

Abstract
Background and Purpose
Insulin‐like peptide 5 (INSL5) is a two‐chain, three‐disulfide‐bonded peptide of the insulin/relaxin superfamily, uniquely expressed in enteroendocrine l‐cells of the colon. It is the cognate ligand of relaxin family peptide RXFP4 receptor that is mainly expressed in the colorectum and enteric nervous system. This study identifies new signalling pathways activated by INSL5 acting on RXFP4 receptors.
Experimental Approach
INSL5/RXFP4 receptor signalling was investigated using AlphaScreen® proximity assays. Recruitment of Gαi/o proteins by RXFP4 receptors was determined by rescue of Pertussis toxin (PTX)‐inhibited cAMP and ERK1/2 responses following transient transfection of PTX‐insensitive Gαi/o C351I mutants. Cell proliferation was studied with bromodeoxyuridine. RXFP4 receptor interactions with β‐arrestins, GPCR kinase 2 (GRK2), KRas and Rab5a was assessed with real‐time BRET. Gene expression was investigated using real‐time quantitative PCR. Insulin release was measured using HTRF and intracellular Ca2 + flux monitored in a Flexstation® using Fluo‐4‐AM.
Key Results
INSL5 inhibited forskolin‐stimulated cAMP accumulation and increased phosphorylation of ERK1/2, p38MAPK, Akt Ser473, Akt Thr308 and S6 ribosomal protein. cAMP and ERK1/2 responses were abolished by PTX and rescued by mGαoA, mGαoB and mGαi2 and to a lesser extent mGαi1 and mGαi3. RXFP4 receptors interacted with GRK2 and β‐arrestins, moved towards Rab5a and away from KRas, indicating internalisation following receptor activation. INSL5 inhibited glucose‐stimulated insulin secretion and Ca2 + mobilisation in MIN6 insulinoma cells and forskolin‐stimulated cAMP accumulation in NCI‐H716 enteroendocrine cells.
Conclusions and Implications
Knowledge of signalling pathways activated by INSL5 at RXFP4 receptors is essential for understanding the biological roles of this novel gut hormone.
Linked Articles
This article is part of a themed section on Recent Progress in the Understanding of Relaxin Family Peptides and their Receptors. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.10/issuetoc
Abbreviations
- BrdU
5‐bromo‐2′‐deoxyuridine
- GLP‐1
glucagon‐like peptide 1
- GRK2
G protein receptor kinase 2
- INSL5
insulin‐like peptide 5
- mTORC
mammalian target of rapamycin complex
- PTX
Pertussis toxin
- S6RP
S6 ribosomal protein
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | GRK‐2 |
| RXFP4 receptors | KRas |
| Enzymes b | MEK1/2 |
| Adenylyl cyclase | mTORC1 |
| Akt (PKB) | p38MAP kinase |
| ERK1 | Rab5a |
| ERK2 | Src |
| LIGANDS |
|---|
| hINSL5 |
| mINSL5 |
| Forskolin |
| Insulin |
| LY294002 |
| Rapamycin (everolimus) |
| S6RP, S6 ribosomal protein |
| U0126 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a, 2015b).
Introduction
Insulin‐like peptide 5 (INSL5) is a two‐chain, three‐disulfide‐bond peptide with a striking structural similarity to insulin. It was identified in 1999 as a member of the insulin/relaxin superfamily by searching the expressed sequence tags database (Conklin et al., 1999; Hsu, 1999). Subsequently, INSL5 was identified as the cognate ligand for the relaxin family peptide receptor 4 (RXFP4; formerly GPR100/GPCR142, Liu et al., 2005; Alexander et al., 2015a) based on matching pharmacology and ligand–receptor distribution in peripheral tissues. Although relaxin‐3, a neuropeptide that is highly expressed in the nucleus incertus, is also a full agonist at RXFP4 receptors (Liu et al., 2005; Smith et al., 2010), it is not regarded as the physiological ligand due to disparate anatomical expression of the peptide and receptor (Boels and Schaller, 2003; Liu et al., 2005; Sutton et al., 2006).
More recently, INSL5 was identified as a novel gut hormone that is expressed in enteroendocrine L‐cells together with glucagon‐like peptide 1 (GLP‐1) and peptide YY, with high levels of expression in the colon and proximal rectum (Mashima et al., 2013; Thanasupawat et al., 2013; Grosse et al., 2014). Mice that were administered INSL5 i.p. displayed a dose‐dependent increase in food intake that was absent in RXFP4−/− mice, suggesting that INSL5 is an orexigenic gut hormone acting via RXFP4 receptors (Grosse et al., 2014). In addition, a recent study has suggested that INSL5 augments glucose‐stimulated insulin secretion from MIN6 pancreatic beta cells and GLP‐1 release from murine enteroendocrine GLUTag cells (Luo et al., 2015), indicating that INSL5–RXFP4 receptors may constitute a novel incretin axis involved in the regulation of metabolism and energy balance.
Relatively little is known of the signal transduction pathways activated by RXFP4 receptors, but these receptors are known to couple to Pertussis toxin (PTX)‐sensitive Gαi/o proteins to inhibit forskolin‐stimulated cAMP accumulation (Liu et al., 2003, 2005; Belgi et al., 2011). RXFP4 receptors also activate ERKs 1 and 2 (ERK1/2) in a heterologous system overexpressing these receptors (Belgi et al., 2013) and in MIN6 and GLUTag cells endogenously expressing RXFP4 receptors (Luo et al., 2015). RXFP4 receptors do not appear to couple to Gαq proteins since INSL5 evokes an intracellular Ca2 + response only in systems co‐transfected with the promiscuous Gα16 (Liu et al., 2003, 2005).
In this study, we present evidence that stimulation of RXFP4 receptors by INSL5 activates intracellular signalling pathways in addition to ERK1/2 and inhibition of cAMP, in particular p38MAPK, Akt and S6 ribosomal protein (S6RP), in CHO cells stably expressing RXFP4 receptors. The pattern of signalling activation suggests that RXFP4 receptors are involved in the control of cellular growth and proliferation, and this was supported by increased 5‐bromo‐2′‐deoxyuridine (BrdU) incorporation following stimulation with INSL5. We were also able to show that INSL5 inhibits glucose‐stimulated insulin secretion and Ca2 + mobilisation in MIN6 insulinoma cells and cAMP accumulation in NCI‐H716 enteroendocrine cells. In CHO‐RXFP4 cells, PTX‐insensitive G protein mutants were used to identify the Gαi/o subunits involved in activation of RXFP4‐mediated ERK1/2 and inhibition of cAMP signalling. BRET was used to show that agonist‐activated RXFP4 receptors interact with G protein receptor kinase 2 (GRK2) and β‐arrestin 1 and 2 and moves towards the early endosome marker Rab5a. Over the same time period RXFP4 moves away from the plasma membrane marker KRas, suggesting that following ligand stimulation, RXFP4 receptors undergo classical GRK and β‐arrestin‐mediated receptor internalisation, followed by sequestration to an early endosomal compartment. Unravelling the signalling profile of drugs acting at RXFP4 receptors is essential for development of therapies targeting this receptor.
Methods
Cell culture and transient transfection
CHO‐K1 cells stably expressing human RXFP4 receptors (CHO‐RXFP4) (Belgi et al., 2013) were grown and maintained in 175 cm2 flasks in DMEM/F‐12 medium supplemented with 5% (v/v) FBS at 37°C in humidified air containing 5% CO2. MIN6 murine insulinoma cells (a gift from A/Professor Helen Thomas, St Vincent's Institute of Medical Research, Melbourne, Australia) were grown and maintained in 175 cm2 flasks in high‐glucose DMEM supplemented with 15% FBS, 55 μM β‐mercaptoethanol, 25 mM HEPES, 100 IU·mL−1 penicillin and 100 μg·mL−1 streptomycin. NCI‐H716 human adenocarcinoma cells, an established model of L‐type enteroendocrine cells, were grown in high‐glucose RPMI supplemented with 10% FBS, 100 IU·mL−1 penicillin and 100 μg·mL−1 streptomycin. For the constructs outlined below, transient transfections were carried out using Lipofectamine 2000 (Thermo Fisher Scientific, Scoresby, VIC, Australia) according to the manufacturer's protocol. After 24 h, transfected cells were trypsinized, seeded onto 96‐well plates and used within 24 h.
Constructs
cDNA constructs for human RXFP4 receptors and PTX‐insensitive Gαi/o subunit mutants were from the cDNA Resource Center (Bloomsberg, PA, USA; http://www.cdna.org). These G protein α‐subunits contain a Cys351 to Ile351 mutation (C351I) that renders them insensitive to ADP‐ribosylation by PTX (Bahia et al., 1998). Human RXFP4 receptor sequence was amplified from RXFP4‐pcDNA3.1+ using the forward primer 5′‐GGGGACAAGTTTGTACAAAAAAGCAGGCTTCCACCATGCCCACACTCAATACTTCT‐3′ (start codon underlined) and reverse primer 5′‐GGGGACCACTTTGTACAAGAAAGCTGGGTCCCCGGGTGTCCCTCTGTC‐3′, containing attB1 and attB2 sites respectively. The PCR product was gel purified and cloned into pDONR201 vector (Thermo Fisher) using BP clonase II to create an entry clone containing attL1‐ and attL2‐flanked RXFP4 sequence. An LR recombination reaction was performed between the entry clone and a destination vector containing attR1, attR2 and Rluc8 sequence to create the RXFP4‐Rluc8‐tagged construct. The RXFP4‐Rluc8 insert was sequenced on both strands (Australian Genome Research Facility, Parkville, VIC, Australia). Human β‐arrestin 1‐Venus, β‐arrestin 2‐Venus and GRK2‐Venus constructs have been described previously (Kocan et al., 2008; Jensen et al., 2013; Kocan et al., 2014). KRas‐Venus and Rab5a‐Venus constructs were kindly provided by Professor Nevin Lambert (Georgia Regents University, Augusta, GA, USA).
Protein phosphorylation
Phosphorylation of ERK1/2 (Thr202/Tyr204), p38MAPK (Thr180/Tyr182), Akt (Thr308 and Ser473) and S6RP (Ser235/236) were determined using AlphaScreen SureFire® kits (PerkinElmer, Glen Waverley, VIC, Australia). Cells were seeded onto 96‐well plates at a density of 50 000–60 000 cells per well and incubated overnight in complete media to allow cell adhesion. The following day, cells were serum starved for 4–6 h to reduce basal phosphorylation levels and then stimulated with human or mouse INSL5 (hINSL5 or mINSL5, 200 nM each) for up to 45 min for time course studies or with increasing concentrations of the peptides (10−11 to 10–6.5 M) for concentration–response studies for the indicated time periods. For inhibitor studies, cells were pre‐incubated with inhibitors for 30 min except PTX where cells were exposed for 18 h. After stimulation, cells were lysed and frozen at −80°C. For assay, 4 μL of thawed cell lysate was added to a white 384‐well microplate (ProxiPlate; PerkinElmer) containing 5 μL acceptor beads mix (40 parts reaction buffer/10 parts activation buffer/1 part protein A acceptor beads) and incubated for 2 h at room temperature. Then, 2 μL donor bead mix (20 parts dilution buffer/1 part streptavidin‐coated donor beads) was added and incubated for 2 h at room temperature. All additions and incubations were carried out under low‐light conditions to avoid photobleaching. Plates were read using an Envision multilabel plate reader (PerkinElmer; excitation wavelength = 680 nm; emission wavelength = 520–620 nm).
Inhibition of cAMP accumulation
The amount of cAMP generated by forskolin and its inhibition by INSL5 peptides was measured using an AlphaScreen cAMP kit (PerkinElmer). Cells were seeded and serum starved as described above for protein phosphorylation assays. Following serum starvation, we changed the medium to HBSS supplemented with 500 μM IBMX, 5 mM HEPES and 0.1% (w/v) BSA and equilibrated for 1 h at 37°C. Cells were stimulated with hINSL5 or mINSL5 (10−11 to 10–6.5 M) for 15 min and then with forskolin (3 μM) for 30 min. Cells were lysed in buffer containing 0.3% Tween‐20, 5 mM HEPES and 0.1% (w/v) BSA and frozen at ‐80°C, followed by fluorescence detection according to the protocol provided by the manufacturer. In brief, 5 μL of cell lysate was added to a white 384‐well microplate (OptiPlate; PerkinElmer) containing 5 μL acceptor beads mix (1 U acceptor beads in 5 μL lysis buffer) and incubated for 30 min at room temperature. Then, 15 μL of donor beads mix (1 U biotinylated cAMP/1 U donor beads in 15 μL lysis buffer) was added and incubated for 1 h at room temperature. Fluorescence was measured using an EnVision plate multilabel plate reader (PerkinElmer; excitation wavelength = 680 nm; emission wavelength = 520–620 nm).
Cell proliferation
Cells were seeded onto 96‐well clear‐bottom black plates at a density of 5000 cells per well in serum‐free DMEM/F‐12 medium. The following day, cells were treated with hINSL5 or mINSL5 (10−12 to 10−8 M) for 24 h, before labelling with BrdU overnight. The amount of BrdU incorporated into cellular DNA, a marker of cell proliferation, was measured using a BrdU cell proliferation (chemiluminescent) elisa kit (Roche Diagnostics, Mannheim, Germany) according to the manufacturer's instructions.
Real‐time kinetic BRET studies
CHO‐K1 cells grown on six‐well plates were transiently co‐transfected with cDNAs encoding RXFP4‐Rluc8 and β‐arrestin 1‐Venus, β‐arrestin 2‐Venus, GRK2‐Venus or KRas‐Venus; 24 h after transfection, cells were harvested and seeded onto 96‐well opaque white plates in phenol red‐free DMEM with 5% FBS and grown overnight. The next day, coelenterazine h (5 μM) was added to cells, followed by stimulation with hINSL5 or mINSL5 (200 nM each). Dual light emission [480 nm (donor wavelength window); 530 nm (acceptor wavelength window)] was simultaneously recorded in real time using a LUMIstar Omega microplate reader (BMG Labtech, Ortenberg, Germany) before and after addition of ligands.
RNA purification and real‐time quantitative PCR
Total RNA was isolated from MIN6 cells using RNeasy mini RNA purification kit and treated with RNase‐free DNase (Qiagen, Hilden, Germany) according to the manufacturer's instruction. Purified RNA (500 ng) was reverse transcribed (iScript Reverse Transcription Supermix; Bio‐Rad, Hercules, CA, USA), the cDNA diluted 1:40 and 4 μL of the resulting solution used for PCR 10 μL reactions containing 0.5 μL Taqman primers and probes (Rxfp4: Mm00731536_s1, Glp1r: Mm00445292; reference gene Actb: Mm00607939; Thermo Fisher Scientific), 5 μL of Taqman Fast Advanced Master Mix (Thermo Fisher Scientific) and 0.5 μL Ultrapure DNase/RNase‐free H2O (Thermo Fisher Scientific). Real‐time quantitative PCR (40 cycles) was performed with a Mastercycler ep Realplex (Eppendorf, Hamburg, Germany). After initial denaturation at 95°C for 30 s, fluorescence was detected over 40 cycles (95°C for 5 s and 60°C for 30 s).
Insulin secretion assay
MIN6 cells were seeded onto 96‐well plates at 50 000–60 000 cells per well and allowed to grow overnight. The next day, the cells were equilibrated for 2 h in KRB buffer (115 mM NaCl, 5 mM KCl, 24 mM NaHCO3, 1 mM MgCl2, 2.5 mM CaCl2), followed by stimulation with either 0 mM or 10 mM glucose in the absence and presence of increasing concentrations of mINSL5 (0.1–200 nM) for 2 h. Supernatants were collected, and the insulin secreted was measured using an HTRF insulin kit (Cisbio, Codolet, France).
Intracellular Ca2 + mobilisation
MIN6 cells were seeded onto 96‐well plates as above. Following serum starvation, we incubated the cells in HBSS supplemented with 2 mM probenecid, 0.5% (w/v) BSA and 1 μM Fluo‐4‐AM (Thermo Fisher Scientific) for 30 min, washed and equilibrated for a further 30 min in buffer without Fluo‐4‐AM. mINSL5 was added in the Flexstation (Molecular Devices, Sunnyvale, CA, USA) and real‐time fluorescence measured every 3 s for 10 min (excitation = 485 nm; emission = 520 nm).
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Time course and concentration–response studies for protein phosphorylation were expressed as fold change of fluorescence over that of vehicle control, for inhibition of cAMP accumulation as % of the forskolin response or for cell proliferation as % of the response to 10% FBS, and plotted using Prism (v6.0; GraphPad software, San Diego, CA, USA). All concentration–response curves were fitted using a three‐parameter Hill equation within Prism. pEC50 values of mINSL5 and hINSL5 were compared using two‐way ANOVA with post hoc Tukey's multiple comparisons test. For inhibitor studies, data were expressed as fold change of fluorescence over that of vehicle control, and statistical analysis was performed using repeated‐measures two‐way ANOVA followed by Dunnett's multiple comparisons test. Ligand‐induced BRET ratio was calculated by subtracting the acceptor/donor wavelength ratio (530 nm/480 nm) of vehicle‐treated cells from the corresponding wavelength ratio of ligand‐treated cells and normalised to the value at t = 0 min (prior to addition of vehicle or ligand) (Kocan and Pfleger, 2011; Kocan et al., 2014). For real‐time quantitative PCR, data were expressed as the relative abundance of the gene of interest relative to the reference gene, actb, using the following equation:
For the Ca2 + mobilisation study, data were expressed as baseline‐subtracted raw fluorescence units, and areas under the Ca2 + curves were calculated using the built‐in function in Prism.
Materials
hINSL5 and mINSL5 were synthesised and purified at The Florey Institute of Neuroscience and Mental Health (Melbourne, Australia) as described previously (Hossain et al., 2008; Belgi et al., 2013). For some experiments, mINSL5 purchased from Phoenix Pharmaceuticals (Burlingame, CA, USA) was used, following verification that it had the same potency as INSL5 prepared in‐house (data not shown). FBS, HEPES, DMEM Ham's/F‐12, HBSS and PTX were all from Life Technologies (Carlsbad, CA, USA). IBMX was from Sigma‐Aldrich (Castle Hill, NSW, Australia). Coelenterazine h was purchased from Nanolight (Pinetop, AZ, USA).
Results
INSL5 activated ERK1/2 phosphorylation in a heterologous system expressing RXFP4 receptors (Belgi et al., 2013) and in pancreatic MIN6 and enteroendocrine GLUTag cells that endogenously express the receptor (Luo et al., 2015). Here, we confirm the activation of ERK1/2 but in addition demonstrate that RXFP4 receptors activate Akt, p38MAPK and S6RP, which are signalling proteins involved in a wide variety of cellular functions including growth, survival, proliferation and/or metabolism.
INSL5 promotes ERK1/2, Akt, p38MAPK and S6RP phosphorylation and inhibits forskolin‐stimulated cAMP accumulation
Treatment times were optimised for each assay (Supporting Information Fig. S1), and neither mINSL5 nor hINSL5 had any effect in any assay carried out with untransfected CHO‐K1 cells (Supporting Information Fig. 2). Previously, it has been shown that mINSL5 has a higher potency than hINSL5 in CHO‐RXFP4 cells in assays measuring the activation of ERK1/2 and the inhibition of cAMP production (Belgi et al., 2013). We confirmed these findings and also showed for phosphorylation of Akt, p38MAPK and S6RP that mINSL5 consistently had greater potency than hINSL5 (Figure 1A–F; Table 1). Potencies (expressed as pEC50) of mINSL5 ranged from 8.86 for inhibition of forskolin‐stimulated cAMP accumulation down to 7.80 and 7.86 for phosphorylation of Akt at Thr308 and Ser473 respectively, reflecting differences in coupling efficiency between the activated receptor and each signalling pathway. The rank order of potency was cAMP inhibition > S6RP = ERK1/2 > p38MAPK > Akt Ser473 = Akt Thr308. No accurate pEC50 values could be determined for hINSL5‐stimulated Akt phosphorylation. The pEC50 values of hINSL5 for cAMP, ERK1/2, p38MAPK and S6RP were not significantly different, clustering between 7.46 and 7.62 (P > 0.05). On the other hand, pEC50 values for mINSL5 were significantly different between Akt Thr308 and cAMP, S6RP and ERK1/2, and Akt Ser473 and cAMP.
Figure 1.
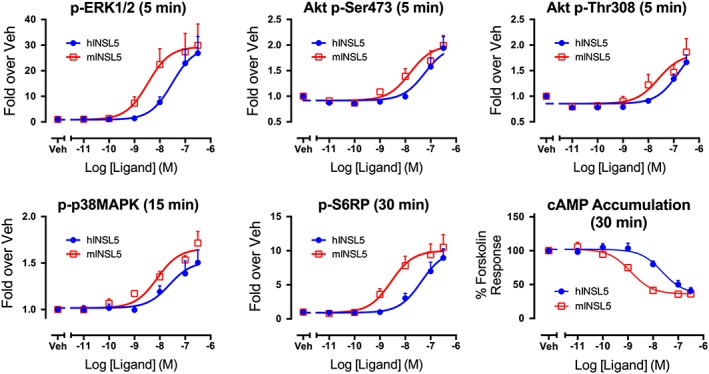
Concentration–response relationships for activation of ERK1/2, Akt, p38MAPK and S6RP and inhibition of cAMP production by hINSL5 and mINSL5 in CHO‐RXFP4 cells. In (A) phosphorylation of ERK1/2 (5 min; n = 6), (B) Akt Ser473 (5 min; n = 6), (C) Akt Thr308 (5 min; n = 6), (D) p38MAPK (15 min; n = 5) and (E) S6RP (30 min; n = 5), with in (F) inhibition of forskolin‐stimulated cAMP accumulation [30 min; n = 5 (mINSL5), n = 7 (hINSL5)]. CHO‐RXFP4 cells were treated with increasing concentrations of hINSL5 or mINSL5 (10−11 to 10–6.5 M). Results are normalized to the maximum response to hINSL5 in protein phosphorylation assays or % of response to 3 μM forskolin in cAMP accumulation assay. Data points represent mean ± SEM of independent experiments. Veh, vehicle.
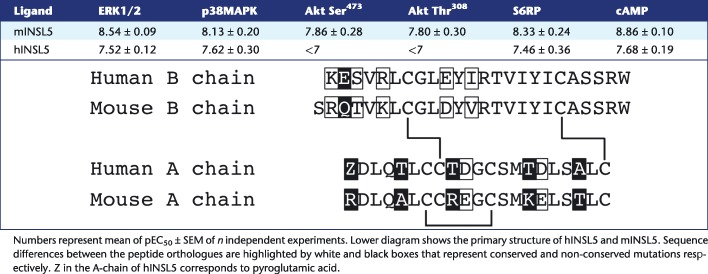
Table 1.
pEC50 values for hINSL5 and mINSL5 activation of ERK1/2 (n = 6), p38MAPK (n = 5), Akt Ser473 (n = 6), Akt Thr308 (n = 6) and S6RP (n = 5) and inhibition of cAMP (n = 5–7) in CHO‐RXFP4 cells
Activation of ERK1/2 by RXFP4 receptors requires Gαi/o, MEK and Src and is partially regulated by PI3K but does not depend on activation of mTOR
Gαi/o proteins and MEK1/2 (a kinase immediately upstream of ERK1/2) have obligatory roles for RXFP4 receptor activation of p‐ERK1/2, because PTX (Gαi/o inhibitor; 100 ng·mL−1; 18 h) or U0126 (MEK inhibitor; 10 μM; 30 min) both completely inhibited the INSL5‐induced p‐ERK1/2 response in CHO‐RXFP4 cells (n = 5; Figure 2A). Src is often involved in GPCR‐mediated ERK1/2 signalling (Luttrell et al., 1996; Cao et al., 2000), and PP2 (selective Src family kinase inhibitor; 10 μM; 30 min) completely inhibited the INSL5‐stimulated ERK1/2 response (n = 5; Figure 2A), suggesting that Src family kinases have a critical role in the activation of ERK1/2 by RXFP4 receptors. The PI3K inhibitor LY294002 (10 μM; 30 min) partly inhibited the ERK1/2 response to INSL5 (n = 5; Figure 2A), suggesting that PI3K may contribute to the ERK1/2 response. Mammalian target of rapamycin complex (mTORC)1 was examined using the mTOR inhibitors KU0063794 (1 μM; 30 min) or rapamycin (100 nM; 30 min), since cross‐regulation can occur between the Ras–ERK and PI3K–Akt pathways (Mendoza et al., 2011). Neither KU0063794 nor rapamycin (n = 5; Figure 2A) inhibited the INSL5‐mediated ERK1/2 response, with KU0063794 in fact causing a slightly, but significantly, elevated response (n = 5; Figure 2A), implying that RXFP4 receptor activation of ERK1/2 does not involve mTORC1.
Figure 2.
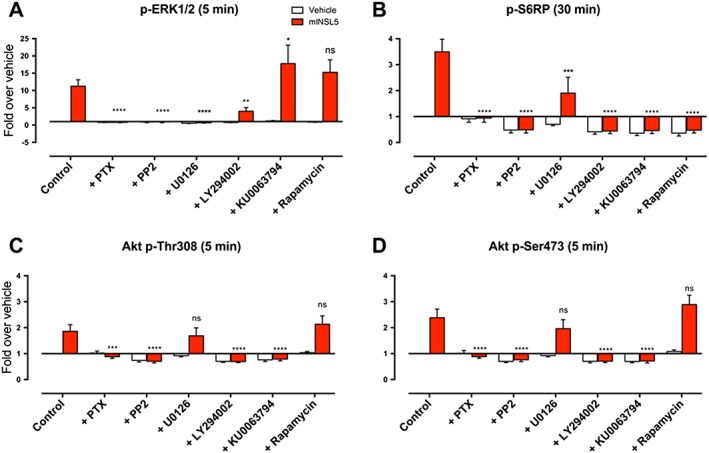
Effects of pathway inhibitors on signalling responses to mINSL5 in CHO‐RXFP4 cells. CHO‐RXFP4 cells were pretreated with the Gαi/o inhibitor (PTX, 100 ng•mL−1), MEK inhibitor (U0126, 10 μM), Src family tyrosine kinase inhibitor (PP2, 10 μM), PI3K inhibitor (LY294002, 10 μM) or the mTOR complex inhibitors (KU0063794, 1 μM; rapamycin, 100 nM) followed by INSL5 (100 nM). Pathways examined were (A) p‐ERK1/2, (B) Akt p‐Ser473 , (C) Akt p‐Thr308 and (D) p‐S6RP. Cells were treated with inhibitors for 30 min before and then during mINSL5 stimulation, except for the PTX treatment where cells were exposed for 18 h. Results are quantified as fold change in fluorescence over that of the vehicle treatment in the control group. Bars represent mean ± SEM of experiments [n = 5 (PTX, LY294002, KU0063794, rapamycin), n = 6 (PP2, U0126), n = 10 (Control)]. *P < 0.05; significantly different from the mINSL5 response in the control group; repeated‐measures two‐way ANOVA followed by Dunnett's multiple comparisons test. ns, non‐significant.
RXFP4 receptor activation of Akt Thr308 and Akt Ser473 requires Gαi/o, Src, PI3K and mTORC2 but does not involve MEK1/2 or mTORC1
Activation of Akt by INSL5 in CHO‐RXFP4 requires Gαi/o proteins and Src, as PTX (100 ng·mL−1; 18 h; n = 5) or PP2 (10 μM; 30 min; n = 6) completely inhibited Akt phosphorylation at Thr308 (Figure 2C) and Ser473 (Figure 2D). PI3K has an obligatory role in the activation of Akt by INSL5 as LY294002 (10 μM; 30 min) completely blocked responses at Thr308 (n = 7; Figure 2C) and Ser473 (n = 7; Figure 2D). Conversely, MAPK signalling does not play a part in the INSL5‐mediated Akt response because inhibition of MEK1/2 with U0126 (10 μM; 30 min) did not alter the response at either phosphorylation site (n = 6; Figure 2C, D). Interestingly, activation of Akt by INSL5 at Thr308 and Ser473 was inhibited by KU0063794 (1 μM; 30 min; n = 5) but completely resistant to rapamycin (100 nM; 30 min; n = 5; Figure 2C, D). This implies that mTORC2, but not mTORC1, is required for the INSL5‐mediated Akt response, as KU0063794 inhibits both mTORC1 and mTORC2 (García‐Martínez et al., 2009), while rapamycin selectively inhibits only mTORC1 (Loewith et al., 2002).
RXFP4 receptors activate S6RP via pathways involving Gαi/o, Src, MEK1/2, PI3K and mTORC1
S6RP, a protein component of the 40S ribosomal subunit, is a downstream effector of the ribosomal S6 kinase S6K, which is activated by the Akt–mTOR pathway (Ruvinsky and Meyuhas, 2006). Activation of S6RP by INSL5 in CHO‐RXFP4 cells absolutely required Gαi/o proteins and Src family kinases as there was complete inhibition of the S6RP response by PTX (100 ng·mL−1; 18 h; n = 5) and PP2 (10 μM; 30 min; n = 5) respectively (Figure 2B). In addition, MAPK signalling can also influence S6RP activation (Roux et al., 2007). Inhibition of MEK1/2 with U0126 (10 μM; 30 min; n = 6) inhibited INSL5‐stimulated S6RP activation by ~50% in CHO‐RXFP4 cells (Figure 2B). Inhibition of PI3K by LY294002 (10 μM; 30 min; n = 6) and mTOR by KU0063794 (1 μM; 30 min; n = 5) or rapamycin (100 nM; 30 min; n = 5) also abolished INSL5‐stimulated S6RP activation (Figure 2B), suggesting that the PI3K–mTORC1 pathway is pivotal in mediating this response in CHO‐RXFP4 cells.
RXFP4 receptors utilise multiple Gαi/o subunits to inhibit forskolin‐stimulated cAMP accumulation and activate ERK1/2 in CHO‐RXFP4 cells
RXFP4 receptors inhibit forskolin‐stimulated cAMP production (Liu et al., 2005; Belgi et al., 2013; Shabanpoor et al., 2013), suggesting that these receptors couple to Gαi/o. However, whether the RXFP4 receptors utilise a selective subset or all of the Gαi/o subunits remains unknown. This was determined for INSL5‐mediated ERK1/2 activation and inhibition of cAMP in CHO‐RXFP4 cells by transiently transfecting cells with PTX‐insensitive Gαi/o mutants (mGαoA, mGαoB, mGαi1, mGαi2 or mGαi3) followed by treatment with PTX. In essence, a rescue of cAMP or ERK1/2 responses normally suppressed by PTX in the presence of exogenous Gαi/o mutants reveals the pattern of Gαi/o activation by RXFP4 receptors.
We first showed that INSL5‐mediated inhibition of forskolin‐stimulated cAMP accumulation was abolished by PTX in mock‐transfected CHO‐RXFP4 cells (n = 7; Figure 3A), confirming the involvement of Gαi/o in the signal transduction from RXFP4 receptors. We then demonstrated that cAMP responses could be rescued in PTX‐treated cells singly transfected with each Gαi/o mutant. The degree of cAMP rescue ranged from 83 ± 16% for mGαoB (n = 6; Figure 3C), 59 ± 4% for mGαoA (n = 7; Figure 3B), 52 ± 6% for mGαi2 (n = 7; Figure 3E), 49 ± 14% for mGαi1 (n = 7; Figure 3D) and 40 ± 5% for mGαi3 (n = 6; Figure 3F). When similar studies were performed for INSL5‐mediated ERK1/2 activation, the degree of rescue was less pronounced across all the Gαi/o mutants tested. ERK1/2 response rescue varied from 25 ± 6% for mGαoB ( n = 5; Figure 3I), 24 ± 6% for mGαoA (n = 5; Figure 3H), 20 ± 5% for mGαi2 (n = 5; Figure 3K), 10 ± 3% for mGαi3 (n = 5; Figure 3L) to 5 ± 2% for mGαi1 (non‐significant, n = 5; Figure 3J). RXFP4 receptors were therefore able to engage multiple Gαi/o isoforms to promote cAMP inhibition and ERK1/2 activation but did indicate a preference for GαoA, GαoB and Gαi2.
Figure 3.
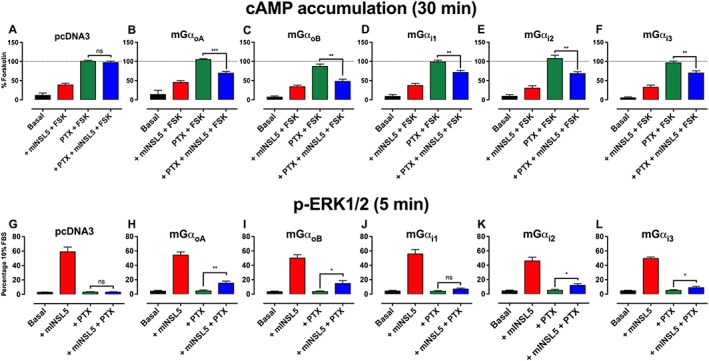
Determination of the Gαi/o isoforms utilise by RXFP4 receptors using PTX‐resistant Gα mutants. cAMP inhibition and ERK1/2 activation following stimulation by mINSL5 in CHO‐RXFP4 cells. RXFP4 receptors inhibit forskolin‐stimulated cAMP accumulation, indicating an interaction with Gαi/o proteins. CHO‐RXFP4 cells were transiently transfected with pcDNA3 (mock transfection) in (A, G) or Gi/o subunit constructs carrying the C351I mutation in (B, H) mGαoA, (C, I) mGαoB, (D, J) mGαi1, (E, K) mGαi2 or (F, L) mGαi3. In the upper panels, cells were incubated with PTX (100 ng•mL−1) for 16 h, and cAMP was production stimulated by forskolin (3 μM) followed by treatment with mINSL5 (100 nM). Results are normalized to the cAMP response stimulated by forskolin. In the lower panels, the ERK1/2 response was measured in cells incubated with PTX (100 ng•mL−1) for 16 h followed by treatment with mINSL5 (100 nM). Results are expressed as percentage of response elicited by 10% FBS. Bars represent mean ± SEM of independent experiments [cAMP: n = 6 (mGαoA, mGαi3), n = 7 (pcDNA3, mGαoB, mGαi1, mGαi2); ERK1/2: n = 5]. *P < 0.05; significantly different from the forskolin response (cAMP inhibition) or the baseline response (ERK1/2 activation) in PTX‐treated cells; repeated‐measures two‐way ANOVA followed by Dunnett's multiple comparisons test. ns, non‐significant.
INSL5 promotes cell proliferation in vitro
As INSL5 activates signalling pathways implicated in cellular growth and proliferation such as ERK1/2 and Akt, we examined whether INSL5 influenced cell proliferation in vitro. hINSL5 and mINSL5 enhanced BrdU incorporation in CHO‐RXFP4 cells in a concentration‐dependent manner (mINSL5: pEC50 = 9.11 ± 0.25; hINSL5: pEC50 = 8.38 ± 0.43; n = 5; Figure 4), indicating that activation of RXFP4 receptors promoted cell proliferation.
Figure 4.
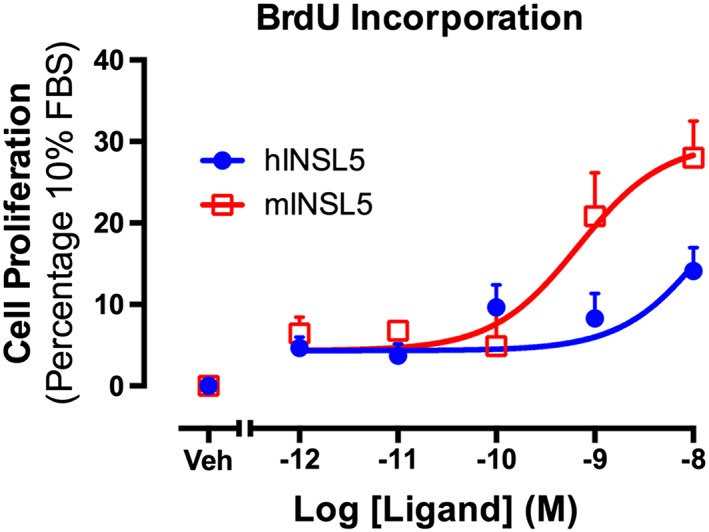
Activation of RXFP4 receptors by hINSL5 or mINSL5 promotes increased cellular BrdU incorporation. Concentration–response relationships are shown for cellular BrdU incorporation stimulated by hINSL5 or mINSL5 (10−12 to 10−8 M) in CHO‐RXFP4 cells. Results are expressed as percentage of response elicited by 10% FBS. Data points represent means ± SEM of 5 independent experiments. Veh, vehicle.
RXFP4 receptors interact with GRK2, β‐arrestin 1, β‐arrestin 2 and the early endosome marker Rab5a and dissociates from membrane‐bound KRas following INSL5 activation
BRET was used to examine Rluc8‐tagged RXFP4 receptor interactions with Venus‐tagged GRK2, β‐arrestin 1, β‐arrestin 2, Rab5a (early endosome marker) or KRas (plasma membrane marker) following activation by INSL5. Treatment with mINSL5 or hINSL5 (200 nM each) promoted RXFP4 receptor interactions with GRK2 [n = 8 (mINSL5), n = 6 (hINSL5); Figure 5A], β‐arrestin 1 [n = 5 (mINSL5), n = 6 (hINSL5); Figure 5B], β‐arrestin 2 (n = 5; Figure 5C) and Rab5a (n = 5; Figure 5E) and caused dissociation of the receptor from KRas [n = 8 (mINSL5), n = 5 (hINSL5); Figure 5D], suggesting that RXFP4 receptors undergo classical GRK phosphorylation and β‐arrestin‐mediated receptor internalisation, followed by sequestration to early endosomal compartment following ligand stimulation.
Figure 5.
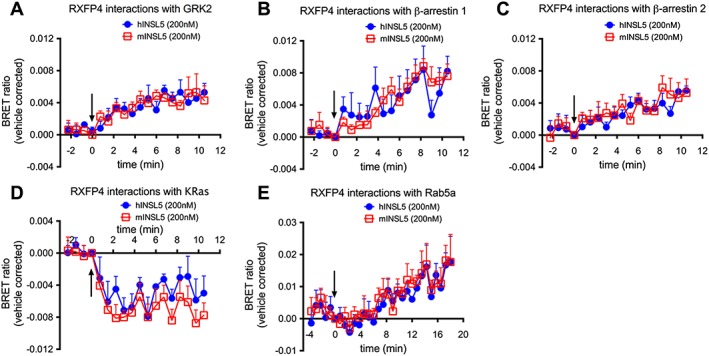
Real‐time kinetic BRET studies of interactions between RXFP4 receptors and (A) GRK2, (B) β‐arrestin 1, (C) β‐arrestin 2, (D) membrane‐bound KRas and (E) early endosome marker Rab5a induced by hINSL5 and mINSL5. CHO‐K1 cells were transiently co‐transfected with RXFP4‐Rluc8 and Venus‐tagged GRK2, β‐arrestin 1, β‐arrestin 2, Rab5a or KRas constructs. Following transfection, we stimulated the cells with hINSL5 or mINSL5 (200 nM). Ligand‐induced BRET ratios were calculated by subtracting the ratio for the vehicle‐treated sample from the BRET ratio for each ligand‐treated sample as described. Data points represent mean ± SEM of independent experiments (n = 5–8).
INSL5 inhibits glucose‐stimulated insulin secretion and calcium mobilisation in MIN6 insulinoma cells and inhibits cAMP accumulation in NCI‐H716 enteroendocrine cells
A previous study showed that INSL5 potentiated glucose‐stimulated insulin secretion in MIN6 insulinoma cells and primary murine islets, suggesting that INSL5–RXFP4 receptors may constitute a novel incretin axis, similar to GLP‐1–GLP‐1 receptor (Luo et al., 2015). We first confirmed that rxfp4 mRNA is expressed in MIN6 cells using real‐time quantitative PCR, though at a markedly lower level than glp1r (n = 6; Figure 6A). Next, we investigated the effects of INSL5 on glucose‐mediated insulin secretion in MIN6 cells. mINSL5 (0.1–200 nM) did not affect the ability of MIN6 to secrete insulin in the absence of glucose stimulation (0 mM glucose; n = 5; Figure 6B). Treatment of MIN6 cells with 10 mM glucose evoked robust insulin secretion that was inhibited by the co‐addition of mINSL5 (0.1 and 1 nM; P < 0.05; n = 5; Figure 6B). There was evidence of a bell‐shaped concentration–response relationship since the inhibitory effect of mINSL5 on glucose‐stimulated insulin secretion was lost at higher concentrations of mINSL5. Because Ca2 + influx typically occurs in beta cells following a depolarising stimulus (e.g. glucose) and is widely considered an important trigger for the exocytosis of insulin granules, we investigated the effect of mINSL5 on glucose‐stimulated Ca2 + mobilisation in MIN6 cells. Co‐addition of mINSL5 (10−9 M) diminished the Ca2 + response evoked by 10 mM glucose, whereas co‐addition of GLP‐1 (10−8 M) potentiated the glucose‐mediated Ca2 + response (Figure 6C). The Ca2 + mobilisation experiment was also performed in parallel with different concentrations of mINSL5, and the AUCs were encompassed by the different treatments quantified (P < 0.05 for mINSL5 [10−9 M]; P < 0.01 for GLP‐1; Figure 6D). Our results obtained using two different approaches (insulin secretion and Ca2 + mobilisation) suggest that INSL5 does not behave like GLP‐1 as it inhibits glucose‐stimulated insulin secretion. In addition, we did not observe p‐ERK1/2 and cAMP inhibition in response to RXFP4 receptor activation in MIN6 cells as observed in the CHO‐RXFP4 system (data not shown). However, in NCI‐H716 cells – an established human cell model of enteroendocrine cells – hINSL5 (10−7 M) inhibited forskolin‐stimulated cAMP accumulation (n = 5; Figure 6E).
Figure 6.
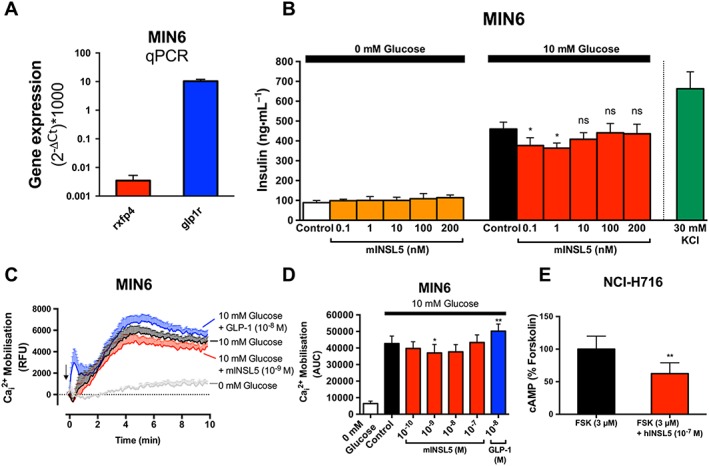
INSL5 inhibits glucose‐stimulated insulin secretion and calcium mobilisation in MIN6 murine insulinoma cells and cAMP accumulation in NCI‐H716 human enteroendocrine (NHI‐H716) cells. In (A), real‐time quantitative PCR (qPCR) assay to determine the expression levels of rxfp4 and glp1r that are expressed as ratios relative to actb, multiplied by 1000 (n = 6). In (B), MIN6 cells equilibrated for 2 h in KRB buffer, followed by stimulation in either 0 or 10 mM glucose by mINSL5 (0.1–200 nM) for 2 h. Supernatants were assayed for insulin (expressed in ng•mL−1; n = 5). In (C), Ca2 + mobilisation in MIN6 cells (n = 5) using Fluo‐4‐AM (1 μM) in response to 10 mM glucose alone or in the presence of mINSL5 (10−9 M) or GLP‐1 (10−8 M). Concentration–response relationships for inhibition of Ca2 + responses (AUC) by INSL5 were bell shaped (D). In (E), NCI‐H716 human enteroendocrine cells stimulated with forskolin (3 μM) showed inhibition of cAMP accumulation with hINSL5 (10−7 M; n = 5). Data points represent mean ± SEM of n independent experiments. *P < 0.05; significantly different from control. RFU, Relative Fluorescence Unit ns, non‐significant.
Discussion
The peptide mINSL5 displayed greater potency than hINSL5 in all of our signalling assays (Table 1), consistent with the higher binding affinity observed for the mouse homologue, compared with hINSL5 (Belgi et al., 2013). Under the assay conditions used here, mINSL5 was most potent for inhibition of cAMP accumulation, as observed previously using a cAMP response element reporter gene assay (Belgi et al., 2011; Belgi et al., 2013). There is high sequence conservation between the human and mouse peptides, particularly in the long B‐chain α‐helix that extends from GlyB8 through to the C‐terminal TrpB24. The INSL5 residues that govern activation of RXFP4 receptors, ArgB13, TyrB17 and ArgB23, are identical between human and mouse peptides, as are residues making up the hydrophobic core of the folded peptide (LeuA3, CysA7, CysA12, LeuA17, CysA21, LeuB6, TyrB11 and ValB15)(Haugaard‐Jonsson et al., 2009). Key differences are found in a series of charged residues that contribute to correct folding of INSL5 via electrostatic interactions and hydrogen bonding. In hINSL5, AspA2 forms hydrogen bonds to amide protons of GlnA4 and ThrA5 and the side‐chain hydroxyl group proton of ThrA5. AspA10 and AspA16 both interact with LysB1, and there is a weaker interaction between GluB2 and ArgB5. These residues also undergo hydrogen bonding with additional side‐chain serine and threonine hydroxyl groups or to amide protons (Haugaard‐Jonsson et al., 2009). While AspA2 and GlnA4 are conserved, ThrA5 is an alanine in mINSL5, AspA10 is glutamate, AspA16 is also glutamate and LysB1 is arginine. The last three differences are conservative in nature, but the longer side chain of glutamate compared with aspartate is likely to produce subtle differences in the overall structure of INSL5 that contribute to the observed higher affinity of the mouse homologue for human RXFP4 receptors. In addition, GluB2 is a glutamine, and ArgB5 is a lysine in mINSL5, so this interaction would be altered. A recent study identifies LysA15 as a key residue in the A‐chain of mINSL5 that is responsible for the improved RXFP4 receptor activity compared with hINSL5 (Patil et al., 2016).
Pharmacological blockade of Gαi/o by PTX and Src by PP2 abolished activation of ERK1/2, Akt and S6RP by mINSL5, indicating that Gαi/o and Src are upstream of these events and obligatory for RXFP4 receptor signalling. In comparison, the closely related RXFP3 receptor shows partial, but not complete, suppression of H3‐relaxin‐stimulated ERK1/2 activation by PP2 (van der Westhuizen et al., 2007), suggesting that RXFP4 and RXFP3 receptors may activate ERK1/2 in a different manner. Interestingly, RXFP4, but not RXFP3, receptors contain a proline‐rich binding motif (PXXP) in intracellular loop 1 (Vroling et al., 2011) that may permit direct binding of the SH3 domain of Src to the receptor. Indeed, direct interaction with Src has been shown to be critical for β3‐adrenoceptor‐mediated ERK1/2 activation (Cao et al., 2000). Whether such an interaction also dictates RXFP4 receptor signalling to ERK1/2 remains to be determined.
Our inhibitor experiments indicate that there is redundancy in some RXFP4 receptor signalling pathways (Figures 2 and 7). For example, the MEK1/2 inhibitor U0126 partly inhibited phosphorylation of S6RP, despite complete inhibition by KU0063794 and rapamycin, indicating that the primary pathway is via activation of mTORC1. Similarly, LY294002 partly inhibited ERK1/2 phosphorylation. mTOR is not upstream of INSL5‐stimulated ERK1/2 activation but is necessary for Akt and S6RP activation. Furthermore, mTORC2, but not mTORC1, is the upstream mediator of Akt activation by RXFP4 receptors, as selective inhibition of mTORC1 by rapamycin had no significant effect on the INSL5‐stimulated Akt response, which was blocked by combined inhibition of mTORC1 and mTORC2 by KU0063794. This is in accord with previous findings showing that rictor‐bound mTOR (mTORC2) is responsible for Akt phosphorylation at Ser473 and facilitates phosphoinositide‐dependent kinase‐1‐mediated Akt phosphorylation at Thr308 (Sarbassov et al., 2005).
Figure 7.
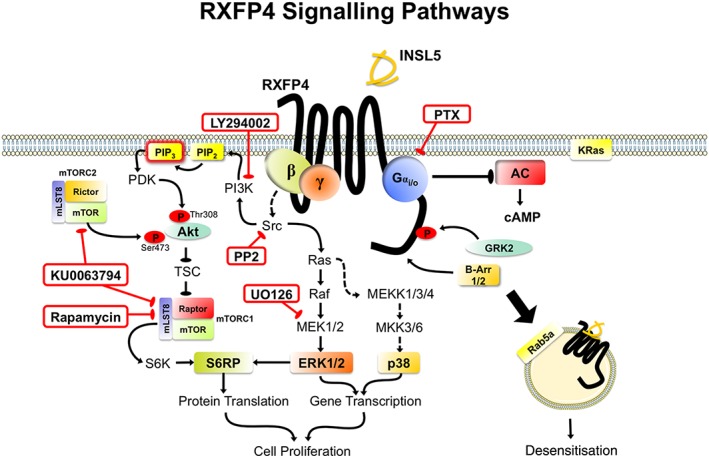
RXFP4 receptor signal transduction pathways. Inhibitors used in studying RXFP4 receptor signalling mechanisms are boxed in red. Following INSL5 stimulation, RXFP4 receptors recruit multiple Gαi/o subunits (predominantly GαoA, GαoB, Gαi2) that activate a range of signalling pathways, including cAMP inhibition, ERK1/2 (Thr202/Tyr204), Akt (Thr308/Ser473) p38MAPK (Thr180/Tyr182) and S6RP (Ser235/236) via intermediary pathways such as mTORC1/C2, Src, PI3K and MEK1/2, leading to enhanced cell proliferation. Activated RXFP4 receptors interact with GRK2 and β‐arrestins (β‐Arr 1/2), which leads to movement of the receptor away from the plasma membrane into early endosome compartments. Note: speculative pathways are shown with dotted lines. mammalian target of rapamycin, mTOR; phosphoinositide‐dependent kinase, PDK.
The recruitment of Gαi/o isoforms to RXFP4 receptors was explored using PTX‐insensitive mutants. The RXFP4 receptors were able to engage multiple Gαi/o isoforms to promote cAMP inhibition and ERK1/2 activation and, in this respect, resemble RXFP3 receptors (Kocan et al., 2014) with which RXFP4 receptors share a high degree of homology. The rescue of responses was greater for cAMP than for ERK1/2 responses, but there was little distinction between the G protein isoforms, suggesting that the RXFP4 receptor is quite promiscuous and that the precise pattern of G protein activation may vary with cell type. For RXFP3 receptors, studies in CHO cells using PTX‐insensitive G protein mutants showed coupling predominantly to GαoB and Gαi2, whereas in HEK cells, GαoA coupling was also observed (van der Westhuizen, 2008). Direct measurement of G protein coupling in CHO cells expressing RXFP3 receptors using BRET showed coupling to GαoA, GαoB, Gαi2 and Gαi3, but interestingly, activation of the receptor by the biased agonists relaxin and R3(BΔ23‐27)R/I5 only caused coupling to GαoB and Gαi2, perhaps indicating that these are preferred (Kocan et al., 2014). In comparison, the relaxin RXFP1 receptor selectively recruits GαoB and Gαi3, in addition to Gαs, to modulate intracellular cAMP levels (Halls et al., 2006). Unlike RXFP1, the pharmacological actions of RXFP4 receptors are solely Gαi/o mediated, as stimulation with INSL5 alone failed to produce an increase in cAMP level with no Gαs component (data not shown).
Many ligand‐activated GPCRs undergo homologous desensitisation involving phosphorylation of serine and threonine residues in the cytoplasmic tail by GRKs, leading to β‐arrestin 1/2 recruitment and receptor internalisation (DeWire et al., 2007). Here, we show that INSL5 treatment was followed by interaction between RXFP4 receptors and GRK2 and β‐arrestin 1/2, associated with receptor internalisation and translocation to the early endosome compartment, an effect also observed with ligand‐activated RXFP3 receptors (Figure 7) (Kocan et al., 2014). This contrasts with RXFP1 and RXFP2 receptors, which lack the ability to recruit β‐arrestins and are poorly internalised following receptor stimulation (Callander et al., 2009). Thus, RXFP4 behaves somewhat similarly to RXFP3 receptors, in BRET interaction studies and in cell signalling assays.
There is evidence to suggest that the mRNA for RXFP4 receptors is expressed in MIN6 pancreatic beta cells and that stimulation by INSL5 activates ERK1/2 (Luo et al., 2015). We confirmed RXFP4 mRNA expression in MIN6, although at a substantially lower level than that of GLP‐1 receptors. Our finding that INSL5 inhibits glucose‐mediated insulin release and Ca2 + response in MIN6 contrasts with the previous study (Luo et al., 2015), which suggested that INSL5 potentiated glucose‐stimulated insulin secretion in MIN6 cells and primary islets. However, it is well accepted that increased cAMP and Ca2 + levels promote exocytosis of insulin granules in beta cells (see Rorsman and Braun, 2013), whereas Gi‐coupled receptors such as the growth hormone secretagogue receptor (Dezaki et al., 2007) or the α2‐adrenoceptor (Gibson et al., 2006) inhibit insulin release. Our finding that RXFP4 receptors are coupled to Gαi/o proteins inhibits cAMP accumulation in the CHO cell system and inhibits Ca2 + release, and insulin release in MIN6 cells would appear to follow that paradigm. We were also able to show inhibition of cAMP accumulation in enteroendocrine NCI‐H716 cells.
INSL5 stimulation of RXFP4 receptors activated MAPK pathways and the PI3K–Akt–mTOR–S6RP signalling cascade that are associated with cell proliferation in CHO cells (Figure 7). The roles of ERK1/2 and PI3K–Akt–mTOR signalling in cellular growth, proliferation and metabolism are well documented (Laplante and Sabatini, 2009; Polak and Hall, 2009). S6RP is a downstream effector of mTOR that is involved in protein synthesis, and has been implicated in pancreatic beta cell homeostasis as shown by insulin deficiency and smaller beta cell size in mice with global knock‐in of a dominant negative S6RP that lacks all five possible phosphorylation sites (Ruvinsky et al., 2005). Interestingly, INSL5 knockout mice also appear to have a reduced number of beta cells in the islet of Langerhans (Burnicka‐Turek et al., 2012). Thus, it is appealing to suggest that the INSL5–RXFP4 receptor axis may be involved in beta cell homeostasis, although more evidence is needed to establish this link.
In this study, CHO‐K1 cells stably transfected with RXFP4 receptors were selected as a model to investigate intracellular signalling pathways. Furthermore, CHO‐K1 cells have been successfully utilised to characterise signalling pathways activated by a number of recently deorphanised GPCRs, including the closely related RXFP3 receptor (van der Westhuizen et al., 2007; Kocan et al., 2014) and the ghrelin receptor growth hormone secretagogue receptor 1a (Mousseaux et al., 2006). Such expression systems can therefore provide a useful guide to signalling pathways likely to be found in endogenously expressing systems. Currently, cells that endogenously express RXFP4 receptors are not yet well characterised. Nevertheless, there is evidence that RXFP4 receptors are mainly expressed in the gastrointestinal tract, particularly in the duodenum, colon and rectum, but interestingly not significantly in the pancreas, as indicated by next generation sequencing of protein coding genes in human tissues (Uhlén et al., 2015). Furthermore, expression of RXFP4 receptors in the enteric nervous system has been demonstrated by in situ hybridisation, with localisation to submucosal and myenteric nerve plexuses of the colon (Grosse et al., 2014). This would suggest that INSL5, released from L‐cells in the gastrointestinal tract, may activate RXFP4 receptors in an autocrine/paracrine manner. Indeed, our cAMP result in the NCI‐H716 enteroendocrine cells and the ERK1/2 result in GLUTag cells (Luo et al., 2015) support this notion.
Altogether, our study demonstrates that INSL5 stimulation of RXFP4 receptors activated a range of signalling cascades which include inhibition of cAMP production, activation of ERK1/2, p38MAPK, Akt and S6RP signalling (Figure 7), which promoted cell proliferation in vitro. Activation of RXFP4 receptors also caused interaction with multiple Gαi/o proteins and subsequent recruitment of GRK2 and β‐arrestins to initiate receptor internalisation. In cells that natively express RXFP4 receptors, INSL5 inhibited insulin release and Ca2 + mobilisation in MIN6 cells and inhibited cAMP production in NCI‐H716 cells. These findings add to our understanding of RXFP4 receptor signal transduction mechanisms that will be essential in the development of novel anti‐obesity, anti‐diabetic and/or appetite‐modulating drugs.
Author contributions
S.Y.A. and M.K. performed the research; S.Y.A., M.K., B.A.E. and R.J.S. analysed and provided critical evaluation of the data; R.A.D.B., N.P. and M.A.H. synthesized and purified hINSL5 and mINSL5; D.S.H. planned and designed inhibitor studies and provided inhibitors; M.L.H. guided and optimized the cAMP studies; S.Y.A., M.K. and R.J.S. conceived the study, designed and critically interpreted the data and wrote the manuscript that was critically evaluated by D.S.H., B.A.E., R.A.D.B., M.L.H. and M.A.H.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Figure S1 Time course of activation of signalling pathways by hINSL5 and mINSL5 in CHO‐RXFP4 cells. In (A), phosphorylation of ERK1/2, (B) Akt Ser473, (C) Akt Thr308, (D) p38MAPK and (E) S6RP. CHO‐RXFP4 cells were treated with hINSL5 or mINSL5 (100 nM each) for up to 45 min, and activation of ERK1/2 (n = 5), Akt Ser473 (n = 5), Akt Thr308 (n = 5), p38MAPK (n = 5) and S6RP (n = 3) was determined using phospho‐specific AlphaScreen SureFire kit. In (F), there was no Ca2+ mobilisation detectable in response to stimulation by mINSL5 (100 nM) even though the same cells produced a substantial response to addition of ATP (10 μM; n = 3). Results are quantified as fold change in fluorescence over that of the vehicle treatment (fold over vehicle) for phosphorylation assays or expressed as raw fluorescence counts for calcium mobilisation. All data points represent mean ± SEM of independent experiments.
Figure S2 INSL5 does not promote signalling in CHO‐K1 cells lacking RXFP4 receptors. Non‐transfected CHO‐K1 cells were treated with increasing concentrations of hINSL5 or mINSL5 (10–12 to 10–6.5 M). Results are quantified as fold change in fluorescence over that of the vehicle treatment (fold over vehicle). Note that none of the signalling pathways tested were activated by INSL5, even though the same cells produced robust responses to 10% FBS treatment.
Figure S3 Time‐course studies (over 45 min) were carried out in CHO‐RXFP4 cells activated by either hINSL5 or mINSL5.
Supporting info item
Supporting info item
Supporting info item
Acknowledgements
This work is supported by an Australian National Health and Medical Research Council (NHMRC) programme grant (1055134; R.J.S.). D.S.H. is supported by an NHMRC Career Development Fellowship (545952). S.Y.A. is supported by a Faculty of Pharmacy and Pharmaceutical Sciences, Monash University Postgraduate Scholarship. Research at the Florey was supported by an ARC Linkage grant to R.A.D.B. and M.A.H. (LP120100654) and by the Victorian Government Operational Infrastructure Support Programme. R.A.D.B. is supported by an NHMRC Research Fellowship.
Ang, S. Y. , Hutchinson, D. S. , Patil, N. , Evans, B. A. , Bathgate, R. A. D. , Halls, M. L. , Hossain, M. A. , Summers, R. J. , and Kocan, M. (2017) Signal transduction pathways activated by insulin‐like peptide 5 at the relaxin family peptide RXFP4 receptor. British Journal of Pharmacology, 174: 1077–1089. doi: 10.1111/bph.13522.
Contributor Information
Roger J Summers, Email: roger.summers@monash.edu.
Martina Kocan, Email: martina.kocan@monash.edu.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahia DS, Wise A, Fanelli F, Lee M, Rees S, Milligan G (1998). Hydrophobicity of residue351 of the G protein Gi1α alpha determines the extent of activation by the α2A‐adrenoceptor. Biochemistry 37: 11555–11562. [DOI] [PubMed] [Google Scholar]
- Belgi A, Bathgate RA, Kocan M, Patil N, Zhang S, Tregear GW et al. (2013). Minimum active structure of insulin‐like peptide 5. J Med Chem 56: 9509–9516. [DOI] [PubMed] [Google Scholar]
- Belgi A, Hossain MA, Shabanpoor F, Chan L, Zhang S, Bathgate RA et al. (2011). Structure and function relationship of murine insulin‐like peptide 5 (INSL5): free C‐terminus is essential for RXFP4 receptor binding and activation. Biochemistry 50: 8352–8361. [DOI] [PubMed] [Google Scholar]
- Boels K, Schaller HC (2003). Identification and characterisation of GPR100 as a novel human G‐protein‐coupled bradykinin receptor. Br J Pharmacol 140: 932–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnicka‐Turek O, Mohamed BA, Shirneshan K, Thanasupawat T, Hombach‐Klonisch S, Klonisch T et al. (2012). INSL5‐deficient mice display an alteration in glucose homeostasis and an impaired fertility. Endocrinology 153: 4655–4665. [DOI] [PubMed] [Google Scholar]
- Callander GE, Thomas WG, Bathgate RA (2009). Prolonged RXFP1 and RXFP2 signaling can be explained by poor internalization and a lack of β‐arrestin recruitment. Am J Physiol Cell Physiol 296: C1058–C1066. [DOI] [PubMed] [Google Scholar]
- Cao W, Luttrell LM, Medvedev AV, Pierce KL, Daniel KW, Dixon TM et al. (2000). Direct binding of activated c‐Src to the β3‐adrenergic receptor is required for MAP kinase activation. J Biol Chem 275: 38131–38134. [DOI] [PubMed] [Google Scholar]
- Conklin D, Lofton‐Day CE, Haldeman BA, Ching A, Whitmore TE, Lok S et al. (1999). Identification of INSL5, a new member of the insulin superfamily. Genomics 60: 50–56. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK (2007). β‐arrestins and cell signaling. Annu Rev Physiol 69: 483–510. [DOI] [PubMed] [Google Scholar]
- Dezaki K, Kakei M, Yada T (2007). Ghrelin uses Galphai2 and activates voltage‐dependent K+ channels to attenuate glucose‐induced Ca2+ signaling and insulin release in islet beta‐cells: novel signal transduction of ghrelin. Diabetes 56: 2319–2327. [DOI] [PubMed] [Google Scholar]
- García‐Martínez JM, Moran J, Clarke RG, Gray A, Cosulich SC, Chresta CM et al. (2009). Ku‐0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem J 421: 29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse J, Heffron H, Burling K, Hossain MA, Habib AM, Rogers GJ et al. (2014). Insulin‐like peptide 5 is an orexigenic gastrointestinal hormone. Proc Natl Acad Sci 111: 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halls ML, Bathgate RA, Summers RJ (2006). Comparison of signaling pathways activated by the relaxin family peptide receptors, RXFP1 and RXFP2, using reporter genes. J Pharmacol Exp Ther 320: 281–290. [DOI] [PubMed] [Google Scholar]
- Haugaard‐Jönsson LM, Hossain MA, Daly NL, Craik DJ, Wade JD, Rosengren KJ, (2009). Structure of human insulin‐like peptide 5 and characterization of conserved hydrogen bonds and electrostatic interactions within the relaxin framework. Biochem J 419: 619–627. [DOI] [PubMed] [Google Scholar]
- Hossain MA, Bathgate RA, Kong CK, Shabanpoor F, Zhang S, Haugaard Jönsson LM et al. (2008). Synthesis, conformation, and activity of human insulin‐like peptide 5 (INSL5). Chembiochem 9: 1816–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu SY (1999). Cloning of two novel mammalian paralogs of relaxin/insulin family proteins and their expression in testis and kidney. Mol Endocrinol 13: 2163–2174. [DOI] [PubMed] [Google Scholar]
- Jensen DD, Godfrey CB, Niklas C, Canals M et al. (2013). The bile acid receptor TGR5 does not interact with β‐arrestins or traffic to endosomes but transmits sustained signals from plasma membrane rafts. J Biol Chem 288: 22942–22960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocan M, Pfleger KD (2011). Study of GPCR–protein interactions by BRET. Methods Mol Biol 746: 357–371. [DOI] [PubMed] [Google Scholar]
- Kocan M, Sarwar M, Hossain MA, Wade JD, Summers RJ (2014). Signalling profiles of H3 relaxin, H2 relaxin and R3(BΔ23‐27)R/I5 acting at the relaxin family peptide receptor 3 (RXFP3). Br J Pharmacol 171: 2827–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocan M, See HB, Seeber RM, Eidne KA, Pfleger KD (2008). Demonstration of improvements to the bioluminescence resonance energy transfer (BRET) technology for the monitoring of G protein‐coupled receptors in live cells. J Biomol Screen 13: 888–898. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM (2009). mTOR signaling at a glance. J Cell Sci 122: 3589–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Chen J, Sutton S, Roland B, Kuei C, Farmer N et al. (2003). Identification of relaxin‐3/INSL7 as a ligand for GPCR142. J Biol Chem 278: 50765–50770. [DOI] [PubMed] [Google Scholar]
- Liu C, Kuei C, Sutton S, Chen J, Bonaventure P, Wu J et al. (2005). INSL5 is a high affinity specific agonist for GPCR142 (GPR100). J Biol Chem 280: 292–300. [DOI] [PubMed] [Google Scholar]
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D et al. (2002). Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell 10: 457–468. [DOI] [PubMed] [Google Scholar]
- Luo X, Li T, Zhu Y, Dai Y, Zhao J, Guo Z et al. (2015). The insulinotrophic effect of insulin‐like peptide 5 in vitro and in vivo. Biochem J 466: 467–473. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Hawes BE, van Biesen T, Luttrell DK, Lansing TJ, Lefkowitz RJ (1996). Role of c‐Src tyrosine kinase in G protein‐coupled receptor‐ and Gβγ subunit‐mediated activation of mitogen‐activated protein kinases. J Biol Chem 271: 19443–19450. [DOI] [PubMed] [Google Scholar]
- Mashima H, Ohno H, Yamada Y, Sakai T, Ohnishi H (2013). INSL5 may be a unique marker of colorectal endocrine cells and neuroendocrine tumors. Biochem Biophys Res Commun 432: 586–592. [DOI] [PubMed] [Google Scholar]
- Mendoza MC, Er EE, Blenis J (2011). The Ras–ERK and PI3K–mTOR pathways: cross‐talk and compensation. Trends Biochem Sci 36: 320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousseaux D, Le Gallic L, Ryan J, Oiry C, Gagne D, Fehrentz J et al. (2006). Regulation of ERK1/2 activity by ghrelin‐activated growth hormone secretagogue receptor 1A involves a PLC/PKCε pathway. Br J Pharmacol 148: 350–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil NA, Hughes RA, Rosengren KJ, Kocan M, Ang SY, Tailhades J et al. (2016). Engineering of a novel simplified human insulin‐like peptide 5 agonist. J Med Chem 59: 2118–2125. [DOI] [PubMed] [Google Scholar]
- Polak P, Hall MN (2009). mTOR and the control of whole body metabolism. Curr Opin Cell Biol 21: 209–218. [DOI] [PubMed] [Google Scholar]
- Rorsman P, Braun M (2013). Regulation of insulin secretion in human pancreatic islets. Annu Rev Physiol 75: 155–179. [DOI] [PubMed] [Google Scholar]
- Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J et al. (2007). RAS/ERK signaling promotes site‐specific ribosomal protein S6 phosphorylation via RSK and stimulates cap‐dependent translation. J Biol Chem 282: 14056–14064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvinsky I, Meyuhas O (2006). Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem Sci 31: 342–348. [DOI] [PubMed] [Google Scholar]
- Ruvinsky I, Sharon N, Lerer T, Cohen H, Stolovich‐Rain M, Nir T et al. (2005). Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev 19: 2199–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005). Phosphorylation and regulation of Akt/PKB by the rictor–mTOR complex. Science 307: 1098–1101. [DOI] [PubMed] [Google Scholar]
- Shabanpoor F, Bathgate RA, Wade JD, Hossain MA (2013). C‐terminus of the B‐chain of relaxin‐3 is important for receptor activity. PLoS One 8: e82567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CM, Shen P, Banerjee A, Bonaventure P, Ma S, Bathgate RA et al. (2010). Distribution of relaxin‐3 and RXFP3 within arousal, stress, affective, and cognitive circuits of mouse brain. J Comp Neurol 518: 4016–4045. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton SW, Bonaventure P, Kuei C, Nepomuceno D, Wu J, Zhu J et al. (2006). G‐protein‐coupled receptor (GPCR)‐142 does not contribute to relaxin‐3 binding in the mouse brain: further support that relaxin‐3 is the physiological ligand for GPCR135. Neuroendocrinology 82: 139–150. [DOI] [PubMed] [Google Scholar]
- Gibson TB, Lawrence MC, Gibson CJ, Vanderbilt CA, McGlynn K, Arnette D et al. (2006). Inhibition of glucose‐stimulated activation of extracellular signal‐regulated protein kinases 1 and 2 by epinephrine in pancreatic beta‐cells. Diabetes 55: 1066–1073. [DOI] [PubMed] [Google Scholar]
- Thanasupawat T, Hammje K, Adham I, Ghia J, Del Bigio MR, Krcek J et al. (2013). INSL5 is a novel marker for human enteroendocrine cells of the large intestine and neuroendocrine tumours. Oncol Rep 29: 149–154. [DOI] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A et al. (2015). Tissue‐based map of the human proteome. Science 347: 1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- van der Westhuizen ET, Werry TD, Sexton PM, Summers RJ (2007). The relaxin family peptide receptor 3 activates extracellular signal‐regulated kinase 1/2 through a protein kinase C‐dependent mechanism. Mol Pharmacol 71: 1618–1629. [DOI] [PubMed] [Google Scholar]
- van der Westhuizen, ET (2008). Molecular characterisation of human and mouse relaxin‐3 receptors (RXFP3) in recombinant and endogenously expressing cell lines. Ph.D. Thesis, Monash University, Melbourne, Australia.
- Vroling B, Sanders M, Baakman C, Borrmann A, Verhoeven S, Klomp J et al. (2011). GPCRDB: information system for G protein‐coupled receptors. Nucleic Acids Res 39: D309–D319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Time course of activation of signalling pathways by hINSL5 and mINSL5 in CHO‐RXFP4 cells. In (A), phosphorylation of ERK1/2, (B) Akt Ser473, (C) Akt Thr308, (D) p38MAPK and (E) S6RP. CHO‐RXFP4 cells were treated with hINSL5 or mINSL5 (100 nM each) for up to 45 min, and activation of ERK1/2 (n = 5), Akt Ser473 (n = 5), Akt Thr308 (n = 5), p38MAPK (n = 5) and S6RP (n = 3) was determined using phospho‐specific AlphaScreen SureFire kit. In (F), there was no Ca2+ mobilisation detectable in response to stimulation by mINSL5 (100 nM) even though the same cells produced a substantial response to addition of ATP (10 μM; n = 3). Results are quantified as fold change in fluorescence over that of the vehicle treatment (fold over vehicle) for phosphorylation assays or expressed as raw fluorescence counts for calcium mobilisation. All data points represent mean ± SEM of independent experiments.
Figure S2 INSL5 does not promote signalling in CHO‐K1 cells lacking RXFP4 receptors. Non‐transfected CHO‐K1 cells were treated with increasing concentrations of hINSL5 or mINSL5 (10–12 to 10–6.5 M). Results are quantified as fold change in fluorescence over that of the vehicle treatment (fold over vehicle). Note that none of the signalling pathways tested were activated by INSL5, even though the same cells produced robust responses to 10% FBS treatment.
Figure S3 Time‐course studies (over 45 min) were carried out in CHO‐RXFP4 cells activated by either hINSL5 or mINSL5.
Supporting info item
Supporting info item
Supporting info item