

Abstract
The cortical actin cytoskeleton has been shown to be critical for the reorganization and heterogeneity of plasma membrane components of many cells, including T cells. Building on previous studies at the T cell immunological synapse, we quantitatively assess the structure and dynamics of this meshwork using live-cell superresolution fluorescence microscopy and spatio-temporal image correlation spectroscopy. We show for the first time, to our knowledge, that not only does the dense actin cortex flow in a retrograde fashion toward the synapse center, but the plasma membrane itself shows similar behavior. Furthermore, using two-color, live-cell superresolution cross-correlation spectroscopy, we demonstrate that the two flows are correlated and, in addition, we show that coupling may extend to the outer leaflet of the plasma membrane by examining the flow of GPI-anchored proteins. Finally, we demonstrate that the actin flow is correlated with a third component, α-actinin, which upon CRISPR knockout led to reduced plasma membrane flow directionality despite increased actin flow velocity. We hypothesize that this apparent cytoskeletal-membrane coupling could provide a mechanism for driving the observed retrograde flow of signaling molecules such as the TCR, Lck, ZAP70, LAT, and SLP76.
Introduction
Upon TCR engagement, T cells form a cell-cell junction with antigen-presenting cells termed the “immunological synapse”. Critical to synapse formation and stability is the cortical actin cytoskeleton (1) as well as signaling proteins residing within, and proximal to, the plasma membrane (PM). T cells actively remodel their actin cytoskeleton during synapse formation leading to cell spreading over the antigen-presenting cells and a characteristic reorganization of molecular components. After remodeling, the outer zone, or distal Supra-Molecular Activation Cluster (dSMAC), contains a dense meshwork of actin filaments that contrasts to the center of the synapse (central Supra-Molecular Activation Cluster; cSMAC), which is actin-sparse (2). Interestingly, in mature synapses, the actin cytoskeleton treads in a retrograde fashion from the periphery to the center without causing cell motility (3), suggesting that actin polymerization during and postsynapse formation may have distinct and overlapping roles.
Cortical actin structure and dynamics are believed to influence signaling at the immune synapse in several ways. Firstly, proximal T cell signaling intermediates are believed to be corralled (gathered together and confined) toward the central region at speeds similar to that of actin flow, where their signaling is downregulated (4, 5). Secondly, polymerized actin creates a dense meshwork that may act as a barrier for intracellular vesicle trafficking and membrane docking, spatially confining signaling to specific regions (5, 6). In natural killer cells, the actin mesh at the cSMAC is depleted, granting cytotoxic granule passage to the membrane where they fuse and are released into the synaptic cleft to kill target cells (7, 8, 9). Thirdly, it has been shown in numerous cell types that the cortical actin meshwork can influence protein diffusion and compartmentalization within the membrane; theories to explain this include the “picket fence” (10) and more recently “active composite” (11) models. This meshwork also regulates the nanoscale clustering of certain membrane proteins (12, 13).
The cortical actin meshwork and the plasma membrane may interact directly (14) or indirectly via transmembrane and scaffolding proteins (4, 15, 16) including TCR (17), which has been observed to cause modulation of protein lateral mobility including trapping, tethering, and corralling (18). These sites of interaction frequently occur at areas of high membrane lipid order (12, 19, 20). Indeed, highly ordered membrane domains have been observed at the synapse periphery where cortical actin is enriched (21). As disrupting the actin cytoskeleton disrupts membrane lipid order and declusters order-dependent signaling molecules such as Lck (22), these studies suggest a relationship between actin, the plasma membrane, and membrane-associated proteins.
To better observe and understand these interactions, superresolution microscopy has provided key advantages (23), including observations and mechanisms of control in nanometer-scale protein cluster formation at the membrane and remodeling of actin structures (7, 24, 25). Superresolution offers a number of significant advantages over conventional fluorescence microscopy. The actin mesh density is known to be extremely high at the synapse periphery, and therefore superresolution allows improved quantification as each pixel is not an ensemble of many individual fibers with diverse flow properties. Here, we employ single-molecule localization microscopy (SMLM) to assess the distribution of actin at the T cell synapse with nanometer precision. In addition, we have previously demonstrated that a combination of total internal reflection (TIRF), superresolution via structured illumination microscopy (SIM), and analysis by spatio-temporal image correlation spectroscopy (STICS (26); Fig. S1) can quantitatively assess cortical actin flow in T cell synapses (27, 28). This is possible even in the presence of a dense meshwork and can be achieved at high spatial and temporal resolutions.
Using these methods, together with their multicolor, cross-correlation variants, we quantitatively analyze F-actin and plasma membrane dynamics in live T cells during immunological synapse formation, to our knowledge, for the first time. After initial T cell spreading, F-actin and the peripheral regions of the plasma membrane itself flow in a retrograde manner, mirroring each other’s directionality and velocity. The overall flows are both dependent on actin polymerization, as demonstrated by the use of actin-modulating drugs. We show through the observation of flow of GPI-anchored proteins in the extracellular leaflet that both leaflets of the plasma membrane may exhibit flow.
There are numerous potential candidate molecules for mediating this correlation. One of these, α-actinin, cross links F-actin and plays a role in T cell synapse formation, as well as linking the cytoskeleton to transmembrane proteins and lipids (29). The α-actinin can interact simultaneously with the cytoskeleton and distinct membrane constituents such as β-integrin directly and indirectly through linkers such as talin and vinculin (30, 31). Via actin and protein binding domains (29, 32), we hypothesized that the scaffolding function of this protein might be in part responsible for the correlation of membrane and actin flow (33).
Using CRISPR knockouts, we show both actin and the plasma membrane exhibit reduced flow directionality. These actin-membrane linkers may help translate F-actin’s retrograde flow to the plasma membrane itself and these dynamics could hold functional importance during T cell stimulation for the translation of signaling proteins to the synapse center, balancing prolonged signaling with signal cessation and downregulation.
Materials and Methods
Tissue culture and transfection
Jurkat E6.1 T cells were cultured in RPMI + Glutamax media supplemented with 10% FBS and 1% PenStrep at 3–4 × 105/mL, and placed in a humidified 37°C environment of air + 5% CO2. Cells were resuspended in fresh media every 2 days and split the day before transfection to ensure cells were at logarithmic growth phase. For live-cell imaging, Jurkat T cells were transfected with the appropriate construct(s) through electroporation (Bio-Rad, Hercules, CA). For α-actinin experiments the plasmid-coded for ACTN1, transcription variant three. Transfected cells were incubated overnight in 10 mL equilibrated RPMI supplement. Primary human T cells were isolated from blood via PBMC and transfected using Amaxa (Lonza, Basel, Switzerland) electroporation.
CRISPR-Cas9 design and realization
The genomic sequence flanking the start codon of the ACTN1 gene (NCBI Gene: 87) was submitted to the Zhang Lab CRISPR Design Tool (http://crispr.mit.edu/) to find target sequences and design guide RNAs using the human genome (hg19) to check for off-target effects. Four CRISPR guide sequences were selected based on high quality aggregate scores (as determined by the Design Tool), locations either close to the start codon or within the first intron, and targeting either the sense or anti-sense strands. The four sgRNA sequences were each assembled into pSpCas9n(BB)-2A-GFP plasmid (PX461, a gift from Feng Zhang; Addgene plasmid No. 48140). These four plasmids were transfected into Jurkat E6.1 cells using an Amaxa Nucleofector IIb device (using 1 × 106 cells and 1 μg of each plasmid DNA in Nucleofector V solution, transfected using the built-in program X-005). Single-cell colonies were generated by serial dilution, expanded, and screened for ACNT1 expression by Western blot with monoclonal mouse anti-ACTN1 antibody (clone OTI7A4; Cambridge Bioscience, Cambridge, UK).
Generating immunological synapses
To engineer T cells to produce synapses, Labtek No. 1.5 8-well-chamber glass coverslips (Nunc, Langenselbold, Germany) were coated with αCD3 and αCD28 antibodies (Cambridge Bioscience and BD Biosciences, Franklin Lakes, NJ) at a concentration of 1 μg/mL. These were either left overnight at 4°C or placed in an incubator for 2 h. Before imaging, coverslips were gently washed with RT PBS to remove any antibody still in suspension and 200 μL of prewarmed HBSS + 20 mM HEPES added to each chamber.
SMLM: SMLM sample preparation
Integrating exchangeable single-molecule localization (IRIS) imaging is an SMLM technique that relies on capturing binding events of a probe to its target as it transiently shifts between bound and unbound states. Here a modified version of the F-actin binding peptide LifeAct, coupled to a small Atto655 molecule dye, was used (34). Fluorophore signal is captured once the molecule is bound and localized. Cells were washed and resuspended in HBSS at 3–4 × 105 cells/mL and dropped onto an antibody-coated coverslip that was returned to the incubator for 5 min to allow cells to form synapses. The HBSS was then removed from the wells and 200 μL of equilibrated cytoskeletal buffer (CB) + 4% PFA added to each well, coverslips were then returned to the incubator for 20 min to fix the cells. CB consisted of 10 mM MES at 6.1 pH, 5 mM MgCl, 5 mM EGTA, 150 mM NaCl, and 5 mM glucose, with a final pH of 7 and stored at 4°C until the day of imaging.
After fixation, cells were washed in CB and Image iT-FX Signal Enhancer (Thermo Fisher Scientific, Waltham, MA) was gently added until covering the wells, which was left for 30 min at room temperature to reduce nonspecific binding. Cells were then permeabilized with CB + 0.1% Triton-X for 5 min at room temperature, washed in CB, and then 0.5 nM of the LifeAct peptide conjugated to the Atto-655 probe was added, providing a binding time of 20–100 ms. Conjugation was achieved through an N-terminal cysteine via maleimide chemistry.
SMLM imaging
IRIS imaging was carried out on the N-STORM microscope (Nikon, Melville, NY) in TIRF mode, using a 100× 1.49 NA CFI Apochromat TIRF objective for a pixel size of 160 nm; to focus the laser intensity, a collimating lens was inserted. After locating a cell using bright-field illumination, the 647 nm laser was switched on and a region of interest (ROI) of 512 × 512 pixels selected. Frame rates of 50 ms were used to match the binding time of the peptide (34), with the laser power set to 50% (≈1.125 kW/cm2), which improved the signal to noise of bound fluorophores within the evanescent wave without increasing the background from unbound fluorophores away from the actin cortex. Electron multiplier (EM) gain was set to 300 with conversion gain set to 3.
SMLM analysis
Image stacks of 50,000–100,000 frames were analyzed using the Nikon Imaging Software analysis package (NIS, v.4.20; Nikon) with the overlapping peaks function enabled and a lower threshold of 3000 applied. Data were reconstructed using the NIS STORM package (Nikon). By scanning the image in 5 × 5 pixel subregions, local background was subtracted, with the signal above this localized by fitting an x and y Gaussian to find the centroid.
Candidate molecules were then filtered by brightness (min and max signal counts). The minimum and maximum molecule width was set to 200–400 nm, to reject noise. The localization precision was then theoretically calculated according to the principle by Thompson et al. (35). The mean of the resulting localization histogram was found to be 14 nm (n = 5). A drift correction was then applied that utilizes autocorrelation to correct for gradual displacement in frames over time. Average localization precision was extracted from the full list of emitter coordinates after filtering.
TIRF-SIM: TIRF-SIM sample preparation
For live-cell imaging, the coated coverslip was placed in the heated incubation chamber of the microscope. A quantity of 200 μL of transfected T cells were pelleted at 268× g for 20 s and resuspended in equilibrated HBSS + 20 mM HEPES before being carefully pipetted into one of the eight-well chambers at 3–4 × 105 cells/mL. Then, 5–10 min after cells landed on the coverslip, TIRF-SIM acquisition of LifeAct-GFP-expressing cells was carried out for 1 min; laser illumination was set to 10% (≈16.5 μW/cm2).
For PM imaging, cells were stained for 5–10 min with 5 μM DiO or DiI added directly to the media before being pelleted at 268× g for 20 s, and resuspended in an equal volume of equilibrated HBSS + 20 mM HEPES. Cells were then immediately pipetted onto the coverslip for imaging.
For drug treatments, cells were added to the coverslips and allowed to form synapses, as above. Dosages were then added directly to the coverslip chamber. For cytochalasin-D and jasplakinolide, final concentrations were 2 μM while 7-ketocholesterol and blebbistatin were added at a final concentration of 10 and 50 μM, respectively. Images were acquired as below.
An MTT assay was performed to establish that drug concentrations used were not cytotoxic; 5 × 106 primary human T cells were incubated with MTT in 500 μL standard media control conditions, with 50:50 v/v DMSO, or the corresponding drug treatments for 1 h at 37°C. Cells were then lysed in 50:50 v/v DMSO before solution absorbance was recorded at 595 nm, using a Victor 1420 plate reader (EG&G Wallac, Gaithersburg, MD). Conditions were performed in triplicate.
TIRF-SIM imaging
For live-cell imaging, the microscope’s incubation chamber (Tokai Hit, Shizuoka-ken, Japan) and heated lens collar were turned on to 37°C >2 h before imaging. For single-channel TIRF-SIM on the N-SIM system (Nikon), 10% 488 nm excitation was used, which equates to 16.5 μW in wide-field mode, with a 100 × 1.49 NA CFI Apochromat TIRF objective. For blebbistatin imaging, the 561-nm channel was used due to blebbistatin’s photoinactivation by 488 nm light (36). After selecting an ROI of 512 × 512, EM gain was set to 300 with conversion gain set to 1. As the microscope relies on a physically shifting illumination pattern to achieve the three orientations and three phase shifts to create a reconstructed image, 1 frame/s was achieved when using a 50 ms frame acquisition.
Two-channel TIRF-SIM images were obtained on an Axio Observer Z1 (Carl Zeiss, Oberkochen, Germany) fitted with a spatial light modulator, producing the structured illumination pattern (37). A 100 × 1.49 NA objective (Olympus, Tokyo, Japan) was used, with 488- and 561-nm laser excitation and the two-channel signal collected serially on two separate sCMOS cameras; to ensure minimal cross talk, a narrow-band GFP filter (515/30 nm) and long-pass orange/red filter (568 nm) were used. A heated chamber including humidifier (Okolab, Pozzuoli, Italy) was set to 37°C before imaging. Images were acquired using the same exposure time settings as the single-channel datasets.
To minimize drift during single- and two-channel live cell imaging, coverslips were placed in the heated chamber for ≈10 min before imaging to ensure temperature equilibrium. Z-plane drift was also reduced for single-channel experiments through the Perfect Focus System (Nikon).
TIRF-SIM image reconstruction
For reconstruction of the single-channel data, the software Analyze (v4.20.01; Nikon) was used, achieving a pixel size of 30 nm. The illumination modulation contrast and high-resolution noise suppression were both set to the default of 1. Illumination modulation contrast distinguishes the stripes from the structured illumination pattern, whereas high-resolution noise suppression can crop the higher resolution information from the forward Fourier transform; for the value of 1, this led to most of the higher frequency information remaining in the final image, for improved resolution.
Two-channel SIM data were reconstructed with a custom-written program (38) and the reconstructed datasets were aligned using an algorithm in the tool set Priism (http://msg.ucsf.edu/IVE).
TIRF-SIM analysis with STIC(C)S
STICS theory has been described in Hebert et al. (26). For our data, we used the tool STICSGUI, v0.29 (Wiseman Research Group, McGill University). For single-channel F-actin datasets, an analysis subregion size of 8 × 8 pixels, with a shift of 1 pixel between subregions, was applied. To remove immobile and diffusive fluorescent populations before analysis, we chose the immobile filter, set to 21 frames, removing fluorescence signal from the correlation function if it remained static for this number of frames. A polygon ROI of the cell periphery was then selected. After running the software, data were saved and subregion velocity information plotted. For single-channel PM and two-channel data, a subregion size of 16 × 16 pixels and a subregion shift of 4 pixels was used to improve the reliability of the more homogeneous signal derived from the PM.
For directionality analysis, a seed point was manually chosen in the central region of the cell; each vector was then assessed against this seed point to deduce its angle of deviation, giving directional information.
Statistical analysis
Analysis was carried out using a two-tailed Student’s t-test using GraphPad Prism version 5.00 for Windows (GraphPad Software, La Jolla, CA). Data shown in text was plotted as mean ± standard deviation.
Results
We first imaged actin using the LifeAct probe, with the IRIS approach (34), giving a mean localization precision of 12 nm. Jurkat T cell synapses were formed using a stimulatory coverslip (Fig. S2), as described in Ashdown et al. (28).
After stimulation by a coverslip coated with antibodies against CD3 and CD28, the data indicated a dense meshwork of actin at the synapse periphery but a relatively actin sparse region toward the center, which contained numerous long actin fibers. Using live-cell superresolution TIRF-SIM and cells transfected with LifeAct-GFP, we imaged the molecular flow of cortical F-actin during T cell immunological synapse formation against an antibody-coated stimulatory coverslip as with fixed cell experiments. Using this system, we were able to generate reconstructed superresolution images of fluorescent beads or actin at spatial resolutions of ≤120 and 100 nm, respectively, in 75-nm optical sections at one frame/s (Fig. S2 b; Movie S1). The velocity and directionality of F-actin dynamics at the observed dense, peripheral regions (Fig. 1 a) were quantified using STICS (26) (Fig. 1 b). In line with previous studies (3, 4, 17), actin flow had an average velocity of 1.63 ± 0.46 μm/min (Fig. 1 c) and was confirmed in primary human T cells 1.86 ± 0.26 μm/min (Fig. S3, a–e). Actin flow in Jurkat T cells was predominantly retrograde in nature, with 72.9% of vectors within a 90° cone directed toward the center (Fig. 1 d).
Figure 1.

F-actin flow characterized using TIRF-SIM and STICS analysis. (a) Images show characterization of F-actin flow in T cells; shown are representative TIRF-SIM images (a, e, i, and m) of cells 5–10 min after contact with a stimulatory coverslip and after different treatments (labeled bottom left of images). ROIs within the blue boxes are to the right (b, f, j, and n), showing the output vector maps, from STICS analysis, representing ≈1 μm2. Scale bars, 5 μm. (b) Graphs show STICS outputs of normalized F-actin flow; any changes after drug treatment in either subregion velocity (c, g, k, and o) or directionality (d, h, l, and p) are plotted. Vector angles are calculated against a seed point (cell center) as shown in (a) as a red dot, where a vector oriented toward a seed point gave an angle of 0° (solid arrow), perpendicular to 90° (dotted arrow), and away from 180° (dashed arrow). (Controls: 1.63 ± 0.46 μm, n = 24. Cytochalasin-D (C-D): 0.99 ± 0.27 μm, n = 10. Jasplakinolide (Jasp): 2.07 ± 1.06 μm, n = 27. 7-ketocholesterol (7KC): 1.61 ± 0.49 μm, n = 24.) To see this figure in color, go online.
To ensure that any high-resolution noise associated with SIM images was not contributing to STICS output vectors, spatial filters were applied to actin images during the reconstruction process, degrading the resolution to conventional microscopy standards of ∼250 nm and thereby removing these artifacts. Flow speeds and directionality are not significantly different between these conditions (Fig. S4, a–c), although some directional information was lost when correlating the lower resolution datasets.
Additionally STICS analysis of standard TIRF images (Fig. S4 d) was quantified using the same absolute subregion size values. This generated spurious vector directionalities (Fig. S4 e), indicating that TIRF-SIM datasets improve the validity of STICS analysis at these length-scales.
Next, drug treatments that affect polymerization rates were used and were shown to disrupt both F-actin flow velocities and directionality. Cytochalasin-D (Fig. 1, e and f), which reduces the rate of polymerization by capping the growing end of F-actin, was found to reduce the velocity of retrograde flow to 0.99 ± 0.27 μm/min (Fig. 1 g, p < 0.001), while also scrambling the directionality (Fig. 1 h, p = 0.01). Jasplakinolide, which increases polymerization and stabilizes actin filaments (Fig. 1, i and j) had no significant effect on velocity (Fig. 1 k, p = 0.06), but did perturb directionality (Fig. 1 l, p < 0.01). As jasplakinolide increases actin polymerization universally, but in an uncontrolled manner, the scrambling of the directionality demonstrates the system relies on balancing polymerization and depolymerization in a regulated fashion to achieve the ordered retrograde flow during synapse formation.
To investigate whether membrane lipid order had an effect on F-actin retrograde flow, we reduced lipid order using the cholesterol analog 7-ketocholesterol (7KC), as previously demonstrated for T cells (39) (Fig. 1, m and n). F-actin flow velocities (Fig. 1 o) and directionality (Fig. 1 p) were not significantly altered (p = 0.88 and 0.32). Finally, we treated cells using blebbistatin, which inhibits Myosin II motors. In this case, no difference in flow velocities or directionality was observed between the treated cells and control cells (Fig. S3 f). The fact that inhibiting Myosin II motors did not slow actin retrograde flow velocity at the dSMAC strongly suggests the actin flow in this region is driven primarily by actin polymerization. These findings agree with previous studies using coated glass coverslips and Jurkat T cells, where flow change was not observed in the dSMAC after Myosin II inhibition, but instead disrupted the actin-arcs of the pSMAC (3, 40).
To clarify the changes all drug treatments had on actin retrograde flow, we plotted the percentage change compared to control conditions (Fig. S5). Positive (negative) y-axis values indicate the cells undergoing drug treatment have a greater (fewer) number of vectors at that range of velocities or angles. As expected, cytochalasin-D reduced F-actin velocity (Fig. S5, a and d), while jasplakinolide had no significant effect (Fig. S5, b and d). 7KC showed no significant differences, indicating actin flow is not dependent on membrane lipid order (Fig. S5, c and d). These treatment conditions were shown not to adversely affect cell viability (Fig. S6 a). It was also shown that T cell F-actin architecture (Fig. S6 b) remained consistent both 1 and 5 min after a 1-min imaging time-course (Fig. S6, c and d) compared to T cells imaged 1 min before and after no laser exposure (Fig. S6, e and f).
LifeAct labels F-actin by transiently binding to multiple monomers, potentially stabilizing the labeled structure. As such, we repeated the experiments of Fig. 1 with monomeric GFP-actin to label actin structures without this transient binding event (Fig. S7). The results were not statistically different compared to the LifeAct results for both control and drug treatments, with CD significantly slowing actin flow (p < 0.003) and disrupting actin directionality (p = 0.0005), while jasplakinolide-treated cells were not significantly different for either flow speed (0.07) or direction (p = 0.1) versus controls.
Studies have shown that the cortical actin meshwork and the plasma membrane may interact directly (14) or indirectly via transmembrane and scaffolding proteins (4, 15, 16). We therefore hypothesized that the flow of the cortical actin mesh might be coupled to the membrane, causing retrograde membrane flow. Cells were stained with the lipophilic membrane dye DiO, synapses were formed on activating coverslips with cells imaged by TIRF-SIM (Movie S2), and membrane flow was quantified by STICS. Our data indicated that the membrane also flows in a retrograde fashion (63.3% inward) with a velocity comparable to that of actin (Fig. 2, a and b), whereas in the actin-poor synapse center, vector directionality exhibited near-homogenous distribution across all angles (Fig. 2, a and b) with 30.7% inward. Plotting individual cell means demonstrated flow directionality was significantly altered between the synapse periphery and center (Fig. 2 c, p < 0.0001).
Figure 2.

Characterization of plasma membrane flow in T cells imaged with TIRF-SIM and analyzed by STICS. (a) The plasma membrane was labeled with the lipophilic dye DiO, and imaged 5 min after contact with an antibody-coated coverslip. Shown is a representative TIRF-SIM image with the magnified ROI within the blue boxes indicating the output vector map from STICS analysis of 1) synapse periphery flow and 2) synapse center flow. Scale bars, 5 μm. (b) Histograms showing the directionality of plasma membrane flow at the periphery (right) and center (left) normalized to total number of vectors. (c) Scatterplots of cell averages, showing speeds for synapse peripheries (4.00 ± 1.61 μm), synapse centers (3.55 ± 1.09 μm), and synapse directionality at the periphery (56.07 ± 17.02°) and center (82.42 ± 12.03°) (n.s., nonsignificant; ∗∗∗∗, p< 0.0001; n = 16). To see this figure in color, go online.
To ensure the detected flow within the membrane was not a SIM-artifact, homogeneously distributed plate-bound fluorophores were imaged and analyzed, showing reduced vector numbers (i.e., reduced detected flow) and similar random characteristics to the cell center regions (Fig. S8). Together, these results demonstrate STICS does not correlate any artifactual features of TIRF-SIM, with fewer vectors generated from samples with static or diffusive signal.
Interference reflection microscopy (IRM), which is a method for detecting the proximity of the membrane to the coverslip, indicates that this flow may be in the form of membrane ruffles. Darker pixels represent regions of the sample closer to the coverslip and are seen progressing with a wavelike motion toward the synapse center. These IRM dynamics exhibit nonsignificant flow speeds versus the membrane (p = 0.19), or actin imaged with GFP-actin (p = 0.43) or LifeAct-GFP (p = 0.24) (Fig. S9 a and b; Movie S3). To our knowledge, this is the first demonstration of retrograde flow of the plasma membrane itself in this system.
To better understand the relationship between F-actin flow and the flow of the plasma membrane, we extended the single-color imaging method to multicolor superresolution microscopy in the form of two-channel TIRF-SIM and analyzed the data using a cross-correlation variant STICCS (41). Using LifeAct-GFP and the red-emitting membrane dye DiI, we were able to detect, image (Fig. 3 a; Movie S4), and analyze actin and the membrane simultaneously. Results are expressed as a ratio of the velocities from channels A and B. Directionality scores can range from −1 if vector A is oriented opposite to vector B, whereas a score of +1 demonstrates vectors are perfectly aligned.
Figure 3.

STICCS analysis of two-channel TIRF-SIM data. (a) Jurkat T cells were imaged using two-color TIRF-SIM with F-actin labeled with EGFP-LifeAct and the plasma membrane stained with DiI. Shown are the single-channel correlation outputs for actin and the plasma membrane (middle images) and the cross-correlation function between the two channels (right). Scale bars, 5 μm. (b) Cross-correlation heatmaps from a toroidal ROI at the dSMAC of the cell from (a) show regions of greater and lesser correlation for subregion velocities (left) and directionality (right). For velocities this ratiometric measure indicates the difference between vectors where channel 1 may be flowing slower than channel 2 (giving 0), the same speed (giving +1), or flowing twice as fast (+2). Directionality is represented in a similar way, but as vector angles are established with reference to a seed point and are bound to a discrete range between 0 and 180, vectors pointing in opposite directions give −1 and those pointing in the same direction give +1. (c) Scatter plots of whole cell mean velocities (left = 1.02 ± 0.39) and directionalities (right = 0.57 ± 0.27; n = 18) are shown. To see this figure in color, go online.
F-actin and plasma membrane flow velocities and directionality were correlated at the immune synapse periphery, indicating coupling between the two flows (Fig. 3, b and c). The mean ratio of the flow velocities at the periphery was 1.02 ± 0.39 (Fig. 3 c, left plot), indicating the flows have a similar velocity. The correlation between the directionalities was also high, 0.57 ± 0.27 (where “1” indicates collinear flow; Fig. 3 c, right plot), whereas it was observed that the correlations were reduced as the flows moved from the periphery toward the synapse center, possibly indicating a decoupling of actin from the plasma membrane.
We next imaged cells transfected with both LifeAct-mCherry and GPI-GFP, an outer-leaflet-residing marker within the plasma membrane that has been shown in vitro to mirror certain lipids of the inner leaflet in an actin-dependent manner (42). The correlated flow showed similar results to the LifeAct-DiI dye imaging for both velocity (1.05 ± 0.35) and directionality (0.39 ± 0.13; Fig. S10; Movie S5). These results may demonstrate that outer-leaflet proteins like GPI-anchored proteins may also correlate with actin flow, in agreement with the active composite model. Finally, as a negative control, analysis of cytosolic-GFP did not show any significant flow, and when correlated with the membrane dye DiI showed no correlated velocities (1.31 ± 0.52) or directional correlation (0.10 ± 0.13; Fig. S11).
After these results, we hypothesized that the protein α-actinin, which can bind to several transmembrane proteins as well as having the ability to cross-link actin fibers (33), might act as a linker between cortical actin and plasma membrane components. The α-actinin mediates F-actin dynamics through transmembrane proteins, including those found in T cell membrane domains containing CD3 (29), and was chosen because it is known to bind to actin and transmembrane proteins such as integrins and causes clustering of transmembrane proteins in lymphocytes (29, 30, 31, 43). It is also enriched at the immunological synapse, involved in actin rearrangement, and acts as a barrier to HIV infection (29).
Again, we used two-color, live-cell superresolution microscopy and quantitative cross-correlation STICCS analysis to investigate the correlation between actin flow and that of α-actinin (Fig. 4; Movie S6). The α-actinin-mCherry was transiently expressed in Jurkat T cells through electroporation, using a plasmid encoding for ACTN1. While overexpressing actin cross-linker proteins could change nanoscale behaviors of actin, actin continued to flow in a retrograde manner akin to untransfected cells.
Figure 4.

STICCS analysis of two-channel TIRF-SIM data. (a) Jurkat T cells were imaged using two-color TIRF-SIM with F-actin labeled with EGFP-LifeAct and α-actinin labeled with mCherry (middle two images) and cross-correlation outputs (right). Scale bars, 5 μm. (b) Cross-correlation heatmaps from a toroidal ROI at the dSMAC of the cell of velocities (left) and directionality (right) are shown, as described in Fig. 3. (c) Scatter plots of whole cell mean velocities (left = 0.78 ± 0.19) and directionalities (right = 0.92 ± 0.07) are given. (d) Given here is a radial profile plot of single cell, showing normalized fluorescent intensity of actin and α-actinin distribution versus distance from cell center (n = 19). To see this figure in color, go online.
Our results showed almost perfect cross correlation between actin and α-actinin, indicating highly similar flows (Fig. 4, b and c). The α-actinin was most prominent at the synapse periphery (Fig. 4 d)—where the correlation between the cytoskeleton flow and that of the plasma membrane was strongest.
To investigate the role of α-actinin further, we generated Jurkat T cell CRISPR knockouts; after confirmation by Western blot that α-actinin was knocked-out (Fig. 5 a), these α-actinin−/− cells were then imaged while forming synapses using TIRF-SIM. We found knocking out this protein significantly reduces the orderedness of cortical actin retrograde flow, with both flow speeds and mean angles increasing (Fig. S12). These results demonstrate the loss of the cross-linking and stabilization function of α-actinin makes it easier for actin fibers to be transported inwards by polymerization (higher velocity), but that these then lack organization with the rest of the cortical mesh (loss of directionality).
Figure 5.

Image correlation of the plasma membrane by TIRF-SIM and STICS analysis in Jurkat T cell forming immunological synapses after α-actinin CRISPR knockout. (a) Image and quantification of α-actinin knockout were normalized to wild-type Jurkat T cells; Western blots show both anti-ACTN1 and anti-GAPDH as a control. (b) The plasma membrane was labeled with the lipophilic dye DiO, and imaged 5 min after contact with an antibody-coated coverslip. Scale bars, 5 μm. (c) STICS output vector maps are shown. (d) Plots of the cell means for membrane flow velocity (2.8 ± 0.67 μm) and membrane flow directionality (80.4 ± 19.7°) are shown. Histograms show the normalized (e) speed within the dSMAC and (f) directionality. n = 16 (controls) and 8 (α-actinin−/−); n.s., nonsignificant; ∗∗p < 0.005. To see this figure in color, go online.
These results demonstrate the cross-linking and stabilizing ability of α-actinin may function as a moderator of this retrograde flow, with unlinked actin fibers potentially leading to increased random distribution and polymerization at the immunological synapse, seen here as higher velocities and scrambled directionality.
Because we hypothesize that α-actinin was mediating observed actin flow to the plasma membrane, we next imaged membrane flow in α-actinin−/− cells (Fig. 5 b) with quantification (Fig. 5 c) and comparison with control cells (Fig. 5 d). This revealed that whereas flow speeds were not significantly reduced (Fig. 5 e, p = 0.06), flow directionality was scrambled (Fig. 5 f, p < 0.005) with 39.9% of vectors exhibiting retrograde flow (n = 8). Whereas in control cells, actin and membrane flows were the same (Fig. 9 b), in the α-actinin−/− cells actin flow velocity is increased although the membrane flow is not, implying a loss of coupling between the two structures.
Discussion
In this study, we have applied STICS and STICCS analysis to superresolution TIRF-SIM data, examining the flow velocities and directionality of the cortical actin cytoskeleton and the plasma membrane at the T cell immunological synapse. This extends previous studies using conventional microscopy (3, 4, 17), resolving dynamic details of the dense actin meshwork with quantification, at scales not previously possible. The use of superresolution imaging is an important advance: the cortical actin mesh is extremely dense at the synapse periphery; therefore, the smaller resolution elements of SIM reduce the ensemble averaging of many independent fibers that occurs with conventional resolution microscopy. Additionally, the correlation function varies approximately as the square root of the number of fluctuations sampled and the number of fluorescent entities within the subregion is inversely proportional to the correlation function amplitude. Therefore, for the same subregion size, the use of superresolution microscopy produces correlation functions with enhanced signal-to-noise. Coupling this mode of imaging with STICS allowed the quantification of flow velocities and directions over an area of the membrane that is not possible in the kymograph analysis used previously, which only computes velocities along a line profile. This is especially important for computing the angle of flow to the center position and therefore quantifying how retrograde this flow is.
Under control conditions, F-actin exhibited retrograde flow only at the periphery of the immune synapse. When actin polymerization was modulated by drug treatments, this flow could be slowed and the highly orientated directionality perturbed. The plasma membrane, when imaged under control conditions, exhibited similar retrograde flow to that of F-actin, and this flow was evident when observing both LifeAct-GFP and actin-GFP. Cytochalasin-D, which inhibits actin polymerization, slowed actin retrograde flow whereas blebbistatin, which inhibits Myosin II motors, had no effect. This strongly indicates the flow is driven primarily by actin polymerization in the dSMAC. To our knowledge, this is the first time that such a net flow of membrane lipid components themselves has been observed.
Using two-color superresolution TIRF-SIM and cross-correlation analysis, it was found that the flows of the cortical actin and the plasma membrane are correlated. This analysis therefore demonstrates a potential linkage between the cortical actin meshwork and the plasma membrane, including GPI-anchored proteins in the outer leaflet.
The correlation between the two flows is apparent in superresolved, nanoscale subregions of the membrane and is therefore not due to the microscale movement of the two structures. IRM data showed that the fast moving F-actin cortex at the dSMAC may drive inward-flowing membrane ruffles; such ruffling has previously been observed (44).
Correlation of the actin and membrane flow was highest in areas enriched with the actin binding protein α-actinin whose retrograde flow was also highly correlated with that of actin. We therefore hypothesize that α-actinin may operate as a linker between the two flowing populations of actin and the plasma membrane. In addition to its actin binding domains, α-actinin can also interact directly with transmembrane proteins (β-integrins) and indirectly (via vinculin and talin) (33), all of which are expressed in T cells. To investigate this further, we used CRISPR gene-editing technology to produce α-actinin knockout T cells. These showed increased actin flow velocities but scrambled actin directionality. This is consistent with the cross-linking ability of α-actinin which, when lost, allows more rapid but uncoordinated polymerization-driven retrograde flow of the actin fibers. Membrane flow was also scrambled, although it did not show a significant decrease in velocity, which, given that actin flow was increased, is consistent with α-actinin acting as a mediator between the two systems. Beyond findings presented here, other actin-membrane linker candidates, such as Ezrin, Radixin, Moesin proteins and Fascin, may also warrant investigation.
Here it is demonstrated the cross linking of actin directly beneath the plasma membrane may also enhance the nonspecific interaction of the cytoskeleton with the membrane, thereby enhancing the picket-fence model of cytoskeletal-membrane interactions (10). This may occur within ordered phase membrane microdomains that are known to facilitate actin-membrane interactions (19) and have been observed to be enriched at the T cell synapse periphery (21).
When characterizing membrane dynamics, particularly for transmembrane proteins, molecules with large extracellular domains, protein segregation, and clustering, it is important to highlight there are potential differences found between the optically favorable but rigid antibody-coated coverslips used in this study, and supported lipid bilayer or cell-cell interactions used by others (reviewed in (45)). When studying transmembrane proteins it is important to remember these occupy both dynamic and quasi-static populations based on their proximity to the cytoskeleton and their ligands. Moreover, morphological changes in three dimensions as seen here with IRM imaging may be different when cells encounter other cells rather than coverslips.
While the role fulfilled by the cytoskeleton in regulating cell shape, organization, and polarization is well understood (46, 47, 48), the role of the plasma membrane, beyond a platform for signal transduction, is less well known. Some reports suggest cell migration does not lead to a flowing of the membrane (49), indicating a slipping of the F-actin cortex relative to the membrane, rather than creating static adhesions that can drive forces through the membrane. However, it is known that many of the membrane and membrane-proximal proteins that are required for the regulation of T cell activation demonstrate microclustering and retrograde flow at the synapse (1, 2, 5, 50). We therefore hypothesize that this might also be facilitated by the retrograde flow of the plasma membrane itself, which is driven by the flow of the cortical actin cytoskeleton.
Author Contributions
G.W.A. and D.M.O. designed the experiments. G.W.A., G.L.B., and D.J.W. carried out the cell culture, transfections, imaging, and analysis. D.J.W. carried out the CRISPR knockouts. E.P., P.W.W., and M.H. provided STICS analysis code and support. R.P. carried out the IRIS imaging and reconstruction. H.E. supplied the IRIS probe. L.S. provided support for the two-channel SIM imaging. G.W.A., G.L.B., D.J.W., and D.M.O. wrote the manuscript.
Acknowledgments
The authors thank Satya Khuon at the Advanced Imaging Center, Janelia Research Campus for her support, along with Debora Keller of the FILM Imaging Centre at Imperial College London for her help with the IRM imaging. Single-channel TIRF-SIM microscopy was carried out in the Nikon Imaging Centre, King’s College London. The α-actinin-mCherry plasmid was a gift from the Davidson Lab.
D.M.O. acknowledges European Research Council grant No. 337187 and Marie-Curie Career Integration grant No. 334303. P.W.W. acknowledges the Natural Sciences and Engineering Research Council of Canada’s Discovery Grant program. Two-channel TIRF-SIM microscopy was performed at the Advanced Imaging Center, Janelia Research Campus, jointly sponsored by the Howard Hughes Medical Institute and the Gordon and Betty Moore Foundation.
Editor: Anne Kenworthy.
Footnotes
Twelve figures and eight movies are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(17)30291-6.
Supporting Material
F-actin is labeled by the actin binding peptide LifeAct-GFP. Scale bar, 5 μm.
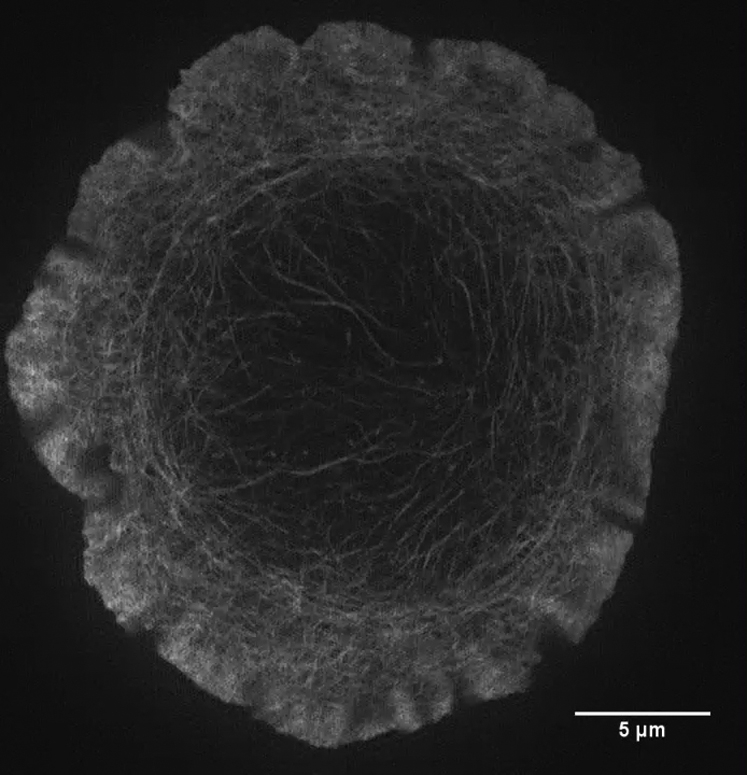{kind=link}
Cell is labeled with DiO membrane dye. Scale bar, 5 μm.
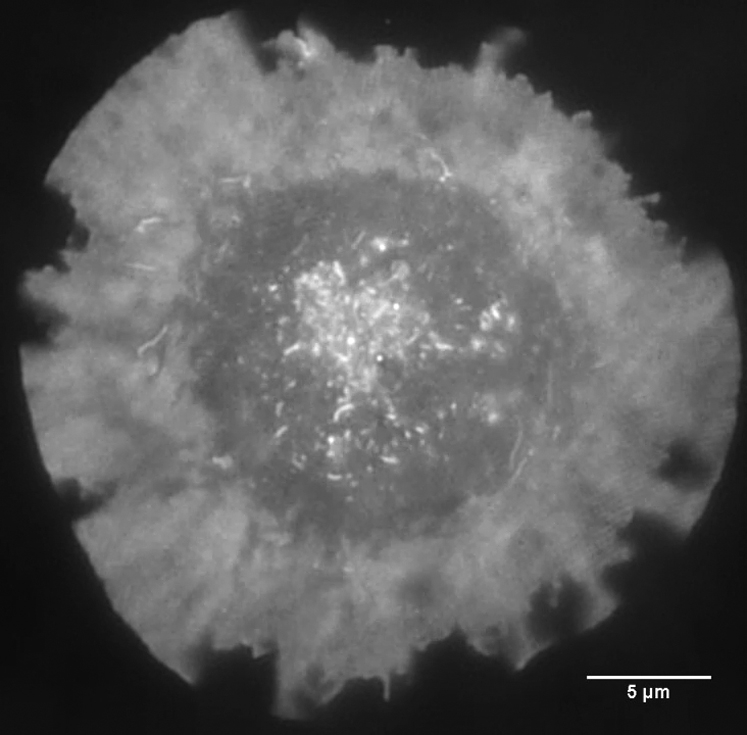{kind=link}
Lighter areas indicate cell components further from the coverslip, darker areas closer to the coverslip. Scale bar, 5 μm.
{kind=link}
F-actin is shown on the left, and the membrane dye DiI is shown on the right. Scale bar, 5 μm.
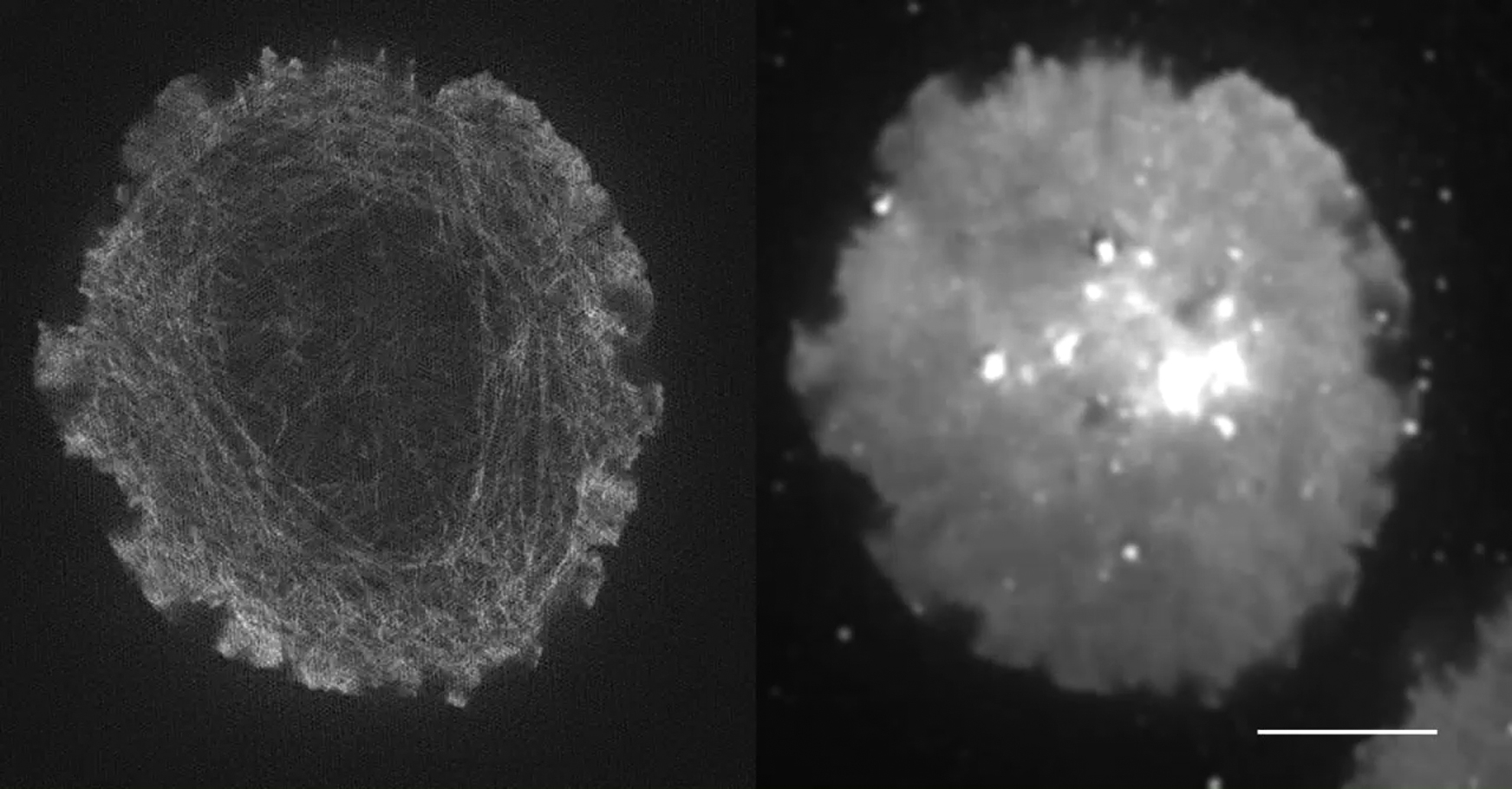{kind=link}
F-actin (left) is labeled with LifeAct-GFP and the PM (right) is labeled with GPI-GFP. Scale bar, 5 μm.
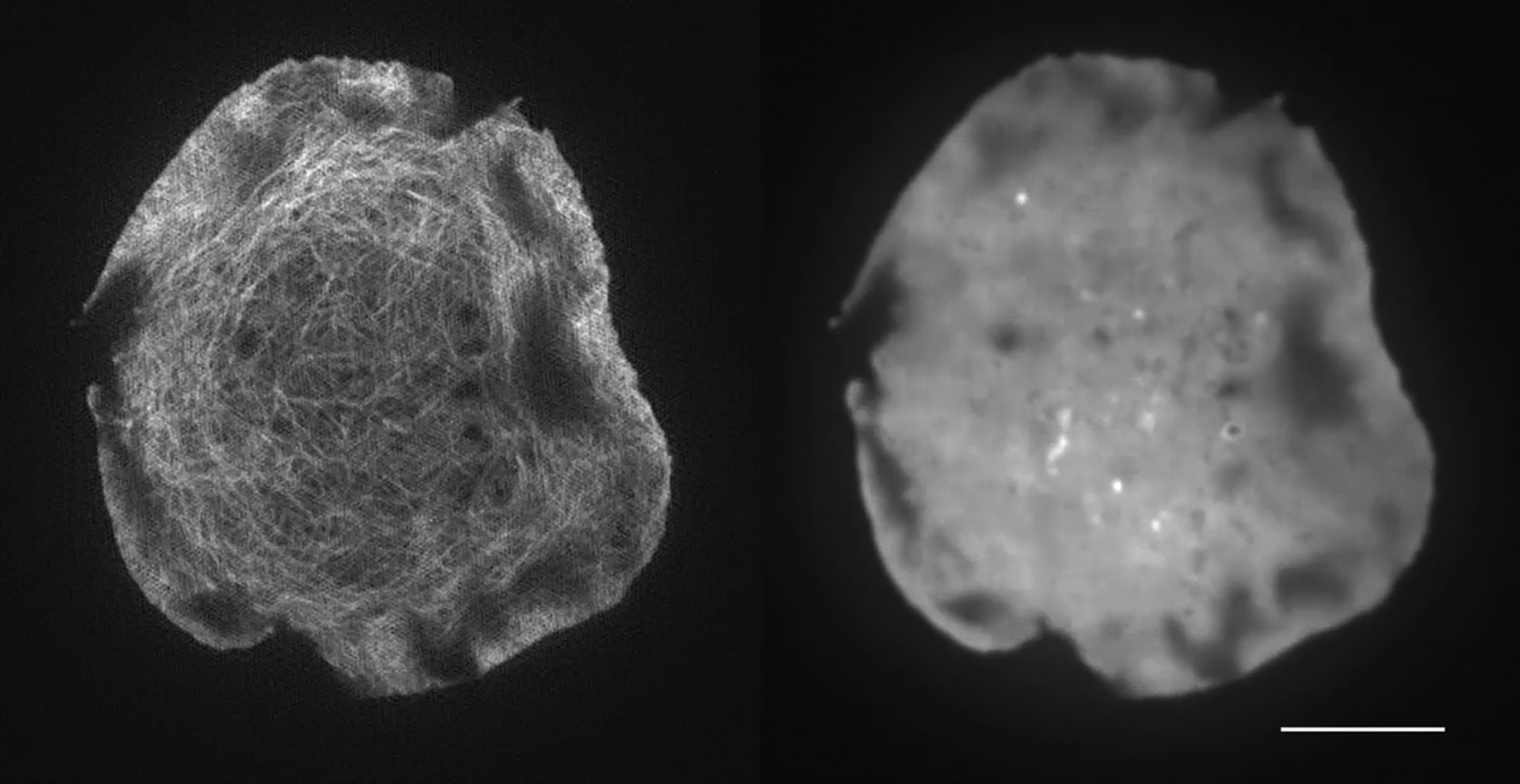{kind=link}
F-actin (left) is labeled with LifeAct-GFP and α-actinin (right) is labeled with mCherry. Scale bar, 5 μm.
{kind=link}
F-actin is labeled by the actin binding peptide LifeAct-GFP. Scale bar, 5 μm.
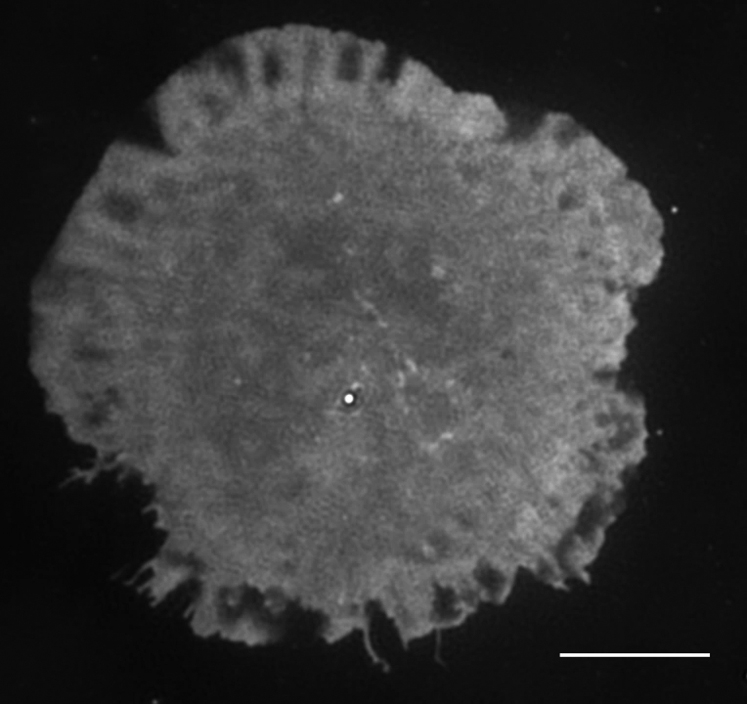{kind=link}
Cell is labeled with DiO membrane dye. Scale bar, 5 μm.
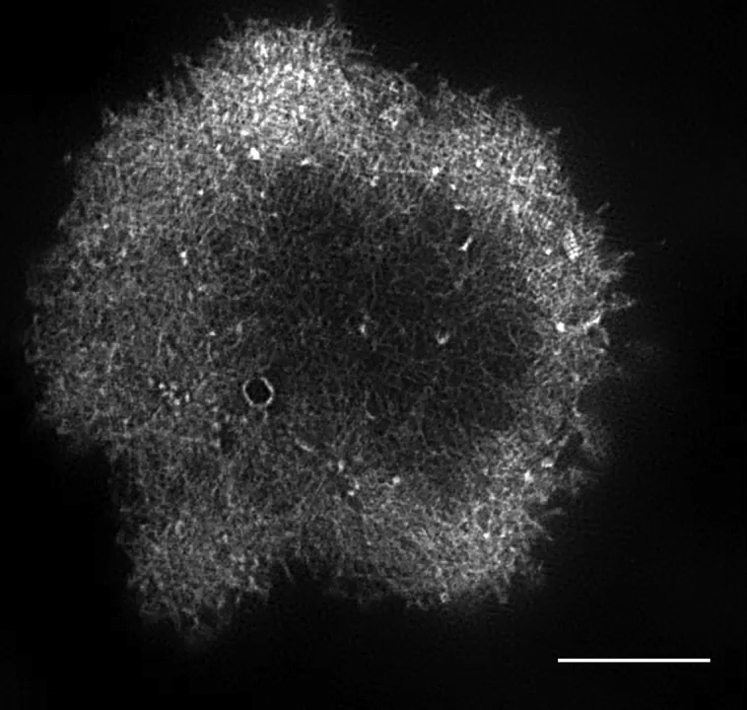{kind=link}
References
- 1.Campi G., Varma R., Dustin M.L. Actin and agonist MHC-peptide complex-dependent T cell receptor microclusters as scaffolds for signaling. J. Exp. Med. 2005;202:1031–1036. doi: 10.1084/jem.20051182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beemiller P., Jacobelli J., Krummel M.F. Integration of the movement of signaling microclusters with cellular motility in immunological synapses. Nat. Immunol. 2012;13:787–795. doi: 10.1038/ni.2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Babich A., Li S., Burkhardt J.K. F-actin polymerization and retrograde flow drive sustained PLCγ1 signaling during T cell activation. J. Cell Biol. 2012;197:775–787. doi: 10.1083/jcb.201201018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yi J., Wu X.S., Hammer J.A., 3rd Actin retrograde flow and actomyosin II arc contraction drive receptor cluster dynamics at the immunological synapse in Jurkat T cells. Mol. Biol. Cell. 2012;23:834–852. doi: 10.1091/mbc.E11-08-0731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Varma R., Campi G., Dustin M.L. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity. 2006;25:117–127. doi: 10.1016/j.immuni.2006.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ritter A.T., Asano Y., Griffiths G.M. Actin depletion initiates events leading to granule secretion at the immunological synapse. Immunity. 2015;42:864–876. doi: 10.1016/j.immuni.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown A.C.N., Oddos S., Davis D.M. Remodelling of cortical actin where lytic granules dock at natural killer cell immune synapses revealed by super-resolution microscopy. PLoS Biol. 2011;9:e1001152. doi: 10.1371/journal.pbio.1001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rak G.D., Mace E.M., Orange J.S. Natural killer cell lytic granule secretion occurs through a pervasive actin network at the immune synapse. PLoS Biol. 2011;9:e1001151. doi: 10.1371/journal.pbio.1001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mace E.M., Orange J.S. Lytic immune synapse function requires filamentous actin deconstruction by Coronin 1A. Proc. Natl. Acad. Sci. USA. 2014;111:6708–6713. doi: 10.1073/pnas.1314975111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kusumi A., Ike H., Fujiwara T. Single-molecule tracking of membrane molecules: plasma membrane compartmentalization and dynamic assembly of raft-philic signaling molecules. Semin. Immunol. 2005;17:3–21. doi: 10.1016/j.smim.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 11.Gowrishankar K., Ghosh S., Rao M. Active remodeling of cortical actin regulates spatiotemporal organization of cell surface molecules. Cell. 2012;149:1353–1367. doi: 10.1016/j.cell.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 12.Chichili G.R., Rodgers W. Clustering of membrane raft proteins by the actin cytoskeleton. J. Biol. Chem. 2007;282:36682–36691. doi: 10.1074/jbc.M702959200. [DOI] [PubMed] [Google Scholar]
- 13.Lillemeier B.F., Pfeiffer J.R., Davis M.M. Plasma membrane-associated proteins are clustered into islands attached to the cytoskeleton. Proc. Natl. Acad. Sci. USA. 2006;103:18992–18997. doi: 10.1073/pnas.0609009103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heinemann F., Vogel S.K., Schwille P. Lateral membrane diffusion modulated by a minimal actin cortex. Biophys. J. 2013;104:1465–1475. doi: 10.1016/j.bpj.2013.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bashour K.T., Tsai J., Kam L.C. Cross talk between CD3 and CD28 is spatially modulated by protein lateral mobility. Mol. Cell. Biol. 2014;34:955–964. doi: 10.1128/MCB.00842-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Comrie W.A., Li S., Burkhardt J.K. The dendritic cell cytoskeleton promotes T cell adhesion and activation by constraining ICAM-1 mobility. J. Cell Biol. 2015;208:457–473. doi: 10.1083/jcb.201406120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu C.H., Wu H.J., Groves J.T. Altered actin centripetal retrograde flow in physically restricted immunological synapses. PLoS One. 2010;5:e11878. doi: 10.1371/journal.pone.0011878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Viola A., Gupta N. Tether and trap: regulation of membrane-raft dynamics by actin-binding proteins. Nat. Rev. Immunol. 2007;7:889–896. doi: 10.1038/nri2193. [DOI] [PubMed] [Google Scholar]
- 19.Dinic J., Ashrafzadeh P., Parmryd I. Actin filaments attachment at the plasma membrane in live cells cause the formation of ordered lipid domains. Biochim. Biophys. Acta. 2013;1828:1102–1111. doi: 10.1016/j.bbamem.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 20.Machta B.B., Papanikolaou S., Veatch S.L. Minimal model of plasma membrane heterogeneity requires coupling cortical actin to criticality. Biophys. J. 2011;100:1668–1677. doi: 10.1016/j.bpj.2011.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Owen D.M., Oddos S., Cebecauer M. High plasma membrane lipid order imaged at the immunological synapse periphery in live T cells. Mol. Membr. Biol. 2010;27:178–189. doi: 10.3109/09687688.2010.495353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chichili G.R., Cail R.C., Rodgers W. Cytoskeletal modulation of lipid interactions regulates Lck kinase activity. J. Biol. Chem. 2012;287:24186–24194. doi: 10.1074/jbc.M111.320747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang B., Bates M., Zhuang X. Super-resolution fluorescence microscopy. Annu. Rev. Biochem. 2009;78:993–1016. doi: 10.1146/annurev.biochem.77.061906.092014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lillemeier B.F., Mörtelmaier M.A., Davis M.M. TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nat. Immunol. 2010;11:90–96. doi: 10.1038/ni.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williamson D.J., Owen D.M., Gaus K. Pre-existing clusters of the adaptor Lat do not participate in early T cell signaling events. Nat. Immunol. 2011;12:655–662. doi: 10.1038/ni.2049. [DOI] [PubMed] [Google Scholar]
- 26.Hebert B., Costantino S., Wiseman P.W. Spatiotemporal image correlation spectroscopy (STICS) theory, verification, and application to protein velocity mapping in living CHO cells. Biophys. J. 2005;88:3601–3614. doi: 10.1529/biophysj.104.054874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ashdown G.W., Cope A., Owen D.M. Molecular flow quantified beyond the diffraction limit by spatiotemporal image correlation of structured illumination microscopy data. Biophys. J. 2014;107:L21–L23. doi: 10.1016/j.bpj.2014.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ashdown G., Pandžić E., Owen D. Cortical actin flow in T cells quantified by spatio-temporal image correlation spectroscopy of structured illumination microscopy data. J. Vis. Exp. 2015;106:e53749. doi: 10.3791/53749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gordón-Alonso M., Sala-Valdés M., Yáñez-Mó M. EWI-2 association with α-actinin regulates T cell immune synapses and HIV viral infection. J. Immunol. 2012;189:689–700. doi: 10.4049/jimmunol.1103708. [DOI] [PubMed] [Google Scholar]
- 30.Kelly D.F., Taylor K.A. Identification of the β1-integrin binding site on α-actinin by cryoelectron microscopy. J. Struct. Biol. 2005;149:290–302. doi: 10.1016/j.jsb.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 31.McGregor A., Blanchard A.D., Critchley D.R. Identification of the vinculin-binding site in the cytoskeletal protein α-actinin. Biochem. J. 1994;301:225–233. doi: 10.1042/bj3010225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hampton C.M., Taylor D.W., Taylor K.A. Novel structures for α-actinin:F-actin interactions and their implications for actin-membrane attachment and tension sensing in the cytoskeleton. J. Mol. Biol. 2007;368:92–104. doi: 10.1016/j.jmb.2007.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sjöblom B., Salmazo A., Djinović-Carugo K. α-actinin structure and regulation. Cell. Mol. Life Sci. 2008;65:2688–2701. doi: 10.1007/s00018-008-8080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kiuchi T., Higuchi M., Watanabe N. Multitarget super-resolution microscopy with high-density labeling by exchangeable probes. Nat. Methods. 2015;12:743–746. doi: 10.1038/nmeth.3466. [DOI] [PubMed] [Google Scholar]
- 35.Thompson R.E., Larson D.R., Webb W.W. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 2002;82:2775–2783. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sakamoto T., Limouze J., Sellers J.R. Blebbistatin, a myosin II inhibitor, is photoinactivated by blue light. Biochemistry. 2005;44:584–588. doi: 10.1021/bi0483357. [DOI] [PubMed] [Google Scholar]
- 37.Fiolka R., Shao L., Gustafsson M.G.L. Time-lapse two-color 3D imaging of live cells with doubled resolution using structured illumination. Proc. Natl. Acad. Sci. USA. 2012;109:5311–5315. doi: 10.1073/pnas.1119262109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gustafsson M.G.L., Shao L., Sedat J.W. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys. J. 2008;94:4957–4970. doi: 10.1529/biophysj.107.120345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rentero C., Zech T., Gaus K. Functional implications of plasma membrane condensation for T cell activation. PLoS One. 2008;3:e2262. doi: 10.1371/journal.pone.0002262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murugesan S., Hong J., Hammer J.A. Formin-generated actomyosin arcs propel T cell receptor microcluster movement at the immune synapse. J. Cell Biol. 2016;215:383–399. doi: 10.1083/jcb.201603080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toplak T., Pandzic E., Wiseman P.W. STICCS reveals matrix-dependent adhesion slipping and gripping in migrating cells. Biophys. J. 2012;103:1672–1682. doi: 10.1016/j.bpj.2012.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raghupathy R., Anilkumar A.A., Mayor S. Transbilayer lipid interactions mediate nanoclustering of lipid-anchored proteins. Cell. 2015;161:581–594. doi: 10.1016/j.cell.2015.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kelly D.F., Taylor D.W., Taylor K.A. Structure of the α-actinin-vinculin head domain complex determined by cryo-electron microscopy. J. Mol. Biol. 2006;357:562–573. doi: 10.1016/j.jmb.2005.12.076. [DOI] [PubMed] [Google Scholar]
- 44.Benninger R.K.P., Vanherberghen B., Onfelt B. Live cell linear dichroism imaging reveals extensive membrane ruffling within the docking structure of natural killer cell immune synapses. Biophys. J. 2009;96:L13–L15. doi: 10.1016/j.bpj.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Balagopalan L., Sherman E., Samelson L.E. Imaging techniques for assaying lymphocyte activation in action. Nat. Rev. Immunol. 2011;11:21–33. doi: 10.1038/nri2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burkhardt J.K., Carrizosa E., Shaffer M.H. The actin cytoskeleton in T cell activation. Annu. Rev. Immunol. 2008;26:233–259. doi: 10.1146/annurev.immunol.26.021607.090347. [DOI] [PubMed] [Google Scholar]
- 47.Dustin M.L., Cooper J.A. The immunological synapse and the actin cytoskeleton: molecular hardware for T cell signaling. Nat Immunol. 2000;1:23–29. doi: 10.1038/76877. [DOI] [PubMed] [Google Scholar]
- 48.Samstag Y., Eibert S.M., Wabnitz G.H. Actin cytoskeletal dynamics in T lymphocyte activation and migration. J. Leukoc. Biol. 2003;73:30–48. doi: 10.1189/jlb.0602272. [DOI] [PubMed] [Google Scholar]
- 49.Kucik D.F., Elson E.L., Sheetz M.P. Cell migration does not produce membrane flow. J. Cell Biol. 1990;111:1617–1622. doi: 10.1083/jcb.111.4.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yokosuka T., Sakata-Sogawa K., Saito T. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat. Immunol. 2005;6:1253–1262. doi: 10.1038/ni1272. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
F-actin is labeled by the actin binding peptide LifeAct-GFP. Scale bar, 5 μm.
Cell is labeled with DiO membrane dye. Scale bar, 5 μm.
Lighter areas indicate cell components further from the coverslip, darker areas closer to the coverslip. Scale bar, 5 μm.
F-actin is shown on the left, and the membrane dye DiI is shown on the right. Scale bar, 5 μm.
F-actin (left) is labeled with LifeAct-GFP and the PM (right) is labeled with GPI-GFP. Scale bar, 5 μm.
F-actin (left) is labeled with LifeAct-GFP and α-actinin (right) is labeled with mCherry. Scale bar, 5 μm.
F-actin is labeled by the actin binding peptide LifeAct-GFP. Scale bar, 5 μm.
Cell is labeled with DiO membrane dye. Scale bar, 5 μm.