1 expression in rat mesenteric arteries (van Drongelen et al., 2011; van Drongelen et al., 2012; van Drongelen et al., 2013). Similarly, the increase in mesenteric artery vascular stiffness associated with ageing in mice was not correlated with decreased Rxfp1 expression in the vessel wall (Jelinic et al., 2015). To conclude, factors that regulate vascular RXFP1 receptors in both healthy animals (male and female), and in the context of disease, are yet to be established.
Cultured endothelial and vascular smooth muscle cells
The signalling mechanisms underlying rhRLX‐RXFP1 interactions are well‐characterized using native cells that overexpress RXFP1 receptors (Bathgate et al., 2013; Halls et al., 2015). Although these cells are important in understanding agonist‐receptor interactions, they have limited physiological relevance to cellular function in endothelial cells (ECs) and VSMCs. Recent work investigated rhRLX ‐RXFP1 receptor interactions using human umbilical vein and artery ECs and VSMCs, which highlighted distinct signalling mechanisms between different cell types (Sarwar et al., 2015). Specifically, rhRLX increased cAMP and cGMP accumulation in a bell‐shaped concentration‐dependent manner in human umbilical vein ECs and VSMCs. Conversely, rhRLX concentration dependently increased cAMP and cGMP levels in human umbilical artery VSMCs, but in a sigmoidal‐shaped manner. These heterogeneous patterns of activation were attributed to involvement of different G‐proteins in the signalling pathways. Poor expression of RXFP1 receptors with no binding to relaxin in human umbilical artery ECs explains why relaxin did not evoke detectable cAMP and cGMP accumulation in these cells (Sarwar et al., 2015).
Earlier work in human coronary artery and aortic ECs demonstrated that rhRLX treatment causes concentration‐dependent phosphorylation of eNOS, leading to increased NO production (McGuane et al., 2011b). However, relaxin had no effect on NO generation in human coronary VSMCs, indicating that it selectively increased NO production in ECs. Furthermore, rhRLX treatment did not affect calcium influx in human coronary and aortic ECs (McGuane et al., 2011b), suggesting that increased NO production by relaxin was not due to increased calcium signalling in ECs. NO production in these cells was attenuated by inhibiting NOS, PI3K, Akt and a heterodynamic G‐protein (Gαi/o), suggesting that relaxin activates NO production via a relaxin‐RXFP1‐PI3K‐Akt‐NOS pathway (McGuane et al., 2011b). Activation of Akt in response to rhRLX treatment was also observed in rat aortic ECs (Dschietzig et al., 2012). In addition to its effects on ECs, porcine relaxin stimulated NO production and cGMP accumulation in bovine aortic VSMCs by increasing iNOS expression and activity (Bani et al., 1998).
An EC‐VSMC co‐culture approach was recently used to assess the effects of relaxin on crosstalk between these vascular cells. In human coronary artery, ECs co‐cultured with VSMCs, rhRLX stimulated cGMP and cAMP accumulation; this was partially blocked by the COX inhibitor, indomethacin, implying the likely involvement of the vasodilator prostanoid, PGI2. However, this only occurred in human coronary artery ECs, and not umbilical vein ECs (Sarwar et al., 2016). These data show heterogeneous cellular signalling responses to relaxin in cells from different origins, and strengthen our idea that in vivo relaxin treatment will produce region‐dependent vascular responses depending on the blood vessel type (arteries vs. veins) or vessel size (large vs. small).
Aorta
In normal healthy rats, rhRLX treatment for 2 but not 3 days augmented ACh‐evoked relaxation, which was accompanied by increased phosphorylation of eNOS and enhanced NO‐mediated relaxation in the aorta (Leo et al., 2016b). rhRLX also inhibited agonist‐evoked contraction. Specifically, ex vivo rhRLX treatment for 6 h suppressed endothelin‐1 (ET1)‐induced contraction in segments of rat isolated aortas. This effect was endothelium‐dependent and involved activation of endothelin ETB receptors. rhRLX treatment also enhanced vascular relaxation mediated by endothelin‐3 (a selective agonist for ETB receptors), an effect that was abolished by the ETB‐selective antagonist A‐192621 (Dschietzig et al., 2003). This work in the rat aorta, along with that which reported rhRLX‐induced ETB‐dependent renal vasodilation in pregnant rats (Novak et al., 2001), is strong evidence that an important mechanism of relaxin action in the aorta is to enhance ETB‐mediated vasodilation.
Other studies have shown that ex vivo treatment with rhRLX for 48 h prevented aortic vascular dysfunction induced by the proinflammatory cytokine TNF‐α or high glucose. The first study involved incubation of rat aortic rings with TNF‐α, which increases oxidative stress and causes endothelial dysfunction. This was accompanied by a reduction in expression of eNOS and increased eNOS phosphorylation at Thr495, leading to impaired eNOS activity (Dschietzig et al., 2012). Co‐incubation of these aortic rings with rhRLX caused PI3K‐dependent eNOS dephosphorylation at Thr495 and eNOS phosphorylation at Ser1177 and Ser633, and attenuated arginase II expression. This results in increased eNOS activity, leading to improved ACh‐mediated endothelium‐dependent relaxation. These effects of rhRLX involved signalling via glucocorticoid receptors because incubation with the antagonist RU486 negated the restoration of vascular function (Dschietzig et al., 2012). rhRLX treatment also attenuated TNF‐α‐induced increases in superoxide and nitrotyrosine formation, as well as restoring SOD expression (Dschietzig et al., 2012).
The second ex vivo model involved incubation of mouse aortic rings with high glucose (30 mm) for 3 days (Ng et al., 2016). This caused endothelial dysfunction (reduced sensitivity to the endothelium‐dependent agonist, ACh) by increasing the contribution of reactive oxygen species and vasoconstrictor prostanoids. Co‐incubation of aortae with rhRLX for 3 days prevented endothelial dysfunction independent of NO availability. The presence of the COX inhibitor indomethacin and the antioxidant, tempol improved endothelium‐dependent relaxation in high glucose‐treated aortae, but not in rhRLX‐treated aortae. This suggests that rhRLX suppressed the vasoconstrictor prostanoid pathway, and increased endothelium‐derived PGI2 production (Ng et al., 2016). However, rhRLX also has vasoprotective effects possibly by reducing free radicals or enhancing the antioxidant capacity in the aorta during acute hyperglycaemia. Importantly, in vivo rhRLX treatment for 4 weeks effectively reversed endothelial dysfunction and reduced atherosclerotic plaque size in the aorta of apolipoprotein‐deficient mice, which was attributed to a reduction in vascular superoxide production (Tiyerili et al., 2016). A recent study extended these ex vivo findings in an in vivo model of endothelial dysfunction. Specifically, rhRLX infusion for 8 weeks prevented endothelial dysfunction in the guinea pig aorta caused by cigarette smoke. There was also a reduction in reactive oxygen species and increased NO production (Pini et al., 2016). Overall, these findings demonstrate that rhRLX is a vasoprotective molecule under conditions of acute inflammation or hyperglycaemia, as well as in chronic settings such as smoking and atherosclerosis by reducing vascular oxidative stress in the aorta.
Femoral arteries and veins
To date, only one study has investigated the effects of rhRLX treatment on endothelial function in femoral arteries and veins (Jelinic et al., 2014). Despite the presence of RXFP1 receptors in these vessels, subcutaneous infusion of rhRLX for 5 days did not enhance endothelial vasodilator function. One explanation is that the RXFP1 receptors are predominantly localized to the VSMCs and to a lesser extent within ECs (Jelinic et al., 2014); thus, rhRLX is not capable of enhancing endothelium‐dependent relaxation. It is also possible that rhRLX modifies vascular function through an action on the VSMC, but this is yet to be explored in these blood vessels.
Small renal arteries
The effects of in vivo rhRLX treatment on small resistance‐size arteries were first demonstrated in small renal arteries (Novak et al., 2002), as continuous relaxin infusion for 5 days reduced myogenic contraction. These effects of rhRLX were obliterated by the NOS inhibitor L‐NAME or in endothelium‐denuded arteries. This was compelling evidence that the vascular effects of rhRLX were mediated by endothelium‐derived NO (Novak et al., 2002). Subsequent studies showed that rhRLX increased arterial gelatinase activity and indirectly activated ETB receptors (Jeyabalan et al., 2003; Jeyabalan et al., 2007) (Figure 1). Consistent with these in vivo studies, ex vivo incubation of vessels with rhRLX for 3 h also inhibited myogenic reactivity in mouse and rat small renal arteries (and in human subcutaneous arteries), effects that were similarly mediated by gelatinases, ETB receptors and NO (McGuane et al., 2011a). Interestingly, the mechanism by which rhRLX inhibits myogenic constriction is time‐dependent and involves differential up‐regulation of gelatinase activity. Chronic infusion of rhRLX for 5 days in rats increased MMP2 activity and inhibited myogenic reactivity (Jeyabalan et al., 2003). In contrast, a shorter duration of rhRLX treatment (4 to 6 h) also inhibited myogenic reactivity but involved a selective increase MMP9 activity (Jeyabalan et al., 2007). Regardless of the activation of either MMP2 or MMP9 by rhRLX, both MMPs can convert big ET into ET1–32 which in turn activates ETB receptors leading to NO production (Figure 1). The involvement of ETB receptors was demonstrated in experiments using ETB receptor antagonists that abolished the ability of rhRLX to reduce myogenic contraction in small renal arteries (Jeyabalan et al., 2003; Jeyabalan et al., 2007). The earlier work of Dschietzig et al. (2003) implied that rhRLX directly increases ETB receptor expression in human umbilical vein ECs, but not human VSMCs. Moreover, ET1 binding to EC membranes was markedly increased after a 6 h exposure of the cells to rhRLX. However, in vivo rhRLX treatment had no effect on ETB receptor expression and/or activity in rat small renal arteries (Kerchner et al., 2005). Although there is strong agreement that rhRLX‐induced vasodilation is mediated by ETB receptors (most likely in endothelium), the ability of rhRLX to alter arterial ETB receptor expression remains to be confirmed.
Figure 1.
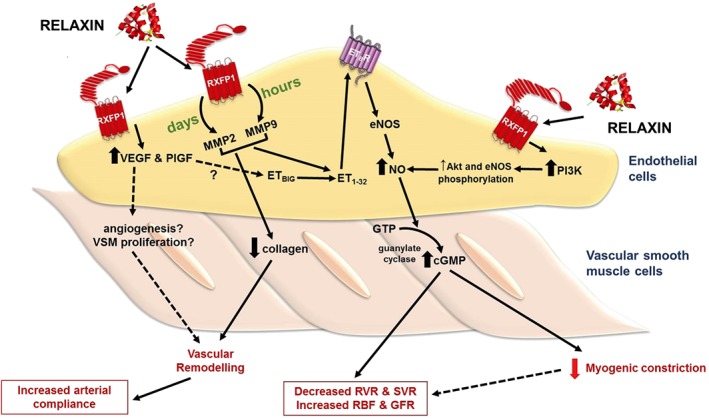
Effects of relaxin treatment in small renal arteries. Relaxin administration for hours and days increases MMP activities in ECs, leading to the conversion of big ET to ET1–32, which activates endothelial ETB receptors. Stimulation of endothelial ETB receptors causes NO production and activates smooth muscle soluble guanylate cyclase, leading to cGMP accumulation and vasodilation. Vasodilation of small renal arteries reduces myogenic reactivity and renal vascular resistance (RVR) and increases renal blood flow (RBF) and GFR. Relaxin also directly acts on endothelial RXFP1 receptors to increase PI3 kinase‐dependent Akt‐eNOS phosphorylation, resulting in NO production. In addition to vasodilation, relaxin treatment causes vascular remodelling in the small renal arteries. Relaxin treatment increases angiogenic factors such as VEGF and PlGF, and reduces collagen content, causing vascular remodelling and increases arterial compliance. SVR, systemic vascular resistance.
Angiogenic growth factors such as VEGF and the placental growth factor (PlGF) are also reported to mediate rhRLX's renal vascular actions (McGuane et al., 2011a). In the presence of VEGF‐ or PlGF‐neutralizing antibodies, rhRLX's ability to reduce myogenic reactivity ex vivo and increase endothelium‐dependent relaxation of rodent renal arteries was abolished. Moreover, the renal vasodilatory effects of in vivo rhRLX were eliminated in conscious female rats co‐treated with the VEGF receptor tyrosine kinase inhibitor, SU5416 (McGuane et al., 2011a). One important point to note is that most of these studies involve rhRLX treatment in either male or non‐pregnant female rats, so it remains a possibility that relaxin may mediate vasodilation through different pathways in the two sexes. There is no question that the most consistent component of rhRLX‐mediated vasodilation in all studies is NO. The involvement of VEGF, PlGF, MMP2/9 and ETB receptors in mediating the vasodilatory effects of rhRLX on small renal arteries is compelling (Figure 1), but it has yet to be shown if they are also involved in mediating the maternal renal vascular adaptations to pregnancy attributed to endogenous relaxin.
Cerebral parenchymal arterioles
The cerebral circulation is unique because the middle cerebral artery (MCA) has significant myogenic tone and contributes ∼50% to cerebrovascular resistance (Faraci et al., 1987). This allows local changes in cerebral blood flow while maintaining cerebrovascular resistance. Pial arteries eventually become brain parenchymal arterioles (PAs) after penetrating into the brain tissue (Hamel, 2006). Cipolla and colleagues investigated the role of rhRLX in both MCAs and PAs by infusing relaxin continuously for 10 days in male rats. rhRLX had no effect on the MCAs, but pressure‐induced myogenic constriction slightly decreased in endothelium‐intact cerebral PAs from rhRLX‐treated rats. Furthermore, the contractile response to the IKCa channel inhibitor, TRAM34, was increased after rhRLX treatment (Chan and Cipolla, 2011). Although these effects were not statistically significant, they indicated that rhRLX may reduce myogenic tone and up‐regulate IKCa‐dependent EDH responses in PAs at a basal level. A subsequent study showed that 14 days of rhRLX infusion in spontaneously hypertensive rats (SHRs) resulted in decreases in cerebral PA myogenic tone. Similarly, rhRLX treatment improved EDH‐mediated relaxation in cerebral PAs of SHRs (Chan et al., 2013). Interestingly, RXFP1 receptors were not detected in the PAs, but rhRLX treatment increased VEGF and MMP2 expression in the brain cortex, which expresses these receptors. Combined treatment of rhRLX, and a VEGF neutralizing antibody abolished the vascular effects of rhRLX in the cerebral PAs. Taken together, these findings suggest that rhRLX exerts its effects on the cerebral PAs through a RXFP1‐VEGF signalling pathway in the adjacent brain tissue (Chan et al., 2013).
Mesenteric arteries and veins
A large body of work has investigated the effects of relaxin treatment in the mesenteric vasculature of healthy rats and to a lesser extent under diseased conditions. Earlier work demonstrated that rhRLX reduced myogenic reactivity in the mesenteric arteries as observed in the small renal arteries (Novak et al., 2002; Jeyabalan et al., 2007). However, more recent studies revealed that exogenous rhRLX augments endothelium‐dependent relaxation that was agonist‐specific and time‐dependent. Specifically, rhRLX administration in rats for 2 to 5 days enhanced bradykinin (BK)‐dependent, but not ACh‐induced relaxation (Jelinic et al., 2014; Leo et al., 2016b). Similarly, acute i.v. injection of rhRLX also enhanced BK‐mediated relaxation (Leo et al., 2014b). Enhancement of BK‐evoked relaxation was not a result of increased expression of BK B2 receptors (Leo et al., 2014b). RXFP1 receptors are able to heterodimerise with angiotensin AT2 receptors in the kidneys, forming RXFP1‐AT2 receptor heterodimer complexes that rhRLX could act on to reduce renal interstitial fibrosis (Chow et al., 2014). As there are no changes to B2 receptor expression, one possibility is that relaxin selectively signals through RXFP1‐B2 receptor heterodimer complexes to enhance endothelial function.
The ability of relaxin to enhance endothelial vasodilator function is abolished by L‐NAME, suggesting that NO is responsible for the vascular effects of rhRLX (Novak et al., 2002; van Drongelen et al., 2011; van Drongelen et al., 2012; van Drongelen et al., 2013; Jelinic et al., 2014). However, 5 days of rhRLX treatment had no effect in mesenteric arteries from aged (40–46 weeks old) (van Drongelen et al., 2011) or obese rats (van Drongelen et al., 2012) but improved flow‐mediated NO‐dependent vasodilation in arteries of SHRs (aged 10–12 weeks) (van Drongelen et al., 2013). It was important to show that the lack of a response to relaxin was not due to a ligand‐mediated reduction in RXFP1 expression nor did age or disease compromise receptor expression in this artery.
A single i.v. injection of rhRLX enhanced basal NOS activity, with increased phosphorylation of Akt at Ser473 and iNOS expression after 3 h (Leo et al., 2014b). In addition, this acute response to rhRLX injection enhanced BK‐mediated relaxation, which was underpinned by IKCa‐dependent EDH, but not NO or PGI2 (Figure 2). Enhancement of EDH‐type relaxation involved a signalling pathway through the BK receptor because opening of IKCa and small‐conductance calcium‐activated K+ channel channels by NS309 was not directly affected by i.v. relaxin injection (Leo et al., 2014b). Interestingly, despite the absence of detectable rhRLX in the circulation 24 h after a single injection of rhRLX, BK‐mediated relaxation remained enhanced. The main mediator of the prolonged vascular response is likely to be PGI2. Vascular Ptgir expression and responses to the PGI2 analogue, iloprost, did not change after i.v. rhRLX injection, suggesting that this prolonged effect of i.v. relaxin relied on the endothelium (Leo et al., 2014b). These acute i.v. experiments provided the first evidence that rhRLX‐mediated endothelium‐dependent relaxation involves the contribution of novel vasodilators, EDH and PGI2 (Figure 2). Subsequent studies continuously infused rhRLX i.v. for 2–3 days; this also enhanced endothelial vasodilator function but activated different mechanisms (Leo et al., 2016b). Specifically, rhRLX infusion for 2 days increased basal NOS activity and potentiated NO‐mediated relaxation. This was attributed to increased eNOS expression (Leo et al., 2016b). However, this enhanced basal NOS activity was not sustained after 3 days of rhRLX infusion because there was a compensatory down‐regulation of eNOS phosphorylation and protein expression (Leo et al., 2016b). Although the BK‐mediated relaxation was sustained, it was dependent on COX2‐derived PGI2. Furthermore, rhRLX infusion for 3 days but not 2 days increased Cox2 expression and BK‐induced production of PGI2, assayed as its metabolite, 6‐keto PGF1α. This is compelling evidence that exogenous rhRLX stimulates PGI2 production in mesenteric arteries (Leo et al., 2016b) and it strongly suggests that the identity of the endothelial‐derived vasorelaxing factors activated after rhRLX treatment is time‐dependent.
Figure 2.
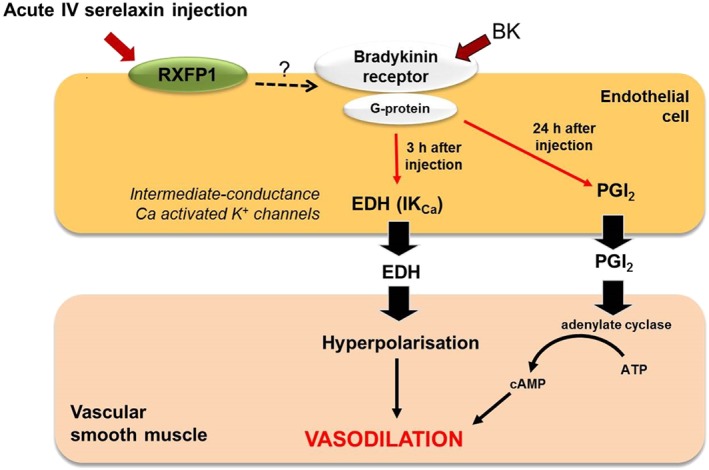
Proposed mechanism(s) of acute i.v. injection of relaxin in rat mesenteric arteries. Relaxin injection causes rapid (3 h) and sustained (24 h) augmentation of BK‐evoked relaxation. The enhanced BK‐mediated relaxation is dependent on the IKCa channels 3 h after acute i.v. relaxin injection. In contrast, the sustained (24 h) relaxation to BK is dependent on PGI2.
The effect of rhRLX treatment in the mesenteric vasculature is limited to arteries as no studies to date have shown changes in mesenteric venous function. Neither myogenic reactivity nor endothelium‐dependent relaxation of mesenteric vein were altered after 2, 3 or 5 days of rhRLX treatment despite the presence of RXFP1 receptors in these blood vessels (Li et al., 2005; Jelinic et al., 2014; Leo et al., 2016b). Therefore, the vascular effects of relaxin on veins cannot be excluded and more studies are required to determine the optimum dose and duration of relaxin treatment needed to elicit changes in venous function.
Remodelling actions of relaxin in the vasculature
The vascular actions of exogenous relaxin extend to modifications of passive mechanical wall properties. Subcutaneous rhRLX infusion in rodents increased circumferential arterial compliance in small renal arteries (Conrad et al., 2004; Debrah et al., 2011). In mice treated with rhRLX for 5 days, the relative increase in small renal artery compliance was mediated by both geometric (outward) and compositional (decreased collagen) remodelling. Outward remodelling was characterized by an increase in unpressurised wall area and wall‐thickness‐to‐lumen area ratio. rhRLX treatment also increased VSMC density and decreased total collagen content in the artery wall, without altering pro‐MMP2 and MMP9 activities (Debrah et al., 2011).
Although there is consensus that relaxin treatment enhances endothelium‐dependent relaxation to vasodilators and reduces myogenic tone in mesenteric arteries, there are substantial differences in the data on passive compliance. Subcutaneous rhRLX treatment in young male Wistar rats for 3–5 days increased arterial compliance (circumferential and longitudinal) and passive volume compliance, and reduced passive circumferential stiffness (Li et al., 2005; Jelinic et al., 2014). The latter was associated with outward remodelling (increased cross‐sectional area and inner diameter) but not changes to the total soluble collagen or elastin content (Jelinic et al., 2014). However, 5 days of rhRLX treatment failed to alter mesenteric artery passive mechanical wall properties in young (10–12 weeks old) or old (40–46 weeks old) non‐pregnant female Wistar Hannover rats (van Drongelen et al., 2011). Recently unpublished data from our laboratory using a different sub‐strain of Wistar rat also demonstrated that neither 3, 5 nor 10 days of relaxin treatment altered passive mechanical properties of mesenteric arteries in young animals (8 weeks old). In this study, plasma rhRLX concentrations were detectable in the range of 57–89 ng·mL−1 and resulted in significant decreases in plasma osmolality, demonstrating that the relaxin was biologically active. One explanation of these data is that the potential beneficial effects of rhRLX on vascular remodelling are hard to distinguish from normal physiological remodelling because these studies used healthy young adult rats with arteries that were in a growth and development phase. Another is that there may be strain differences in the expression of receptors.
Consistent with the data in mesenteric arteries, relaxin treatment has subtle or no remodelling effects in other vascular beds in healthy animals. For example, rhRLX increased wall thickness and lumen diameter of brain PAs, indicative of outward remodelling, without affecting passive compliance in non‐pregnant female rats (Chan and Cipolla, 2011). These effects on remodelling were mediated by PPAR‐γ. Conversely, rhRLX treatment (for 5 or more days) had no effect on passive mechanical wall properties in the external iliac arteries and veins (Debrah et al., 2011; Jelinic et al., 2014), middle cerebral arteries (Chan and Cipolla, 2011) or mesenteric veins (Li et al., 2005; Jelinic et al., 2014). Thus, the effects of relaxin on passive compliance appear to be highly region‐specific. Table 2 highlights the differences between studies in rats including sex and age of the animals, and duration of relaxin treatment. These are all factors that could contribute to the regional effects of relaxin on different vascular beds and need to be considered when undertaking work on vascular remodelling. As mentioned previously, strain differences in responsiveness to relaxin may also contribute to differences between studies.
Table 2.
Summary of the effects of exogenous in vivo relaxin treatment on passive mechanical wall properties in rats
| Sex | Age (weeks) | Treatment duration | Vessel | Effect of serelaxin | Reference |
|---|---|---|---|---|---|
| F | 12–14 | 5 days | small renal artery | ↑ passive compliance, outward hypertrophic remodelling | Conrad et al. (2004) |
| M | 12 | 5 days | mesenteric artery | ↓ circumferential stiffness & ↑ passive compliance | Jelinic et al. (2014) |
| M | 8–12 | 3, 5 and 10 days | no effect | Jelinic et al., unpublished | |
| F | 20–24 | 3 days | ↑ passive compliance | Li et al. (2005) | |
| F | 10–12; 40–46 | 5 days | no effect | – | |
| F | 10–12 (control) (obese) | Van Drongelen et al. (2011, 2012, 2013) | |||
| F | 10–12 (control) (SHR) | – | |||
| M | 12 | 5 days | FA, FV, MV | no effect | Li et al. (2005); Jelinic et al. (2014) |
| F | 14–16 | 10 days | brain parenchymal artery middle cerebral artery | outward remodelling no effect | Chan and Cipolla (2011) |
| 14 days (WKY and SHR) | brain parenchymal artery middle cerebral artery | ↑ distensibility, outward remodelling no effect | Chan et al. (2013) | ||
| M | 68 (SHR) | 14 days +7 days washout | carotid artery | ↑ distensibility, outward remodelling | Xu et al. (2010) |
FA, femoral artery; FV, femoral vein; MV, mesenteric vein; WKY, Wistar Kyoto rats; M, male; F, female.
The potential effects of relaxin on vascular remodelling are more pronounced in animal studies where vascular structure and distensibility have been compromised by disease. Treatment of aged (17 months old) male SHRs for 14 days with rhRLX, followed by a 7 day washout period, increased vessel diameter and elastin content and reduced collagen content of the aorta. It also enhanced passive compliance (circumferential) in the carotid artery (Xu et al., 2010). Similarly, rhRLX treatment for 14 days in young (14–16 weeks old) SHRs reversed the hypertension – induced inward remodelling to increase passive distensibility in brain PAs but had no effect on middle cerebral arteries (Chan et al., 2013). This remodelling was associated with increased MMP2 and VEGF expression in the brain cortex. These factors are hypothesized to interact with brain PAs to enhance distensibility (Chan et al., 2013). Conversely, a shorter duration of rhRLX infusion for 5 days appeared to have no effect on the circumferential remodelling of mesenteric arteries from old, obese and hypertensive (SHR) male rats (van Drongelen et al., 2011; van Drongelen et al., 2012; van Drongelen et al., 2013). This suggests that a longer duration of relaxin may be necessary to affect passive mechanical properties of mesenteric arteries in disease models. Therefore, we conclude that vascular remodelling is one of the beneficial effects of relaxin but this may limited to specific vessel beds and under disease conditions, and of course, in response to pregnancy.
Conclusion and future directions
During the last decade, a large body of research has not only identified an important vasodilatory role for endogenous relaxin, but has also uncovered novel mechanisms of relaxin action in the vasculature that involve prostanoids, EDH and antioxidant activity. Of importance, the detrimental effects of a lack of endogenous relaxin on the maternal renal, mesenteric and uterine vasculature during pregnancy are now established. Pregnancy‐associated hypertensive disease and placental insufficiency (leading to fetal growth restriction) stem from inadequate maternal vascular adaptations, thus relaxin deficiency should be considered when assessing potential causes of these disease entities. The use of Rln −/− animal models (rats and mice) has enabled the discovery of novel vascular phenotypes in males and non‐pregnant females and propose a new role for endogenous relaxin as mediator of vascular homeostasis. This work also emphasized the heterogeneous actions of relaxin between vascular beds. The major development in this area of research has been the breadth of in vivo relaxin treatment studies in conscious animals to assess relaxin actions in different vascular beds. Clearly, relaxin acts through endothelium‐dependent NO‐mediated vasodilator pathways involving MMPs, eNOS and NO to modulate myogenic reactivity in small renal arteries. But analysis of agonist‐evoked endothelium‐dependent relaxation that is enhanced by relaxin has also uncovered the involvement of EDH (IKCa channel activity) and vasodilator prostanoids (synthesis of PGI2) pathways.
Much of this review has focused on the endothelium, but RXFP1 receptors are also located in VSMCs so future research needs to investigate the direct effects of relaxin on these cells. The rationale for this is best illustrated in recent work in late pregnant Rln −/− mice in which the normal pregnancy‐associated attenuation of Ang II‐mediated vasoconstriction in mesenteric arteries did not occur (Marshall et al., 2016). This adaptive failure was endothelium‐independent and was likely due to reduced smooth muscle‐derived vasodilator prostanoids. It will also be important to revisit the work of Bani et al. (1998), which showed porcine relaxin stimulates NO production and cGMP accumulation in bovine aortic VSMCs by increasing iNOS expression and activity. Decreases in intracellular Ca2 + concentrations, increases in myosin light chain phosphatase activity and calcium‐activated K+ channel activity all contribute to smooth muscle cell relaxation. Previous work on uterine smooth muscle has shown that porcine relaxin targets each of these pathways to cause relaxation (Nishikori et al., 1983; Rao and Sanborn, 1986; Meera et al., 1995). Based on this evidence, future research needs to investigate if relaxin acts directly on RXFP1 receptors in VSMCs and stimulates similar intracellular mechanisms to cause relaxation.
The evidence that relaxin can remodel the vasculature and improve arterial compliance is less compelling outside the setting of pregnancy. The inconsistency between studies is hampered by different experimental approaches (duration of relaxin treatment, age and sex of animals). Most studies in young rats report that rhRLX treatment fails to alter passive mechanical wall properties or circumferential wall stiffness, suggesting that rhRLX's beneficial effects are not evident in healthy animals. However, work in SHRs treated with rhRLX for 14 days revealed reduced collagen content and changes in aorta vessel diameter, improvements in carotid artery distensibility (Xu et al., 2010) and increased passive distensibility in brain PAs (Chan et al., 2013). Thus, we conclude that relaxin is capable of vascular remodelling, but further work will be needed to address the mechanisms of action as it is not clear that relaxin acts directly on the extracellular matrix to reduce arterial stiffness. Moreover, it needs to be established if the actions of relaxin on vascular remodelling are limited to disease conditions and specific vessel beds.
Conflict of interest
The authors disclose that this project was partially funded by Novartis Pharma AG, who also provided the serelaxin as a condition of an Australian Research Council Linkage Grant. L.J.P. was also a paid consultant for Novartis Pharma AG. K.P.C. is an inventor or co‐inventor of use patents for relaxin, and has served as a paid or unpaid consultant to Connetics, BAS Medical, Corthera, Novartis and other companies concerning the use of relaxin.
Acknowledgements
C. H. L. received the JN Peter's Research Fellowship and an Early Career Grant (Faculty of Science, The University of Melbourne). M. J. and S. M. received an Australian Postgraduate Award, and H. H. N. a Melbourne International Fee Remission Scholarship and a Melbourne International Research Scholarship. The research was funded by an Australian Research Council Linkage Grant (L. J. P. and M. T.), the National Health and Medical Research Council (L. J. P. and M. T.), Investigator‐Initiated Trials (L. J. P. and C. H. L.) from Novartis Pharmaceuticals Australia and the NIH (K. P. C. and J. N.). We thank Dr Dennis Stewart and Dr Elaine Unemori for their guidance and constant encouragement over the last decade, and Novartis Pharma AG for providing the relaxin.
Leo, C. H. , Jelinic, M. , Ng, H. H. , Marshall, S. A. , Novak, J. , Tare, M. , Conrad, K. P. , and Parry, L. J. (2017) Vascular actions of relaxin: nitric oxide and beyond. British Journal of Pharmacology, 174: 1002–1014. doi: 10.1111/bph.13614.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anumba DO, El Gelany S, Elliott SL, Li TC (2009). Serum relaxin levels are reduced in pregnant women with a history of recurrent miscarriage, and correlate with maternal uterine artery Doppler indices in first trimester. Eur J Obstet Gynecol Reprod Biol 147: 41–45. [DOI] [PubMed] [Google Scholar]
- Bani D, Failli P, Bello MG, Thiemermann C, Bani Sacchi T, Bigazzi M et al. (1998). Relaxin activates the L‐arginine‐nitric oxide pathway in vascular smooth muscle cells in culture. Hypertension 31: 1240–1247. [DOI] [PubMed] [Google Scholar]
- Bani‐Sacchi T, Bigazzi M, Bani D, Mannaioni PF, Masini E (1995). Relaxin‐induced increased coronary flow through stimulation of nitric oxide production. Br J Pharmacol 116: 1589–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bathgate RA, Halls ML, van der Westhuizen ET, Callander GE, Kocan M, Summers RJ (2013). Relaxin family peptides and their receptors. Physiol Rev 93: 405–480. [DOI] [PubMed] [Google Scholar]
- Bramham K, Seed PT, Lightstone L, Nelson‐Piercy C, Gill C, Webster P et al. (2016). Diagnostic and predictive biomarkers for pre‐eclampsia in patients with established hypertension and chronic kidney disease. Kidney Int 89: 874–885. [DOI] [PubMed] [Google Scholar]
- Chan SL, Cipolla MJ (2011). Relaxin causes selective outward remodeling of brain parenchymal arterioles via activation of peroxisome proliferator‐activated receptor‐γ. FASEB J 25: 3229–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SL, Sweet JG, Cipolla MJ (2013). Treatment for cerebral small vessel disease: effect of relaxin on the function and structure of cerebral parenchymal arterioles during hypertension. FASEB J 27: 3917–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow BS, Kocan M, Bosnyak S, Sarwar M, Wigg B, Jones ES et al. (2014). Relaxin requires the angiotensin II type 2 receptor to abrogate renal interstitial fibrosis. Kidney Int 86: 75–85. [DOI] [PubMed] [Google Scholar]
- Cipolla M, Osol G (1994). Hypertrophic and hyperplastic effects of pregnancy on the rat uterine arterial wall. Am J Obstet Gynecol 171: 805–811. [DOI] [PubMed] [Google Scholar]
- Conrad KP (2010). Unveiling the vasodilatory actions and mechanisms of relaxin. Hypertension 56: 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KP (2011a). Emerging role of relaxin in the maternal adaptations to normal pregnancy: implications for preeclampsia. Semin Nephrol 31: 15–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KP (2011b). Maternal vasodilation during pregnancy: emerging role of relaxin. Am J Physiol Regul Integr Comp Physiol 301: R267–R275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KP (2016). G‐Protein‐coupled receptors as potential drug candidates in preeclampsia: targeting the relaxin/insulin‐like family peptide receptor 1 for treatment and prevention. Hum Reprod 22: 647–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KP, Shroff SG (2011). Effects of relaxin on arterial dilation, remodeling, and mechanical properties. Curr Hypertens Rep 13: 409–420. [DOI] [PubMed] [Google Scholar]
- Conrad KP, Debrah DO, Novak J, Danielson LA, Shroff SG (2004). Relaxin modifies systemic arterial resistance and compliance in conscious, nonpregnant rats. Endocrinology 145: 3289–3296. [DOI] [PubMed] [Google Scholar]
- Cooke C‐LM, Davidge ST (2003). Pregnancy‐induced alterations of vascular function in mouse mesenteric and uterine arteries. Biol Reprod 68: 1072–1077. [DOI] [PubMed] [Google Scholar]
- Debrah DO, Debrah JE, Haney JL, McGuane JT, Sacks MS, Conrad KP et al. (2011). Relaxin regulates vascular wall remodeling and passive mechanical properties in mice. J Appl Physiol 111: 260–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debrah DO, Novak J, Matthews JE, Ramirez RJ, Shroff SG, Conrad KP (2006). Relaxin is essential for systemic vasodilation and increased global arterial compliance during early pregnancy in conscious rats. Endocrinology 147: 5126–5131. [DOI] [PubMed] [Google Scholar]
- Dschietzig T, Bartsch C, Richter C, Laule M, Baumann G, Stangl K (2003). Relaxin, a pregnancy hormone, is a functional endothelin‐1 antagonist: attenuation of endothelin‐1‐mediated vasoconstriction by stimulation of endothelin type‐B receptor expression via ERK‐1/2 and nuclear factor‐kappaB. Circ Res 92: 32–40. [DOI] [PubMed] [Google Scholar]
- Dschietzig T, Brecht A, Bartsch C, Baumann G, Stangl K, Alexiou K (2012). Relaxin improves TNF‐α‐induced endothelial dysfunction: the role of glucocorticoid receptor and phosphatidylinositol 3‐kinase signalling. Cardiovasc Res 95: 97–107. [DOI] [PubMed] [Google Scholar]
- Du XJ, Samuel CS, Gao XM, Zhao L, Parry LJ, Tregear GW (2003). Increased myocardial collagen and ventricular diastolic dysfunction in relaxin deficient mice: a gender‐specific phenotype. Cardiovasc Res 57: 395–404. [DOI] [PubMed] [Google Scholar]
- Edwards G, Feletou M, Weston AH (2010). Endothelium‐derived hyperpolarising factors and associated pathways: a synopsis. Pflugers Arch 459: 863–879. [DOI] [PubMed] [Google Scholar]
- Faraci FM, Mayhan WG, Heistad DD (1987). Segmental vascular responses to acute hypertension in cerebrum and brain stem. Am J Physiol 252: H738–H742. [DOI] [PubMed] [Google Scholar]
- Fisher C, MacLean M, Morecroft I, Seed A, Johnston F, Hillier C et al. (2002). Is the pregnancy hormone relaxin also a vasodilator peptide secreted by the heart? Circulation 106: 292–295. [DOI] [PubMed] [Google Scholar]
- Gooi JH, Richardson ML, Jelinic M, Girling JE, Wlodek ME, Tare M et al. (2013). Enhanced uterine artery stiffness in aged pregnant relaxin mutant mice is reversed with exogenous relaxin treatment. Biol Reprod 89: 18. [DOI] [PubMed] [Google Scholar]
- Halls ML, Bathgate RA, Sutton SW, Dschietzig TB, Summers RJ (2015). International Union of Basic and Clinical Pharmacology. XCV. Recent advances in the understanding of the pharmacology and biological roles of relaxin family peptide receptors 1‐4, the receptors for relaxin family peptides. Pharmacol Rev 67: 389–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel E (2006). Perivascular nerves and the regulation of cerebrovascular tone. J Appl Physiol 100: 1059–1064. [DOI] [PubMed] [Google Scholar]
- Hermsteiner M, Zoltan DR, Künzel W (2001). The vasoconstrictor response of uterine and mesenteric resistance arteries is differentially altered in the course of pregnancy. Eur J Obstet Gynecol Reprod Biol 100: 29–35. [DOI] [PubMed] [Google Scholar]
- Jelinic M, Leo CH, Post Uiterweer ED, Sandow SL, Gooi JH, Wlodek ME et al. (2014). Localization of relaxin receptors in arteries and veins, and region‐specific increases in compliance and bradykinin‐mediated relaxation after in vivo serelaxin treatment. FASEB J 28: 275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelinic M, Tare M, Conrad KP, Parry LJ (2015). Differential effects of relaxin deficiency on vascular aging in arteries of male mice. Age 37: 9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyabalan A, Novak J, Danielson LA, Kerchner LJ, Opett SL, Conrad KP (2003). Essential role for vascular gelatinase activity in relaxin‐induced renal vasodilation, hyperfiltration, and reduced myogenic reactivity of small arteries. Circ Res 93: 1249–1257. [DOI] [PubMed] [Google Scholar]
- Jeyabalan A, Novak J, Doty KD, Matthews J, Fisher MC, Kerchner LJ et al. (2007). Vascular matrix metalloproteinase‐9 mediates the inhibition of myogenic reactivity in small arteries isolated from rats after short‐term administration of relaxin. Endocrinology 148: 189–198. [DOI] [PubMed] [Google Scholar]
- Johal T, Lees CC, Everett TR, Wilkinson IB (2014). The nitric oxide pathway and possible therapeutic options in pre‐eclampsia. Br J Clin Pharamacol 78: 244–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerchner LJ, Novak J, Hanley‐Yanez K, Doty KD, Danielson LA, Conrad KP (2005). Evidence against the hypothesis that endothelial endothelin B receptor expression is regulated by relaxin and pregnancy. Endocrinology 146: 2791–2797. [DOI] [PubMed] [Google Scholar]
- Kim TH, Weiner CP, Thompson LP (1994). Effect of pregnancy on contraction and endothelium‐mediated relaxation of renal and mesenteric arteries. Am J Physiol 267: H41–H47. [DOI] [PubMed] [Google Scholar]
- Lafayette RA, Hladunewich MA, Derby G, Blouch K, Druzin ML, Myers BD (2011). Serum relaxin levels and kidney function in late pregnancy with or without preeclampsia. Clin Nephrol 75: 226–232. [DOI] [PubMed] [Google Scholar]
- Leo CH, Jelinic M, Gooi JH, Tare M, Parry LJ (2014a). A vasoactive role for endogenous relaxin in mesenteric arteries of male mice. PLoS One 9: e107382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo CH, Jelinic M, Ng HH, Tare M, Parry LJ (2016a). Serelaxin: a novel therapeutic for vascular diseases. Trends Pharmacol Sci 37: 498–507. [DOI] [PubMed] [Google Scholar]
- Leo CH, Jelinic M, Ng HH, Tare M, Parry LJ (2016b). Time‐dependent activation of prostacyclin and nitric oxide pathways during continuous intravenous infusion of serelaxin (recombinant human H2 relaxin). Br J Pharmacol 173: 1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo CH, Jelinic M, Parkington HC, Tare M, Parry LJ (2014b). Acute intravenous injection of serelaxin (recombinant human relaxin‐2) causes rapid and sustained bradykinin‐mediated vasorelaxation. J Am Heart Assoc 3: e000493–e000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Brookes ZL, Kaufman S (2005). Acute and chronic effects of relaxin on vasoactivity, myogenic reactivity and compliance of the rat mesenteric arterial and venous vasculature. Regul Pept 132: 41–46. [DOI] [PubMed] [Google Scholar]
- Luksha L, Agewall S, Kublickiene K (2009). Endothelium‐derived hyperpolarizing factor in vascular physiology and cardiovascular disease. Atherosclerosis 202: 330–344. [DOI] [PubMed] [Google Scholar]
- Luksha L, Luksha N, Kublickas M, Nisell H, Kublickiene K (2010). Diverse mechanisms of endothelium‐derived hyperpolarizing factor‐mediated dilatation in small myometrial arteries in normal human pregnancy and preeclampsia. Biol Reprod 83: 728–735. [DOI] [PubMed] [Google Scholar]
- Majed BH, Khalil RA (2012). Molecular mechanisms regulating the vascular prostacyclin pathways and their adaptation during pregnancy and in the newborn. Pharmacol Rev 64: 540–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall SA, Leo CH, Senadheera SN, Girling JE, Tare M, Parry LJ (2016). Relaxin deficiency attenuates pregnancy‐induced adaptation of the mesenteric artery to angiotensin II in mice. Am J Physiol Regul Integr Comp Physiol 310: R847–R857. [DOI] [PubMed] [Google Scholar]
- Massicotte G, Parent A, St‐Louis J (1989). Blunted responses to vasoconstrictors in mesenteric vasculature but not in portal vein of spontaneously hypertensive rats treated with relaxin. Proc Soc Exp Biol Med 190: 254–259. [DOI] [PubMed] [Google Scholar]
- McGuane JT, Danielson LA, Debrah JE, Rubin JP, Novak J, Conrad KP (2011a). Angiogenic growth factors are new and essential players in the sustained relaxin vasodilatory pathway in rodents and humans. Hypertension 57: 1151–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuane JT, Debrah JE, Sautina L, Jarajapu YP, Novak J, Rubin JP et al. (2011b). Relaxin induces rapid dilation of rodent small renal and human subcutaneous arteries via PI3 kinase and nitric oxide. Endocrinology 152: 2786–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meera P, Anwer K, Monga M, Oberti C, Stefani E, Toro L et al. (1995). Relaxin stimulates myometrial calcium‐activated potassium channel activity via protein kinase A. Am J Physiol 269: C312–C317. [DOI] [PubMed] [Google Scholar]
- Ng HH, Jelinic M, Parry LJ, Leo CH (2015). Increased superoxide production and altered nitric oxide‐mediated relaxation in the aorta of young but not old male relaxin‐deficient mice. Am J Physiol Heart Circ Physiol 309: H285–H296. [DOI] [PubMed] [Google Scholar]
- Ng HH, Leo CH, Parry LJ (2016). Serelaxin (recombinant human relaxin‐2) prevents high glucose‐induced endothelial dysfunction by ameliorating prostacyclin production in the mouse aorta. Pharmacol Res 107: 220–228. [DOI] [PubMed] [Google Scholar]
- Nishikori K, Weisbrodt NW, Sherwood OD, Sanborn BM (1983). Effects of relaxin on rat uterine myosin light chain kinase activity and myosin light chain phosphorylation. J Biol Chem 258: 2468–2474. [PubMed] [Google Scholar]
- Novak J, Danielson LA, Kerchner LJ, Sherwood OD, Ramirez RJ, Moalli PA et al. (2001). Relaxin is essential for renal vasodilation during pregnancy in conscious rats. J Clin Invest 107: 1469–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak J, Parry LJ, Matthews JE, Kerchner LJ, Indovina K, Hanley‐Yanez K et al. (2006). Evidence for local relaxin ligand‐receptor expression and function in arteries. FASEB J 20: 2352–2362. [DOI] [PubMed] [Google Scholar]
- Novak J, Ramirez RJ, Gandley RE, Sherwood OD, Conrad KP (2002). Myogenic reactivity is reduced in small renal arteries isolated from relaxin‐treated rats. Am J Physiol Regul Integr Comp Physiol 283: R349–R355. [DOI] [PubMed] [Google Scholar]
- Novak J, Reckelhoff J, Bumgarner L, Cockrell K, Kassab S, Granger JP (1997). Reduced sensitivity of the renal circulation to angiotensin II in pregnant rats. Hypertension 30: 580–584. [DOI] [PubMed] [Google Scholar]
- O'Sullivan KP, Marshall SA, Cullen S, Saunders T, Hannan NJ, Senadheera SN et al. (2016). Evidence of proteinuria in relaxin‐deficient mice but no other characteristics of preeclampsia. Reprod Fertil Dev. doi: 10.1071/RD16056. [DOI] [PubMed] [Google Scholar]
- Osol G, Mandala M (2009). Maternal uterine vascular remodeling during pregnancy. Physiology 24: 58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pini A, Boccalini G, Baccari MC, Becatti M, Garella R, Fiorillo C et al. (2016). Protection from cigarette smoke‐induced vascular injury by recombinant human relaxin‐2 (serelaxin). J Cell Mol Med 20: 891–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MR, Sanborn BM (1986). Relaxin increases calcium efflux from rat myometrial cells in culture. Endocrinology 119: 435–437. [DOI] [PubMed] [Google Scholar]
- Sandow SL, Senadheera S, Bertrand PP, Murphy TV, Tare M (2012). Myoendothelial contacts, gap junctions, and microdomains: anatomical links to function? Microcirculation 19: 403–415. [DOI] [PubMed] [Google Scholar]
- Sarwar M, Samuel CS, Bathgate RA, Stewart DR, Summers RJ (2015). Serelaxin‐mediated signal transduction in human vascular cells: bell‐shaped concentration‐response curves reflect differential coupling to G proteins. Br J Pharmacol 172: 1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarwar M, Samuel CS, Bathgate RA, Stewart DR, Summers RJ (2016). Enhanced serelaxin signalling in co‐cultures of human primary endothelial and smooth muscle cells. Br J Pharmacol 173: 484–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MC, Murdoch AP, Danielson LA, Conrad KP, Davison JM (2006). Relaxin has a role in establishing a renal response in pregnancy. Fertil Steril 86: 253–255. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart DR, Celniker AC, Taylor CAJ, Cragun JR, Overstreet JW, Lasley BL (1990). Relaxin in the peri‐implantation period. J Clin Endocrinol Metab 70: 1771–1773. [DOI] [PubMed] [Google Scholar]
- Tiyerili V, Beiert T, Schatten H, Camara B, Jehle J, Schrickel JW et al. (2016). Anti‐atherosclerotic effects of serelaxin in apolipoprotein E‐deficient mice. Atherosclerosis . doi:10.1016/j.atherosclerosis.2016.06.008. [DOI] [PubMed] [Google Scholar]
- Uiterweer EDP, Koster MP, Jeyabalan A, Kuc S, Siljee J, Conrad KP et al. (2014). First trimester serum relaxin concentration and prediction of early and late onset preeclampsia [Abstract]. Reprod Sci 21: 394A. [Google Scholar]
- van der Heijden OWH, Essers YPG, Fazzi G, Peeters LLH, De Mey JGR, van Eys GJJM (2005). Uterine artery remodeling and reproductive performance are impaired in endothelial nitric oxide synthase‐deficient mice. Biol Reprod 72: 1161–1168. [DOI] [PubMed] [Google Scholar]
- van Drongelen J, Ploemen IH, Pertijs J, Gooi JH, Sweep FC, Lotgering FK et al. (2011). Aging attenuates the vasodilator response to relaxin. Am J Physiol Heart Circ Physiol 300: H1609–H1615. [DOI] [PubMed] [Google Scholar]
- van Drongelen J, van Koppen A, Pertijs J, Gooi JH, Parry LJ, Sweep FC et al. (2012). Impaired vascular responses to relaxin in diet‐induced overweight female rats. J Appl Physiol 112: 962–969. [DOI] [PubMed] [Google Scholar]
- van Drongelen J, van Koppen A, Pertijs J, Gooi JH, Sweep FC, Lotgering FK et al. (2013). Impaired effect of relaxin on vasoconstrictor reactivity in spontaneous hypertensive rats. Peptides 49: 41–48. [DOI] [PubMed] [Google Scholar]
- Vodstrcil LA, Tare M, Novak J, Dragomir N, Ramirez RJ, Wlodek ME et al. (2012). Relaxin mediates uterine artery compliance during pregnancy and increases uterine blood flow. FASEB J 26: 4035–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Chakravorty A, Bathgate RA, Dart AM, Du XJ (2010). Relaxin therapy reverses large artery remodeling and improves arterial compliance in senescent spontaneously hypertensive rats. Hypertension 55: 1260–1266. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Vanhoutte PM, Leung SW (2015). Vascular nitric oxide: beyond eNOS. J Pharmacol Sci 129: 83–94. [DOI] [PubMed] [Google Scholar]