

Abstract
The insulin‐like peptide relaxin, originally identified as a hormone of pregnancy, is now known to exert a range of pleiotropic effects including vasodilatory, anti‐fibrotic, angiogenic, anti‐apoptotic and anti‐inflammatory effects in both males and females. Relaxin produces these effects by binding to a cognate receptor RXFP1 and activating a variety of signalling pathways including cAMP, cGMP and MAPKs as well as by altering gene expression of TGF‐β, MMPs, angiogenic growth factors and endothelin receptors. The peptide has been shown to be effective in halting or reversing many of the adverse effects including fibrosis in animal models of cardiovascular disease including ischaemia/reperfusion injury, myocardial infarction, hypertensive heart disease and cardiomyopathy. Relaxin given to humans is safe and produces favourable haemodynamic changes. Serelaxin, the recombinant form of relaxin, is now in extended phase III clinical trials for the treatment of acute heart failure. Previous clinical studies indicated that a 48 h infusion of relaxin improved 180 day mortality, yet the mechanism underlying this effect is not clear. This article provides an overview of the cellular mechanism of effects of relaxin and summarizes its beneficial actions in animal models and in the clinic. We also hypothesize potential mechanisms for the clinical efficacy of relaxin, identify current knowledge gaps and suggest new ways in which relaxin could be useful therapeutically.
Linked Articles
This article is part of a themed section on Recent Progress in the Understanding of Relaxin Family Peptides and their Receptors. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.10/issuetoc
Abbreviations
- ADHF
acute decompensated heart failure
- AHF
acute heart failure
- Ang II
angiotensin II
- AC
arterial compliance
- b‐FGF
basic FGF
- BMDECs
bone marrow‐derived endothelial cells
- CI
cardiac index
- CO
cardiac output
- ET‐1
endothelin‐1
- ECM
extracellular matrix
- iCa2+
intracellular calcium
- IR
ischaemia–reperfusion
- mRen2
murine renin gene
- MI
myocardial infarction
- PGF
placental growth factor
- PVR
pulmonary vascular resistance
- SHR
spontaneously hypertensive rat
- SVR
systemic vascular resistance
- TIMP
tissue inhibitors of metalloproteinase
- TGF‐β1
transforming growth factor β1
- VSMC
vascular smooth muscle cells
- VAS
visual analogue scale
Tables of Links
| TARGETS | |
|---|---|
| Enzymes a | PI3K |
| ACE | PKA |
| Adenylyl cyclase | PKC‐ζ |
| Akt | GPCRs b |
| Connexins | AT2 receptor |
| eNOS | ETB receptors |
| ERK1/2 | RXFP1 receptors |
| Guanylyl cyclase | Nuclear hormone receptors c |
| iNOS | Glucocorticoid receptors |
| nNOS |
| LIGANDS | |
|---|---|
| Ang II, angiotensin II | NO |
| cAMP | PD123319 |
| CCL2 (MCP‐1) | Relaxin |
| cGMP | SU5416 (semaxanib) |
| Enalapril | TGF‐β1 |
| ET‐1, endothelin‐1 | TIMP‐1 |
| Indomethacin | TNF‐α |
| Isoprenaline |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a, 2015b, 2015c).
Introduction
Relaxin is a heterodimeric peptide hormone closely related structurally to insulin. The major circulating form of relaxin is that produced by the RLN2 gene and is the cognate ligand for the RXFP1 receptor, a classical GPCR containing seven transmembrane spanning regions as well as a large extracellular domain with 10 leucine‐rich repeats and a unique N‐terminal LDL receptor type A module (Hsu et al., 2002). Relaxin is closely related to insulin‐like peptide 3, relaxin‐3 and insulin‐like peptide 5, which are the cognate ligands for the relaxin family peptide receptors RXFP2, RXFP3 and RXFP4 respectively (Alexander et al., 2015b). RXFP1 mRNA and protein are found in mammalian reproductive tissues but also in the heart, arteries, kidney, lung, liver and blood cells as well as in a number of areas of the brain (Dschietzig et al., 2001b; Bathgate et al., 2013; Halls et al., 2015). It is now recognized that relaxin acting through RXFP1 receptors has pleiotropic effects including vasodilation, anti‐fibrotic, angiogenic, anti‐apoptotic and anti‐inflammatory effects (Figure 1).
Figure 1.
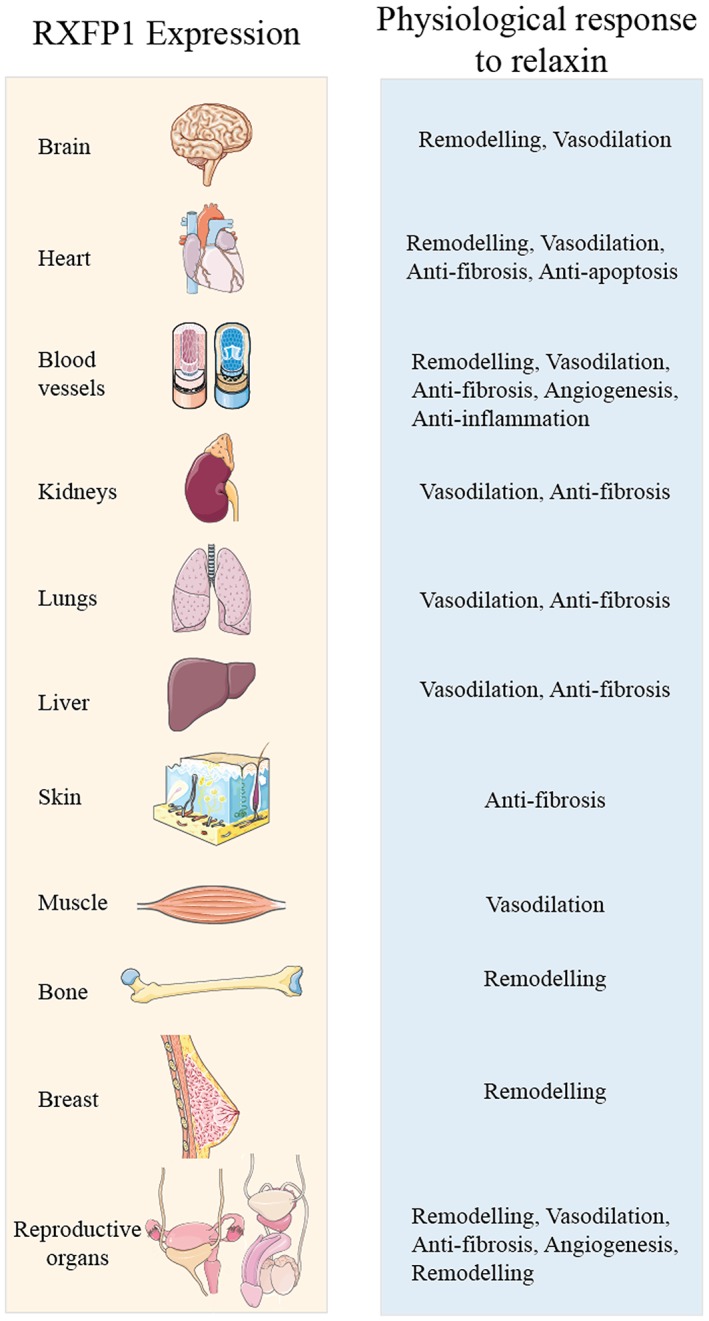
The tissue distribution of RXFP1 receptors and the physiology of relaxin in humans. Relaxin has vasodilatory, anti‐fibrotic and remodelling effects in various reproductive and non‐reproductive organs.
The pleiotropic effects of relaxin
Relaxin, originally identified as a hormone of pregnancy, mediates many of the cardiovascular and renal adaptations of pregnancy (Conrad and Shroff, 2011). Relaxin‐neutralizing antibodies or ovariectomy prevents the rise in arterial compliance (AC) and cardiac output (CO) and the fall in systemic vascular resistance (SVR) seen in pregnant rats (Debrah et al., 2006) and prevents the renal vasodilation normally observed during pregnancy (Novak et al., 2001). These effects are also observed in non‐pregnant animals (Danielson et al., 1999; Conrad and Novak, 2004; Debrah et al., 2005a).
Other chronic effects of relaxin include important and consistent inhibitory effects on stimulated extracellular matrix (ECM) deposition. It inhibits proliferation and differentiation of fibroblasts into myofibroblasts (activated fibroblasts that promote ECM/collagen deposition and fibrosis) in rat cardiac (Samuel et al., 2004a), renal (Masterson et al., 2004), hepatic (Williams et al., 2001) and human pulmonary tissues (Unemori et al., 1996; Huang et al., 2001); it also activates various ECM/collagen‐degrading MMPs and inhibits tissue inhibitors of metalloproteinase (TIMP) activity (Samuel et al., 2007; Bennett, 2009). Additionally, the peptide represents an important driver of VEGF‐related angiogenesis and wound healing (Unemori et al., 1992; Unemori et al., 2000).
Relaxin has anti‐apoptotic and anti‐hypertrophic effects in human trophoblasts (Lodhi et al., 2013), human osteosarcomas (Ma et al., 2013a), epithelial and stromal cells (Lee et al., 2005) and rodent cardiomyocytes (Moore et al., 2007; Samuel et al., 2011). These anti‐apoptotic effects of relaxin are dependent on RXFP1 receptors as the peptide has no apoptotic effects in RXFP1−/− mice (Yao et al., 2009).
Relaxin has anti‐inflammatory effects in humans and animals. It inhibits vascular inflammation by inhibiting TNF‐α‐induced expression of vascular cell adhesion molecule, platelet endothelial cell adhesion molecule and the chemokine CCL2 (Brecht et al., 2011). It furthermore inhibits markers of inflammation in an NO‐dependent manner by attenuating activation of neutrophils (Bani et al., 2002; Masini et al., 2004) and basophils (Bani et al., 2002) and by inhibiting histamine release from mast cells (Masini et al., 1995). Relaxin also inhibits the aggregation of isolated human and rabbit platelets (Bani et al., 1995a) and decreases the number of circulating platelets in rats.
Cellular basis for the actions of relaxin
Signal transduction pathways activated by relaxin
cAMP
cAMP is involved in many physiological responses including inotropy and chronotropy in the heart and vasodilation in the vasculature (Houslay et al., 2007). Acute administration of relaxin increases cAMP levels in HEK‐293, THP‐1 and MCF‐7 cells; rat renal and cardiac fibroblasts; rat heart; human endothelial and smooth muscle cells; and fibroblasts from human arteries and veins (Kompa et al., 2002; Nguyen and Dessauer, 2005a; Halls et al., 2009; Horton et al., 2011). The characteristics of relaxin‐mediated cAMP accumulation are dependent on the cellular background. In HEK‐RXFP1 cells, there is increased cAMP accumulation associated with coupling to Gαs modulated by GαOB and a delayed increase in cAMP mediated through Gαi3 (Halls et al., 2006), βγ subunits, PI3K and translocation of PKC‐ζ to the cell membrane to activate AC (Nguyen and Dessauer, 2005b; Halls et al., 2006). The Gαi3–Gβγ–PI3K–PKC‐ζ pathway also operates in THP‐1 cells, in human endothelial and smooth muscle cells and in rat and human cardiac fibroblasts but is missing in rat renal fibroblasts and Colo16 cells, where Pertussis toxin pretreatment enhances relaxin‐mediated cAMP accumulation (Halls et al., 2009).
NO and cGMP
Relaxin utilizes NO to produce vasodilation in vitro (Bani‐Sacchi et al., 1995; Bani et al., 1998a; McGuane et al., 2011b) and in vivo (Jeyabalan et al., 2003; Conrad and Shroff, 2011). In vitro, relaxin rapidly activates the NO/cGMP pathway in vascular smooth muscle, coronary artery endothelium, bovine aortic smooth muscle, human breast cancer cells, perfused guinea pig and rat hearts and mouse uterus (Bani‐Sacchi et al., 1995; Bani et al., 1995b; Bani et al., 1998a, 1998b; Failli et al., 2002; Masini et al., 2002) as well as rat cardiac (Samuel et al., 2004a) and renal (Mookerjee et al., 2009; Chow et al., 2014) fibroblasts. This is achieved by increasing the activity and/or expression of NOS. Relaxin interacts with all three NOS subtypes: endothelial NOS (eNOS) (McGuane et al., 2011b), inducible NOS (iNOS) (Failli et al., 2002; Quattrone et al., 2004) and neuronal NOS (nNOS) (Chow et al., 2012; Mookerjee et al., 2009), to increase NO production, although the interaction is not well understood and is highly species‐ and tissue‐dependent. Vasodilatory responses and phosphorylation of eNOS in human coronary artery endothelial cells to acutely administered relaxin are attenuated by inhibition of Gαi/o coupling to PI3K, PKB/Akt, indicating that NO/cGMP production is dependent on Gαi/o, PI3K, Akt and NOS (Figure 2) (McGuane et al., 2011b). However, in rat renal fibroblasts, chronic relaxin over 72 h promotes nNOS‐derived NO/cGMP signalling through Gαs and GαOB (Mookerjee et al., 2009; Chow et al., 2014). In vivo, the NOS/NO/cGMP pathway is critical for relaxin‐induced vasodilation and cardioprotection. Relaxin protects rat lungs after ischaemia–reperfusion (IR) injury through iNOS induction (Alexiou et al., 2013), reduces infarct size in rat brains by eNOS activation (Wilson et al., 2006) and reduces cardiac infarct size in the pig through a NOS‐dependent mechanism (Masini et al., 1997) but also reduces fibrosis when given chronically in a bleomycin‐induced lung fibrosis mouse model through an NO/cGMP‐dependent pathway (Huang et al., 2011).
Figure 2.
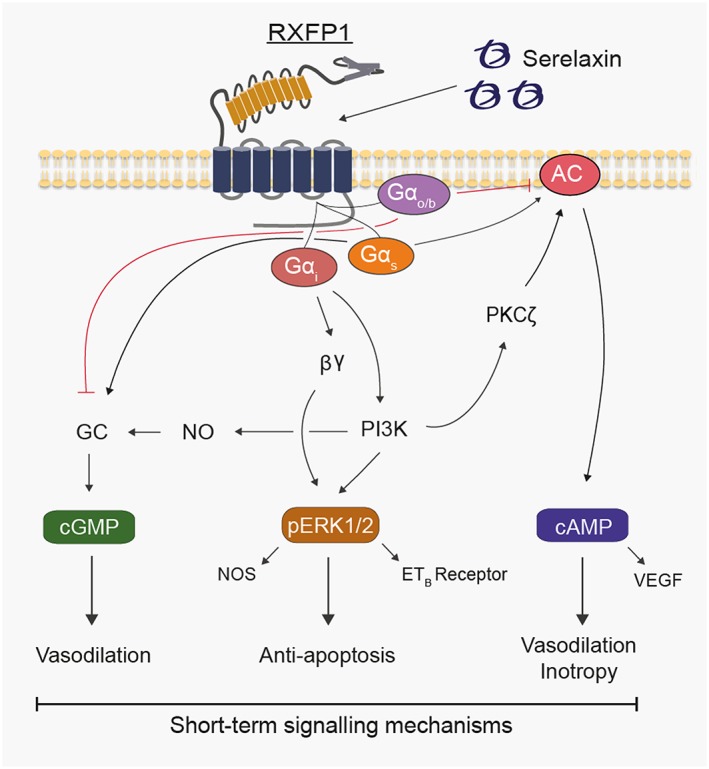
Mechanisms involved in short‐term (minutes) relaxin‐mediated signal transduction in human primary vascular cells.
MAPKs
The MAPK ERK1/2 inhibits apoptosis and promotes cell survival and is likely to play key roles in cardiomyocyte survival in heart failure (HF) (Bueno and Molkentin, 2002). Relaxin stimulation causes a transient increase in ERK1/2 phosphorylation (pERK1/2) in HEK‐RXFP1 cells, THP‐1 cells (Nguyen and Dessauer, 2005a), HeLa cells (Dschietzig et al., 2003), human endometrial stromal cells (Zhang et al., 2002), rat renal fibroblasts (Chow et al., 2012), human smooth muscle cells (Zhang et al., 2002) and fibrocartilaginous cells (Ahmad et al., 2012). A more prolonged pERK1/2 response occurs in HUVECs and immortalised HUVECs (Eahy926 cells) (Dschietzig et al., 2003; Dschietzig et al., 2009a). Relaxin rapidly increases pERK1/2 in human vascular cells in a Gαi‐ and PI3K‐dependent manner (Sarwar et al., 2015; Figure 2), and this likely mediates the anti‐apoptotic and cardiac cell survival effects of relaxin in humans. Other MAPKs such as p38MAPK are also activated, but this response is dependent on the cellular background. Thus, relaxin increases p38MAPK in human vascular smooth muscle cells (VSMC) but not in HUVEC or HeLa cells (Dschietzig et al., 2003), whereas in human renal fibroblasts, relaxin does not affect pERK1/2, p38MAPK or pJNK (Heeg et al., 2005).
Calcium
Relaxin has no direct effect on basal intracellular calcium (iCa2+) levels but inhibits agonist (angiotensin II (Ang II) and thrombin)‐mediated iCa2+ in rat VSMC and endothelial cells, rat coronary artery endothelial cells, bovine aortic VSMC and guinea pig hearts (Bani et al., 1998b; Failli et al., 2002; Masini et al., 2002), perhaps reflecting that RXFP1 receptors are not Gαq coupled. Although Gαi coupling increases iCa2+ (Kiselyov et al., 2003), this does not occur with the relaxin/RXFP1receptor system, as relaxin does not affect iCa2+ signalling. Relaxin most likely acts as a physiological antagonist by producing vasodilator effects that counteract the vasoconstrictor actions of Ang II and endothelin‐1 (ET‐1) that cause iCa2+ release via their respective receptors.
Chronic changes produced by relaxin
Matrix metalloproteinases
The anti‐fibrotic effects observed with chronically administered relaxin include inhibition of transforming growth factor β1 (TGF‐β1), promotion of the expression and/or activity of ECM/collagen‐degrading MMPs and inhibition of TIMP activity (Samuel et al., 2007). In vitro relaxin increases MMP‐2 activity in human renal (Heeg et al., 2005) and rat cardiac (Samuel et al., 2004a; Heeg et al., 2005) and renal (Chow et al., 2014) fibroblasts, whereas it increases MMP‐9 activity in THP‐1 cells, rat renal fibroblasts and temporomandibular joint (TMJ) cells (Heeg et al., 2005; Ho et al., 2007; Ahmad et al., 2012; Chow et al., 2014). Relaxin‐induced activation of MMP‐2, MMP‐9 and MMP‐13 in human and rat renal fibroblasts is associated with iNOS‐induced NO production (Chow et al., 2012) and inhibition of Smad‐2 phosphorylation and thus TGF‐β1 activation (Heeg et al., 2005; Mookerjee et al., 2009). Relaxin also interacts with other MMPs in a tissue‐dependent manner because in TMJ fibrocartilaginous cells, relaxin increases MMP‐1, MMP‐9 and MMP‐13 activity but only increases MMP‐13 activity in rat ventricular fibroblasts (Kapila et al., 2009; Hossain et al., 2011; Ahmad et al., 2012). In human dermal fibroblasts and rat renal myofibroblasts, relaxin increased MMP‐2 and MMP‐9 by a mechanism involving RXFP1 receptors, pERK1/2 and NO/cGMP (Figures 2 and 3) (Chow et al., 2012). Similarly, in fibrocartilaginous cells, relaxin increased MMP‐9 and MMP‐13 by stimulating RXFP1 receptors and activating PI3K, Akt, ERK and PKC‐ζ and the transcription factors Elk‐1 and c‐fos (Ahmad et al., 2012). NF‐kB also has a role in relaxin‐mediated MMP‐9 activation in human THP‐1 and fibrocartilaginous cells (Ho et al., 2007; Ahmad et al., 2012).
Figure 3.
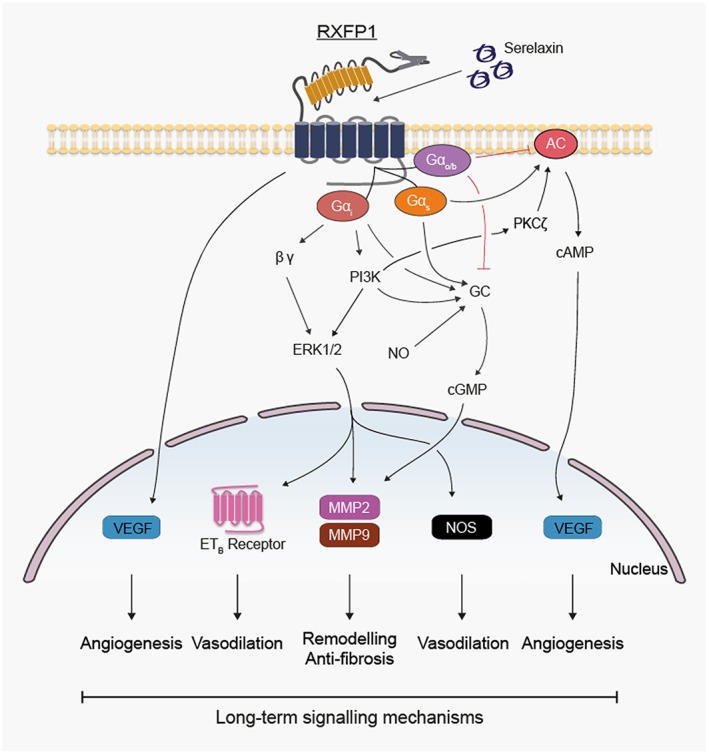
Effects of longer‐term (hours to days) relaxin administration on expression of genes likely to influence signal transduction in human primary vascular cells.
Angiogenic growth factors
Involvement of growth factors and cytokines in the angiogenic effects of chronic relaxin is conflicting and likely to be tissue and cell specific. In THP‐1 cells, relaxin increases the expression of VEGF (VEGF165 and VEGF121) and basic FGF (b‐FGF) and causes angiogenesis at ischaemic wound sites in rodents (Unemori et al., 2000). Relaxin stimulates VEGF production in human endometrial cells (Unemori et al., 1999), Saos‐2 cells (Ma et al., 2013b), THP‐1 cells (Unemori et al., 2000) and human endothelial cells (Unemori et al., 1992). The mechanism involves cAMP in normal human endometrial cells where relaxin induces VEGF expression in a cAMP‐dependent manner without affecting b‐FGF, TGF‐β1 and placental growth factor (PGF) (Figures 2 and 3) (Unemori et al., 1999). Although VEGF receptor inhibitors prevent relaxin‐induced increases in GFR and decreases in renal vascular resistance without affecting mean arterial pressure (McGuane et al., 2011a), in rat small renal arteries, SU5416 increases relaxin‐mediated vasodilation (McGuane et al., 2011b). These apparently contradictory effects may relate to actions of SU5416 other than VEGF receptor blockade that include inhibition of the tyrosine kinases FLT‐3 and RET and other protein kinases such as c‐Kit and Met (Mologni et al., 2006). However, VEGF and PGF neutralization prevents relaxin‐mediated inhibition of myogenic constriction in mouse and rat small renal and human subcutaneous arteries (McGuane et al., 2011a), suggesting that VEGF and PGF may have a role in relaxin‐mediated vasodilation.
In mice, relaxin increases circulating levels of bone marrow‐derived endothelial cells (BMDECs) that facilitate angiogenesis and contribute to repair of vascular endothelium. BMDECs are recruited to the site of angiogenesis where relaxin promotes vasculogenesis by RXFP1 receptor activation, leading to NO generation and PI3K and Akt signalling (Segal et al., 2012; Ma et al., 2013b). This may explain the healing properties of relaxin because chronic administration in diabetic mice increases VEGF levels in the wound area and promotes skin recovery, an effect reversed by administration of VEGF antibodies (Bitto et al., 2013).
Endothelin receptors
Endothelin receptors are secondary mediators in the vasodilatory responses to relaxin. In HeLa cells, HUVECs and bovine aortic endothelial cells, but not in human VSMC, relaxin increases mRNA and protein of the ETB receptor by a Ras‐independent Raf‐1–MEK‐1–ERK1/2 kinase cascade that activates NF‐kB (Figures 2 and 3), and the vasoconstrictor responses to ET‐1 and ET‐3 in rat aorta strips are inhibited by relaxin treatment in an ERK1/2‐ and ETB‐dependent manner (Dschietzig et al., 2003). The role of ETB receptors in vasodilatory responses to relaxin is strongly affected by cell and tissue background. In small renal arteries from virgin, midterm pregnant and non‐pregnant rats, relaxin has no effect on ETB receptor levels (Kerchner et al., 2005). In lung IR injury, relaxin is protective, but the effect does not involve ETB receptors (Alexiou et al., 2013). However, in human coronary artery endothelial cells and HUVECs but not in human aortic endothelial cells or human dermal microvascular endothelial cells, relaxin produced small and inconsistent increases expression of ETB receptors (Kerchner et al., 2005). This apparent discrepancy was explained by the use of small rather than large arteries, no pulmonary endothelial cells and the significantly lower relaxin concentrations used in the latter study. Furthermore, small renal arteries from relaxin‐treated non‐pregnant rats showed reduced myogenic reactivity, an effect not observed in mice lacking ETB receptors (Jeyabalan et al., 2003). More recent studies with 24 and 48 h exposure to relaxin (1.68 nM) show modest but consistent increases in ETB receptors in HUVEC, human umbilical arterial smooth muscle cells and human cardiac fibroblasts and marked increases in human umbilical venous smooth muscle cells (Sarwar et al., 2015).
Actions of relaxin in the cardiovascular system
Relaxin targets vascular cells
There is evidence from preclinical and clinical studies that relaxin targets the vasculature. In rats, chronic relaxin increases CO, global AC, renal plasma flow and GFR and reduces SVR and myogenic constriction without affecting mean arterial pressure (Danielson et al., 1999; Conrad et al., 2004; Debrah et al., 2005a). Given acutely, relaxin specifically vasodilates rat mesenteric arteries, rodent small renal and mesenteric arteries (McGuane et al., 2011b) and human small gluteal and subcutaneous arteries but not pulmonary resistance arteries (McGuane et al., 2011b). Relatively little is known about the effects of relaxin on veins despite relaxant effects on mesenteric veins without affecting myogenic reactivity (Li et al., 2005) and powerful effects on signalling in human umbilical venous smooth muscle cells (Sarwar et al., 2015). The regionally specific effects of relaxin can be explained by differential RXFP1 receptor expression in different vascular beds. Thus, in rats, RXFP1 receptors are highly expressed in endothelial cells of the aorta, vena cava, mesenteric artery and vein as well as in VSMC of the femoral artery and vein and small pulmonary arteries (Jelinic et al., 2014).
Signal transduction pathways activated by relaxin in vascular cells
Relaxin given acutely causes cAMP, cGMP and pERK1/2 signalling in endothelial cells, VSMC and fibroblasts from human arteries and veins (Sarwar et al., 2015), mediated by Gαs, Gαi/o and GαOB G‐proteins and involving PI3K downstream of G‐protein activation (Figure 2). These pathways likely mediate the rapid dilation observed in human subcutaneous arteries and rodent renal and mesenteric arteries where acute relaxin rapidly activates Gαi, PI3K and NOS signalling (McGuane et al., 2011b). Longer‐term relaxin also changes gene expression in endothelial cells, VSMC and fibroblasts from human arteries and veins with increased gelatinases (MMP‐2 and MMP‐9), ETB receptors, VEGF and NOS in most but not all cell types (Sarwar et al., 2015) (Figure 3). These changes may account for the haemodynamic changes observed during pregnancy and in vivo, particularly the increase in AC and decrease in SVR (thereby improving CO) observed in pregnant (Debrah et al., 2006) and non‐pregnant animals (Conrad et al., 2004). More importantly, these changes are likely to account for the widespread haemodynamic changes observed in patients with acute HF (AHF) treated with serelaxin (Ponikowski et al., 2014).
Relaxin promotes vascular cell crosstalk
The endothelium secretes vasoactive substances including NO, PGs and endothelium‐derived hyperpolarizing factor that regulate smooth muscle tone. Responses to relaxin were abolished in human gluteal arteries denuded of endothelium (Fisher, 2009), and in an in vitro co‐culture model, the relaxin responses in VSMC were endothelium dependent and governed by expression of RXFP1 receptors in endothelial cells (Figure 4) (Sarwar et al., 2016).
Figure 4.
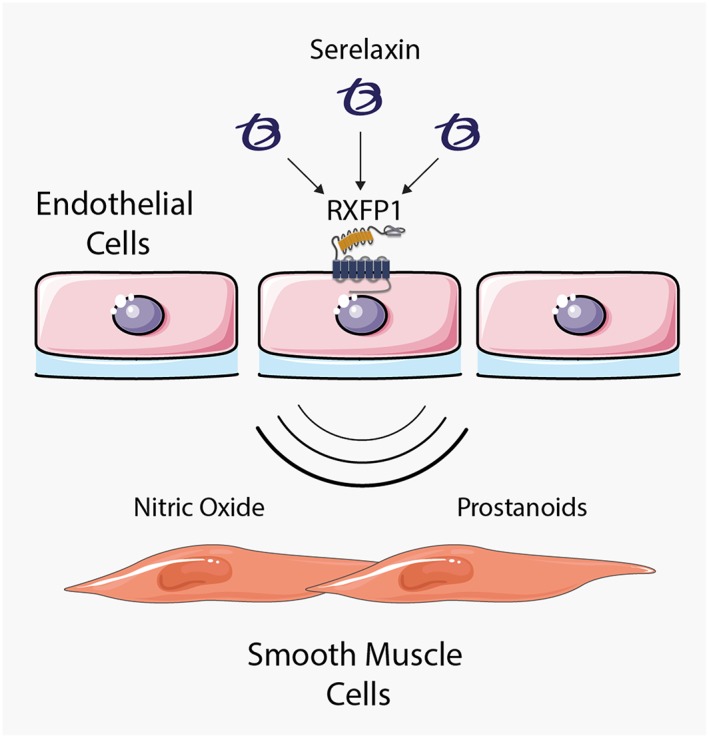
Relaxin only activates endothelial cells that express cell surface RXFP1 receptors to promote cellular crosstalk between endothelial cells and smooth muscle cells involving NO and, in some endothelial cells, prostanoids.
NO synthesis regulates relaxin‐mediated crosstalk between endothelial cells and VSMC in the human vasculature (Figure 4). This is observed in vitro in human arterial/venous endothelial cells and VSMC (Sarwar et al., 2016), in vivo in conscious rats where NO inhibition abolished relaxin‐mediated increases in renal vasodilation (Danielson et al., 1999) and in rat and guinea pig hearts where NO inhibition abolished relaxin‐mediated increases in coronary flow (Bani‐Sacchi et al., 1995).
Prostanoids are also involved in specific circumstances. In human coronary artery endothelial cells but not HUVEC, relaxin promotes COX activity to cause an increase in cAMP accumulation in co‐cultured smooth muscle cells (Sarwar et al., 2016). In ex vivo human gluteal arteries, indomethacin (a non‐specific COX inhibitor) almost abolished relaxin‐mediated vasodilation (Fisher, 2009), suggesting that, in some circumstances, there is a role for prostanoids in relaxin signal transduction.
Actions of relaxin on the heart
The mammalian heart contains relaxin binding sites that are exclusively found in the atrium (Osheroff et al., 1992; Tan et al., 1999). Acutely administered relaxin has positive chronotropic and inotropic effects in the rat isolated perfused heart and isolated atria, with a potency greater than ET‐1, Ang II or isoprenaline (Kakouris et al., 1992; Coulson et al., 1996; Toth et al., 1996).
Relaxin inhibits injury‐induced hypertrophy and apoptosis in neonatal rat cardiomyocytes and increases the growth and maturation of neonatal mouse cardiomyocytes. RXFP1 receptors are expressed in cardiac fibroblasts from several species including rodents and humans where they cause short‐term and long‐term changes in signal transduction (Sarwar et al., 2016) (Figures 2 and 3). Relaxin also acts on the pacemaker cells of the rabbit sino‐atrial node by modulating L‐type Ca2 + current to increase heart rate. The effect of relaxin was not observed when administered following the β‐adrenoceptor agonist isoprenaline or the cell permeable cAMP analogue 8‐Br‐cAMP that produced similar effects. The responses to relaxin, isoprenaline and 8‐Br‐cAMP were blocked by the PKA inhibitor PKI, suggesting that they were all mediated by a cAMP/PKA‐dependent pathway (Han et al., 1994).
Effects of relaxin in human tissues
Human blood vessels
In general, studies on human vessels are in concordance with mechanisms elucidated in rodents (McGuane et al., 2011a, 2011b). Relaxin has both acute and longer‐term vasodilatory properties. Acutely (within minutes), relaxin dilates arteries by activating eNOS (Bani et al., 1998a; McGuane et al., 2011b; Dschietzig et al., 2012), PI3K, Akt and PKA with subsequent eNOS phosphorylation at Ser1195, Ser613 and Thr495 and NO generation (McGuane et al., 2011b; Dschietzig et al., 2012). Over a longer period (h), the ETB receptor mediates NO‐dependent vasodilation (Fernandez‐Patron et al., 1999; Dschietzig et al., 2003). In mesenteric and renal resistance vessels of ~100–300 μm diameter, relaxin activates MMP‐9 and later MMP‐2 to generate the ET‐1 peptide, ET1–32 from its precursor, big ET‐1 (Jeyabalan et al., 2003). ET1–32, though a more potent vasoconstrictor than ET1–21 in endothelium‐denuded rat mesenteric arteries, preferentially interacts with the endothelial ETB receptors due to its site of generation in intact vessels (Fernandez‐Patron et al., 2000) and precipitates the release of NO (Fernandez‐Patron et al., 1999; McGuane et al., 2011b). VEGF and PGF are also involved in sustained vasodilation, but the precise mechanism involved is still obscure; the VEGF and PGF effects appear to be located downstream of MMPs and initiated by MMP‐induced release of matrix‐bound angiogenic factors (McGuane et al., 2011b).
An alternative mechanism in larger vessels and the pulmonary circulation is relaxin‐induced up‐regulation of endothelial ETB receptors (see above), which expression of RXFP1 receptors mediates NO production and, equally importantly, ET‐1 clearance (Dschietzig et al., 2001a, 2001b; Dschietzig et al., 2003). Finally, relaxin promotes angiogenesis both in humans and in rodents (Unemori et al., 1999; Unemori et al., 2000) by inducing the synthesis of VEGF and b‐FGF. This may serve to improve tissue and organ perfusion in the long term.
Human heart
In contrast to the evidence in rodents, little information is available regarding human myocardial tissue. Immunohistochemical studies (Dschietzig et al., 2001b) of left ventricle from patients with end‐stage HF indicate relaxin expression in interstitial cells and cardiomyocytes. The expression of RXFP1 receptors has been described in human cardiac fibroblasts (Sarwar et al., 2015) and cardiomyocytes. In human atrial and ventricular samples obtained from donor and terminally failing explanted hearts (Dschietzig et al., 2011), relaxin addition evoked a potent inotropic effect in atrial (EC50 below 1 nM), but not ventricular, myocardium from both normal and failing hearts. This effect critically involves PKA and inhibition of the rapidly inactivating potassium outward current, I to. Additionally, the contribution of Gαi3–PI3K signalling clearly increases in failing human atria, in accord with Gαi/o up‐regulation in chronic HF (CHF) (Eschenhagen et al., 1992). RXFP1 receptors were expressed in atrial, but not ventricular, myocardium from both control and failing hearts.
Many questions remain regarding the role of the relaxin system in human myocardium including whether there is expression of RXFP1 receptors in adult ventricular cardiomyocytes and which of the canonical and non‐canonical signalling pathways, apart from Gαi3–PI3K, are active and how they are regulated.
Haemodynamic effects of relaxin in the cardiovascular system
Haemodynamic effects in vivo: vascular actions
Relaxin appears to have a facilitatory role in cardiovascular adaptation during pregnancy but is not essential at least in the mouse since a majority of relaxin‐1 knockout (KO) mice produce litters (Zhao et al., 1999). Reduction in SVR in pregnant women is partially attributable to the vasodilatory action of elevated circulatory levels of relaxin (Sherwood, 2004). In rodents, acute or chronic relaxin induces vasodilatation, reduced BP, a decline in arterial stiffness, reduction in SVR and an increase in CO and renal blood flow (Conrad and Novak, 2004). Haemodynamic changes seen during pregnancy and with relaxin administration are similar. There is a sustained reduction of vascular resistance following a bolus injection of relaxin, involving bradykinin‐ and prostacyclin‐mediated vasodilatation (Leo et al., 2014b). Relaxin (up to 6 h) reduced SVR and increased CO in hypertensive rats chronically infused with Ang II, while in an spontaneously hypertensive rat (SHR) model, similar haemodynamic effects were evident only after relaxin therapy for several days (Debrah et al., 2005b). Whereas increased CO was not reported by other studies, chronic relaxin therapy for 2 weeks reduced large artery stiffness by lowering vascular collagen content in aged SHR (Xu et al., 2010). Collectively, these preclinical findings indicate that several mechanisms are responsible for the reduction in vascular resistance following acute and chronic relaxin therapy (Du et al., 2014).
Haemodynamic effects in vivo: cardiac actions
In humans, unlike rodents, administration of relaxin even at high doses did not increase heart rate (Ponikowski et al., 2014), perhaps reflecting lower expression of RXFP1 receptors in human heart, relative to rodents. The inotropic action of relaxin involves both Gαs modulated by GαOB (Kompa et al., 2002), which also facilitates PKA signalling by activation of the Gαi3–PI3K pathway (Halls et al., 2015). In isolated myofilaments, relaxin activates PKC/Ca2 + signalling, leading to positive inotropy (Shaw et al., 2009). In failing and non‐failing human atria, relaxin increased contractile force, by signalling that is PKA‐dependent and negatively regulated by Gαi (Dschietzig et al., 2011). There is no evidence for a ventricular inotropic action of relaxin in rodents in vivo (Lekgabe et al., 2005; Xu et al., 2010; Samuel et al., 2011). Collectively, studies in atrial tissues from rodents have convincingly shown chronotropic and inotropic actions of relaxin. In humans, the cardiac index (CI) was unchanged by relaxin treatment although afterload was reduced (Ponikowski et al., 2014) and, apparently, relaxin does not produce inotropy in ventricular myocardium nor increase heart rate in human hearts.
Studies in models lacking RXFP1 receptors or relaxin (KO models)
The male but not the female relaxin KO mouse displays widespread organ fibrosis involving the reproductive system, nipples, skin, lungs, heart and kidney, with treatment with relaxin for up to 2 weeks able to reverse established fibrosis in many organs and tissues, including the heart (Zhao et al., 2000; Du et al., 2003; Samuel et al., 2003; Samuel et al., 2004a, 2004b). Relaxin influences vascular proliferation in the endometrium, is associated with increased regional blood flow during pregnancy and promotes uterine and placental growth. These adaptive changes are significantly attenuated in relaxin‐1 KO mice and reversed by relaxin treatment (Gooi et al., 2013). Male relaxin KO mice display reduced compliance of resistance arteries (renal, uterus, mesentery and femoral arteries) and an attenuated vasodilatory response, implying that endogenous relaxin is required to maintain normal vascular structure and function (Gooi et al., 2013; Leo et al., 2014a; Jelinic et al., 2015; Ng et al., 2015).
RXFP1 receptors comprise the major type of relaxin receptor in the cardiovascular system (Bathgate et al., 2013; Halls et al., 2015). In RXFP1 receptor KO mice, relaxin‐induced increases in NO production by endothelial cells were abolished (Segal et al., 2012). No reduction in tissue collagen content and remodelling of the reproductive tract was seen in conditional RXFP1 receptor KO mice, a phenotype similar to that of relaxin KO mice (Krajnc‐Franken et al., 2004; Kaftanovskaya et al., 2015).
Relaxin in animal models of cardiovascular disease
IR and myocardial infarction (MI)
Following cardiac IR, the injured myocardium displays sequential cell death, microvascular damage, innate immune response and subsequent reparative and/or reactive myocardial fibrosis which is limited by relaxin. In guinea pigs, rats and pigs, reduction in myocardial injury by relaxin administered prior to reperfusion was associated with attenuated Ca2 + overload, oxidative stress, histamine release or inflammatory cell infiltration (Masini et al., 1997; Bani et al., 1998b; Formigli et al., 2007). In the porcine IR model, treatment with relaxin or regional delivery of myoblasts transfected with the relaxin gene improved viability of engrafted myoblasts and improved healing (Formigli et al., 2005; Formigli et al., 2007). Interestingly, the onset of ventricular arrhythmias during acute IR was reduced in relaxin‐treated rat or porcine hearts, likely to be due to reduced myocardial injury (Bani et al., 1998b; Nistri et al., 2008). In vitro studies show that relaxin protects against cardiomyocyte apoptotic death induced by oxidative stress (Moore et al., 2007), hypoxia–reoxygenation (Boccalini et al., 2015) or high glucose exposure (Zhang et al., 2015) and involved activation of Akt together with increased Bcl‐2/Bax ratio, Notch‐1 activation or suppressed endoplasmic reticulum (ER) stress.
In myocardial ischaemia, microvascular damage associated with regional inflammation and regional leukocyte interaction with vascular endothelial cells represents a pivotal part of overall cardiac injury (Kloner, 2011). In cultured endothelial cells, exposure to cytokines, inflammatory signalling and leukocyte adhesion were attenuated by relaxin (Brecht et al., 2011). In the pig IR model (Formigli et al., 2007), the anti‐fibrotic effect was associated with a reduction in infarct size, while in the mouse MI model (without reperfusion) (Samuel et al., 2011), infarct size was unchanged, and yet fibrotic healing was attenuated by relaxin without adverse influence on the extent of post‐infarct ventricular remodelling and function.
Hypertensive heart disease
The effects of relaxin have been studied in rodent models of hypertension such as SHR, drug‐induced renal injury, transgenic expression of murine renin gene (mRen2) plus diabetes or hypertension induced by chronic infusion of Ang II. In SHR or rats with Ang II‐induced hypertension, relaxin therapy (1–7 days) reduced pulmonary vascular resistance (PVR) and vascular stiffness and increased renal blood flow and CO, leading to unchanged mean BP (Debrah et al., 2005b) with the response to relaxin being quicker in Ang II than in SHR models. In SHR rats with left ventricular hypertrophy, endogenous myocardial relaxin mRNA and protein levels were higher than in age‐matched Wistar control rats and correlated inversely with left atrial and left ventricular mass index (Dschietzig et al., 2005). While this was supported by in vitro findings showing inhibition of cardiomyocyte hypertrophy induced by a fibroblast‐conditioned medium (Moore et al., 2007), other in vivo studies on elderly SHR did not show changes in heart weight following chronic relaxin treatment for 2 weeks (Lekgabe et al., 2005; Xu et al., 2010).
Studies in 9‐month‐old SHRs provided strong evidence for anti‐fibrotic effects of relaxin, and treatment for 2 weeks (at 500 μg · kg−1 · day−1) effectively reduced cardiac and renal fibrosis (Lekgabe et al., 2005). Reduced atrial fibrosis following 2 week relaxin therapy (Parikh et al., 2013) and, in 17‐month‐old SHRs, improved large artery compliance, measured in vitro and in vivo, together with a significant reduction in arterial collagen content were observed (Xu et al., 2010).
In the salt‐sensitive Dahl rat, relaxin attenuated the rise in BP and increased renal nNOS and eNOS (Yoshida et al., 2011), and selective inhibition of the three NOS isoforms blocked the anti‐hypertensive effects of relaxin. Relaxin for 6 weeks significantly decreased systolic BP, glomerular and tubulointerstitial changes and TGF‐β signalling. Relaxin also ameliorated renal ischaemic reperfusion injury, improved renal function and reduced circulating and renal expression of TNF‐α (Yoshida et al., 2013). In rats with renal injury caused by bromoethylamine or reduced renal mass, relaxin for 4 weeks preserved renal function, improved renal creatinine clearance and ameliorated proteinuria, oxidative stress and renal fibrosis, which occur together with a reduction or absence of renal hypertension (Garber et al., 2001; Garber et al., 2003). Ang II infusion into rats caused hypertension, albuminuria, glomerular sclerosis and enhanced oxidative stress, which were improved by relaxin for 2 weeks. These effects were abolished by L‐NAME, indicating an NO‐dependent mechanism (Sasser et al., 2011).
Chronic relaxin was ineffective in rats with dual transgenic expression of mRen2 and angiotensinogen, early onset of hypertension, rapid development of organ damage (cardiomyocyte hypertrophy, loss of glomeruli, fibrosis, renal damage and severe diastolic dysfunction with preserved systolic function) and early premature death (over 70% by week 7) (Haase et al., 2014). This severe and rapidly developing model makes testing therapeutic efficacy difficult, and in addition, the plasma levels of relaxin in this model may have been suboptimal. Considering the reciprocal effects of cardiac–renal injury or dysfunction (Braam et al., 2014), further research is warranted to simultaneously investigate effects of relaxin on cardiac and renal injury in animal models of disease.
Cardiomyopathy
Preclinical studies in rodents have used cardiomyopathy induced by chronic treatment with the β‐adrenoceptor agonist isoprenaline or transgenic overexpression of human β2‐adrenoceptors (Bathgate et al., 2008; Hossain et al., 2011; Samuel et al., 2014). In the isoprenaline model in rats, relaxin administered with or after isoprenaline administration protected hearts against injury measured by levels of circulating biomarkers and decreased cardiac fibrosis (Zhang et al., 2005; Samuel et al., 2014). Comparison of the anti‐fibrotic efficacy of relaxin and the ACE inhibitor enalapril in mice with cardiac fibrosis induced by isoprenaline showed that co‐administered relaxin or enalapril for 17 days effectively reduced cardiac fibrosis with relaxin displaying better efficacy. Delayed treatment with relaxin was also effective.
In mRen2 hypertensive rats with streptozotocin‐induced diabetes, relaxin therapy for 2 weeks reduced cardiac fibrosis and suppressed fibroblast proliferation and TIMP‐1 activity (Samuel et al., 2008). In mice with type 2 diabetes by high‐fat diet feeding for 13 weeks, relaxin administration over the last 3 weeks reduced fasting glucose level, improved insulin sensitivity, restored endothelial‐dependent vasodilatation and reduced cardiac and hepatic fibrosis (Bonner et al., 2013).
Clinical studies with relaxin
CHF and AHF
The first dose‐finding trial in patients with stable systolic CHF (Dschietzig et al., 2009b) indicated that relaxin infusion (10–960 μg · kg−1 · day−1) for up to 24 h was safe and evoked favourable haemodynamic changes. Thus, it elevated CI without affecting heart rate; decreased pulmonary capillary wedge pressure, SVR and PVR; and did not significantly alter systolic BP/mean BP or right atrial pressure. Haemodynamic changes generally started within 45–60 min. Pharmacokinetics in stable patients with systolic CHF and impaired renal function (Voors et al., 2014) showed that relaxin (30 μg · kg−1 · day−1 over 24 h) did not change systolic BP compared with placebo. In AHF patients, who usually present with systolic BP higher than in stable CHF patients (Gheorghiade et al., 2006), systolic BP was decreased by relaxin with patients with higher initial BP experiencing larger effects (Teerlink et al., 2013; Voors et al., 2011). This suggests a self‐limiting effect on BP – advantageous for organ protection during AHF (Voors et al., 2011).
In a haemodynamic study in AHF patients (Ponikowski et al., 2014), the changes with relaxin (30 μg · kg−1 · day−1) confirmed the findings of the pilot trial in stable CHF (Dschietzig et al., 2009b), with the significant exception of CI being unchanged. One explanation may be that the high adrenergic drive usually seen in dyspnoeic AHF patients would subside as soon as pulmonary congestion is relieved by relaxin, so introducing a confounder regarding CI. In addition, in AHF patients, urine output and fluid balance are not controlled, and as these are improved by relaxin, the more negative volume balance may have affected CI. The Pre‐RELAX‐AHF trial (Teerlink et al., 2009) identified the optimal dose of serelaxin, 30 μg · kg−1 · day−1, and confirmed the safety of the drug as well as several trends towards beneficial outcomes.
To date, the phase III RELAX‐AHF trial involving more than 1100 patients is the largest cardiovascular relaxin trial (Teerlink et al., 2013). Major inclusion criteria included dyspnoea at rest or on minimal exertion, pulmonary congestion on chest radiograph, elevated levels of B‐type natriuretic peptides, mild to moderate renal dysfunction, systolic BP exceeding 125 mmHg, enrolment within 16 h of hospital admission and administration of ≥40 mg of furosemide before enrolment. Patients with acute coronary syndrome were excluded, and dose reductions of relaxin upon excessive systolic BP drops were pre‐specified. The primary end points were dyspnoea relief measured as improvement on visual analogue scale (VAS) on day 5, as well as on the Likert scale after 6, 12, and 24 h of therapy. The secondary end points included mortality and re‐hospitalization rates within 60 days. Among the tertiary end points, 180‐day mortality and different safety assessments were pre‐specified. Of note, mean systolic BP at baseline was 142 mmHg, higher than in any previous AHF study, ~40% of all patients had atrial fibrillation, and ~45% showed an ejection fraction >40%. The mean time to inclusion was uniquely short (8 h). RELAX met one primary end point, the improvement on VAS, but failed to meet the second, related to the Likert scale. All secondary end points showed a neutral outcome. Multiple, statistically unadjusted, comparisons indicated less administration of intravenous diuretics; less worsening of renal function and worsening of HF before day 14; and less dyspnoea, oedema and rales in the rhRlx group. The effects of relaxin were not dependent on certain subgroups of the study population (Metra et al., 2013b).
A major and rather unexpected finding was the relaxin‐related benefit in terms of 180 day mortality, with an improvement from 9.6 to 6.1% for cardiovascular and from 11.3 to 7.3% for all‐cause mortality and a number needed to treat of approximately 25. The safety analysis showed a significant decrease in systolic BP at different time points (by 4–6 mmHg), more frequent dose modifications (29 vs. 18%), but similar rates of hypotensive adverse events and no systolic BP rebound after drug cessation in the serelaxin, as compared with the placebo, group. Of note, a combined analysis of the Pre‐RELAX and RELAX trials (Teerlink et al., 2009; Teerlink et al., 2013) suggests that relaxin primarily reduced deaths classified as ‘non‐HF cardiovascular cause’ and ‘sudden death’, though this conclusion is merely hypothesis generating, because of the overall small number of events (Greene and Gheorghiade, 2014). As RELAX‐AHF was not powered to detect mortality differences, a second and larger mortality trial in AHF patients is underway.
The encouraging results obtained in RELAX were also corroborated in the (much smaller) Japanese Serelaxin trial (Sato et al., 2015) underlining the efficacy of the peptide in an Asian population.
Current knowledge gaps
Mechanism of 48 h treatments having effects after 180 days
Certainly, the study design of RELAX considered the lessons from the many failed AHF studies by enrolling earlier after AHF onset and by focusing exclusively on patients with rather high BP. But what are the critical roles of relaxin in reversing or reducing damage associated with HF? The rather moderate and self‐limiting effect on BP should be considered because it is most likely to prevent renal adverse events (Gheorghiade et al., 2006; Voors et al., 2011).
With regard to signal transduction, it is worth mentioning the inhibitory effect of relaxin on ET‐1 that has a pivotal role in AHF‐related systemic and pulmonary afterload mismatch, neurohumoral activation and inflammation (Webb, 1995; Stangl et al., 2000; Kawanabe and Nauli, 2011). Relaxin inhibits the stimulation of ET‐1 release, reduces ET‐1 gene expression, promotes endothelial expression of its clearance receptor, ETB, and acts thereby as a functional ET‐1 antagonist (Dschietzig et al., 2001a, 2001b; Dschietzig et al., 2003). Also, activation of the cGMP‐PKG pathway is highly beneficial in many pathological cardiovascular states (Paulus and Tschope, 2013). Relaxin feeds into this pathway at many points, by stimulating NO production and/or NOS expression (Bani et al., 1998a; Alexiou et al., 2013) and by increasing NO bioavailability through its anti‐inflammatory (Masini et al., 2004; Brecht et al., 2011), antioxidant and endothelium‐protective actions (Dschietzig et al., 2012).
The anti‐fibrotic effects of relaxin may play a role in organ protection, and reversal of established cardiac fibrosis by relaxin is desirable particularly in patients with HF with preserved ejection fraction (Figure 5). The peptide also exerts a broad range of vascular and organ‐protective (Alexiou et al., 2013; Collino et al., 2013) actions mediated mainly via NO and the glucocorticoid receptors (Dschietzig et al., 2004; Dschietzig et al., 2012). Numerous in vitro and in vivo studies show cardiac protection following brief administration of relaxin against acute injury due to ischaemia, oxidative stress, inflammation or drug toxicity (Du et al., 2014). That these factors precipitate cardiac decompensation or acute worsening of HF has been well established (Cotter et al., 2010; Januzzi et al., 2012; Metra et al., 2013a). Thus, the cardiac protective property of relaxin might result in suppression of reactive and/or reparative fibrosis (Figure 5).
Figure 5.
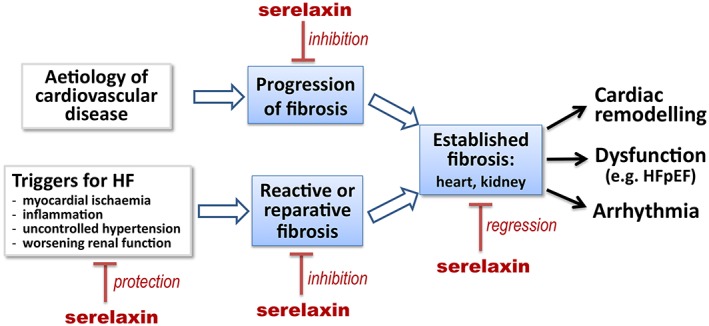
Therapeutic potential of serelaxin in heart disease and HF. Relaxin therapy mediates fibrosis reversal in rodent models of cardiac fibrosis. There is also strong experimental evidence and clinical evidence for the cardiac protective property of relaxin against factors that function as triggers for cardiac decompensation or acute worsening of HF. These actions would be expected to ameliorate cardiac fibrosis.
A key finding in the RELAX‐AHF trial is that the dynamic changes in the first couple of days of biomarkers that reflect damage of multiple organs (heart, kidney and liver) and have predictive value for 180 day prognosis in patients with acute decompensated HF (ADHF) are reduced by relaxin (Metra et al., 2013a). In fact, in addition to these pivotal organs, there is also evidence for damage or malfunction of other organs such as intestine, lung and skeletal muscle, in the setting of ADHF (Peschel et al., 2003; Lauten et al., 2011; Hogan et al., 2012a, 2012b). Thus, we suggest that a compromised tissue microcirculation underlies the multiple organ damage or malfunction (Du et al., 2014). Considering the present strong evidence for organ and endothelial protection by relaxin in a variety of disease settings (Du et al., 2014), we find it likely that damage and dysfunction of the microvasculature in many organs in response to acute decompensation of HF are also exacerbated in clinical conditions following ADHF. In this context, acute relaxin administration protects against microvascular injury, poor microcirculation and organ damage. The amelioration of organ damage and dysfunction by short‐term administration of relaxin would affect long‐term prognosis by limitation of cardiovascular injury and reduction of reparative and reactive fibrosis (Figure 5).
Role of venodilation by relaxin in treating AHF
The effects of relaxin on the venous circulation could potentially account for the haemodynamic effects of relaxin in AHF and CHF. One recent study in rat mesenteric veins showed that, in contrast to arteries, they are not remodelled and do not show reduced myogenic reactivity in response to relaxin (Li et al., 2005; Jelinic et al., 2014). Both studies were performed in vitro and may have been influenced by venous tone being significantly more dependent than arterial tone on sympatho‐adrenergic innervation and pacemaker cells (Cajal cells) (Gelman, 2008). Contraction of splanchnic veins by relaxin (as seen in pregnancy; Slangen et al., 1996; Edouard et al., 1998) or dilation of central veins would increase venous return and explain the baseline‐dependent, ‘self‐limiting’ drop of BP observed in AHF and CHF patients treated with relaxin. The issue is therefore still open for interesting and important investigations.
Anti‐arrhythmic potential
Reduction of 180 day mortality by relaxin treatment was observed in the RELAX‐AHF trial (Teerlink et al., 2013). With some of the sudden deaths likely to be attributable to arrhythmias (Felker et al., 2014), there is emerging evidence for an anti‐arrhythmic action of relaxin. In an elderly SHR model (Parikh et al., 2013), relaxin (0.5 mg · kg−1 · day−1) over 2 weeks followed by programmed electrical stimulation of the isolated atria reduced atrial fibrillation, an effect associated with significant reduction in atrial fibrosis and increased Na+ current density (Parikh et al., 2013). It has been suggested that there is a reciprocal influence between HF and arrhythmias and that arrhythmias act as a facilitating factor for decompensation or worsening of HF (Dukes et al., 2015; Kotecha and Piccini, 2015). Fibrosis reversal and anti‐arrhythmic effects are observed after relaxin for 14 days, although it is not known if this could be achieved by a 48 h treatment. Further studies are necessary to explore the time course of anti‐fibrotic and potentially anti‐arrhythmic actions by relaxin either as a monotherapy or in conjunction with other cardiac medications.
There is also evidence that suggests that relaxin influences myoendothelial coupling in humans. Gap junctions play an important role in vessel structure, function and therefore myoendothelial coupling. Gap junctions are composed of connexin (Cx) proteins, and in the cardiovascular system, the main connexins expressed are Cx40, Cx37, Cx43 and Cx45 (de Wit et al., 2008). Relaxin increases the expression of Cx43 in several tissues, including mouse neonatal immature cardiomyocytes, rodent cardiomyocytes and skeletal myoblasts (Formigli et al., 2005) and hearts from SHR (Parikh et al., 2013). There is also a role of Cx proteins in cellular crosstalk because relaxin promotes the coupling between mouse skeletal muscle myoblasts and adult rat ventricular cardiomyocytes in an in vitro co‐culture model that involves Cx43, suggesting a potential role for gap junction communication that is enhanced by relaxin (Formigli et al., 2005). Thus, there is a need to thoroughly investigate the effects of relaxin on myoendothelial coupling to better understand its cardiovascular effects in humans.
Regulation of RXFP1 receptors or of relaxin in the cardiovascular system under diseased conditions
Endogenous expression of relaxin is elevated in heart disease (Dschietzig et al., 2001b; Xu et al., 2008) although the extent of up‐regulation seems insufficient to exert compensatory action (Xu et al., 2008). Regulation of relaxin expression is poorly defined. The rat relaxin gene promoter region contains guanine (G)‐rich repeats, allowing formation of G‐quadruplexes that can be stabilized by ligands such as berberine (Gu et al., 2012). These sequences are adjacent to the binding motif of STAT3 that negatively regulates relaxin expression. In cardiac fibroblasts and in rats treated with Ang II, berberine suppressed fibroblast activation, collagen synthesis and cardiac fibrosis by up‐regulating relaxin (Gu et al., 2012).
There is limited information on gene regulation of RXFP1 receptors in disease. The expression of RXFP1 receptors is moderately decreased in failing human myocardium (Dschietzig et al., 2011). In reproductive tissues, expression of RXFP1 receptors is up‐regulated by oestrogen and progesterone (Yan et al., 2008; Vodstrcil et al., 2010), but oestrogen treatment has no effect or may even reduce expression in atria (Tan et al., 1999). It remains unknown whether expression of RXFP1 receptors in the cardiovascular system is gender‐dependent or varies during pregnancy. While mRNA for RXFP1 receptors was up‐regulated in the infarcted rat heart (Kompa et al., 2002), expression of these receptors was unchanged in a mouse model of chronic pressure overload (Xu et al., 2008). The expression of RXFP1 receptors in cultured cardiomyocytes or intact heart is down‐regulated by activation of β1‐adrenoceptors but up‐regulated by activation of α1‐AR (Moore et al., 2014). This may influence the efficacy of serelaxin in patients treated with β‐blockers. This was not observed in the RELAX‐AHF trial (Metra et al., 2013a), perhaps due to small sample size. This question, however, could be addressed in the current RELAX‐AHF2 trial.
Could the standard therapy given to patients in RELAX‐AHF be confounding the results, that is, could ACE inhibitors/AT receptor antagonists /β‐blockers affect the response to relaxin?
The pleiotropic actions of relaxin make it unique in AHF therapy. Cardiac remodelling is fundamental in heart disease and failure, and several key factors emerge regardless of aetiology including hypertrophy, inflammation, interstitial fibrosis and continuous loss of cardiomyocytes. These components orchestrate functional decline and development of complications, thereby influencing short‐ and long‐term prognosis of AHF. In theory, changes in some of these key components would be expected to have a beneficial influence on patients with AHF and thus confound the effects of relaxin. On the other hand, ACE inhibitors, Ang II receptor antagonists and β‐blockers have not been shown to be effective in AHF.
The evidence that RXFP1 receptors form heterodimers with the Ang II AT2 receptor and that these are essential for the anti‐fibrotic effects of relaxin in the kidney (Chow et al., 2014) raises important questions that are relevant to its clinical use. The anti‐fibrotic effects of relaxin are completely lost in mice treated with the AT2 receptor antagonist PD123319 or in AT2 receptor KO mice. These findings provide an explanation for the observation that the anti‐fibrotic effects of relaxin are only seen in pathological conditions, with no effect on normal tissue. AT2 receptors are typically expressed at very low levels in healthy tissue, but their expression dramatically increases after injury or disease (Matsubara, 1998; Carey, 2005). RXFP1 receptors are also poorly expressed at the cell surface, but at higher levels within the ER (Kern et al., 2007). Enhanced heterodimerization facilitated by the injury‐induced increase in AT2 receptor expression may increase trafficking of RXFP1 receptors (as an AT2 –RXFP1 receptor heterodimer) to the cell surface, thus explaining why relaxin treatment is more effective under pathological conditions. While the AT2 receptor is not currently regarded as a therapeutic target, there is emerging evidence that the RXFP1 receptor forms heterodimers with other GPCRs that are therapeutically important. It is also interesting to note that, in an earlier study, arteries obtained from patients on ACE inhibitors showed marked attenuation of the vasodilator response to relaxin, effects that appeared to be further enhanced by inhibition of COX (Fisher et al., 2002). However, patients not receiving ACE inhibitors showed little effect of inhibition of COX on the response to relaxin (Fisher et al., 2002).
Conclusions
After a long gestation period, the peptide hormone relaxin is now emerging as an effective treatment for AHF. Although a great deal is now known regarding the cellular actions of relaxin, it is still far from clear which of its pleiotropic effects – vasodilation, anti‐fibrotic, angiogenic, anti‐apoptotic or anti‐inflammatory – is responsible for the beneficial effects. This review has summarized current knowledge of the cardiovascular effects of relaxin, identified knowledge gaps and has suggested possible mechanisms of action.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
T.B.D. was supported by grants 89874634 and 89844575 from BAS Medical Inc. and grants KF2181501AJ9 and KF2318301 from Zentrales Innovationsprogramm Mittelstand. R.J.S was supported by NHMRC Programme grant 1055134 and ARC Linkage grant LP110100288. X.J.D. was supported by NHMRC fellowship (1043026) and project grants (1004235 and 1081710).
Sarwar, M. , Du, X.‐J. , Dschietzig, T. B. , and Summers, R. J. (2017) The actions of relaxin on the human cardiovascular system. British Journal of Pharmacology, 174: 933–949. doi: 10.1111/bph.13523.
References
- Ahmad N, Wang W, Nair R, Kapila S (2012). Relaxin induces matrix‐metalloproteinases‐9 and ‐13 via RXFP1: induction of MMP‐9 involves the PI3K, ERK, Akt and PKC‐zeta pathways. Mol Cell Endocrinol 363: 46–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G Protein‐Coupled Receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexiou K, Wilbring M, Matschke K, Dschietzig T (2013). Relaxin protects rat lungs from ischemia–reperfusion injury via inducible NO synthase: role of ERK‐1/2, PI3K, and forkhead transcription factor FKHRL1. PLoS One 8: e75592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bani D, Baronti R, Vannacci A, Bigazzi M, Sacchi TB, Mannaioni PF et al. (2002). Inhibitory effects of relaxin on human basophils activated by stimulation of the Fc epsilon receptor. The role of nitric oxide. Int Immunopharmacol 2: 1195–1204. [DOI] [PubMed] [Google Scholar]
- Bani D, Bigazzi M, Masini E, Bani G, Sacchi TB (1995a). Relaxin depresses platelet aggregation: in vitro studies on isolated human and rabbit platelets. Lab Invest 73: 709–716. [PubMed] [Google Scholar]
- Bani D, Failli P, Bello MG, Thiemermann C, Bani Sacchi T, Bigazzi M et al. (1998a). Relaxin activates the L‐arginine‐nitric oxide pathway in vascular smooth muscle cells in culture. Hypertension 31: 1240–1247. [DOI] [PubMed] [Google Scholar]
- Bani D, Masini E, Bello MG, Bigazzi M, Sacchi TB (1995b). Relaxin activates the L‐arginine‐nitric oxide pathway in human breast cancer cells. Cancer Res 55: 5272–5275. [PubMed] [Google Scholar]
- Bani D, Masini E, Bello MG, Bigazzi M, Sacchi TB (1998b). Relaxin protects against myocardial injury caused by ischemia and reperfusion in rat heart. Am J Pathol 152: 1367–1376. [PMC free article] [PubMed] [Google Scholar]
- Bani‐Sacchi T, Bigazzi M, Bani D, Mannaioni PF, Masini E (1995). Relaxin‐induced increased coronary flow through stimulation of nitric oxide production. Br J Pharmacol 116: 1589–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bathgate RA, Halls ML, van der Westhuizen ET, Callander GE, Kocan M, Summers RJ (2013). Relaxin family peptides and their receptors. Physiol Rev 93: 405–480. [DOI] [PubMed] [Google Scholar]
- Bathgate RA, Lekgabe ED, McGuane JT, Su Y, Pham T, Ferraro T et al. (2008). Adenovirus‐mediated delivery of relaxin reverses cardiac fibrosis. Mol Cell Endocrinol 280: 30–38. [DOI] [PubMed] [Google Scholar]
- Bennett RG (2009). Relaxin and its role in the development and treatment of fibrosis. Transl Res 154: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitto A, Irrera N, Minutoli L, Calo M, Lo Cascio P, Caccia P et al. (2013). Relaxin improves multiple markers of wound healing and ameliorates the disturbed healing pattern of genetically diabetic mice. Clin Sci 125: 575–585. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Boccalini G, Sassoli C, Formigli L, Bani D, Nistri S (2015). Relaxin protects cardiac muscle cells from hypoxia/reoxygenation injury: involvement of the Notch‐1 pathway. FASEB J 29: 239–249. [DOI] [PubMed] [Google Scholar]
- Bonner JS, Lantier L, Hocking KM, Kang L, Owolabi M, James FD et al. (2013). Relaxin treatment reverses insulin resistance in mice fed a high‐fat diet. Diabetes 62: 3251–3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braam B, Joles JA, Danishwar AH, Gaillard CA (2014). Cardiorenal syndrome–current understanding and future perspectives. Nat Rev Nephrol 10: 48–55. [DOI] [PubMed] [Google Scholar]
- Brecht A, Bartsch C, Baumann G, Stangl K, Dschietzig T (2011). Relaxin inhibits early steps in vascular inflammation. Regul Pept 166: 76–82. [DOI] [PubMed] [Google Scholar]
- Bueno OF, Molkentin JD (2002). Involvement of extracellular signal‐regulated kinases 1/2 in cardiac hypertrophy and cell death. Circ Res 91: 776–781. [DOI] [PubMed] [Google Scholar]
- Carey RM (2005). Cardiovascular and renal regulation by the angiotensin type 2 receptor: the AT2 receptor comes of age. Hypertension 45: 840–844. [DOI] [PubMed] [Google Scholar]
- Chow BS, Chew EG, Zhao C, Bathgate RA, Hewitson TD, Samuel CS (2012). Relaxin signals through a RXFP1–pERK–nNOS–NO–cGMP‐dependent pathway to up‐regulate matrix metalloproteinases: the additional involvement of iNOS. PLoS One 7: e42714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow BS, Kocan M, Bosnyak S, Sarwar M, Wigg B, Jones ES et al. (2014). Relaxin requires the angiotensin II type 2 receptor to abrogate renal interstitial fibrosis. Kidney Int 86: 75–85. [DOI] [PubMed] [Google Scholar]
- Collino M, Rogazzo M, Pini A, Benetti E, Rosa AC, Chiazza F et al. (2013). Acute treatment with relaxin protects the kidney against ischaemia/reperfusion injury. J Cell Mol Med 17: 1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KP, Debrah DO, Novak J, Danielson LA, Shroff SG (2004). Relaxin modifies systemic arterial resistance and compliance in conscious, nonpregnant rats. Endocrinology 145: 3289–3296. [DOI] [PubMed] [Google Scholar]
- Conrad KP, Novak J (2004). Emerging role of relaxin in renal and cardiovascular function. Am J Physiol Regul Integr Comp Physiol 287: R250–R261. [DOI] [PubMed] [Google Scholar]
- Conrad KP, Shroff SG (2011). Effects of relaxin on arterial dilation, remodeling, and mechanical properties. Curr Hypertens Rep 13: 409–420. [DOI] [PubMed] [Google Scholar]
- Cotter G, Voors AA, Weatherley BD, Pang PS, Teerlink JR, Filippatos G et al. (2010). Acute heart failure clinical drug development: from planning to proof of activity to phase III. Cardiology 116: 292–301. [DOI] [PubMed] [Google Scholar]
- Coulson CC, Thorp JM Jr, Mayer DC, Cefalo RC (1996). Central hemodynamic effects of recombinant human relaxin in the isolated, perfused rat heart model. Obstet Gynecol 87: 610–612. [DOI] [PubMed] [Google Scholar]
- Danielson LA, Sherwood OD, Conrad KP (1999). Relaxin is a potent renal vasodilator in conscious rats. J Clin Invest 103: 525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit C, Boettcher M, Schmidt VJ (2008). Signaling across myoendothelial gap junctions–fact or fiction? Cell Commun Adhes 15: 231–245. [DOI] [PubMed] [Google Scholar]
- Debrah DO, Conrad KP, Danielson LA, Shroff SG (2005a). Effects of relaxin on systemic arterial hemodynamics and mechanical properties in conscious rats: sex dependency and dose response. J Appl Physiol 98: 1013–1020. [DOI] [PubMed] [Google Scholar]
- Debrah DO, Conrad KP, Jeyabalan A, Danielson LA, Shroff SG (2005b). Relaxin increases cardiac output and reduces systemic arterial load in hypertensive rats. Hypertension 46: 745–750. [DOI] [PubMed] [Google Scholar]
- Debrah DO, Novak J, Matthews JE, Ramirez RJ, Shroff SG, Conrad KP (2006). Relaxin is essential for systemic vasodilation and increased global arterial compliance during early pregnancy in conscious rats. Endocrinology 147: 5126–5131. [DOI] [PubMed] [Google Scholar]
- Dschietzig T, Alexiou K, Kinkel HT, Baumann G, Matschke K, Stangl K (2011). The positive inotropic effect of relaxin‐2 in human atrial myocardium is preserved in end‐stage heart failure: role of G(i)‐phosphoinositide‐3 kinase signaling. J Card Fail 17: 158–166. [DOI] [PubMed] [Google Scholar]
- Dschietzig T, Bartsch C, Baumann G, Stangl K (2009a). RXFP1‐inactive relaxin activates human glucocorticoid receptor: further investigations into the relaxin‐GR pathway. Regul Pept 154: 77–84. [DOI] [PubMed] [Google Scholar]
- Dschietzig T, Bartsch C, Kinkel T, Baumann G, Stangl K (2005). Myocardial relaxin counteracts hypertrophy in hypertensive rats. Ann N Y Acad Sci 1041: 441–443. [DOI] [PubMed] [Google Scholar]
- Dschietzig T, Bartsch C, Richter C, Laule M, Baumann G, Stangl K (2003). Relaxin, a pregnancy hormone, is a functional endothelin‐1 antagonist: attenuation of endothelin‐1‐mediated vasoconstriction by stimulation of endothelin type‐B receptor expression via ERK‐1/2 and nuclear factor‐kB. Circ Res 92: 32–40. [DOI] [PubMed] [Google Scholar]
- Dschietzig T, Bartsch C, Stangl V, Baumann G, Stangl K (2004). Identification of the pregnancy hormone relaxin as glucocorticoid receptor agonist. FASEB J 18: 1536–1538. [DOI] [PubMed] [Google Scholar]
- Dschietzig T, Brecht A, Bartsch C, Baumann G, Stangl K, Alexiou K (2012). Relaxin improves TNF‐alpha‐induced endothelial dysfunction: the role of glucocorticoid receptor and phosphatidylinositol 3‐kinase signalling. Cardiovasc Res 95: 97–107. [DOI] [PubMed] [Google Scholar]
- Dschietzig T, Richter C, Bartsch C, Bohme C, Heinze D, Ott F et al. (2001a). Flow‐induced pressure differentially regulates endothelin‐1, urotensin II, adrenomedullin, and relaxin in pulmonary vascular endothelium. Biochem Biophys Res Commun 289: 245–251. [DOI] [PubMed] [Google Scholar]
- Dschietzig T, Richter C, Bartsch C, Laule M, Armbruster FP, Baumann G et al. (2001b). The pregnancy hormone relaxin is a player in human heart failure. FASEB J 15: 2187–2195. [DOI] [PubMed] [Google Scholar]
- Dschietzig T, Teichman S, Unemori E, Wood S, Boehmer J, Richter C et al. (2009b). Intravenous recombinant human relaxin in compensated heart failure: a safety, tolerability, and pharmacodynamic trial. J Card Fail 15: 182–190. [DOI] [PubMed] [Google Scholar]
- Du XJ, Hewitson TD, Nguyen MN, Samuel CS (2014). Therapeutic effects of serelaxin in acute heart failure. Circ J 78: 542–552. [DOI] [PubMed] [Google Scholar]
- Du XJ, Samuel CS, Gao XM, Zhao L, Parry LJ, Tregear GW (2003). Increased myocardial collagen and ventricular diastolic dysfunction in relaxin deficient mice: a gender‐specific phenotype. Cardiovasc Res 57: 395–404. [DOI] [PubMed] [Google Scholar]
- Dukes JW, Dewland TA, Vittinghoff E, Mandyam MC, Heckbert SR, Siscovick DS et al. (2015). Ventricular ectopy as a predictor of heart failure and death. J Am Coll Cardiol 66: 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edouard DA, Pannier BM, London GM, Cuche JL, Safar ME (1998). Venous and arterial behavior during normal pregnancy. Am J Physiol 274: H1605–H1612. [DOI] [PubMed] [Google Scholar]
- Eschenhagen T, Mende U, Nose M, Schmitz W, Scholz H, Haverich A et al. (1992). Increased messenger RNA level of the inhibitory G protein alpha subunit Gi alpha‐2 in human end‐stage heart failure. Circ Res 70: 688–696. [DOI] [PubMed] [Google Scholar]
- Failli P, Nistri S, Quattrone S, Mazzetti L, Bigazzi M, Sacchi TB et al. (2002). Relaxin up‐regulates inducible nitric oxide synthase expression and nitric oxide generation in rat coronary endothelial cells. FASEB J 16: 252–254. [DOI] [PubMed] [Google Scholar]
- Felker GM, Teerlink JR, Butler J, Hernandez AF, Miller AB, Cotter G et al. (2014). Effect of serelaxin on mode of death in acute heart failure: results from the RELAX‐AHF study. J Am Coll Cardiol 64: 1591–1598. [DOI] [PubMed] [Google Scholar]
- Fernandez‐Patron C, Radomski MW, Davidge ST (1999). Vascular matrix metalloproteinase‐2 cleaves big endothelin‐1 yielding a novel vasoconstrictor. Circ Res 85: 906–911. [DOI] [PubMed] [Google Scholar]
- Fernandez‐Patron C, Stewart KG, Zhang Y, Koivunen E, Radomski MW, Davidge ST (2000). Vascular matrix metalloproteinase‐2‐dependent cleavage of calcitonin gene‐related peptide promotes vasoconstriction. Circ Res 87: 670–676. [DOI] [PubMed] [Google Scholar]
- Fisher C, MacLean M, Morecroft I, Seed A, Johnston F, Hillier C et al. (2002). Is the pregnancy hormone relaxin also a vasodilator peptide secreted by the heart? Circulation 106: 292–295. [DOI] [PubMed] [Google Scholar]
- Fisher CJ (2009). Relaxin: a new cardiovascular hormone in humans? Comparative potency and mechanisms of action. MD thesis Dept Medicine and Therapeutics. Glasgow University.
- Formigli L, Francini F, Tani A, Squecco R, Nosi D, Polidori L et al. (2005). Morphofunctional integration between skeletal myoblasts and adult cardiomyocytes in coculture is favored by direct cell–cell contacts and relaxin treatment. Am J Physiol Cell Physiol 288: C795–C804. [DOI] [PubMed] [Google Scholar]
- Formigli L, Perna AM, Meacci E, Cinci L, Margheri M, Nistri S et al. (2007). Paracrine effects of transplanted myoblasts and relaxin on post‐infarction heart remodelling. J Cell Mol Med 11: 1087–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber SL, Mirochnik Y, Brecklin C, Slobodskoy L, Arruda JA, Dunea G (2003). Effect of relaxin in two models of renal mass reduction. Am J Nephrol 23: 8–12. [DOI] [PubMed] [Google Scholar]
- Garber SL, Mirochnik Y, Brecklin CS, Unemori EN, Singh AK, Slobodskoy L et al. (2001). Relaxin decreases renal interstitial fibrosis and slows progression of renal disease. Kidney Int 59: 876–882. [DOI] [PubMed] [Google Scholar]
- Gelman S (2008). Venous function and central venous pressure: a physiologic story. Anesthesiology 108: 735–748. [DOI] [PubMed] [Google Scholar]
- Gheorghiade M, Abraham WT, Albert NM, Greenberg BH, O'Connor CM, She L et al. (2006). Systolic blood pressure at admission, clinical characteristics, and outcomes in patients hospitalized with acute heart failure. JAMA 296: 2217–2226. [DOI] [PubMed] [Google Scholar]
- Gooi JH, Richardson ML, Jelinic M, Girling JE, Wlodek ME, Tare M et al. (2013). Enhanced uterine artery stiffness in aged pregnant relaxin mutant mice is reversed with exogenous relaxin treatment. Biol Reprod 89: 18. [DOI] [PubMed] [Google Scholar]
- Greene SJ, Gheorghiade M (2014). Matching mechanism of death with mechanism of action: considerations for drug development for hospitalized heart failure. J Am Coll Cardiol 64: 1599–1601. [DOI] [PubMed] [Google Scholar]
- Gu HP, Lin S, Xu M, Yu HY, Du XJ, Zhang YY et al. (2012). Up‐regulating relaxin expression by G‐quadruplex interactive ligand to achieve antifibrotic action. Endocrinology 153: 3692–3700. [DOI] [PubMed] [Google Scholar]
- Haase N, Rugor J, Przybyl L, Qadri F, Muller DN, Dechend R (2014). Relaxin does not improve angiotensin II‐induced target‐organ damage. PLoS One 9: e93743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halls ML, Bathgate RA, Summers RJ (2006). Relaxin family peptide receptors RXFP1 and RXFP2 modulate cAMP signaling by distinct mechanisms. Mol Pharmacol 70: 214–226. [DOI] [PubMed] [Google Scholar]
- Halls ML, Bathgate RA, Sutton SW, Dschietzig TB, Summers RJ (2015). International Union of Basic and Clinical Pharmacology. XCV. Recent advances in the understanding of the pharmacology and biological roles of relaxin family peptide receptors 1–4, the receptors for relaxin family peptides. Pharmacol Rev 67: 389–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halls ML, Hewitson TD, Moore XL, Du XJ, Bathgate RA, Summers RJ (2009). Relaxin activates multiple cAMP signaling pathway profiles in different target cells. Ann N Y Acad Sci 1160: 108–111. [DOI] [PubMed] [Google Scholar]
- Han X, Habuchi Y, Giles WR (1994). Relaxin increases heart rate by modulating calcium current in cardiac pacemaker cells. Circ Res 74: 537–541. [DOI] [PubMed] [Google Scholar]
- Heeg MHJ, Koziolek MJ, Vasko R, Schaefer L, Sharma K, Muller GA et al. (2005). The antifibrotic effects of relaxin in human renal fibroblasts are mediated in part by inhibition of the Smad2 pathway. Kidney Int 68: 96–109. [DOI] [PubMed] [Google Scholar]
- Ho TY, Yan W, Bagnell CA (2007). Relaxin‐induced matrix metalloproteinase‐9 expression is associated with activation of the NF‐kB pathway in human THP‐1 cells. J Leukoc Biol 81: 1303–1310. [DOI] [PubMed] [Google Scholar]
- Hogan CJ, Ward KR, Franzen DS, Rajendran B, Thacker LR (2012a). Sublingual tissue perfusion improves during emergency treatment of acute decompensated heart failure. Am J Emerg Med 30: 872–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan CJ, Ward KR, Kontos MC, Thacker LR, Pittman R (2012b). Peripheral tissue oxygenation improves during ED treatment of acute heart failure. Am J Emerg Med 30: 196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JS, Yamamoto SY, Bryant‐Greenwood GD (2011). Relaxin modulates proinflammatory cytokine secretion from human decidual macrophages. Biol Reprod 85: 788–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain MA, Man BC, Zhao C, Xu Q, Du XJ, Wade JD et al. (2011). H3 relaxin demonstrates antifibrotic properties via the RXFP1 receptor. Biochemistry 50: 1368–1375. [DOI] [PubMed] [Google Scholar]
- Houslay MD, Baillie GS, Maurice DH (2007). cAMP‐specific phosphodiesterase‐4 enzymes in the cardiovascular system: a molecular toolbox for generating compartmentalized cAMP signaling. Circ Res 100: 950–966. [DOI] [PubMed] [Google Scholar]
- Hsu SY, Nakabayashi K, Nishi S, Kumagai J, Kudo M, Sherwood OD et al. (2002). Activation of orphan receptors by the hormone relaxin. Science 295: 671–674. [DOI] [PubMed] [Google Scholar]
- Huang X, Cheng Z, Sunga J, Unemori E, Zsebo K (2001). Systemic administration of recombinant human relaxin (RHRLX) ameliorates the acute cyclosporine nephrotoxicity in rats. J Heart Lung Transplant 20: 253. [DOI] [PubMed] [Google Scholar]
- Huang X, Gai Y, Yang N, Lu B, Samuel CS, Thannickal VJ et al. (2011). Relaxin regulates myofibroblast contractility and protects against lung fibrosis. Am J Pathol 179: 2751–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Januzzi JL Jr, Filippatos G, Nieminen M, Gheorghiade M (2012). Troponin elevation in patients with heart failure: on behalf of the third Universal Definition of Myocardial Infarction Global Task Force: Heart Failure Section. Eur Heart J 33: 2265–2271. [DOI] [PubMed] [Google Scholar]
- Jelinic M, Leo CH, Post Uiterweer ED, Sandow SL, Gooi JH, Wlodek ME et al. (2014). Localization of relaxin receptors in arteries and veins, and region‐specific increases in compliance and bradykinin‐mediated relaxation after in vivo serelaxin treatment. FASEB J 28: 275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelinic M, Tare M, Conrad KP, Parry LJ (2015). Differential effects of relaxin deficiency on vascular aging in arteries of male mice. Age 37: 9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyabalan A, Novak J, Danielson LA, Kerchner LJ, Opett SL, Conrad KP (2003). Essential role for vascular gelatinase activity in relaxin‐induced renal vasodilation, hyperfiltration, and reduced myogenic reactivity of small arteries. Circ Res 93: 1249–1257. [DOI] [PubMed] [Google Scholar]
- Kaftanovskaya EM, Huang Z, Lopez C, Conrad K, Agoulnik AI (2015). Conditional deletion of the relaxin receptor gene in cells of smooth muscle lineage affects lower reproductive tract in pregnant mice. Biol Reprod 92: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakouris H, Eddie LW, Summers RJ (1992). Cardiac effects of relaxin in rats. Lancet 339: 1076–1078. [DOI] [PubMed] [Google Scholar]
- Kapila S, Xie Y, Wang W (2009). Induction of MMP‐1 (collagenase‐1) by relaxin in fibrocartilaginous cells requires both the AP‐1 and PEA‐3 promoter sites. Orthod Craniofac Res 12: 178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawanabe Y, Nauli SM (2011). Endothelin. Cell Mol Life Sci 68: 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerchner LJ, Novak J, Hanley‐Yanez K, Doty KD, Danielson LA, Conrad KP (2005). Evidence against the hypothesis that endothelial endothelin B receptor expression is regulated by relaxin and pregnancy. Endocrinology 146: 2791–2797. [DOI] [PubMed] [Google Scholar]
- Kern A, Agoulnik AI, Bryant‐Greenwood GD (2007). The low‐density lipoprotein class A module of the relaxin receptor (leucine‐rich repeat containing G‐protein coupled receptor 7): its role in signaling and trafficking to the cell membrane. Endocrinology 148: 1181–1194. [DOI] [PubMed] [Google Scholar]
- Kiselyov K, Shin DM, Muallem S (2003). Signalling specificity in GPCR‐dependent Ca2+ signalling. Cell Signal 15: 243–253. [DOI] [PubMed] [Google Scholar]
- Kloner RA (2011). No‐reflow phenomenon: maintaining vascular integrity. J Cardiovasc Pharmacol Ther 16: 244–250. [DOI] [PubMed] [Google Scholar]
- Kompa AR, Samuel CS, Summers RJ (2002). Inotropic responses to human gene 2 (B29) relaxin in a rat model of myocardial infarction (MI): effect of pertussis toxin. Br J Pharmacol 137: 710–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha D, Piccini JP (2015). Atrial fibrillation in heart failure: what should we do? Eur Heart J 36: 3250–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajnc‐Franken MA, van Disseldorp AJ, Koenders JE, Mosselman S, van Duin M, Gossen JA (2004). Impaired nipple development and parturition in LGR7 knockout mice. Mol Cell Biol 24: 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauten A, Ferrari M, Goebel B, Rademacher W, Schumm J, Uth O et al. (2011). Microvascular tissue perfusion is impaired in acutely decompensated heart failure and improves following standard treatment. Eur J Heart Fail 13: 711–717. [DOI] [PubMed] [Google Scholar]
- Lee HY, Zhao S, Fields PA, Sherwood OD (2005). The extent to which relaxin promotes proliferation and inhibits apoptosis of cervical epithelial and stromal cells is greatest during late pregnancy in rats. Endocrinology 146: 511–518. [DOI] [PubMed] [Google Scholar]
- Lekgabe ED, Kiriazis H, Zhao C, Xu Q, Moore XL, Su Y et al. (2005). Relaxin reverses cardiac and renal fibrosis in spontaneously hypertensive rats. Hypertension 46: 412–418. [DOI] [PubMed] [Google Scholar]
- Leo CH, Jelinic M, Gooi JH, Tare M, Parry LJ (2014a). A vasoactive role for endogenous relaxin in mesenteric arteries of male mice. PLoS One 9: e107382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo CH, Jelinic M, Parkington HC, Tare M, Parry LJ (2014b). Acute intravenous injection of serelaxin (recombinant human relaxin‐2) causes rapid and sustained bradykinin‐mediated vasorelaxation. J Am Heart Assoc 3: e000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Brookes ZL, Kaufman S (2005). Acute and chronic effects of relaxin on vasoactivity, myogenic reactivity and compliance of the rat mesenteric arterial and venous vasculature. Regul Pept 132: 41–46. [DOI] [PubMed] [Google Scholar]
- Lodhi RS, Nakabayashi K, Suzuki K, Yamada AY, Hazama R, Ebina Y et al. (2013). Relaxin has anti‐apoptotic effects on human trophoblast‐derived HTR‐8/SV neo cells. Gynecol Endocrinol 29: 1051–1054. [DOI] [PubMed] [Google Scholar]
- Ma JF, Liu L, Yang WJ, Zang LN, Xi YM (2013a). RNAi‐mediated knockdown of relaxin decreases in vitro proliferation and invasiveness of osteosarcoma MG‐63 cells by inhibition of MMP‐9. Eur Rev Med Pharmacol Sci 17: 1102–1109. [PubMed] [Google Scholar]
- Ma JF, Von Kalle M, Plautz Q, ‐M Xu F, Singh L, Wang L (2013b). Relaxin promotes in vitro tumour growth, invasion and angiogenesis of human Saos‐2 osteosarcoma cells by AKT/VEGF pathway. Eur Rev Med Pharmacol Sci 17: 1345–1350. [PubMed] [Google Scholar]
- Masini E, Bani D, Bello MG, Bigazzi M, Mannaioni PF, Sacchi TB (1997). Relaxin counteracts myocardial damage induced by ischemia–reperfusion in isolated guinea pig hearts: evidence for an involvement of nitric oxide. Endocrinology 138: 4713–4720. [DOI] [PubMed] [Google Scholar]
- Masini E, Di Bello MG, Bani D, Bigazzi M, Bani Sacchi T, Mannaioni PF (1995). Relaxin inhibits histamine release from mast cells: involvement of nitric oxide production. Inflamm Res 44 (Suppl 1): S12–S13. [DOI] [PubMed] [Google Scholar]
- Masini E, Nistri S, Vannacci A, Bani Sacchi T, Novelli A, Bani D (2004). Relaxin inhibits the activation of human neutrophils: involvement of the nitric oxide pathway. Endocrinology 145: 1106–1112. [DOI] [PubMed] [Google Scholar]
- Masini E, Zagli G, Ndisang JF, Solazzo M, Mannaioni PF, Bani D (2002). Protective effect of relaxin in cardiac anaphylaxis: involvement of the nitric oxide pathway. Br J Pharmacol 137: 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson R, Hewitson TD, Kelynack K, Martic M, Parry L, Bathgate R et al. (2004). Relaxin down‐regulates renal fibroblast function and promotes matrix remodelling in vitro. Nephrol Dial Transplant 19: 544–552. [DOI] [PubMed] [Google Scholar]
- Matsubara H (1998). Pathophysiological role of angiotensin II type 2 receptor in cardiovascular and renal diseases. Circ Res 83: 1182–1191. [DOI] [PubMed] [Google Scholar]
- McGuane JT, Danielson LA, Debrah JE, Rubin JP, Novak J, Conrad KP (2011a). Angiogenic growth factors are new and essential players in the sustained relaxin vasodilatory pathway in rodents and humans. Hypertension 57: 1151–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuane JT, Debrah JE, Sautina L, Jarajapu YP, Novak J, Rubin JP et al. (2011b). Relaxin induces rapid dilation of rodent small renal and human subcutaneous arteries via PI3 kinase and nitric oxide. Endocrinology 152: 2786–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metra M, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH et al. (2013a). Effect of serelaxin on cardiac, renal, and hepatic biomarkers in the Relaxin in Acute Heart Failure (RELAX‐AHF) development program: correlation with outcomes. J Am Coll Cardiol 61: 196–206. [DOI] [PubMed] [Google Scholar]
- Metra M, Ponikowski P, Cotter G, Davison BA, Felker GM, Filippatos G et al. (2013b). Effects of serelaxin in subgroups of patients with acute heart failure: results from RELAX‐AHF. Eur Heart J 34: 3128–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mologni L, Sala E, Cazzaniga S, Rostagno R, Kuoni T, Puttini M et al. (2006). Inhibition of RET tyrosine kinase by SU5416. J Mol Endocrinol 37: 199–212. [DOI] [PubMed] [Google Scholar]
- Mookerjee I, Hewitson TD, Halls ML, Summers RJ, Mathai ML, Bathgate RA et al. (2009). Relaxin inhibits renal myofibroblast differentiation via RXFP1, the nitric oxide pathway, and Smad2. FASEB J 23: 1219–1229. [DOI] [PubMed] [Google Scholar]
- Moore XL, Su Y, Fan Y, Zhang YY, Woodcock EA, Dart AM et al. (2014). Diverse regulation of cardiac expression of relaxin receptor by α1‐ and β1‐adrenoceptors. Cardiovasc Drugs Ther 28: 221–228. [DOI] [PubMed] [Google Scholar]
- Moore XL, Tan SL, Lo CY, Fang L, Su YD, Gao XM et al. (2007). Relaxin antagonizes hypertrophy and apoptosis in neonatal rat cardiomyocytes. Endocrinology 148: 1582–1589. [DOI] [PubMed] [Google Scholar]
- Ng HH, Jelinic M, Parry LJ, Leo CH (2015). Increased superoxide production and altered nitric oxide‐mediated relaxation in the aorta of young but not old male relaxin‐deficient mice. Am J Physiol Heart Circ Physiol 309: H285–H296. [DOI] [PubMed] [Google Scholar]
- Nguyen BT, Dessauer CW (2005a). Relaxin stimulates cAMP production in MCF‐7 cells upon overexpression of type V adenylyl cyclase. Ann N Y Acad Sci 1041: 296–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen BT, Dessauer CW (2005b). Relaxin stimulates protein kinase C z translocation: requirement for cyclic adenosine 3′,5′‐monophosphate production. Mol Endocrinol 19: 1012–1023. [DOI] [PubMed] [Google Scholar]
- Nistri S, Cinci L, Perna AM, Masini E, Mastroianni R, Bani D (2008). Relaxin induces mast cell inhibition and reduces ventricular arrhythmias in a swine model of acute myocardial infarction. Pharmacol Res 57: 43–48. [DOI] [PubMed] [Google Scholar]
- Novak J, Danielson LA, Kerchner LJ, Sherwood OD, Ramirez RJ, Moalli PA et al. (2001). Relaxin is essential for renal vasodilation during pregnancy in conscious rats. J Clin Invest 107: 1469–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osheroff PL, Cronin MJ, Lofgren JA (1992). Relaxin binding in the rat heart atrium. Proc Natl Acad Sci U S A 89: 2384–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh A, Patel D, McTiernan CF, Xiang W, Haney J, Yang L et al. (2013). Relaxin suppresses atrial fibrillation by reversing fibrosis and myocyte hypertrophy and increasing conduction velocity and sodium current in spontaneously hypertensive rat hearts. Circ Res 113: 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus WJ, Tschope C (2013). A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62: 263–271. [DOI] [PubMed] [Google Scholar]
- Peschel T, Schonauer M, Thiele H, Anker SD, Schuler G, Niebauer J (2003). Invasive assessment of bacterial endotoxin and inflammatory cytokines in patients with acute heart failure. Eur J Heart Fail 5: 609–614. [DOI] [PubMed] [Google Scholar]
- Ponikowski P, Mitrovic V, Ruda M, Fernandez A, Voors AA, Vishnevsky A et al. (2014). A randomized, double‐blind, placebo‐controlled, multicentre study to assess haemodynamic effects of serelaxin in patients with acute heart failure. Eur Heart J 35: 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quattrone S, Chiappini L, Scapagnini G, Bigazzi B, Bani D (2004). Relaxin potentiates the expression of inducible nitric oxide synthase by endothelial cells from human umbilical vein in in vitro culture. Mol Hum Reprod 10: 325–330. [DOI] [PubMed] [Google Scholar]
- Samuel CS, Bodaragama H, Chew JY, Widdop RE, Royce SG, Hewitson TD (2014). Serelaxin is a more efficacious antifibrotic than enalapril in an experimental model of heart disease. Hypertension 64: 315–322. [DOI] [PubMed] [Google Scholar]
- Samuel CS, Cendrawan S, Gao XM, Ming Z, Zhao C, Kiriazis H et al. (2011). Relaxin remodels fibrotic healing following myocardial infarction. Lab Invest 91: 675–690. [DOI] [PubMed] [Google Scholar]
- Samuel CS, Hewitson TD, Zhang Y, Kelly DJ (2008). Relaxin ameliorates fibrosis in experimental diabetic cardiomyopathy. Endocrinology 149: 3286–3293. [DOI] [PubMed] [Google Scholar]
- Samuel CS, Lekgabe ED, Mookerjee I (2007). The effects of relaxin on extracellular matrix remodeling in health and fibrotic disease. Adv Exp Med Biol 612: 88–103. [DOI] [PubMed] [Google Scholar]
- Samuel CS, Parry LJ, Summers RJ (2003). Physiological or pathological–a role for relaxin in the cardiovascular system? Curr Opin Pharmacol 3: 152–158. [DOI] [PubMed] [Google Scholar]
- Samuel CS, Unemori EN, Mookerjee I, Bathgate RA, Layfield SL, Mak J et al. (2004a). Relaxin modulates cardiac fibroblast proliferation, differentiation, and collagen production and reverses cardiac fibrosis in vivo. Endocrinology 145: 4125–4133. [DOI] [PubMed] [Google Scholar]
- Samuel CS, Zhao C, Bond CP, Hewitson TD, Amento EP, Summers RJ (2004b). Relaxin‐1‐deficient mice develop an age‐related progression of renal fibrosis. Kidney Int 65: 2054–2064. [DOI] [PubMed] [Google Scholar]
- Sarwar M, Samuel CS, Bathgate RA, Stewart DR, Summers RJ (2016). Enhanced serelaxin signalling in co‐cultures of human primary endothelial and smooth muscle cells. Br J Pharmacol 173: 484–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarwar M, Samuel CS, Bathgate RA, Stewart DR, Summers RJ (2015). Serelaxin‐mediated signal transduction in human vascular cells: bell‐shaped concentration–response curves reflect differential coupling to G proteins. Br J Pharmacol 172: 1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasser JM, Molnar M, Baylis C (2011). Relaxin ameliorates hypertension and increases nitric oxide metabolite excretion in angiotensin II but not N(omega)‐nitro‐l‐arginine methyl ester hypertensive rats. Hypertension 58: 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Takahashi W, Hirayama A, Ajioka M, Takahashi N, Okishige K et al. (2015). Multicenter, randomized, double‐blinded, placebo‐controlled phase II study of serelaxin in Japanese patients with acute heart failure. Circ J 79: 1237–1247. [DOI] [PubMed] [Google Scholar]
- Segal MS, Sautina L, Li S, Diao Y, Agoulnik AI, Kielczewski J et al. (2012). Relaxin increases human endothelial progenitor cell NO and migration and vasculogenesis in mice. Blood 119: 629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw EE, Wood P, Kulpa J, Yang FH, Summerlee AJ, Pyle WG (2009). Relaxin alters cardiac myofilament function through a PKC‐dependent pathway. Am J Physiol Heart Circ Physiol 297: H29–H36. [DOI] [PubMed] [Google Scholar]
- Sherwood OD (2004). Relaxin's physiological roles and other diverse actions. Endocr Rev 25: 205–234. [DOI] [PubMed] [Google Scholar]
- Slangen BF, Out IC, Verkeste CM, Peeters LL (1996). Hemodynamic changes in early pregnancy in chronically instrumented, conscious rats. Am J Physiol 270: H1779–H1784. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stangl K, Dschietzig T, Richter C, Laule M, Stangl V, Tanis E et al. (2000). Pulmonary release and coronary and peripheral consumption of big endothelin and endothelin‐1 in severe heart failure: acute effects of vasodilator therapy. Circulation 102: 1132–1138. [DOI] [PubMed] [Google Scholar]
- Tan YY, Wade JD, Tregear GW, Summers RJ (1999). Quantitative autoradiographic studies of relaxin binding in rat atria, uterus and cerebral cortex: characterization and effects of oestrogen treatment. Br J Pharmacol 127: 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teerlink JR, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH et al. (2013). Serelaxin, recombinant human relaxin‐2, for treatment of acute heart failure (RELAX‐AHF): a randomised, placebo‐controlled trial. Lancet 381: 29–39. [DOI] [PubMed] [Google Scholar]
- Teerlink JR, Metra M, Felker GM, Ponikowski P, Voors AA, Weatherley BD et al. (2009). Relaxin for the treatment of patients with acute heart failure (Pre‐RELAX‐AHF): a multicentre, randomised, placebo‐controlled, parallel‐group, dose‐finding phase IIb study. Lancet 373: 1429–1439. [DOI] [PubMed] [Google Scholar]
- Toth M, Taskinen P, Ruskoaho H (1996). Relaxin stimulates atrial natriuretic peptide secretion in perfused rat heart. J Endocrinol 150: 487–495. [DOI] [PubMed] [Google Scholar]
- Unemori EN, Erikson ME, Rocco SE, Sutherland KM, Parsell DA, Mak J et al. (1999). Relaxin stimulates expression of vascular endothelial growth factor in normal human endometrial cells in vitro and is associated with menometrorrhagia in women. Hum Reprod 14: 800–806. [DOI] [PubMed] [Google Scholar]
- Unemori EN, Ferrara N, Bauer EA, Amento EP (1992). Vascular endothelial growth factor induces interstitial collagenase expression in human endothelial cells. J Cell Physiol 153: 557–562. [DOI] [PubMed] [Google Scholar]
- Unemori EN, Lewis M, Constant J, Arnold G, Grove BH, Normand J et al. (2000). Relaxin induces vascular endothelial growth factor expression and angiogenesis selectively at wound sites. Wound Repair Regen 8: 361–370. [DOI] [PubMed] [Google Scholar]
- Unemori EN, Pickford LB, Salles AL, Piercy CE, Grove BH, Erikson ME et al. (1996). Relaxin induces an extracellular matrix‐degrading phenotype in human lung fibroblasts in vitro and inhibits lung fibrosis in a murine model in vivo. J Clin Invest 98: 2739–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodstrcil LA, Shynlova O, Westcott K, Laker R, Simpson E, Wlodek ME et al. (2010). Progesterone withdrawal, and not increased circulating relaxin, mediates the decrease in myometrial relaxin receptor (RXFP1) expression in late gestation in rats. Biol Reprod 83: 825–832. [DOI] [PubMed] [Google Scholar]
- Voors AA, Dahlke M, Meyer S, Stepinska J, Gottlieb SS, Jones A et al. (2014). Renal hemodynamic effects of serelaxin in patients with chronic heart failure: a randomized, placebo‐controlled study. Circ Heart Fail 7: 994–1002. [DOI] [PubMed] [Google Scholar]
- Voors AA, Davison B, Felker GM, Ponikowski P, Unemori E, Cotter G et al. (2011). Early drop in systolic blood pressure and worsening renal function in acute heart failure: renal results of Pre‐RELAX‐AHF. Eur J Heart Fail 13: 961–967. [DOI] [PubMed] [Google Scholar]
- Webb DJ (1995). Evidence for endothelin‐1‐mediated vasoconstriction in severe chronic heart failure. Endothelin antagonism in heart failure. Circulation 92: 3372. [DOI] [PubMed] [Google Scholar]
- Williams EJ, Benyon RC, Trim N, Hadwin R, Grove BH, Arthur MJ et al. (2001). Relaxin inhibits effective collagen deposition by cultured hepatic stellate cells and decreases rat liver fibrosis in vivo. Gut 49: 577–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BC, Connell B, Saleh TM (2006). Relaxin‐induced reduction of infarct size in male rats receiving MCAO is dependent on nitric oxide synthesis and not estrogenic mechanisms. Neurosci Lett 393: 160–164. [DOI] [PubMed] [Google Scholar]
- Xu Q, Chakravorty A, Bathgate RA, Dart AM, Du XJ (2010). Relaxin therapy reverses large artery remodeling and improves arterial compliance in senescent spontaneously hypertensive rats. Hypertension 55: 1260–1266. [DOI] [PubMed] [Google Scholar]
- Xu Q, Lekgabe ED, Gao XM, Ming Z, Tregear GW, Dart AM et al. (2008). Endogenous relaxin does not affect chronic pressure overload‐induced cardiac hypertrophy and fibrosis. Endocrinology 149: 476–482. [DOI] [PubMed] [Google Scholar]
- Yan W, Chen J, Wiley AA, Crean‐Harris BD, Bartol FF, Bagnell CA (2008). Relaxin (RLX) and estrogen affect estrogen receptor alpha, vascular endothelial growth factor, and RLX receptor expression in the neonatal porcine uterus and cervix. Reproduction 135: 705–712. [DOI] [PubMed] [Google Scholar]
- Yao L, Agoulnik AI, Cooke PS, Meling DD, Sherwood OD (2009). Relative roles of the epithelial and stromal tissue compartment(s) in mediating the actions of relaxin and estrogen on cell proliferation and apoptosis in the mouse lower reproductive tract. Ann N Y Acad Sci 1160: 121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Kumagai H, Kohsaka T, Ikegaya N (2013). Relaxin protects against renal ischemia–reperfusion injury. Am J Physiol Renal Physiol 305: F1169–F1176. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Kumagai H, Suzuki A, Kobayashi N, Ohkawa S, Odamaki M et al. (2011). Relaxin ameliorates salt‐sensitive hypertension and renal fibrosis. Nephrol Dial Transplant 27: 2190–2197. [DOI] [PubMed] [Google Scholar]
- Zhang J, Qi YF, Geng B, Pan CS, Zhao J, Chen L et al. (2005). Effect of relaxin on myocardial ischemia injury induced by isoproterenol. Peptides 26: 1632–1639. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Liu SH, Erikson M, Lewis M, Unemori E (2002). Relaxin activates the MAP kinase pathway in human endometrial stromal cells. J Cell Biochem 85: 536–544. [DOI] [PubMed] [Google Scholar]
- Zhang X, Ma X, Zhao M, Zhang B, Chi J, Liu W et al. (2015). H2 and H3 relaxin inhibit high glucose‐induced apoptosis in neonatal rat ventricular myocytes. Biochimie 108: 59–67. [DOI] [PubMed] [Google Scholar]
- Zhao L, Roche PJ, Gunnersen JM, Hammond VE, Tregear GW, Wintour EM et al. (1999). Mice without a functional relaxin gene are unable to deliver milk to their pups. Endocrinology 140: 445–453. [DOI] [PubMed] [Google Scholar]
- Zhao L, Samuel CS, Tregear GW, Beck F, Wintour EM (2000). Collagen studies in late pregnant relaxin null mice. Biol Reprod 63: 697–703. [DOI] [PubMed] [Google Scholar]