

Abstract
Background and Purpose
Dihydrotanshinone I (DHI), a lipophilic component of traditional Chinese medicine Salvia miltiorrhiza Bunge, has various therapeutic effects. We investigated the anti‐fibrotic effect of DHI and its underlying mechanisms in vitro and in vivo.
Experimental Approach
Rats subjected to bile duct ligation (BDL) were treated with DHI (25 mg·kg−1·day−1, i.p.) for 14 days. Serum biochemical and liver tissue morphological analyses were performed. The human hepatic stellate cell line LX‐2 served as a liver fibrosis model in vitro. Liver fibrogenic genes, yes‐associated protein (YAP) downstream genes and autophagy markers were examined using western blot and real‐time PCR analyses. Similar analyses were done in rat primary hepatic stellate cells (pHSCs). Autophagy flux was assessed by immunofluorescence.
Key Results
In BDL rats, DHI administration attenuated liver necrosis, bile duct proliferation and collagen accumulation and reduced the expression of genes associated with fibrogenesis, including Tgfb1, Mmp‐2, Acta2 and Col1a1. DHI (1, 5, 10 μmol·L−1) time‐ and dose‐dependently suppressed the protein level of COL1A1, TGFβ1 and α‐SMA in LX‐2 cells and rat pHSCs. Furthermore, DHI blocked the nuclear translocation of YAP, which inhibited the YAP/TEAD2 interaction and its downstream fibrogenic genes, connective tissue growth factor, SOX4 and survivin. This stimulated autophagic flux and accelerated the degradation of liver collagen.
Conclusions and Implications
DHI exerts anti‐fibrotic effects in BDL rats, LX‐2 cells and rat pHSCs by inhibiting the YAP and TEAD2 complex and stimulating autophagy. These findings indicate that DHI may be a potential therapeutic for the treatment of liver fibrosis.
Abbreviations
- γ‐GT
γ‐glutamyl‐transferase
- ALP
alkaline phosphatase
- ALT
alanine transaminase
- AST
aspartate transaminase
- BDL
bile duct ligation
- COL1A1
collagen type I α 1 chain
- CTGF
connective tissue growth factor
- DHI
dihydrotanshinone I
- ECM
extracellular matrix
- MAP1LC3B
microtubule‐associated proteins 1 light chain 3
- PC III
procollagen type III
- TEAD2
TEA domain family member 2
- YAP
yes‐associated protein
- α‐SMA
α‐smooth muscle actin
Tables of Links
| TARGETS | |
|---|---|
| Other protein targets a | Enzymes b |
| Survivin (BICR5) | MMP‐2 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a,b).
Introduction
Hepatic fibrosis is a wound‐healing response to various liver injuries, such as hepatitis viral infection, cholestasis, drug toxicity, non‐alcoholic steatohepatitis and lipid oxidation (Kocabayoglu and Friedman, 2013). It is a common pathological change found in many types of chronic liver diseases and is characterized by the excessive accumulation of extracellular matrix (ECM), which exceeds the degradation capacity of MMP. This results in an imbalance between the accumulation and degradation of ECM constituents and damages the structure and function of normal liver (Bigg et al., 2007). Hepatic stellate cells (HSCs) are quiescent in healthy liver and are continuously activated and converted into myofibroblasts, which are the major source of ECM and the principal cell type involved in liver fibrogenesis during chronic liver damage (Friedman, 2008). Numerous studies have elucidated the various factors and cell signalling pathways that regulate HSC activation. In addition to classical TGFβ1 signalling, Wnt signalling and other pathways, the Hippo pathway has been shown to be closely associated with HSC activation (Mannaerts et al., 2015) and biliary epithelial cell proliferation (Bai et al., 2012). The Hippo signalling pathway was primarily identified in Drosophila and is highly conserved in mammals. This pathway plays a critical role in the control of organ size by regulating both cell proliferation and apoptosis, which, when dysregulated, mediate the progression of various diseases (Mohamed et al., 2016). When the Hippo pathway is inactivated, the transcriptional co‐activator yes‐associated protein (YAP, Yki in Drosophila) is dephosphorylated and translocates to the nucleus, where it interacts with other transcription factors, such as TEA domain‐containing protein (TEAD)1–4 (Ota and Sasaki, 2008), and promotes the expression of downstream target genes, such as connective tissue growth factor (CTGF) (Zhao et al., 2008), TGFβ (Yi et al., 2013). Most of these target genes have important functions in liver fibrogenesis.
Autophagy is a self‐digestion process of long‐lasting proteins, misfolded proteins and damaged intracellular organelles that maintains cellular homeostasis and provides material and energy for cell survival (Levine and Kroemer, 2008). Autophagy consists of several steps, including phagosome formation, autophagosome formation, autophagosome and lysosome fusion to autophagolysosomes and degradation of autophagolysosomes. Autophagy is increasingly recognized as a protective mechanism against a number of human diseases. Several studies have suggested that autophagy is closely associated with the pathophysiology of various liver diseases, such as alcoholic hepatitis (Song et al., 2015), nonalcoholic fatty liver disease (Mao et al., 2016) and liver fibrosis (Seo et al., 2014).
Liver fibrosis is a characteristic of a number of pathological conditions, including cholestasis, hepatitis and nonalcoholic fatty liver disease. Persistent liver fibrosis results in cirrhosis and hepatic carcinoma. To date, the treatment of liver fibrosis is quite limited. Some etiotropic treatments have demonstrated partial anti‐fibrosis effects of chronic liver disease, but the choice of treatment is limited (Trautwein et al., 2015). Although many compounds with potential antifibrotic activity are being tested in clinical studies, none has yet been validated clinically or commercially as a therapy for hepatic fibrosis. Therefore, there is an urgent need to identify novel and effective agents to treat cholestasis and subsequent liver fibrosis. Our laboratory is dedicated to the screening and research of natural antifibrotic compounds. We have used a high‐throughput drug‐screening model based on the collagen type I α 1 chain (COL1A1) promoter to identify potential anti‐hepatic fibrosis agents from 160 natural substances. Dihydrotanshinone I (DHI, Figure 1G), a lipophilic component from one of the best‐known traditional Chinese medicinal plants, Salvia miltiorrhiza Bunge (Tanshen), was identified as a potential therapeutic for liver fibrosis. Previous studies have described the biological functions of DHI, including liver protection (Xu et al., 2006), anti‐inflammatory activities (Choi et al., 2004) and an ability to reduce adiposity (Zhang et al., 2010), but the effects of DHI on liver fibrosis have, thus far, not been reported. Therefore, our study, for the first time, investigated the anti‐fibrotic effects of DHI on human stellate cell line LX‐2, rat primary HSCs and bile duct ligation (BDL)‐induced hepatic fibrosis rats and elucidated the potential mechanisms underlying its function in vitro and in vivo.
Figure 1.
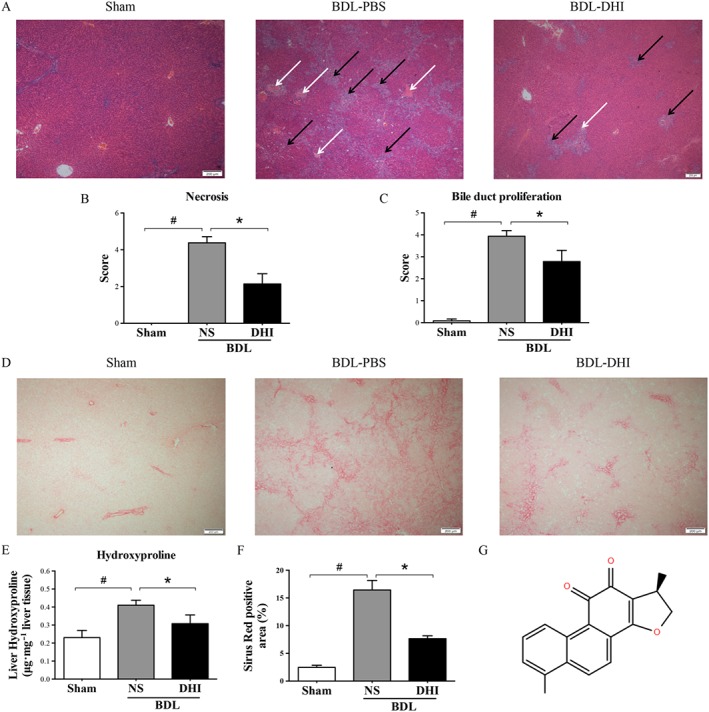
(A) Liver pathological changes were detected by H&E staining (magnification × 40). The necrosis and bile duct proliferation areas have been indicated by white and black arrows respectively. Double‐blinded quantitative assessment of (B) hepatic necrosis and (C) liver bile duct proliferation showed that DHI ameliorated BDL‐induced histopathological changes. (D) The degree of liver collagen accumulation was determined by Sirius red straining (magnification × 40). (E) Percentage of Sirius red positively strained areas and (F) liver hydroxyproline concentration assays demonstrated that DHI reduced liver fibrosis in BDL rats. (G) The structure of DHI. The values are expressed as the mean ± SEM (n = 6 in sham group; n = 7 in BDLNS/BDL‐DHI group), #P < 0.05; significantly different from sham group, * P < 0.05; significantly different from BDL‐NS group; ANOVA followed by Tukey's test.
Methods
Animal experiments
Male adult Sprague–Dawley rats (8 weeks old; 180–200 g, SPF class) were acquired from the Institute of Laboratory Animal Science (Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China). The research protocols were consistent with the regulations of Good Laboratory Practice for non‐clinical laboratory studies of drugs issued by the National Scientific and Technological Committee of People's Republic of China. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). The rats were raised in an environmentally controlled room (22 ± 2°C, 40–60% relative humidity, 12 h alternating light/dark cycle) and provided with a standard rat feedstuff and water ad libitum. They were housed in ventilated plastic cages. Rats were randomly assigned into three groups (sham, BDL‐NS and BDL‐DHI group) followed by a randomization procedure (http://www.randomizer.org/), with seven animals per group.
A total of 14 rats were prepared for BDL. These rats were anaesthetized with isoflurane. We opened the abdomen, found and isolated the choledochal duct and then ligated the bile duct using surgical sutures. Twenty‐four hours after surgery, the BDL rats received daily i.p. injections of either normal saline solution (NS) or 25 mg·kg−1 DHI suspended in NS for 14 days. A sham operation served as a healthy control. But one rat from the sham group died after the operation probably because of an overdose of anaesthetic. The sham group was one less than two other groups (n = 6 in sham group; n = 7 in BDL‐NS/BDL‐DHI group). After 14 days, after an overnight fast, blood samples were collected from the abdominal aorta and, after the animal had been killed, liver samples were also obtained and used for further analyses.
Serum biochemistry and liver histology
Serum alanine transaminase (ALT), aspartate transaminase (AST), γ‐glutamyl‐transferase (γ‐GT) and alkaline phosphatase (ALP) were analysed using a Hitachi 7100 analyser with kits obtained from Zhongsheng Beikong Biotechnology (Beijing, China). Hyaluronic acid (HA), laminin and procollagen type III (PC III) were detected by radioimmunology assays, using kits purchased from Beijing North Institute of Biological Technology (Beijing, China) according to the manufacturer's instructions. Liver tissue paraffin sections were stained with haematoxylin and eosin (H&E) and sirius red. Liver necrosis and bile duct proliferation were quantified on a 1 to 5 scale in a blinded manner using a Leica DM1000 microscope. Histological sections from each animal were observed at low magnification (10 × objective lens, Olympus, IX73) and analysed using ImageJ to calculate the percentage of the fibrotic area. Liver hydroxyproline concentration was evaluated utilizing a kit purchased from Nanjing Jiancheng Company (Nanjing, China) according to the manufacturer's instructions.
Cell culture and treatment
The human HSC line LX‐2 was cultured in DMEM/GlutaMAX I (DMEM; Invitrogen) with 10% FBS and 1% penicillin/streptomycin (P/S, Invitrogen, Carlsbad, CA, USA). Rat primary HSCs (rat pHSCs) were isolated and prepared by Procell Life Technologies, Inc (Wuhan, China). Briefly, the liver was perfused with HBSS solution without Ca2+ and Mg2+ and collagenase IV and DNAse through the hepatic portal vein. The livers were removed and forced through a 200‐gauge mesh. Parenchymal cells were separated by centrifugation. The supernatant was transferred to a 50 mL centrifuge tube and centrifuged. The cell pellet was then subjected to the discontinuous density gradient centrifugation in the presence of 18% Nycodenz stock solution. Cell purity was observed by fluorescence microscopy and determined by immunofluorescence staining for desmin. Rat pHSCs were cultured in DMEM (High Glucose) with 10% FBS and 1% antibiotics. HEK293T cells were stored by our laboratory and maintained in DMEM with 10% FBS and 1% antibiotics. All of these cells were grown in a 5% CO2 humidified atmosphere at 37°C. Rat pHSCs were cultured in untreated flasks, which induces them exhibit an activated phenotype and display a fibroblast‐like morphology; so they do not need to be starved or stimulated by TGFβ1.
Real‐time PCR
Total RNA from the cell and liver tissue samples was extracted and purified using an AZfresh™ Total RNA MiniPrep Kit (R0302C, Azanno, Sweden). Complementary DNA was generated using a Transcriptor First Strand cDNA Synthesis Kit (Roche), and relative expression levels of specific genes were determined with an ABI 7500 Fast Real‐Time PCR system. The GAPDH/Gapdh gene was used as an internal reference to normalize target gene expressions. Fold changes in the mRNA levels of target genes related to the invariant control were calculated as described previously (Yu et al., 2015).
Immunoprecipitation and western blot analysis
As described previously (Liu et al., 2016), cells were washed with PBS, and protein was extracted in RIPA buffer. Total cell lysates were immunoprecipitated with the appropriate antibodies overnight at 4°C and then incubated with Protein A/G Plus‐agarose (Santa Cruz Biotechnology, sc‐2003) for 4 h at 4°C. After five washes at 4°C, the immunocomplex was mixed with 2 × SDS loading buffer and boiled for 5 min. Equal amounts of coprecipitates or lysates were electrophoresed by SDS‐PAGE and transferred to PVDF membranes. The membranes were blocked with 5% skimmed milk in PBS‐T buffer at room temperature for 1 h and then immunoblotted overnight with primary antibodies. The membranes were subsequently incubated for 1 h with an appropriate secondary antibody and then developed using a ProteinSimple FluorChem HD2 imaging system.
Whole genome oligonucleotide microarray analysis
TRIzol reagent and a NucleoSpin RNA clean‐up kit (Macherey‐Nagel, Germany) were used to isolate and purify total RNA respectively. Each RNA sample from different groups was mixed with the same amount of solution. These samples were then labelled with Cy3 and Cy5 during the reverse transcription process using labelling kits (Genesphere Inc., Hatfield, PA, USA). These labelled cDNA samples were hybridized with the microarrays (Phanlanx. Taiwan) overnight at 45°C. After hybridization and the subsequent washing process, the arrays were analysed using a LuxScan 10 K/A dual channel laser scanner (CapitalBio, Beijing). Genes that exhibited consistent alterations (both ≥1.5 fold and ≤0.67 fold) in both microarrays were selected as differentially expressed genes.
Nuclear and cytoplasmic protein extraction
Nuclear and cytoplasmic proteins from LX‐2 cells were extracted using a Nuclear‐Cytosol Extraction Kit (Applygen Technologies Inc., Beijing, China). The extracts were mixed with 2 × SDS loading buffer and analysed by western blotting as previously described.
Immunofluorescence analysis
Cells were fixed in 4% formaldehyde for 15 min and then permeabilized with 0.5% Triton X‐100 for 30 min at room temperature. After incubation with a primary antibody at 4°C overnight and a fluorescent secondary antibody (Alexa Fluor‐488 anti‐rabbit antibody, Invitrogen) for 1 h at room temperature, the cells were imaged using a fluorescence microscope (OLYMPUS, IX73) (Liu et al., 2015b).
Statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). The results are presented as the mean ± SEM. Differences among experimental groups were assessed for significance using one‐way ANOVA with Tukey's multiple comparison tests using SPSS software. P < 0.05 was regarded as statistically significant. Tukey's methods were run only if F achieved P < 0.05 and there was no significant variance in homogeneity. For data that did not pass normality testing, non‐parametric statistics were used (Kruskal–Wallis test).
Materials
TGFβ1 (240‐B) was acquired from R&D System (Minneapolis, MN, USA). DHI was purchased from Spring & Autumn Biological Engineering Co., Ltd. (Nanjing, China). Antibodies were obtained from Cell Signalling Technology (Beverly, MA, USA). Real‐time PCR master mix was purchased from Roche (Indianapolis, IN, USA). ABI TaqMan primers/probes were purchased from Applied Biosystems (Foster City, CA, USA). The YAP/TEA domain family member 2 (TEAD2)/CTGFluc luciferase system has been described previously (Tian and Yu, 2010a). Transient transfection was performed using Lipofectamine2000 (Invitrogen, 11668019) according to the manufacturer's instructions.
Results
DHI ameliorated BDL‐induced liver injury and reduced hepatic fibrosis indices
The ALT, AST, γ‐GT and ALP levels were significantly increased in the BDL rats compared with those of the sham rats, and statistical reductions in these values were observed when the BDL rats were treated with DHI. Radioimmunoassays showed that serum liver fibrosis markers, such as HA and PC III, were statistically elevated in BDL rats; DHI decreased these markers, but only PC III was significantly decreased (Table 1).
Table 1.
Serum biochemical parameters of BDL rats
| Sham‐PBS (n = 6) | BDL‐NS (n = 7) | BDL‐DHI (n = 7) | |
|---|---|---|---|
| Serum ALT (U·L−1) | 45.64 ± 2.15 | 131.7 ± 12.67a | 84.28 ± 15.68b |
| Serum AST (U·L−1) | 160.88 ± 8.61 | 780.63 ± 63.03a | 509.11 ± 99.75b |
| Serum γ‐GT (U·L−1) | 0.38 ± 0.06 | 64.45 ± 4.79a | 38.58 ± 10.82b |
| Serum ALP (U·L−1) | 232.92 ± 14.36 | 407.36 ± 37.35a | 277.2 ± 32.34b |
| Serum HA (ng·mL−1) | 33.27 ± 6.55 | 611.06 ± 147.64a | 389.77 ± 93.61 |
| Serum LN (ng·mL−1) | 61.91 ± 11.81 | 77.15 ± 3.67 | 69.29 ± 5.62 |
| Serum PCIII (ng·mL−1) | 15.82 ± 1.44 | 31.62 ± 1.86a | 23.96 ± 1.34b |
The values are expressed as the mean ± SEM (n = 6 in sham group; n = 7 in BDL‐NS/BDL‐DHI group),
P < 0.05; significantly different from sham group,
P < 0.05; significantly different from BDL‐NS group; ANOVA followed by Tukey's test.
H&E staining assays demonstrated that the histological structure of liver from the BDL group was severely damaged. Extensive liver parenchyma necrosis and newly formed bile ducts were observed. DHI administration substantially reduced these pathological changes compared with those of the BDL control group (Figure 1A). In a blinded assessment, the DHI‐treated group had significantly lower scores for parenchymal necrosis and bile duct proliferation than those of the BDL rats (Figure 1B, C). Sirius red staining visualizes collagen, which is used to evaluate the degree and characteristics of fibrosis. In the BDL group, Sirius red‐stained collagen fibrils extended not only to the portal areas but also to the hepatic parenchyma. DHI treatment strongly attenuated the accumulation of collagen in the liver, as shown by the substantial decrease in positively stained areas (Figure 1D, E). Hydroxyproline is characteristically found in collagen fibres, and its levels indicate the severity of hepatic fibrosis. DHI dramatically decreased the increase in liver hydroxyproline concentration caused by BDL (Figure 1F). These data indicate that DHI can ameliorate the BDL‐induced liver injury and may play a therapeutic role in hepatic fibrosis.
DHI significantly reduced hepatic fibrosis‐related gene expression in BDL rats
To verify the anti‐fibrotic activity of DHI in vivo, we measured the expression of fibrogenic markers, including Tgfb1, Acta2, Col1a1, Mmp‐2, Timp1 and Timp2. TGFβ1 plays an important role in the activation of HSCs. α‐smooth muscle actin (α‐SMA), encoded by Acta2 gene, is a biomarker of HSCs. COL1A1 is a very significant component of ECM. MMP2, TIMP1 and TIMP2 contribute to the balance between synthesis and degradation of ECM. Our results showed that DHI significantly lowered the mRNA expression of these fibrogenic markers (Figure 2A). As shown in Figure 2B, the results of western blot assay confirmed that DHI has a marked anti‐fibrotic effect; the protein levels of TGFβ1, α‐SMA and COL1A1 were reduced by DHI (Figure 2C). These findings indicate that DHI treatment inhibits the progression of fibrosis in this rat model of hepatic fibrosis.
Figure 2.
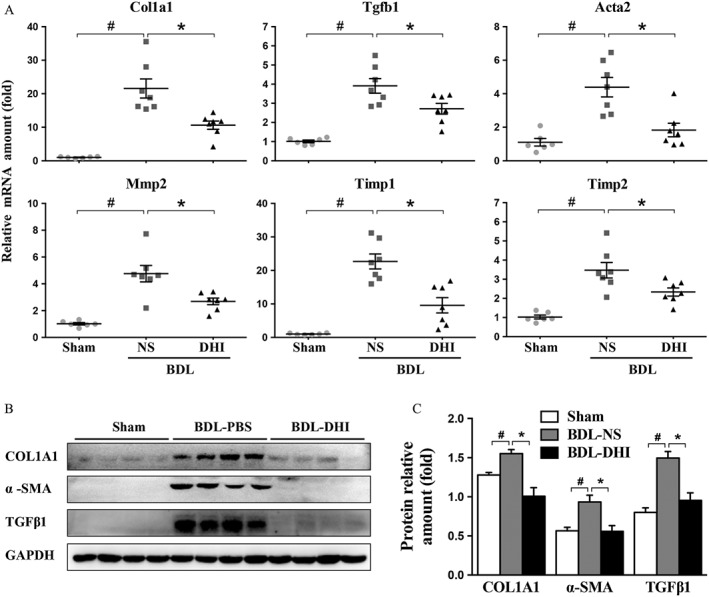
DHI significantly inhibited the expression of genes involved in hepatic fibrosis in BDL rat livers. (A) mRNA expressions of Col1a1, Mmp‐2, Acta2, Timp1/2 and Tgfβ1 in the sham, BDL‐NS and BDL‐DHI livers were normalized against Gapdh. (B) Western blot analysis of α‐SMA, collagen 1a1 (COL1A1) and TGFβ1 expressions in rat liver samples. GAPDH served as the loading control. (C) Western blot results for each animal are quantified in the three groups. The data are expressed as the mean ± SEM (n = 6 in sham group; n = 7 in BDL‐NS/BDL‐DHI group), #P < 0.05; significantly different from sham group, *P < 0.05; significantly different from BDL‐NS group ANOVA followed by Tukey's test.
DHI strongly repressed hepatic fibrogenic gene expression in HSCs
We further evaluated the in vitro anti‐fibrotic effects of DHI. The human HSC line LX‐2 was starved in serum‐free DMEM/Glutamax I for 24 h before being treated with TGFβ1. DHI was also in the same serum‐free medium and in the presence of TGFβ1. The rat HSCs did not need starvation or TGFβ1 stimulation because they can self‐activate during cultivation. DHI dose‐dependently inhibited the mRNA expression of various fibrogenic genes, including TGFβ1, α‐SMA and COL1A1, in both LX‐2 cells (Figure 3A) and rat pHSCs (Figure 3B). The protein expressions also showed a corresponding decrease after treatment with DHI in a dose‐ and time‐dependent manner in both LX‐2 cells (Figure 3C, E) and rat pHSCs (Figure 3D, F). These results suggested that DHI is a potent anti‐hepatic fibrosis agent for human/rat liver stellate cells.
Figure 3.
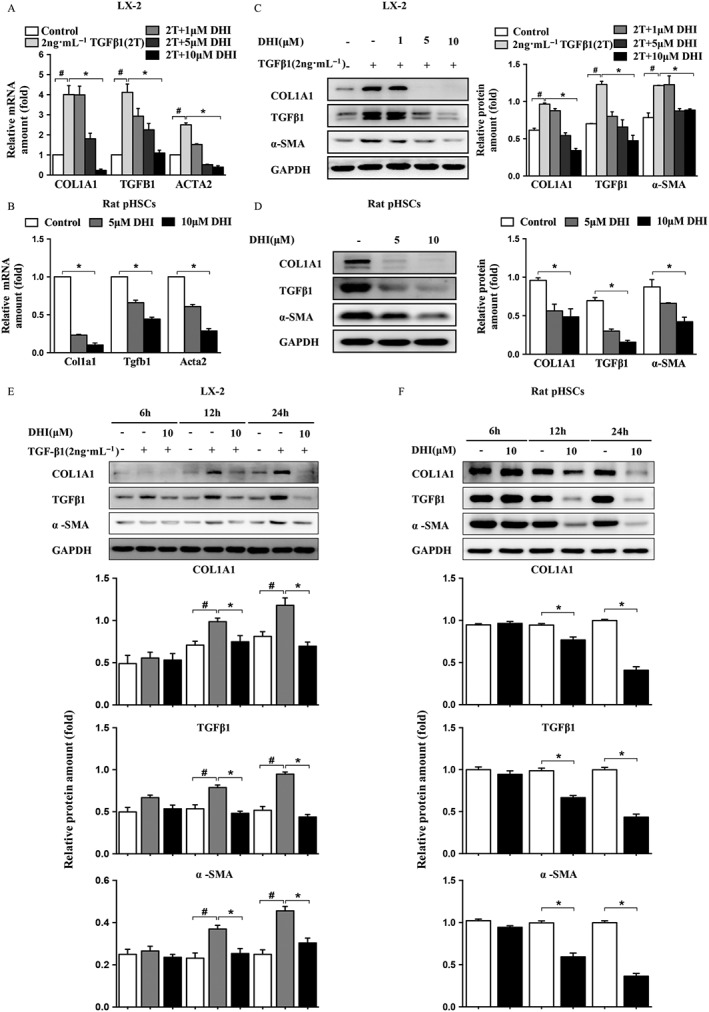
Identification of the DHI anti‐fibrotic effect in vitro. DHI suppressed the mRNA expressions of COL1A1, TGFB1 and ACTA2 in a dose‐dependent manner in (A) LX‐2 cells and (B) rat pHSCs. DHI dose‐ and time‐dependently reduced the protein expressions of COL1A1, TGF‐β1, α‐SMA in (C and E) LX‐2 cells and (D and F) rat pHSCs. The values are expressed as the mean ± SEM of five independent assays, #P < 0.05; significantly different from the control group and *P < 0.05; significantly different from the TGFβ1 treatment group in LX‐2 cells; *P < 0.05; significantly different from the control group in rat pHSCs; ANOVA followed by Tukey's test. The mRNA/protein expression levels were normalized against GAPDH.
To analyse the genome‐wide alterations in gene expression, we extracted total RNA from DHI‐treated LX‐2 cells for microarray experiments. Genes with a 1.5‐fold variation in the signal intensity were defined as significant altered genes. We identified 1573 genes that were down‐regulated and 1066 genes that were up‐regulated in 2 ng·mL−1 TGFβ1‐treated LX‐2 cells compared with untreated. In addition, 1785 genes were down‐regulated and 1690 genes were up‐regulated in 10 μM DHI‐treated LX‐2 cells compared with 2 ng·mL−1 TGFβ1 treated cells (Figure 4A). We found that a total of 806 genes were included in both sets of significantly changed genes. Among these genes, 60 were reported to be involved in hepatic fibrosis encompassing COL1A1 and ACTA2 (Figure 4B and Table 2). In addition, CTGF, a well‐known downstream gene of the Hippo pathway, was down‐regulated 0.48 fold, and a key marker of autophagy, microtubule‐associated proteins 1 light chain 3 (MAP1LC3B; LC3B), showed a 1.98‐fold increase in expression with the DHI treatment compared with that of TGFβ1‐induced cells, which indicated that DHI may inhibit the Hippo pathway and stimulate autophagy (red arrows indicate CTGF and MAP1LC3B genes in Figure 4B).
Figure 4.
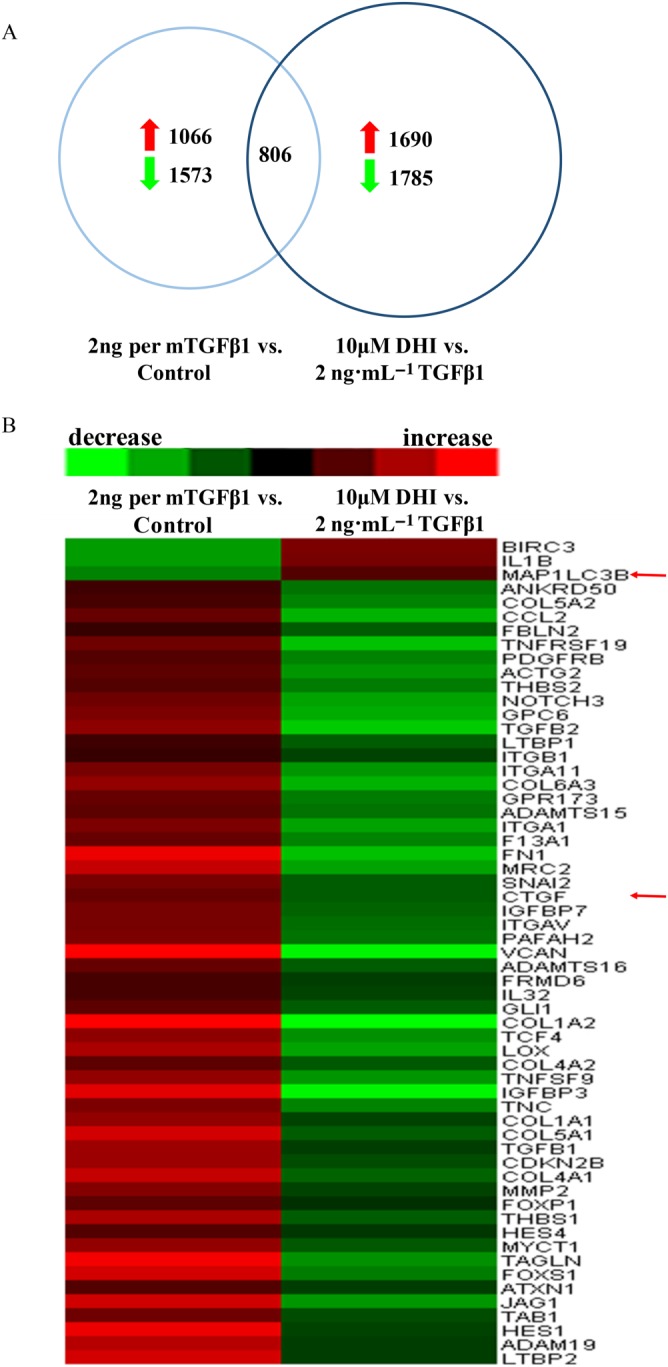
Microarray analysis of LX‐2 cells. Total RNA was isolated from five independent LX‐2 cell samples, and then each group of total RNA was mixed with the same amount of total RNA from each group then labelled with Cy3/Cy5 and measured by microarray assay. Red: increase in gene expression; green: decrease in gene expression in LX‐2 cells. (A) Number of genes significantly altered in microarray assays. (B) Expression profiling and heat map of the above‐mentioned grouping of 60 fibrogenesis‐related genes.
Table 2.
Seven processes classified by DAVID Functional Annotation Bioinformatics Microarray Analysis of significantly expressed genes
| # | Gene ontology | P‐value |
|---|---|---|
| 1 | ECM‐receptor interaction | 8.89E‐08 |
| 2 | Focal adhesion | 1.25E‐07 |
| 3 | Pathways in cancer | 3.60E‐06 |
| 4 | Cytokine‐cytokine receptor interaction | 0.002352617 |
| 5 | NOD‐like receptor signalling pathway | 0.003154988 |
| 6 | TGFβ signalling pathway | 0.017566481 |
| 7 | MAPK signalling pathway | 0.040245167 |
DHI down‐regulated the Hippo pathway and inhibited the interaction of YAP and TEAD2
Based on the results of the genome‐wide analysis, we investigated whether the Hippo pathway was involved in the anti‐hepatic fibrosis activity of DHI. To address the above question, we investigated the effect of DHI on the Hippo pathway activity by performing a dual‐luciferase assay. The results showed that DHI strongly repressed the relative luciferase activity of YAP/TEAD2/CTGFluc (Figure 5A). This effect is not the result of altered protein levels as we observed no significant protein variations in both YAP and TEAD2 after DHI treatment in LX‐2 cells (Figure 5B). It is well known that the transfer of YAP to the nucleus is critical for its growth‐promoting function. Therefore, determined whether DHI has an effect on the YAP nuclear localization. Indeed, our results showed that the translocation of YAP from the cytoplasm to the nucleus was blocked by DHI in a dose‐dependent manner (Figure 5C). If DHI prevents YAP from entering into the nucleus, we would expect a reduced YAP/TEAD2 interaction, as such an interaction requires the nuclear localization of YAP. Co‐immunoprecipitation analyses showed that DHI strongly inhibited the YAP/TEAD2 complex formation at a concentration of 10 μM (Figure 5D, E). The mRNA expression of well‐known downstream genes regulated by YAP in the Hippo pathway, including CTGF, survivin and SOX4, was dramatically down‐regulated in DHI‐treated LX‐2 cells, rat pHSCs and rat liver samples (Figure 5F–H). These results indicate that DHI supresses the translocation of YAP from the cytoplasm to the nucleus, interrupting the YAP/TEAD2 interactions and, consequently, decreasing the expression of downstream target genes, such as CTGF.
Figure 5.
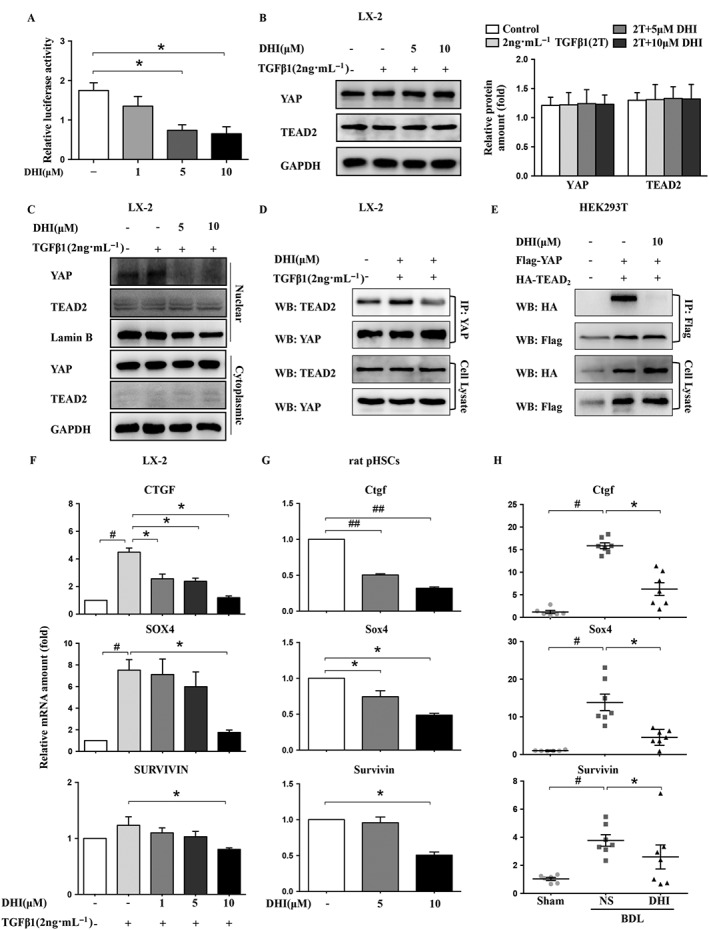
DHI interrupted the YAP/TEAD2 complex and suppressed the expression of its downstream fibrogenic genes in HSCs. LX‐2 cells were co‐transfected with pcDNA3.1‐YAP, pcDNA3.1‐TEAD2 and pGL4.17‐CTGF (A) with Renilla, and then, the luciferase activity was detected using a dual‐luciferase reporter assay, and normalized against the activity of Renilla. Western blot analysis of YAP and TEAD2 expression in LX‐2 cells (B). GAPDH served as a loading control. (C) The nuclear and cytoplasmic proteins of LX‐2 cells were separated after DHI treatment and analysed by western blotting. (D) The interaction between YAP and TEAD2 was investigated using immunoprecipitation assays of LX‐2 cells. (E) YAP‐Flag was co‐transfected with TEAD2‐HA into HEK293T cells. Whole cell extracts were immunoprecipitated with anti‐Flag and blotted with an anti‐HA antibody. The mRNA expressions of the YAP downstream genes CTGF/Ctgf, survivin and SOX4/Sox4 in LX‐2 cells (F), rat pHSCs (G) and rat liver samples (H) were detected by real‐time PCR assays and normalized against GAPDH/Gapdh. The in vitro data are expressed as the mean ± SEM of five independent assays, #P < 0.05; significantly different from the control group and *P < 0.05; significantly different from the TGFβ1 treatment group in LX‐2 cells; *P < 0.05; significantly different from the control group in rat pHSCs; the in vivo values are expressed as the mean ± SD (n = 6 in sham group; n = 7 in BDL‐NS/BDL‐DHI group), #P < 0.05; significantly different from sham group, *P < 0.05; significantly different from BDL‐NS group; ANOVA followed by Tukey's test.
DHI stimulated autophagy through the Hippo pathway in HSCs
Our genome‐wide analysis also indicated that DHI may stimulate autophagy. To further probe the effect of DHI on autophagy, two important biomarkers of autophagy were monitored: the conversion of the soluble form of LC3 (LC3‐I) to a lipidated form (LC3‐II) and the expression of the sequestosome 1 protein. Interestingly, DHI significantly increased the conversion to LC3‐II and reduced the p62 level in a dose‐ and time‐dependent manner in both LX‐2 cells (Figure 6A, B) and rat pHSCs (Figure 6C, D). Immunofluorescence showed that the endogenous LC3 expression pattern transformed from diffuse fluorescence to puncta accumulation after the treatment of DHI, which indicated the stimulation of autophagy (Figure 6E). Chloroquine, a lysosomal protease inhibitor, also enhanced the DHI‐induced accumulation of LC3B‐II in LX‐2 cells (Supporting Information Fig. S1). Based on these results, we concluded that DHI stimulates autophagy.
Figure 6.
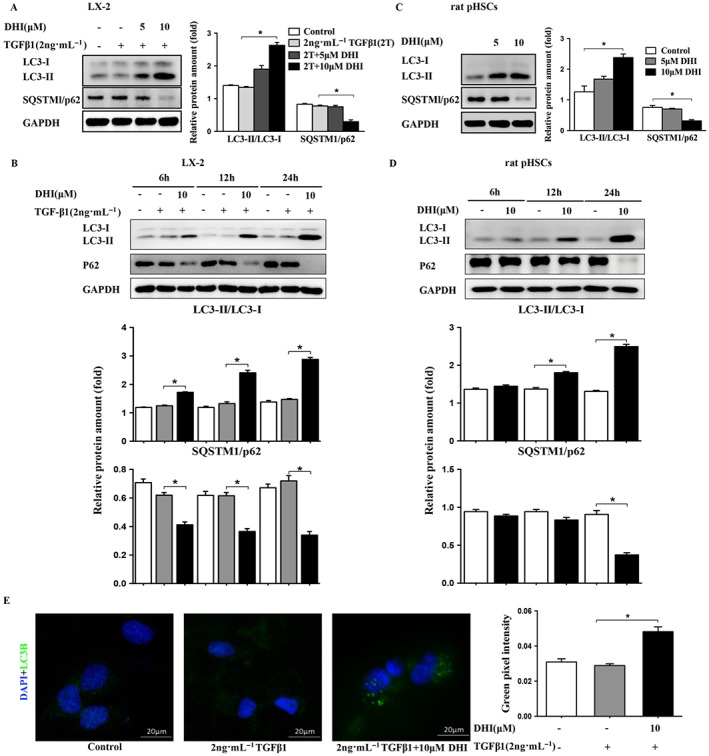
DHI enhanced autophagy in HSCs. DHI dose‐dependently and time‐dependently enhanced the conversion of LC3‐I to LC3‐II and reduced the protein expressions of SQSTMl/p62 in (A and B) LX‐2 cells and (C and D) rat pHSCs. (E) LX2 cells were treated with DHI, and LC3 puncta formation was analysed by immunofluorescence; original magnification ×400. Quantitative analysis of green pixel intensity was also performed by image J. The data are expressed as the mean ± SEM of five independent assays, #P < 0.05; significantly different from the control group and *P < 0.05; significantly different from the TGFβ1 treatment group in LX‐2 cells; *P < 0.05; significantly different from the control group of rat pHSCs; ANOVA followed by Tukey's test.
Next, we determined whether the observed autophagy phenotypes were regulated by YAP, the key regulator of Hippo pathway. We knocked down the YAP protein expression using RNA interference technology to inhibit the YAP/TEAD2 complex and found that YAP knockdown further promoted the conversion of LC3‐I to LC3‐II and enhanced the anti‐fibrotic effect of DHI by promoting collagen degradation (Figure 7A). YAP overexpression by transfecting Flag‐YAP plasmids exerted an opposite effect on autophagy activity in HSCs and the anti‐fibrotic effect of DHI, which inhibited the conversion to LC3‐II and increased collagen accumulation (Figure 7B). Taken together, our results indicate that DHI prevents the YAP/TEAD2 complex formation and then stimulates autophagy, which would have a synergistic effect on liver fibrosis.
Figure 7.
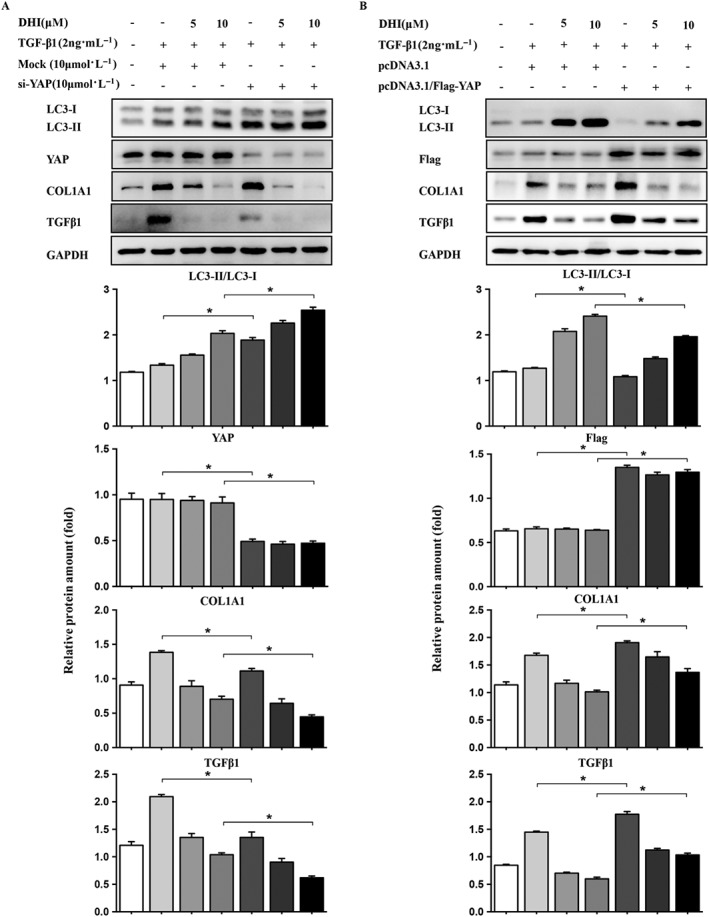
The relationship between autophagy and YAP. LX‐2 cells were transfected with (A) 15 nM YAP‐siRNA or control siRNA (Mock) or (B) Flag‐YAP or vector. The protein expressions of LC3 and liver fibrosis markers were determined by immunoblotting with the indicated antibodies. GAPDH/Gapdh was used as a loading control for all western blot assays. The data are expressed as the mean ± SEM of five independent assays, #P < 0.05; significantly different from the control group and *P < 0.05; significantly different from the TGFβ1 treatment group in LX‐2 cells; ANOVA followed by Tukey's test.
Discussion
Persistent activation of HSCs and chronic hepatic parenchymal damage contribute to liver dysfunction. Various stimuli promote the transformation of HSCs to myofibroblasts, which synthesize ECM components and damage the structure of the liver. The initiation of fibrogenesis by activated HSCs has been shown using various experimental models; the results of these studies indicate the importance of preventing HSC activation in the treatment of hepatic fibrosis. Traditional Chinese medicine is a valuable part of Chinese culture, and monomeric compounds from Chinese herbs are potential therapeutics for liver fibrosis. In previous studies, we reported that (–)‐Epigallocatechin‐3‐gallate (EGCG) (Yu et al., 2015), gastrodin (Zhao et al., 2015) and bicyclol (Zhen et al., 2015) had protective a effect on liver fibrosis, but their mechanisms were not fully elucidated. Here, we report that DHI, the lipophilic component of S. miltiorrhiza Bunge, shows excellent anti‐fibrotic efficacy. Its mechanism may be associated with suppression of the YAP and TEAD2 complex and stimulation of autophagy. This conclusion is based on four major observations: (i) BDL rats administered DHI showed significant improvements in their hepatic histological structure and a decreased accumulation of collagen. (ii) The mRNA and protein levels of fibrosis‐related genes, such as ACTA2 and COL1A1, were dramatically suppressed by DHI in vitro and in vivo. (iii) DHI blocked the nuclear translocation of YAP, disturbing the YAP/TEAD2 complex and inhibiting downstream genes, such as CTGF, SOX4 and survivin, which had a therapeutic effect on hepatic fibrosis. (iv) DHI enhanced autophagy flux to accelerate liver collagen degradation, which is associated with inhibition of YAP nuclear translocation and the YAP/TEAD2 complex.
The Hippo pathway downstream effector YAP has been characterized as a significant regulator of liver size and regeneration by promoting proliferation and inhibiting apoptosis. Mice with overexpressed YAP showed liver hyperplasia, resulting in an anomalous fivefold increase in liver/body weight ratio and an increased expression of anti‐apoptotic genes, such as survivin. YAP also plays an important role in activation of HSCs (Mannaerts et al., 2015), which is a general reaction to various types of liver damage and produces ECM to protect hepatic tissues. The overexpression of YAP can also improve cardiac regeneration after ischaemia in heart tissues (Xin et al., 2013). Incremental matrix stiffness accelerates the nuclear translocation of YAP/TAZ in lung fibrosis, which can promote fibroblast adhesion and amplify fibrosis (Liu et al., 2015a). YAP is a key regulator of tissue fibrosis because it stimulates downstream fibrogenesis and apoptosis‐related genes. Nevertheless, this function requires nuclear translocation and the participation of the transcription factors TEA domain‐containing proteins (TEADs) (Tian et al., 2010b). Inhibiting the nuclear translocation of YAP and subsequently disrupting the YAP/TEAD2 complex had anti‐fibrotic effects, which is consistent with our findings.
As demonstrated previously, DHI can induce autophagy in colon cancer cells, but the molecular mechanism remains unknown (Hu et al., 2015). In accordance with these studies, our research also showed that DHI triggered autophagy and diminished the expression of COL1A1 in HSCs, preventing hepatic fibrosis. Previous investigates have shown that activated HSCs treated with bafilomycin A1 to inhibit autophagy can increase type I collagen levels compared with those of the non‐treatment group, implying that stimulating autophagy in activated HSCs suppresses the accumulation of type I collagen (Seo et al., 2014). Autophagy in the kidney facilitates type I collagen degradation induced by TGFβ1 stimulation without changing the mRNA level, which indicates that autophagy exerts a cytoprotective effect against renal fibrosis (Kim et al., 2012). The β2‐adrenoceptor also expedites type I collagen degradation by increasing autophagy flux in cardiac fibroblasts, indicating that autophagy might mitigate cardiac fibrosis (Aranguiz‐Urroz et al., 2011). In contrast to these results, several studies reported that autophagy is increased during HSC activation and promotes the loss of cytoplasmic lipid droplets to provide energy for the activation (Hernandez‐Gea et al., 2012). Although the reasons for this discrepancy remain unclear, it is likely that autophagy causes diverse effects on HSCs during different periods of activation. When quiescent HSCs are stimulated, autophagy accelerates hepatic fibrosis and HSC activation by altering lipid metabolism. However, in activated HSCs, autophagy has the opposite effect on the development of liver fibrosis by promoting the degradation of type I collagen.
The relationship between YAP and autophagy has not been fully elucidated. However, YAP enhanced autophagic flux in human ovarian and breast cancer cell lines and decreased the sensitivity of cancer cells to chemotherapeutic drugs, such as cisplatin (Song et al., 2015; Xiao et al., 2016). Contrasting results were found in human hepatic stellate LX‐2 cells in this study. Increased conversion of LC3‐II and reduced expression of fibrogenic genes were detected along with YAP gene knockout, showing that inhibiting YAP expression or blocking YAP/TEAD2 complex can both lead to activation of autophagy and inhibition of liver fibrosis. In contrast, the overexpression of YAP decreased the autophagy level in HSCs and anti‐fibrotic effect of DHI. Based on these results, YAP can down‐regulate autophagy activity and then inhibit collagen degradation. The effects of DHI on this gene results in a synergistic effect on fibrosis, as it promoted its anti‐fibrotic effects by stimulating autophagy to accelerate collagen degradation and diminish the TGFβ1 protein levels. However, the mechanism underlying the relationship between YAP and autophagy still needs to be explored further.
Despite the fact that significant findings were revealed by this investigation, it also has limitations. We hypothesized that the anti‐fibrotic effects of DHI are mediated by disrupting the YAP/TEAD2 complex and then down‐regulating the expression of CTGF, which plays a crucial role in liver fibrosis. Other genes downstream of the Hippo pathway have been directly or indirectly implicated in hepatic fibrosis, but these results need to be confirmed. SOX4 is a well‐known epithelial‐mesenchymal transition (EMT)‐associated transcription factor, and inhibiting SOX4 using siRNA blocked EMT (Song et al., 2015). Survivin, which is overexpressed in HSCs isolated from fibrotic liver, has been identified as a target of NF‐κB and can prevent caspase‐induced apoptosis (Czech et al., 2013), which is associated with the apoptosis resistance of activated HSCs. Few reports have examined the correlation between liver fibrosis and these genes, except CTGF. We expect that this issue will be further elucidated by future studies.
Conclusion
Hepatic fibrosis is a common pathological process found in most chronic liver diseases. The terminal stage of hepatic fibrosis is known as cirrhosis and has a high mortality rate and is associated with multiple complications. The present study showed that DHI, a monomeric compound from a traditional Chinese medicinal plant, can improve liver function and alleviate hepatic fibrosis in vivo in BDL rats and in vitro in a human HSC line LX‐2 and rat pHSCs. The underlying mechanism is associated with blocking YAP nuclear localization and subsequently disrupting the YAP/TEAD2 complex, which stimulates autophagy and down‐regulates the expression of fibrogenic genes (Figure 8). Our findings suggest that DHI displays therapeutic efficacy on liver fibrosis in vitro and in vivo and may be a novel therapeutic strategy for patients with hepatic fibrosis in the near future.
Figure 8.
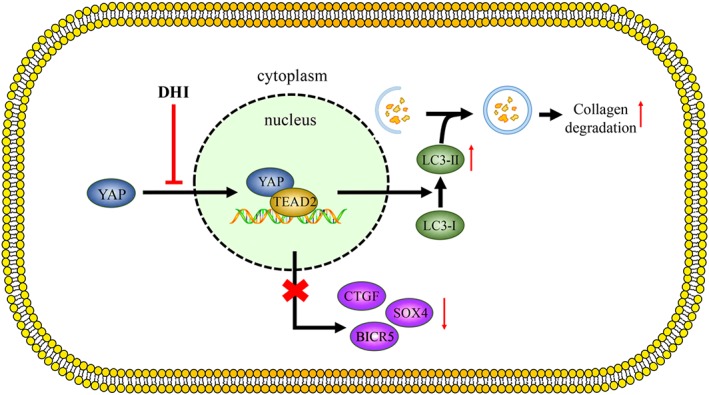
The beneficial effects of DHI in vitro and in vivo. DHI can inhibit the nuclear translocation of YAP and then disrupt the YAP/TEAD2 interaction, which down‐regulates the expression of its downstream fibrogenic genes and might be associated with autophagy stimulation.
Author contributions
M.G., H.L., H.H. and R.S. designed the research project; M.G., Y.Z., N.L. and S.Z. performed the experiment; M.G. analysed the data; H.H. and R.S. contributed the reagents and materials; and M.G., W.Z., Y.Z. and J.Y. wrote and modified the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Treatment with CQ augmented the expression of LC3B‐II in LX‐2 cells. The expression of LC3B‐II was measured by western blotting. Data are presented as the mean ± SD of 3 independent assays. *P < 0.05, significantly different from the TGFβ1 treatment group in LX‐2 cells; ANOVA followed by Tukey's test.
Acknowledgements
This work was supported by the financial support from the following: CAMS Initiative for Innovative Medicine of China (CAMS‐I2M), the National Nature Scientific Foundation of China (81673497, 81473249 and 81321004), the National Mega‐project for Innovative Drugs of China (2014ZX09201042) and the Beijing Nature Scientific Foundation (7162130).
Ge, M. , Liu, H. , Zhang, Y. , Li, N. , Zhao, S. , Zhao, W. , Zhen, Y. , Yu, J. , He, H. , and Shao, R. (2017) The anti‐hepatic fibrosis effects of dihydrotanshinone I are mediated by disrupting the yes‐associated protein and transcriptional enhancer factor D2 complex and stimulating autophagy. British Journal of Pharmacology, 174: 1147–1160. doi: 10.1111/bph.13766.
Contributor Information
Hongwei He, Email: hehwei@imb.pumc.edu.cn.
Rong‐guang Shao, Email: shaor@imb.pumc.edu.cn.
References
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The concise guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranguiz‐Urroz P, Canales J, Copaja M, Troncoso R, Vicencio JM, Carrillo C et al. (2011). Beta(2)‐adrenergic receptor regulates cardiac fibroblast autophagy and collagen degradation. Biochim Biophys Acta 1812: 23–31. [DOI] [PubMed] [Google Scholar]
- Bai H, Zhang N, Xu Y, Chen Q, Khan M, Potter JJ et al. (2012). Yes‐associated protein regulates the hepatic response after bile duct ligation. Hepatology 56: 1097–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigg HF, Rowan AD, Barker MD, Cawston TE (2007). Activity of matrix metalloproteinase‐9 against native collagen types I and III. FEBS J 274: 1246–1255. [DOI] [PubMed] [Google Scholar]
- Choi HS, Cho DI, Choi HK, Im SY, Ryu SY, Kim KM (2004). Molecular mechanisms of inhibitory activities of tanshinones on lipopolysaccharide‐induced nitric oxide generation in RAW 264.7 cells. Arch Pharm Res 27: 1233–1237. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech B, Dettmer K, Valletta D, Saugspier M, Koch A, Stevens AP et al. (2013). Expression and function of methylthioadenosine phosphorylase in chronic liver disease. PLoS One 8: e80703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman SL (2008). Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 88: 125–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez‐Gea V, Ghiassi‐Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z et al. (2012). Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 142: 938–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu T, Wang L, Zhang L, Lu L, Shen J, Chan RL et al. (2015). Sensitivity of apoptosis‐resistant colon cancer cells to tanshinones is mediated by autophagic cell death and p53‐independent cytotoxicity. Phytomedicine 22: 536–544. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SI, Na HJ, Ding Y, Wang Z, Lee SJ, Choi ME (2012). Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)‐beta1. J Biol Chem 287: 11677–11688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocabayoglu P, Friedman SL (2013). Cellular basis of hepatic fibrosis and its role in inflammation and cancer. Front Biosci (Schol Ed) 5: 217–230. [DOI] [PubMed] [Google Scholar]
- Levine B, Kroemer G (2008). Autophagy in the pathogenesis of disease. Cell 132: 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Lagares D, Choi KM, Stopfer L, Marinkovic A, Vrbanac V et al. (2015a). Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol 308: L344–L357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Ma Y, He HW, Wang JP, Jiang JD, Shao RG (2015b). SLC9A3R1 stimulates autophagy via BECN1 stabilization in breast cancer cells. Autophagy 11: 2323–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Zhang CX, Ma Y, He HW, Wang JP, Shao RG (2016). SphK1 inhibitor SKI II inhibits the proliferation of human hepatoma HepG2 cells via the Wnt5A/beta‐catenin signaling pathway. Life Sci 151: 23–29. [DOI] [PubMed] [Google Scholar]
- Mannaerts I, Leite SB, Verhulst S, Claerhout S, Eysackers N, Thoen LF et al. (2015). The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J Hepatol 63: 679–688. [DOI] [PubMed] [Google Scholar]
- Mao Y, Yu F, Wang J, Guo C, Fan X (2016). Autophagy: a new target for nonalcoholic fatty liver disease therapy. Hepat Med 8: 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed A, Sun C, De Mello V, Selfe J, Missiaglia E, Shipley J et al. (2016). The Hippo effector TAZ (WWTR1) transforms myoblasts and its abundance is associated with reduced survival in embryonal rhabdomyosarcoma. J Pathol 240: 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota M, Sasaki H (2008). Mammalian Tead proteins regulate cell proliferation and contact inhibition as transcriptional mediators of Hippo signaling. Development 135: 4059–4069. [DOI] [PubMed] [Google Scholar]
- Seo HY, Jang BK, Jung YA, Lee EJ, Kim HS, Jeon JH et al. (2014). Phospholipase D1 decreases type I collagen levels in hepatic stellate cells via induction of autophagy. Biochem Biophys Res Commun 449: 38–43. [DOI] [PubMed] [Google Scholar]
- Song Q, Mao B, Cheng J, Gao Y, Jiang K, Chen J et al. (2015). YAP enhances autophagic flux to promote breast cancer cell survival in response to nutrient deprivation. PLoS One 10 e0120790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander S et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W, Yu J (2010a). Structural and functional analysis of the YAP‐binding domain of human TEAD2. Proc Natl Acad Sci U S A 107: 7293–7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W, Yu J, Tomchick DR, Pan D, Luo X (2010b). Structural and functional analysis of the YAP‐binding domain of human TEAD2. Proc Natl Acad Sci U S A 107: 7293–7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautwein C, Friedman SL, Schuppan D, Pinzani M (2015). Hepatic fibrosis: concept to treatment. J Hepatol 62: S15–S24. [DOI] [PubMed] [Google Scholar]
- Xiao L, Shi XY, Zhang Y, Zhu Y, Zhu L, Tian W et al. (2016). YAP induces cisplatin resistance through activation of autophagy in human ovarian carcinoma cells. Onco Targets Ther 9: 1105–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J et al. (2013). Hippo pathway effector YAP promotes cardiac regeneration. Proc Natl Acad Sci U S A 110: 13839–13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu JK, Hiroshi K, Zheng JJ, Jiang T, Yao XS (2006). Protective effect of tanshinones against liver injury in mice loaded with restraint stress. Acta Pharm Sin 41: 631–635. [PubMed] [Google Scholar]
- Yi C, Shen Z, Stemmer‐Rachamimov A, Dawany N, Troutman S, Showe LC et al. (2013). The p130 isoform of angiomotin is required for YAP‐mediated hepatic epithelial cell proliferation and tumorigenesis. Sci Signal 6: ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu DK, Zhang CX, Zhao SS, Zhang SH, Zhang H, Cai SY et al. (2015). The anti‐fibrotic effects of epigallocatechin‐3‐gallate in bile duct‐ligated cholestatic rats and human hepatic stellate LX‐2 cells are mediated by the PI3K/Akt/Smad pathway. Acta Pharmacol Sin 36: 473–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J et al. (2010). The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell 19: 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Ye X, Yu J, Li L, Li W, Li S et al. (2008). TEAD mediates YAP‐dependent gene induction and growth control. Genes Dev 22: 1962–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Li N, Zhen Y, Ge M, Li Y, Yu B et al. (2015). Protective effect of gastrodin on bile duct ligation‐induced hepatic fibrosis in rats. Food Chem Toxicol 86: 202–207. [DOI] [PubMed] [Google Scholar]
- Zhen Y‐Z, Li N‐R, He H‐W, Zhao S‐S, Zhang G‐L, Hao X‐F et al. (2015). Protective effect of bicyclol against bile duct ligation‐induced hepatic fibrosis in rats. World J Gastroenterol 21: 7155–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Treatment with CQ augmented the expression of LC3B‐II in LX‐2 cells. The expression of LC3B‐II was measured by western blotting. Data are presented as the mean ± SD of 3 independent assays. *P < 0.05, significantly different from the TGFβ1 treatment group in LX‐2 cells; ANOVA followed by Tukey's test.