

Abstract
Relaxin is a small heterodimeric peptide hormone of the insulin/relaxin superfamily produced mainly in female and male reproductive organs. It has potent antifibrotic, vasodilatory and angiogenic effects and regulates the normal function of various physiological systems. Preclinical studies and recent clinical trials have shown the promise of recombinant relaxin as a therapeutic agent in the treatment of cardiovascular and fibrotic diseases. However, there are the universal drawbacks of peptide‐based pharmacology that apply to relaxin: a short half‐life in vivo requires its continuous delivery, and there are high costs of production, storage and treatment, as well as the possibility of immune responses. All these issues can be resolved by the development of low non‐peptide MW agonists of the relaxin receptors which are stable, bioavailable, easily synthesized and specific. In this review, we describe the discovery and characterization of the first series of such compounds. The lead compound, ML290, binds to an allosteric site of the relaxin GPCR, RXFP1. ML290 shows high activity and efficacy, measured by cAMP response, in cells expressing endogenous or transfected RXFP1. Relaxin‐like effects of ML290 were shown in various functional cellular assays in vitro. ML290 has excellent absorption, distribution, metabolism and excretion properties and in vivo stability. The identified series of low MW agonists does not activate rodent RXFP1 receptors and thus, the production of a RXFP1 humanized mouse model is needed for preclinical studies. The future analysis and clinical perspectives of relaxin receptor agonists are discussed.
Linked Articles
This article is part of a themed section on Recent Progress in the Understanding of Relaxin Family Peptides and their Receptors. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.10/issuetoc
Abbreviations
- 7TM
GPCR seven transmembrane domain
- INSL3
insulin‐like peptide 3
- LRR
leucine‐rich repeat
- RLN2
relaxin 2
Tables of Links
| TARGETS | |
|---|---|
| Other protein targets a | Enzymes c |
| VEGF | A2denylate cyclase |
| GPCRs b | Akt (PKB) |
| β2‐adrenoceptors | ERK1 |
| RXFP1 receptors | ERK2 |
| RXFP2 receptors | NOS |
| PI3K | |
| PKA |
| LIGANDS |
|---|
| cAMP |
| cGMP |
| INSL3, insulin‐like peptide 3 |
| NO |
| Relaxin |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a,b,c).
Relaxin in basic science and clinical trials
Relaxin, a small 6kD peptide, signals through its cognate GPCR named relaxin/insulin‐like family peptide receptor 1 (RXFP1) (Halls et al., 2015). The peptide is produced mainly in the ovarian corpora lutea and placenta in females and in prostate in males and was initially identified as a hormone affecting pubic symphysis relaxation during parturition in rodents (see Sherwood, 2004). Further roles of relaxin on female reproduction were demonstrated in the maintenance of myometrial quiescence and softening and hypertrophy of the cervix during pregnancy, as well as endometrial, nipple and possibly mammary gland development. In recent years, it became clear that relaxin has pleiotropic effects on physiological systems beyond reproduction (Halls et al., 2015). The common features of relaxin action in different organs are potent vasodilatory properties (Conrad and Shroff, 2011) and antifibrotic effects associated with the regulation of collagen remodelling (Samuel et al., 2017). Relaxin has wound healing and organ protective effects due to its induction of cellular proliferation and the activation of NOS and NO. Relaxin also promotes angiogenesis by modifying expression of VEGF and other cytokines (Unemori et al., 1999).
It is worth noting that the majority of studies were performed with rodents, and relaxin effects still are not always well established in humans (Marshall et al., 2016). However, the therapeutic effects of relaxin in the treatment of liver, renal, cardiac, skin and pulmonary fibrosis, as well as inflammation and wound healing, are now well documented in animal models (Samuel et al., 2017). Several clinical trials have been conducted or proposed with recombinant human relaxin peptide (often called serelaxin) as a treatment for systemic sclerosis (Seibold et al., 2000), cervical ripening (Brennand et al., 1997; Kelly et al., 2001; Weiss et al., 2009; Weiss et al., 2016), preeclampsia (Conrad, 2016) and the latest and most advanced trial of serelaxin in acute heart failure (Teerlink et al., 2013; Sato et al., 2015; Leo et al., 2016) (Table 1). Clinical trials for systemic sclerosis and cervical ripening did not produce any notable therapeutic benefits. However, it was determined that relaxin injections were safe and well tolerated in patients. The data for preeclampsia and other conditions have not been reported yet. In Phase III clinical trial in acute heart failure, 1161 patients were involved (Teerlink et al., 2013). After 48 h intravenous infusion of relaxin, there was an improvement of one of the two primary dyspnoea (shortness of breath) endpoints. No significant effects were detected for the secondary endpoints of cardiovascular death or readmission to hospital for heart failure or renal failure or days alive out of the hospital up to day 60 (Teerlink et al., 2013). The most significant result was the drastic reduction of overall death at day 180 (placebo, 65 deaths; serelaxin, 42; hazard ratio 0·63, 95% confidence interval 0·42–0·93; P = 0·019). Currently, a large‐scale confirmatory Phase III clinical trial for acute heart failure with modified primary and secondary outcomes and a number of other studies for various cardio‐vasculatory abnormalities are being conducted by Novartis Pharmaceutical (Table 1). This clinical programme will include more than 10 000 patients. The range of pleotropic effects and possible clinical applications underlines the importance of the relaxin signalling system as a therapeutic target in various human diseases (Figure 1).
Table 1.
Relaxin clinical trials (source: ClinicalTrials.gov)
| Status and phase | Study title and dates | Sponsor and identifier | ||
|---|---|---|---|---|
| 1 | Completed, Phase II and III, n = 1161 | Efficacy and safety of relaxin for the treatment of acute heart failure/2007–2012 | Corthera/Novartis NCT00520806 | |
| Condition | Heart failure, congestive | |||
| Intervention | Drug: rhRLN | |||
| 2 | Terminated, Phase II, n = 11 | Recombinant human relaxin for the treatment of decompensated CHF/2006–2007 | Corthera/Novartis NCT00406575 | |
| Condition | Heart failure, congestive | |||
| Intervention | Drug: rhRLN | |||
| 3 | Completed, Phase II, n = 18 | A pilot study of recombinant human relaxin (rhRlx) in compensated CHF/2005– | Corthera/Novartis NCT00259116 | |
| Condition | Heart failure, congestive | |||
| Intervention | Drug: rhRLN | |||
| 4 | Suspended, Phase I, n = 18 (estimated) | Evaluation of the safety of relaxin in preeclampsia/2005– | Corthera/Novartis CT00333307 | |
| Condition | Pre‐eclampsia | |||
| Intervention | Drug: rhRLN | |||
| 5 | Terminated, Phase II, n = 3 | Safety and efficacy of RLX030 in pregnant women with pre‐eclampsia/2013–2014 | Novartis Pharmaceuticals NCT01566630 | |
| Condition | Pre‐eclampsia | |||
| Intervention | RLX030 | |||
| 6 | Completed, Phase III, n = 231 | Recombinant human relaxin in the treatment of diffuse scleroderma/1998–2001 | University of Medicine and Dentistry of New Jersey NCT00704665 | |
| Condition: | Systemic sclerosis | |||
| Intervention | Drug: rhRLN | |||
| 7 | Recruiting, Observational n = 30 (estimated) | Relaxin in multiple sclerosis (MS)/2013– | Providence Health & Services NCT01909492 | |
| Condition | Multiple sclerosis, relapsing‐remitting | |||
| Interventions | Procedure: blood draw; | |||
| Procedure: lumbar puncture | ||||
| 8 | Completed, Phase II, n = 1 | Phase II study of recombinant relaxin for progressive systemic sclerosis/1991–1997 | National Center for Research Resources (NCRR) NCT00004380 | |
| Condition | Systemic Sclerosis | |||
| Intervention | Drug: rhRLN | |||
| 9 | Completed, Phase II, n = 72 | Recombinant human relaxin (rhRlx) in pregnant women scheduled for induction of labour/2005–2006 | Corthera/Novartis NCT00259103 | |
| Condition | Labour, induced | |||
| Intervention | Drug: rhRLN | |||
| 10 | Recruiting, Phase II n = 60 (estimated) | Study of the vascular effects of serelaxin/2014– | Novartis Pharmaceuticals NCT01979614 | |
| Condition | Coronary artery disease | |||
| Intervention | Drug: rhRLN | |||
| 11 | Not yet recruiting Phase II, n = 20 (estimated) | Serelaxin to lower portal pressure/2016– | University of Edinburgh, Novartis Pharmaceuticals and NHS Lothian NCT02669875 | |
| Conditions | Liver cirrhosis; hypertension, portal | |||
| Intervention | Drug: rhRLN | |||
| 12 | Completed, Phase II, n = 47 | An exploratory haemodynamic study in patients with compensated cirrhosis and portal hypertension/2012–2014 | Novartis Pharmaceuticals NCT01640964 | |
| Condition | Compensated cirrhosis and portal hypertension | |||
| Intervention | Drug: terlipressin acetate; serelaxin | |||
| 13 | Completed, Phase II, n = 321 | Safety of repeat doses of IV serelaxin in subjects with chronic heart failure/2014–2015 | Novartis Pharmaceuticals NCT01982292 | |
| Condition | Chronic heart failure | |||
| Intervention | Drug: serelaxin | |||
| 14 | Recruiting, Phase II, n = 125 (estimated) | Study of the effect of serelaxin on high‐sensitivity cardiac tropin I release in patients with chronic heart Failure/2016‐ | Novartis Pharmaceuticals NCT02625922 | |
| Condition | Chronic heart failure | |||
| Intervention | Drug: serelaxin | |||
| 15 | Completed, Phase II, n = 46 | Study of safety, tolerability and pharmacokinetics of serelaxin in Japanese acute heart failure (AHF) patients/2014 | Novartis Pharmaceuticals NCT02002702 | |
| Condition | Acute heart failure | |||
| Intervention | Drug: serelaxin | |||
| 16 | Recruiting, Phase III, n = 2685 (estimated) | Effect of serelaxin versus standard of care in acute heart failure (AHF) patients/2014– | Novartis Pharmaceuticals NCT02064868 | |
| Condition | Acute heart failure; systolic blood pressure ≥125 mmHg,; mild‐to‐moderate renal impairment | |||
| Intervention | Drug: serelaxin; standard of care | |||
| 17 | Completed, Phase I, n = 36 | PK of Serelaxin in severe renal impairment and end stage renal disease/2013 | Novartis Pharmaceuticals NCT01875523 | |
| Condition | Renal failure, chronic; end‐stage renal disease | |||
| Intervention | Drug: serelaxin | |||
| 18 | Recruiting, Phase III, n = 1520 (estimated) | Efficacy, safety and tolerability of sexelaxin when added to standard therapy in AHF/2014– | Novartis Pharmaceuticals NCT02007720 | |
| Condition | Acute heart failure | |||
| Intervention | Drug: serelaxin | |||
| 19 | Recruiting, Phase II, n = 36 (estimated) | Pharmacokinetics and safety of serelaxin on top of standard of care therapy in pediatric patients with acute heart failure/2014– | Novartis Pharmaceuticals NCT02151383 | |
| Condition | Acute Heart Failure | |||
| Intervention | Drug: serelaxin | |||
| 20 | Recruiting, Phase III, n = 6800 (estimated) | Efficacy, safety and tolerability of serelaxin when added to standard therapy in AHF/2013‐ | Novartis Pharmaceuticals NCT01870778 | |
| Condition | Acute Heart failure | |||
| Intervention | Drug: serelaxin | |||
| 21 | Completed, Phase II, n = 71 | Haemodynamic responses to RLX030 infusion in subjects with acute heart failure/2012–2014 | Novartis Pharmaceuticals NCT01543854 | |
| Condition | Acute heart failure | |||
| Intervention | Drug: serelaxin | |||
rhRLN, Serelaxin or RLX30 is recombinant human relaxin 2. n, enrolment.
Figure 1.
Relaxin receptor as a drug target. IVF, in vitro ferilization.
There are ongoing attempts to find an alternative to the full‐length recombinant peptide based on existing models of hormone–receptor interactions. Synthetic and recombinant forms and derivatives of the relaxin hormone with truncated A‐ and B‐chains were tested for activity and stability. This resulted in the identification of a single‐chain derivative of the B‐chain, B7‐33, as a biased RXFP1 receptor agonist that preferentially activates the pERK pathway over cAMP in cells that endogenously expressed RXFP1 receptors (Hossain et al., 2016). There were also reports of short linear peptides derived from a naturally occurring protein containing a collagen‐like repeat (Shemesh et al., 2009; Pini et al., 2010). However, due to a short half‐life and low plasma stability, peptide derivatives still require delivery through continuous intravenous injections or by an osmotic subcutaneous pump. This becomes especially challenging in treatment of chronic conditions such as organ fibrosis. An additional problem, especially in animal models where human relaxin or its derivatives are used, is the mounting immune response to injected peptide (Samuel et al., 2017). Antibody production in patients were studied but not reported yet (study 13 in Table 1). An obvious alternative to peptide ligand is a low MW, non‐peptide agonist. Such compounds are often stable, easy to synthesize and adaptable for oral delivery. It is clear however that a small molecule cannot bind to the multiple orthosteric sites that the peptide ligand uses in the ectodomain and extracellular loops of the GPCR seven transmembrane domain (7TM) (Halls et al., 2005; Scott et al., 2012; Diepenhorst et al., 2014; Halls et al., 2015; Sethi et al., 2016). Often, the downstream signalling profile is biased, resulting in preferential activation of a subset of cellular responses. The structure‐based design of low MW agonists is challenging as the receptor interaction sites are difficult to predict, and, as is the case for RXFP1 receptors, the structure of inactive/active forms of 7TM can only be modelled based on the solved crystal structures of related GPCRs. The alternative approach of unbiased, high throughput screening of large‐sized libraries of low MW compounds, using cell‐based cAMP response was attempted by several groups. The only reported series of low MW RXFP1 receptor agonists came from our screening campaign at the National Center for Advancing Translational Sciences, NIH (Chen et al., 2013; Xiao et al., 2013). Here, we review the available data on the screening, structure, suggested mode of interaction with the receptor and functional and pharmacological properties of these agonists.
Relaxin receptor RXFP1 structure and signalling cascade
Relaxin is a peptide hormone in the relaxin/insulin‐like family. In primates, there are two closely related genes encoding relaxin 1 and relaxin 2 (RLN2) peptide; however, only RLN2 is believed to be the main circulating form of the hormone (Garibay‐Tupas et al., 2000). Similar to insulin, it is translated as a preprohormone containing a signal peptide, B‐chain, C‐peptide and A‐chain. Upon delivery to the surface, the N‐terminal signal peptide and interconnecting C‐peptide are removed, and the mature 6 kDa heterodimer is produced. The heterodimer consists of two peptide chains (A and B) tethered by two disulphide bonds, with an additional disulfide bridge within the A‐chain. Thus, relaxin structurally is very similar to insulin hormone. Accumulating data show that the B‐chain α‐helix contains the receptor binding site (summarized in Halls et al., 2015). The cognate relaxin receptor, RXFP1 (previously called LGR7), is a member of the subfamily of GPCRs that contain leucine‐rich repeats (Hsu et al., 2002). The RXFP2 receptor is another member of this family (previously GREAT and LGR8), and shares 60% sequence identity and similar structure (Overbeek et al., 2001; Gorlov et al., 2002). RXFP2 is a cognate receptor for insulin‐like 3 peptide (INSL3), a member of the same peptide hormone family as relaxin (Kumagai et al., 2002; Bogatcheva et al., 2003).
Both RXFP1 and RXFP2 receptors have the same structure that features a large ectodomain containing a single LDL class A (LDLa) module, an LDLa linker and 10 leucine‐rich repeats (LRRs). LRRs form a horse shoe‐shaped structure consisting of a β‐sheet on the concave side and an array of α‐helices on the convex side of the LRRs (Kobe and Deisenhofer, 1993). The 7TM domain has three extracellular loops and three intracellular loops that play a role in the induction of cellular signalling responses upon peptide binding. The proposed model of relaxin binding to RXFP1 receptors involves binding of peptide to the LRRs via B‐chain residues and to the LDLa linker via A‐chain residues that causes relaxin to stabilize and extend the linker in a helical conformational state. This results in interactions between the linker, LDLa and relaxin with the receptor 7TM domain, leading to activation of the receptor and the appropriate signalling pathways (Hopkins et al., 2007; Kong et al., 2013; Diepenhorst et al., 2014; Halls et al., 2015; Sethi et al., 2016). Both RXFP1 and RXFP2 receptors couple to Gɑ s and Gα oB proteins, which upon receptor activation stimulate AC to generate cAMP, followed by PKA activation. In addition, RXFP1 receptors also couple to Gα i3 proteins to further modulate cAMP production. Stimulation of RXFP1 receptors activates NO/NOS/cGMP, NF‐κB and Akt and the phosphorylation of ERK1/2 and PI3K pathways with subsequent activation of two protein kinase C variants, PKCζ and PKCδ, and other signalling pathways. These various responses were described in RXFP1‐transfected HEK293T cells or in cells expressing endogenous relaxin receptors (Halls et al., 2015). It should be noted that the detected downstream cellular signalling varies in various RXFP1 receptor expressing cells in different organs. In summary, relaxin generates a complex pattern of cellular signalling and gene expression, resulting in a complex physiological response.
Discovery of low MW agonists of RXFP1 receptors
THP1 cells and HEK293T cells stably expressing RXFP1 receptors (HEK293T‐RXFP1) are two well‐described cell lines that exhibit strong response to relaxin treatment. The most robust response to relaxin treatment in HEK293T‐RXFP1 is through classical Ga s‐coupled signalling, resulting in elevated intracellular and extracellular cAMP concentrations (Hsu et al., 2002). In addition, treatment of HEK293T‐RXFP1 with relaxin rapidly changes cell–cell interactions that can be detected using an xCelligence Analyzer (Roche Diagnostics, Indianapolis, IN) (Xiao et al., 2013). Human THP1 monocytes express high levels of RXFP1 receptors and respond to relaxin by activation of cAMP synthesis and increased expression of VEGF (Unemori et al., 2000; Bartsch et al., 2001). These cell lines were used for primary and secondary screens for low MW RXFP1 receptor agonists (Figure 2). HEK293T‐RXFP1 cells were used in a homogeneous cAMP assay to screen for low MW agonists of human RXFP1 receptors in a quantitative high‐throughput screening format (Chen et al., 2013; Xiao et al., 2013). The assay was successfully miniaturized and used to screen a 365 677‐compound NIH library. Because a low hit rate was expected, relatively lenient selection criteria were adopted in this screen. At the same time, a 4‐point concentration screening scheme allowed the selection of the compounds based on the quality of dose–response curves (Chen et al., 2013). Following confirmation and counter screening against INSL3‐stimulated HEK293T‐RXFP2 and vasopressin‐stimulated HEK293T‐V1b receptors, two low MW agonists that were >100‐fold more selective towards RXFP1 over RXFP2 receptors were identified. While their reported potencies in the cAMP assay were low (EC50 4–6 μM, 60–80% efficacy), both compounds shared molecular features, suggesting the significance of their common scaffold (Chen et al., 2013). Extensive medicinal chemistry efforts were undertaken to improve potency and selectivity through modification and replacement of the side groups of the original scaffold (Xiao et al., 2013). Optimized compounds showed a remarkable increase in activity from the micromolar EC50 values of the initial hits to potencies <50 nM with the same efficacy as relaxin (Figure 3). In secondary screens, we determined that these compounds were able to increase cAMP levels and VEGF gene expression in THP1 cells. Similar to relaxin, the small molecules increased cellular impedance in HEK293T‐RXFP1 but not in parental HEK293T cells. While additional cell‐specific activity assays might be needed to select the best compound for a particular application, the low MW compound ML290 was selected for further validation (Table 2).
Figure 2.
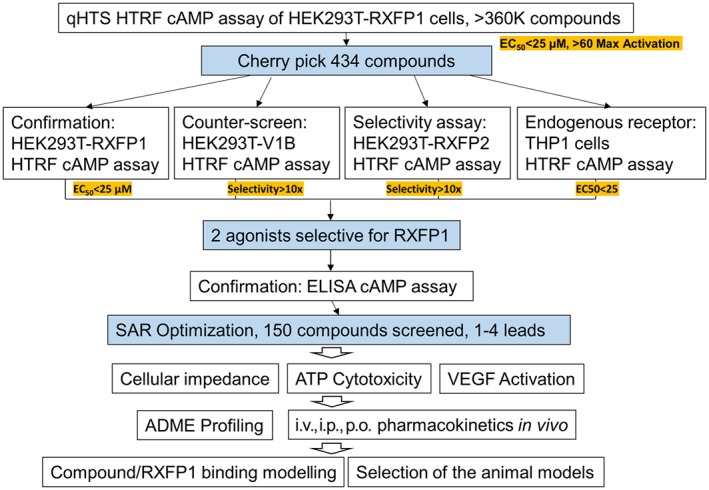
Summary of high‐throughput screening (HTS) screening and follow‐up studies of low MW agonists of RXFP1 receptors. Selection criteria are highlighted in yellow. HTFR, homogeneous time resolved fluorescence; SAR, structure activity relationship; ADME, absorption, distribution, metabolism, and excretion.
Figure 3.
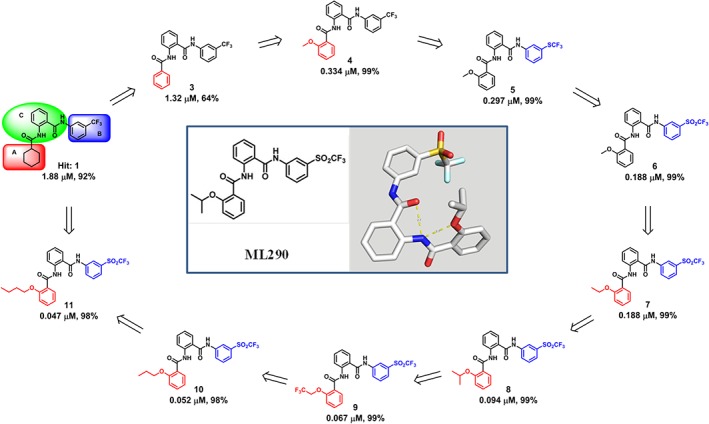
Heat‐to‐Lead optimization of lowMW relaxin receptor agonist. Shown are EC50 and efficacy (% of maximal response) of compounds in cAMP assay.
Table 2.
Low MW relaxin receptor agonist ML290
| Molecular formula (MW) | RXFP1 EC50 (μM; Max. Resp.) | ATP Tox. EC50 (μM; Max. resp.) | Aqueous kinetic solubility in PBS (pH = 7) | Liver microsomal stability (t1/2 in min; at 2 μM) | Plasma stability (% remaining after 2 h) | PK t1/2 in mice (h) at 30 mg·kg−1 i.p. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| μg·mL−1 | μM | M | H | M | H | Plasma | Heart | |||
| C24H21F3N2O5S (506.5) | 0.094 (98%) | 9.4 (−85%) | 3.5 | 7 | 122 | 161 | 99 | 100 | 8.5 | 7.5 |
H, human serum; M, mouse serum. For intraperitoneal injections (i.p.) in pharmacokinetics (PK) studies the following formulation was used: 10% NMP (N‐Methyl‐2‐pyrrolidone) + 10% SolutolHS15 + 10% PEG400 + 70% Saline.
There were two major factors that suggested that ML290 might be a good lead for in vivo studies. First, ML290 cytotoxicity was low, with EC50 just below 10 μM in an ATP toxicity assay. Second, our data indicated that ML290 has excellent stability both in vitro and in vivo. In vitro studies were performed with mouse and human plasma resulting in half‐life stability over 2 h (Table 2). It is comparable with the reported native relaxin stability in male human serum (Nair et al., 2015). Structural studies, which determined the minimum energy conformation of ML290 in solid state analysed by X‐ray crystallography and in solution by variable temperature NMR and nuclear Overhauser effect spectroscopy, suggested that the two intramolecular hydrogen binding interactions found in this molecule appear to stabilise its 3D conformation.
Our published and unpublished pharmacokinetics studies in mice and Rhesus monkeys suggest that various routes of delivery result in a remarkable stability of the compound with a half‐life of about 8 h in both plasma and heart. This contrasts drastically with a in vivo half‐life of approximately 10 min for human recombinant relaxin (Chen et al., 1993). There were no significant adverse effects of ML290 on animals used in these experiments. Even oral gavage was effective, although further optimization of the vehicle because of the limited solubility of ML290 (3.3–17.0 μM) might be required (Xiao et al., 2013).
Structural insights into agonist–receptor interactions
Unlike the typical orthosteric site of the 7TM pocket for most druggable GPCRs, relaxin binds to RXFP1 receptors through interactions predominantly within the interface between the LRRs domain and extracellular loops 1 and 2 (Halls et al., 2015). Initially, three important observations were made concerning the possible binding site for ML290. First, ML290 activated human RXFP1 receptors with a mutation in the LDLa domain rendering it unresponsive to relaxin stimulation (Xiao et al., 2013). This suggested that, unlike the natural ligand, the LDLa domain is not required for ML290 activation of RXFP1 receptors. Second, a binding assay showed that ML290 did not compete with relaxin for RXFP1 receptor binding, and therefore, relaxin and ML290 receptor binding sites are different (Xiao et al., 2013). Finally, cells transfected with mouse RXFP1 receptors did not respond to ML290. Subsequent analysis of human‐mouse RXFP1 chimeric receptors and site‐specific mutagenesis demonstrated that the ML290 species‐specificity was defined by its binding to the variable amino acids in the third extracellular loop of the 7TM domain. Importantly, these changes in amino acid sequences between mouse and human receptors did not affect the activity of relaxin (Xiao et al., 2013). Therefore, ML290 appears to bind to a different site in the receptor from the cognate ligand relaxin.
A putative binding model of the low MW agonists to the receptor was generated using an approach combining homology modelling, docking, molecular dynamics simulations and binding free energy calculations of various ML290 derivative compounds (Figure 3) (Hu et al., 2016). Currently, the transmembrane domain structure of RXFP1 receptors has not been solved and thus the human β2 adrenoceptor was used as a template to model inactive and activated states of the receptor. Clustering analysis of docking poses suggested that ML290 binds to the internal region within the 7TM of human RXFP1 receptors, at an allosteric site of the receptor. The core 2‐acetamido‐N‐phenylbenzamide is positioned in the pocket by forming extensive van der Waals' and hydrophobic interactions mainly with residues from transmembrane domains 5 and 7 (TM5 and TM7). The lack of polar and hydrogen bonding interactions within the binding site is less common compared with other GPCR agonist binding (Lebon et al., 2012). Interestingly, the trifluoromethylsulfonyl group of ML290 interacts with the two hallmark residues G659/T660 at the C‐terminus of extracellular loop 3 (ECL3) of TM7. The G659/T660 to D659/S660 (as in mouse RXFP1 receptors) substitution within ECL3 of human RXFP1 receptors abolished the activation by ML290 (Xiao et al., 2013). Because the flexibility of ECL3 is associated with conformational changes in TM6 and TM7, the close interactions of the low MW compound with ECL3 and the G659/T660 motif suggest its important functional role in triggering activation of RXFP1 receptors.
The theoretical binding model was validated by comparison of the experimental data of known ML290 derivatives and their structure–activity relationship analysis (Xiao et al., 2013; Hu et al., 2016). The most active compound in the cAMP assay had the following essential features: (a) an intramolecular H‐bond that was retained within the binding model, which was in agreement with the crystal structure of the isolated compund and solution NMR; (b) an alkoxy group in ortho‐position stabilizing the binding conformation of the compound in the pocket; and (c) an aniline ring at the meta‐position, oriented to ECL3 and interacting closely with the G659/T660 motif. The binding interaction showed that the aliphatic chain was inserted into a hydrophobic pocket region formed by residues I493, Y579 and I583, between TM5 and TM6, that provided a structural basis of the increased binding affinities of compounds with longer alkoxy aliphatic chains at the ortho‐position.
The ML290 binding model was further confirmed by our extensive site‐directed mutagenesis of the human RXFP1 receptor. The results indicated that amino acids in TM3 (I493, T496), TM5 (I583), TM6 (I644, F645), TM7 (W664, I667, F668, L670), ECL2 (F564) and ECL3 (G659) significantly affected ML290 induction of cAMP (Figure 4). Remarkably, all amino acids but G659 were conserved between human and mouse RXFP1 receptor sequences. These data, together with the binding model, suggest why residues G659/T660 within ECL3 of human RXFP1 receptors are important in receptor activation and selectivity by the low MW agonist. In contrast, human RXFP2 and mouse and rat RXFP1 receptors have the residue motif D/T or D/S. Glycine 659 in the RXFP1 receptor allows a flexible conformation in which the backbone of threonine is capable of forming a hydrogen bond with ML290. Remarkably, substitution of D/S to G/T in the mouse RXFP1 ECL3 sequence fully restored the ability of ML290 to activate the mutant mouse receptor (Xiao et al., 2013; Hu et al., 2016). It should be noted however that some of the substitutions also reduced receptor activation by RLN. The latter can be explained by poor cell surface expression of some of these constructs, and further studies to clarify the binding of ML290 are needed to confirm the proposed model of its interaction with RXFP1 receptors.
Figure 4.
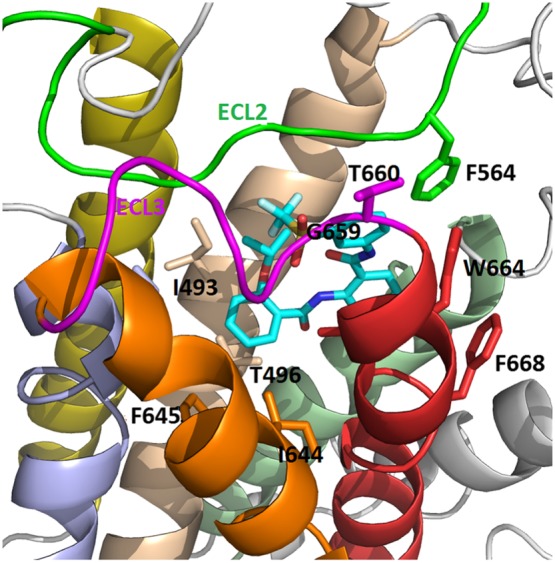
Model of ML290 binding with human RXFP1 receptors. The agonist ML290 is shown in stick form (C atoms in cyan, O and N atoms are shown in red and blue). Shown are key residues within the RXFP1 receptor involved in binding interactions. TM3 (brown), TM5 (blue), TM6 (orange), TM7 (red), ECL2 (green) and ECL3 (magenta) are shown in different colors.
It is worth mentioning that ML290 behaves as an allosteric inverse agonist (Christopoulos et al., 2014) of relaxin in the cAMP assay when mouse RXFP1 receptors are used (Hu et al., 2016). This suggests that ML290 binds to the mouse receptor and prevents its conformational change to active state upon stimulation with relaxin. The question remains whether such a structural lock prevents activation of all G proteins bound to RXFP1 receptors or only the Gα s subunit required for an activation of adenylate cyclase. It is possible that other signalling pathways generated by RXFP1 receptor activation might still be induced by ML290 upon binding to the mouse receptor.
Animal models for preclinical testing of low MW compounds
The vast majority of experiments testing the effects of relaxin in live animals were performed in mice and rats. The inability of ML290 to activate RXFP1 receptors from these species creates a need to find a suitable animal model for preclinical studies. To this end, expression constructs with cDNA sequences of RXFP1 receptors from Rhesus macaque, pig, guinea pig and rabbit were cloned and their response to relaxin and ML290 was analysed (Huang et al., 2015). Comparison of amino acid sequences revealed that all amino acids identified in modelling studies, apart from ECL3, that were important for ML290 binding were conserved between species. The high sequence homology and the presence of the crucial Gly Thr amino acid pair at the end of ECL3 in macaque and pig RXFP1 receptors correlated with a full cAMP response upon ML290 and relaxin stimulation. Guinea pig RXFP1 receptors were activated by relaxin but responded to ML290 only marginally at very high doses of compound. Surprisingly, the rabbit receptors was not activated by relaxin but treatment with ML290 caused a significant increase in cAMP production. Chimeric receptors containing the rabbit or guinea pig receptor ectodomain and the human 7TM domain were better activated by ML290 than rabbit RXFP1 receptors, indicating that additional sites within human 7TM participate in agonist binding (Huang et al., 2015).
The analysis of receptors from various species highlighted the importance of careful selection of an animal model. Apart from known differences in the biology of relaxin (Sherwood, 2004), the rabbit and mouse RXFP1 receptors are unsuitable for testing responses to ML290. Existing techniques to modify the genome of laboratory animals using both traditional embryonic stem cell gene targeting in mouse and rats or the use of CRISPR‐Cas9 gene editing in any mammals offer an opportunity to create ‘optimized’ animal models. In this situation, the preferred animal model would be to make a ‘humanized’ mouse with the human receptor gene under control of the endogenous mouse RXFP1 promoter and other regulatory elements. The human gene would then be expressed at the same level as the mouse receptor in different organs and tissues. Importantly, the mouse gene should be disrupted and the human orthologue should fully complement its absence in homozygotes. Such a mutant was recently produced by our team and soon will be available for testing of ML290 or other RXFP1 receptor modulators in vivo.
Future directions
The identification of new ligands for RXFP1 receptors raises the question of whether the complex effects of relaxin upon cell cultures or in live animals can be faithfully reproduced by these compounds. The dramatic size difference between relaxin and ML290, the allosteric mode of low MW compound interaction with RXFP1 receptors and potentially different receptor activation dynamics indicate that the resulting downstream signalling may be biased between the two ligands. As noted above, the activation of RXFP1 receptors by relaxin produces pleotropic response in different cells, tissues and organs. Consequently, biased signalling from ML290‐activated RXFP1 receptors would have important implications therapeutically with an activation of desired effects and less side effects (Leach et al., 2015). Indeed, even the derivative of the relaxin B‐peptide, B7‐33 peptide, preferentially activates the pERK pathway over cAMP in cells with endogenously expressed receptor (Hossain et al., 2016). As a result, B7‐33 had anti‐fibrotic effects in three different preclinical rodent lung or heart fibrosis models but did not promote prostate cancer cell tumour growth (Hossain et al., 2016). It is not clear now how ML290 will behave in various disease models where relaxin has demonstrated therapeutic effects. Detailed comparison of proteomic and gene expression profiles in specific cell types may be useful for the prediction of potential physiological effects of low MW agonists in live animals. It should be noted however that due to the pleotropic effects of relaxin, it is still not always clear which RXFP1 receptor‐regulated signalling pathway is therapeutically significant in different diseases.
Another interesting but not yet studied question is the effect of ML290 on RXFP1 signalosome formation, receptor coupling to various G‐proteins, β‐arrestins and other proteins (Halls and Cooper, 2010; Halls, 2012). It was shown that constitutive assembly of the RXFP1‐signalosome in a higher‐order protein complex facilitates receptor sensitivity to attomolar concentrations of relaxin (Halls and Cooper, 2010). Such complex formation produces constitutive activity and dual coupling to G‐proteins and β‐arrestins and reveals a concentration‐biased agonism mediated by natural ligand. Whether ML290 will affect the formation and signalling of such complexes, thus interfering with endogenous relaxin in vivo and disrupting signalosomes is not known. The effect of various concentrations of ML290 should be further tested in future discovery programmes.
Further long‐term in vivo toxicity and receptor specificity studies should define the potential clinical utility of the compounds. To address this issue, it is important to use an appropriate animal model. Unfortunately, RXFP1 receptors from the small laboratory animals we tested do not respond to either ML290 or relaxin. Moreover, as we have shown, while ML290 does not activate cAMP in cells transfected with rodent receptors, it behaves as an antagonist at mouse RXFP1 receptors (Hu et al., 2016) and hence can potentially affect endogenous relaxin signalling in this species. Thus, the use of both RXFP1 receptor‐deficient mutant mice (Kamat et al., 2004; Kaftanovskaya et al., 2015) and a humanized RXFP1 receptor mouse model might be necessary in preclinical testing.
Understanding of the structural basis of the interactions of low MW compounds with the receptor opens the possibility of structure‐based drug design and lead optimization, as well as in silico screening of millions of chemical databases for novel and efficient low MW agonists. Such improvements should also address solubility, the prospects of oral delivery and an establishment of safe dosage. Injected relaxin has a short half‐life in serum, although signalosome‐mediated RXFP1 receptors may respond to very low concentration of the peptide. In HEK293T and Cos‐7 transfected cells, activation of RXFP1 receptors does not result in significant receptor phosphorylation, desensitization or internalization (Callander et al., 2009). However, in human primary decidual cells and in a second line of HEK293T cells stably expressing RXFP1 receptors, an internalization of these receptors enhanced by overexpression of β‐arrestin‐2 has been demonstrated (Kern and Bryant‐Greenwood, 2009). As the low (low MW agonists) MW agonists have much higher stability, will treatment with them lead to increased desensitization of RXFP1 receptors?
The ability of ML290 to partially antagonize relaxin‐induced mouse receptor signalling via cAMP indicates that it is possible to also identify human RXFP1 receptor antagonists. Suppression of relaxin/RXFP1 receptor signalling in a range of cancer cells and in mouse cancer models can reduce relaxin‐mediated tumour growth, invasiveness and vascularization, as well as increasing apoptosis (Feng et al., 2007; Klonisch et al., 2007; Silvertown et al., 2007; Feng et al., 2010; Feng and Agoulnik, 2011; Glogowska et al., 2013; Neschadim et al., 2014). Targeting of RXFP1 receptors with low MW antagonists might offer a new approach in lessening the tumour burden. The identification of RXFP1 receptor agonists indicates that activation of the INSL3 receptor RXFP2 by low MW compounds is also feasible. Therapeutically, RXFP2 receptor agonists may be used to help prevent bone loss (Ferlin et al., 2008; Ferlin et al., 2017) or for treatment of reproductive abnormalities (Agoulnik, 2007).
In summary, the first‐in‐class, low MW, agonists of the relaxin receptor offer not only a tool to study various basic questions of relaxin endocrinology but may also be directly tested to evaluate the therapeutic benefits of activation of RXFP1 receptors in preclinical and clinical studies.
Conflict of interest
The authors are inventors in the Patent 9452973, modulators of relaxin receptor 1, US patent and trademark office.
Acknowledgements
The authors thank Courtney Myhr for editorial comments. The work on low MW modulators of relaxin family receptors was supported by National Institute of Health grants R03MH085705, 1U01CA177711 and 1R01AR070093 (A.I.A) and Molecular Libraries Initiative of the NIH Roadmap for Medical Research (U54MH084681).
Agoulnik, A. I. , Agoulnik, I. U. , Hu, X. , and Marugan, J. (2017) Synthetic non‐peptide low molecular weight agonists of the relaxin receptor 1. British Journal of Pharmacology, 174: 977–989. doi: 10.1111/bph.13656.
References
- Agoulnik AI (2007). Relaxin and related peptides in male reproduction. Adv Exp Med Biol 612: 49–64. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5734–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch O, Bartlick B, Ivell R (2001). Relaxin signalling links tyrosine phosphorylation to phosphodiesterase and adenylyl cyclase activity. Mol Hum Reprod 7: 799–809. [DOI] [PubMed] [Google Scholar]
- Bogatcheva NV, Truong A, Feng S, Engel W, Adham IM, Agoulnik AI (2003). GREAT/LGR8 is the only receptor for insulin‐like 3 peptide. Mol Endocrinol 17: 2639–2646. [DOI] [PubMed] [Google Scholar]
- Brennand JE, Calder AA, Leitch CR, Greer IA, Chou MM, MacKenzie IZ (1997). Recombinant human relaxin as a cervical ripening agent. Br J Obstet Gynaecol 104: 775–780. [DOI] [PubMed] [Google Scholar]
- Callander GE, Thomas WG, Bathgate RA (2009). Prolonged RXFP1 and RXFP2 signaling can be explained by poor internalization and a lack of beta‐arrestin recruitment. Am J Physiol Cell Physiol 296: C1058–C1066. [DOI] [PubMed] [Google Scholar]
- Chen CZ, Southall N, Xiao J, Marugan JJ, Ferrer M, Hu X et al. (2013). Identification of small‐molecule agonists of human relaxin family receptor 1 (RXFP1) by using a homogenous cell‐based cAMP assay. J Biomol Screen 18: 670–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SA, Perlman AJ, Spanski N, Peterson CM, Sanders SW, Jaffe R et al. (1993). The pharmacokinetics of recombinant human relaxin in nonpregnant women after intravenous, intravaginal, and intracervical administration. Pharm Res 10: 834–838. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Changeux JP, Catterall WA, Fabbro D, Burris TP, Cidlowski JA et al. (2014). International union of basic and clinical pharmacology. XC. multisite pharmacology: recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol Rev 66: 918–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KP (2016). G‐Protein‐coupled receptors as potential drug candidates in preeclampsia: targeting the relaxin/insulin‐like family peptide receptor 1 for treatment and prevention. Hum Reprod Update 22: 647–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KP, Shroff SG (2011). Effects of relaxin on arterial dilation, remodeling, and mechanical properties. Curr Hypertens Rep 13: 409–420. [DOI] [PubMed] [Google Scholar]
- Diepenhorst NA, Petrie EJ, Chen CZ, Wang A, Hossain MA, Bathgate RA et al. (2014). Investigation of interactions at the extracellular loops of the relaxin family peptide receptor 1 (RXFP1). J Biol Chem 289: 34938–34952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Agoulnik AI (2011). Expression of LDL‐A module of relaxin receptor in prostate cancer cells inhibits tumorigenesis. Int J Oncol 39: 1559–1565. [DOI] [PubMed] [Google Scholar]
- Feng S, Agoulnik IU, Bogatcheva NV, Kamat AA, Kwabi‐Addo B, Li R et al. (2007). Relaxin promotes prostate cancer progression. Clin Cancer Res 13: 1695–1702. [DOI] [PubMed] [Google Scholar]
- Feng S, Agoulnik IU, Truong A, Li Z, Creighton CJ, Kaftanovskaya EM et al. (2010). Suppression of relaxin receptor RXFP1 decreases prostate cancer growth and metastasis. Endocr Relat Cancer 17: 1021–1033. [DOI] [PubMed] [Google Scholar]
- Ferlin A, De Toni L, Sandri M, Foresta C (2017). Relaxin and insulin-like peptide 3 in the musculoskeletal system: from bench to bedside. Br J Pharmacol 174: 1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlin A, Pepe A, Gianesello L, Garolla A, Feng S, Giannini S et al. (2008). Mutations in the insulin‐like factor 3 receptor are associated with osteoporosis. J Bone Miner Res 23: 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garibay‐Tupas JL, Bao S, Kim MT, Tashima LS, Bryant‐Greenwood GD (2000). Isolation and analysis of the 3'‐untranslated regions of the human relaxin H1 and H2 genes. J Mol Endocrinol 24: 241–252. [DOI] [PubMed] [Google Scholar]
- Glogowska A, Kunanuvat U, Stetefeld J, Patel TR, Thanasupawat T, Krcek J et al. (2013). C1q‐tumour necrosis factor‐related protein 8 (CTRP8) is a novel interaction partner of relaxin receptor RXFP1 in human brain cancer cells. J Pathol 231: 466–479. [DOI] [PubMed] [Google Scholar]
- Gorlov IP, Kamat A, Bogatcheva NV, Jones E, Lamb DJ, Truong A et al. (2002). Mutations of the GREAT gene cause cryptorchidism. Hum Mol Genet 11: 2309–2318. [DOI] [PubMed] [Google Scholar]
- Halls ML (2012). Constitutive formation of an RXFP1‐signalosome: a novel paradigm in GPCR function and regulation. Br J Pharmacol 165: 1644–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halls ML, Bathgate RA, Sutton SW, Dschietzig TB, Summers RJ (2015). International union of basic and clinical pharmacology. XCV. Recent advances in the understanding of the pharmacology and biological roles of relaxin family peptide receptors 1‐4, the receptors for relaxin family peptides. Pharmacol Rev 67: 389–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halls ML, Bond CP, Sudo S, Kumagai J, Ferraro T, Layfield S et al. (2005). Multiple binding sites revealed by interaction of relaxin family peptides with native and chimeric relaxin family peptide receptors 1 and 2 (LGR7 and LGR8). J Pharmacol Exp Ther 313: 677–687. [DOI] [PubMed] [Google Scholar]
- Halls ML, Cooper DM (2010). Sub‐picomolar relaxin signalling by a pre‐assembled RXFP1, AKAP79, AC2, beta‐arrestin 2, PDE4D3 complex. EMBO J 29: 2772–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins EJ, Layfield S, Ferraro T, Bathgate RA, Gooley PR (2007). The NMR solution structure of the relaxin (RXFP1) receptor lipoprotein receptor class A module and identification of key residues in the N‐terminal region of the module that mediate receptor activation. J Biol Chem 282: 4172–4184. [DOI] [PubMed] [Google Scholar]
- Hossain MA, Kocan M, Yao ST, Royce SG, Nair VB, Siwek C et al. (2016). A single‐chain derivative of the relaxin hormone is a functionally selective agonist of the G protein‐coupled receptor, RXFP1. Chemical Science 7: 3805–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu SY, Nakabayashi K, Nishi S, Kumagai J, Kudo M, Sherwood OD et al. (2002). Activation of orphan receptors by the hormone relaxin. Science 295: 671–674. [DOI] [PubMed] [Google Scholar]
- Hu X, Myhr C, Huang Z, Xiao J, Barnaeva E, Ho BA et al. (2016). Structural insights into the activation of human relaxin family peptide receptor 1 by small‐molecule agonists. Biochemistry 55: 1772–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Myhr C, Bathgate RA, Ho BA, Bueno A, Hu X et al. (2015). Activation of relaxin family receptor 1 from different mammalian species by relaxin peptide and small‐molecule agonist ML290. Front Endocrinol 6: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaftanovskaya EM, Huang Z, Lopez C, Conrad K, Agoulnik AI (2015). Conditional deletion of the relaxin receptor gene in cells of smooth muscle lineage affects lower reproductive tract in pregnant mice. Biol Reprod 92: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat AA, Feng S, Bogatcheva NV, Truong A, Bishop CE, Agoulnik AI (2004). Genetic targeting of relaxin and insulin‐like factor 3 receptors in mice. Endocrinology 145: 4712–4720. [DOI] [PubMed] [Google Scholar]
- Kelly AJ, Kavanagh J, Thomas J (2001). Relaxin for cervical ripening and induction of labour. Cochrane Database Syst Rev (2): CD003103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern A, Bryant‐Greenwood GD (2009). Characterization of relaxin receptor (RXFP1) desensitization and internalization in primary human decidual cells and RXFP1‐transfected HEK293 cells. Endocrinology 150: 2419–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klonisch T, Bialek J, Radestock Y, Hoang‐Vu C, Hombach‐Klonisch S (2007). Relaxin‐like ligand‐receptor systems are autocrine/paracrine effectors in tumor cells and modulate cancer progression and tissue invasiveness. Adv Exp Med Biol 612: 104–118. [DOI] [PubMed] [Google Scholar]
- Kobe B, Deisenhofer J (1993). Crystal structure of porcine ribonuclease inhibitor, a protein with leucine‐rich repeats. Nature 366: 751–756. [DOI] [PubMed] [Google Scholar]
- Kong RC, Petrie EJ, Mohanty B, Ling J, Lee JC, Gooley PR et al. (2013). The relaxin receptor (RXFP1) utilizes hydrophobic moieties on a signaling surface of its N‐terminal low density lipoprotein class A module to mediate receptor activation. J Biol Chem 288: 28138–28151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai J, Hsu SY, Matsumi H, Roh JS, Fu P, Wade JD et al. (2002). INSL3/Leydig insulin‐like peptide activates the LGR8 receptor important in testis descent. J Biol Chem 277: 31283–31286. [DOI] [PubMed] [Google Scholar]
- Leach K, Conigrave AD, Sexton PM, Christopoulos A (2015). Towards tissue‐specific pharmacology: insights from the calcium‐sensing receptor as a paradigm for GPCR (patho)physiological bias. Trends Pharmacol Sci 36: 215–225. [DOI] [PubMed] [Google Scholar]
- Lebon G, Warne T, Tate CG (2012). Agonist‐bound structures of G protein‐coupled receptors. Curr Opin Struct Biol 22: 482–490. [DOI] [PubMed] [Google Scholar]
- Leo CH, Jelinic M, Ng HH, Tare M, Parry LJ (2016). Serelaxin: a novel therapeutic for vascular diseases. Trends Pharmacol Sci 37: 498–507. [DOI] [PubMed] [Google Scholar]
- Marshall SA, Senadheera SN, Parry LJ, Girling JE (2016). The role of relaxin in normal and abnormal uterine function during the menstrual cycle and early pregnancy. Reprod Sci. doi:10.1177/1933719116657189. [DOI] [PubMed] [Google Scholar]
- Nair VB, Bathgate RA, Separovic F, Samuel CS, Hossain MA, Wade JD (2015). Synthetic covalently linked dimeric form of H2 relaxin retains native RXFP1 activity and has improved in vitro serum stability. Biomed Res Int 2015: 731852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neschadim A, Pritzker LB, Pritzker KP, Branch DR, Summerlee AJ, Trachtenberg J et al. (2014). Relaxin receptor antagonist AT‐001 synergizes with docetaxel in androgen‐independent prostate xenografts. Endocr Relat Cancer 21: 459–471. [DOI] [PubMed] [Google Scholar]
- Overbeek PA, Gorlov IP, Sutherland RW, Houston JB, Harrison WR, Boettger‐Tong HL et al. (2001). A transgenic insertion causing cryptorchidism in mice. Genesis 30: 26–35. [DOI] [PubMed] [Google Scholar]
- Pini A, Shemesh R, Samuel CS, Bathgate RA, Zauberman A, Hermesh C et al. (2010). Prevention of bleomycin‐induced pulmonary fibrosis by a novel antifibrotic peptide with relaxin‐like activity. J Pharmacol Exp Ther 335: 589–599. [DOI] [PubMed] [Google Scholar]
- Samuel CS, Royce SG, Hewitson TD, Denton KM, Cooney TE, Bennett RG (2017). Anti‐fibrotic actions of relaxin. Br J Pharmacol 174: 962–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Takahashi W, Hirayama A, Ajioka M, Takahashi N, Okishige K et al. (2015). Multicenter, Randomized, double‐blinded, placebo‐controlled phase II study of serelaxin in japanese patients with acute heart failure. Circ J 79: 1237–1247. [DOI] [PubMed] [Google Scholar]
- Scott DJ, Rosengren KJ, Bathgate RA (2012). The different ligand‐binding modes of relaxin family peptide receptors RXFP1 and RXFP2. Mol Endocrinol 26: 1896–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibold JR, Korn JH, Simms R, Clements PJ, Moreland LW, Mayes MD et al. (2000). Recombinant human relaxin in the treatment of scleroderma. A randomized, double‐blind, placebo‐controlled trial. Ann Intern Med 132: 871–879. [DOI] [PubMed] [Google Scholar]
- Sethi A, Bruell S, Patil N, Hossain MA, Scott DJ, Petrie EJ et al. (2016). The complex binding mode of the peptide hormone H2 relaxin to its receptor RXFP1. Nat Commun 7: 11344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemesh R, Hermesh C, Toporik A, Levine Z, Novik A, Wool A et al. (2009). Activation of relaxin‐related receptors by short, linear peptides derived from a collagen‐containing precursor. Ann N Y Acad Sci 1160: 78–86. [DOI] [PubMed] [Google Scholar]
- Sherwood OD (2004). Relaxin's physiological roles and other diverse actions. Endocr Rev 25: 205–234. [DOI] [PubMed] [Google Scholar]
- Silvertown JD, Symes JC, Neschadim A, Nonaka T, Kao JC, Summerlee AJ et al. (2007). Analog of H2 relaxin exhibits antagonistic properties and impairs prostate tumor growth. FASEB J 21: 754–765. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teerlink JR, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH et al. (2013). Serelaxin, recombinant human relaxin‐2, for treatment of acute heart failure (RELAX‐AHF): a randomised, placebo‐controlled trial. Lancet 381: 29–39. [DOI] [PubMed] [Google Scholar]
- Unemori EN, Erikson ME, Rocco SE, Sutherland KM, Parsell DA, Mak J et al. (1999). Relaxin stimulates expression of vascular endothelial growth factor in normal human endometrial cells in vitro and is associated with menometrorrhagia in women. Hum Reprod 14: 800–806. [DOI] [PubMed] [Google Scholar]
- Unemori EN, Lewis M, Constant J, Arnold G, Grove BH, Normand J et al. (2000). Relaxin induces vascular endothelial growth factor expression and angiogenesis selectively at wound sites. Wound Repair Regen 8: 361–370. [DOI] [PubMed] [Google Scholar]
- Weiss G, Teichman S, Stewart D, Nader D, Wood S, Breining P et al. (2016). Recombinant human relaxin versus placebo for cervical ripening: a double‐blind randomised trial in pregnant women scheduled for induction of labour. BMC Pregnancy Childbirth 16: 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss G, Teichman S, Stewart D, Nader D, Wood S, Unemori E (2009). A randomized, double‐blind, placebo‐controlled trial of relaxin for cervical ripening in post‐delivery date pregnancies. Ann N Y Acad Sci 1160: 385–386. [DOI] [PubMed] [Google Scholar]
- Xiao J, Huang Z, Chen CZ, Agoulnik IU, Southall N, Hu X et al. (2013). Identification and optimization of small‐molecule agonists of the human relaxin hormone receptor RXFP1. Nat Commun 4: 1953. [DOI] [PMC free article] [PubMed] [Google Scholar]