

Abstract
Background and Purpose
Gastric cancer is one of the leading causes of morbidity and mortality worldwide. Akt is an anti‐apoptotic kinase that plays a dynamic role in cell survival and is implicated in the pathogenesis of gastric cancer. MK‐2206, the first allosteric inhibitor of Akt, is in clinical trials for a number of cancers. Although preclinical studies showed promise, clinical trials reported it had no effect when given alone at tolerated doses. The aim of our study was to delineate the effects of MK‐2206 on gastric cancer cells and explore the ability of combination treatments to enhance the anti‐tumour activity of MK‐2206.
Experimental Approach
SGC‐7901, BGC‐823 cells and immunodeficient mice were chosen as a model to study the treatment effects. Changes in cell viability, apoptosis and ROS, endoplasmic reticulum stress and mitochondrial dysfunction in the cells were analysed by MTT assays, ROS imaging and FACSCalibur, electron microscopy, JC‐1 staining and western blotting.
Key Results
MK‐2206 induced apoptotic cell death through the generation of ROS. We utilized ROS production to target gastric cancer cells by combining MK‐2206 and an ROS inducer EF24. Our in vitro and in vivo xenograft studies showed that combined treatment with MK‐2206 and EF24 synergistically induced apoptosis in gastric cancer cells and caused cell cycle arrest. These activities were mediated through ROS generation and the induction of endoplasmic reticulum stress and mitochondrial dysfunction.
Conclusion and Implications
Targeting ROS generation by using a combination of an Akt inhibitor and EF24 could have potential as a therapy for gastric cancer.
Abbreviations
- ATF‐4
activating transcription factor 4
- Cdc2
cyclin‐dependent kinase 1 (cell division cycle protein 2)
- CHOP
CAAT/enhancer‐binding protein homologous protein
- DCFH‐DA
2′,7′‐dichlorodihydrofluorescein diacetate
- EIF2
eukaryotic initiation factor 2
- ER
endoplasmic reticulum
- HRP
horseradish peroxidase
- JC‐1
cationic carbocyanine dye
- Ki‐67
nuclear protein associated with cell proliferation
- MDA
malondialdehyde
- MDM‐2
murine double minute 2
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- NAC
N‐acetyl cysteine
- PARP
poly (ADP‐ribose) polymerase
- PI
propidium iodide
Tables of Links
| TARGETS | |
|---|---|
| Other protein targets a | Enzymes b |
| Bax | Akt (PKB) |
| Bcl‐2 | Caspase‐3 |
| EIF2α | |
| JNK | |
| mTOR | |
| PARP | |
| PI3K | |
| PTEN |
| LIGANDS |
|---|
| MK‐2206 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a,b).
Introduction
Gastric cancer is the fourth most common malignancy and is ranked as the second leading cause of cancer death worldwide (Siegel et al., 2015). With an ageing population, the incidence of gastric cancer is gradually increasing (Ferlay et al., 2010). The aetiology of gastric cancer is multifactorial and associated with socio‐economic status, poor dietary habits, helicobacter pylori infection, genetic variations, age and family history (Meitzler et al., 2014). Chemotherapy is an extremely important therapeutic strategy for advanced gastric cancer; however, chemotherapy resistance is a problem, particularly for patients who have progressive cancer and/or had it reoccur subsequent to their first‐line treatment (Kim and Park, 2015; Marin et al., 2016).
A promising target for cancer treatment, including gastric cancer, is the Akt protein in the PI3K pathway. In this signalling pathway, stimulation of receptor tyrosine kinases or GPCRs activates PI3K, which in turn activates Akt. Activated/phosphorylated Akt modulates signals from phosphatase and tensin homologue (PTEN) and the mammalian target of rapamycin (mTOR) to induce various effects on cells. In the context of tumourigenesis, Akt signalling mediates proliferation and cell metastasis, suppression of cell apoptosis and angiogenesis (Tokunaga et al., 2008). Gene mutations and amplifications of PIK3CA (PI3K, catalytic α polypeptide), amplification of Akt and a loss of PTEN can all cause activation of the PI3K‐Akt–mTOR pathway. In gastric cancer, such mutations or amplifications are present in 30–60% of cases (Markman et al., 2010). A recent study also showed Akt expression in 74% of the gastric tumours and p‐Akt expression in 78% using tissue microarrays (Nam et al., 2003). These findings suggest that Akt may be targeted for cancer therapy. In fact, the first allosteric inhibitor of Akt MK‐2206, with nanomolar inhibitory potency against Akt1, Akt2 and Akt3, is now being tested in adult cancers (Yan, 2009; Yap et al., 2011). MK‐2206 inhibits Akt phosphorylation at both Thr308 and Ser473 (Yan, 2009; Yap et al., 2011). In vitro and in vivo studies examining the effect of MK‐2206 on gastric cancer cells are essentially lacking. MK‐2206 has only been shown to reduce gastric cancer cell growth and induce apoptosis when used in combination with platinum‐based chemotherapeutic drugs and mTOR inhibitors (Almhanna et al., 2013; Ji et al., 2014; Tao et al., 2016). This may be due to the fact that like many other cancer treatments, beneficial effects are not seen with maximum tolerated doses. Indeed, this is the case in acute myelogenous leukaemia (Konopleva et al., 2014). Therefore, research efforts are directed at finding combinations of treatments, which enhance the effect of MK‐2206.
Cancer cells seem to function with higher levels of endogenous oxidative stress in culture and in vivo compared with their normal counterparts (Perry et al., 2000; Boonstra and Post, 2004). Several intrinsic and extrinsic mechanisms are believed to induce oxidative stress during cancer development and disease progression (Trachootham et al., 2009). Interestingly, ROS and oxidative stress can also be utilized to suppress cancer growth (Kardeh et al., 2014). Numerous studies show that an elevation of ROS production in cancer cells may lead to growth inhibition. We have shown this anti‐tumour mechanism in gastric cancer cells (Zou et al., 2015a; Zou et al., 2016a; Zou et al., 2016b). Therefore, ROS production could be an important target for combination treatment with MK‐2206. One inducer of ROS that may be utilized is a novel curcumin analogue EF24 (Thomas et al., 2008) that inhibits the proliferation of a variety of cancer cells in vitro and in vivo (Subramaniam et al., 2008). In our previous study, we showed that EF24 inhibits gastric cancer cell growth by inducing an increase in ROS (Zou et al., 2016b), which supports the use of EF24 in combination with MK‐2206 to treat gastric cancer.
In the present study, we examined the anti‐tumour activity of MK‐2206 in gastric cancer cells and in a human gastric cancer xenograft model. We also tested the effect of combining MK‐2206 and EF24 and investigated the underlying mechanisms of their anti‐tumour activities. Our results show that MK‐2206 and EF24 increase ROS production in gastric cancer cells. Combining the two treatments caused significant cell cycle arrest and the induction of apoptosis. We also showed that these effects were mediated through ROS‐induced endoplasmic reticulum (ER) stress and mitochondrial dysfunction in gastric cancer cells.
Methods
Cell culture and reagents
Normal human gastric epithelial cell line GES‐1 and human gastric cancer cell lines SGC‐7901 and BGC‐ 823 were purchased from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). All cell types were cultured in RPMI 1640 (Gibco, Eggenstein, Germany) supplemented with 10% heat‐inactivated FBS (Hyclone, Logan, UT, USA), 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin. N‐acetyl cysteine (NAC) was purchased from Sigma‐Aldrich (St. Louis, MO, USA). FITC Annexin V Apoptosis Detection Kit I and propidium iodide (PI) were purchased from BD Pharmingen (Franklin Lakes, NJ, USA). ROS probe 2′,7′‐dichlorodihydrofluorescein diacetate (DCFH‐DA) and mitochondrial integrity probe JC‐1 were purchased from Thermo Fisher (Carlsbad, CA, USA). Antibodies against cell division cycle protein 2 (Cdc2), B‐cell lymphoma 2 (Bcl‐2), Bax, cyclin B1, cleaved PARP, murine double minute 2 (MDM‐2), Ki67 and GAPDH were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against activating transcription factor‐4 (ATF‐4), eukaryotic initiation factor 2 (EIF2α and phospho/p‐EIF2α), p‐Akt, CCAAT/enhancer‐binding protein homologous protein (CHOP), JNK and p‐JNK and cleaved caspase 3 were purchased from Cell Signalling Technology (Danvers, MA, USA). HRP‐conjugated secondary antibodies were obtained from Santa Cruz.
Cell viability assay
To measure viability following MK‐2206 treatment, cells were seeded on 96‐well plates at a density of 8 × 103 per well. Cells were allowed to attach overnight in complete growth media and then treated with MK‐2206 (dissolved in DMSO; diluted in RPMI medium) for 24 h before performing the 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay. Combined treatments with MK‐2206 and EF24 were carried out at the same time.
Determination of intracellular ROS
Intracellular ROS contents were measured by flow cytometry utilizing DCFH‐DA. Briefly, 5 × 105 cells were plated on 60 mm dishes, allowed to attach overnight and then treated with MK‐2206 (5 or 10 μM) and EF24 (2 μM) for 2 h. NAC pretreatment, where indicated, was carried out for 2 h. Cells were stained with 10 μM DCFH‐DA at 37°C for 30 min in the dark. DCF fluorescence (produced in the presence of ROS) was analysed using flow cytometry and Nikon epifluorescence microscope equipped with a digital camera (Nikon, Japan).
Cell cycle and apoptosis analysis
For cell cycle analysis, SGC‐7901 and BGC‐823 cells were treated with MK‐2206 (5 or 10 μM) and EF24 (2 μM) for 15 h. Cells were then stained with PI at a final concentration of 0.05 mg·mL−1 and incubated at 4°C for 20 min in the dark. Cell cycle analysis was performed in FACSCalibur. For apoptosis determination, SGC‐7901 and BGC‐823 cells were treated with MK‐2206 (5 or 10 μM) and EF24 (2 μM) for 24 h. Cells were then harvested and apoptotic cell death was evaluated by double staining cells with FITC‐conjugated Annexin V and PI for 30 min.
Colony formation assay
Cells were seeded at 500 cells per well in 6‐well plates and treated with either MK‐2206, EF24 or a combination of the two. Cells were allowed to grow for 8 days and stained with crystal violet solution (0.5 in 25% methanol) to assess colony growth.
Western blot analysis
Lysates from cells and tumour tissues were prepared and protein levels determined using the Bradford assay (Bio‐Rad, Hercules, CA, USA). Proteins were separated by 10% SDS‐PAGE and transferred to PVDF transfer membranes. The blots were blocked for 2 h at room temperature with freshly prepared 5% nonfat milk in TBST and then incubated with specific primary antibodies overnight at 4°C. HRP‐conjugated secondary antibodies and ECL substrate (Bio‐Rad) were used for detection.
Transient transfection of small interfering RNA (siRNA)
The siRNA duplexes against the human Akt gene used in this study were purchased from Invitrogen (Carlsbad, CA, USA) and Sigma‐Aldrich (St. Louis, MO, USA). Sequence 1: (Sense: 5′‐GCACUUUCGGCAAGGUGAUdTdT‐3′, antisense: 5′‐AUCACCUUGCCGAAAGUGCdTdT‐3′). Sequence 2: (Sense: 5′‐GACGGGCACAUUAAGAUCAdTdT‐3′, antisense: 5′‐UGAUCUUAAUGUGCCCGUCdTdT‐3′). SGC‐7901 and BGC‐823cells (3 × 105 per well) were seeded into 6‐well plates and cultured for 24 h, and then were transfected with siRNA duplexes (100 nM) or control siRNA by lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Cells were further incubated for 48 h before harvest for detection of Akt expression by western blots.
Electron microscopy
SGC‐7901 cells were treated with vehicle control (DMSO) or 10 μM MK‐2206 in combination with 2 μM EF24. NAC pretreatment where applicable was carried out for 2 h. Following treatment, cells were fixed with 2.5% glutaraldehyde overnight at 4°C. The cells were then post‐fixed in 1% OsO4 at room temperature for 60 min, stained with 1% uranyl acetate, dehydrated through graded acetone solutions, and embedded in epon. Areas containing cells were block‐mounted and cut into 70 nm sections and examined with an electron microscope (H‐7500, Hitachi, Ibaraki, Japan).
Evaluation of mitochondrial membrane potential (Δψm)
The effects of MK‐2206 and EF24 on the cell mitochondrial membrane potential (Δψm) were examined by fluorescence microscopy using JC‐1 (Thermo Fisher). JC‐1 is a cationic carbocyanine dye that accumulates in the mitochondria. Upon changes to membrane potential, JC‐1 leaks out into the cytosol. Cells were exposed 10 μM MK‐2206 in combination with 2 μM EF24 for 12 h, with or without 2 h pretreatment with NAC. Fluorescence images were acquired by using a Nikon epi‐fluorescence microscope.
MitoSOX™ red mitochondrial superoxide assay
SGC‐7901 or BGC‐823 cells were seeded in 6‐well plates and cultured for 24 h, and then treated with MK‐2206 (10 μM) and EF24 (2 μM). Mitochondrial superoxide was measured using the MitoSOX™ Red mitochondrial superoxide indicator (Thermo Fisher Scientific #M36008, Waltham, MA, USA) according to the manufacturer's protocol.
ATP assay
SGC‐7901 or BGC‐823 cells were seeded in 6‐well plates and cultured for 24 h, and then treated with MK‐2206 (10 μM) and EF24 (2 μM). Cellular ATP content was measured using a commercial kit (Cat No. #S0026, Beyotime Biotech, Nanjing, China) according to the manufacturer's protocol.
Gastric cancer xenografts
Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015). All experimental procedures were approved by the Institutional and Local Committee on the Care and Use of Animals of Wenzhou Medical College, and all animals received humane care according to the National Institutes of Health (USA) guidelines. Five‐week‐old athymic BALB/c ν/ν female mice (18–22 g; total n = 32) were purchased from Vital River Laboratories (Beijing, China). Animals were housed at a constant room temperature with a 12/12 h light/dark cycle and fed a standard rodent diet. The mice were divided into four experimental groups on randomization and blinding with eight mice in each group. SGC‐7901 cells (1 × 106) in 0.1 mL PBS was injected s.c. into the right flank of each mouse. When tumours reached a volume of 100–200 mm3, mice were treated by i.p. injection of 10 mg·kg−1 MK‐2206, 3 mg·kg−1 EF24 or a combination of MK‐2206 and EF24 (in a solution of PBS with 6% castor oil) once a day. Control mice received the same vehicle. All mice were treated for 14 days. The tumour volumes were determined by measuring length (l) and width (w) and calculating volume (V = 0.5 × l × w2) at the indicated time points. At the end of the experiments, the mice were killed after being anaesthetised by i.p. injection with pentobarbital (50 mg·kg−1), and their tumours were isolated by surgery in a room separated from the other animals, then weighed for in vitro experiments. Samples were prepared for histology and protein assays.
Immunohistochemistry and histology
The harvested tumour tissues were fixed in 10% formalin and embedded in paraffin. Five‐micrometre‐thick sections were placed on positively charged slides. Tissue sections were stained using routine immunohistochemical techniques and primarily antibodies against p‐Akt (1:100), cleaved caspase 3 (1:100) or Ki‐67 (1:200) overnight. The signal was detected using 3,3'N‐Diaminobenzidine Tertrahydrochloride (DAB). Heart, kidney and liver sections were also stained with H&E for histological analysis and assessment of potential toxicity.
Malondialdehyde (MDA) assay
Tumour samples from mice were homogenized and sonicated. Tissue lysates were then centrifuged at 12 000 × g for 10 min at 4°C to collect the supernatant. Total protein content was determined by using the Bradford assay. MDA levels were measured by using a Lipid Peroxidation MDA assay kit (Beyotime Institute of Biotechnology).
Statistical analysis
All experiments are randomized and blinded. The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data are reported as mean ± SEM. Statistical analysis was performed with GraphPad Prism 5.0 software (San Diego, CA, USA). In accordance with journal policy, statistical analysis was performed only when a minimum of n = 5 independent samples was acquired. We used one‐way ANOVA followed by Dunnett's post hoc test when comparing more than two groups of data and one‐way ANOVA, non‐parametric Kruskal–Wallis test followed by Dunn's post hoc test when comparing multiple independent groups. When comparing two groups, the unpaired t‐test was used. Differences between group means were considered statistically significant when a value of at least P < 0.05 was achieved. Post‐tests were run only if F achieved P < 0.05 and there was no significant variance in homogeneity.
Results
MK‐2206 effectively suppresses Akt phosphorylation and gastric cancer cell growth, and induces apoptosis
MK‐2206 has been shown to block Akt phosphorylation in hepatic carcinoma cells (Grabinski et al., 2012). We first tested whether MK‐2206 inhibits Akt phosphorylation in gastric cancer cells by examining the levels of p‐Akt (Ser473) in SGC‐7901 and BGC‐823 cells. As show in Figure 1A, gastric cancer cells treated with MK‐2206 exhibited significantly reduced levels of p‐Akt (Ser473), and this was achieved with a low concentration of 0.1 μM. Moreover, total Akt levels did not change upon MK‐2206 treatment. We then treated cells with increasing concentrations of MK‐2206 and assessed cell viability using the MTT assay. Treatment of SGC‐7901 and BGC‐823 with MK‐2206 reduced cell viability, as shown in Figure 1B, C (IC50 = 23.62 μM for BGC‐823 and 25.72 μM for SGC‐7901). MK‐2206 also showed the same effect on normal gastric epithelial cells (IC50 = 18.13 μM; Figure 1C). It is important to note that MK‐2206 inhibited Akt phosphorylation at a concentration that produced little, if any, effect on cell viability in both normal and gastric cancer cells. This suggests that the mechanism underlying reduced cell viability by MK‐2206 may be uncoupled from its activity on Akt phosphorylation. Uncovering the mechanisms employed at high concentrations of MK‐2206 is essential in order to enhance this activity for effective cancer treatment.
Figure 1.
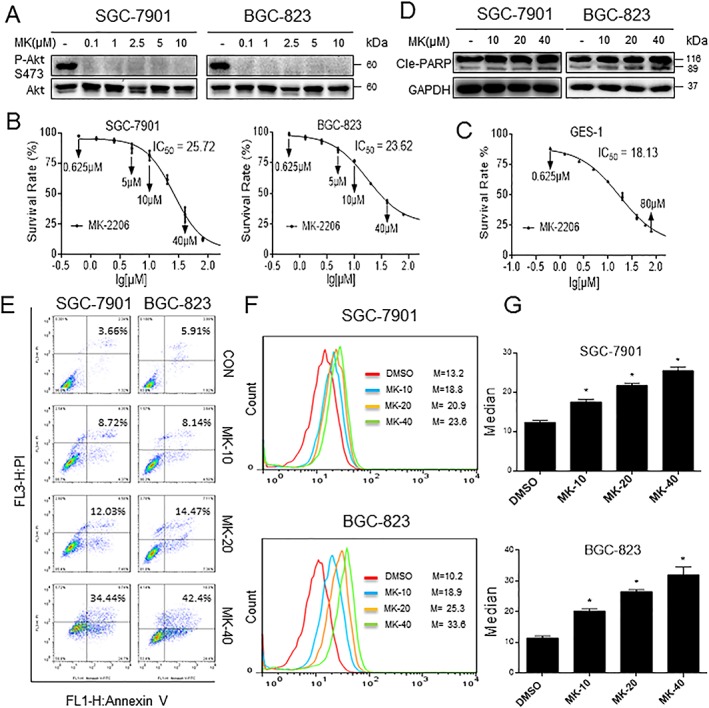
MK‐2206 reduces gastric cancer cell viability and generates ROS. (A) Effect of MK‐2206 treatment on gastric cancer cell lines showing effective inhibition of Akt phosphorylation. (B–C) Cells were treated with MK‐2206 for 24 h, and cell viability was measured. Figure showing reduced viability of human gastric cancer cells (B) and normal cells (C) upon MK‐2206 exposure. (D) Gastric cancer cells were treated with MK‐2206 for 16 h, and cleaved‐PARP levels were determined by western blot. (E) Induction of apoptosis in human gastric cancer cells was determined by Annexin V/PI staining following treatment with MK‐2206 for 24 h (CON = control; MK‐10, −20 and −40 = 10, 20 and 40 μM MK‐2206). (F) Intracellular ROS generation by MK‐2206 was measured in SGC‐7901 and BGC‐823 cells by redox‐sensitive dye DCFH‐DA. (G) Quantification of DCF data from (F). All representative images are from five independent experiments. Data are reported as mean ± SEM and analysed by Student's t‐test; n = 5 independent experiments; *P < 0.05 compared with DMSO control.
We determined if apoptosis contributed by assessing cleaved PARP (marker of apoptosis) levels by western blotting and Annexin V/PI staining of cells. Both gastric cancer cell lines showed concentration‐dependent cell apoptosis following MK‐2206 exposure as evidenced by increased cleaved PARP levels and Annexin/PI staining (Figure 1D, E). A higher concentration of MK‐2206 than IC50 values was needed to induce a significant apoptosis with PI/Annexin V, indicating multiple mechanisms are involved in the effect of this drug on cancer cells. Previous studies have shown that ROS are critical regulators of apoptosis, and reports have also indicated that MK‐2206 increases ROS levels (Cheng et al., 2011). Therefore, we sought to determine whether MK‐2206‐induced apoptosis was mediated through increased ROS production. We measured ROS levels in both gastric cancer cell lines by use of the redox‐sensitive fluorescence probe DCFH‐DA. DCFH‐DA is deacteylated once inside the cell and is oxidized to DCF in the presence of ROS. As shown in Figure 1F, G, treatment of cells with MK‐2206 for 2 h caused a dose‐dependent increase in ROS levels. These findings show that MK‐2206 led to increased ROS production in addition to its expected role as an Akt inhibitor. To determine whether MK‐2206‐induced ROS production is due to Akt inhibition or not, we constructed two separate Akt siRNA to knockdown the Akt expression in SGC‐7901 or BGC‐823 cells (Supporting Information Fig. S1a). As shown in Supporting Information Fig. S2b). The knockdown of Akt did not increase ROS accumulation, and MK‐2206 still increased the level of ROS in both Akt‐negative BGC and SGC cells, indicating that the ability of MK‐2206 to increase ROS is not dependent on Akt.
EF24 enhances ROS production by MK‐2206
We have previously shown that EF24 induces ROS generation in gastric cancer cell lines through modulation of thioredoxin reductase (Zou et al., 2016b). We wanted to know whether the addition of EF24 to the MK‐2206 treatment potentiates ROS generation. Indeed, the combination of EF24 and MK‐2206 led to significantly higher ROS levels compared with single treatments alone (Figure 2A and Supporting Information Fig. S2a). As expected, this increased level of ROS was not seen when cells were pretreated with NAC (Figure 2B and Supporting Information Fig. S2b). A representative image of DCF‐positive cells is provided in Figure 2C. Collectively, our results revealed that the intracellular ROS levels were significantly higher following the combined treatment of MK‐2206 and EF24 than those obtained after exposure to a single agent in both cancer cell lines. We further analysed mitochondrial superoxide generation using MitoSOX™ fluorescence. Data in Supporting Information Fig. S3a–b show that the combination treatment did not induce mitochondrial superoxide generation either in SGC‐7901 or BGC‐823 cells, indicating that the MK‐2206/EF24‐induced ROS is not from mitochondria.
Figure 2.
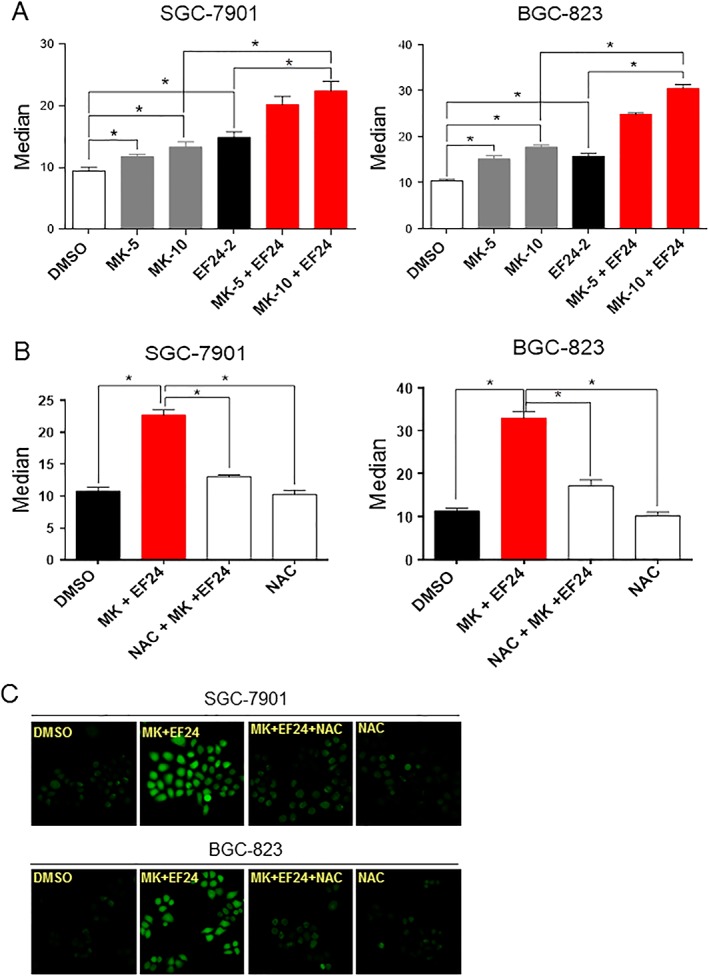
EF24 potentiates ROS production by MK‐2206. (A) Quantification of DCF flow cytometry data of intracellular ROS levels in SGC‐7901 and BGC‐823 cells treated with different concentrations of MK‐2206 (5, 10 and 20 μM) with or without 2 μM EF24 (Median = mean fluorescence intensity). (B) Quantification of DCF flow cytometry data showing the effect of NAC pretreatment for 2 h on ROS levels. Data are reported as mean ± SEM and analysed by Student's t‐test; n = 6 independent experiments; *P < 0.05. (C) Representative DCF fluorescence images of cells exposed to MK‐2206 and EF24 with or without NAC pretreatment. All images are representative of six independent experiments.
The combined treatment of MK‐2206 and EF24 suppresses the proliferation and induces apoptosis in human gastric cancer cells through ROS
Intracellular ROS levels are known to be higher in cancer cells compared with normal cells suggesting ROS have an important role in tumourigenesis. Emerging data also show that further increases in ROS in cancer cells may act to suppress cancer growth (Kardeh et al., 2014). We have also recently shown that an elevation in ROS levels in gastric cancer cells leads to cell apoptosis (Zou et al., 2015a; 2016a,b). It is, therefore, plausible that the combined treatment with MK‐2206 and EF24 enhances cell death through the induction of ROS levels (Figure 2). To test this, we treated gastric cancer cells with increasing concentrations of MK‐2206 in combination with 2 μM EF24 for 24 h. This co‐treatment of cells resulted in significantly reduced viability compared to the single agent treatments (IC50 = 6.19 μM for BGC‐823, 6.27 μM for SGC‐7901; Figure 3A). Annexin V/PI revealed that the combined treatment had a synergistic effect on cell death (Figure 3B). These flow cytometry data were mirrored in western blot analysis of cleaved caspase 3 and cleaved PARP (Figure 3C). Western blot analysis also showed that co‐treatment of gastric cancer cells with MK‐2206 and EF24 dose‐dependently decreased the levels of anti‐apoptotic protein Bcl‐2 and increased the levels of pro‐apoptotic protein Bax compared with single‐agent treatment (Figure 3C). In addition, colony formation by gastric cancer cells was significantly reduced when the cells were treated with both MK‐2206 and EF24 (Figure 3D). These results clearly show that MK‐2206 and EF24 act synergistically to induce cell death in gastric cancer cells. Furthermore, the induction of apoptosis by the combined treatment was mediated through an elevation in intracellular ROS levels as NAC completely reversed MK‐2206/EF24‐induced cell death (Figure 3E–G and Supporting Information Fig. S4a). To test which type of oxidative stress is involved in killing cells, the effects of other anti‐oxidants containing catalase, Butyl hydroxy anisd (BHA) and ROS scavenger Trolox were investigated. As shown in Supporting Information Fig. S4b, catalase reversed MK‐2206/EF24‐induced cell death, while BHA and Trolox failed to reverse the effects of the combined treatment. In Supporting Information Fig. S1, we showed that MK‐2206‐induced ROS production was Akt‐independent. Thus, we further speculated that knockdown of Akt expression does not increase apoptosis induced by EF24. Lipo2000 (LP2000) is a vehicle for siRNA transfection. As shown in Supporting Information Fig. S4c, a combination of Akt knockdown and EF24 failed to increase apoptosis when compared with the EF24 + LP2000 group in both SGC‐7901 and BGC‐823 cells, suggesting that the effect of the combination of EF24 and MK‐2206 is also not dependent on Akt.
Figure 3.
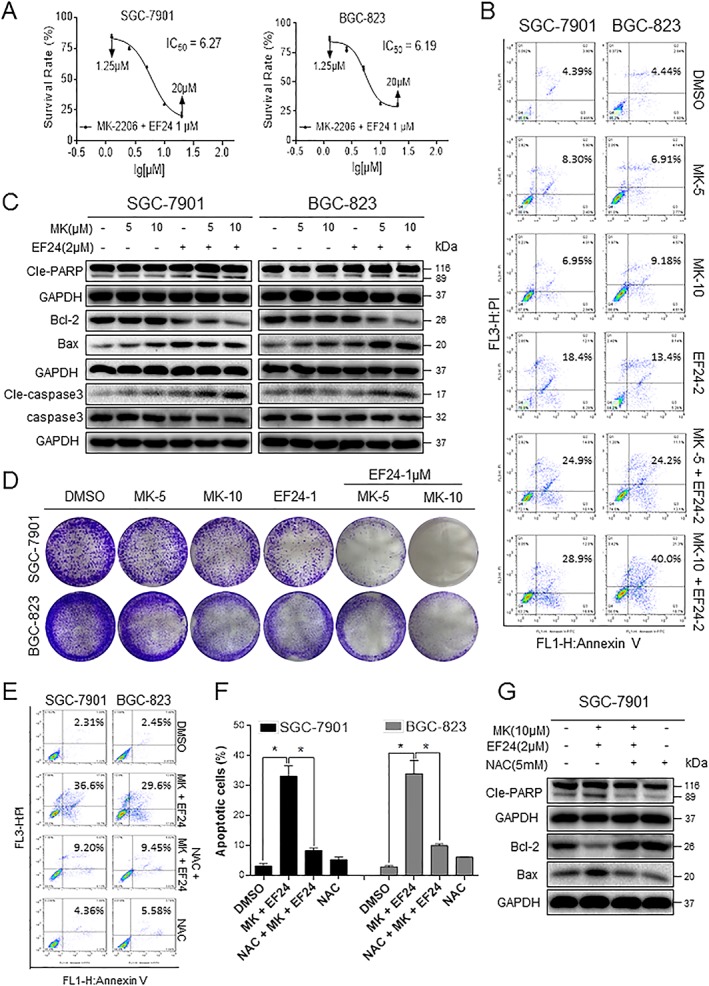
The combination treatment of MK‐2206 and EF24 induces apoptosis in human gastric cancer cells. (A) The effects of combined MK‐2206 and EF24 treatment on the viability of human gastric cancer cells. Cells were treated with increasing concentrations of MK‐2206 and 2 μM EF24 for 24 h. (B) The induction of apoptosis as assessed by Annexin V/PI staining following combined treatment with MK‐2206 and EF24 for 24 h. (C) Western blot analysis of apoptosis‐related proteins in cells treated with MK‐2206 and EF24 for 16 h. Figure showing levels of Bcl‐2, Bax, cleaved‐PARP and cleaved‐caspase 3. (D) Effect of combined treatment on colony formation. Cells were treated for 24 h and stained with crystal violet on day 8. (E) Gastric cancer cells were pretreated with 5 mM NAC for 2 h before exposure to MK‐2206 and EF24 for 24 h. Apoptosis was detected by Annexin V/PI staining. (F) Quantification of data presented in panel (E). Data are reported as mean ± SEM and analysed by Student's t‐test; n = 5 independent experiments; *P < 0.05. (G) Analysis of cleaved‐PARP, Bcl‐2 and Bax levels in SGC‐7901 cells pretreated with NAC. All representative images are from five independent experiments.
Elevated ROS through MK‐2206 and EF24 co‐treatment causes G2/M cell cycle arrest
We also analysed the effect of combined MK‐2206 and EF24 treatment on cell cycle distribution. For these studies, we first treated cells with increasing concentrations of MK‐2206 together with 2 μM EF24 for 15 h and performed cell cycle analysis through PI staining. Our results showed that the addition of EF24 increases MK‐2206‐induced G2/M phase arrest in both gastric cancer cell lines (Figure 4A, B and Supporting Information Fig. S5). Western blot analysis of G2/M phase‐related proteins also showed significantly reduced levels of Cdc2, MDM‐2 and cyclin B1 when cells were co‐treated with MK‐2206 and EF24 (Figure 4C and Supporting Information Figs S6–S7). Similar to our findings on cell death, cell cycle arrest by the combined treatment also involved ROS production as NAC was able to prevent G2/M arrest (Figure 4D–F and Supporting Information Figs S8–S10).
Figure 4.
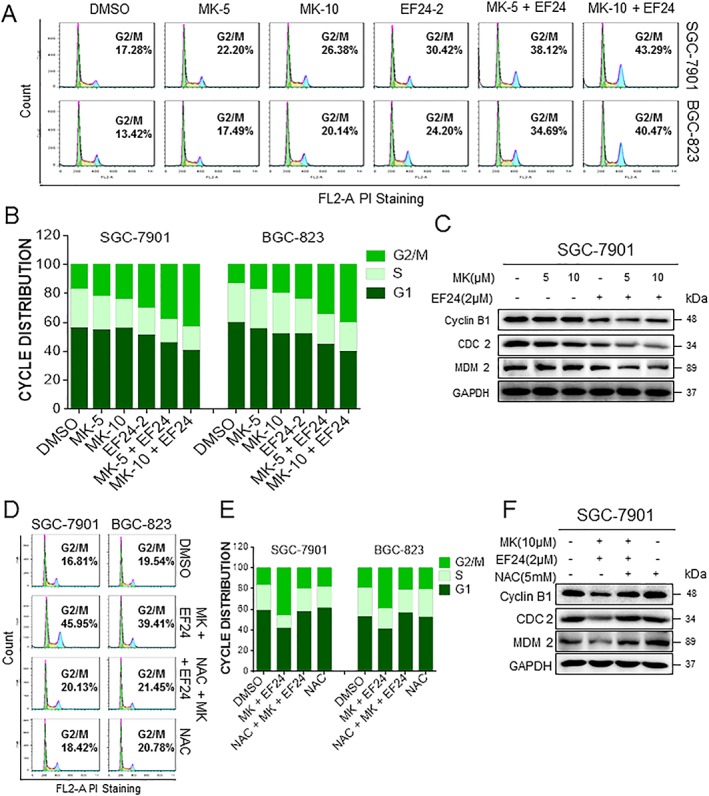
EF24 enhances MK‐2206‐mediated cell cycle arrest. (A) SGC‐7901 and BGC‐823 cells (SGC and BGC, respectively) were treated with a combination of MK‐2206 and EF24 for 18 h. The number of cells in the G2/M phase was determined by PI staining. (B) Representative histogram from cell cycle analysis presented in panel (A). (C) Expression of G2/M phase‐related proteins MDM‐2, cyclin B1 and Cdc2 in cells exposed to MK‐2206 and EF24 for 15 h. (D) Effect of NAC pretreatment on cell cycle distribution as determined by PI staining. Cells were pretreated with 5 mM NAC for 2 h before exposure to MK‐2206 and EF24 for 18 h. (E) Histogram generated from PI flow cytometry data presented in panel (D). (F) Total lysates from cells exposed to MK‐2206 and EF24, with or without NAC pretreatment, were subjected to analysis of cell cycle‐related proteins. All images are representative of five independent experiments.
ROS‐dependent mitochondrial dysfunction and ER stress activation in human gastric cancer cells
Previous studies have reported that ROS generation activates ER stress‐related apoptosis in hepatoma cells (Moon et al., 2011). We speculated that the increase in ROS induced by co‐treatment of gastric cancer cells with MK‐2206 and EF24 may also lead to cell death through ER stress. To test this, we examined ER morphology by electron microscopy. We found that a short 6 h combined treatment resulted in swollen ER, which was not observed in DMSO‐treated SGC‐7901 cells (Figure 5A). Furthermore, these structural alterations were not seen in cells treated with NAC prior to MK‐2206 and EF24 implicating a role for ROS in ER stress. To confirm these results, we performed western blotting for ER‐stress associated proteins including EIF2, ATF‐4 and CHOP. We showed significantly increased levels of CHOP and ATF‐4 and increased phosphorylated EIF2α in gastric cancer cells exposed to the combined treatment (Figure 5B and Supporting Information Fig. S11a). These readouts of ER‐stress were also normalized upon NAC pretreatment (Figure 5C and Supporting Information Fig. S11b).
Figure 5.
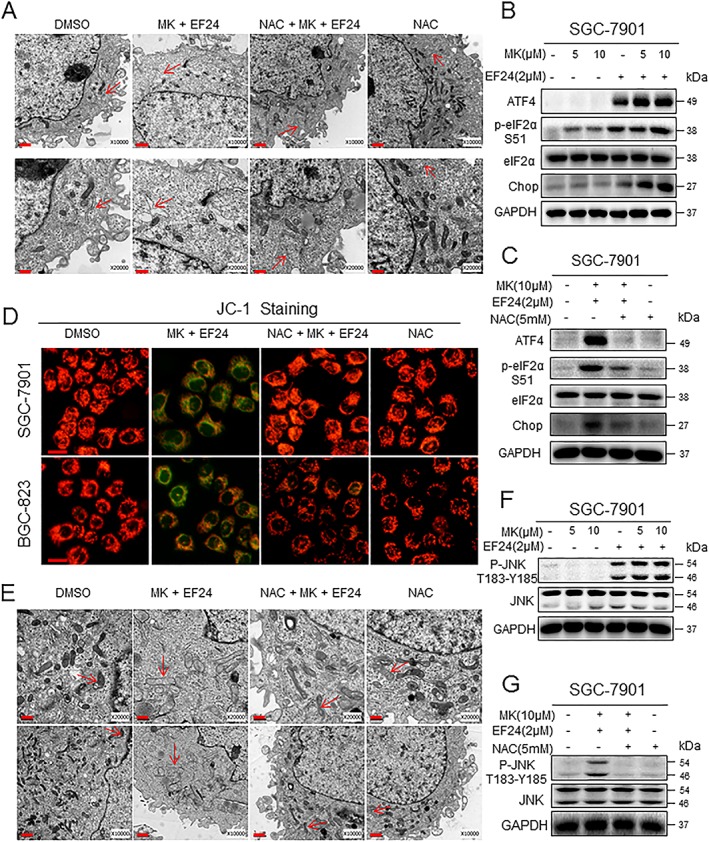
ER stress and mitochondrial dysfunction induced by MK‐2206 and EF24 treatment. (A) Effect of combined treatment with MK‐2206 and EF24 on ER in SGC‐7901 cells as assessed by electron microscopy. Exposure of cells to 10 μM MK‐2206 in combination with 2 μM EF24 for 6 h induced ER alterations (arrows indicate swollen ER). However, the ER appears normal (indicated by arrows) in the DMSO control group, NAC‐treated cells and cells treated with NAC prior to MK‐2206 and EF24. Lower panel of (A) shows higher magnification. (B) Western blot analysis of ER stress pathway markers in cells treated with MK‐2206 and EF 24 for 3 h (ATF‐4 and p‐EIF2α) or 12 h (CHOP). (C) ER stress pathway proteins in cells exposed to MK‐2206 and EF24 as in panel (B) but with NAC pretreatment of 2 h. For panels (B) and (C), GAPDH and eIF2α served as internal controls. (D) Mitochondrial membrane potential (Δψm) was detected by JC‐1 dye. SGC‐7901 and BGC‐823 cells were exposed to 10 μM MK‐2206 in combination with 2 μM EF24 for 12 h, with or without 2 h pretreatment with NAC (scale bar = 20 μM). (E) Effect of MK‐2206 in combination with EF24 on the morphology of mitochondria in SGC‐7901 cells. Exposure of cells to 10 μM MK‐2206 in combination with 2 μM EF24 for 12 h induced mitochondrial dysfunction as illustrated by swollen mitochondria (arrows). These changes were not seen in cells treated with DMSO (control), NAC or MK and EF24 following NAC pretreatment (arrows indicate normal mitochondria). Lower panel of (E) shows higher magnification. (F) Western blot analysis of JNK phosphorylation in SGC‐7901 cells exposed to MK‐2206 in combination with EF24 for 6 h. (G) p‐JNK levels in cells cultured as in (F) but with NAC pretreatment. All images are representative of five independent experiments.
We next examined mitochondrial alterations, as studies have also implicated mitochondria in both cell cycle regulation and apoptosis (Wang, 2001). Alterations in mitochondria are also intimately associated with ROS generation. Loss of mitochondrial membrane potential (△ψ m) is catastrophic for cells and leads to the release of cytochrome c into the cytosol. We measured mitochondrial membrane potential through fluorescence microscopy by using JC‐1 dye. JC‐1 is a cationic carbocyanine dye that accumulates in the mitochondria of living cells. The dye leaks out yielding green fluorescence upon changes to mitochondrial membrane potential. In our studies, the co‐treatment reduced mitochondrial membrane potential as evidenced by a change in red to green fluorescence in gastric cancer cells (Figure 5D and Supporting Information Fig. S12). This loss of mitochondrial membrane integrity was prevented in NAC‐pretreated cells. We also examined electron micrographs to evaluate mitochondrial structural changes and showed abnormally enlarged and swollen mitochondria with shredded cristae and disruption of outer membranes in cells co‐treated with MK‐2206 and EF24 (Figure 5E). These structural aberrations were not seen in control cells, cells treated with NAC alone or cells pretreated with NAC prior to exposure to MK‐2206 and EF24. To better understand the severity of mitochondrial damage, we determined the ATP levels in SGC‐7901 or BGC‐823 cells. As shown in Supporting Information Fig. S13, the combined treatment with MK‐2206 and EF24 led to a decrease in ATP levels in cells, while pretreatment with NAC prevented this decrease in ATP levels. JNK has been reported to mediate mitochondrial signal transduction in response to extracellular stimulation including ROS (Si et al., 2016). JNK activation induced by increased ROS levels also leads to ER stress (Zou et al., 2015b). Analysis of phosphorylated JNK revealed significantly increased levels in cells treated with MK‐2206 and EF24 (Figure 5F and Supporting Information Fig. S14a). These enhanced p‐JNK levels were reduced after NAC pretreatment (Figure 5G and Supporting Information Fig. S14b). These results suggest that co‐treatment with MK‐2206 and EF24 induces cytoplasmic ROS‐mediated ER stress, mitochondrial dysfunction and apoptosis. Furthermore, these activities may potentially be mediated through p‐JNK.
EF24 enhances the anti‐tumour activity of MK‐2206 in human gastric cancer xenografts
Our last objective was to assess the effect of combining EF24 with MK‐2206 in a human gastric cancer xenograft model. We injected SGC‐7901 cells in immunodeficient mice and treated the mice with 10 mg·kg−1 MK‐2206, 3 mg·kg−1 EF24 or a combination of the two. After 14 days, we observed significant inhibition of tumour growth with MK‐2206 and EF24 alone (Figure 6A–C). The combination of EF24 and MK‐2206 enhanced this inhibitory effect on tumour growth compared with single treatments. Body weight record (Supporting Information Fig. S15a) and histological analysis of heart, liver and kidney tissues (Supporting Information Fig. S15b) failed to show any alterations suggesting that the combined treatment did not produce any toxic effects. To determine whether the same anti‐tumour mechanisms are at play in the xenograft model as our in vitro studies, we assessed oxidative stress and cell death. The levels of lipid peroxidation product MDA were significantly elevated in tumour samples from mice treated with a combination of MK‐2206 and EF24 (Figure 6D). Single treatments failed to reach significance. However, MK‐2206 treatment did increase cleaved caspase 3 levels and decreased p‐AKT and Ki‐67 immunoreactivity (Figure 6E and Supporting Information Fig. S15c). These findings indicate increased apoptosis and reduced cell proliferation in tumour tissues. Although EF24 treatment alone failed to induce any appreciable effect on p‐Akt levels, Ki‐67 levels were clearly reduced. In addition, EF24 treatment induced apoptosis, as evident by increased cleaved caspase 3 levels. Combining MK‐2206 and EF24 produced more pronounced effects, which can be easily seen in the Ki‐67 images. We confirmed these findings by western blot analysis (Figure 6F) and showed that treatment with MK‐2206 and EF24 increased the levels of p‐EIF2α, cleaved PARP and cleaved caspase 3 when compared with single‐agent groups.
Figure 6.
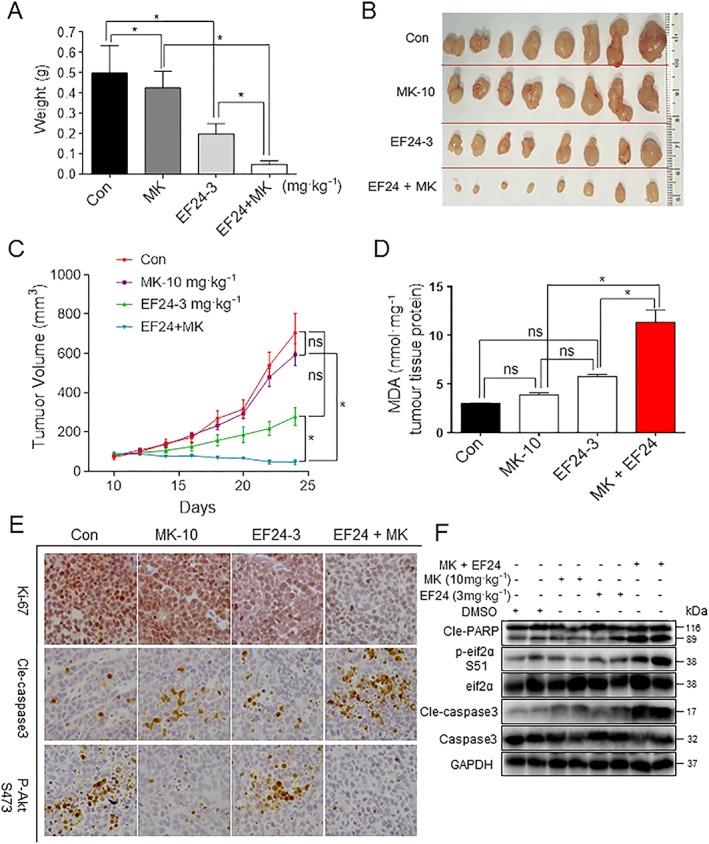
Anti‐tumour activity is enhanced by combining EF24 and MK‐2206 in gastric cancer xenograft model. (A–C) Tumour volume changes, harvested tumour specimens and tumour weight from mice treated with 10 mg·kg−1 MK‐2206 (MK‐10) or 10 mg·kg−1 MK‐2206 in combination with 3 mg·kg−1 EF24. (D) Levels of oxidative stress marker MDA in the tumour tissues. (E) Immunohistochemical staining of tumour specimens for cell proliferation marker Ki‐67, apoptosis marker cleaved caspase 3 and phospho‐Akt (scale bar =50 μm). (F) Western blot analysis of cleaved‐PARP cleavage, cleaved‐caspase 3 and p‐EIF2α using tumour tissue lysates. Data are reported as mean ± SEM and analysed by Student's t‐test; n = 8 mice per group; *P < 0.05 and ns = not significant. All images are representative of eight mice per group.
Discussion
Targeted therapy is gaining momentum for gastric cancer treatment. One of the pathways that has been reported to be dysregulated is the PI3K‐Akt‐mTOR signalling pathway. MK‐2206 is a highly selective, allosteric Akt inhibitor, with higher potency for Akt1 and Akt2 isoforms than Akt3 (Yan, 2009). MK‐2206 is currently being tested in phase II trials as a second‐line therapy for gastric and gastroesophageal cancer (Ramanathan et al., 2015). However, MK‐2206 has not shown meaningful clinical activity as a single agent in patients with refractory biliary cancer (Ahn et al., 2015) or acute myelogenous leukaemia (Konopleva et al., 2014) at tolerated doses. Because of the role of Akt in basic cell activities such as growth, proliferation and migration, these findings are not unexpected. In our study, we found that MK‐2206 effectively inhibits Akt phosphorylation in gastric cancer cells even at concentrations as low as 0.1 μM. However, an increase in cell death was only observed with higher concentrations in both gastric cancer and normal gastric cells. These results suggest that inhibition of Akt phosphorylation and activity may not be sufficient to induce cell death. Furthermore, increasing the dose of Akt inhibitors to achieve effective cell death may lead to cytotoxicity in normal cells. A potential solution to this issue is to combine MK‐2206 with other cancer drugs. In breast and lung cancer cell lines, addition of MK‐2206 enhanced the effects of other chemotherapeutic and targeted agents (Hirai et al., 2010). Therefore, this combination approach seems feasible for gastric cancer.
Under physiological conditions, the maintenance of an appropriate level of intracellular ROS is important for redox balance and cell growth (Martin and Barrett, 2002). However, excessive ROS generation overcomes cellular antioxidant defences and triggers apoptosis. Interestingly, cancer cells are more sensitive to rapid increases in ROS levels than normal cells (Trachootham et al., 2009). Our previous study has shown that increased ROS generation might be an effective strategy for treating human gastric cancer (Zou et al., 2015a,b; 2016a).
In the present study, we found that MK‐2206 increased ROS levels in a concentration‐dependent manner (from 10 to 40 μM). Although the mechanism of this increased ROS is not clear, we found that the ROS‐inducing ability of MK‐2206 is not dependent on Akt. Knockdown of Akt by genetic silencing did not induce or prevent ROS generation in cancer cells (Supporting Information Fig. S1), indicating that MK‐2206 activates ROS via another target and signalling pathways. More importantly, silencing Akt by siRNA failed to affect gastric cancer cell apoptosis (Supporting Information Fig. S4b), verifying that Akt inhibition induced by a low‐concentration MK‐2206 did not reduce cell viability (Figure 1A–D). Thus, our findings imply a new effect of relatively high concentrations of MK‐2206 at. It will be very interesting to investigate this new mechanism underlying the anti‐cancer effect of MK‐2206 in the future.
Recently, combined (rather than single‐agent) chemotherapy has been found to be a superior treatment method (Almhanna et al., 2013). Notably, in our previous study, a combination treatment with low‐doses of ROS inducers produces abundant ROS generation and triggers cell apoptosis (Zou et al., 2015a). We have also demonstrated that EF24 does not cause any adverse effect on GES‐1 (normal) gastric epithelial cells at a concentration of 2 μM (Zou et al., 2016b). Thus, ROS production by EF24 may be utilized in combination with MK‐2206 in gastric cancer. Our studies do show that EF24 has a synergistic effect on MK‐2206‐induced apoptotic cell death. Therefore, EF24 could be a promising anti‐cancer molecule and a sensitizing agent. Combined treatment may also prevent side effects associated with high‐doses of single agents.
On delving into the mechanism behind the enhanced anti‐tumour effect of MK‐2206 and EF24, we found that an elevation in ROS caused mitochondrial dysfunction and ER stress. Both the mitochondria and ER are vital intracellular organelles, which are also involved in inducing cell apoptosis. The accumulation of high levels of ROS has been shown to induce ER stress, which leads to unfolded protein response in order to promote the survival of tumour cells (Wu, 2006). Our findings show that co‐treatment of gastric cancer cells with MK‐2206 and EF24 induces ROS‐mediated structural changes and stress response in ER. This stress response is mediated by a number of proteins, but the key ones involved are CHOP and ATF‐4. The basal level of CHOP expression is low to almost undetectable in most cell types. However, its expression is rapidly increased through ATF‐4‐dependent transcription (Ron and Habener, 1992; Averous et al., 2004). CHOP then induces cell cycle arrest and apoptosis in response to ER stress (Zinszner et al., 1998). In addition to ER stress, we found that ROS production by MK‐2206 and EF24 caused mitochondrial integrity deficits. Previous studies have reported accelerated opening of mitochondrial permeability transition pore by ROS (Kowaltowski et al., 2001; Mashayekhi et al., 2014). Mitochondrial membrane permeability is regulated by Bcl‐2 family members via multiple molecular mechanisms. By means of electron microscopy, we observed that the mitochondria in the co‐treated gastric cancer cells were abnormally enlarged or swollen. Moreover, co‐treatment with MK‐2206 and EF24 decreased Δψm and the antiapoptotic/proapoptotic (Bcl‐2/Bax) protein ratio in gastric cancer cells. Notably, NAC fully reversed these changes. Based on these results, we conclude that excessive ROS production caused the activation of ER stress and mitochondrial apoptotic pathways in gastric cancer cells.
In summary, the results presented here demonstrate that the pro‐apoptotic effects of MK‐2206 are mediated by an Akt‐independent mechanism in gastric cancer cells. We also showed that EF24 enhances the anti‐tumour effects of MK‐2206 both in cultured cells and in an animal model of gastric cancer. Both MK‐2206 and EF24 induced and increase in ROS in gastric cancer cells. The combination led to the induction of ER stress, mitochondrial dysfunction and apoptosis. We confirmed that all these activities were dependent on ROS generation through the use of ROS scavenger at all steps. Based on these results, we conclude that ROS generation may be a significant target for the development of new anti‐cancer drugs. Furthermore, our results also suggest the combination of a low‐dose of MK‐2206 and EF24 could have potential as a therapy for the treatment of human gastric cancer in the clinic.
Author contributions
X.C. performed the conception and design, collection, analysis and interpretation of data and manuscript writing; P.Z., W.C. and V.R. did the collection and interpretation of data and revised the manuscript; X.D., C.F., W.Z., C.Q. and Q.Y. carried out the collection and analysis of data. X.Z. and G.L. performed the conception and design and interpretation of data and manuscript revision. All authors approved final version of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 The effects of MK‐2206 on ROS production and apoptosis in a gastric cancer cell line that is negative for Akt. (A) The Akt expression was determined by Western blotting after knockdown with two different siRNAs for 48 h in SGC‐7901 or BGC‐823 cells. (B) Knockdown of Akt in SGC‐7901 or BGC‐823 cells could not increase the ROS levels. All representative images are from 5 independent experiments.
Figure S2 EF24 potentiates ROS production by MK‐2206. (A) Intracellular ROS levels in SGC‐7901 and BGC‐823 cells treated with different concentrations of MK‐2206 (5, 10, and 20 μM) with or without 2 μM EF24 [M = mean fluorescence intensity]. (B) Effect of N‐acetyl cysteine (NAC) pretreatment of 2 h on ROS levels [M = mean fluorescence intensity]. Relative fluorescence intensity was assayed by flow cytometer. All representative images are from 6 independent experiments.
Figure S3 (A) Relative fluorescence intensity for MitoSOX™ of SGC‐7901 or BGC‐823 cells treatmented with MK‐2206 (10 μM) and EF24 (2 μM) at the the indicated concentrations. (B) SGC‐7901 or BGC‐823 cells were pretreated with 5 mM NAC for 2 h before exposure to MK‐2206 (10 μM) and EF24 (2 μM) for 2 h. Relative fluorescence intensity for MitoSOX™ was assayed by flow cytometer. All representative images are from 6 independent experiments.
Figure S4 MK‐2206 in combination with EF24 induces apoptosis in BGC‐823 gastric cancer cells. (A) Levels of cleaved PARP in BGC‐823 cells treated with MK‐2206, EF24, or a combination of the two for 16 h. NAC treatment, where indicated, was carried out for 2 h prior to exposure to MK‐2206 and/or EF24. (B) Gastric cancer cells were pretreated with NAC (5 mM), Catalse (2 kU/mL), Trolox (0.5 mM) and BHA (0.1 mM) for 2 h before exposure to MK‐2206 and EF24 for 24 h. Apoptosis was detected by Annexin‐V/PI staining. (C) Knockdown of Akt in SGC‐7901 or BGC‐823 cells could not enhance apoptosis induced by EF24. All representative images are from 5 independent experiments.
Figure S5 MK‐2206 in combination with EF24 induces cell cycle arrest in BGC‐823 gastric cancer cells. Quantification of cell cycle analysis presented in Figure 4A. All representative images are from 5 independent experiments. [Data are reported as mean ± s.e.m. and analysed by Student's t‐test; n = 5 independent experiments; * P < 0.05, ** P < 0.01].
Figure S6 Quantification of G2/M phase‐related proteins MDM‐2, cyclin B1 and Cdc2 presented in Figure 4C. All representative images are from 5 independent experiments. [Data are reported as mean ± s.e.m. and analysed by Student's t‐test; n = 5 independent experiments; * P < 0.05, ** P < 0.01].
Figure S7 Expression of G2/M phase‐related proteins MDM‐2, cyclin B1 and Cdc2 in BGC‐823 cells exposed to MK‐2206 and EF24 for 15 h. All representative images are from 5 independent experiments.
Figure S8 Quantification of cell cycle analysis presented in Figure 4D. All representative images are from 5 independent experiments. [Data are reported as mean ± s.e.m. and analysed by Student's t‐test; n = 5 independent experiments; * P < 0.05, ** P < 0.01].
Figure S9 Quantification of G2/M phase‐related proteins MDM‐2, cyclin B1 and Cdc2 presented in Figure 4F. All representative images are from 5 independent experiments. [Data are reported as mean ± s.e.m. and analyzed by Student's t‐test; n=5 independent experiments; * p< 0.05, ** p< 0.01].
Figure S10 Effect of NAC pretreatment on cell cycle proteins in BGC‐823 cells. All representative images are from 5 independent experiments.
Figure S11 MK‐2206 and EF24 induce ER stress in BGC‐823 cells. (A) Western blot analysis of ER stress pathway proteins in BGC‐823 cells treated with MK‐2206 and EF24 for 3 h (ATF4 and p‐EIF2α) or 12 h (CHOP). (B) ER stress pathway proteins in cells exposed to MK‐2206 and EF24 as in panel A but with NAC pretreatment of 2 h. For panels A and B, GAPDH and eIF2α served as internal controls. All representative images are from 5 independent experiments.
Figure S12 Quantification of JC‐1 staining analysis presented in Figure 5D. [Data are reported as mean ± s.e.m. and analysed by Student's t‐test; n = 5 independent experiments; * P < 0.05, ** P < 0.01].
Figure S13 Gastric cancer cells were pre‐treated with 5 mM NAC for 2 h before exposure to MK‐2206 (10 μM) and EF24 (2 μM) for 12 h. ATP levels were analysed by commercial kit. [Data are reported as mean ± s.e.m. and analyzed by Student's t‐test; n=6 independent experiments; *P < 0.05, **P < 0.01, ***P < 0.001].
Figure S14 MK‐2206 and EF24 induce JNK phosphorylation in BGC‐823 cells. (A) Western blot analysis of JNK phosphorylation in BGC‐823 cells exposed to MK‐2206 in combination with EF24 for 6 h. (B) p‐JNK levels in cells cultured as in E but with 2 h NAC pretreatment. All representative images are from 5 independent experiments.
Figure S15 MK‐2206 and EF24 treatment does not cause significant toxicity in mice. (A) Mouse body weight record. (B) Histology of heart, liver, and kidney tissues from mice treated with 10 mg/kg MK‐2206 and/or 3 mg/kg EF24 showing representative H & E stained sections. Data and representative images are from 8 mice per group. (C) Quantification of IHC analysis presented in Figure 6E. Data and representative images are from 8 mice per group. [Data are reported as mean ± s.e.m. and analyzed by Student's t‐test; n=8 mice per group; *P < 0.05, **P < 0.01].
Acknowledgements
The work was supported by the National Natural Science Foundation of China (81503107 and 81573657), the Zhejiang Province Natural Science Funding of China (LY13H160022 and LY12H16003), the Technology Foundation for Medical Science of Zhejiang Province (2012KYA129) and Wenzhou Science and Technology project (2014Y0344).
Chen, X. , Dai, X. , Zou, P. , Chen, W. , Rajamanickam, V. , Feng, C. , Zhuge, W. , Qiu, C. , Ye, Q. , Zhang, X. , and Liang, G. (2017) Curcuminoid EF24 enhances the anti‐tumour activity of Akt inhibitor MK‐2206 through ROS‐mediated endoplasmic reticulum stress and mitochondrial dysfunction in gastric cancer. British Journal of Pharmacology, 174: 1131–1146. doi: 10.1111/bph.13765.
Contributor Information
Xiaohua Zhang, Email: xiaohuazhang2015@sina.com.
Guang Liang, Email: wzmcliangguang@163.com.
References
- Ahn DH, Li J, Wei L, Doyle A, Marshall JL, Schaaf LJ et al. (2015). Results of an abbreviated phase‐II study with the Akt inhibitor MK‐2206 in patients with advanced biliary cancer. Sci Rep 5: 12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The concise guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almhanna K, Cubitt CL, Zhang S, Kazim S, Husain K, Sullivan D et al. (2013). MK‐2206, an Akt inhibitor, enhances carboplatinum/paclitaxel efficacy in gastric cancer cell lines. Cancer Biol Ther 14: 932–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Averous J, Bruhat A, Jousse C, Carraro V, Thiel G, Fafournoux P (2004). Induction of CHOP expression by amino acid limitation requires both ATF4 expression and ATF2 phosphorylation. J Biol Chem 279: 5288–5297. [DOI] [PubMed] [Google Scholar]
- Boonstra J, Post JA (2004). Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene 337: 1–13. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Ren X, Zhang Y, Patel R, Sharma A, Wu H et al. (2011). EEF‐2 kinase dictates cross‐talk between autophagy and apoptosis induced by Akt inhibition, thereby modulating cytotoxicity of novel Akt inhibitor MK‐2206. Cancer Res 71: 2654–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM (2010). Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127: 2893–2917. [DOI] [PubMed] [Google Scholar]
- Grabinski N, Ewald F, Hofmann BT, Staufer K, Schumacher U, Nashan B et al. (2012). Combined targeting of AKT and mTOR synergistically inhibits proliferation of hepatocellular carcinoma cells. Mol Cancer 11: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K et al. (2010). MK‐2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo . Mol Cancer Ther 9: 1956–1967. [DOI] [PubMed] [Google Scholar]
- Ji D, Zhang Z, Cheng L, Chang J, Wang S, Zheng B et al. (2014). The combination of RAD001 and MK‐2206 exerts synergistic cytotoxic effects against PTEN mutant gastric cancer cells: involvement of MAPK‐dependent autophagic, but not apoptotic cell death pathway. PLoS One 9: e85116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardeh S, Ashkani‐Esfahani S, Alizadeh AM (2014). Paradoxical action of reactive oxygen species in creation and therapy of cancer. Eur J Pharmacol 735: 150–168. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SM, Park SH (2015). Chemotherapy beyond second‐line in advanced gastric cancer. World J Gastroenterol 21: 8811–8816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopleva MY, Walter RB, Faderl SH, Jabbour EJ, Zeng Z, Borthakur G et al. (2014). Preclinical and early clinical evaluation of the oral AKT inhibitor, MK‐2206, for the treatment of acute myelogenous leukemia. Clin Cancer Res 20: 2226–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowaltowski AJ, Castilho RF, Vercesi AE (2001). Mitochondrial permeability transition and oxidative stress. FEBS Lett 495: 12–15. [DOI] [PubMed] [Google Scholar]
- Marin JJ, Al‐Abdulla R, Lozano E, Briz O, Bujanda L, Banales JM et al. (2016). Mechanisms of resistance to chemotherapy in gastric cancer. Anticancer Agents Med Chem 16: 318–334. [DOI] [PubMed] [Google Scholar]
- Markman B, Atzori F, Perez‐Garcia J, Tabernero J, Baselga J (2010). Status of PI3K inhibition and biomarker development in cancer therapeutics. Ann Oncol: official journal of the European Society for Medical Oncology/ESMO 21: 683–691. [DOI] [PubMed] [Google Scholar]
- Martin KR, Barrett JC (2002). Reactive oxygen species as double‐edged swords in cellular processes: low‐dose cell signaling versus high‐dose toxicity. Hum Exp Toxicol 21: 71–75. [DOI] [PubMed] [Google Scholar]
- Mashayekhi V, Eskandari MR, Kobarfard F, Khajeamiri A, Hosseini MJ (2014). Induction of mitochondrial permeability transition (MPT) pore opening and ROS formation as a mechanism for methamphetamine‐induced mitochondrial toxicity. Naunyn Schmiedebergs Arch Pharmacol 387: 47–58. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meitzler JL, Antony S, Wu Y, Juhasz A, Liu H, Jiang G et al. (2014). NADPH oxidases: a perspective on reactive oxygen species production in tumor biology. Antioxid Redox Signal 20: 2873–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon DO, Park SY, Choi YH, Ahn JS, Kim GY (2011). Guggulsterone sensitizes hepatoma cells to TRAIL‐induced apoptosis through the induction of CHOP‐dependent DR5: involvement of ROS‐dependent ER‐stress. Biochem Pharmacol 82: 1641–1650. [DOI] [PubMed] [Google Scholar]
- Nam SY, Lee HS, Jung GA, Choi J, Cho SJ, Kim MK et al. (2003). Akt/PKB activation in gastric carcinomas correlates with clinicopathologic variables and prognosis. APMIS 111: 1105–1113. [DOI] [PubMed] [Google Scholar]
- Perry G, Raina AK, Nunomura A, Wataya T, Sayre LM, Smith MA (2000). How important is oxidative damage? Lessons from Alzheimer's disease. Free Radic Biol Med 28: 831–834. [DOI] [PubMed] [Google Scholar]
- Ramanathan RK, McDonough SL, Kennecke HF, Iqbal S, Baranda JC, Seery TE et al. (2015). Phase 2 study of MK‐2206, an allosteric inhibitor of AKT, as second‐line therapy for advanced gastric and gastroesophageal junction cancer: a SWOG cooperative group trial (S1005). Cancer 121: 2193–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Habener JF (1992). CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant‐negative inhibitor of gene transcription. Genes Dev 6: 439–453. [DOI] [PubMed] [Google Scholar]
- Si L, Zheng L, Xu L, Yin L, Han X, Qi Y et al. (2016). Dioscin suppresses human laryngeal cancer cells growth via induction of cell‐cycle arrest and MAPK‐mediated mitochondrial‐derived apoptosis and inhibition of tumor invasion. Eur J Pharmacol 774: 105–117. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A (2015). Cancer statistics, 2015. CA Cancer J Clin 65: 5–29. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam D, May R, Sureban SM, Lee KB, George R, Kuppusamy P et al. (2008). Diphenyl difluoroketone: a curcumin derivative with potent in vivo anticancer activity. Cancer Res 68: 1962–1969. [DOI] [PubMed] [Google Scholar]
- Tao K, Yin Y, Shen Q, Chen Y, Li R, Chang W et al. (2016). Akt inhibitor MK‐2206 enhances the effect of cisplatin in gastric cancer cells. Biomed Rep 4: 365–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SL, Zhong D, Zhou W, Malik S, Liotta D, Snyder JP et al. (2008). EF24, a novel curcumin analog, disrupts the microtubule cytoskeleton and inhibits HIF‐1. Cell Cycle 7: 2409–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga E, Oki E, Egashira A, Sadanaga N, Morita M, Kakeji Y et al. (2008). Deregulation of the Akt pathway in human cancer. Curr Cancer Drug Targets 8: 27–36. [DOI] [PubMed] [Google Scholar]
- Trachootham D, Alexandre J, Huang P (2009). Targeting cancer cells by ROS‐mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 8: 579–591. [DOI] [PubMed] [Google Scholar]
- Wang X (2001). The expanding role of mitochondria in apoptosis. Genes Dev 15: 2922–2933. [PubMed] [Google Scholar]
- Wu WS (2006). The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev 25: 695–705. [DOI] [PubMed] [Google Scholar]
- Yan L (2009). MK‐2206: a potent oral allosteric AKT inhibitor. Cancer Res 69 (9 Supplement): DDT01‐01–DDT01‐01. [Google Scholar]
- Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K et al. (2011). First‐in‐man clinical trial of the oral pan‐AKT inhibitor MK‐2206 in patients with advanced solid tumors. J Clin Oncol Off J Am Soc Clin Oncol 29: 4688–4695. [DOI] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H et al. (1998). CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 12: 982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou P, Chen M, Ji J, Chen W, Chen X, Ying S et al. (2015a). Auranofin induces apoptosis by ROS‐mediated ER stress and mitochondrial dysfunction and displayed synergistic lethality with piperlongumine in gastric cancer. Oncotarget 6: 36505–36521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou P, Zhang J, Xia Y, Kanchana K, Guo G, Chen W et al. (2015b). ROS generation mediates the anti‐cancer effects of WZ35 via activating JNK and ER stress apoptotic pathways in gastric cancer. Oncotarget 6: 5860–5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou P, Xia Y, Chen T, Zhang J, Wang Z, Chen W et al. (2016a). Selective killing of gastric cancer cells by a small molecule targeting ROS‐mediated ER stress activation. Mol Carcinog 55: 1073–1086. [DOI] [PubMed] [Google Scholar]
- Zou P, Xia Y, Chen W, Chen X, Ying S, Feng Z et al. (2016b). EF24 induces ROS‐mediated apoptosis via targeting thioredoxin reductase 1 in gastric cancer cells. Oncotarget 7: 18050–18064. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The effects of MK‐2206 on ROS production and apoptosis in a gastric cancer cell line that is negative for Akt. (A) The Akt expression was determined by Western blotting after knockdown with two different siRNAs for 48 h in SGC‐7901 or BGC‐823 cells. (B) Knockdown of Akt in SGC‐7901 or BGC‐823 cells could not increase the ROS levels. All representative images are from 5 independent experiments.
Figure S2 EF24 potentiates ROS production by MK‐2206. (A) Intracellular ROS levels in SGC‐7901 and BGC‐823 cells treated with different concentrations of MK‐2206 (5, 10, and 20 μM) with or without 2 μM EF24 [M = mean fluorescence intensity]. (B) Effect of N‐acetyl cysteine (NAC) pretreatment of 2 h on ROS levels [M = mean fluorescence intensity]. Relative fluorescence intensity was assayed by flow cytometer. All representative images are from 6 independent experiments.
Figure S3 (A) Relative fluorescence intensity for MitoSOX™ of SGC‐7901 or BGC‐823 cells treatmented with MK‐2206 (10 μM) and EF24 (2 μM) at the the indicated concentrations. (B) SGC‐7901 or BGC‐823 cells were pretreated with 5 mM NAC for 2 h before exposure to MK‐2206 (10 μM) and EF24 (2 μM) for 2 h. Relative fluorescence intensity for MitoSOX™ was assayed by flow cytometer. All representative images are from 6 independent experiments.
Figure S4 MK‐2206 in combination with EF24 induces apoptosis in BGC‐823 gastric cancer cells. (A) Levels of cleaved PARP in BGC‐823 cells treated with MK‐2206, EF24, or a combination of the two for 16 h. NAC treatment, where indicated, was carried out for 2 h prior to exposure to MK‐2206 and/or EF24. (B) Gastric cancer cells were pretreated with NAC (5 mM), Catalse (2 kU/mL), Trolox (0.5 mM) and BHA (0.1 mM) for 2 h before exposure to MK‐2206 and EF24 for 24 h. Apoptosis was detected by Annexin‐V/PI staining. (C) Knockdown of Akt in SGC‐7901 or BGC‐823 cells could not enhance apoptosis induced by EF24. All representative images are from 5 independent experiments.
Figure S5 MK‐2206 in combination with EF24 induces cell cycle arrest in BGC‐823 gastric cancer cells. Quantification of cell cycle analysis presented in Figure 4A. All representative images are from 5 independent experiments. [Data are reported as mean ± s.e.m. and analysed by Student's t‐test; n = 5 independent experiments; * P < 0.05, ** P < 0.01].
Figure S6 Quantification of G2/M phase‐related proteins MDM‐2, cyclin B1 and Cdc2 presented in Figure 4C. All representative images are from 5 independent experiments. [Data are reported as mean ± s.e.m. and analysed by Student's t‐test; n = 5 independent experiments; * P < 0.05, ** P < 0.01].
Figure S7 Expression of G2/M phase‐related proteins MDM‐2, cyclin B1 and Cdc2 in BGC‐823 cells exposed to MK‐2206 and EF24 for 15 h. All representative images are from 5 independent experiments.
Figure S8 Quantification of cell cycle analysis presented in Figure 4D. All representative images are from 5 independent experiments. [Data are reported as mean ± s.e.m. and analysed by Student's t‐test; n = 5 independent experiments; * P < 0.05, ** P < 0.01].
Figure S9 Quantification of G2/M phase‐related proteins MDM‐2, cyclin B1 and Cdc2 presented in Figure 4F. All representative images are from 5 independent experiments. [Data are reported as mean ± s.e.m. and analyzed by Student's t‐test; n=5 independent experiments; * p< 0.05, ** p< 0.01].
Figure S10 Effect of NAC pretreatment on cell cycle proteins in BGC‐823 cells. All representative images are from 5 independent experiments.
Figure S11 MK‐2206 and EF24 induce ER stress in BGC‐823 cells. (A) Western blot analysis of ER stress pathway proteins in BGC‐823 cells treated with MK‐2206 and EF24 for 3 h (ATF4 and p‐EIF2α) or 12 h (CHOP). (B) ER stress pathway proteins in cells exposed to MK‐2206 and EF24 as in panel A but with NAC pretreatment of 2 h. For panels A and B, GAPDH and eIF2α served as internal controls. All representative images are from 5 independent experiments.
Figure S12 Quantification of JC‐1 staining analysis presented in Figure 5D. [Data are reported as mean ± s.e.m. and analysed by Student's t‐test; n = 5 independent experiments; * P < 0.05, ** P < 0.01].
Figure S13 Gastric cancer cells were pre‐treated with 5 mM NAC for 2 h before exposure to MK‐2206 (10 μM) and EF24 (2 μM) for 12 h. ATP levels were analysed by commercial kit. [Data are reported as mean ± s.e.m. and analyzed by Student's t‐test; n=6 independent experiments; *P < 0.05, **P < 0.01, ***P < 0.001].
Figure S14 MK‐2206 and EF24 induce JNK phosphorylation in BGC‐823 cells. (A) Western blot analysis of JNK phosphorylation in BGC‐823 cells exposed to MK‐2206 in combination with EF24 for 6 h. (B) p‐JNK levels in cells cultured as in E but with 2 h NAC pretreatment. All representative images are from 5 independent experiments.
Figure S15 MK‐2206 and EF24 treatment does not cause significant toxicity in mice. (A) Mouse body weight record. (B) Histology of heart, liver, and kidney tissues from mice treated with 10 mg/kg MK‐2206 and/or 3 mg/kg EF24 showing representative H & E stained sections. Data and representative images are from 8 mice per group. (C) Quantification of IHC analysis presented in Figure 6E. Data and representative images are from 8 mice per group. [Data are reported as mean ± s.e.m. and analyzed by Student's t‐test; n=8 mice per group; *P < 0.05, **P < 0.01].