

Summary
Aging is attended by a progressive decline in protein homeostasis (proteostasis), aggravating the risk for protein aggregation diseases. To understand the coordination between proteome imbalance and longevity, we addressed the mechanistic role of the quality-control ubiquitin ligase CHIP, which is a key regulator of proteostasis. We observed that CHIP deficiency leads to increased levels of the insulin receptor (INSR) and reduced lifespan of worms and flies. The membrane-bound INSR regulates the insulin and IGF1 signaling (IIS) pathway and thereby defines metabolism and aging. INSR is a direct target of CHIP, which triggers receptor monoubiquitylation and endocytic-lysosomal turnover to promote longevity. However, upon proteotoxic stress conditions and during aging, CHIP is recruited toward disposal of misfolded proteins, reducing its capacity to degrade the INSR. Our study indicates a competitive relationship between proteostasis and longevity regulation through CHIP-assisted proteolysis, providing a mechanistic concept for understanding the impact of proteome imbalance on aging.
Keywords: C. elegans, Drosophila, ubiquitin, proteostasis, chaperone, insulin signaling, aging, longevity, DAF-2, CHIP
Graphical Abstract
Highlights
-
•
The ubiquitin ligase CHIP triggers insulin receptor turnover
-
•
Insulin receptor level is linked to insulin and IGF1 signaling and longevity
-
•
Engagement of CHIP in protein quality control limits insulin receptor degradation
-
•
Proteotoxic stress aggravates insulin receptor stability, drives aging, and shortens lifespan
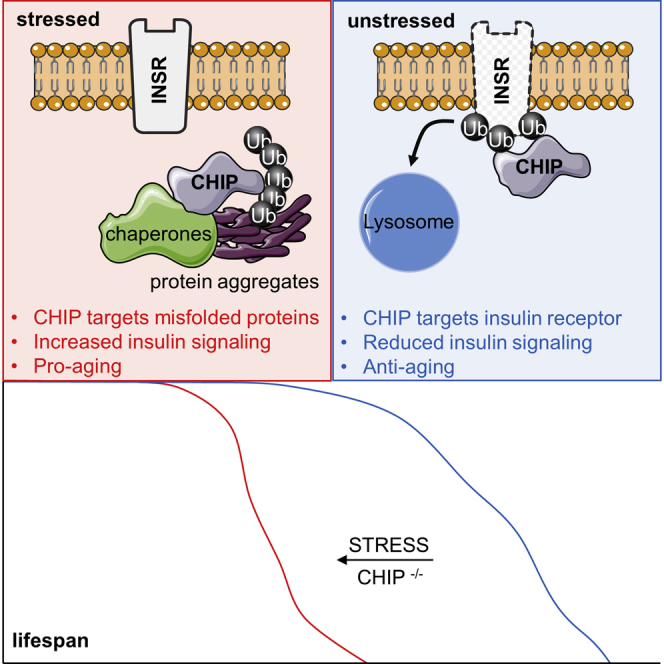
A ubiquitin ligase promotes either longevity or proteostasis, depending on the cellular status.
Introduction
Proteostasis represents the functional balance of the proteome, which is permanently challenged by environmental stress and alterations in physiology. A decline in proteostasis leads to progressive aggregation of misfolded proteins, overwhelming the cellular protein quality control (PQC) networks. The maintenance of PQC mechanisms provided by molecular chaperones and proteolytic pathways is not only a long-term challenge for individual cells but also for the entire organism, given that damaged proteins dramatically accumulate with progressive aging (López-Otín et al., 2013).
The quality-control E3 ubiquitin ligase CHIP acts along with molecular chaperones and degradation machineries to keep the cellular balance between protein folding and degradation (Arndt et al., 2007, Connell et al., 2001, Cyr et al., 2002, Murata et al., 2003). CHIP provokes the ubiquitylation of damaged proteins consigned by chaperone partners to induce disposal through endocytic-lysosomal pathways (Okiyoneda et al., 2010, Slotman et al., 2012), proteasomal degradation (Connell et al., 2001, Cyr et al., 2002, Murata et al., 2003), and autophagy (Arndt et al., 2010). In agreement with its role in protein quality control, CHIP prevents an age-related pathologic accumulation of protein aggregates (Connell et al., 2001, Petrucelli et al., 2004). Surprisingly, however, CHIP−/− deletion mice exhibit normal embryonic development and unaffected turnover of many known CHIP substrates, suggesting functional redundancy among quality-control ubiquitin ligases (Min et al., 2008, Morishima et al., 2008). On the other hand, CHIP deficiency accelerates aging (Min et al., 2008), which points to the existence of at least one critical CHIP-specific substrate that controls longevity. However, the mechanistic link between proteostasis decline and aging has remained unexplained so far.
Insulin and insulin-like growth factor 1 (IGF1) signaling (IIS) represents an evolutionarily conserved pathway that senses nutrient supply and determines longevity in multicellular organisms (Barbieri et al., 2003). IIS defines various cellular processes, including innate immunity and stress response. Reduced insulin signaling extends lifespan and improves proteotoxic stress resistance via the activation of multiple transcription factors (Cohen et al., 2010, Hesp et al., 2015, Kenyon, 2010, Kevei and Hoppe, 2014). DAF-2 has been identified as a membrane bound receptor regulating the IIS pathway in Caenorhabditis elegans. Mechanistically, downregulation of DAF-2 function activates the FOXO transcription factor DAF-16, causing dramatic lifespan extension (Kenyon et al., 1993). Besides extensive genetic analyses of the IIS pathway, however, the impact of DAF-2 protein level regulation on aging has not been addressed.
Here, we reveal an important function of CHIP-mediated proteolysis in IIS. We identify a degradation pathway that controls the level of active DAF-2/INSR in C. elegans, Drosophila melanogaster, and human cells. The key regulator of this conserved mechanism is the ubiquitin ligase CHIP, which fine-tunes IIS via monoubiquitylation and subsequent endocytic-lysosomal degradation of the insulin receptor. Upon proteotoxic stress and during aging, however, CHIP is preferentially shuttled towards quality-control pathways for degradation of damaged proteins, causing a stabilization of the INSR. Accordingly, proteotoxic accumulation of damaged proteins or aberrant CHIP function attenuates INSR degradation and affects metabolism and longevity through increased IIS. In conclusion, our results demonstrate a competition between proteostasis and lifespan regulation through CHIP-assisted proteolysis, providing an evolutionarily conserved concept for understanding the impact of proteome imbalance on aging.
Results
Lack of the E3 Ligase CHIP Increases IIS Activity
To obtain insight into the physiological function of CHIP, we examined its role in aging, using C. elegans as a model organism. Interestingly, the loss-of-function alleles chn-1(by155) and chn-1(tm2692), both lacking the single CHIP worm ortholog CHN-1, showed reduced lifespan (Figures 1A and S1A; Table S1). The short lifespan phenotype of both deletion mutants was also recapitulated by RNAi depletion of chn-1 (Figure S1B; Table S1). chn-1(by155) worms exhibited reduced body size, which often reflects limited nutrient uptake (Figure 1B). Therefore, we investigated the effect of chn-1 depletion in context of the well-established longevity pathway triggered by dietary restriction (DR). DR can be reproduced genetically by using eat-2(ad465) mutants, which exhibit reduced pharyngeal pumping rates, causing lower food intake, smaller body size, and extended lifespan (Lakowski and Hekimi, 1998). However, deletion of chn-1 only partially shortened the lifespan of eat-2(ad465) mutant worms, suggesting that CHN-1 acts parallel to DR in a different longevity pathway (Figure 1C; Table S1).
Figure 1.

CHIP Modulates Lifespan via Increased Insulin Signaling
(A) C. elegans chn-1 mutants (by155 and tm2692) exhibit short lifespans.
(B) Body size of chn-1 deficient worms is reduced, compared to wild-type worms (mean values ± SEM were obtained by measuring at least n = 20 worms). ∗∗∗p < 0.001.
(C) Deletion of chn-1 shortens lifespan of eat-2(ad465) worms.
(D and E) DAF-16::GFP overexpression extends the lifespan of chn-1(by155) and chn-1(tm2692) worms.
(F) Depletion of daf-16 by RNAi shortens the lifespan of chn-1(by155) worms.
(G) chn-1 deficient worms exhibit delayed nuclear localization of DAF-16::GFP triggered by heat stress. DAF-16::GFP localization in wild-type, chn-1(by155) mutant worms, and worms overexpressing chn-1 (CHN-1::FLAG) was scored as nuclear or diffuse cytoplasmic (nuclear and cytoplasmic). Individual worms were classified based on the intracellular distribution of the DAF-16::GFP fluorescence. Data shown are from one representative experiment (n = 100).
(H) CHN-1 is important for efficient activation of DAF-16. Real-time PCR identified reduced levels of sod-3 and sip-1 mRNAs in worms lacking chn-1, similar to the daf-16 loss-of-function mutant (mean values were obtained in n = 3 independent biological replicates).
(I) Ubiquitous depletion of dCHIP through RNAi causes reduced lifespan of flies.
(J) Loss of dCHIP activates AKT signaling (S505, p-AKT) and stabilizes dINSR in flies (mean values were obtained in n = 5 independent experiments).
(K) Depletion of CHIP in HEK293 cells activates AKT signaling (phosphorylated AKT [S473, p-AKT] was quantified from n = 5 independent experiments).
(H, J, and K) Data are means SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See Table S1 for lifespan statistics.
See also Figure S1.
Figure S1.

CHN-1 Modulates IIS in C. elegans, Related to Figure 1
(A) The loss-of-function alleles chn-1(by155) and chn-1(tm2692) lack CHN-1 protein. To detect CHN-1 protein indicated lysates of adult worms were subjected to immunoblot with anti-CHN-1 antibodies.
(B) Ubiquitous depletion of chn-1 by RNAi results in reduced lifespan of wild-type worms (n = 6 independent experiments).
(C) Representative images of cytoplasmic and nuclear DAF-16::GFP localization in worms. Scale bar represents 100 μm.
IIS is well known to regulate both lifespan and metabolism in C. elegans (Perez and Van Gilst, 2008). Hence, the phenotypes we observed in the chn-1 deletion mutants could also be linked to this conserved signaling pathway (Kimura et al., 1997). Reduced activity of the DAF-2/INSR mobilizes the downstream FOXO transcription factor DAF-16, resulting in enhanced metabolism and extended lifespan of nematodes (Lin et al., 2001, Ogg et al., 1997). Nuclear localization of DAF-16 can be triggered by its overexpression to support transcriptional activity and longevity (Henderson and Johnson, 2001). In fact, transgenically expressed DAF-16::GFP was able to suppress both chn-1(by155) and chn-1(tm2692) deletion mutants and extended lifespan (Figures 1D and 1E; Table S1). Conversely, short lifespan caused by RNAi-mediated downregulation of daf-16 was not further reduced by chn-1 deletion. The combined depletion of both genes rather resulted in almost identical lifespan curves, suggesting an epistatic relationship (Figure 1F; Table S1). In light of this genetic interaction, we further monitored DAF-16 nuclear localization as visual readout to test whether insulin signaling is increased in the absence of chn-1. In fact, localization of DAF-16::GFP into the nucleus was delayed in the chn-1(by155) deletion mutant (Figures 1G and S1C; Table S2), whereas it was enhanced by transgenic overexpression of chn-1::FLAG (Figures 1G, S1C, and S2A; Table S2). We wondered whether the reduced lifespan and DAF-16 mobility of chn-1 deletion worms might reflect reduced transcriptional activity of DAF-16. Recent studies identified several target genes of DAF-16 important for lifespan regulation (Lee et al., 2003). Following mRNA expression levels by real-time PCR, we identified reduced amounts of sod-3 and sip-1 mRNAs in worms lacking chn-1, similar to daf-16 loss-of-function, suggesting that CHN-1 is important for efficient activation of DAF-16 (Figure 1H).
Figure S2.

CHN-1/CHIP Overexpression Affects Lifespan, Related to Figure 2
(A) Level of CHN-1::FLAG overexpression strain was analyzed by immunoblotting of indicated worm lysates (day 1 and 10 old animals).
(B) Overexpression of chn-1 does not enhance lifespan of wild-type worms (n = 4 independent experiments).
(C) Overexpression of dCHIP was driven by the use of the midgut specific TIGS-2 driver line and dCHIP levels were verified in gut extracts. Equal loading was verified by ponceau S staining (ponceau).
(D) Flies of the genotype TIGS-2/ UAS-dCHIP were either placed on ethanol or RU486 (1 μg/ml or 5 μg/ml) containing food to induce dCHIP expression. dCHIP expression leads to toxicity in a dose-dependent manner and results in reduced lifespan.
(E) Depletion of CHIP in HEK293 cells does not affect the expression level of PDK1, AKT, mTOR, or Hsp90.
(F) Specificity of anti-DAF-2 antibodies was evaluated using daf-2(e1370) worms expressing thermolabile DAF-2 protein. Worms were grown at 25°C until day 1 of adulthood, lysed, and analyzed by western blotting using DAF-2 specific antibodies.
Given the evolutionarily conserved function of insulin signaling (Barbieri et al., 2003), we wondered whether CHIP depletion provides similar short-lived phenotypes in D. melanogaster. In fact, RNAi-mediated downregulation of Drosophila CHIP (dCHIP) strongly reduced the lifespan of flies (Figure 1I; Table S1). Whereas the reduction of CHN-1 and dCHIP levels diminished longevity, overexpression did not enhance lifespan of worms and was even toxic in flies (Figures S2A–S2D; Table S1). Overexpression of the chaperone-associated ubiquitin ligase might cause an excessive routing of chaperone clients onto degradation pathways and might lead to non-physiological changes of the chaperone network given the HSF-1 regulating activity of CHIP (Dai et al., 2003, Qian et al., 2006).
Lifespan regulation critically depends on IIS signaling, which is activated upon ligand binding to the INSR at the plasma membrane and involves a cascade of intracellular phosphorylation events (van der Horst and Burgering, 2007). Given the impact of CHIP depletion on lifespan regulation and genetic correlation with IIS, we wondered whether any of the IIS downstream targets are regulated by CHIP-mediated degradation. In fact, depletion of CHIP did not influence the protein level of PDK1, AKT, or mTOR; also, the level of the chaperone Hsp90 remained unchanged in human embryonic kidney (HEK293) cells (Figure S2E). However, it increased AKT kinase phosphorylation in flies (S505, p-AKT) and human cells (S473, p-AKT) (Figures 1J and 1K), providing further evidence for increased insulin signaling upon depletion of CHIP (Su et al., 2011). Collectively, these results suggest that CHIP provides a conserved role in the regulation of IIS activity.
CHIP Activity Correlates with DAF-2/INSR Protein Level
Given our genetic epistasis data placing CHN-1 function upstream of DAF-2, we performed western blot analysis to monitor the DAF-2 protein level in vivo (Figure S2F). Intriguingly, DAF-2 abundance increased specifically later in life and was significantly enhanced in chn-1(by155) mutants especially at day 10 of adulthood (Figures 2A and 2B). Given that CHN-1 teams up with the related ubiquitin ligase UFD-2 in another degradation pathway regulating turnover of the myosin chaperone UNC-45 (Hoppe et al., 2004), we tested whether UFD-2 would be additionally involved in DAF-2 regulation. Loss-of-function ufd-2(tm1380) mutants displayed reduced UNC-45 turnover (Figure 2C). Importantly, however, in contrast to chn-1(by155), ufd-2(tm1380) mutants did not exhibit a stabilization of DAF-2 (Figure 2C). To rule out unspecific consequences of CHN-1 depletion on overall protein turnover, we compared the total amount of ubiquitylated proteins in wild-type and chn-1(by155) mutant worms at days 1 and 10 of adulthood. Since there was no increase of ubiquitylated proteins detected in worm lysates lacking CHN-1 (Figure 2D), the stabilization of DAF-2 suggests a substrate-specific role of CHN-1 in IIS regulation rather than a general defect in ubiquitin-dependent protein degradation. In flies, the observed induction of AKT kinase phosphorylation upon loss of dCHIP was also accompanied by an increase of the dINSR level (Figures 1J and 2E). In agreement with increased insulin signaling activity, the stabilized receptor did not accumulate in detergent-insoluble aggregates (Figure 2F). Finally, depletion of human CHIP in HEK293 cells resulted in a significant increase in the protein amount of human INSR, whereas transcript levels remained unchanged (Figure 2G). Conversely, overexpression of CHIP caused reduced protein levels of INSR (Figure 2H). These data demonstrate that CHIP plays a conserved role in limiting INSR levels.
Figure 2.

CHIP Regulates DAF-2/INSR Protein Level
(A, B) chn-1(by155) shows increased levels of DAF-2 protein, analyzed by immunoblotting of worm lysates of indicated age (day 0; L4 worms) (mean values were obtained in n = 6 independent experiments).
(C) Lack of UFD-2 does not stabilize DAF-2.
(D) chn-1(by155) mutation does not increase the global level of ubiquitylated proteins in young or aged adult worms.
(E) Ubiquitous depletion of dCHIP increases levels of dINSR (detected in whole fly extracts) (mean values were obtained in n = 5 independent experiments).
(F) dINSR stabilized upon dCHIP depletion does not accumulate in detergent-insoluble aggregates. Fly heads were lysed in SDS-containing buffer and the resultant extract was separated in an SDS-soluble and -insoluble fraction by centrifugation at 100,000 × g.
(G) Depletion of CHIP in human HEK293 cells causes increased levels of INSR as determined by western blot, whereas mRNA levels remain unchanged (mean values were obtained in n = 5 independent experiments).
(H) Overexpression of CHIP results in decreased level of INSR in HEK293 cells (mean values were obtained in n = 5 independent experiments).
(B, E, G, and H) Data are means ± SEM. ∗p < 0.05, ∗∗p < 0.01.
See also Figure S2.
CHIP Binds and Monoubiquitylates the INSR
Regarding the correlation between CHN-1 and DAF-2 protein levels, we wondered whether the two proteins are directly associated. Indeed, we found that FLAG-tagged CHN-1 binds DAF-2 in immunoprecipitation assays by using lysates of transgenic worms expressing CHN-1::FLAG (Figures 3A, 3B, and S3A). Intriguingly, the interaction was most obvious at day 10 of adulthood, which correlates with the timing of DAF-2 protein level increase in worms lacking CHN-1 (Figures 2A and 2B). This interaction is evolutionarily conserved given that we also detected binding between the INSR and CHIP in human cell lysates (Figure 3C).
Figure 3.

CHIP Binds and Monoubiquitylates DAF-2/INSR
(A and B) CHN-1 interacts with DAF-2. Receptor complexes were isolated by immunoprecipitation from wild-type worms expressing CHN-1::FLAG (1st or 10th day of adulthood).
(C) CHIP interacts with INSR in human cells. After transient transfection of HEK293 cells with a FLAG::CHIP expressing plasmid, INSR complexes were isolated by immunoprecipitation.
(D and E) Ubiquitylation reactions of DAF-2 and dINSR were carried out as indicated using CHN-1(WT), CHN-1(U-box), CHIP, dCHIP, WWP-1, and UFD-2 as ubiquitin ligases.
(F and G) DAF-2/INSR undergoes monoubiquitylation. In vitro ubiquitylation assays were carried out as indicated using wild-type ubiquitin or a lysine-lacking variant (UbK0). Samples from experiments analyzing mono vs. polyubiquitylation (G) were run on separate gels and are presented here separated by a dashed line.
(H) CHIP does not ubiquitylate IGF1R. Ubiquitylation reactions of IGF1R and INSR were carried out as indicated using CHIP as ubiquitin ligase. Samples from experiments analyzing IGF1R and INSR ubiquitylation were run on separate gels and are presented here separated by a dashed line.
(I) Depletion of CHIP in human HEK293 cells does not affect protein levels of IGF1R. Data are means ± SEM obtained in n = 4 independent experiments.
See also Figure S3.
Figure S3.

CHN-1/CHIP-Dependent Ubiquitylation of the INSR, Related to Figure 3
(A) CHN-1 protein level does not significantly change during aging. CHN-1 protein level was evaluated by western blotting using CHN-1 specific antibodies in whole worm lysates of adult worms (representative immunoblots are shown of n = 3 experiments).
(B and C) Ubiquitylation of INSR was carried out as indicated using CHN-1, CHN-1(ΔU-box), CHN-1(U-box), (TPR)CHN-1, CHIP, UFD-2, and WWP-1 as ubiquitin ligases.
(D) Mutations in the TPR domain of CHN1 ((TPR)CHN-1) abrogate INSR ubiquitylation. The ubiquitin ligase WWP-1 is unable to ubiquitylate INSR. Ubiquitylation reaction was carried out using FLAG-tagged ubiquitin (FLAGUb) in the presence of E. coli BL21 lysates. Activity of used E3 enzymes was evaluated by western blotting using anti-FLAG specific antibodies showing ubiquitylated bacterial proteins.
(E) In vitro ubiquitylated DAF-2, INSR were subjected to mass spectrometry to identify the lysine residues used for ubiquitin attachment. Conserved residues are indicated in red. Previously published residues are marked as follows: #O’Rahilly et al., 1991; +Kim et al., 2011; ∗Udeshi et al., 2013.
Regarding the direct interaction, we tested whether the INSR is a target for CHIP ligase activity and performed in vitro ubiquitylation assays with recombinantly expressed proteins. Indeed, we detected efficient ubiquitylation of DAF-2, dINSR, or INSR either by CHN-1, dCHIP, or human CHIP, respectively (Figures 3D–3G). This reaction was completely blocked by a H218Q point mutation within the conserved U-box domain of CHN-1, essential for E3 ligase catalytic activity (U-box), or complete deletion of the entire domain (ΔU-box) (Figures 3D, S3B, and S3C) (Rosser et al., 2007). The amino-terminal tetratricopeptide repeat (TPR) domain of CHIP mediates interaction with Hsp70 and Hsp90, and is required for the intrinsic ability of CHIP to recognize substrate proteins (Rosser et al., 2007). Mutation of the TPR domain also inhibited receptor ubiquitylation, revealing that the domain contributes to the direct recognition of INSR by CHIP in the chaperone-free reaction (Figures S3C and S3D). In contrast to the role of CHN-1/CHIP, DAF-2 was not modified by the related ubiquitin ligases UFD-2 (Hoppe et al., 2004) or WWP-1, a HECT domain protein implicated in control of DR-dependent longevity (Figures 3D and S3B–S3D) (Carrano et al., 2009).
Different lysine (K) residues of ubiquitin are used for isopeptide bond formation between single ubiquitin molecules to form polyubiquitin chains with distinct linkages (Komander and Rape, 2012). Regardless of the use of wild-type (Ub) or a mutant form of ubiquitin without lysines (UbK0), we obtained a similar pattern of ubiquitylation on DAF-2 or the INSR in our defined in vitro studies, indicating that CHN-1/CHIP mediates multiple monoubiquitylation of the INSR rather than polyubiquitylation (Figures 3F and 3G). These experiments suggest a conserved role of CHIP in monoubiquitylation of the INSR in worms, flies, and human cells. The lysine residues that are ubiquitylated were identified by mass spectrometric analyses of modified DAF-2/INSR obtained from independent in vitro experiments. K1047 was preferentially used for INSR ubiquitylation in vitro and clusters with the additional conserved conjugation site K1057 (Figure S3E). In support of our findings, recent proteomic studies also identified K1047 as a major residue targeted for receptor ubiquitylation in human cells (Kim et al., 2011). In contrast to the INSR, CHIP is not able to ubiquitylate the insulin-like growth factor receptor 1 (IGF1R) (Figure 3H). Accordingly, depletion of CHIP did not stabilize IGF1R (Figure 3I). Together, these findings highlight the substrate-specific role of CHIP in INSR degradation both in vitro and in vivo.
Ubiquitylation Provokes Endocytic-Lysosomal Turnover of the INSR
Notably, proteasomal inhibition by MG132 did not affect INSR stability in HEK293 cells (Figure 4A). In contrast, the amount of INSR increased upon treatment of human cells with the lysosomal inhibitors bafilomycin A1 (BafA1) and chloroquine, or a dynamin inhibitor that blocks endocytosis (Figure 4A). Moreover, dynamin inhibition reversed the reduced INSR level boosted by CHIP overexpression (Figure 4B). These findings would be consistent with an endocytic-lysosomal or autophagic degradation pathway for INSR. To distinguish between these possibilities, we depleted the essential autophagy factor ATG7 from human HEK293 cells by siRNA and monitored INSR level (Komatsu et al., 2005). Efficient depletion of ATG7 significantly inhibited autophagy, as evident from p62 accumulation and reduced levels of lipidated LC3 (LC3-II) (Figure 4C). Yet, ATG7 depletion did not affect INSR stability, thus excluding a critical contribution of autophagy to INSR degradation. Importantly, the CHIP-targeted lysine K1047 of the INSR is crucial for receptor degradation in human cells given that the conservative K1047R amino acid substitution stabilized the INSR to the same extent as inhibition of lysosomal degradation by chloroquine treatment (Figures 4D, 4E, and S3E). Taken together, our data reveal an endocytic and lysosomal pathway for INSR turnover initiated by CHIP-dependent monoubiquitylation.
Figure 4.

CHIP-Mediated Ubiquitylation Initiates Endocytic INSR Degradation
(A) INSR level is sensitive to inhibition of endocytosis and lysosomal degradation. HEK293 cells were treated with 10 μM MG132, 200 nM Bafilomycin A1, 100 μM chloroquine, or 10 μM dynamin inhibitor V34-2 for 16 hr before lysis and levels of indicated proteins were determined by immunoblotting.
(B) CHIP overexpression causes diminished amounts of INSR in HEK293 cells, which is reversed by dynamin inhibition.
(C) Depletion of ATG7 in HEK293 does not affect INSR level. HEK293 cells were transiently transfected for 48 hr with siRNA specific for ATG7 (+) or control siRNA (−).
(D and E) Lysine residue 1047 (K1047), located in the cytoplasmic domain of INSR, is required for endocytic turnover of the receptor. HEK293 cells were transiently transfected for 48 hr with INSR and INSR-K1047R expressing plasmids or empty vector, and treated with 100 μM chloroquine for 16 hr prior to lysis as indicated.
(A–C and E) Data are means ± SEM obtained in n = 5 independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
CHIP-Dependent Ubiquitylation of DAF-2 Is Linked to Longevity
We further assessed the physiological relevance of the ubiquitin ligase activity by expressing wild-type and mutated variants of chn-1 in the chn-1(by155) deletion strain. Unlike wild-type CHN-1, the catalytically inactive CHN-1(U-box) mutant defective in DAF-2 ubiquitylation was not able to rescue the shortened lifespan phenotype of chn-1(by155) worms (Figures 5A, 5B, and S3A). This result supports the idea that CHN-1-dependent ubiquitylation of DAF-2 is tightly linked to regulation of longevity. To further clarify whether the level of DAF-2 strictly correlates with lifespan, we analyzed its protein amount in different C. elegans daf-2 mutant alleles (Figure 5C) (Kimura et al., 1997, Malone and Thomas, 1994, Patel et al., 2008, Scott et al., 2002). The hypomorphic daf-2(e1368) mutant promotes longevity and is able to extend the short lifespan of chn-1-deficient worms (Figure 5D). This genetic suppression further supports the idea that the shortened lifespan of chn-1 depletion is caused by DAF-2 stabilization (Figures 2A and 2B). In contrast to e979 and e1391 temperature-sensitive mutant proteins, the DAF-2e1368 protein amount is elevated at day 5 of adulthood (Figure 5C). When combined with chn-1(by155), daf-2(e1368) lifespan was slightly reduced, which is in line with further increase of DAF-2e1368 level in the chn-1(by155) mutant background (Figures 5D and 5E). Contrary to all other long-lived loss-of-function alleles, the daf-2(gk390525) allele (Thompson et al., 2013, Bulger et al., 2017) displayed increased DAF-2 level already at day 1 of adulthood and shortened lifespan (Figures 5C and 5F). Intriguingly, daf-2(gk390525) harbors a single point mutation in K1614E, one of the lysine residues in DAF-2 targeted for CHN-1-dependent ubiquitylation (Figures 3D and S3E). Thus, defective ubiquitylation seems to result in an elevated DAF-2gk390525 protein level and premature aging. Indeed, the reduced lifespan of daf-2(gk390525) worms can be extended again by RNAi-mediated depletion of daf-2 (Figure 5G). Moreover, the DAF-2gk390525 level was not further increased and lifespan of daf-2(gk390525) is not more diminished when combined with the chn-1(by155) mutation, again highlighting the substrate-specific role of CHN-1 in DAF-2 ubiquitylation and turnover (Figures 5H, 5I, and S4A). These observations further indicate that CHN-1-mediated ubiquitylation regulates DAF-2 stability, insulin signaling, and longevity.
Figure 5.

CHN-1 Ligase Activity Regulates DAF-2 Degradation and Longevity
(A and B) CHN-1::FLAG expression rescues the short lifespan of chn-1(by155) mutant worms; catalytic inactive CHN-1(U-box)::FLAG does not.
(C) DAF-2 protein level increases during aging in analyzed daf-2 mutant alleles. Importantly, daf-2(gk390525) allele displays increased DAF-2 level already at day 1 of adulthood.
(D) Short lifespan of chn-1(by155) is suppressed by daf-2(e1368) loss-of-function mutant.
(E) Deletion of chn-1 increases level of DAF-2e1368 mutant protein, analyzed by immunoblotting of worm lysates of indicated age. (F) daf-2(gk390525) worms exhibit short lifespan.
(G) Depletion of DAF-2 by RNAi extends the short lifespan of daf-2(gk390525) allele.
(H) Deletion of chn-1 does not further increase the level of DAF-2K1614R mutant protein (daf-2(gk390525) allele), analyzed by immunoblotting of worm lysates of indicated age.
(I) Loss of chn-1 shortens lifespan of daf-2(gk390525) allele to the level of chn-1(by155) worms. See Table S1 for lifespan statistics.
See also Figures S4 and S6.
Figure S4.

Defective Ubiquitylation Contributes to Elevated DAF-2gk390525 Protein Levels, Related to Figure 5
(A) Loss of chn-1 does not significantly increase the level of DAF-2gk390525 protein (mean values were obtained in n = 3 independent experiments).
Proteolytic Regulation of IIS Is Diminished by Proteotoxic Stress
To address the physiological relevance of CHN-1/CHIP-dependent INSR degradation, CHN-1 was downregulated either during larval development of worms or from day 1 of adulthood onward. It turned out that chn-1 is most important in adult life because its depletion specifically shortened life expectancy when applied post-developmentally (Figures S5A and S5B). chn-1 mutants are sensitive to heat stress, which might be linked to the age-related decline in proteostasis known to cause high abundance of misfolded proteins (Figure S5C) (Murata et al., 2003, Walther et al., 2015). In contrast to molecular chaperones, CHN-1 levels remained unchanged during aging and were not upregulated in response to different proteotoxic conditions (Figures 2A and S5D). A decline in proteostasis would thus redirect CHN-1 activity toward degradation of misfolded proteins and limit monoubiquitylation of DAF-2. Indeed, worms shifted to high temperature at the beginning of adulthood (D1) showed elevated levels of DAF-2 protein similar to those of chn-1 lacking mutants grown at normal conditions (Figures 2A, 6A, and 6B). In line with a central role in proteostasis, depletion of CHN-1/dCHIP in worms and flies caused high sensitivity to oxidative stress induced by paraquat treatment (Figures 6C and 6D). Moreover, in flies and human cells we also detected elevated level of the INSR upon oxidative stress (Figures 6E and 6F). This increase most likely reflects reduced receptor turnover caused by limited disposability of CHIP under proteotoxic stress. In support of this hypothesis, INSR levels could be restored in paraquat-treated human cells by overexpression of CHIP (Figure 6F). Thus, ubiquitin ligase activity of CHIP seems to be limited under proteotoxic stress conditions. In addition, we also tested the impact of canavanine on dINSR stability in flies. Incorporation of this non-proteinogenic amino acid leads to aberrantly folded polypeptides and increased proteotoxicity (Dasuri et al., 2011, Rosenthal et al., 1987). Similar to increased oxidative stress, canavanine treatment strongly stabilized dINSR, providing additional support for the correlation between proteotoxic stress and INSR stability (Figure S5E).
Figure S5.

Role of CHIP in Proteostasis, Related to Figure 6
(A and B) CHN-1 function during development is not important for lifespan regulation.
(C) Post developmental knockdown of chn-1 results in higher sensitivity against heat stress (35°C) as compared to wild-type worms (mean values were obtained in n = 4 independent experiments).
(D) CHN-1 protein level does not change when exogenous stress is applied to day 1 adult worms.
(E) Treatment with L-canavanine (20 mM) leads to a strong stabilization of dINSR in adult head extracts of aged wild-type flies (3 weeks old).
(F) polyQ40 exerts mild toxicity in the body-wall muscle cells of chn-1 depleted worms assessed by counting body bending of individual animals (1st or 5th day of adulthood).
(G) Expression of aggregation prone polyQ-103 in HEK293 cells reduces INSR turnover rate as determined by mRNA/protein ratio (mean values were obtained in n = 3 independent experiments).
(C and G) Data are means ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Figure 6.

Regulation of INSR Stability Competes with Degradation of Damaged Proteins
(A and B) Temperature-shifted (15°C to 25°C at day 1 of adulthood) wild-type worms show an age-related increase in DAF-2 protein level (mean values were obtained in n = 6 independent experiments).
(C) chn-1(by155) mutant worms are sensitive to oxidative stress. Young adult wild-type and chn-1(by155) mutant worms were treated with 5 mM paraquat, and their survival rates were measured for the indicated time (mean values were obtained in n = 4 independent experiments).
(D) dCHIP depleted flies show increased sensitivity to paraquat treatment, causing reduced lifespan. Control and RNAi depleted flies were treated with food containing 20 mM paraquat, and their survival rates were determined (n = 5 independent experiments).
(E) Paraquat treatment leads to a significant stabilization of dINSR as determined in adult head extracts (mean values were obtained in n = 3 independent experiments).
(F) Oxidative stress induced by paraquat causes stabilization of INSR in HEK293 cells, which is compensated by CHIP overexpression (mean values were obtained in n = 5 independent experiments).
(G) chn-1 deletion enhances the age-dependent aggregation of the polyQ protein Q44::YFP. Worms with diffuse or aggregated intestinal staining of Q44::YFP were scored and plotted against the age of the worms (mean values were obtained in n = 6 independent experiments).
(H) CHIP is recruited to inclusion bodies formed by aggregation prone polyQ-103 in HeLa cells as detected by confocal microscopy. Scale bar represents 10 μm.
(I) Expression of aggregation prone polyQ-103 in HEK293 cells causes INSR but not IGF1R stabilization (mean values were determined in n = 5 independent experiments).
(B, C, E, F, H, and J) Data are means ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See Table S1 for lifespan statistics.
See also Figure S5.
The age-related collapse of proteostasis networks often induces aggregation of damaged and metastable proteins (López-Otín et al., 2013, Walther et al., 2015). By transgenic expression of aggregation-prone fluorescently tagged polyglutamine (polyQ) expansions in intestinal and muscle cells (Morley et al., 2002, Mohri-Shiomi and Garsin, 2008), we found that acute depletion of chn-1 by RNAi caused early-onset protein aggregation and cellular toxicity in C. elegans (Figures 6G and S5F). This observation is in line with recent findings (Kalia et al., 2011, Matsumura et al., 2013, Min et al., 2008, Rosser et al., 2007), reflecting an important role of CHIP in maintenance of the cellular proteome. We next addressed the role of CHIP in protein aggregation by using human cells expressing an extended polyglutamine repeat sequence fused to GFP (polyQ103-GFP). PolyQ103-GFP formed large, intracellular inclusion bodies that efficiently recruited cytosolic CHIP (Figure 6H). The recruitment toward protein aggregates diminished the pool of CHIP throughout the cytosol, which did not affect the amount of IGF1R, but specifically led to a strong increase of INSR level (Figure 6I). The observed increase could be attributed to reduced INSR turnover by monitoring the mRNA to protein ratio (Figure S5G). Taken together, our findings illustrate that CHIP integrates proteolysis of damaged proteins, which accumulate under proteotoxic stress conditions or during aging, with regulation of INSR turnover and IIS activity (Figure 7).
Figure 7.

Model for the Reciprocal Regulation of Proteostasis and Longevity
The membrane bound DAF-2/INSR triggers IIS and thereby adjusts stress response and lifespan. Under normal conditions (unstressed), CHIP binds DAF-2/INSR to mediate monoubiquitylation and initiate the endocytic-lysosomal turnover of the receptor. Upon proteotoxic stress conditions, CHIP activity is recruited toward quality-control pathways for degradation of damaged proteins. Thus, proteome maintenance competes with ubiquitylation and degradation of the INSR, which in turn affects IIS, metabolism, and lifespan.
Discussion
This study uncovered a balanced crosstalk between proteostasis maintenance and longevity, which paves the way toward understanding how proteome instability contributes to aging. A central node of this conserved mechanism is the quality-control E3 ubiquitin ligase CHIP, which tightly cooperates with the molecular chaperones Hsp70 and Hsp90 in the degradation of damaged proteins (Arndt et al., 2007, Connell et al., 2001, Cyr et al., 2002, Murata et al., 2003, Petrucelli et al., 2004).
We demonstrate here that CHIP mediates ubiquitin-dependent degradation of the INSR and thereby modulates insulin signaling and longevity. CHIP ubiquitylated the INSR in vitro in the absence of its partner chaperones, pointing to a specific and direct recognition of the receptor, distinct from the reported role in chaperone-dependent processing of misfolded proteins (Zhang et al., 2005, Stankiewicz et al., 2010) (Figures 3D–3G). The substrate specificity of the observed interaction is further highlighted by the fact that CHIP does not ubiquitylate or induce the degradation of the closely related IGF1R (Figures 3H, 3I, and 6I). Moreover, INSR ubiquitylation could not be induced by other ubiquitin ligases such as WWP-1 or UFD-2, which cooperates with CHN-1/CHIP on muscle-specific degradation pathways. In agreement with a specific functional interplay, DAF-2/INSR was stabilized by CHN-1/CHIP depletion or by mutations of DAF-2/INSR ubiquitylation sites utilized by CHIP (Figures 2A, 2B, 2E, 2G, 4E, 5C, and S3E). In all cases, receptor stabilization significantly shortened lifespan, illustrating the close interdependence of CHIP-mediated INSR degradation and longevity regulation.
In contrast to the reduced lifespan caused by CHN-1 or dCHIP depletion, overexpression did not enhance longevity of worms and was even toxic in flies (Figures S2A–2D; Table S1). CHIP is known to modulate the proteotoxic stress response by activating the transcription factor HSF-1 (heat shock factor 1) and by reducing the level of Hsp70 after heat shock (Dai et al., 2003, Qian et al., 2006). Thus, the rather toxic effect of elevated CHIP level might be caused by non-physiological changes of the chaperone network with deleterious consequences (Yan et al., 2003, Stankowski et al., 2011). Accordingly, endogenous CHN-1 protein amounts also remained unchanged during aging or in response to proteotoxicity (Figures 2A and S5D). CHN-1-dependent DAF-2 turnover was also not affected by dietary interventions (Figure S6A). These observations suggest an intricately balanced regulation of CHN-1/CHIP activity to avoid excessive rerouting of chaperone substrates toward degradation. Irrespective of organismal changes, however, overexpression of CHIP directly affected INSR protein levels and compensated the stress-induced stabilization of the receptor in human cells (Figures 4B and 6F).
Figure S6.
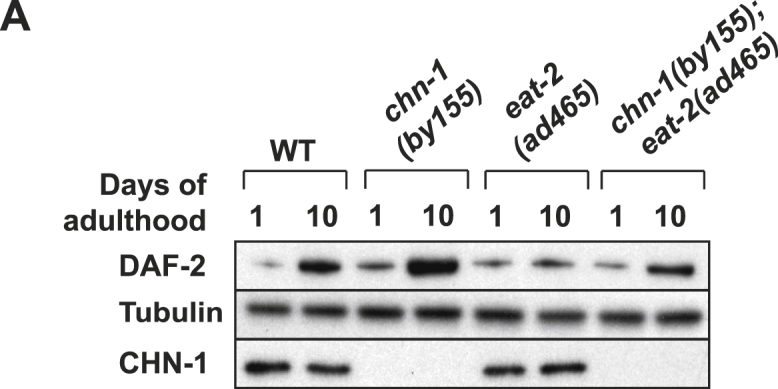
DAF-2 Level Upon Dietary Restriction, Related to Figure 5
(A) The age-related stabilization of DAF-2 in chn-1 mutants is not further increased in eat-2(ad465) worms. Western blot analysis of DAF-2 protein was carried out by using anti-DAF-2 antibodies on whole-worm lysates of indicated age.
chn-1(by155) mutants were sensitive to heat stress, oxidative stress, and protein aggregation, similar to what we observed upon inactivation of CHIP (Figures 6C, 6D, 6G, S5C, and S5F). Moreover, these proteotoxic conditions also caused elevated DAF-2/INSR levels (Figures 6A, 6E, 6F, 6I, S5E, and S5G). Given these findings, it is intriguing to speculate that CHIP integrates proteolysis of damaged proteins with INSR degradation to control metabolism and longevity (Figure 7). Accumulation of damaged proteins upon proteotoxic conditions and during aging challenges the dynamic equilibrium of quality-control networks and shuttles CHIP into PQC pathways to degrade damaged proteins, causing stabilization of the functional INSR and consequently lifespan reduction (Figure 7). From the organismal perspective, the prioritization of proteome stability is highly beneficial and ensures reproduction early in life, but this is provided at the expense of aging. CHN-1 activity seems to be most important later in life (Figures 2A, 2B, 6A–6C, 6G, S5A, and S5B), suggesting that other recently identified quality-control ubiquitin ligases might help to maintain proteostasis during development (Fang et al., 2014, Heck et al., 2010).
Like C. elegans and Drosophila, CHIP-deficient mice develop normally but exhibit reduced lifespan associated with age-related pathophysiological defects (Min et al., 2008). With regard to the conserved regulation, it is intriguing that mutations in the orthologous human gene STUB1 are linked to accelerated aging (Heimdal et al., 2014). Given the importance of insulin signaling for metabolic regulation, it is also interesting that liver cells of CHIP-null mutant mice develop insulin resistance (Kim et al., 2016). Moreover, a single point mutation in K1095E, a lysine residue in the human INSR that we identified to be ubiquitylated, is also associated with severe insulin resistance of patients suffering from diabetes (Ardon et al., 2014, O’Rahilly et al., 1991). These strong functional similarities suggest an evolutionarily conserved coordination of proteostasis and aging regulated by CHIP-assisted degradation. A detailed understanding of how INSR degradation is adjusted to metabolic insults might lead to efficient therapies for age-related diseases, including obesity and diabetes.
STAR★Methods
Key Resources Table
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-His | QIAGEN | Cat# 34660 |
| Rabbit polyclonal anti-DAF-2 | This Study | N/A |
| Rabbit polyclonal anti-CHN-1 | This Study | N/A |
| Mouse monoclonal anti-FLAG | Sigma-Aldrich | Cat# F3165; RRID: AB_259529 |
| Rabbit anti-UNC-45 | Gazda et al., 2013 | N/A |
| Mouse Monoclonal anti-alpha Tubulin (Clone DM 1A) | Sigma-Aldrich | Cat# T9026; RRID: AB_477593 |
| Mouse Monoclonal anti-alpha Tubulin (Clone B-5-1-2) | Sigma-Aldrich | Cat# T6074; RRID: AB_477582 |
| Peroxidase-conjugated AffiniPure Goat Anti-Mouse IgG+IgM | Jackson Immuno Research | Cat# 115-035-003; RRID: AB_10015289 |
| Peroxidase-conjugated AffiniPure Mouse Anti-Rabbit IgG+IgM | Jackson Immuno Research | Cat# 211-035-109; RRID: AB_2339150 |
| Donkey anti-rabbit IRDye 800CW/680 | Li-Cor | Cat# 926-32223; RRID: AB_621845 |
| Donkey anti-mouse IRDye 800CW/680 | Li-Cor | Cat# 926-32212; RRID: AB_621847 |
| Mouse monoclonal anti-Actin | Abcam | Cat# ab8224; RRID: AB_449644 |
| Rabbit polyclonal anti-AKT | Cell Signaling | Cat# 9272; RRID:AB_329827 |
| Rabbit monoclonal anti-Phospho AKT (Ser473) | Cell Signaling | Cat# 4060S; RRID: AB_2315049 |
| Rabbit polyclonal anti-phospho Drosophila AKT (Ser505) | Cell Signaling | Cat# 4054S; RRID: AB_331414 |
| Rabbit monoclonal anti-ATG7 | Novus Biologicals | Cat# NBP1-95872; RRID: AB_11011190 |
| Rabbit polyclonal anti-Chicken IgG | Sigma-Aldrich | Cat# C2288; RRID: AB_258791 |
| Rabbit polyclonal anit-CHIP | Milipore | Cat# PC711; RRID: AB_2198058 |
| Rat monoclonal anti-GFP | Chromotek | Cat# 3h9, ABIN398304; RRID: AB_10773374 |
| Mouse monoclonal anti-Histidine-Tag | Serotec | Cat# MCA1396; RRID: AB_322084 |
| Rat monoclonal anti-Hsp90 | Abcam | Cat# ab13494; RRID: AB_300398 |
| Rabbit polyclonal anti-LC3B | Novus Biologicals | Cat# NB100-2220; RRID: AB_10003146 |
| Mouse monoclonal anti-p62 | Santa Cruz | Cat# sc-28359; RRID: AB_628279 |
| Rabbit anti-PDK1 | Cell Signaling | Cat# 3062; RRID: AB_2236832 |
| Rabbit polyclonal anti-mTOR | Abcam | Cat# ab45989; RRID: AB_944296 |
| Rabbit monoclonal anti-IGF1R | Cell Signaling | Cat# 9750S; RRID: AB_10950969 |
| Rabbit polyclonal anti-INSR | Santa Cruz Biotechnology | Cat# sc-711; RRID: AB_631835 |
| Goat polyclonal anti-gamma Tubulin | Santa Cruz Biotechnology | Cat# sc-7396; RRID: AB_2211262 |
| Rabbit polyclonal anti-Stub1 (CHIP) | Abcam | Cat# 69451; RRID: AB_2198071 |
| Rabbit polyclonal anti-dINSR | This Study | N/A |
| Bacterial and Virus Strains | ||
| BL21-CodonPlus (DE3)-RIL | New England Biolabs | Cat# C2530H |
| OP50 | CGC | N/A |
| HT115 | CGC | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| completeTablets EDTA free | Sigma-Aldrich | Cat# 4693132001 |
| UbcH5b | Boston Biochem | Cat# E2-622 |
| FLAG::Ubiquitin | Boston Biochem | Cat# U-120 |
| E1 activating Enzyme | Enzo Life Sciences | Cat# BML-UW9410-0050 |
| Ubiquitin Conjugation Reaction Buffer | Enzo Life Sciences | Cat# BML-KW9885-0001 |
| ATP Regeneration Solution | Enzo Life Sciences | Cat# BML-EW9810-0100 |
| Ubiquitin | Sigma-Aldrich | Cat# U6253 |
| UbK0 | Boston Biochem | Cat# UM-Nok |
| FLAG M2 Affinity Gel | Sigma-Aldrich | Cat# A2220 |
| 3X FLAG peptide | Sigma-Aldrich | Cat# F4799 |
| Bafilomycin A1 | LC-Labs | Cat# B1080 |
| Chloroquine diphosphate salt | Sigma-Aldrich | Cat# C6628 |
| Dynamin Inhibitor V-34-2 | Calbiochem | Cat# 324414 |
| Methylviologen dichloride hydrate | Sigma-Aldrich | Cat# 856177-CAS75365-73-0 |
| MG132 | Peptanova | Cat# 3175-V |
| L-Canavanine | Sigma-Aldrich | Cat# C1625 |
| Paraquat dichloride hydrate | Sigma-Aldrich | Cat# 36541 |
| RU486 (Mifepristone) | Sigma-Aldrich | Cat# M8046 |
| VSV::CHN-1::6His | This Study | N/A |
| VSV::CHN-1 H218Q::6His | This Study | N/A |
| VSV::CHN-1 ΔU-box::6His | This Study | N/A |
| VSV: CHN-1 N9D, K13A, K73A::6His | This Study | N/A |
| UFD-2::6His | This Study | N/A |
| 6His::DAF-2 C terminus (aa 1205-1928) | This Study | N/A |
| WWP-1::6His | This Study | N/A |
| CHIP | Arndt et al., 2005 | N/A |
| INSR Full Length | This Study | N/A |
| INSR K1047R | This Study | N/A |
| dINSR (aa 1815-2144) | This Study | N/A |
| INSR C terminus (aa 987-1428) | This Study | N/A |
| TRIzol | Invitrogen | Cat# 15596026 |
| Critical Commercial Assays | ||
| ECL kit | GE Healthcare | Cat# RPN2232 |
| QuickChange Lightning Site-Directed Mutagenesis Kit | Agilent Technologies | Cat# 210519 |
| InviTrap Spin Universal RNA Mini Kit | Stratec | Cat# 1060100300 |
| jetPRIME transfection reagent | Peqlab | Cat# 114-75 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 4368814 |
| Brilliant III Ultra-Fast SYBR Green QPCR Master Mix | Agilent Technologies | Cat# 600882 |
| Sso Fast EvaGreen supermix | Bio-Rad | Cat# 1725201 |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat# 1708891 |
| RNeasy Mini Kit | QIAGEN | Cat# 74104 |
| Experimental Models: Cell Lines | ||
| HEK293 (Human embryonic kidney cells) | Sigma-Aldrich | Cat# 85120602 |
| HeLa (Epitheloid cervix carcinoma cells (female)) | Sigma-Aldrich | Cat# 93021013 |
| Experimental Models: Organisms/Strains | ||
| C. elegans: unc-119(ed4)III; hhIs136[unc-119(+); chn-1p::chn-1::FLAG] | This study | N/A |
| C. elegans:unc-119(ed4)III; hhIs145[unc-119(+); chn-1p::chn-1(U-box)::FLAG] | This study | N/A |
| C. elegans: Bristol (N2) strain as wild-type (WT) | CGC | N2 |
| C. elegans: chn-1(by155)I | CGC | WormBase ID: WBVar00000641 |
| C. elegans:chn-1(tm2692)I | CGC | WormBase ID: WBVar00251538 |
| C. elegans:daf-16(mgDf47)I | CGC | WormBase ID: WBVar00275574 |
| C. elegans:daf-16(m26)I | CGC | WormBase ID: WBVar00088538 |
| C. elegans:ufd-2(tm1380)II | CGC | WormBase ID: WBVar00250374 |
| C. elegans:daf-2(e1370)III | CGC | WormBase ID: WBVar00143949 |
| C. elegans:daf-2(e1368)III | CGC | WormBase ID: WBVar00143947 |
| C. elegans:daf-2(m212)III | CGC | WormBase ID: DR608 |
| C. elegans:daf-2(m596)III | CGC | WormBase ID: WBVar00088714 |
| C. elegans:daf-2(m579)III | CGC | WormBase ID: WBVar00088710 |
| C. elegans:daf-2(sa193)III | CGC | WormBase ID: WBVar00242513 |
| C. elegans:daf-2(e979)III | CGC | WormBase ID: WBVar00143653 |
| C. elegans:daf-2(e1391)III | CGC | WormBase ID: WBVar00143967 |
| C. elegans:daf-2(m577)III | CGC | WormBase ID: WBVar00088709 |
| C. elegans:daf-2(gk390525)III | CGC | WormBase ID:WBVar00813221 |
| C. elegans:eat-2(ad465) | CGC | WormBase ID: WBVar00000014 |
| C.elegans: unc-119(ed4)III | CGC | WormBase ID: WBVar00145094 |
| C. elegans:dgEx80[pAMS66 vha-6p::Q44::YFP + rol-6(su1006)] | Mohri-Shiomi and Garsin, 2008 | N/A |
| C. elegans:zIs356(daf-16p::daf-16::GFP; rol-6) | Lee et al., 2001 | N/A |
| C. elegans:rmIs133(unc-54::Q40::YFP) | Morley et al., 2002 | N/A |
| D. melanogaster: w1118 | Bloomington Stock Center | RRID: BDSC_3605 |
| D. melanogaster: actin-Gal4 | Bloomington Stock Center | RRID: BDSC_3954 |
| D. melanogaster: RNAi-dCHIP | VDRC | RRID: FlyBase_FBst0460471 |
| D. melanogaster: TIGS-2 | Scott Pletscher | N/A |
| D. melanogaster: UAS-dCHIP | Verena Arndt | N/A |
| Oligonucleotides | ||
| See Table S3 for primer sequences | This Study | N/A |
| siRNA human CHIP 5 | QIAGEN | Cat# SI00081963 |
| siRNA human CHIP 5 | QIAGEN | Cat# SI03109659 |
| siRNA human ATG7 1 | QIAGEN | Cat# SI04194883 |
| siRNA human ATG7 3 | QIAGEN | Cat# SI04231360 |
| Allstars negative control siRNA | QIAGEN | Cat# SI03650318 |
| Recombinant DNA | ||
| pET21a-VSV::CHN-1::6His | This Study | N/A |
| pET21a- VSV::CHN-1 H218Q::6His | This Study | N/A |
| pET21a- VSV::CHN-1 ΔU-box::6His | This Study | N/A |
| pET21a- VSV: CHN-1 N9D, K13A, K73A::6His | This Study | N/A |
| pET21a-UFD-2::6His | This Study | N/A |
| pT7-6His::DAF-2 C terminus (aa 1205-1928) | This Study | N/A |
| pET21a-WWP-1::6His | This Study | N/A |
| pCMV-tag2b-CHIP | Arndt et al., 2005 | N/A |
| pCMV-tag2b-INSR Full Length | This Study | N/A |
| pCMV-tag2b-INSR K1047R | This Study | N/A |
| pETM-11-dINSR (aa 1815-2144) | This Study | N/A |
| pMAL-C2-INSR C terminus (aa 987-1428) | This Study | N/A |
| pEGFP-polyQ-25 | Westhoff et al., 2005 | N/A |
| pEGFP-polyQ-103 | Westhoff et al., 2005 | N/A |
| Software and Algorithms | ||
| GraphPad Prism 5 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Image Studio 4.0 | LI-COR Biosciences | https://www.licor.com/bio/products/software/image_studio/ |
| Excel 2013 | Microsoft | https://products.office.com/en-us/excel |
| XLSTAT | XLSTAT | https://www.xlstat.com/en/ |
| ImageJ 1.48v | Wayne Rasband (NIH) | https://imagej.nih.gov/ij/ |
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to and will be fulfilled by Thorsten Hoppe (thorsten.hoppe@uni-koeln.de).
Experimental Model and Subject Details
Caenorhabditis elegans Maintenance and Transgenic Lines
The following strains were used in this study: Bristol (N2) strain as wild-type (WT) strain, chn-1(by155)I, chn-1(tm2692)I, daf-16(mgDf47)I, daf-16(mu86)I, daf-16(m26)I, ufd-2(tm1380)II, eat-2(ad465)II, daf-2(e1368)III, daf-2(e1370)III, daf-2(m212)III, daf-2(m596)III, daf-2(m579)III, daf-2(sa193)III, daf-2(e979)III, daf-2(e1391)III, daf-2(m577)III, daf-2(gk390525)III, unc-119(ed4)III, zIs356[daf-16p::daf-16::GFP], dgEx80[pAMS66 vha-6p::Q44::YFP + rol-6(su1006)], rmIs133[unc- 54p::Q40::YFP]. Unless otherwise stated mutant strains were at least six times outcrossed against the wild-type strain to provide isogenic conditions. Nematodes were grown at 20°C (unless otherwise stated) on nematode growth medium (NGM) agar plates seeded with Escherichia coli OP50 spread over the surface as a food source (Brenner, 1974). Transgenic animals expressing the chn-1p::chn-1::FLAG were constructed by PCR amplification (Table S3) of the wild-type genomic DNA (567 bp chn-1 promoter and 2783 bp chn-1 genomic DNA) and the unc-54 3′UTR (877 bp) and cloned into the pBSK vector containing the unc-119(+) marker for selection of transgenic worms. FLAG tag was fused to the C terminus of the gene by using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies) (Table S3). Deletion of the U-box domain, mutation of the U-box (H218Q), or TPR (N9D, K13A, K73A) domain was introduced by QuikChange Lightning Site-Directed Mutagenesis Kit (Table S3). The constructs were particle bombarded into unc-119(ed4)III mutants as described previously (Praitis et al., 2001). Microparticle bombardment of C. elegans unc-119(ed4) hermaphrodites was carried out using a BioRad Biolistic PDS-1000/HE with 1/4″gap distance, 9 mm macrocarrier to screen distance, 28 inches of Hg vacuum, and 1350 p.s.i. rupture disc. For each bombardment, 1 μl of 1–2 μg/μl plasmid DNA was coupled to 0.6 mg of 1.0-μm microcarrier gold beads, and bombarded onto a monolayer of ∼10,000 unc-119(ed4) L4 and adult hermaphrodites (a-70-80 μl pellet) placed on 90-mm NGM plates. Worms were allowed to recover for 1 hr after bombardment and were then transferred onto ten 90-mm NGM plates seeded with OP50 E. coli and grown at 20°C. Because unc-119 mutants cannot form dauers, they die in the absence of food (Maduro and Pilgrim, 1995), making it easy to identify the non-Unc rescued transformants 14 days after bombardment. From each plate containing animals rescued for the unc-119(ed4) mutation, individual transformed animals were cloned and their F1 progeny scored for presence of unc-119(ed4) mutants. Homozygous stable lines were identified by the complete absence of unc-119(ed4)III mutant progeny over several generations. Heterozygous lines were identified based on the presence of three distinct classes of progeny: heterozygous transformed animals, homozygous untransformed animals, and a third class of sterile or inviable animals. To ensure that each line was the result of an independent transformation event, we retained only one transformed line from each NGM plate. Transgenic strains generated in this study are as following: unc-119(ed4)III; hhIs136[unc-119(+), chn-1p::chn-1::FLAG], unc-119(ed4)III; hhIs145[unc-119(+); chn-1p::chn-1(U-box)::FLAG].
Drosophila melanogaster
The following strains were used in this study: w1118 were used as wild-type flies, dCHIP-RNAi (VDRC stock number 10538), actin-GAL4 (#BL-3954, Bloomington stock center), UAS-dCHIP (kindly provided by Verena Arndt, University of Bonn), TIGS-2 (kindly provided by Scott Pletscher, University of Michigan). Flies were maintained on standard Drosophila Jazz-Mix instant food (Fisher Scientific) at 25°C and 65% humidity in a 12 hr light/ 12 hr dark incubator. For ubiquitous depletion RNAi flies were crossed to actin-Gal4 driver line. Control flies are of the genotype actin-Gal4/w1118. For overexpression of dCHIP in adult midguts UAS-dCHIP flies were crossed to the TIGS-2 driver line. Expression of dCHIP was induced by placing flies of the same genetic background on RU486-containing (induced) or ethanol-containing (non-induced) food. For food preparation, a 25 mg/ml stock solution of RU486 (Mifepristone, Sigma) was made in 100% ethanol. Appropriate volumes of the 25 mg/ml RU486 stock solution were diluted with water and ethanol to a final ethanol concentration of 2% to avoid RU486 precipitation. 500 μl of the diluted solution with the desired concentration of RU486 was added onto the surface of standard fly food. The vials were then allowed to dry at room temperature for 24–36 hr depending on the ambient humidity. For paraquat or canavanine food the indicated concentration was solved in 1 ml water and applied onto the surface of standard fly food and further processed as mentioned above. Flies were flipped every other day.
Human Cells
HEK293 cells were cultured at 37°C under 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, L-glutamine, penicillin/streptomycin (PS), sodium pyruvate and non-essential amino acids (NEAA). HeLa cells were cultured at 37°C under 5% CO2 in Dulbecco’s modified Eagles’s medium (DMEM) supplemented with 10% fetal bovine serum and penicillin/streptomycin (PS).
Method Details
Lifespan Analysis
In lifespan assays, first day of adulthood was defined as day 1. From day 0 on, synchronized worm populations (50 worms per plate for a total of 200 individuals per experiment) were transferred daily to fresh NGM plates seeded with OP50 E. coli until egg laying ceased. For RNAi based lifespan assays 50 synchronized L1 stage worms were fed with OP50 E. coli or with gene-specific RNAi expressing HT115 E. coli bacteria. Worms were transferred daily to fresh plates until egg laying ceased. Worms were examined every day for touch-provoked movement and pharyngeal pumping, until death. Animals that crawled off the plates or died owing to internal hatching of progeny or extruded organs were censored. For lifespan measurement in D. melanogaster, male flies were collected within 24 hr from eclosion and maintained at standard density (up to 30 flies per vial) on Drosophila Jazz-Mix instant food or in case of induced expression on either RU486-containing (induced) or ethanol-containing (non-induced) food. Dead flies were counted and food was changed every other day. For each genotype, at least 5 independent cohorts of flies raised at different times from at least 5 independent crosses were analyzed. Statistical data of individual lifespan experiments is presented in Table S1. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment. No statistical methods were used to predetermine sample size.
RNA Interference
RNAi in C. elegans was performed using the RNAi feeding method (Timmons and Fire, 1998). L1 or L4 larvae of wild-type worms were placed on HT115 E. coli bacteria expressing daf-2 or daf-16-specific double-stranded RNA (dsRNA). As wild-type control, bacteria transformed with the empty pPD129.36 vector were used as food source. For developmental or post-developmental silencing of chn-1 L1 larvae or day 1 adult worms were placed on chn-1 dsRNA expressing HT115. RNAi in flies was induced by binary UAS-GAL4 system (Brand and Perrimon, 1993). For ubiquitous depletion RNAi flies were crossed to actin-Gal4 driver line. Control flies are of the genotype actin-Gal4/w1118.
C. elegans Body Length Measurements
Worms were maintained at 20°C until the first day of adulthood. Relative body length of worms was measured by taking images of worms with a Leica M165FC stereomicroscope (10X eye piece, 0.75 - 6.0 zoom ratio) using 50 X magnification. Images were transformed to jpeg format and body length of worms was determined by drawing a line on the longitudinal axis of each worm in ImageJ 1.48v software. Relative values of body length was calculated by using the mean body length value of at least 20 single worms of each genotype and normalizing these values against WT worms’ mean body length.
C. elegans Body Bends Measurements
Body-bend assays were performed in a M9 buffer at room temperature. Individual day-1 adult worm was placed in 50 μl M9 buffer on a microscope slide and the number of left and rightward body bends in 30 s were counted, where one leftward and one rightward bend is equal to one stroke.
DAF-16::GFP Localization Assay
The synchronized DAF-16::GFP containing strains were maintained at 20°C. 100 day-1 old adult worms were transferred to small plates (35x10mm) seeded with OP50 and subjected to 35°C heat shock for indicated time in water bath and immediately visualized using Zeiss Axio Zoom.V16 microscope. Worms were classified into two categories based on the extent of DAF-16::GFP nuclear-cytoplasmic distribution.
Microscopy
Fluorescence and DIC images of worms were taken with an Axio Imager Z1 microscope mounted with Axiocam 503 mono camera (Carl Zeiss), and processed with the analysis software Zen 2012 SP1 (Carl Zeiss). HeLa cells were transfected with pCMV-tag2b-CHIP and pEGFP-PolyQ-25 or pEGFP-PolyQ-103 for 48 hr. Cells were then washed two times with PBS, followed by fixation in 4% paraformaldehyde for 15 min. After another washing step, cells were permeabilized for 10 min in PBS containing 0.2% Triton-100. Cells were blocked in 3% bovine serum albumin for 30 min and afterward washed 3 times in PBS. Primary antibody incubation was performed in 1:200 dilution in PBS for 1 hr. Alexa-labeled secondary antibodies (Invitrogen) were used in a 1∶500 dilution for 1 hr. Cells were washed again for 3 times in PBS and afterward mounted in Mowiol. Images were acquired using a Zeiss Axiovert 510 microscope.
RNA Isolation and Real-Time PCR
Total RNA was isolated using TRIzol (Invitrogen) and QIAGEN RNeasy kit. Worms were washed off the plates using M9 buffer and 1 ml TRIzol, and silica beads (1 mm diameter) were added to the samples and homogenized by Precellys tissue homogenizer. Chloroform was added and samples were vortexed vigorously before phase separation through centrifugation. The aqueous phase was transferred on the QIAGEN RNeasy Mini Kit and RNA was isolated according to manufacturer’s instructions. cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Gene expression levels were determined by real-time PCR using Brilliant III Ultra-Fast SYBR Green QPCR Master Mix (Agilent Technologies) and Biorad CFX96 Real-Time PCR Detection System. Relative gene expressions were normalized to act-3 (T04C12.4) and tbg-1 (F58A4.8). mRNA levels. RNAi isolation from HEK293 cells was performed by the use of InviTrap Spin Universal RNA Mini Kit (Stratec). cDNA was synthesized from 0.5 μg RNA using iScript cDNA synthesis kit from Bio-Rad. Quantitative PCR (qPCR) was performed using SsoFast EvaGreen Supermix (Bio-Rad) with transcript levels for GAPDH and beta-2-microglobulin (B2M) being monitored as reference transcripts. Relative Gene expression was calculated using the ΔΔCT method. The specificity of the PCR amplification was verified by melting curve analysis of the final products using Bio-Rad CFX 3.1 software. In each experiment three biological and three technical replicate samples were analyzed. The primer sequences used in the RT–PCR reactions are shown in Table S3.
Protein Extraction, Binding Assays, and Immunotechniques
Co-immunoprecipitations were performed on sonicated lysates from wild-type or mutant C. elegans that were resuspended in IP buffer [50 mM Tris pH 8.0, 100 mM NaCl, 0.5 mM EDTA, 4% Glycerol and 1% Triton X-100, 2 mM PMSF and cOmplete EDTA-free Protease Inhibitor (Roche)]. For immunoprecipitation of CHN-1::FLAG we used anti-FLAG M2 Affinity Gel (Sigma) incubated with 1 mg of worm extract for 2 hr at room temperature. The immunoprecipitates were washed with 50 mM Tris pH 8.0, 50 mM NaCl, eluted with 3X FLAG peptide (Sigma) and resolved by SDS-PAGE followed by immunoblotting. To prepare Drosophila protein extracts, 10 male flies per genotype were shock frozen in liquid nitrogen and grinded with a pestle until white powder remained. 200 μl of RIPA buffer [25 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.5% sodium deoxycholate, 1% Nonidet P-40, 0.1% SDS, 10% glycerol, complete EDTA-free Protease Inhibitor (Roche)] was added and lysates were incubated for 15 min on ice. Afterward lysates were centrifuged at 16,000 × g for 30 min at 4°C. Protein concentration of supernatant was measured by Bradford protein assay (Bio-Rad) and 40 μg protein per genotype were analyzed by SDS-PAGE and immunoblotting. For gut preparation and evaluation of Akt signaling, dCHIP depleted flies and control flies were aged for 5 weeks respectively and 20 guts per genotype were collected in icecold PBS buffer. PBS buffer was removed and guts were resuspended in 50 μl RIPA for proper lysis. Guts were grinded with a pestle until a homogeneous solution remained and immediately sonicated 3 × 20 s at 100% amplitude. Gut lysates were incubated for 15 min on ice and afterward centrifuged at 8,000 × g for 20 min at 4°C. Supernatants were collected and protein concentrations were measured by Bradford protein assay (Bio-Rad). 30 μg protein per genotype was analyzed by SDS-PAGE and immunoblotting. For monitoring dINSR protein level in paraquat and canavanine treated adult flies (3 weeks old), 20 heads each were collected in 60 μl RIPA buffer and prepared as described above. For differential centrifugation dCHIP depleted flies and control flies were aged for 5 weeks and 20 heads per genotype were lysed in 200 μl RIPA buffer containing 1% SDS, followed by centrifugation at 3,000 × g for 5 min to discard tissue debris. The supernatant was subjected to centrifugation at 100,000 × g for 1 hr at 4°C to separate soluble and insoluble protein fractions. The SDS-insoluble pellet fraction was resuspended in 100 mM Tris, pH 8, 8 M urea, sonicated three times for 20 s at 100% amplitude, and afterward boiled at 97°C for 10 min. To prepare human cell extracts HEK293 cells were lysed 48h after transfection in RIPA and incubated for 5 min on ice. Lysates were sonicated and after additional 15 min incubation on ice lysates were centrifuged at 16,000 × g for 20 min at 4°C. Supernatants were collected, protein concentration was measured by Bradford protein assay (Bio-Rad) and samples were analyzed by SDS-PAGE (40 μg protein loaded per lane) and immunoblotting using specific antibodies. If not indicated otherwise, cells were treated with 10 μM MG132 (Peptanova), 200 nM Bafilomycin A1 (LC-Labs), 100 μM chloroquine (Sigma-Aldrich), or 10 μM dynamin inhibitor V34-2 (Calbiochem) for 16 hr before lysis. Paraquat (Sigma-Aldrich) treatment (20 mM) was done for 20 hr. For immunoprecipitation of INSR, HEK293 cells were lysed in RIPA buffer without SDS. After 20 min of incubation on ice, the lysate was sonicated and centrifuged at 16,000 × g for 20 min at 4°C. Supernatant was then adjusted to a protein concentration of 10 mg/ml using MOPS/KCl buffer [20 mM MOPS-KOH, pH 7.2, 100 mM KCl, complete EDTA-free Protease Inhibitor (Roche)] at a ratio 1:1 with RIPA buffer. The supernatant was used as a soluble cell extract for immunoprecipiation. Antibodies directed against INSR or control antibody (rabbit anti-chicken IgG, Sigma) were added to a final concentration of 4 μg/ml extract and incubated for 1 hr at 4°C. 20 μl protein G-Sepharose was added and samples were incubated for an additional 1 hr at 4°C. Sepharose was washed six times with washing buffer (20 mM MOPS-KOH, pH 7.2, 100 mM KCl, 0.5% Tween) and 2 times with washing buffer without Tween. Sepharose-bound proteins were eluted using 0.1 M glycine-HCl, pH 3.5, precipitated with 10% TCA (trichloroacetic acid) and resolved in SDS sample buffer. Co-immunoprecipitated proteins were analyzed by SDS-PAGE and immunoblotting.
Protein Expression and siRNA in Human Cells
HEK293 or HeLa cells were seeded at approximately 30%–40% confluency and transfected 24 hr after plating. Transfection was done using jetPRIME (Peqlab) transfection reagent. After incubation over-night, cell medium was removed and fresh medium was added to the cells. 48 hr after transfection cells were lysed either for protein extraction or RNA purification. For expression of wild-type INSR in HEK293 cells pRT3-INSR-WT (Addgene) was used as template for cloning full-length cDNA of human INSR into pCMV-tag2b vector (Clonetech) (Table S3). The K1047R mutation was introduced into pCMV-tag2b-INSR by site-directed mutagenesis (Table S3). For overexpression of CHIP pCMV-tag2b-CHIP vector was used (Arndt et al., 2005). Expression of poly-Q proteins was induced by transient transfection with plasmids pEGFP-polyQ-25 and pEGFP-polyQ-103 (Westhoff et al., 2005). For efficient depletion two different siRNAs were used per gene (FlexiTube siRNAs CHIP 1+5 and FlexiTube siRNAs ATG7 1+3 (QIAGEN)). Allstars negative control siRNA (QIAGEN) was used as control siRNA.
In Vitro Ubiquitylation
10 μg of purified recombinant protein: His::CHIP, His::CHN-1, His::CHN-1(U-box), His::CHN-1(ΔU-box), His::(TPR)CHN-1, WWP-1::His or UFD-2::His was mixed with 1 μg of recombinant protein corresponding to the C terminus of human INSR (amino acids 987-1428), dINSR (amino acids 1815-2144) or DAF-2 (amino acids 1205-1928), and E1 (25 ng), E2 (UbcH5b; 400 ng), 2 μg of FLAG::ubiquitin or lysine lacking variant (UbK0), energy regenerating solution (Boston Biochem) and Ubiquitin Conjugation Reaction Buffer (Enzo Life Sciences). Samples were incubated at 30°C for 1 hr, terminated by boiling for 5 min with SDS-sample buffer, and resolved by SDS-PAGE followed by immunoblotting using anti-insulin Rβ antibody (Santa Cruz) to monitor ubiquitylation of INSR, anti-DAF-2 antibody to monitor ubiquitylation of DAF-2, and anti-FLAG antibody (Sigma) to monitor E3 ligase activity.
Stress Assays
For oxidative stress assay, young adult worms were transferred to OP50-seeded NGM plates containing 5 mM paraquat. Survival of worms was monitored every day after transfer and scored as described for the lifespan assays. In heat shock experiments day 1 adult worms were transferred on chn-1 dsRNA expressing or control empty vector expressing HT115 bacteria seeded plates and incubated at 35°C in water bath for the indicated time. Survival of worms was scored as described for the lifespan assays. For ubiquitous dCHIP depletion RNAi flies were crossed to actin-Gal4 driver line and collected after 2 days of adulthood and placed on 20 mM paraquat (methylviologen (Sigma-Aldrich)) containing food. Survival rates were measured every other day. Wild-type flies were collected after 2 days of adulthood and placed on either H2O or 20 mM L-Canavanine (Sigma-Aldrich) containing food to induce protein misfolding.
Statistical Analysis
Kaplan and Meier survival curves of the lifespan analysis and median lifespan values were calculated using GraphPad Prism 5 software and Xlstat for Excel. The log-rank (Mantel–Cox) test was used to evaluate differences in survival and determine p values. Quantification of immunoblots was done with ImageJ 1.48v or Image Studio 4.0 software. Data obtained for changes in protein level are presented as fold change compared to the control with the control value determined in each independent experiment set to 1. Statistical significance was assessed by two-tailed paired Student’s t test using Excel software (Microsoft). Stress resistance assays and polyQ-aggregation assays were evaluated by two-tailed paired Student’s t test.
Author Contributions
R.T., W.P., and E.K. designed, performed, and analyzed the experiments. R.T. and S.A. performed Drosophila and cell culture studies; W.P., E.K., M.E.A., and V.B., performed C. elegans studies. W.P. performed in vitro ubiquitylation assays, mass spectrometric analyses, and most final experiments for the revisions. T.H. and J.H. supervised the design and data interpretation; T.H. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Acknowledgments
We thank Y. Kohara, M. Marr, the Caenorhabditis Genetics Center (funded by the NIH National Center for Research Resources), the Bloomington Stock Center, the Vienna Drosophila RNAi Center, the Dana-Farber Cancer Institute, Addgene, and Geneservice for antibodies, plasmids, cDNAs, and strains; Agata Lisowski, Ela Stellbrink, and Karen Himmelberg for technical help; Gerrit Loch for help with the fly work; and Hendrik Nolte and Marcus Krüger for mass spectrometric analyses. We also thank André Franz and Robin Lorenz for reading the manuscript. This work is supported by grants from the Deutsche Forschungsgemeinschaft (DFG) to J.H (SFB635 and SFB645) and to T.H. (CECAD, SFB635, and DIP8 grant 2014376) and from the European Research Council (consolidator grant 616499) to T.H.
This work is dedicated to the memory of our wonderful mentor and dear friend Stefan Jentsch, who unfortunately passed away much too soon (J.H. and T.H.).
Published: April 20, 2017
Footnotes
Supplemental Information includes six figures and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2017.04.003.
Contributor Information
Jörg Höhfeld, Email: hoehfeld@uni-bonn.de.
Thorsten Hoppe, Email: thorsten.hoppe@uni-koeln.de.
Supporting Citations
The following references appear in the Supplemental Information: Ulbricht et al. (2015).
Supplemental Information
References
- Ardon O., Procter M., Tvrdik T., Longo N., Mao R. Sequencing analysis of insulin receptor defects and detection of two novel mutations in INSR gene. Mol. Genet. Metab. Rep. 2014;1:71–84. doi: 10.1016/j.ymgmr.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt V., Daniel C., Nastainczyk W., Alberti S., Höhfeld J. BAG-2 acts as an inhibitor of the chaperone-associated ubiquitin ligase CHIP. Mol. Biol. Cell. 2005;16:5891–5900. doi: 10.1091/mbc.E05-07-0660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt V., Rogon C., Höhfeld J. To be, or not to be--molecular chaperones in protein degradation. Cell. Mol. Life Sci. 2007;64:2525–2541. doi: 10.1007/s00018-007-7188-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt V., Dick N., Tawo R., Dreiseidler M., Wenzel D., Hesse M., Fürst D.O., Saftig P., Saint R., Fleischmann B.K. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr. Biol. 2010;20:143–148. doi: 10.1016/j.cub.2009.11.022. [DOI] [PubMed] [Google Scholar]
- Barbieri M., Bonafè M., Franceschi C., Paolisso G. Insulin/IGF-I-signaling pathway: an evolutionarily conserved mechanism of longevity from yeast to humans. Am. J. Physiol. Endocrinol. Metab. 2003;285:E1064–E1071. doi: 10.1152/ajpendo.00296.2003. [DOI] [PubMed] [Google Scholar]
- Brand A.H., Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulger D.A., Fukushige T., Yun S., Semple R.K., Hanover J.A., Krause M.W. Caenorhabditis elegans DAF-2 as a model for human insulin receptoropathies. G3 (Bethesda) 2017;7:257–268. doi: 10.1534/g3.116.037184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrano A.C., Liu Z., Dillin A., Hunter T. A conserved ubiquitination pathway determines longevity in response to diet restriction. Nature. 2009;460:396–399. doi: 10.1038/nature08130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E., Du D., Joyce D., Kapernick E.A., Volovik Y., Kelly J.W., Dillin A. Temporal requirements of insulin/IGF-1 signaling for proteotoxicity protection. Aging Cell. 2010;9:126–134. doi: 10.1111/j.1474-9726.2009.00541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell P., Ballinger C.A., Jiang J., Wu Y., Thompson L.J., Höhfeld J., Patterson C. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 2001;3:93–96. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- Cyr D.M., Höhfeld J., Patterson C. Protein quality control: U-box-containing E3 ubiquitin ligases join the fold. Trends Biochem. Sci. 2002;27:368–375. doi: 10.1016/s0968-0004(02)02125-4. [DOI] [PubMed] [Google Scholar]
- Dai Q., Zhang C., Wu Y., McDonough H., Whaley R.A., Godfrey V., Li H.H., Madamanchi N., Xu W., Neckers L. CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J. 2003;22:5446–5458. doi: 10.1093/emboj/cdg529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasuri K., Ebenezer P.J., Uranga R.M., Gavilán E., Zhang L., Fernandez-Kim S.O., Bruce-Keller A.J., Keller J.N. Amino acid analog toxicity in primary rat neuronal and astrocyte cultures: implications for protein misfolding and TDP-43 regulation. J. Neurosci. Res. 2011;89:1471–1477. doi: 10.1002/jnr.22677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y., Jaiseng W., Ma Y., Hu L., Yamazaki S., Zhang X., Hayafuji T., Shi L., Kuno T. E3 ubiquitin ligase Pub1 is implicated in endocytosis of a GPI-anchored protein Ecm33 in fission yeast. PLoS ONE. 2014;9:e85238. doi: 10.1371/journal.pone.0085238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazda L., Pokrzywa W., Hellerschmied D., Löwe T., Forné I., Mueller-Planitz F., Hoppe T., Clausen T. The myosin chaperone UNC-45 is organized in tandem modules to support myofilament formation in C. elegans. Cell. 2013;152:183–195. doi: 10.1016/j.cell.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck J.W., Cheung S.K., Hampton R.Y. Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc. Natl. Acad. Sci. USA. 2010;107:1106–1111. doi: 10.1073/pnas.0910591107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimdal K., Sanchez-Guixé M., Aukrust I., Bollerslev J., Bruland O., Jablonski G.E., Erichsen A.K., Gude E., Koht J.A., Erdal S. STUB1 mutations in autosomal recessive ataxias - evidence for mutation-specific clinical heterogeneity. Orphanet J. Rare Dis. 2014;9:146. doi: 10.1186/s13023-014-0146-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson S.T., Johnson T.E. daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr. Biol. 2001;11:1975–1980. doi: 10.1016/s0960-9822(01)00594-2. [DOI] [PubMed] [Google Scholar]
- Hesp K., Smant G., Kammenga J.E. Caenorhabditis elegans DAF-16/FOXO transcription factor and its mammalian homologs associate with age-related disease. Exp. Gerontol. 2015;72:1–7. doi: 10.1016/j.exger.2015.09.006. [DOI] [PubMed] [Google Scholar]
- Hoppe T., Cassata G., Barral J.M., Springer W., Hutagalung A.H., Epstein H.F., Baumeister R. Regulation of the myosin-directed chaperone UNC-45 by a novel E3/E4-multiubiquitylation complex in C. elegans. Cell. 2004;118:337–349. doi: 10.1016/j.cell.2004.07.014. [DOI] [PubMed] [Google Scholar]
- Kalia L.V., Kalia S.K., Chau H., Lozano A.M., Hyman B.T., McLean P.J. Ubiquitinylation of α-synuclein by carboxyl terminus Hsp70-interacting protein (CHIP) is regulated by Bcl-2-associated athanogene 5 (BAG5) PLoS ONE. 2011;6:e14695. doi: 10.1371/journal.pone.0014695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon C.J. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- Kenyon C., Chang J., Gensch E., Rudner A., Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Kevei É., Hoppe T. Ubiquitin sets the timer: impacts on aging and longevity. Nat. Struct. Mol. Biol. 2014;21:290–292. doi: 10.1038/nsmb.2806. [DOI] [PubMed] [Google Scholar]
- Kim W., Bennett E.J., Huttlin E.L., Guo A., Li J., Possemato A., Sowa M.E., Rad R., Rush J., Comb M.J. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell. 2011;44:325–340. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.M., Grenert J.P., Patterson C., Correia M.A. CHIP(-/-)-Mouse Liver: Adiponectin-AMPK-FOXO-Activation Overrides CYP2E1-Elicited JNK1-Activation, Delaying Onset of NASH: Therapeutic Implications. Sci. Rep. 2016;6:29423. doi: 10.1038/srep29423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K.D., Tissenbaum H.A., Liu Y., Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- Komander D., Rape M. The ubiquitin code. Annu. Rev. Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- Komatsu M., Waguri S., Ueno T., Iwata J., Murata S., Tanida I., Ezaki J., Mizushima N., Ohsumi Y., Uchiyama Y. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowski B., Hekimi S. The genetics of caloric restriction in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA. 1998;95:13091–13096. doi: 10.1073/pnas.95.22.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee R.Y., Hench J., Ruvkun G. Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr. Biol. 2001;11:1950–1957. doi: 10.1016/s0960-9822(01)00595-4. [DOI] [PubMed] [Google Scholar]
- Lee S.S., Kennedy S., Tolonen A.C., Ruvkun G. DAF-16 target genes that control C. elegans life-span and metabolism. Science. 2003;300:644–647. doi: 10.1126/science.1083614. [DOI] [PubMed] [Google Scholar]
- Lin K., Hsin H., Libina N., Kenyon C. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat. Genet. 2001;28:139–145. doi: 10.1038/88850. [DOI] [PubMed] [Google Scholar]
- López-Otín C., Blasco M.A., Partridge L., Serrano M., Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maduro M., Pilgrim D. Identification and cloning of unc-119, a gene expressed in the Caenorhabditis elegans nervous system. Genetics. 1995;141:977–988. doi: 10.1093/genetics/141.3.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone E.A., Thomas J.H. A screen for nonconditional dauer-constitutive mutations in Caenorhabditis elegans. Genetics. 1994;136:879–886. doi: 10.1093/genetics/136.3.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura Y., Sakai J., Skach W.R. Endoplasmic reticulum protein quality control is determined by cooperative interactions between Hsp/c70 protein and the CHIP E3 ligase. J. Biol. Chem. 2013;288:31069–31079. doi: 10.1074/jbc.M113.479345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J.N., Whaley R.A., Sharpless N.E., Lockyer P., Portbury A.L., Patterson C. CHIP deficiency decreases longevity, with accelerated aging phenotypes accompanied by altered protein quality control. Mol. Cell. Biol. 2008;28:4018–4025. doi: 10.1128/MCB.00296-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohri-Shiomi A., Garsin D.A. Insulin signaling and the heat shock response modulate protein homeostasis in the Caenorhabditis elegans intestine during infection. J. Biol. Chem. 2008;283:194–201. doi: 10.1074/jbc.M707956200. [DOI] [PubMed] [Google Scholar]
- Morishima Y., Wang A.M., Yu Z., Pratt W.B., Osawa Y., Lieberman A.P. CHIP deletion reveals functional redundancy of E3 ligases in promoting degradation of both signaling proteins and expanded glutamine proteins. Hum. Mol. Genet. 2008;17:3942–3952. doi: 10.1093/hmg/ddn296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley J.F., Brignull H.R., Weyers J.J., Morimoto R.I. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA. 2002;99:10417–10422. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata S., Chiba T., Tanaka K. CHIP: a quality-control E3 ligase collaborating with molecular chaperones. Int. J. Biochem. Cell Biol. 2003;35:572–578. doi: 10.1016/s1357-2725(02)00394-1. [DOI] [PubMed] [Google Scholar]
- O’Rahilly S., Choi W.H., Patel P., Turner R.C., Flier J.S., Moller D.E. Detection of mutations in insulin-receptor gene in NIDDM patients by analysis of single-stranded conformation polymorphisms. Diabetes. 1991;40:777–782. doi: 10.2337/diab.40.6.777. [DOI] [PubMed] [Google Scholar]
- Ogg S., Paradis S., Gottlieb S., Patterson G.I., Lee L., Tissenbaum H.A., Ruvkun G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- Okiyoneda T., Barrière H., Bagdány M., Rabeh W.M., Du K., Höhfeld J., Young J.C., Lukacs G.L. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science. 2010;329:805–810. doi: 10.1126/science.1191542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D.S., Garza-Garcia A., Nanji M., McElwee J.J., Ackerman D., Driscoll P.C., Gems D. Clustering of genetically defined allele classes in the Caenorhabditis elegans DAF-2 insulin/IGF-1 receptor. Genetics. 2008;178:931–946. doi: 10.1534/genetics.107.070813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez C.L., Van Gilst M.R. A 13C isotope labeling strategy reveals the influence of insulin signaling on lipogenesis in C. elegans. Cell Metab. 2008;8:266–274. doi: 10.1016/j.cmet.2008.08.007. [DOI] [PubMed] [Google Scholar]
- Petrucelli L., Dickson D., Kehoe K., Taylor J., Snyder H., Grover A., De Lucia M., McGowan E., Lewis J., Prihar G. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet. 2004;13:703–714. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- Praitis V., Casey E., Collar D., Austin J. Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics. 2001;157:1217–1226. doi: 10.1093/genetics/157.3.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian S.B., McDonough H., Boellmann F., Cyr D.M., Patterson C. CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature. 2006;440:551–555. doi: 10.1038/nature04600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal G.A., Berge M.A., Bleiler J.A., Rudd T.P. Aberrant, canavanyl protein formation and the ability to tolerate or utilize L-canavanine. Experientia. 1987;43:558–561. doi: 10.1007/BF02143585. [DOI] [PubMed] [Google Scholar]
- Rosser M.F., Washburn E., Muchowski P.J., Patterson C., Cyr D.M. Chaperone functions of the E3 ubiquitin ligase CHIP. J. Biol. Chem. 2007;282:22267–22277. doi: 10.1074/jbc.M700513200. [DOI] [PubMed] [Google Scholar]
- Scott B.A., Avidan M.S., Crowder C.M. Regulation of hypoxic death in C. elegans by the insulin/IGF receptor homolog DAF-2. Science. 2002;296:2388–2391. doi: 10.1126/science.1072302. [DOI] [PubMed] [Google Scholar]
- Slotman J.A., da Silva Almeida A.C., Hassink G.C., van de Ven R.H., van Kerkhof P., Kuiken H.J., Strous G.J. Ubc13 and COOH terminus of Hsp70-interacting protein (CHIP) are required for growth hormone receptor endocytosis. J. Biol. Chem. 2012;287:15533–15543. doi: 10.1074/jbc.M111.302521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz M., Nikolay R., Rybin V., Mayer M.P. CHIP participates in protein triage decisions by preferentially ubiquitinating Hsp70-bound substrates. FEBS J. 2010;277:3353–3367. doi: 10.1111/j.1742-4658.2010.07737.x. [DOI] [PubMed] [Google Scholar]
- Stankowski J.N., Zeiger S.L.H., Cohen E.L., DeFranco D.B., Cai J., McLaughlin B. C-terminus of heat shock cognate 70 interacting protein increases following stroke and impairs survival against acute oxidative stress. Antioxid. Redox Signal. 2011;14:1787–1801. doi: 10.1089/ars.2010.3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C.H., Wang C.Y., Lan K.H., Li C.P., Chao Y., Lin H.C., Lee S.D., Lee W.P. Akt phosphorylation at Thr308 and Ser473 is required for CHIP-mediated ubiquitination of the kinase. Cell. Signal. 2011;23:1824–1830. doi: 10.1016/j.cellsig.2011.06.018. [DOI] [PubMed] [Google Scholar]
- Thompson O., Edgley M., Strasbourger P., Flibotte S., Ewing B., Adair R., Au V., Chaudhry I., Fernando L., Hutter H. The million mutation project: a new approach to genetics in Caenorhabditis elegans. Genome Res. 2013;23:1749–1762. doi: 10.1101/gr.157651.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons L., Fire A. Specific interference by ingested dsRNA. Nature. 1998;395:854. doi: 10.1038/27579. [DOI] [PubMed] [Google Scholar]
- Udeshi N.D., Svinkina T., Mertins P., Kuhn E., Mani D.R., Qiao J.W., Carr S.A. Refined preparation and use of anti-diglycine remnant (K-ε-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol. Cell. Proteomics. 2013;12:825–831. doi: 10.1074/mcp.O112.027094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbricht A., Gehlert S., Leciejewski B., Schiffer T., Bloch W., Höhfeld J. Induction and adaptation of chaperone-assisted selective autophagy CASA in response to resistance exercise in human skeletal muscle. Autophagy. 2015;11:538–546. doi: 10.1080/15548627.2015.1017186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Horst A., Burgering B.M. Stressing the role of FoxO proteins in lifespan and disease. Nat. Rev. Mol. Cell Biol. 2007;8:440–450. doi: 10.1038/nrm2190. [DOI] [PubMed] [Google Scholar]
- Walther D.M., Kasturi P., Zheng M., Pinkert S., Vecchi G., Ciryam P., Morimoto R.I., Dobson C.M., Vendruscolo M., Mann M., Hartl F.U. Widespread proteome remodeling and aggregation in aging C. elegans. Cell. 2015;161:919–932. doi: 10.1016/j.cell.2015.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhoff B., Chapple J.P., van der Spuy J., Höhfeld J., Cheetham M.E. HSJ1 is a neuronal shuttling factor for the sorting of chaperone clients to the proteasome. Curr. Biol. 2005;15:1058–1064. doi: 10.1016/j.cub.2005.04.058. [DOI] [PubMed] [Google Scholar]
- Yan J., Wang J., Li Q., Hwang J.R., Patterson C., Zhang H. AtCHIP, a U-box-containing E3 ubiquitin ligase, plays a critical role in temperature stress tolerance in Arabidopsis. Plant Physiol. 2003;132:861–869. doi: 10.1104/pp.103.020800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M., Windheim M., Roe S.M., Peggie M., Cohen P., Prodromou C., Pearl L.H. Chaperoned ubiquitylation-crystal structures of the CHIP U box E3 ubiquitin ligase and a CHIP-Ubc13-Uev1a complex. Mol. Cell. 2005;20:525–538. doi: 10.1016/j.molcel.2005.09.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.